

Abstract
Defects in the pancreatic β-cell’s secretion system are well-described in type 2 diabetes (T2D) and include impaired proinsulin processing and a deficit in mature insulin-containing secretory granules; however, the cellular mechanisms underlying these defects remain poorly understood. To address this, we used an in situ fluorescent pulse-chase strategy to study proinsulin trafficking. We show that insulin granule formation and the appearance of nascent granules at the plasma membrane are decreased in rodent and cell culture models of prediabetes and hyperglycemia. Moreover, we link the defect in insulin granule formation to an early trafficking delay in endoplasmic reticulum (ER) export of proinsulin, which is independent of overt ER stress. Using a ratiometric redox sensor, we show that the ER becomes hyperoxidized in β-cells from a dietary model of rodent prediabetes and that addition of reducing equivalents restores ER export of proinsulin and insulin granule formation and partially restores β-cell function. Together, these data identify a critical role for the regulation of ER redox homeostasis in proinsulin trafficking and suggest that alterations in ER redox poise directly contribute to the decline in insulin granule production in T2D.
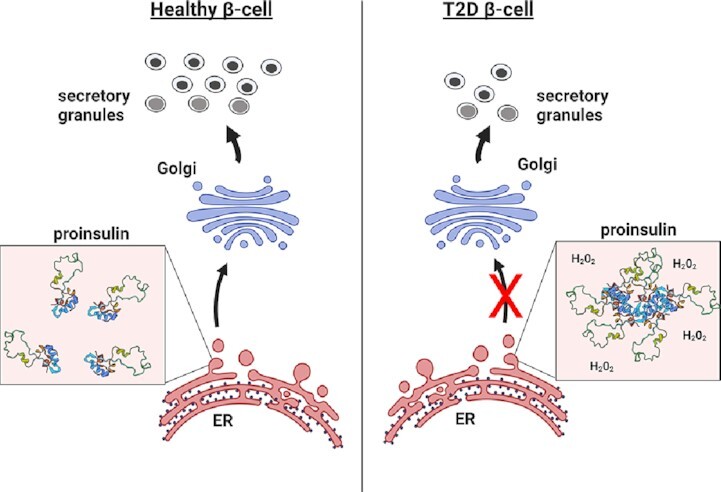
This model highlights a critical link between alterations in ER redox and ER function with defects in proinsulin trafficking in T2D. Hyperoxidation of the ER lumen, shown as hydrogen peroxide, impairs proinsulin folding and disulfide bond formation that prevents efficient exit of proinsulin from the ER to the Golgi. This trafficking defect limits available proinsulin for the formation of insulin secretory granules during the development of T2D.
Keywords: endoplasmic reticulum, protein folding, redox, proinsulin trafficking, insulin granule formation, β-cell function
Graphical Abstract
Graphical Abstract.
Introduction
Loss of islet β-cell secretory function and insulin insufficiency have been identified as critical events in the progression of insulin resistance to type 2 diabetes (T2D).1,2 Overt changes in the β-cell’s secretory pathway accompany the onset of T2D and include decreased numbers of insulin-containing secretory granules, proinsulin processing deficiencies, and hyperproinsulinemia,3–7 yet the underlying cellular mechanisms are not well understood. While increased insulin exocytosis and proinsulin/insulin degradation likely contribute to the overall deficit of the available insulin supply in T2D,8–10 perturbations in the ultrastructure of the β-cell secretory organelles (ER, Golgi, and autophagosomes) suggest that alterations in early stages of proinsulin sorting and trafficking also contribute to the insulin insufficiency in T2D.3,4,7 Studies on early stages of proinsulin trafficking and insulin granule formation have been challenging, in part, due to the prolonged half-life of proteins within the mature secretory granule, such as insulin (t ½ ∼2.7 d),11 which are also highly abundant. Nevertheless, defects in proinsulin trafficking and insulin granule formation can directly decrease insulin secretory capacity12–14 and may occur in T2D15; however direct measures of insulin granule formation in T2D have been lacking.
Insulin is initially synthesized as the prohormone precursor, proinsulin, which is folded via the assistance of endoplasmic reticulum (ER) resident chaperones, such as BiP16 and GRP94.17,18 To maintain proinsulin folded structure, protein disulfide isomerases (PDIs), including Pdia1 and Prdx4, facilitate the formation of the three critical disulfide bonds in the A and B chains of proinsulin at positions A7–B7, A20–B19, and A6–A11.19–21 Within the oxidizing environment of the ER, disulfide bonds rapidly form as cysteine pairs emerge from the protein conducting channel during translocation19; however, disulfide bonds arising from nonsequential cysteines, such as the three disulfides present in proinsulin, are likely to mis-pair and require isomerization by resident PDIs to correct.20,22 Mutations in proinsulin that perturb native disulfide bond formation have been defined as insulinopathies and result in the syndrome known as mutant INS-gene-induced diabetes of youth (MIDY).23,24 These mutations are characterized by the buildup of proteotoxic proinsulin aggregates in the ER, which ultimately result in β-cell loss from apoptosis.25 Recent work in human and rodent T2D β-cells has also identified the accumulation of oligomeric aggregates of proinsulin arising from the formation of mismatched intermolecular disulfide bonds.21,22 These observations suggest that defects in proinsulin disulfide bond isomerization are prominent in T2D; however, unlike the known mutations of MIDY, the underlying mechanisms that impair disulfide bond formation in T2D are not known. Increased interactions of proinsulin with PDIs have been noted in T2D β-cells and is consistent with defects in disulfide bond formation21; however, significant changes in PDI expression have not been observed, suggesting that PDI availability is not a limiting factor driving proinsulin folding defects in T2D. Furthermore, while defects in proinsulin folding in the ER could limit the supply of insulin necessary to maintain the insulin granule pool, studies directly examining proinsulin trafficking and insulin granule formation in T2D models are lacking.
In the present study, we evaluated proinsulin trafficking and insulin granule formation in rodent and cell culture models of prediabetes and hyperglycemia. Using a fluorescent pulse-chase proinsulin reporter, we found a significant decrease in the production of insulin granules and delayed trafficking to the plasma membrane, which coincided with impaired β-cell function. Further examination using our trafficking model revealed a significant delay in proinsulin export from the ER in both animal and cell culture models of prediabetes and hyperglycemia, which was accompanied by a significant increase in the ER oxidative potential, but without overt ER stress. Addition of reducing equivalents to attenuate ER oxidative poise re-established successful proinsulin export from the ER and generation of insulin granules. Hydrogen peroxide scavenging also rebalanced ER redox and partially restored β-cell function. Together, our data suggest that the decline in insulin granule formation during the development of T2D stems from a failure of the β-cell to maintain reducing equivalents necessary to support the critical ER function of proinsulin folding and export.
Materials and Methods
Cell Culture, Islet Isolation, and Reagents
INS1 832/3 cells (a gift from Dr. Christopher Newgard) were cultured as previously described.26 INS1 832/3 cells stably expressing proCpepSNAP have been described previously.13 For introduction of adenoviral reporters, cells were transduced with ∼1–5 × 107 IFU/mL adenovirus for 18 h and assayed 72–96 h post-treatment. Cell culture reagents were from Thermo Life Technologies unless specified otherwise. Chemical reagents were from Sigma-Aldrich unless specified otherwise. BSA-conjugated fatty acid solution was prepared as follows: Oleate/palmitate (2:1; 10 m m) was dissolved in water at 95°C, cooled to 50°C, BSA (Sigma, fatty acid free) added to a final concentration of 10%, and maintained at 37°C for an additional 1 h. Mouse islets were isolated via collagenase V digestion and purified using Histopaque 1 077 and 1 119. Islets were cultured in RPMI supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin and maintained at 37°C in 5% CO2. Pools of islets were transduced with ∼1–5 × 108 IFU/mL adenovirus for 18 h and assayed 72–96 h post-treatment.
Animal Studies
Male C57BL6/J mice (8–10 wks old; Jackson Laboratories) were placed on a Western diet (Research Diets, D12079B) or standard chow for up to 29 wks. Body weight and ad lib fed blood glucose were measured weekly. BKS.Cg-Dock7m (C57BLKS/J) Leprdb/+ and Leprdb/db mice (db/+, db/db) were either generated by heterozygous cross and genotypes confirmed via real time PCR according to Jackson Laboratories or directly purchased from Jackson Laboratories. Hyperglycemic db/db mice (ad lib fed blood glucose >220 mg/dl) were compared to age-matched (10–14 wks old), normoglycemic littermate controls (db/+). Glucose tolerance was measured in 4–6 h fasted mice given a 1 mg/g body weight glucose (i.p.) challenge. Blood glucose was determined using a One Touch Ultra 2 glucometer. Plasma insulin was determined by ELISA (ALPCO). All animal protocols were approved by the University of Iowa Institutional Animal Use and Care Committee.
Plasmids and Viruses
The shuttle plasmid, pENTR-RIP, containing a rat insulin promoter (RIP), multiple cloning site, chloramphenicol resistance marker, ccdB, and a bovine growth hormone (bGH) polyA signal was generated by Gibson assembly (IDT) and subcloned into pENTR2b (Thermo Life) replacing the entire cassette between the attP1 and attP2 sites. PCR fragments of CgB (DNASU) and CLIP (gift from Eric Campeau; RRID:Addgene_29 650) were subcloned into pENTR-RIP via Gibson assembly. ERroGFP containing a human ERp57 signal sequence followed by a superfolder roGFPiE and a C-terminal KDEL tag was a gift from Neil Bulleid, University of Glasgow27 and subcloned into pENTR-RIP via PCR and Gibson assembly. RIP-CgB-CLIP, RIP-ERroGFP were assembled into a modified pAd-PL/DEST via Gateway cloning using LR Clonase II. Recombinant adenoviruses were generated in HEK293 cells and purified by cesium chloride gradient. All sequences were verified by the Iowa Institute of Human Genetics, University of Iowa.
Glucose-Stimulated Insulin Secretion
Insulin secretion in INS1 832/3 cells was measured by static incubation as previously described.12 Cells were lysed in RIPA buffer and total protein determined by BCA (Pierce). Insulin secretion using isolated islets was performed by perifusion using Biorep Perifusion System as previously described.28 Insulin (secreted and content) was measured by ELISA (rodent 80-INSMR-CH10; ALPCO) or Alphalisa (AL204C; Perkin-Elmer Insulin).
Immunoblot Analysis
Clarified cell lysates were resolved on 4–12% NuPAGE gels (Thermo Life Technologies) and transferred to supported nitrocellulose membranes (BioRad). Membranes were probed with diluted antibodies raised against chromogranin B (goat, Santa Cruz C-19 1:1000), CHOP (mouse, 1:1000 Cell Signaling 2895), cleaved Caspase 3 (rabbit, R&D systems MAB835, 1:500), CPE (rabbit, Proteintech 13710–1-AP, 1:1000), PC2 (rabbit, Proteintech 10552–1-AP, 1:1000), and gamma-tubulin (mouse, Sigma T5326, 1:10 000). Donkey anti-mouse, anti-rabbit, or anti-goat antibodies (Licor) coupled to IR-dye 680 or 800 were used to detect primary antibodies. Blots were developed using an Odyssey CLx Licor Instrument. Original uncropped blots are provided as a supplemental data file.
Fluorescence Microscopy and Imaging
Isolated islets expressing proCpepSNAP (AdRIP) were dispersed into monolayers using Accutase (Sigma-Aldrich) and plated onto HTB9 coated coverslips or 6 cm glass bottom dishes (Mattek) as previously described.13,29 INS1 832/3 cells stably expressing proCpepSNAP were plated on HTB9 coated coverslips at low density and cultured overnight as previously described.13 For SNAPtag labeling, cells were initially incubated with SNAPcell block (10 µm; NEB) diluted in culture media for 20 min, washed 3 times for 5 min each. To allow for de novo protein synthesis of new proCpepSNAP, cells were cultured for an additional 2 h in experiments examining post-Golgi granule formation or 45 min in experiments examining ER-Golgi transport. Cells were pulse-labeled with SNAPcell-505 (1 µm; NEB) for 20 min in media, washed 3 times for 5 min each in culture media with glucose (5 m m) and chased as indicated. For studies utilizing CgB-CLIP, an identical labeling scheme was followed using CLIPcell block (10 µm; NEB) and CLIPcell-TMR (1 µm; NEB) where appropriate. Following treatments, cells were fixed in 10% neutral-buffered formalin. For immunostaining, cells were incubated overnight with antibodies raised against GM130 (mouse, BD Transduction 610 823, 1:200), GRP94 (rabbit, kind gift of Dr. Christopher Nicchita, Duke University, 1:500), or TGN38 (mouse, Novus Biologicals NB300-575, 1:200) as indicated. Highly cross-adsorbed fluorescent conjugated secondary antibodies (whole IgG, donkey anti-rabbit rhodamine red-X, donkey anti-mouse AlexaFluor 647, Jackson ImmunoResearch) were used for detection. Cells were counterstained with DAPI and mounted using Fluorosave (Calbiochem).
For granule counting and distance measures, images were captured on a Leica SP8 confocal microscope using a HC PL APO CS2 63x/1.40 oil objective with 3x zoom as z-stacks (5 per set, 0.3 μm step, 0.88 μm optical section) and deconvolved (Huygen’s Professional). Total granule number was calculated using Imaris (Bitplane) from spot-rendered granules (SNAP-505 labeled) defined as 150–300 nm objects and normalized to the total number of proCpepSNAP-labeled cells. Total fluorescence intensity was measured using Fiji/ImageJ as the raw integrated density following thresholding to remove background and normalized to the total number of proCpepSNAP-labeled cells as a measure of SNAP labeling. Granule distance measurements from the Golgi were determined using a distance transformation module in Imaris (Bitplane) from spot-rendered granules (SNAP-505 labeled) and surface rendering of the Golgi identified by GM130 or TGN38 immunostaining. Granule distances were binned as indicated and expressed as a percentage of the total to normalize between cells. For islet studies, we used a cutoff of less than 0.5 μm to identify granules proximal to the Golgi as attached, and granules >2 μm as successful Golgi export based on time course studies of granule distance clustering.13 Golgi/ER fluorescence of SNAP labeling was determined from thresholded masks defining Golgi and ER area from immunostaining using macros written for Fiji NIH software.
For plasma membrane detection, SNAP-labeled immunostained (fixed) cells were maintained in PBS and imaged using a Leica TIRF AM microscope (100× oil objective) in TIRF mode with a penetration depth of 110–150 nm. In some experiments, SNAP25 immunostaining was used to define the plasma membrane. Granule numbers from TIRF images, cell area and or cell number (defined by nuclei) were determined using Fiji/ImageJ.
For ER redox measurements, INS1 832/3 cells or primary islets were treated with AdRIP-ERroGFP and imaged on an inverted Olympus IX83 microscope with a 20× objective (HC PL APO CS2; 0.75 NA) using Chroma 49 002 GFP/CY2 Bandpass (470/535) and custom Chroma BX3-mounted (395/510) filters. Cells were subsequently cultured and reimaged following dithiothreitol (DTT, 10 m m) and diamide (5 m m) treatment, for 12 min each.28 Fluorescent intensities of each channel were calculated from masked images (to remove background) of whole cells using macros written for Fiji NIH software. Ratiometric intensities (395/470 nm) were normalized by comparing to DTT (0%) and diamide (100%) treated samples using GraphPad Prism software.
Ultrastructure
All EM related reagents were from Electron Microscopy Sciences (EMS; Hatfield, PA, USA). Isolated islets were PBS washed and fixed in 2.5% glutaraldehyde, 4% formaldehyde cacodylate buffer overnight (16–18 h) at 4°C. Tissue was post-fixed in 1% OsO4 for 1 h, dehydrated using a graded alcohol series followed by propylene oxide and embedded in Epon resin as previously described.30 Resin blocks were cut to ultrathin (50–70 nm) sections with a diamond knife and mounted on Formvar-coated copper grids. Grids were double contrasted with 2% uranyl acetate then with lead citrate. Images were captured at 1,500x, 3,000x, 6,000x, and 8,000x magnifications by a JEOL JEM-1400 transmission electron microscope.
Quantitative RT-PCR
RNA was harvested using a Zymo RNA minikit and cDNA synthesized in an iScript reaction (Bio Rad). Real-time PCRs were performed using the QuantStudio-7 FLEX or QuantStudio-7 PRO detection system and Design and Analysis Software (Applied Biosystems). All primer sequences are provided as a supplemental table.
Statistical Analysis
Data are presented as the mean ± SD for statistical significance determinations, data were analyzed by the two-tailed unpaired, Student’s t-test or by ANOVA with post-hoc analysis for multiple group comparisons as indicated (GraphPad Prism). A P-value <0.05 was considered significant.
Results
Impaired Proinsulin Trafficking Occurs in Models of β-Cell Dysfunction
In T2D, β-cell dysfunction is accompanied by a decline in insulin-containing secretory granules.3,4,7 Whether alterations to insulin granule formation contribute to this deficit is not known. To address this, we evaluated proinsulin trafficking and insulin granule formation in a rodent model of prediabetes. For these studies, C57BL6/J mice were placed on a Western diet (WD; 40% fat/kcal, 43% carbohydrate/kcal) or standard chow (SC) for 8–10 wks. Ad lib fed hyperglycemia (Figure 1A) and increased body weight (Supplemental Figure S1) were apparent within 4 wks of dietary intervention and persistent for the duration of diet. Reduced glucose tolerance (Figure 1B), fasting hyperinsulinemia, and impaired glucose-stimulated insulin release (Figure 1C) were observed by 8 wks of WD. To directly evaluate β-cell function at similar glucose levels, isolated islets from SC and WD fed mice were examined by perifusion. As shown in Figure 1D, islets from WD fed mice exhibited a significant impairment in insulin secretion at 16.7 m m glucose. Collectively, these data are consistent with defects in β-cell function in T2D.1,2
Figure 1.

Insulin granule formation is impaired in a rodent model of islet dysfunction. 8–10 wks old male C57BL6/J mice (n = 5–9 per group) were placed on a standard chow (SC) or Western diet (WD). Ad lib fed blood glucose (A) were recorded weekly. After 8 wks of diet, 4 h fasted mice were injected i.p. with 1 mg/g body weight glucose and blood glucose monitored for 2 h as indicated (B) and plasma sampled for insulin (C). (D) β-cell function was examined in isolated mouse islets by perifusion. (E) Model of the proCpepSNAP reporter with SNAPtag inserted within the C-peptide region of human preproinsulin. (F) Schematic of proCpepSNAP pulse-chase labeling timeline. Islets from SC vs. WD C57BL6/J mice (8 wks, G–J; 14 wks, K–L) were treated with AdRIP-proCpepSNAP. 48 h post-infection, islets were pulse-labeled with SNAP-505 (green), chased for 2 h, immunostained for GM130 (magenta), and counterstained with DAPI (blue). Confocal images were collected (G), and the total number of proCpepSNAP-labeled nascent granules quantified (H) (n = 4 mice per group; 22–25 cells/mouse) and total SNAP fluorescence per cell quantified (I). (J) Frequency distribution of binned proCpepSNAP-labeled granule distances (microns) from the Golgi were quantified (n = 4 mice per group). (K–L) proCpepSNAP pulse-chase labeling of β-cells from C57BL6/J mice (14 wks, SC or WD) were imaged by TIRF microscopy (K) and the total number of plasma membrane (PM)-localized proCpepSNAP-labeled granules are quantified (L) (n = 5–7 animals per group; 26–57 cells/mouse). (G and K) Scale bar = 5 μm. (A–D, H–J, and L) Data represent the mean ± SD * P < 0.05, ** P < 0.01, *** P< 0.005, or not significant (ns) by two-way ANOVA with Sidak post-test analysis (A–D, and J) or Student’s t-test (H, I, and L).
We previously developed an in situ pulse-chase fluorescent-labeling strategy based on the modified DNA repair enzyme, SNAPtag, to track nascent proinsulin transit through the secretory system and measure insulin granule formation.13 We inserted SNAPtag within the C-peptide region of human preproinsulin (Figure 1E), termed proCpepSNAP, and demonstrated that the trafficking, processing, and secretion of proCpepSNAP/CpepSNAP are consistent with the regulation of endogenous proinsulin/insulin.13 Note that this reporter is under control of the RIP to ensure β-cell specific expression. In this system, proCpepSNAP labeling is a terminal event; once labeled, the SNAP reporter is not available for subsequent labeling. Taking advantage of this, our group and others previously used this system to examine proinsulin trafficking.13,31,32 ProCpepSNAP expressing β-cells can be initially prelabeled with a nonfluorescent SNAP-tag probe to mask the existing pool of (pro)CpepSNAP followed by a recovery time to allow de novo protein synthesis of proCpepSNAP (Supplemental Figure S2A). Subsequent fluorescent (SNAP-505) pulse labeling of the nascent proCpepSNAP pool followed by a 30 min chase in media (no label) revealed an initial accumulation of labeled proCpepSNAP coincident with the Golgi marker, GM130 (Supplemental Figure S2B) as previously shown.13 Following a longer chase (2 h) in media (no label) at substimulatory glucose to limit insulin exocytosis, numerous puncta were readily apparent in the cytosol and represent newly formed insulin granules (Supplemental Figure S2B).11,13,31,32
Using the proCpepSNAP pulse-chase system, we focused on the 2 h chase time point to determine whether defects in insulin granule formation occur in islet β-cells from SC and WD fed mice. In the following studies, we defined proCpepSNAP-labeled puncta of approximately 150–300 nm in diameter as newly formed insulin granules. In primary β-cells from healthy, SC fed animals, we observed the appearance of numerous nascent proCpepSNAP-containing granules throughout the cell body (Figure 1G and H). In contrast, β-cells from hyperglycemic/pre-diabetic WD fed animals displayed a substantial reduction in the total number of newly formed proCpepSNAP-labeled granules (Figure 1G and H). Importantly, the total fluorescence intensity of labeling was not different between diets indicating that the difference in granule number was not due to reporter expression or labeling efficiency (Figure 1I). The accumulation of proCpepSNAP label surrounding the Golgi in β-cells from WD mice, which was outside of our cutoff range for granule size (150–300 nm), likely accounts for the remaining label not detected as granules. Based on these observations. we speculated that a trafficking delay occurred in the β-cells from WD fed mice. In support of this, distance quantitation of the granule data revealed a significant increase in the frequency of nascent proCpepSNAP-labeled granules within 0.5 µm of the Golgi (GM130 marker) in β-cells from WD compared to SC fed mice and a corresponding decrease in granules >2 µm from the Golgi (Figure 1J). Note that the change in granule localization (ie distance) in β-cells from WD mice was not due to a change in cell size or Golgi area (Supplemental Figure S3A and B). Further analysis by TIRF microscopy demonstrated a substantial decrease in the arrival of nascent proCpepSNAP-labeled granules localized adjacent to the plasma membrane (≤150 nm) in the WD model (Figure 1K and L). Together, these data demonstrate an impairment in the formation and trafficking of nascent insulin granules in a rodent model of diet-induced obesity and prediabetes.
To further investigate alterations to insulin granule formation, we examined an insulinoma cell culture model of β-cell dysfunction. In these studies, INS1 832/3 cells were cultured with either BSA alone or an oleate/palmitate mixture conjugated to BSA (1 m m; OP) at either normal (7.5 m m) or elevated (20 m m) Glc. The addition of the nonsaturated fatty acid, oleate, which is similarly abundant to palmitate in circulation, allows for long-term exposure (>72 h) to fatty acid without loss of viability so that we can dissociate β-cell dysfunction from β-cell death.33–35 Following 72 h culture, we measured insulin secretion after 1 h static incubation at basal (2.5 m m) and stimulatory (12 m m) glucose (Figure 2A). We observed no effect of fatty acid treatment alone (OP) on β-cell function, whereas elevated glucose culture alone resulted in increased basal insulin secretion. In contrast, oleate/palmitate and elevated glucose together (OPG) resulted in both increased basal insulin secretion and impaired glucose-stimulated insulin secretion. Additionally, OPG culture resulted in decreased expression of proinsulin processing enzymes, PC2 and CPE (Supplemental Figure S4A–B), which has been previously reported in T2D.8 Using this model, we evaluated insulin granule trafficking via our fluorescent proCpepSNAP reporter system. Here, we demonstrate that OPG-cultured INS1 832/3 cells displayed increased retention of nascent proCpepSNAP-labeled granules proximal to the Golgi (<3 µm) and decreased numbers of granules distal to the Golgi (>5 µm; Figure 2B and C) with no alterations in cell size or Golgi area (Supplemental Figure S4C and D). Analysis by TIRF microscopy confirmed the decrease in nascent insulin (proCpepSNAP-labeled) granules localized adjacent to the plasma membrane in INS1 832/3 cells cultured at high glucose (HG; 20 m m) compared to normal glucose (NG; 11 m m) conditions (Figure 2D and E). Together, these data are consistent with a proinsulin trafficking delay in a cell culture model of β-cell dysfunction.
Figure 2.

Elevated glucose and fatty acid culture impair β-cell function and insulin trafficking. INS1 832/3 cells were cultured for 72 h in control media supplemented with BSA, oleate: palmitate (2:1, 1 m m OP), oleate: palmitate (2:1, 1 m m), and elevated glucose (20 m m; OPG), 11 m m glucose (NG), or 20 m m glucose (HG) as indicated. (A) Glucose-stimulated insulin secretion (GSIS) was measured by static incubation in media containing 2.5 m m Glc followed by 12 m m Glc for 1 h each. (B–G) INS1 832/3 cells stably expressing proCpepSNAP were pulse-labeled with SNAP-505 (green), then chased for 2 h (B–C), or 4 h (D and E), immunostained for TGN38 (magenta) and counterstained with DAPI (blue) as indicated. Representative images of 2 h chase are shown (B) and frequency distribution of binned proCpepSNAP-labeled nascent granule distance from the Golgi determined (C) (n = 6 independent experiments; 35–37 cells per condition; scale bar = 5 μm). The total number of nascent (proCpepSNAP-labeled) insulin granules localized to the plasma membrane (PM) via TIRF microscopy after a 4 h chase is shown (D) and normalized to cell area (E) (n = 3 independent experiments; 60–65 cells per condition). (A, C, and E) Data represent the mean ± SD * P < 0.05 or not significant (ns) by two-way ANOVA with Sidak post-test analysis (A and C) or Student’s t-test (E). (A) * comparison of secretion response at stimulatory glucose to control (BSA, 7.5 m m); # comparison of secretion response at basal glucose to control (BSA, 7.5 m m).
Delayed ER Export of Proinsulin in Pre-Diabetes Models of Hyperglycemia
Recent studies have identified proinsulin oligomeric aggregates in T2D β-cells arising from mis-paired intermolecular disulfide bonds.21,22 Based on this, we reasoned that proinsulin aggregation in T2D may cause an early defect in ER export of proinsulin that would explain the impaired insulin granule formation we observed (Figures 1,2). To test this, we used our pulse-chase labeling protocol to capture the early arrival of proCpepSNAP at the Golgi. As expected, examination of control (BSA-cultured) cells demonstrated successful trafficking of newly synthesized proinsulin (proCpepSNAP-labeled) to the Golgi (TGN38), which was largely absent from the ER (GRP94) (Figure 3A). In contrast, pulse-chase labeling in OPG-cultured β-cells revealed a substantial retention of nascent proCpepSNAP coincident with the ER and very little detection within the Golgi. Quantitation revealed a significant decrease in the ratio of Golgi to ER proCpepSNAP fluorescence intensity in OPG-cultured cells (Figure 3B), whereas total labeling was not different between conditions (Figure 3C). We also examined ER-Golgi proinsulin trafficking in β-cells from overtly diabetic C57BLKS/J-db/db mice. Notably, these mice displayed a profound impairment in glucose tolerance (Supplemental Figure S5A) and fasting hyperinsulinemia (Supplemental Figure S5B), which is likely driven by a combination of insulin resistance and reduced metabolic clearance of insulin36. Furthermore, these mice exhibit a poor insulin secretory response to glucose challenge, consistent with β-cell dysfunction (Supplemental Figure S5B; compare t = 0 vs. t = 15 min). Using this model, pulse-chase labeling of β-cells from normoglycemic db/+ controls revealed accumulation of newly synthesized proinsulin (proCpepSNAP) in the Golgi (GM130) (Figure 3E); however, in β-cells from db/db diabetic mice, nascent-labeled proCpepSNAP was primarily localized within the ER (Figure 3E) with a corresponding decrease in the Golgi to ER proCpepSNAP fluorescence (Figure 3D). No differences were observed in total labeling, cell area, or Golgi area between genotypes (Supplemental Figure S5C–E) suggesting that the difference in proCpepSNAP localization was due to alterations in trafficking rather than reporter expression or labeling.
Figure 3.

Delayed ER export of proinsulin in animal and cell culture models of hyperglycemia. (A–C) INS1 832/3 cells stably expressing proCpepSNAP were cultured for up to 72 h in control media supplemented with BSA or media containing oleate/palmitate (2:1, 1 m m) and elevated glucose (20 m m; OPG) as indicated. Cells were pulse-labeled with SNAP-505 (green), chased for 15 min, immunostained for GRP94 (red), TGN38 (magenta), and counterstained with DAPI (blue). Representative images (A) are shown (scale bar = 5 μm) and the ratio of proCpepSNAP fluorescence coincident with the Golgi compared to ER quantified (B) (n = 6–8 independent experiments; 48–64 cells per condition). (C) Total SNAP fluorescence per cell was quantified. (D and E) Isolated mouse islets (C57BLKS/J db/+ vs. db/db) were treated with AdRIP-proCpepSNAP. 48 h post-infection, islets were pulse-labeled with SNAP-505 (green), chased for 10 min, immunostained for GM130 (magenta), and counterstained with DAPI (blue). Representative images (E) are shown (scale bar = 3 μm) and the ratio of proCpepSNAP fluorescence coincident with the Golgi compared to ER quantified (D) (n = 4 mice per group; 6–36 cells/mouse). (B and D) Data represent the mean ± SD * P < 0.05, *** P < 0.0005, or not significant (ns) by Student’s t-test.
The accumulation of proinsulin within the ER could stem from activation of the ER stress response, which has been suggested to contribute to the development of β-cell dysfunction in T2D.37 While expansion of ER membranes have been demonstrated in both human and rodent T2D β-cells, dilation of the ER lumen, which is a key hallmark of proteotoxic stress, is not commonly observed.3,7,38,39 Consistent with these latter data, the increased retention of nascent proinsulin in the ER was not accompanied by distension of the ER lumen or other gross morphological changes in ER ultrastructure in β-cells from diabetic db/db mice (Figure 4A). In INS1 832/3 cells cultured with fatty acid alone (OP) or fatty acid plus elevated glucose (OPG), we did not detect upregulation of ER stress markers, ATF4, CHOP, XBP-1(s/u), GADD34, or BiP by qRT-PCR following overnight culture (Supplemental Figure S6A) despite impaired insulin secretion (Supplemental Figure S6B). Moreover, increased expression of ER stress markers was not evident after 72 h OP or OPG culture (Figure 4B; compare to 18 h thapsigargin treatment), nor did we detect significant expression of CHOP and cleaved caspase 3 by immunoblot (Supplemental Figure S6C). Last, pulse-chase analysis using our fluorescent proCpepSNAP reporter revealed that the chemical chaperone, TUDCA, could not rescue the proinsulin ER export delay observed in OPG-cultured INS1 832/3 cells (Supplemental Figure S6D), which is consistent with the lack of ER stress in our OPG model.
Figure 4.

Delayed ER export of proinsulin is specific and independent of overt ER stress. (A) Islets isolated from 10–14 wks old C57BLKS/J db/+ vs. db/db (n = 3) were examined for ultrastructure by electron microscopy and representative micrographs depicting the ER are shown. (B and E–G) INS1 832/3 cells were cultured for 72 h in control media supplemented with BSA, media containing oleate: palmitate (2:1 and 1 m m OP) or media containing oleate: palmitate (2:1 and 1 m m) and elevated glucose (20 m m; OPG) as indicated. (B) mRNA expression was examined by qRT-PCR (n = 4–6). Cells were treated for 18 h with thapsigargin (50 n m) as indicated. (C) INS1 832/3 cells treated with AdRIP-CgB-CLIP were analyzed by immunoblot with antibodies raised against CLIP or endogenous CgB as indicated. (D) INS1 832/3 cells co-expressing proCpepSNAP and CgB-CLIP were pulse-labeled with SNAP-505 (green) and CLIP-TMR (red) following a 2 h synthesis period, and chased for 2 h prior to fixation. Cells were immunostained for TGN38 (magenta), and counterstained with DAPI (blue). Representative images are shown (scale bar = 5 μm). (E–G) INS1 832/3 cells expressing proCpepSNAP and CgB-CLIP were pulse-labeled with SNAP-505 (green) and CLIP-TMR (red), chased for 15 min, fixed and immunostained for TGN38 (magenta), and counterstained with DAPI (blue). (E) Representative images are shown (scale bar = 5 μm). Mander’s correlation coefficient (MCC) was used to determine colocalization of labeled proCpepSNAP (SNAP) vs CgB-CLIP (CLIP) with TGN38 (F) or proCpepSNAP (SNAP) with CgB-CLIP (CLIP) as indicated (G) (n = 3 independent experiments; 53–70 cells per condition). (B, F, and G) Data represent the mean ± SD * P < 0.05, ** P < 0.005, *** P< 0.0001 by two-way ANOVA with Sidak post-test analysis (B and F) or Student’s t-test (G).
To examine if the ER export delay was specific to proinsulin or instead was a general defect in ER protein export, we examined ER–Golgi trafficking of another granule protein, CgB, using an analogous pulse-chase system, CLIPtag. Notably, CLIPtag uses a distinct substrate from SNAPtag allowing us to perform dual pulse-chase studies of both proteins in the same cell.40 We generated a C-terminal CLIP-tagged CgB reporter, which was expressed at levels similar to endogenous CgB (Figure 4C) and co-trafficked with proCpepSNAP into punctate structures following a 2 h pulse-chase (Figure 4D), likely representing maturing insulin granules. Using this dual pulse-chase system to examine ER-Golgi trafficking, we demonstrated that in BSA-cultured (control) INS1 832/3 cells, nascent proinsulin (proCpepSNAP) and CgB (CgB-CLIP) similarly trafficked to the Golgi following pulse-chase labeling (Figure 4E and F). In OPG-cultured INS1 832/3 cells, CgB-CLIP trafficking from the ER to Golgi occurred without delay and mirrored control, BSA alone cultured cells (Figure 4E) with similar colocalization to the Golgi marker, TGN38 (Figure 4F). This was in stark contrast to proinsulin, which was retained in the ER in OPG-cultured INS1 832/3 cells (Figure 4E), displaying reduced colocalization with both CgB-CLIP (Figure 4G) and the Golgi marker TGN38 (Figure 4F). No differences in labeling efficiency of either proCpepSNAP or CgB-CLIP were detected between culture conditions (Supplemental Figure S6E and F). Collectively, these data demonstrate a specific delay in ER export of proinsulin accompanying chronic exposure to hyperglycemic and hyperlipidemic-like conditions, which is independent of overt ER stress or a general impairment in ER protein export.
Regulation of ER Redox Homeostasis
Potentially, altered redox potential of the ER lumen in T2D may impair proinsulin folding and thereby delay proinsulin ER export. To examine this, we first used an ER-targeted redox sensitive GFP, ERroGFP,27 driven by the RIP using a recombinant adenovirus expressed in islet β-cells isolated from mice on SC or WD. Ratiometric imaging revealed clear differences in the redox status of the ER in β-cells between dietary conditions (Figure 5A) with a significant shift to a more oxidized state in islet β-cells from the WD (Figure 5B). Based on these data, we reasoned that addition of chemical reducing equivalents may re-balance the ER’s redox poise and restore proinsulin trafficking in the WD model. To test this, islets from SC and WD mice were pretreated with DTT (0.5 m m) for 4 h prior to SNAP-tag pulse-chase labeling and the formation of nascent (proCpepSNAP-labeled) insulin granules examined as the primary endpoint for successful proinsulin trafficking. Consistent with our previous data (Figure 1G and H), we observed a substantial decrease in the formation of proCpepSNAP-labeled nascent insulin granules in islet β-cells from WD compared to SC mice (Figure 5C and D). In contrast, pretreatment with reducing agent (DTT) resulted in a substantial increase in the number of newly formed proCpepSNAP-labeled insulin granules in islet β-cells from WD mice but did not affect the appearance of proCpepSNAP-labeled granules in SC β-cells. No difference in total fluorescent labeling was observed between dietary groups or DTT treatment indicating that reporter expression and labeling efficiency were similar across conditions (Figure 5E).
Figure 5.

Insulin granule deficit can be rescued by chemical reducing agent. 8–10 wks old male C57BL6/J mice were placed on a standard chow (SC) or Western diet (WD) for 29 wks. (A) Isolated islets treated with AdRIP-ERroGFP were imaged at 470/535 nm and 395/510 nm to measure ER oxidation. (B) Normalized ratiometric intensities were quantified to determine ER oxidation (n = 4 mice/group). (C-E) Mouse islets were treated with AdRIP-proCpepSNAP. Prior to SNAP labeling, islets were treated with vehicle (control) or DTT (0.5 m m) for 4 h. Islets were pulse-labeled with SNAP-505 (green), chased for 2 h prior to fixation, and counterstained with DAPI (blue). Confocal images were collected (C), and the total number of proCpepSNAP-labeled nascent granules quantified (D) (n = 6–10 mice/group; 20–100 cells/mouse). Total SNAP fluorescence was quantified (E). (B, D, and E) Data represent the mean ± SD * P < 0.05 or not significant (ns) by Student’s t-test (B) or two-way ANOVA with Sidak post-test analysis (D and E).
To more closely examine if addition of reducing equivalents restored ER-Golgi trafficking of proinsulin, we used our OPG cell culture model of β-cell dysfunction. In control (BSA cultured) cells, 4 h pretreatment with DTT had no discernible impact on ER to Golgi trafficking of newly synthesized (labeled) proCpepSNAP, as compared to the vehicle control with similar levels of Golgi accumulation present (Figure 6A and B). In contrast, OPG-cultured cells retained proCpepSNAP-labeled proinsulin in the ER, whereas DTT pretreatment resulted in accumulation of labeled proCpepSNAP in the Golgi, similar to BSA control cells. These data suggest that ER-Golgi transport was restored by addition of reducing equivalents. Note that no difference in total fluorescent labeling was observed between conditions (Figure 6C).
Figure 6.

Proinsulin ER export delay can be rescued by chemical reducing agent. INS1 832/3 cells stably expressing proCpepSNAP were cultured in control media supplemented with BSA or media containing oleate: palmitate (2:1 and 1 m m) and elevated glucose (20 m m; OPG) as indicated. (A-C) Prior to SNAP labeling, cells were treated with vehicle (control) or DTT (0.5 m m) for 4 h. Cells were then pulse-labeled with SNAP-505 (green), fixed, and immunostained for GRP94 (red), TGN38 (magenta), and counterstained with DAPI (blue). Representative images (A) are shown (scale bar = 5 μm) and Mander’s correlation coefficient (B) was used to determine the colocalization of labeled proCpepSNAP (SNAP) with TGN38 (n = 3–4 independent experiments; 71–91 cells/condition). Total SNAP fluorescence was quantified (C). (D) Cells expressing ERroGFP (AdRIP) were cultured overnight as indicated with vehicle (control) or ebselen (Eb, 10 μm) and imaged at 470/535 nm and 395/510 nm. Normalized ratiometric intensities were quantified to determine ER oxidation (n = 4 independent experiments). (E) Cells were cultured overnight as indicated with vehicle (-) or ebselen (Eb, 10 μM). GSIS was measured by static incubation in media containing 2.5 m m Glc followed by 12 m m Glc for 1 h each. (B–E) Data represent the mean ± SD * P < 0.05 by two-way ANOVA with Sidak post-test analysis (B, D, and E), or ns by two-way ANOVA (C).
Next, we investigated the expression of ER oxidoreductases involved in disulfide bond formation over the course of 72 h (Supplemental Figure S7). While no change was observed for several of the most abundantly expressed PDIs in response to OPG culture, including Pdia3, 4, and 6 and peroxiredoxin 4,41 Pdia1 and Ero1α were increased following 48 and 72 h culture in OPG (Supplementary Figure S7A–F). Because hydrogen peroxide is the final byproduct of protein disulfide bond formation via the electron relay between PDIs and Ero1α,42,43 the increased ER oxidation may stem from the inability of Prdx4 to effectively neutralize elevated ER-derived hydrogen peroxide production.44–46 To test this, INS1 832/3 cells cultured in either BSA (control) or OPG-containing media were treated overnight with the hydrogen peroxide scavenger, ebselen (Eb). Using the redox sensitive ERroGFP, we observed an increase in ER oxidation of OPG-cultured cells compared to BSA-cultured control cells (Figure 6D), similar to the observation in rodents fed a Western diet (Figure 5 A and B). In response to Eb, ER oxidation was restored in INS1 832/3 cells cultured in OPG to the level of BSA control cells, whereas Eb had no additional effect on BSA control cells. Furthermore, Eb partially restored GSIS (50% increase) in OPG cultured cells, with a trend to increased GSIS in BSA control cells (Figure 6E), but did not alter expression of PDIs (Figure Supplemental Figure S7G). Collectively, our data highlight a critical link between alterations in ER redox and ER function with defects in proinsulin trafficking in T2D.
Discussion
Defects in the β-cell’s secretory pathway, including changes to insulin trafficking, reduced insulin storage, impaired proinsulin processing, and hyperproinsulinemia, have been known for decades to occur in T2D.1–7 Increased exocytosis of immature granules8 as well as enhanced degradation of newly synthesized insulin granules in T2D9,10 contribute to the overall decrease in insulin storage and hyperproinsulinemia; however, the underlying mechanisms for these defects are poorly understood and the links to overnutrition and hyperglycemia remain vague. Our current study focuses on an emerging concept that oxidative protein folding in the ER is perturbed in the pathogenesis of β-cell dysfunction.20–22 We show that an increase in the oxidative state of the ER in T2D models corresponds to a delay in ER export of proinsulin and decreased insulin granule formation. Furthermore, we demonstrate that proinsulin trafficking and insulin granule formation can be restored by the addition of chemical reducing equivalents and that antioxidant treatment can improve ER redox homeostasis and β-cell function. Collectively, our data highlight a direct link between the regulation of ER redox poise and insulin granule biosynthesis in the demise of β-cell function in T2D.
Following the emergence of nascent proteins from the ER protein-conducting channel, Sec61, disulfide bonds readily form in the oxidizing environment of the ER lumen. In the case of disulfides arising from nonsequential cysteine residues, such as the three disulfide bonds in proinsulin, rearrangement of non-native, or mis-paired disulfides, requires reduction via PDIs and subsequent re-oxidation of cysteine thiols to form the correct disulfide linkages.19,43 Based on our data, we propose that the ER lumen becomes hyperoxidizing during the pathogenesis of β-cell dysfunction, which impedes the necessary re-shuttling of proinsulin’s disulfide linkages and thereby delays ER export of proinsulin. Potentially, increased expression of Pdia1 and Ero1α lead to increased levels of hydrogen peroxide produced during successive rounds of disulfide bond isomerization.42,43 This may overcome the scavenging capabilities of Prdx4 and contribute to hyperoxidation of the ER lumen.44–46 In support of this mechanism, the delay in ER export can be reversed by the addition of chemical reducing equivalents, suggesting that proinsulin disulfide bond isomerization is a kinetic determinant in proinsulin export from the ER and fails to efficiently form if the ER environment is too oxidizing. Similarly, antioxidant treatment has been shown to decrease ER stress and attenuate β-cell dysfunction and diabetes onset in genetic mouse models of ER dysfunction.47–49 Interestingly, we observed no defect in ER to Golgi transit of CgB, which contains a single pair of sequential cysteines forming its lone disulfide linkage. Thus, CgB may be less sensitive to changes in ER redox poise for correct disulfide bond formation and ER export. Together, our data provide evidence linking hyperoxidation of the ER lumen to the decrease in insulin granule formation in T2D models. The observation that Eb treatment restores ER redox balance yet does not prevent Pdia1 and Ero1α upregulation, suggests that the increased Pdia1 and Ero1α expression is independent of ER redox status and instead may be a response to increased client protein demand for disulfide bond formation. Despite the improvement in ER redox with Eb, the partial restoration of GSIS indicates that additional pathways, including metabolic and exocytic defects, further contribute to the demise of β-cell dysfunction in T2D.1,50
Distinct from other cell types, the β-cell is uniquely positioned to sense physiological fluctuations in glucose via the generation of metabolic signals38. Metabolic signaling is well-described to control both proinsulin biosynthesis and insulin release38; however, a role for metabolic activity in the regulation of ER redox poise in the β-cell is not known, but could be an elegant mechanism to facilitate efficient folding of proinsulin during nutrient stimulation and support insulin granule biogenesis. In support of this mechanism, recent data in hepatocytes demonstrates that ER oxidative protein folding can be regulated via nutrient signals through the redox cycling of glutathione and/or thioredoxin51, which are necessary to reset PDIs for sequential rounds of client protein disulfide bond isomerization.43,52 Through this pathway, metabolic supply of redox signals may be a critical pathway to promote proinsulin disulfide bond isomerization in the β-cell by maintaining redox balance of the ER lumen. During the pathogenesis of T2D, changes in metabolic activity include diminished recycling of glutathione/thioredoxin reducing equivalents53,54 and increased production of reactive oxygen species,55 which contribute to overall cellular oxidative stress. As further insult to the β-cell, alteration of these metabolic signals (glutathione, thioredoxin, ROS) may also increase ER oxidative potential and lead to defects in proinsulin disulfide bond isomerization. Thus failing mitochondrial function may not only limit ATP production for exocytosis, but also directly contribute to ER hyperoxidation via an insufficient supply of reducing equivalents and increased generation of reactive oxygen species, such as hydrogen peroxide.56 In further support of this, we show that Eb treatment can modestly improve GSIS in a cell culture model of β-cell dysfunction; however additional studies are needed to confirm these findings in human models. Future studies may investigate the links between mitochondrial dysfunction and ER redox status as a possible mechanism contributing to secretory defects in T2D.
Numerous studies have demonstrated that induction of ER stress can perturb β-cell function and may contribute to the development of β-cell dysfunction in T2D.37 Loss of critical ER stress response sensors, such as PERK and Ire1α, can sensitize β-cells to nutrient stresses and/or directly impair β-cell function leading to the development of diabetes.48,57–59 Similarly, loss of the ER chaperone, GRP94, the BiP co-chaperone, p58IPK, and PDIs, Pdia1 and Prdx4, compromise proinsulin folding, resulting in ER stress and loss of insulin content.17,18,20 In humans, monogenic forms of diabetes arising from INS mutations can prevent proper proinsulin disulfide bond formation acting as dominant negative suppressors of wild type proinsulin trafficking.24,60,61 While these studies demonstrate that perturbations in ER functions and proinsulin folding can compromise β-cell health and insulin production, direct evidence for ER stress as a causative feature in the development of T2D is less clear.7,39 Increased PERK and Ire1α activity have been described in pre-diabetic β-cells,37,62,63 yet diminished, rather than increased, expression of ATF6 and XBP-1(s) occur in β-cells from humans with long-standing T2D62. Moreover, human β-cells are known to cycle between varying levels of UPR protein expression,64 which may represent the natural β-cell adaptation to changes in nutrient load. Indeed, activation of the ER stress pathway is necessary for the β-cell proliferative response to diet-induced obesity.65 Expansion of ER membranes has been demonstrated in both human and rodent T2D β-cells, which may be necessary to support increased proinsulin biosynthesis3,7,38,39; however distension and/or dilation of the ER lumen, a classic marker of proteotoxic ER stress, is not commonly observed. Consistent with these data, we failed to detect pathological changes in ER morphology from diabetic db/db mice and saw little evidence for overt ER stress response activation in our cell culture model, despite clear effects on ER retention of proinsulin in both models. These unresolved issues encourage continued investigation in defining how physiological stresses manifest in the β-cell and determining when the adaptive responses become insufficient to mitigate cellular damage and/or become maladaptive.
Abbreviations
dithiothreitol (DTT); ebselen (Eb); glucose (Glc); glucose-stimulated insulin secretion (GSIS); infectious units (IFU); ) oleate/palmitate (OP); oleate/palmitate + Glc (OPG); protein disulfide isomerase (PDI); rat insulin promoter (RIP); Type 2 diabetes (T2D); and untranslated region (UTR)
Funding
This work was supported by startup funds provided by the Fraternal Order of Eagles Diabetes Research Center, University of Iowa (to S.B.S.), Department of Defense, Congressionally-Directed Medical Research Program grant W81XWH-20–1–0200 (to S.B.S.), National Institutes of Health grant R35GM130331 (to Y.W.), and a National Institutes of Health Predoctoral Training Grants T32GM008629 and T32GM145441.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr. Thomas Rutkowski, Dr. Erica Gansemer, and Dr. Ling Yang for helpful comments and discussion and McKenzie Becker for expert technical assistance. We would like to acknowledge the use of the University of Iowa Central Microscopy Research Facility. Samuel Stephens is the guarantor of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Contributor Information
Kristen E Rohli, Fraternal Order of Eagles Diabetes Research Center, University of Iowa, Iowa City, IA 52242, USA; Interdisciplinary Graduate Program in Genetics, University of Iowa, Iowa City, IA 52242, USA; Department of Internal Medicine, Division of Endocrinology and Metabolism, University of Iowa, Iowa City, IA 52242, USA.
Cierra K Boyer, Fraternal Order of Eagles Diabetes Research Center, University of Iowa, Iowa City, IA 52242, USA; Department of Pharmacology, University of Iowa, Iowa City, IA 52242, USA.
Shelby C Bearrows, Fraternal Order of Eagles Diabetes Research Center, University of Iowa, Iowa City, IA 52242, USA; Department of Internal Medicine, Division of Endocrinology and Metabolism, University of Iowa, Iowa City, IA 52242, USA.
Marshall R Moyer, Fraternal Order of Eagles Diabetes Research Center, University of Iowa, Iowa City, IA 52242, USA; Department of Internal Medicine, Division of Endocrinology and Metabolism, University of Iowa, Iowa City, IA 52242, USA.
Weston S Elison, Department of Nutrition, Dietetics, and Food Science, Brigham Young University, Provo, UT 84602, USA.
Casey J Bauchle, Fraternal Order of Eagles Diabetes Research Center, University of Iowa, Iowa City, IA 52242, USA; Department of Internal Medicine, Division of Endocrinology and Metabolism, University of Iowa, Iowa City, IA 52242, USA.
Sandra E Blom, Fraternal Order of Eagles Diabetes Research Center, University of Iowa, Iowa City, IA 52242, USA; Department of Internal Medicine, Division of Endocrinology and Metabolism, University of Iowa, Iowa City, IA 52242, USA.
Jianchao Zhang, Department of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI 48103, USA.
Yanzhuang Wang, Department of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI 48103, USA; Department of Neurology, School of Medicine, University of Michigan, Ann Arbor, MI 48103, USA.
Samuel B Stephens, Fraternal Order of Eagles Diabetes Research Center, University of Iowa, Iowa City, IA 52242, USA; Interdisciplinary Graduate Program in Genetics, University of Iowa, Iowa City, IA 52242, USA; Department of Internal Medicine, Division of Endocrinology and Metabolism, University of Iowa, Iowa City, IA 52242, USA.
Author Contributions
K.E.R., C.K.B., S.C.B. M.R.M., W.S.E., and S.B.S conceived and designed studies. K.E.R., C.K.B., S.C.B. M.R.M., W.S.E., C.J.B., S.E.B., and S.B.S performed the experiments and analyzed the data. J.Z. and Y.W. performed TEM. K.E.R. and S.B.S. wrote the manuscript.
Conflict of Interest
No competing interests declared.
Data Availability
All primer sequences are provided as a supplemental table. Uncropped versions of all immunoblots are provided as a supplemental file. All data are available upon reasonable request.
References
- 1. Halban PA, Polonsky KS, Bowden DWet al. beta-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabet Care. 2014;37(6):1751–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kahn SE, Zraika S, Utzschneider KM, Hull RL.. The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia. 2009;52(6):1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Masini M, Marselli L, Bugliani Met al. Ultrastructural morphometric analysis of insulin secretory granules in human type 2 diabetes. Acta Diabetol. 2012;49:S1:S247–252. [DOI] [PubMed] [Google Scholar]
- 4. Like AA, Chick WL.. Studies in the diabetic mutant mouse. II. Electron microscopy of pancreatic islets. Diabetologia. 1970;6(3):216–242. [DOI] [PubMed] [Google Scholar]
- 5. Kahn SE, Halban PA.. Release of incompletely processed proinsulin is the cause of the disproportionate proinsulinemia of NIDDM. Diabetes. 1997;46(11):1725–1732. [DOI] [PubMed] [Google Scholar]
- 6. Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D.. Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest. 1984;74(4):1318–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alarcon C, Boland BB, Uchizono Yet al. Pancreatic β-cell adaptive plasticity in obesity increases insulin production but adversely affects secretory function. Diabetes. 2016;65(2):438–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rhodes CJ, Alarcon C.. What beta-cell defect could lead to hyperproinsulinemia in NIDDM? Some clues from recent advances made in understanding the proinsulin-processing mechanism. Diabetes. 1994;43(4):511–517. [DOI] [PubMed] [Google Scholar]
- 9. Pasquier A, Vivot K, Erbs Eet al. Lysosomal degradation of newly formed insulin granules contributes to β cell failure in diabetes. Nat Commun. 2019;10(1):3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shrestha N, Liu T, Ji Yet al. Sel1L-Hrd1 ER-associated degradation maintains β cell identity via TGF-β signaling. J Clin Invest. 2020;130(7):3499–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Muller A, Neukam M, Ivanova Aet al. A global approach for quantitative super resolution and electron microscopy on cryo and epoxy sections using self-labeling protein tags. Sci Rep. 2017;7(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stephens SB, Edwards RJ, Sadahiro Met al. The prohormone vgf regulates beta cell function via insulin secretory granule biogenesis. Cell Rep. 2017;20(10):2480–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bearrows SC, Bauchle CJ, Becker M, Haldeman JM, Swaminathan S, Stephens SB.. Chromogranin B regulates early stage insulin granule trafficking from the Golgi in pancreatic islet beta-cells. J Cell Sci. 2019;132(13):jcs231373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cao M, Mao Z, Kam Cet al. PICK1 and ICA69 control insulin granule trafficking and their deficiencies lead to impaired glucose tolerance. PLoS Biol. 2013;11(4):e1001541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gandasi NR, Yin P, Omar-Hmeadi M, Ottosson Laakso E, Vikman P, Barg S. Glucose-dependent granule docking limits insulin secretion and is decreased in human type 2 diabetes. Cell Metab. 2018;27(2):470–478. e4. [DOI] [PubMed] [Google Scholar]
- 16. Arunagiri A, Haataja L, Cunningham CNet al. Misfolded proinsulin in the endoplasmic reticulum during development of beta cell failure in diabetes. Ann NY Acad Sci. 2018;1418(1):5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim DS, Song L, Wang Jet al. GRP94 is an essential regulator of pancreatic β-cell development, mass, and function in male mice. Endocrinology. 2018;159(2):1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ghiasi SM, Dahlby T, Hede Andersen Cet al. Endoplasmic reticulum chaperone glucose-regulated protein 94 is essential for proinsulin handling. Diabetes. 2019;68(4):747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Winter J, Klappa P, Freedman RB, Lilie H, Rudolph R.. Catalytic activity and chaperone function of human protein-disulfide isomerase are required for the efficient refolding of proinsulin. J Biol Chem. 2002;277(1):310–317. [DOI] [PubMed] [Google Scholar]
- 20. Jang I, Pottekat A, Poothong Jet al. PDIA1/P4HB is required for efficient proinsulin maturation and ß cell health in response to diet induced obesity. Elife. 2019;8:e44528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tran DT, Pottekat A, Mir SAet al. Unbiased profiling of the human proinsulin biosynthetic interaction network reveals a role for peroxiredoxin 4 in proinsulin folding. Diabetes. 2020;69(8):1723–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arunagiri A, Haataja L, Pottekat Aet al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. Elife. 2019;8:e44532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu M, Sun J, Cui Jet al. INS-gene mutations: from genetics and beta cell biology to clinical disease. Mol Aspects Med. 2015;42:3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Støy J, Steiner DF, Park SY, Ye H, Philipson LH, Bell GI.. Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev Endocr Metab Disord. 2010;11(3):205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun J, Cui J, He Q, Chen Z, Arvan P, Liu M.. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol Aspects Med. 2015;42:105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB.. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000;49(3):424–430. [DOI] [PubMed] [Google Scholar]
- 27. Cao X, Lilla S, Cao Zet al. The mammalian cytosolic thioredoxin reductase pathway acts via a membrane protein to reduce ER-localised proteins. J Cell Sci. 2020;133(8):jcs241976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bauchle CJ, Rohli KE, Boyer CKet al. Mitochondrial efflux of citrate and isocitrate is fully dispensable for glucose-stimulated insulin secretion and pancreatic islet β-cell function. Diabetes. 2021;70(8):1717–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayes HL, Peterson BS, Haldeman JM, Newgard CB, Hohmeier HE, Stephens SB.. Delayed apoptosis allows islet beta-cells to implement an autophagic mechanism to promote cell survival. PLoS One. 2017;12(2):e0172567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Joshi G, Chi Y, Huang Z, Wang Y.. Aβ-induced Golgi fragmentation in Alzheimer’s disease enhances Aβ production. Proc Natl Acad Sci. 2014;111(13):E1230–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ivanova A, Kalaidzidis Y, Dirkx Ret al. Age-dependent labeling and imaging of insulin secretory granules. Diabetes. 2013;62(11):3687–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoboth P, Muller A, Ivanova Aet al. Aged insulin granules display reduced microtubule-dependent mobility and are disposed within actin-positive multigranular bodies. Proc Natl Acad Sci. 2015;112(7):E667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chu KY, O’Reilly L, Mellet N, Meikle PJ, Bartley C, Biden TJ. Oleate disrupts cAMP signaling, contributing to potent stimulation of pancreatic beta-cell autophagy. J Biol Chem. 2019;294(4):1218–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abdelmagid SA, Clarke SE, Nielsen DEet al. Comprehensive profiling of plasma fatty acid concentrations in young healthy canadian adults. PLoS One. 2015 Feb 12;10(2):e0116195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boucher A, Lu D, Burgess SCet al. Biochemical mechanism of lipid-induced impairment of glucose-stimulated insulin secretion and reversal with a malate analogue. J Biol Chem. 2004;279(26):27263–27271. [DOI] [PubMed] [Google Scholar]
- 36. Watada H, Tamura Y.. Impaired insulin clearance as a cause rather than a consequence of insulin resistance. J Diabetes Investig. 2017;8(6):723–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yong J, Johnson JD, Arvan P, Han J, Kaufman RJ.. Therapeutic opportunities for pancreatic β-cell ER stress in diabetes mellitus. Nat Rev Endocrinol. 2021;17(8):455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boland BB, Rhodes CJ, Grimsby JS.. The dynamic plasticity of insulin production in β-cells. Mol Metab. 2017;6(9):958–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marchetti P, Bugliani M, Lupi Ret al. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia. 2007;50(12):2486–2494. [DOI] [PubMed] [Google Scholar]
- 40. Gautier A, Juillerat A, Heinis Cet al. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15(2):128–136. [DOI] [PubMed] [Google Scholar]
- 41. DiGruccio MR, Mawla AM, Donaldson CJet al. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol Metab. 2016;5(7):449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zito E. ERO1: a protein disulfide oxidase and H2O2 producer. Free Radical Biol Med. 2015;83:299–304. [DOI] [PubMed] [Google Scholar]
- 43. Ellgaard L, Sevier CS, Bulleid NJ.. How are proteins reduced in the endoplasmic reticulum?. Trends Biochem Sci. 2018;43(1):32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martin RE, Cao Z, Bulleid NJ.. Regulating the level of intracellular hydrogen peroxide: the role of peroxiredoxin IV. Biochem Soc Trans. 2014;42(1):42–46. [DOI] [PubMed] [Google Scholar]
- 45. Tavender TJ, Bulleid NJ.. Peroxiredoxin IV protects cells from oxidative stress by removing H2O2 produced during disulphide formation. J Cell Sci. 2010;123(15):2672–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tavender TJ, Springate JJ, Bulleid NJ.. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010;29(24):4185–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Malhotra JD, Miao H, Zhang K, et al. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci. 2008;105(47):18525–18530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hassler JR, Scheuner DL, Wang Set al. The IRE1α/XBP1s pathway is essential for the glucose response and protection of β cells. PLoS Biol. 2015;13(10):e1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Han J, Song B, Kim Jet al. Antioxidants complement the requirement for protein chaperone function to maintain β-cell function and glucose homeostasis. Diabetes. 2015;64(8):2892–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Campbell JE, Newgard CB.. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat Rev Mol Cell Biol. 2021;22(2):142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gansemer ER, McCommis KS, Martino Met al. NADPH and glutathione redox link tca cycle activity to endoplasmic reticulum homeostasis. iScience. 2020;23(5):101116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Birk J, Meyer M, Aller Iet al. Endoplasmic reticulum: reduced and oxidized glutathione revisited. J Cell Sci. 2013;126(7):1604–1617. [DOI] [PubMed] [Google Scholar]
- 53. Ferdaoussi M, Dai X, Jensen MVet al. Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. J Clin Invest. 2015;125(10):3847–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Haythorne E, Rohm M, van de Bunt M, et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat Commun. 2019;10(1):2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tanaka Y, Tran POT, Harmon J, Robertson RP.. A role for glutathione peroxidase in protecting pancreatic cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci. 2002;99(19):12363–12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rohli KE, Boyer CK, Blom SE, Stephens SB.. Nutrient regulation of pancreatic islet β-cell secretory capacity and insulin production. Biomolecules. 2022;12(2):335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Harding HP, Zeng H, Zhang Yet al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7(6):1153–1163. [DOI] [PubMed] [Google Scholar]
- 58. Xu T, Yang L, Yan Cet al. The IRE1α-XBP1 pathway regulates metabolic stress-induced compensatory proliferation of pancreatic β-cells. Cell Res. 2014;24(9):1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Delépine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C.. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25(4):406–409. [DOI] [PubMed] [Google Scholar]
- 60. Liu M, Hodish I, Rhodes CJ, Arvan P.. Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci. 2007;104(40):15841–15846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Haataja L, Arunagiri A, Hassan Aet al. Distinct states of proinsulin misfolding in MIDY. Cell Mol Life Sci. 2021;78(16):6017–6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Engin F, Nguyen T, Yermalovich A, Hotamisligil GS.. Aberrant islet unfolded protein response in type 2 diabetes. Sci Rep. 2015;4(1): 4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lipson KL, Fonseca SG, Ishigaki Set al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006;4(3):245–254. [DOI] [PubMed] [Google Scholar]
- 64. Xin Y, Dominguez Gutierrez G, Okamoto Het al. Pseudotime ordering of single human β-cells reveals states of insulin production and unfolded protein response. Diabetes. 2018;67(9):1783–1794. [DOI] [PubMed] [Google Scholar]
- 65. Sharma RB, O’Donnell AC, Stamateris REet al. Insulin demand regulates β cell number via the unfolded protein response. J Clin Invest. 2015;125(10):3831–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All primer sequences are provided as a supplemental table. Uncropped versions of all immunoblots are provided as a supplemental file. All data are available upon reasonable request.