

Abstract
Background
Brugada syndrome (Brs) and long QT syndrome (LQTs) are the most observed “inherited primary arrhythmia syndromes” and “channelopathies”, which lead to sudden cardiac death.
Methods
Detailed clinical information of Brs and LQTs patients was collected. Genomic DNA samples of peripheral blood were conducted for whole-exome sequencing on the Illumina HiSeq 2000 platform. Then, we performed bioinformatics analysis for 200 genes susceptible to arrhythmias and cardiomyopathies. Protein interaction and transcriptomic co-expression were analyzed using the online website and GTEx database.
Results
All sixteen cases of Brs and six cases of LQTs were enrolled in the current study. Four Brs carried known pathogenic or likely pathogenic of single-point mutations, including SCN5A p.R661W, SCN5A p.R965C, and KCNH2 p.R692Q. One Brs carried the heterozygous compound mutations of DSG2 p.F531C and SCN5A p.A1374S. Two Brs carried the novel heterozygous truncated mutations (MAF < 0.001) of NEBL (p.R882X) and NPPA (p.R107X), respectively. Except for the indirect interaction between NEBL and SCN5A, NPPA directly interacts with SCN5A. These gene expressions had a specific and significant positive correlation in myocardial tissue, with high degrees of co-expression and synergy. Two Brs carried MYH7 p.E1902Q and MYH6 p.R1820Q, which were predicted as "damaging/possibly damaging" and "damaging/damaging" by Polyphen and SIFT algorithm. Two LQTs elicited the pathogenic single splicing mutation of KCNQ1 (c.922-1G > C). Three LQTs carried a single pathogenic mutation of SCN5A p.R1880H, KCNH2 p.D161N, and KCNQ1 p.R243S, respectively. One patient of LQTs carried a frameshift mutation of KCNH2 p. A188Gfs*143.
Conclusions
The truncated mutations of NEBL (p.R882X) and NPPA (p.R107X) may induce Brugada syndrome by abnormally affecting cardiac sodium channel. SCN5A (p.R661W, p.R965C and p.A1374S) and KCNH2 (p.R692Q) may cause Brugada syndrome, while SCN5A (p.R1880H), KCNQ1 (c.922-1G > C and p.R243S) and KCNH2 (p.D161N and p.A188Gfs*143) may lead to long QT syndrome.
Keywords: Arrhythmia, Brugada syndrome, Long QT syndrome, Natriuretic peptide precursor A, Nebulette
Background
Inherited primary arrhythmia syndromes (IPAS), a rare disease (prevalence < 1/1,000) also called “channelopathies,” are commonly induced by genetic disorders and result in ventricular tachycardia (VT), torsade de pointe (TdP) and ventricular fibrillation (VF), consequently leading to sudden cardiac death (SCD) and even sudden unexplained death [1, 2]. The Brugada syndrome (Brs) and long QT syndrome (LQTs) are the most frequently observed IPAS in the general population. The prevalence of Brs and type-2/3 Brugada pattern electrocardiogram (ECG) is 0.5/1,000 and 6.1/1,000 and is reported to be the highest in Southeast Asia [3]. According to a study enrolling 44, 596 infants 15 to 25 days old (43, 080 whites) from 18 maternity hospitals, the prevalence of LQT between 451 and 470 ms of QTc might be close to 1:2000 [4]. A literature search reported that the prevalence of LQTs-induced sudden infant death syndrome (SIDS) ranged from approximately 3.9 to 20.6%, with an average of 12% [5]. The poor prognostic factors for mixed populations described in the series of Brs and LQTs, including sex (men for Brs, type-2 LQTs for female), symptoms, ECG characteristics, family history of SCD, genetic mutation, and inducibility of ventricular arrhythmia during the cardiac electrophysiological examination [6, 7].
For drug therapies of Brs, quinidine, blocking Ito and IKr channels reduces the arrhythmias incidence, including arrhythmic storms and multiple shocks, or as an alternative to an implantable defibrillator (ICD) in children at risk of arrhythmias. Additionally, isoproterenol, increasing the ICaL inflow current, has been used successfully in cases of electrical storms [6]. Syncope in patients with LQTs is often triggered by periods of high sympathetic activity, including stress and exercise, for example, swimming. The type-1 LQTs patients should not be allowed to participate in competitive sports, especially swimming, or only cautiously with supervision. Type-2 and -3 LQTs patients are more susceptible to events during sleep. Type-2 LQTs patients are particularly sensitive to startle or sudden noises while sleeping, such as alarm clocks and telephones, and thus should avoid unexpected noises during sleep. ß blockers are recommended as the first line of therapy for all patients with LQTs. Patients with type-1 LQTs appear to benefit most from β blockers and should be started on β blockers as the first-line therapy [8]. According to the 2017 AHA/ACC/HRS guidelines, the ICD is the most important treatment for Brs and LQTs [9, 10]. The left cardiac sympathetic denervation should be considered for LQTs patients with β blockers therapy who continue to have syncope, TdP, recurrent appropriate ICD shocks despite antiarrhythmic drug therapy, or cardiac arrest [8].
Approximately 25 genes associated with Brs have been identified, of which eighteen genes are responsible for encoding ion channel subunits and seven genes for encoding regulatory proteins. Mutations on SCN5A are the most dominant for Brs and have more than 300 mutations related to Brs [11]. More than 20 disease-causing genes have been reported in almost 70% LQTs patients, including KCNQ1 (30.1%, type-1 of LQTs), KCNH2 (23.2%, type-2 of LQTs) and SCN5A (5.7%, type-3 of LQTs). However, the genetic causes for about one-third of LQTs remain unknown [12]. Notably, the genotype of SCN5A is a crucial component of the scheme for risk stratification of Brs and LQTs. It encodes Nav1.5, a sodium channel protein, wherein type-1 LQTs with mutations affecting the transmembrane domain or C-loop and type-2 and -3 LQTs with missense mutations on the S5-pore-S6 region have a considerably higher risk for cardiac events. Brs with pore-SCN5A mutation has a higher event risk than SCN5A-negative variants [1]. Based on these researches, the pathogenic genotypes of IPAS, for example, the Brs and LQTs are tightly associated with the risk of malignant cardiac events, especially ventricular arrhythmia and SCD. In this study, we enrolled twenty-two unrelated cases of Brs and LQTs. The potential pathogenic mutations carried by these patients will be identified by Whole Exome Sequencing (WES) to analyze the correlations among pathogenic mutations, clinical phenotypes and their risks. Interestingly, in these cases, we found that some common pathogenic genetic mutations may be related to Brs and LQTs. At the same time, we also first found and speculated that truncated NEBL and NPPA mutations might lead to Brs by aberrantly affecting the function of the cardiac sodium channels.
Methods
Study population and diagnostic criteria
Twenty-two cases of Brs and LQTs were enrolled from June 2015 to June 2017. Detailed clinical information was collected. The clinical information included family history, age of presentation, initial symptoms of VT, physical examination, ECGs, and monitoring of ICD based on their informed consent. The clinical diagnosis of Brs was based on the presence of typical type I Brugada pattern on the ECGs, characterized by a coved ST-segment and J-point elevation ≥ 0.2 mV in the right precordial leads [13]. The ECG's QTc (corrected for heart rate) can be calculated (QTc = QT interval + square root of the RR interval). The QTc interval helps us diagnose LQT. A QTc is prolonged if exceeding 0.47 s in women and 0.45 s in men [14, 15]. According to the Schwartz score, a definite LQTS is defined by an LQTS score ≥ 3.5 points [16].
Ethics approval
This study was approved by the Guangdong Medical Institutional Review Board and Medical Ethics Committees [No. GDREC2016001H (R1)]. With the consent of the ethics committee, we followed up with the patients under the condition of informed consent and obtained blood samples for genetic analysis.
Whole exome sequencing
Peripheral bloods from the patients were extracted for WES. Genomic DNA samples were isolated from peripheral blood using a standard DNA extraction protocol. The isolated genomic DNA was then fragmented into 150-200 bp and subjected to DNA library preparation using established Illumina paired-end protocols. Adaptor-ligated libraries were amplified via PCR. A portion of each library was used to create an equimolar pool. Each pool was amplified to enrich targets sequenced by the Agilent SureSelectXT Target Enrichment System (Agilent Technologies Inc., Santa Clara, CA, USA). According to the manufacturer's protocol, whole-exome capture was performed with the Agilent SureSelectXT Human All Exon 50 Mb Kit (Agilent Technologies Inc.). According to the manufacturer's instructions, the exome-enriched libraries were sequenced with the Illumina Hiseq 2000 platform (Illumina, San Diego, CA, USA), and 100 bp paired-end sequencing reads were generated. Each sample was sequenced per lane to obtain an average theoretical depth of 100 × [17, 18].
Read mapping, variant detection, and functional annotation
After WES, raw reads were collected for quality control, in which low-quality reads were filtered, and 3′/5′ adapters were trimmed using the Trim Galore program (version 0.4.4). Clean reads were aligned to the human reference genome (University of California Santa Cruz, UCSC build hg19) using the Burrows-Wheeler Aligner (BWA, version: 0.7.17-r1188) program. The quality scores were recalibrated, and reads were realigned to the reference genome using the Genome Analysis Toolkit (GATK, version: 3.5-0-g36282e4) software package. Following the exclusion of duplicate reads, insertion-deletions (InDels) and single-nucleotide polymorphisms (SNPs) were called using the GATK or Sequence Alignment/Map tools (SAM tools, Version: 1.3.1). The quality value of variants detected by GATK was 99 (the highest value), and the variant abundance was more than 30% [17, 18].
Pathogenic risk classification
The SNPs and Indels were annotated using a pipeline, in which all insertion and deletion variants occurring at coding regions were considered damaging, and nonsynonymous SNPs were predicted by SIFT (http://sift.jcvi.org/www/) and PolyPhen-2 (Polymorphism Phenotyping v2, http://genetics.bwh.harvard.edu/pph2/) [19]. Subsequently, the common risk genes associated with cardiomyopathies and arrhythmias, as reported in our previous research [18, 20], were detected in the patients. These variants were screened with the following filtering criteria: (1) same variants in the WES data; (2) missense, nonsense, insertion and deletion variants; (3) SNPs with minor allele frequency, not ≥ 0.01 according to the SNP database of National Center; excluded variants with allele frequency in 1000genomes (2015 version) higher than 1%, or higher than 5% in house frequency. The potential risk variants were classified as “pathogenic (P)”, “likely pathogenic (LP)”, “uncertain significance (VUS)”, “likely benign (LB)” or “benign (B)” by the Clinvar database [17, 18] and InterVar tool [21] following the 2015 ACMG/ACP guidelines [22]. The detailed ACMG classification was shown in our previous research [18].
Protein interaction analysis
Using the online website https://string-db.org/, the target gene was input for protein–protein interaction analysis. The combined score between proteins with interaction records was scored by combining other database records, experimental verification, gene fusion, co-localization, co-expression and homology analysis. It is currently the mainstream and high-reliability database of protein-interaction information.
Transcriptomic co-expression analysis
In the Genotype-Tissue Expression (GTEx) database [23], the TPM matrix of ventricular tissue, spleen, whole blood, ovary, lung and liver were used for co-expression analysis. The GTEx version was GTEx analysis V8 (dbgap access phs000424.v8.p2). The "Cor" function in the R language was used to calculate the gene correlation matrix. The method parameter used Spearman correlation, in which the correlation threshold was above 0.7, indicating a very close relationship; 0.4–0.7 indicated a close relationship; 0.2–0.4 indicated a general relationship.
Results
Genotype–phenotype relationship
In all, sixteen cases of Brs (median onset-age, 46-year-old; IQR 21.5-year-old; 22 to 65-year-old) and six cases of LQTs (median onset-age, 15-year-old; IQR 18-year-old; 6 to 55-year-old) were enrolled in the current study (Table 1). The echocardiograms (ECGs) of these patients showed normal cardiac structure. VT or VF was detected in 19 cases. Two cases of Brs were induced VF by electrophysiological examination (EPS). These patients suffered from clinical symptoms, including dizziness, syncope, palpitation, amaurosis, and chest distress. Nineteen cases were implanted with ICD, while three Brs refused ICD implantation. Two cases of Brs and one case of LQTs had a familial history of SCD. One case of Brs was the dominant familial inheritance because three siblings had Brugada-like ECGs without clinical symptoms.
Table 1.
The clinical characteristics of patients with Brugada syndrome and long QT syndrome
| No | DS | Sex | Age (years) | Onset of age (years) | Ventricular arrhythmia | Symptoms | ICD therapy | Drugs | Familial history |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Brs | F | 40 | 38 | VT | Dizzy, syncope | refused | Beta blocker | No |
| 2 | LQTs | F | 61 | 55 | VF | Syncope | ICD | Beta blocker | No |
| 3 | Brs | M | 72 | 65 | VT | Syncope | refused | Beta blocker | Brother (SCD, 31-year-old) |
| 4 | Brs | M | 48 | 47 | VF | Amaurosis, syncope | ICD | No | No |
| 5 | Brs | M | 46 | 45 | VT, VF | Dizzy, amaurosis, syncope | ICD | No | No |
| 6 | Brs | M | 60 | 57 | VT, VF | Syncope | ICD | Beta blocker, mexiletine | No |
| 7 | Brs | M | 57 | 47 | No | Syncope | refused | No | Three brothers (Brs) |
| 8 | Brs | M | 41 | 13 | VF | Syncope | ICD | No | No |
| 9 | Brs | M | 49 | 47 | VT | Palpitation, chest distress | ICD | No | No |
| 10 | Brs | M | 63 | 53 | EPS induced VF | Dizzy, amaurosis, | ICD | No | No |
| 11 | Brs | M | 22 | 22 | VF | Syncope | ICD | No | No |
| 12 | Brs | F | 51 | 51 | VF | Syncope when wake up | ICD | No | No |
| 13 | LQTs | F | 13 | 6 | Tdp, VF | Sleeping syncope | ICD | Mexiletine | No |
| 14 | LQTs | F | 19 | 16 | VF | Palpitation, amaurosis, Syncope | ICD | Beta blocker, potassium magnesium aspartate | No |
| 15 | Brs | M | 41 | 34 | VF | Syncope | ICD | Beta blocker | No |
| 16 | Brs | M | 54 | 53 | EPS induced VF | Amaurosis, palpitation | ICD | No | No |
| 17 | Brs | M | 32 | 31 | VT, VF | Syncope, convulsion | ICD | Beta blocker, potassium | No |
| 18 | Brs | M | 33 | 23 | No | No | ICD | No | No |
| 19 | LQTs | F | 35 | 30 | VF | Syncope | ICD | Beta blocker | No |
| 20 | Brs | M | 41 | 30 | VF, AF | Amaurosis, syncope | ICD | No | No |
| 21 | LQTs | F | 16 | 14 | VF | Amaurosis, syncope | ICD | Beta blocker, pacing rate of 95 bpm | No |
| 22 | LQTs | F | 16 | 12 | VF | Syncope, chest distress | ICD | No | Mother (SCD, 33-year-old) |
DS diseases, M male, F female, AF atrial fibrillation, VT ventricular tachycardia, VF ventricular fibrillation, Tdp torsades de pointes, EPS electrophysiology study, SCD sudden cardiac death, ICD Implantable Cardioverter-Defibrillator,- loss of follow-up or lack of clinical data due to refuse of hospitalization
The WES detected some known and pathogenic/likely-pathogenic (P/LP) mutations. Four cases of Brs demonstrated single mutations with known or likely pathogenicity, including p.A1374S (Clinic/ACMG = LP/VUS, No.5, VT/VF, ICD therapy), p.R661W (Clinic/ACMG = P/VUS, No.8, VF, ICD therapy), and p.R965C (Clinic/ACMG = LP/VUS, No.10, VF induced by EPS, ICD therapy) on SCN5A, and p.R692Q (Clinic/ACMG = LP/VUS, No.18, ICD therapy) on KCNH2. One case of Brs carried the compound heterozygous and pathogenic mutations of DSG2 p.F531C (Clinic/ACMG = LP/LP) and SCN5A p.A1374S (Clinic/ACMG = LP/VUS, No.11, VF, ICD therapy). Two cases of LQTs elicited the pathogenic and single splicing mutation of KCNQ1 c.922-1G > C (Clinic/ACMG = P/P, No.19, VF, ICD therapy). Three cases of LQTs carried a single pathogenic mutation of SCN5A p.R1880H (Clinic/ACMG = P/VUS, No.13, TdP and VF, ICD therapy), KCNH2 p.D161N (Clinic/ACMG = P/LP, No.21, VF, ICD therapy), and KCNQ1 p.R243S (Clinic/ACMG = P/LP, No.22, familial history of SCD, VF, ICD therapy), respectively (Table 2).
Table 2.
The known and likely pathogenic mutations of Brugada syndrome and long QT syndrome
| ID | DS | Chr | Start | Gene | Amino acid change | Het | 1000 g/Esp | SIFT | Polyphen | Clinic | ACMG | Evidence | dbSNP |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | LQTs | chr11 | 2,604,664 | KCNQ1 | NM_000218:exon7:c.922-1G > C | ± | – | – | – | P | P | PVS1, PM2_Supporting, PP4, PP1 | rs387906290 |
| 5 | Brs | chr3 | 38,598,739 | SCN5A | NM_001160161:exon23:c.G4120T:p.A1374S | ± | 0.001 | 0.00(D) | 1.00(D) | LP | VUS | PM2_Supporting, PM1, PP3 | rs200034939 |
| 8 | Brs | chr3 | 38,640,451 | SCN5A | NM_000335:exon13:c.C1981T:p.R661W | ± | < 0.001 | 0.00(D) | 1.00(D) | P | VUS | PM2_Supporting, PP3 | rs199473139 |
| 10 | Brs | chr3 | 38,622,757 | SCN5A | NM_000335:exon17:c.C2893T:p.R965C | ± | 0.001 | 0.00(D) | 1.00(D) | LP | VUS | PM2_Supporting, PS4_M, PS3_Supporting, PP3 | rs199473180 |
| 11 | Brs | chr18 | 29,116,333 | DSG2 | NM_001943:exon11:c.T1592G:p.F531C | ± | – | 0.00(D) | 1.00(D) | LP | LP | PM2_Supporting, PM3_Strong, PS3_Supporting | rs200484060 |
| chr3 | 38,598,739 | SCN5A | NM_001160161:exon23:c.G4120T:p.A1374S | ± | 0.001 | 0.00(D) | 1.00(D) | LP | VUS | PM2_Supporting, PS4_Supporting, PP3 | rs200034939 | ||
| 13 | LQTs | chr3 | 38,592,170 | SCN5A | NM_001099405:exon27:c.G5639A:p.R1880H | ± | < 0.001 | 0.06(T) | 0.99(D) | P | VUS | PM2_Supporting, PS4_Supporting, PP3 | rs370694515 |
| 18 | Brs | chr7 | 150,644,473 | KCNH2 | NM_172057:exon9:c.G2075A:p.R692Q | ± | 0.001 | 0.58(T) | 1.00(D) | LP | VUS | PM2_Supporting, PP2 | rs199473020 |
| 19 | LQTs | chr11 | 2,604,664 | KCNQ1 | NM_000218:exon7:c.922-1G > C | ± | – | – | – | P | P | PVS1, PM2_Supporting, PP4, PP1 | rs387906290 |
| 21 | LQTs | chr7 | 150,649,569 | KCNH2 | NM_001204798:exon2:c.G481A:p.D161N | ± | – | 0.00(D) | 1.00(D) | P | LP | PM2_Supporting, PS4_M, PS3_Supporting, PP2, PP3 | rs199472912 |
| 22 | LQTs | chr11 | 2,593,286 | KCNQ1 | NM_000218:exon5:c.C727A:p.R243S | ± | – | 0.00(D) | 1.00(D) | P | LP | PM2_Supporting, PM5, PM1, PP3 | rs199472713 |
DS diseases, LQTs long QT syndrome, Brs Brugada syndrome, Chr chromosome, 1000G/Esp 1000genomes (2015 version) or Esp6500 database, SNP single nucleotide polymorphism, PP polyphen-2, D damaging, B benign, T tolerated, ± heterozygous carrier, P pathogenic, LP likely pathogenic, – no report
We also found several novel mutations potentially associated with Brs and LQTs. In two Brs patients, we first found the heterozygous p.R882X (Clinic/ACMG = VUS/VUS, No.1, VT, refused ICD therapy) of the NEBL gene (at the rs151012132 locus) and p.R107X (Clinic/ACMG = -/LP, No.3, VT, family history of SCD, refused ICD therapy) of the NPPA gene (Table 3), respectively, as truncating mutations, which were absent from or found with MAF (minor allele frequency) < 0.001 in the 1000genomes population. NEBL p.R882X may induce the loss of domains of partial linker and SH3 in NEBL protein (Fig. 1A, B). NPPA p.R107X only expressed the pro-peptide (Fig. 1C) but lost the effective structure of atrial natriuretic peptide (ANP).
Table 3.
The risk mutations of Brugada syndrome and long QT syndrome
| ID | DS | Chr | Start | Gene | Amino acid change | Het | 1000 g/Esp | SIFT | Polyphen | Clinic | ACMG | Evidence | dbSNP |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Brs | chr1 | 3,342,629 | PRDM16 | PNM_199454:exon14:c.G3124A:p.G1042R | ± | – | 0.00(D) | 1.00(D) | – | VUS | PM2_Supporting | – |
| chr10 | 21,097,556 | NEBL | NM_006393:exon26:c.C2644T:p.R882X | ± | < 0.001 | – | – | VUS | VUS | PM2_Supporting | rs151012132 | ||
| chr2 | 179,447,747 | TTN | NM_003319:exon141:c.G38588A:p.R12863Q | ± | – | 0.04(D) | 1.00(D) | VUS | VUS | PM2_Supporting | – | ||
| chr2 | 179,460,249 | TTN | NM_003319:exon123:c.A30637G:p.I10213V | ± | – | 0.36(T) | 0.95(D) | – | VUS | PM2_Supporting | rs56025724 | ||
| chr21 | 18,919,405 | CXADR | NM_001207063:exon2:c.A104G:p.E35G | ± | – | 0.05(T) | 1.00(D) | – | VUS | PM2_Supporting | – | ||
| 3 | Brs | chr1 | 11,907,301 | NPPA | NM_006172:exon2:c.C319T:p.R107X | ± | – | – | – | – | LP | PM2_Supporting, PVS1 | – |
| chr1 | 228,467,100 | OBSCN | NM_001098623:exon27:c.T7351G:p.F2451V | ± | – | 0.27(T) | 0.93(D) | – | VUS | PM2_Supporting | – | ||
| chr1 | 228,547,344 | OBSCN | NM_052843:exon81:c.C18751T:p.R6251W | ± | – | 0.02(D) | 0.74(P) | – | VUS | PM2_Supporting | – | ||
| chr1 | 228,559,174 | OBSCN | NM_001098623:exon94:c.C20695T:p.R6899W | ± | – | 0.00(D) | 0.64(P) | – | VUS | PM2_Supporting | – | ||
| chr2 | 179,640,347 | TTN | NM_003319:exon27:c.G6106A:p.E2036K | ± | – | 0.27(T) | 1.00(D) | – | VUS | PM2_Supporting | – | ||
| 4 | Brs | chr12 | 114,793,662 | TBX5 | NM_080717:exon8:c.C1082T:p.T361I | ± | < 0.001 | 0.13(T) | 0.46(P) | – | VUS | PM2_Supporting | rs267603320 |
| chr2 | 179,432,053 | TTN | NM_003319:exon154:c.C51611T:p.S17204F | ± | – | 0.00(D) | 0.84(P) | – | VUS | PM2_Supporting | – | ||
| 6 | Brs | chr15 | 39,885,760 | THBS1 | NM_003246:exon19:c.C3158T:p.T1053M | ± | < 0.001 | 0.02(D) | 0.71(P) | – | VUS | PM2_Supporting | rs267604168 |
| chr17 | 39,915,014 | JUP | NM_002230:exon9:c.C1606G:p.Q536E | ± | – | 0.03(D) | 0.30(B) | – | VUS | PM2_Supporting | – | ||
| chr3 | 71,015,109 | FOXP1 | NM_001244813:exon14:c.C1521G:p.N507K | ± | – | 0.04(D) | 0.83(P) | – | VUS | PM2_Supporting | – | ||
| 7 | Brs | chr19 | 16,593,346 | CALR3 | NM_145046:exon7:c.G833A:p.R278H | ± | – | 0.03(D) | 0.00(B) | – | VUS | PM2_Supporting | – |
| chr21 | 18,937,961 | CXADR | NM_001338:exon7:c.C1049T:p.A350V | ± | – | 0.102(T) | 0.949(D) | – | VUS | PM2_Supporting | – | ||
| chr4 | 111,539,442 | PITX2 | NM_000325:exon3:c.G814A:p.A272T | ± | – | 0.41(T) | 0.95(D) | – | VUS | PM2_Supporting | – | ||
| chr5 | 251,519 | SDHA | NM_001294332:exon12:c.A1586C:p.Q529P | ± | – | 0.02(D) | 0.99(D) | – | VUS | PM2_Supporting | – | ||
| 9 | Brs | chr5 | 37,333,576 | NUP155 | NM_001278312:exon13:c.C1507T:p.L503F | ± | – | 0.01(D) | 1.00(D) | – | VUS | PM2_Supporting | – |
| chr7 | 128,481,334 | FLNC | NM_001127487:exon12:c.G1924A:p.V642I | ± | < 0.001 | 0.82(T) | 0.67(P) | – | VUS | PM2_Supporting | rs369387744 | ||
| 12 | Brs | chr10 | 112,581,622 | RBM20 | NM_001134363:exon11:c.T3245G:p.L1082R | ± | – | 0.00(D) | 0.08(B) | – | VUS | PM2_Supporting | – |
| chr20 | 33,345,504 | NCOA6 | NM_001242539:exon7:c.G1047C:p.L349F | ± | – | 0.02(D) | 0.89(P) | – | VUS | PM2_Supporting | – | ||
| 14 | LQTs | chr1 | 228,467,732 | OBSCN | NM_001098623:exon28:c.G7607C:p.G2536A | ± | – | 0.01(D) | 1.00(D) | – | VUS | PM2_Supporting | – |
| chr3 | 38,739,348 | SCN10A | NM_001293307:exon26:c.T5069C:p.M1690T | ± | – | 0.00(D) | 0.99(D) | – | VUS | PM2_Supporting | – | ||
| chr3 | 38,770,058 | SCN10A | NM_001293307:exon14:c.C2321T:p.T774M | ± | – | 0.93(T) | 0.02(B) | – | VUS | PM2_Supporting | – | ||
| chr7 | 150,655,499 | KCNH2 | NM_000238:exon4:c.563_564del:p.A188Gfs*143 | ± | – | – | – | – | LP | PM2_Supporting, PVS1 | – | ||
| 15 | Brs | chr14 | 23,853,757 | MYH6 | NM_002471:exon36:c.G5459A:p.R1820Q | ± | < 0.001 | 0.01(D) | 1.00(D) | – | VUS | PM2_Supporting | rs371222772 |
| chr19 | 39,406,284 | SARS2 | NM_017827:exon16:c.C1519T:p.R507W | ± | < 0.001 | 0.01(D) | 0.54(P) | – | VUS | PM2_Supporting | rs143316017 | ||
| chr6 | 152,472,791 | SYNE1 | NM_033071:exon134:c.C24134T:p.A8045V | ± | – | 0.12(T) | 0.98(D) | – | VUS | PM2_Supporting | – | ||
| 16 | Brs | chr14 | 74,970,636 | LTBP2 | NM_000428:exon31:c.4573_4575del:p.1525_1525del | ± | – | – | – | – | VUS | PM2_Supporting, PM4_Supporting | – |
| chr2 | 179,453,729 | TTN | NM_003319:exon132:c.G35528A:p.R11843Q | ± | 0.001 | 0.10(T) | 1.00(D) | – | VUS | PM2_Supporting | rs377203669 | ||
| chr2 | 179,455,524 | TTN | NM_003319:exon132:c.C33733T:p.R11245C | ± | 0.001 | 0.00(D) | 1.00(D) | – | VUS | PM2_Supporting | rs200898955 | ||
| chr7 | 140,624,425 | BRAF | NM_004333:exon1:c.G79A:p.A27T | ± | – | 0.57(T) | 0.48(P) | – | VUS | PM2_Supporting | – | ||
| chr8 | 106,573,686 | ZFPM2 | NM_012082:exon4:c.A397G:p.M133V | ± | – | 0.60(T) | 0.01(B) | – | VUS | PM2_Supporting | rs77117583 | ||
| 17 | Brs | chr1 | 228,547,680 | OBSCN | NM_052843: exon81:c.G19087A:p.G6363S | ± | 1.00(T) | 0.02(B) | – | VUS | PM2_Supporting | – | |
| chr14 | 23,883,054 | MYH7 | NM_000257: exon39:c.G5704C:p.E1902Q | ± | < 0.001 | 0.08(D) | 0.61(P) | – | VUS | PM2_Supporting | rs187073962 | ||
| 20 | Brs | chr6 | 112,506,509 | LAMA4 | NM_001105206:exon9:c.A1007G:p.K336R | ± | – | 0.57(T) | 0.52(P) | – | VUS | PM2_Supporting | – |
DS diseases, LQTs long QT syndrome, Brs Brugada syndrome, Chr chromosome, 1000G/Esp 1000genomes (2015 version) or Esp6500 database, SNP single nucleotide polymorphism, PP polyphen-2, D damaging, B benign, T tolerated, ± heterozygous carrier, P pathogenic, LP likely pathogenic, – no report
Fig. 1.

The changes in amino acids of NEBL and NPPA proteins induced by the truncated mutations
The variants of MYH7 (p.E1902Q, rs187073962, Clinic/ACMG = -/VUS, No.17, VT/VF, ICD therapy) and MYH6 (p.R1820Q, rs371222772, Clinic/ACMG = -/VUS, No.15, VF, ICD therapy), predicted as “damaging/possibly damaging” and “damaging/damaging” by Polyphen and SIFT algorithms, were demonstrated in patients of Brs. A patient of LQTs carried a frameshift mutation of KCNH2 p.A188Gfs*143 (Clinic/ACMG = -/LP, No.13, TdP and VF, ICD therapy), which did not exist in the 1000genomes population.
Literature summary of NEBL and SCN5A interaction
According to previous studies, abnormal desmosome genetic expressions, including desmocollin-2 (DSC2), desmoglein-2 (DSG2), plakophilin-2 (PKP2), desmoplakin (DSP), plakoglobin (JUP) and desmin (DES) participate in the pathogenic mechanism of arrhythmogenic cardiomyopathy (ACM) [17, 18, 20, 24–27]. Interestingly, loss-of-function of SCN5A mutations induced complex arrhythmia, including Brs, atrial fibrillation (AF), atrial standstill, VT and sick sinus syndrome [28]. In this study, we first discovered some interesting interactions among desmosome proteins and cardiac sodium channels in cardiomyocytes, including DSG2 and Nav1.5 (α subunit of the sodium channel, encoded by SCN5A), PKP2 and Nav1.5, DES and Nav1.5, NEBL and DES in the cardiac desmosomes, through literature research using “NEBL and SCN5A (or Nav1.5, or sodium channel), nebulette and SCN5A (or Nav1.5, or sodium channel), each protein of desmosomes (including DSG2, DSC2, PKP2 and DSP) and NEBL (or nebulette), each protein of desmosomes (including DSG2, DSC2, PKP2 and DSP) and SCN5A (or Nav1.5, or sodium channel), NEBL (or nebulette) and Brugada syndrome, each protein of desmosomes (including DSG2, DSC2, PKP2 and DSP) and Brugada syndrome” in the NCBI PubMed database. We summarized these literatures related to NEBL, desmosome proteins and Nav1.5 as follows (shown in Fig. 2A, B).
Fig. 2.
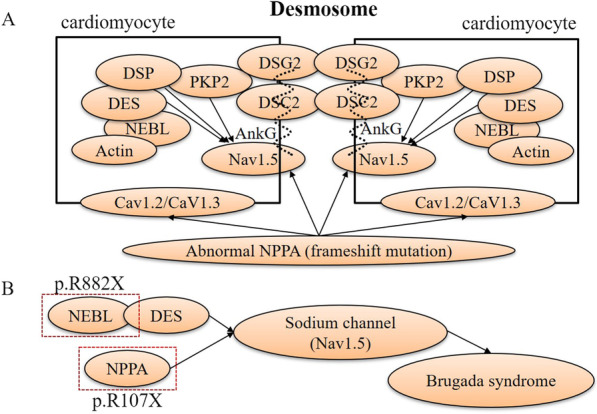
The interactions among NEBL, NPPA and SCN5A associated with Brugada syndrome. The desmosome proteins of cardiomyocytes include desmoglein-2 (DSG2), desmocollin-2 (DSC2), plakophilin-2 (PKP2), desmoplakin (DSP), desmin (DES). SCN5A encoded Nav1.5 protein, as a subunit of the cardiac sodium channel. Ankyrin-G (AnkG) promotes the Nav1.5 anchoring and localizing to the cell membrane. Cav1.2 and Cav1.3 are the subunits of the L-type calcium channel. According to previous research, the arrows illustrate that DSG2, PKP2, DSP, and DES dysfunction would abnormally regulate sodium channel function (Nav1.5). NEBL, nebulin-like protein. NPPA, natriuretic peptide precursor A
NEBL encodes a nebulin-like protein expressed in cardiac muscle. This protein binds to actin, interacting with thin filaments and Z-line-associated proteins in striated muscle and cardiac myofibril assembly. NEBL plays a vital role in the dynamics of the DES-NEBL-actin complex in cardiac myocytes and maintains the relaxation–contraction cycles of the heart. The NEBL exhibits high-affinity interaction and synergic action with DES filaments and is a direct linker between actin and DES. The pathogenic mutations of NEBL will induce dilated cardiomyopathy, hypertrophic cardiomyopathy, left ventricular non-compaction cardiomyopathy, and endocardial fibroelastosis [24–27].
Additionally, the pathogenic mutants E245D, T453I, and knockout of DES increase binding affinity for NEBL, delay filament assembly kinetics, and cause significant attenuation and disruption of cardiac actin-NEBL- DES-Z lines filament network as dynamic DES assembly [29]. The pathogenic mutations of DES can cause severe impairment of filament formation and induce ACM, consequently complicating rhythm disorder, conduction disease, and heart failure [30]. Therefore, the underlying mechanism of NEBL-inducing cardiomyopathies may be comparable to DES. According to previous studies, some cases of ACM overlap the phenotype of Brs [31, 32]. Like DES, PKP2 is one of the critical components in desmosomes of the intercalated disk. It is necessary to maintain gap junction integrity and formation through the DES-DSP-PKP2 complex in desmosomes. The lost expression of PKP2 decreases and disrupts the expression and trafficking of the sodium channel (Nav1.5) at the intercalated disc, which can degrade cardiac sodium current and subsequently lead to overlapped phenotypes of ACM and Brs [33–36]. Based on these evidences, we proposed that the mutation of NEBL might theoretically associate with Brs through the interaction of abnormal NEBL protein with the sodium channel, which has not been demonstrated yet.
Literature summary of NPPA and SCN5A interaction
We also first discovered obvious interactions between natriuretic peptide precursor A (NPPA) and Nav1.5 through current summating research from the NCBI PubMed database using “NPPA and SCN5A (or Nav1.5, or sodium channel), natriuretic peptide precursor A and SCN5A (or Nav1.5, or sodium channel), ANP and SCN5A (or Nav1.5, or sodium channel), Brugada syndrome and NPPA (or ANP, or natriuretic peptide precursor A)”. The literature summary related to NPPA and Nav1.5 was as follows (shown in Fig. 2A, B).
NPPA encodes ANP, expressed in the embryo's atrial and ventricular myocardium. NPPA is also expressed in the adult heart but is downregulated in the ventricles around birth to become restricted to the atria and the ventricular conduction system. In a previous study, for atrial myocyte of transgenic mice carrying a frameshift mutation of NPPA, the expression, currents (INa and ICaL) and action potential duration of cardiac sodium (Nav1.5) and L type calcium (Cav1.2/Cav1.3) channels were significantly reduced. In contrast, the rectifier potassium channel current (IKs) markedly increased compared to the wild type of NPPA. The malignant changes induced by the frameshift NPPA mutation create an atrial substrate of recurrent AF. It is worth noting that ANP is expressed in the atrium and the ventricle. Especially, ANP expression is more significantly re-induced in the ventricles in response to pathological cardiac stress, such as cardiac hypertrophy or myocardial infarction [37]. There were also obvious interactions among NPPA, Nav1.5 and CaV1.2/CaV1.3 (ICaL). The truncated NPPA may induce Brs through the impact on the function of the sodium channel.
NEBL and NPPA interact and co-expressed with SCN5A
The genes of cardiac desmosome components include DSG2, DSC2, PKP2, DSP, JUP and DES. We analyzed the protein interactions corresponding to these genes to test our hypothesis. In the existing interaction database, PPI interaction network analysis shows that these genes have significant interaction (Fig. 3A). The genes including DSG2, PKP2, DSP and JUP directly interact with SCN5A. There is indirect interaction between NEBL/DES and SCN5A, while DES has indirect interaction with SCN5A through DSG2. VCL connects the indirect interaction between NEBL and SCN5A. Vinculin protein encoded by VCL is a cytoskeleton protein related to extracellular matrix adhesion and connection, and its mutation may lead to dilated and hypertrophic cardiomyopathy.
Fig. 3.

Protein interaction and transcriptomic co-expression analysis
In addition, we downloaded the expression data from six different tissue sources (including ventricular tissue, whole blood, spleen, ovary, lung and liver) from the public database of GTEx, and calculated the correlation of the expression of these genes (including SCN5A, NEBL, NPPA, DSP, DES, DSG2 and PKP2) in each tissue (Fig. 3B–G). It was found that the expression of these genes had the highest correlation in cardiac tissue. The correlation between SCN5A and NEBL reached 0.83, and the correlation between SCN5A and NPPA also reached 0.53. The correlations between SCN5A and other genes (including DSP, DES, DSG2 and PKP2) are significantly positive between 0.41 and 0.94, with high degrees of co-expression and synergy. In other tissues, these genes' co-expression has low or no correlation. Therefore, we verified significant co-expression and protein interaction between NPPA, NEBL, SCN5A, DSP, DES, DSG2 and PKP2 genes.
Discussion
Our study enrolled twenty-two cases of Brs and LQTs and conducted WES for these cases to explore the potential pathogenic mutations. Interestingly, according to genotype-phenotype, protein interaction and transcriptomic co-expression analysis, we first found that the truncated mutations of NEBL and NPPA might induce Brs through the abnormal impact on the function of the cardiac sodium channel. Additionally, SCN5A (p.R661W, p.R965C and p.A1374S) and KCNH2 (p.R692Q) may cause Brs, while SCN5A (p.R1880H), KCNQ1 (c.922-1G > C and p.R243S) and KCNH2 (p.D161N and p.A188Gfs*143) may lead to LQTs.
NEBL and NPPA mutations may induce Brugada syndrome by aberrantly affecting the cardiac sodium channel
The cardiac actin-NEBL-DES-Z lines filament network participates in the maintenance of the desmosome junction and the stability of the myocardial structure. As reported before, NEBL p.G202R can augment desmosome separation. The NEBL p.A592E presents abnormal ultrastructure changes and DES downregulation [38]. A GWAS analysis has revealed that NEBL p.A219D (rs2296610) is significantly correlated with AF [39], suggesting that the NEBL mutation may probably associate with an increased risk of arrhythmia. NPPA mutation has been disclosed to link with familial AF, increasing the risk of AF [40] and stroke (NPPA p.V32M) [41]. The heterozygous mutation of NPPA p.S64R caused refractory AF due to the augmented potassium current and shortened atrial action potential [42, 43]. The homozygous mutation of NPPA p.R150Q is associated with dilated cardiomyopathy with atrial standstill [44]. NPPA p.I138T causes AF by activating TNF-α, NF-κB, and IL-1β signaling, inflammation, and fibrosis [45]. The mice with frameshift NPPA mutation elicited the most dramatic prolongation of QRS wave, slightly attenuated atrioventricular conduction and ventricular repolarization through the downregulation of the sodium channel in the atrium, ventricle, and atrioventricular junction [46]. In addition, ANP can reduce mRNA expression of Nav1.5 in the epithelium [47] and modulate KCNQ1 expression [48]. Loss-of-function of Nav1.5 induced by its abnormalities of expression, trafficking, and location to the membrane, will lead to decreased sodium current, delayed activation, or earlier/faster inactivation, which can thus cause Brugada-like ECG or Brugada syndrome [49]. NPPA (p.R107X) and NEBL (p.R882X) mutations were identified in Brs patients. Our further analysis showed the indirect interaction between NEBL and SCN5A and the direct interaction between NPPA and SCN5A. Interestingly, there are high degrees of co-expressions among NEBL, NPPA and SCN5A in myocardial tissue. Therefore, we proposed that truncated mutations of NPPA (p.R107X) and NEBL (p.R882X) may induce Brugada syndrome by aberrantly affecting the cardiac sodium channel, similar to loss-of-function of the sodium channel.
The common ionic-channel genetic mutations caused Brugada syndrome and Long QT syndrome
Our study also identified several pathogenic or likely pathogenic mutations of SCN5A, KCNH2, and KCNQ1 in Brs and LQTs. The mutations of SCN5A (p.A1374S, p.R661W, and p.R965C) and KCNH2 p.R692Q may be associated with Brs, which is consistent with previous studies [50–54]. SCN5A p.R965C can cause hyperpolarized inactivation and slower recovery from the inactivation of the sodium channel [55]. However, the mechanisms of how the mutations of SCN5A (p.A1374S and p.R661W) and KCNH2 p.R692Q induce Brs are still unknown. Up to date, there is no functional research on the splicing mutation (c.922-1G > C) [56] and p.R243S [57–62] of KCNQ1 demonstrated in LQTs. KCNQ1 p.R243C can induce slower activation and the voltage dependence of activation and inactivation, which may shift to more positive potentials in the IKs channel. It can also impair the regulation by PKA and IKs channel-PIP2 (phosphatidylinositol 4, 5-bisphosphate) interactions. Therefore, it increases the risk of life-threatening events while having pronounced benefits from β-blocker treatment [57, 59, 60]. SCN5A p.R1880H (or p.R1898H), predicted to be a pathogenic mutation, has been previously reported in LQTs and Brs. It can dramatically reduce the sodium channel current [63, 64] and the abundance of Nav1.5 and N-Cadherin clusters at the intercalated disc, which is associated with ACM [65]. KCNH2 p.D161N (similar to D501N) has been reported in cases of LQTs, even in a five-year-old boy of the ventricular non-compaction with LQTs [63, 66–69]. The KCNH2 encodes 1159 amino acids of the α-subunit of voltage-dependent potassium channel mediator for the rapid component of delayed rectifying IKr current. For one LQTs case in our study, we also detected a novel and pathogenic frameshift mutation of KCNH2 (p.A188Gfs*143). However, more than sixty patterns of frameshift mutations in KCNH2 have been reported in LQTs [69]. For example, KCNH2 p.G1006fs*49 can cause a significant delay in the voltage-sensitive transition to the channel open state, faster-inactivating kinetics, and quicker recovery from the inactivation for the delayed rectifying IKr current [70].
MYH7 and MYH6 variants were identified in Brugada syndrome
According to a previous report, DSG2 and MYH7 have been identified as new potential Brs candidates [71]. The mutations of MYH7 have been demonstrated in approximately 25% of patients with the overlap of hypertrophic cardiomyopathy and LQTs. Meanwhile, rare mutations of MYH6 have also been identified in these patients [72]. In our study, MYH7 (p.E1902Q) and MYH6 (p.R1820Q) were predicted as "damaging/possibly damaging" and "damaging/damaging" by Polyphen and SIFT algorithms and were also identified in cases of Brs. However, whether these two variants cause Brs remains unclear, which needs further confirmation by more research center data and functional research.
Limitations
The WES of blood DNA from these patients was completed before June 2017. This study was a retrospective study. We did not carry out the verification by Sanger sequencing for these mutations and variants. Our study needs further family genotype–phenotype co-segregation analysis and cell/animal research to investigate how the Brs and LQTs are associated with potential pathogenic mutations of NEBL, NPPA, SCN5A, KCNH2 and KCNQ1.
Conclusions
In our study, we first reported the indirect interaction between NEBL and SCN5A and the direct interaction between NPPA and SCN5A. There are high degrees of co-expressions among NEBL, NPPA and SCN5A in myocardial tissue. The truncated mutations of NEBL (p.R882X) and NPPA (p.R107X) may induce Brs by abnormally affecting the cardiac sodium channel. SCN5A (p.R661W, p.R965C and p.A1374S) and KCNH2 (p.R692Q) may cause Brs, while SCN5A (p.R1880H), KCNQ1 (c.922-1G > C and p.R243S) and KCNH2 (p.D161N and p.A188Gfs*143) may lead to LQTs. Additionally, MYH7 (p.E1902Q) and MYH6 (p.R1820Q) were identified in Brs. However, further pedigree and functional research related to these mutations and variants are needed.
Acknowledgements
We thank Shulin Wu, Chunyu Deng, Zhixin Shan and Fang Rao from Guangdong Cardiovascular Institute. We also appreciate Jinsheng Tao, Huaming Lin (Daan Gene, Guangzhou, China), and Zhipeng Cao for their assistance in data analysis and visualization.
Abbreviations
- IPAS
Inherited primary arrhythmia syndromes
- VT
Ventricular tachycardia
- VF
Ventricular fibrillation
- SCD
Sudden cardiac death
- Brs
Brugada syndrome
- LQTs
Long QT syndrome
- WES
Whole exome Sequencing
- ECGs
Electrocardiograms
- ICD
Implantable cardioverter-defibrillators
- SNPs
Single-nucleotide polymorphisms
- ACM
Arrhythmogenic cardiomyopathy
- PKP2
Plakophilin 2
- DES
Desmin
- DSP
Desmoplakin
- NPPA
Natriuretic peptide precursor A
- ANP
Atrial natriuretic peptide
- AF
Atrial fibrillation
- NEBL
Nebulette
- KCNQ1
Potassium voltage-gated channel subfamily Q member 1
- SCN5A
Sodium voltage-gated channel alpha subunit 5
- MYH6
Myosin heavy chain 6
- MYH7
Myosin heavy chain 7
- KCNH2
Potassium voltage-gated channel subfamily H member 2
Author contributions
JC, HL, ZY and YBL: whole-exome sequencing, bioinformatics analysis and writing; SCG, YTM and YCC: protein interaction and transcriptomic co-expression analysis; JJZ, HJG, YPL and FW: case collection and follow up; SPS, KH, HY and YBL: quality control of clinical data and clinical design. All authors read and approved the final manuscript.
Funding
These fundings supported this study: Talent Development Foundation and High-Level Talent Research Program of The First Dongguan Affiliated Hospital of Guangdong Medical University (GCC2022002), the Science Project of the Second People's Hospital of Guangdong Province [TQ2019-005 and YQ2017-003] and the Medical Science and Technology Research Project of Guangdong Province [A2020069].
Availability of data and materials
The data used in this study is not publicly available, but it might be available from the corresponding author upon reasonable request and permission from relevant Chinese Authorities.
Declarations
Ethics approval and consent to participate
This study was approved by the Guangdong Medical Institutional Review Board and Medical Ethics Committees [No. GDREC2016001H (R1)]. With the consent of the ethics committee, we followed up with the patients and their family members under the condition of informed consent and obtained blood samples for genetic analysis.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jia Chen, Hong Li and Sicheng Guo contributed equally
Contributor Information
Hong Yue, Email: txyyyh@163.com.
Yuting Ma, Email: 709548458@qq.com.
Yubi Lin, Email: linyubi88@qq.com, Email: linyb23@mail.sysu.edu.cn.
References
- 1.Aiba T. Recent understanding of clinical sequencing and gene-based risk stratification in inherited primary arrhythmia syndrome. J Cardiol. 2019;73(5):335–342. doi: 10.1016/j.jjcc.2019.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Gando I, Yang HQ, Coetzee WA. Functional significance of channelopathy gene variants in unexplained death. Forensic Sci Med Pathol. 2019;15(3):437–444. doi: 10.1007/s12024-018-0063-y. [DOI] [PubMed] [Google Scholar]
- 3.Vutthikraivit W, Rattanawong P, Putthapiban P, Sukhumthammarat W, Vathesatogkit P, Ngarmukos T, et al. Worldwide prevalence of brugada syndrome: a systematic review and meta-analysis. Acta Cardiol Sin. 2018;34(3):267–277. doi: 10.6515/ACS.201805_34(3).20180302B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120(18):1761–1767. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ioakeimidis NS, Papamitsou T, Meditskou S, Iakovidou-Kritsi Z. Sudden infant death syndrome due to long QT syndrome: a brief review of the genetic substrate and prevalence. J Biol Res. 2017;24:6. doi: 10.1186/s40709-017-0063-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arbelo E, Brugada J. Risk stratification and treatment of brugada syndrome. Curr Cardiol Rep. 2014;16(7):508. doi: 10.1007/s11886-014-0508-1. [DOI] [PubMed] [Google Scholar]
- 7.Zareba W. Sex and genotype in long QT syndrome risk stratification. JAMA Cardiol. 2019;4(3):254–255. doi: 10.1001/jamacardio.2018.4947. [DOI] [PubMed] [Google Scholar]
- 8.Aleong RG, Milan DJ, Ellinor PT. The diagnosis and treatment of cardiac ion channelopathies: congenital long QT syndrome and Brugada syndrome. Curr Treat Options Cardiovasc Med. 2007;9(5):364–371. doi: 10.1007/s11936-007-0056-8. [DOI] [PubMed] [Google Scholar]
- 9.Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of cardiology/American heart association task force on clinical practice guidelines and the heart rhythm society. J Am Coll Cardiol. 2018;72(14):e91–91e220. doi: 10.1016/j.jacc.2017.10.054. [DOI] [PubMed] [Google Scholar]
- 10.Mascia G, Bona RD, Ameri P, Canepa M, Porto I, Parati G, et al. Brugada syndrome and syncope: a practical approach for diagnosis and treatment. Europace. 2021;23(7):996–1002. doi: 10.1093/europace/euaa370. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Elias A, Benito B. Ion channel disorders and sudden cardiac death. Int J Mol Sci. 2018;19(3):692. doi: 10.3390/ijms19030692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Sherif N, Turitto G, Boutjdir M. Congenital long QT syndrome and torsade de pointes. Ann Noninvasive Electrocardiol. 2017;22(6):e12481. doi: 10.1111/anec.12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilde AA, Behr ER. Genetic testing for inherited cardiac disease. Nat Rev Cardiol. 2013;10(10):571–583. doi: 10.1038/nrcardio.2013.108. [DOI] [PubMed] [Google Scholar]
- 14.Steinberg C. Diagnosis and clinical management of long-QT syndrome. Curr Opin Cardiol. 2018;33(1):31–41. doi: 10.1097/HCO.0000000000000465. [DOI] [PubMed] [Google Scholar]
- 15.Shah SR, Park K, Alweis R. Long QT syndrome: a comprehensive review of the literature and current evidence. Curr Probl Cardiol. 2019;44(3):92–106. doi: 10.1016/j.cpcardiol.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 16.Wilde A, Amin AS, Postema PG. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart. 2022;108(5):332–338. doi: 10.1136/heartjnl-2020-318259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Ma Y, Li H, Lin Z, Yang Z, Zhang Q, et al. Rare and potential pathogenic mutations of LMNA and LAMA4 associated with familial arrhythmogenic right ventricular cardiomyopathy/dysplasia with right ventricular heart failure, cerebral thromboembolism and hereditary electrocardiogram abnormality. Orphanet J Rare Dis. 2022;17(1):183. doi: 10.1186/s13023-022-02348-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin Y, Huang J, Zhu Z, Zhang Z, Xian J, Yang Z, et al. Overlap phenotypes of the left ventricular noncompaction and hypertrophic cardiomyopathy with complex arrhythmias and heart failure induced by the novel truncated DSC2 mutation. Orphanet J Rare Dis. 2021;16(1):496. doi: 10.1186/s13023-021-02112-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24:2125–2137. doi: 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin Y, Zhang Q, Zhong ZA, et al. Whole genome sequence identified a rare homozygous pathogenic mutation of the DSG2 gene in a familial arrhythmogenic cardiomyopathy involving both ventricles. Cardiology. 2017;138(1):41–54. doi: 10.1159/000462962. [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017;100:267–280. doi: 10.1016/j.ajhg.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Human genomics The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin Y, He S, Liao Z, Feng R, Liu R, Peng Y, et al. Whole exome sequencing identified a pathogenic mutation in RYR2 in a Chinese family with unexplained sudden death. J Electrocardiol. 2018;51(2):309–315. doi: 10.1016/j.jelectrocard.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Yang Z, Li T, Xian J, Chen J, Huang Y, Zhang Q, et al. SGLT2 inhibitor dapagliflozin attenuates cardiac fibrosis and inflammation by reverting the HIF-2α signaling pathway in arrhythmogenic cardiomyopathy. FASEB J. 2022;36(7):e22410. doi: 10.1096/fj.202200243R. [DOI] [PubMed] [Google Scholar]
- 26.Lin Y, Huang J, Zhao T, He S, Huang Z, Chen X, et al. Compound and heterozygous mutations of DSG2 identified by whole exome sequencing in arrhythmogenic right ventricular cardiomyopathy/dysplasia with ventricular tachycardia. J Electrocardiol. 2018;51(5):837–843. doi: 10.1016/j.jelectrocard.2018.06.012. [DOI] [PubMed] [Google Scholar]
- 27.Lin Y, Huang J, He S, Feng R, Zhong Z, Liu Y, et al. Case report of familial sudden cardiac death caused by a DSG2 p.F531C mutation as genetic background when carrying with heterozygous KCNE5 p.D92E/E93X mutation. BMC Med Genet. 2018;19(1):148. doi: 10.1186/s12881-018-0580-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin Y, Qin J, Shen Y, Huang J, Zhang Z, Zhu Z, et al. Identification of rare heterozygous linkage R965C–R1309H mutations in the pore-forming region of SCN5A gene associated with complex arrhythmia. Mol Genet Genomic Med. 2021;9(5):e1613. doi: 10.1002/mgg3.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hernandez DA, Bennett CM, Dunina-Barkovskaya L, et al. Nebulette is a powerful cytolinker organizing desmin and actin in mouse hearts. Mol Biol Cell. 2016;27(24):3869–3882. doi: 10.1091/mbc.E16-04-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klauke B, Kossmann S, Gaertner A, et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet. 2010;19(23):4595–4607. doi: 10.1093/hmg/ddq387. [DOI] [PubMed] [Google Scholar]
- 31.Kataoka S, Serizawa N, Kitamura K, et al. An overlap of Brugada syndrome and arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Arrhythm. 2016;32(1):70–73. doi: 10.1016/j.joa.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoogendijk MG. Diagnostic dilemmas: overlapping features of brugada syndrome and arrhythmogenic right ventricular cardiomyopathy. Front Physiol. 2012;3:144. doi: 10.3389/fphys.2012.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novelli V, Malkani K, Cerrone M. Pleiotropic phenotypes associated With PKP2 variants. Front Cardiovasc Med. 2018;5:184. doi: 10.3389/fcvm.2018.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cerrone M, Lin X, Zhang M, et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation. 2014;129(10):1092–1103. doi: 10.1161/CIRCULATIONAHA.113.003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sato PY, Musa H, Coombs W, et al. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009;105(6):523–526. doi: 10.1161/CIRCRESAHA.109.201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moncayo-Arlandi J, Guasch E, Sanz-de M, et al. Molecular disturbance underlies to arrhythmogenic cardiomyopathy induced by transgene content, age and exercise in a truncated PKP2 mouse model. Hum Mol Genet. 2016;25(17):3676–3688. doi: 10.1093/hmg/ddw213. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Z, Zhang Q, Lal H, Nam YJ. Generation of Nppa-tagBFP reporter knock-in mouse line for studying cardiac chamber specification. Genesis. 2019;57(6):e23294. doi: 10.1002/dvg.23294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maiellaro-Rafferty K, Wansapura JP, Mendsaikhan U, et al. Altered regional cardiac wall mechanics are associated with differential cardiomyocyte calcium handling due to nebulette mutations in preclinical inherited dilated cardiomyopathy. J Mol Cell Cardiol. 2013;60:151–160. doi: 10.1016/j.yjmcc.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Low SK, Takahashi A, Ebana Y, et al. Identification of six new genetic loci associated with atrial fibrillation in the Japanese population. Nat Genet. 2017;49(6):953–958. doi: 10.1038/ng.3842. [DOI] [PubMed] [Google Scholar]
- 40.Gudbjartsson DF, Arnar DO, Helgadottir A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448(7151):353–357. doi: 10.1038/nature06007. [DOI] [PubMed] [Google Scholar]
- 41.Pereira NL, Tosakulwong N, Scott CG, et al. Circulating atrial natriuretic peptide genetic association study identifies a novel gene cluster associated with stroke in whites. Circ Cardiovasc Genet. 2015;8(1):141–149. doi: 10.1161/CIRCGENETICS.114.000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Disertori M, Masè M, Narula N, et al. Atrial fibrillation and NPPA gene p.S64R mutation: are cardiologists helpless spectators of healthy mutation carriers? J Cardiovasc Med. 2016;17(3):177–180. doi: 10.2459/JCM.0000000000000302. [DOI] [PubMed] [Google Scholar]
- 43.Abraham RL, Yang T, Blair M, Roden DM, Darbar D. Augmented potassium current is a shared phenotype for two genetic defects associated with familial atrial fibrillation. J Mol Cell Cardiol. 2010;48(1):181–190. doi: 10.1016/j.yjmcc.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Disertori M, Quintarelli S, Grasso M, et al. Autosomal recessive atrial dilated cardiomyopathy with standstill evolution associated with mutation of natriuretic peptide precursor A. Circ Cardiovasc Genet. 2013;6(1):27–36. doi: 10.1161/CIRCGENETICS.112.963520. [DOI] [PubMed] [Google Scholar]
- 45.Cheng C, Liu H, Tan C, Tong D, Zhao Y, Liu X, Si W, Wang L, Liang L, Li J, Wang C, Chen Q, Yimei D, Wang QK, Ren X. Mutation in NPPA causes atrial fibrillation by activating inflammation and cardiac fibrosis in a knock‐in rat model. FASEB J. 2019;33(8):8878–8891. doi: 10.1096/fj.201802455RRR. [DOI] [PubMed] [Google Scholar]
- 46.Menon A, Hong L, Savio-Galimberti E, et al. Electrophysiologic and molecular mechanisms of a frameshift NPPA mutation linked with familial atrial fibrillation. J Mol Cell Cardiol. 2019;132:24–35. doi: 10.1016/j.yjmcc.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo Y, Xia Q, Xia Z, Tang Y. Atrial natriuretic peptide reduces the α-subunit of the epithelial sodium channel mRNA expression in the mouse stria vascularis. Biomed Rep. 2015;3(2):159–162. doi: 10.3892/br.2014.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, Zhao Z, Zu C, et al. Atrial natriuretic peptide modulates the proliferation of human gastric cancer cells via KCNQ1 expression. Oncol Lett. 2013;6(2):407–414. doi: 10.3892/ol.2013.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aam W, Amin AS. Clinical spectrum of SCN5A mutations: long QT syndrome, Brugada syndrome, and cardiomyopathy. JACC Clin Electrophysiol. 2018;4(5):569–579. doi: 10.1016/j.jacep.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 50.Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7(1):33–46. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sommariva E, Pappone C, Martinelli BF, et al. Genetics can contribute to the prognosis of Brugada syndrome: a pilot model for risk stratification. Eur J Hum Genet. 2013;21(9):911–917. doi: 10.1038/ejhg.2012.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshikane Y, Yoshinaga M, Hamamoto K, Hirose S. A case of long QT syndrome with triple gene abnormalities: digenic mutations in KCNH2 and SCN5A and gene variant in KCNE1. Heart Rhythm. 2013;10(4):600–603. doi: 10.1016/j.hrthm.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 53.Silva D, Martins FM, Cavaco D, et al. Natural history of Brugada syndrome in a patient with congenital heart disease. Rev Port Cardiol. 2015;34(7–8):493.e1–4. doi: 10.1016/j.repc.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 54.Priori SG, Napolitano C, Gasparini M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: a prospective evaluation of 52 families. Circulation. 2000;102(20):2509–2515. doi: 10.1161/01.cir.102.20.2509. [DOI] [PubMed] [Google Scholar]
- 55.Hsueh C-H, Chen W-P, Lin J-L, Tsai C-T, Liu Y-B, Juang J-M, Tsao H-M, Ming-Jai S, Lai L-P. Distinct functional defect of three novel Brugada syndrome related cardiac sodium channel mutations. J Biomed Sci. 2009 doi: 10.1186/1423-0127-16-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murray A, Donger C, Fenske C, et al. Splicing mutations in KCNQ1: a mutation hot spot at codon 344 that produces in frame transcripts. Circulation. 1999;100(10):1077–1084. doi: 10.1161/01.cir.100.10.1077. [DOI] [PubMed] [Google Scholar]
- 57.Matavel A, Medei E, Lopes CM. PKA and PKC partially rescue long QT type 1 phenotype by restoring channel-PIP2 interactions. Channels. 2010;4(1):3–11. doi: 10.4161/chan.4.1.10227. [DOI] [PubMed] [Google Scholar]
- 58.Kapa S, Tester DJ, Salisbury BA, et al. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120(18):1752–1760. doi: 10.1161/CIRCULATIONAHA.109.863076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Franqueza L, Lin M, Shen J, Splawski I, Keating MT, Sanguinetti MC. Long QT syndrome-associated mutations in the S4–S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J Biol Chem. 1999;274(30):21063–21070. doi: 10.1074/jbc.274.30.21063. [DOI] [PubMed] [Google Scholar]
- 60.Barsheshet A, Goldenberg I, O-Uchi J, et al. Mutations in cytoplasmic loops of the KCNQ1 channel and the risk of life-threatening events: implications for mutation-specific response to β-blocker therapy in type 1 long-QT syndrome. Circulation. 2012;125(16):1988–1996. doi: 10.1161/CIRCULATIONAHA.111.048041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amin AS, Giudicessi JR, Tijsen AJ, et al. Variants in the 3′ untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur Heart J. 2012;33(6):714–723. doi: 10.1093/eurheartj/ehr473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu JF, Goldenberg I, Moss AJ, et al. Phenotypic variability in Caucasian and Japanese patients with matched LQT1 mutations. Ann Noninvasive Electrocardiol. 2008;13(3):234–241. doi: 10.1111/j.1542-474X.2008.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Napolitano C, Priori SG, Schwartz PJ, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA. 2005;294(23):2975–2980. doi: 10.1001/jama.294.23.2975. [DOI] [PubMed] [Google Scholar]
- 64.Hermida JS, Dassonvalle E, Six I, et al. Prospective evaluation of the familial prevalence of the brugada syndrome. Am J Cardiol. 2010;106(12):1758–1762. doi: 10.1016/j.amjcard.2010.07.049. [DOI] [PubMed] [Google Scholar]
- 65.Te RAS, Agullo-Pascual E, James CA, et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc Res. 2017;113(1):102–111. doi: 10.1093/cvr/cvw234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jongbloed R, Marcelis C, Velter C, Doevendans P, Geraedts J, Smeets H. DHPLC analysis of potassium ion channel genes in congenital long QT syndrome. Hum Mutat. 2002;20(5):382–391. doi: 10.1002/humu.10131. [DOI] [PubMed] [Google Scholar]
- 67.Ogawa K, Nakamura Y, Terano K, Ando T, Hishitani T, Hoshino K. Isolated non-compaction of the ventricular myocardium associated with long QT syndrome: a report of 2 cases. Circ J. 2009;73(11):2169–2172. doi: 10.1253/circj.cj-08-0339. [DOI] [PubMed] [Google Scholar]
- 68.Nagaoka I, Shimizu W, Itoh H, et al. Mutation site dependent variability of cardiac events in Japanese LQT2 form of congenital long-QT syndrome. Circ J. 2008;72(5):694–699. doi: 10.1253/circj.72.694. [DOI] [PubMed] [Google Scholar]
- 69.Kapplinger JD, Tester DJ, Salisbury BA, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6(9):1297–1303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.De Zio R, Gerbino A, Forleo C, et al. Functional study of a KCNH2 mutant: novel insights on the pathogenesis of the LQT2 syndrome. J Cell Mol Med. 2019;23(9):6331–6342. doi: 10.1111/jcmm.14521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Di Resta C, Pietrelli A, Sala S, et al. High-throughput genetic characterization of a cohort of Brugada syndrome patients. Hum Mol Genet. 2015;24(20):5828–5835. doi: 10.1093/hmg/ddv302. [DOI] [PubMed] [Google Scholar]
- 72.Allegue C, Gil R, Blanco-Verea A, et al. Prevalence of HCM and long QT syndrome mutations in young sudden cardiac death-related cases. Int J Legal Med. 2011;125(4):565–572. doi: 10.1007/s00414-011-0572-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used in this study is not publicly available, but it might be available from the corresponding author upon reasonable request and permission from relevant Chinese Authorities.