

Abstract
Purpose of Review
Kidney disease is a strong modulator of the composition and metabolism of the intestinal microbiome that produces toxins and inflammatory factors. The primary pathways for these harmful factors are blood vessels and nerves. Although lymphatic vessels are responsible for clearance of interstitial fluids, macromolecules, and cells, little is known about whether and how kidney injury impacts the intestinal lymphatic network.
Recent Findings
Kidney injury stimulates intestinal lymphangiogenesis, activates lymphatic endothelial cells, and increases mesenteric lymph flow. The mesenteric lymph of kidney-injured animals contains increased levels of cytokines, immune cells, isolevuglandin (IsoLG), a highly reactive dicarbonyl, and of apolipoprotein AI (apoAI). IsoLG is increased in the ileum of kidney injured animals, and intestinal epithelial cells exposed to myeloperoxidase produce more IsoLG. IsoLG-modified apoAI directly increases lymphatic vessel contractions and activates lymphatic endothelial cells. Inhibition of IsoLG by carbonyl scavenger treatment reduces intestinal lymphangiogenesis in kidney-injured animals. Research from our group and others suggests a novel mediator (IsoLG-modified apoAI) and a new pathway (intestinal lymphatic network) in the cross talk between kidneys and intestines and heart.
Summary
Kidney injury activates intestinal lymphangiogenesis and increases lymphatic flow via mechanisms involving intestinally generated IsoLG. The data identify a new pathway in the kidney gut–heart axis and present a new target for kidney disease-induced intestinal disruptions that may lessen the major adverse consequence of kidney impairment, namely cardiovascular disease.
Keywords: Chronic kidney disease, Intestines, Lymphatics, Immune activation, Isolevuglandins
Introduction
Inflammation is a Key Mediator of Cardiovascular Disease
Cardiovascular disease (CVD) is the major cause of mortality worldwide, and atherosclerotic disease resulting in heart attack and stroke is its most common presentation. Low-density lipoprotein cholesterol (LDL-C) is central in the pathogenesis of atherosclerosis and CVD. However, while lipid-lowering therapies provide a large benefit for atherosclerosis and effectively reduce CV events, a sizable residual risk persists [1]. Over the last two decades, the classic “residual cholesterol risk” has been supplanted by “residual inflammatory risk”. The shifting concept reflects abundant experimental, epidemiologic, and clinical trial data, demonstrating that inflammation is a critical driver of atherosclerotic CVD. Animal studies and clinical findings show that inflammatory cells have a key role in the initiation and progression of CVD [2]. Accumulation of cholesterol-laden macrophages in the arterial intima is the hallmark of atherosclerotic fatty streak formation [3]. Subsequent infiltration of neutrophils and T and B cell lymphocytes determine vulnerability to plaque rupture and thus acute CVD events. The inflammatory cells release various factors that determine plaque growth and stability, and proinflammatory cytokines that activate endothelial cells and recruit additional leukocytes that perpetuate the local inflammatory response. The cytokines also stimulate smooth muscle cell proliferation and deposition of extracellular matrix, as well as collagenases, which help degrade fibrous caps and reduce collagen synthesis, thus increasing plaque susceptibility to rupture presenting as an acute clinical CVD event.
Studies of intensive treatment with lipid-lowering statins demonstrated better cardiovascular outcomes and also uncovered their ability to lower inflammatory markers, specifically C-reactive protein (CRP) and interleukins (IL), including the IL-1 [2]. CRP is a sensitive indicator of systemic inflammation that acts both as a driver and marker of increased inflammation. Serum levels of CRP are highly correlated with risk for future atherosclerotic CVD events. Interleukin-1 stimulates production of IL-6, which in turn stimulates production of CRP. The landmark Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) clinical trial in patients with myocardial infarction and a CRP level > 2 mg/L demonstrated that the administration of the monoclonal antibody Canakinumab that targets IL-1β innate immunity pathway for 4 years led to a significantly lower rate of recurrent cardiovascular events compared to placebo [4]. Patients achieving on-treatment high sensitivity (hs)-CRP levels < 2 mg/L benefited the most. The beneficial effect was observed with no reduction in lipid levels from baseline, thus advancing the “inflammatory hypothesis of atherosclerotic CVD”. Studies using other anti-inflammatory agents further solidified the inflammatory hypothesis. The Colchicine Cardiovascular Outcomes Trial (COLCOT) tested the anti-inflammatory drug colchicine and showed significantly reduced cardiovascular events, particularly decreases in risk of stroke and angina requiring coronary revascularization [5]. The Low-Dose Colchicine 2(LoDoCo2) trial showed that low-dose colchicine significantly reduced the primary composite end point of CV death, non-procedural myocardial infarction (MI), ischemic stroke, or ischemia-driven coronary revascularization [6]. Although canakinumab and colchicine are very different drugs, both target the NLRP3 inflammasome, a multiprotein complex mediating inflammation, including in atherosclerotic plaques [7]. The assembly and activation of NLRP3 are primed by several proatherogenic stimuli, including modified lipoproteins, cholesterol crystals, lipopolysaccharide, and reactive oxygen species. Although canakinumab and colchicine act at different levels of the NLRP3 pathways, the central relevance of NLRP3 is underscored by failed clinical trials that utilized anti-inflammatory agents targeting other inflammatory pathways, such as inhibitors of p38 mitogen-associated protein (MAP) kinase, the secretory or lipoprotein-associated forms of phospholipase A2 (sPLA2 and Lp-LPA2), and inhibition of purinergic signaling with methotrexate [8]. Cumulatively, these findings support the idea that mediators of the NLRP3 inflammasome pathway, including, IL-1β, IL-18, and the downstream effector IL-6, are attractive targets to reduce CVD.
In addition to atherosclerotic coronary artery disease, CVD due to heart failure resulting from ischemia or hemodynamic overload also has an important contribution from the inflammatory/immune systems [9]. Biomarkers of sub-clinical inflammation, including IL-6, TNF-α, and CRP, predict incident heart failure in the community and also predict outcome in patients with established but stable heart failure as well as in patients who present with acutely decompensated heart failure [10, 11]. Animal models and patients with both acute and chronic heart failure have increased myocardial infiltration with cellular components, including macrophages, mast cells, B cells, as well as non-cellular components of the immune response including proinflammatory cytokines (TNF, IL-1ß and IL-6) [12]. Heart failure also increases pattern recognition receptors, including Toll-like receptors (TLRs), RIG-I-like receptors (retinoic acid-inducible), NOD-like receptors (NLRs), and the NLRP3 inflammasome on cardiomyocytes, endothelial cells and tissue-resident immune cells. Ongoing preclinical trials targeting inflammation are using anti-cytokine therapies, anti-inflammatory therapies, immunomodulatory therapies and strategies directed at autoimmune responses, including small-molecule CCR2 antagonists, an anti-CCR2 antibody 128,129, CXCR3–CXCL9/CXCL10 pathway antagonists, as well as antibody-based therapeutics to target T cells [9]. Interestingly, a sub-analysis of the CANTOS study revealed that canakinumab caused a dose-dependent reduction in clinical outcomes of heart failure, supporting the idea that IL-1ß inhibition may benefit patients with heart failure [13]. Like patients with atherosclerotic CVD, the greatest reduction in risk of hospitalization for heart failure or heart failure-related mortality was observed in those with greatest reduction in hsCRP.
The Lipid Oxidation Product IsoLG is Downstream of Inflammation and Contributes to CVD
A pathophysiologically important consequence of inflammation in CVD involves generation of oxidative molecules that react with lipoproteins, causing peroxidation. Lipid peroxidation leads to the formation of prostaglandin H2, which is also stimulated during inflammation, and subsequent spontaneous rearrangement of intermediates forming reactive dicarbonyls, including 4-oxo-nonenal (4-ONE), malondialdehyde (MDA), and isolevuglandins (IsoLGs). These reactive lipid dicarbonyls covalently bind to DNA, proteins, and phospholipid, thus disrupting lipoprotein and cellular functions. For example, HDL particles become dysfunctional by modification with IsoLG, which impairs HDL’s capacity to facilitate cholesterol efflux from macrophages and not only reduces HDL’s ability to inhibit cytokine induction, but also potentiates LPS-induced IL-1β expression [14]. These lipid modifications have been linked to oxidative damage in a variety of diseases, including hypertension, Alzheimer’s disease, macular degeneration, gastrointestinal carcinogenesis, and atherosclerosis [15–17]. Importantly, although antioxidant treatment predicts benefit, antioxidants like vitamin C or E have proven to be relatively ineffective suppressors of oxidative injury and lipid peroxidation [18, 19]. Potential reasons for the disappointing results of these therapies are that high doses of antioxidants are needed to suppress lipid peroxidation and because reactive oxygen species (ROS) play critical roles in a number of cell signaling pathways in normal physiology, including protection against bacterial infection. A recent study used small molecule scavengers that selectively react with lipid dicarbonyl species without altering ROS levels, thereby preventing reactive lipid dicarbonyls from modifying cellular macromolecules and without disrupting normal ROS signaling and function [17]. The study used 2-hydroxybenzylamine (2-HOBA), which rapidly reacts with lipid dicarbonyls such as IsoLG and MDA. 2-HOBA treatment significantly attenuated atherosclerosis in hypercholesterolemic Ldlr−/− mice by inhibiting several proatherogenic pathways. 2-HOBA inhibited cell death and necrotic core formation in lesions, leading to the formation of more stable plaques, reflected in greater collagen content and increased fibrous cap thickness. Consistent with the beneficial effects of 2-HOBA on atherosclerosis, the atherosclerotic lesion IsoLG and MDA adduct content was markedly reduced in treated vs control mice, reflecting the scavenging of reactive dicarbonyls. Both light-density lipoprotein (LDL) and high-density lipoprotein (HDL) contained less MDA. The study also showed that 2-HOBA treatment promoted more efficient HDL function in reducing macrophage cholesterol stores. Interestingly, HDL from humans with severe familial hypercholesterolemia (FH), who are at great risk for early CVD events, contained increased MDA adducts compared to control subjects. Moreover, HDL in FH subjects was extremely ineffective in reducing macrophage cholesterol stores. These observations in animal models and humans support the possibility that reactive dicarbonyl scavenging is a novel therapeutic approach for atherosclerotic CVD. Especially interesting is the possible application of these small molecule scavengers in the setting of low-grade chronic inflammation and oxidant stress, as in individuals with chronic kidney disease.
IsoLG in Immune Cells Stimulate Cytokines that Promote Vascular Dysfunction
Vascular dysfunction and stiffening accompany multiple pathologies, including hypertension, atherosclerosis, aging, obesity, and diabetes [20–22]. Excess production of ROS is a common feature of vascular injury and dysfunction. Studies performed in experimental animals implicate ROS production in fibrosis of many tissues and organs including blood vessels, kidney, and lung [23, 24]. In hypertension, antigen-presenting dendritic cells (DCs) develop NADPH oxidase-dependent increases in superoxide formation, leading to IsoLG-modified proteins. These IsoLG-adducted proteins act as neoantigens that are then presented to T cells, promoting differentiation of naïve T cells into IL17-A-producing cells [16, 25–28]. A study by Wu et al. found that chronic oxidative stress leads to accumulation of IsoLG-adducted proteins in DCs and promotes T cell activation, polarization, and proliferation [29]. Immune cells infiltrate into the adventitia and perivascular fat, activating T cells to release the proinflammatory cytokines IL-17A, TNF-α, and IFN-γ [26, 27]. IFN-γ production underlies hypertension-associated fibrosis in the kidney, arteries, and heart [28]. The presence of IsoLG-adducts and T cells in human aortas has been significantly correlated with aortic fibrosis [29]. Release of cytokines by activated T cells in the perivascular space promotes collagen deposition and aortic stiffening, which enhances pulse wave velocity and eventually culminates in vascular inflammation, fibrosis, and dysfunction. IsoLG modification of self-proteins may be a potential mechanism linking oxidative stress to immune activation and may provide insight into how oxidative stress leads to vascular inflammation and injury.
Role of Lymphatics in Development and Progression of CVD
The lymphatic system is comprised of lymphatic vessels, lymph nodes, and lymphoid organs; the lymphatic system plays a critical role in immune regulation, fat absorption, and tissue fluid homeostasis. From early embryonic stages through adulthood, the lymphatic system is closely associated with the cardiovascular system. By 5–6 weeks of development in humans, veinous tissue gives rise to a primitive plexus of lymphatic capillaries. Postnatally, these capillaries continue to develop into a mature network of highly permeable blind-ended vessels that drain interstitial fluid, macromolecules, and immune cells, collectively referred to as lymph, into larger collecting vessels. These vessels are significantly less permeable, have unidirectional valves, and are wrapped in layers of smooth muscle that pump lymph toward lymph nodes. Here, lymph is filtered, and foreign particles are taken up by antigen-presenting cells to initiate specific immune responses. The remaining fluid and macromolecules are then transported through the thoracic duct and back to the systemic circulation via the subclavian vein [30]. Historically, the lymphatic system has been understudied, in part due to the technical challenges of visualizing transparent vessels. However, as a result of advancements made in the discovery of genetic markers, it is now evident that the lymphatics play a critical role in regulating normal organ system physiology and, importantly, can contribute to the progression of numerous diseases if compromised.
Congenital Heart Disease Can Lead to Lymphatic Abnormalities
Pressure gradients dictate interstitial fluid formation as well as lymphatic-mediated fluid return to the cardiovascular system. Congenital heart disease often increases central venous pressure, which can inhibit lymphatic drainage from the thoracic duct. These structural anomalies can also give rise to abnormal hemodynamics that result in increased hydrostatic pressure in the vascular network that in turn can increase interstitial fluid accumulation in an already impaired drainage system. Consequently, patients with congenital heart defects, particularly those with single ventricle defects can develop lymphatic complications that profoundly affect their short- and long-term outcomes [31]. Notably, approximately 13% of patients with congenital heart diseases undergoing palliative procedures, e.g., Fontan, develop protein-losing enteropathy (PLE), a life-threatening condition characterized by leakage of lymphatic fluid and protein into the intestine [32]. It is hypothesized that elevated central venous pressure increases lymph production and impairs intrathoracic lymphatic drainage, leading to dilation of intestinal lymphatics and leakage of lymphatic fluid and protein into the intestinal lumen [33]. Interestingly, the majority of Fontan patients who develop PLE have elevated levels of IFN-γ and TNF-α, cytokines known to disrupt tight junctions in intestinal epithelium and contribute to protein leakage [34]. Similarly, plastic bronchitis, a rare but significant complication in Fontan patients, features dilated pulmonary lymphatic vessels and inappropriate accumulation of protein-rich lymph in the lungs, which solidifies, forming plastic-like casts that plug airway lumens. Inflammation may also contribute to the progression of this disease, as it is speculated that inflammatory mediators could disrupt the pulmonary epithelium, making it easier for lymphatic fluid to leak into the bronchial tree [32].
Lymphatic Regulation of Inflammation and CVD
As above, chronic inflammation is a major risk factor for CVD. Lymphangiogenesis and vessel remodeling are reactivated in response to inflammation [30], and the lymphatic system has an integral role in mediating the inflammatory response by regulating interstitial fluid drainage and trafficking of macromolecules including cytokines, tissue fragments, hormones, and foreign antigens [35]. Thus, the lymphatic system is a major influencer of the progression of CVD and may be an untapped therapeutic target. Lymphatic vessels play an integral role in the progression of atherosclerosis as demonstrated in animal studies, which showed that mice with impaired lymphatic function crossed with atherosclerotic mice had elevated levels of atherogenic lipoproteins and accelerated atherosclerosis, compared to hypercholesterolemic controls with functional lymphatics [36]. Plaque destabilization and cessation of disease progression involve removal and excretion of cholesterol from macrophage stores within the arterial wall, a process termed reverse cholesterol transport. Cholesterol is first hydrolyzed and then mobilized to lipoprotein acceptors such as apoAI, which results in the formation of HDL. Lymphatic vessels are then required to facilitate the transport of HDL from the arterial wall to the bloodstream where it flows to the liver and is excreted [37, 38]. Although different approaches to disrupt the growth and function of lymphatics accelerate atherosclerosis, the exact role of ineffective reverse cholesterol transport remains to be determined.
Lymphatic vessels are also abundant throughout the myocardium, subendocardial space, and even in the atrioventricular and semilunar valves, and significant lymphangiogenesis follows MI in regions adjacent to the infarct as well as in uninjured areas [39]. The lymphatic contribution to the inflammatory response following injury involves the removal of dead cardiomyocytes and initiation of tissue repair and remodeling [40]. This appears to be a critical step for tissue repair as demonstrated in animal studies where lymphangiogenesis was amplified. In these studies, mice with increased cardiac lymphangiogenesis had reduced scar formation and improved cardiac function compared to control mice [41]. Moreover, in mouse models where lymphangiogenesis was inhibited, cardiac injury and dysfunction were exacerbated after myocardial ischemia-reperfusion [42]. Based on the interplay of lymphatic vessels and myocardial infarction, targeted induction of lymphangiogenesis has been proposed as a novel therapeutic strategy for this form of CVD [43].
Hypertension is a major a CVD risk factor that clusters with other risk factors such as increased age, BMI, and diabetes. Studies have shown that lymphangiogenesis in the skin and muscle is initiated in response to salt-induced hypertension and involves macrophage secretion of vascular endothelial growth factor-C (VEGF-C) and that blocking lymphangiogenesis results in increased blood pressure in response to salt loading [44]. Interestingly, recent studies showed that selective upregulation of lymphangiogenesis in the kidney protected against salt- and angiotensin II- induced hypertension [45, 46]. Together, these observations underscore a critical role of the lymphatics in cardiac and extracardiac tissues in promoting CVD.
Role of Inflammation and Lymphatics in Amplifying CVD in Chronic Kidney Disease
Kidney Disease Accelerates CVD
Chronic kidney disease (CKD), defined by the Kidney Disease: Improving Global Outcomes (KDIGO) as abnormalities in kidney structure or function that persist for more than three months, affects 15–20% of the world population [47]. The overarching consequence of CKD is CVD. CKD patients are more likely to die from CVD than to progress to end-stage CKD [48]. In the last five years, both the American College of Cardiology/American Heart Association (ACC/AHA) and the National Kidney Foundation (NKF) have recommended that CKD be considered equivalent to preexisting coronary artery disease (CAD) as a risk predictor. The increased cardiovascular risk is apparent with even modest kidney impairment, and measurable increases in risk have been identified when GFR falls to < 60 mL/min/1.73m2 [49]. CVD risk continues to increase as kidney function declines, becoming especially pronounced in patients requiring dialysis who are at >15 times greater risk of dying from CVD than the general population without CKD [50]. The exaggerated prevalence of CVD in the CKD population is further complicated by the fact that the predictive value of traditional risk factors, including hyperlipidemia, hypertension, diabetes, smoking, and obesity, become attenuated with declining kidney function, and some established risks, such as BMI and hyperlipidemia, may reverse [51]. Moreover, lipid-lowering therapies have shown little to no benefit in several large clinical trials of advanced CKD, including patients on dialysis, despite robust reductions in serum LDL cholesterol [52]. Further complicating the approach to CVD in CKD patients is the fact that as kidney function deteriorates, the type of CVD changes, with non-atherosclerotic disease becoming more important. Thus, in contrast to myocardial infarction and stroke, which are central events of atherosclerotic CVD, arterial calcification, heart failure, left-ventricular hypertrophy, arrhythmias, peripheral artery disease, and sudden cardiac death are more common in individuals with severe kidney impairment compared to patients with modest renal dysfunction or individuals with intact kidneys [53]. Thus, the CKD population presents a unique human circumstance of remarkable excess of CVD with limited responsiveness to lipid-lowering treatment that offers the opportunity and challenge to develop more comprehensive concepts of mechanisms and innovative therapeutic approaches for CVD.
Inflammation in Kidney Disease Reflects Disrupted Intestinal Integrity and the Microbiome
Although CVD is the major consequence of CKD, traditional risk cardiovascular factors, e.g., dyslipidemia, diabetes, obesity, may be more or less important at different stages of CKD. In contrast, inflammation and oxidative stress are consistently increased across the entire spectrum of renal dysfunction are likely key in the pathogenesis of CKD-associated CVD [54, 55]. A sub-analysis of the CANTOS trial that included 1875 patients with GFR < 60 ml/min followed for 48 months found that Canakinumab significantly lowered cardiovascular events compared to placebo in patients with CKD [56]. This benefit was observed in the absence of any effects on atherogenic lipids. As in the main CANTOS study, the beneficial effects on CKD-associated CVD paralleled reduced hsCRP underscoring that inflammation characterizing CKD may be especially relevant to the role of “inflammatory hypothesis of atherosclerotic CVD” in the setting of kidney disease. Interestingly, post hoc analysis of two randomized clinical trials, IL-1 trap in patients with moderate CKD and IL-1 receptor antagonist in patients on maintenance hemodialysis, showed IL-1 blockade improved HDL functionality including its anti-inflammatory activity, e.g., blockade of IL-6, TNFα and NLRP3, and antioxidant function, e.g., lessening superoxide production, which may contribute to the benefits of this therapeutic intervention [57].
The proinflammatory and high oxidative state prevailing across the spectrum of renal disease is, at least in part, related to abnormalities in the integrity of the intestinal barrier and unaltered microbiome [58]. Several factors prevailing in CKD contribute to the barrier dysfunction, including gut dysbiosis, slow intestinal transit time, low dietary fiber intake, metabolic acidosis, gut ischemia and edema, iron therapy, and frequent exposure to antibiotics. The resulting increase in permeability promotes translocation of gut-derived factors such as bacterial components, endotoxins, intestinal metabolites that leak into the circulation, and then initiate immune activation and proinflammatory signaling. Endotoxin stimulation of TNF and NF-kB involves Toll-like receptor 4, which activates inflammatory response in endothelial cells, transforms macrophages into foam cells and promotes procoagulant activity. Impaired intestinal integrity promotes leakage of gut metabolites including metabolites of carbohydrates, e.g., short-chain free fatty acids and proteins, e.g., trimethylamine N-oxide, p-cresol sulfate and indoxyl sulfate and lipid peroxidation products. Each of these metabolites can directly disrupts cholesterol metabolism and increases expression of scavenger receptors, which promotes foam cell formation. Together, these observations indicate that intestines are an important source for inflammatory and oxidative factors and that kidney disease augments the generation of these potentially harmful compounds.
Kidney Disease Stimulates Intestinal Lymphangiogenesis
Traditionally, blood vessels and nerves have been considered the primary conduits by which bacterial components and endotoxins initiate systemic immune activation and proinflammatory signaling. Little attention has been given to lymphatics, whose main function is transport of fluid, solutes, macromolecules, lipids, and cells. Inflammatory injuries and disease increase lymphatic growth and lymph flow in the affected organ. Our groups found that kidney injury not only causes intrarenal lymphangiogenesis but also stimulates lymphangiogenesis in the gut [59]. Using two models of renal damage, we demonstrated that proteinuric kidney injury in mice as well as a proteinuric model in rats augments intestinal lymphangiogenesis, evidenced by increased mRNA and immunostaining for podoplanin, LYVE-1, and VEGF receptor 3. The intestinal lymphangiogenesis was accompanied by macrophage infiltration, which colocalized with VEGF-C protein, suggesting that intestinal macrophages are sources of the increased VEGF-C levels that have been documented in intestinal lymphatics of proteinuric animals. Since kidney injury is known to stimulate VEGF-C production by the proximal tubules, the kidney may be an additional source for VEGF-C in this setting. The expanded lymphatic network showed increased rate of lymph flow and lymph volume was > threefold higher in mesenteric lymphatics of proteinuric rats compared to normal controls. These findings support the idea that intestinal lymphatics are a pathway that delivers gut-generated metabolites into the circulation and distant organs.
IsoLG Originating in Gut is a Mediator of Mesenteric Lymphatic Dysfunction and Activation of Lymphatic Endothelial Cells
Aside from lymphangiogenesis and increase in lymph flow, kidney injury also modifies the composition of the mesenteric lymph. Our study showed that cytokines, including IL-6, IL-10, and IL-17, were elevated in mesenteric lymph of proteinuric animals compared to lymph of uninjured rats. The proteinuric injury also increased intestinal generation of the reactive peroxidation product IsoLG. These observations complement other studies documenting IsoLG along the gastrointestinal tract. For example, increased IsoLG adducts were reported in gastric epithelial cells of patients with gastritis, precancerous intestinal metaplasia, colitis-associated dysplasia, and colitis-associated carcinoma, as well as in mice with colitis-associated carcinoma [60]. Demonstration of IsoLG adducts in human gastric organoids infected with H. pylori supports the idea that intestinal epithelial cells can generate IsoLG. Animals with kidney injury showed increased IsoLG adducts in mesenteric lymph but not in concurrently collected plasma, suggesting that the intestines are the source of these potentially harmful particles. Further, cultured intestinal epithelial cells exposed to myeloperoxidase (MPO), a peroxidase enzyme elevated in many chronic diseases, including CKD, and shown to be enriched in the intestinal wall of proteinuric rats, stimulated production of IsoLG. It is notable that in addition to gastrointestinal epithelial cells, immune cells infiltrating the intestinal wall can form IsoLG adducts as demonstrated in intestines of mice fed a high-salt diet [61]. Thus, both parenchymal intestinal epithelial cells and infiltrating immune cells can augment IsoLG synthesis in the intestines. Interestingly, IsoLG can directly modulate lymphatic vessel dynamics and activate lymphatic endothelial cells. Lymphatic endothelial cells exposed to IsoLG have significantly increased production of ROS Nos3. Isolated mesenteric lymphatic vessels exposed to IsoLG manifest altered functionality, including blunted vasoactivity but greater contraction frequency. The pathophysiologic impact of these lymphatic changes is supported by in vivo studies, showing that inhibition of IsoLG by small molecule scavengers significantly lessens injury-induced intestinal lymphangiogenesis in proteinuric mice [59].
Conclusion and Future Perspectives
Recent studies suggest that the mesenteric lymphatics are a novel pathway linking intestinally generated inflammatory and oxidative metabolites with cardiovascular diseases. Kidney injury amplifies this pathway by stimulating intestinal lymphangiogenesis and increasing lymphatic flow via mechanisms involving intestinally generated IsoLG (Fig. 1). The net effect is greater delivery of intestinally derived molecules such as IsoLG that may contribute to the adverse systemic effects of kidney injury. More research is needed to investigate the specific mechanisms by which kidney disease causes intestinal lymphangiogenesis and IsoLG generation. Blocking intestinally generated IsoLG could become a future therapeutic target to reduce the CVD burden of individuals with renal disease.
Fig. 1.
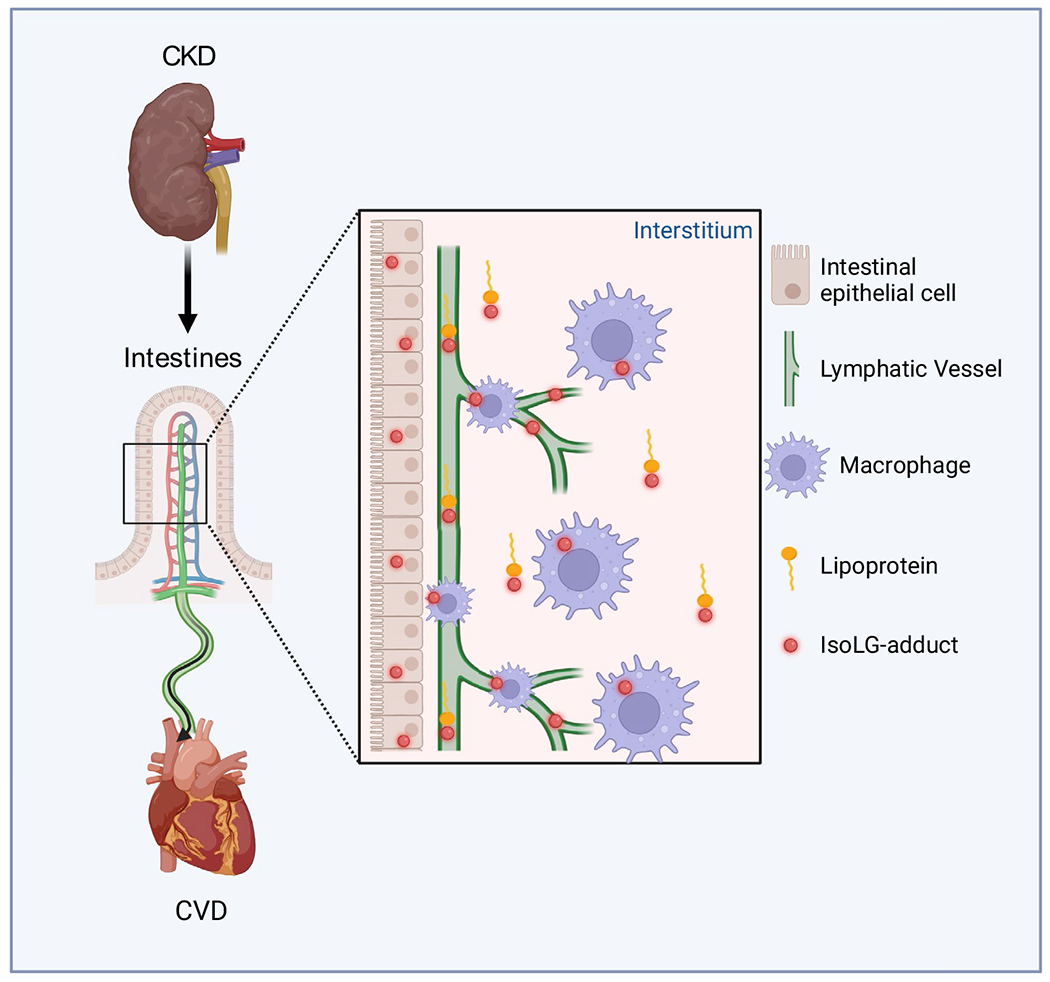
Schematic illustrating of a new mechanism for adverse kidney–intestine–heart cross talk. Chronic kidney disease leads to accumulation of reactive dicarbonyls, namely isolevuglandin (IsoLG). IsoLG is produced by intestinal epithelial cells and carried by lipoproteins. IsoLG produced by infiltrating immune cells adds to the intestinal accumulation. Concurrently, kidney disease activates intestinal lymphangiogenesis and increases mesenteric lymphatic flow via mechanisms involving IsoLG. The net effect is greater delivery of deleterious elements of the intestinal microbiome/metabolome that amplify cardiovascular diseases
Funding
The work was supported by NIH 1P01HL116263 (VK), K01HL13049 (AK), R03HL155041 (AK), R01HL144941 (AK) and R01HD099777 (ELS).
Footnotes
Ethics Approval All reported studies/experiments with human or animal subjects performed by the authors were performed in accordance with all applicable ethical standards, including the Declaration of Helsinki and its amendments, institutional/national research committee standards, and international/national/institutional guidelines.
Conflict of Interest Valentina Kon, Elaine L. Shelton, Ashley Pitzer, and Hai-Chun Yang declare that they have no conflict of interest. Annett Kirabo has a US Patent # 14/232,615 Methods for Treating Inflammation and Hypertension with Gamma-Ketoaldehyde Scavengers.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500–9. 10.1056/NEJMoal500858. [DOI] [PubMed] [Google Scholar]
- 2.Libby P, Nahrendorf M, Swirski FK. Leukocytes link local and systemic inflammation in ischemic cardiovascular disease: an expanded “cardiovascular continuum.” J Am Coll Cardiol. 2016;67(9):1091–103. 10.1016/j.jacc.2015.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res. 2015;107(3):321–30. 10.1093/cvr/cvv147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–31. 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 5.Tardif JC, Kouz S, Waters DD, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. 2019;381(26):2497–505. 10.1056/NEJMoa1912388. [DOI] [PubMed] [Google Scholar]
- 6.Nidorf SM, Fiolet ATL, Mosterd A, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. 2020;383(19):1838–47 10.1056/NEJMoa2021372. [DOI] [PubMed] [Google Scholar]
- 7.Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15(4):203–14. 10.1038/nrcardio.2017.161. [DOI] [PubMed] [Google Scholar]
- 8.Liberale L, Montecucco F, Schwarz L, Luscher TF, Camici GG. Inflammation and cardiovascular diseases: lessons from seminal clinical trials. Cardiovasc Res. 2021;117(2):411–22. 10.1093/cvr/cvaa211. [DOI] [PubMed] [Google Scholar]
- 9.Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. 2020;17(5):269–85. 10.1038/s41569-019-0315-x. [DOI] [PubMed] [Google Scholar]
- 10.Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation. 2001;103(16):2055–9. 10.1161/01.cir.103.16.2055. [DOI] [PubMed] [Google Scholar]
- 11.Kalogeropoulos A, Georgiopoulou V, Psaty BM, et al. Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J Am Coll Cardiol. 2010;55(19):2129–37. 10.1016/j.jacc.2009.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abernethy A, Raza S, Sun JL, et al. Pro-inflammatory biomarkers in stable versus acutely decompensated heart failure with preserved ejection fraction. J Am Heart Assoc. 2018. 10.1161/JAHA.117.007385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Everett BM, Cornel JH, Lainscak M, et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019;139(10):1289–99. 10.1161/CIRCULATIONAHA.118.038010. [DOI] [PubMed] [Google Scholar]
- 14.May-Zhang LS, Yermalitsky V, Huang J, et al. Modification by isolevuglandins, highly reactive gamma-ketoaldehydes, deleteriously alters high-density lipoprotein structure and function. J Biol Chem. 2018;293(24):9176–87. 10.1074/jbc.RA117.001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boutaud O, Ou JJ, Chaurand P, Caprioli RM, Montine TJ, Oates JA. Prostaglandin H2 (PGH2) accelerates formation of amyloid beta1-42 oligomers. J Neurochem. 2002;82(4):1003–6. 10.1046/j.1471-4159.2002.01064.x. [DOI] [PubMed] [Google Scholar]
- 16.Kirabo A, Fontana V, de Faria AP, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. 2014;124(10):4642–56. 10.1172/JCI74084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao H, Huang J, Yancey PG, et al. Scavenging of reactive dicarbonyls with 2-hydroxybenzylamine reduces atherosclerosis in hypercholesterolemic Ldlr(−/−) mice. Nat Commun. 2020;11(1):4084. 10.1038/s41467-020-17915-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leopold JA. Antioxidants and coronary artery disease: from pathophysiology to preventive therapy. Coron Artery Dis. 2015;26(2):176–83. 10.1097/MCA.0000000000000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts LJ 2nd , Oates JA, Linton MF, et al. The relationship between dose of vitamin E and suppression of oxidative stress in humans. Free Radic Biol Med. 2007;43(10):1388–93. 10.1016/j.freeradbiomed.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kals J, Kampus P, Kals M, et al. Inflammation and oxidative stress are associated differently with endothelial function and arterial stiffness in healthy subjects and in patients with atherosclerosis. Scand J Clin Lab Invest. 2008;68(7):594–601. 10.1080/00365510801930626. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell GF, Guo CY, Benjamin EJ, et al. Cross-sectional correlates of increased aortic stiffness in the community: the Framingham Heart Study. Circulation. 2007;115(20):2628–36. 10.1161/CIRCULATIONAHA.106.667733. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell GF, Parise H, Benjamin EJ, et al. Changes in arterial stiffness and wave reflection with advancing age in healthy men and women: the Framingham Heart Study. Hypertension. 2004;43(6):1239–45. 10.1161/01.HYP.0000128420.01881.aa. [DOI] [PubMed] [Google Scholar]
- 23.Stephen EA, Venkatasubramaniam A, Good TA, Topoleski LD. The effect of oxidation on the mechanical response and microstructure of porcine aortas. J Biomed Mater Res A. 2014;102(9):3255–62. 10.1002/jbm.a.34998. [DOI] [PubMed] [Google Scholar]
- 24.Soskel NT, Watanabe S, Sandberg LB. Mechanisms of lung injury in the copper-deficient hamster model of emphysema. Chest. 1984;85(6 Suppl):70S–73S. 10.1378/chest.85.6_supplement.70s. [DOI] [PubMed] [Google Scholar]
- 25.Barbaro NR, Foss JD, Kryshtal DO, et al. dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017;21(4):1009–20. 10.1016/j.celrep.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guzik TJ, Hoch NE, Brown KA, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10):2449–60. 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madhur MS, Lob HE, McCann LA, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55(2):500–7. 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marko L, Kvakan H, Park JK, et al. Interferon-gamma signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension. 2012;60(6):1430–6. 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- 29.Wu J, Saleh MA, Kirabo A, et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. 2016;126(1):50–67. 10.1172/JCI80761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010;140(4):460–76. 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 31.Itkin M, Pizarro C, Radtke W, Spurrier E, Rabinowitz DA. Lymphatic management in single-ventricle patients. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2020;23:41–7. 10.1053/j.pcsu.2020.03.001. [DOI] [PubMed] [Google Scholar]
- 32.Larue M, Gossett JG, Stewart RD, Backer CL, Mavroudis C, Jacobs ML. Plastic bronchitis in patients with fontan physiology: review of the literature and preliminary experience with fontan conversion and cardiac transplantation. World J Pediatr Congenit Heart Surg. 2012;3(3):364–72. 10.1177/2150135112438107. [DOI] [PubMed] [Google Scholar]
- 33.Hess J, Kruizinga K, Bijleveld CM, Hardjowijono R, Eygelaar A. Protein-losing enteropathy after Fontan operation. J Thorac Cardiovasc Surg. 1984;88(4):606–9. [PubMed] [Google Scholar]
- 34.Bode L, Murch S, Freeze HH. Heparan sulfate plays a central role in a dynamic in vitro model of protein-losing enteropathy. J Biol Chem. 2006;281(12):7809–15. 10.1074/jbc.M510722200. [DOI] [PubMed] [Google Scholar]
- 35.Abouelkheir GR, Upchurch BD, Rutkowski JM. Lymphangiogenesis: fuel, smoke, or extinguisher of inflammation’s fire? Exp Biol Med (Maywood). 2017;242(8):884–95. 10.1177/1535370217697385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vuorio T, Nurmi H, Moulton K, et al. Lymphatic vessel insufficiency in hypercholesterolemic mice alters lipoprotein levels and promotes atherogenesis. Arterioscler Thromb Vasc Biol. 2014;34(6):1162–70. 10.1161/ATVBAHA.114.302528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim HY, Thiam CH, Yeo KP, et al. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SRBI-mediated transport of HDL. Cell Metab. 2013;17(5):671–84. 10.1016/j.cmet.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 38.Martel C, Li W, Fulp B, et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Invest. 2013;123(4):1571–9. 10.1172/JCI63685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ratajska A, Gula G, Flaht-Zabost A, et al. Comparative and developmental anatomy of cardiac lymphatics. Sci World J. 2014;2014:183170. 10.1155/2014/183170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204(12):3037–47. 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klotz L, Norman S, Vieira JM, et al. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature. 2015;522(7554):62–7. 10.1038/nature14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimizu Y, Polavarapu R, Eskla KL, et al. Impact of lymphangiogenesis on cardiac remodeling after ischemia and reperfusion injury. J Am Heart Assoc. 2018;7(19):e009565. 10.1161/JAHA.118.009565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brakenhielm E, Alitalo K. Cardiac lymphatics in health and disease. Nat Rev Cardiol. 2019;16(1):56–68. 10.1038/s41569-018-0087-8. [DOI] [PubMed] [Google Scholar]
- 44.Wiig H, Schroder A, Neuhofer W, et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J Clin Invest. 2013;123(7):2803–15. 10.1172/JCI60113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lopez Gelston CA, Balasubbramanian D, Abouelkheir GR, et al. Enhancing renal lymphatic expansion prevents hypertension in mice. Circ Res. 2018;122(8):1094–101. 10.1161/CIRCRESAHA.118.312765. [DOI] [PubMed] [Google Scholar]
- 46.Balasubbramanian D, Gelston CAL, Lopez AH, et al. Augmenting renal lymphatic density prevents angiotensin ii-induced hypertension in male and female mice. Am J Hypertens. 2020;33(1):61–9. 10.1093/ajh/hpz139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hill NR, Fatoba ST, Oke JL, et al. Global prevalence of chronic kidney disease - a systematic review and meta-analysis. PLoS ONE. 2016;11(7):e0158765. 10.1371/journal.pone.0158765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reiss AB, Miyawaki N, Moon J, et al. CKD, arterial calcification, atherosclerosis and bone health: Inter-relationships and controversies. Atherosclerosis. 2018;278:49–59. 10.1016/j.atherosclerosis.2018.08.046. [DOI] [PubMed] [Google Scholar]
- 49.Matsushita K, Coresh J, Sang Y, et al. Estimated glomerular filtration rate and albuminuria for prediction of cardiovascular outcomes: a collaborative meta-analysis of individual participant data. Lancet Diabetes Endocrinol. 2015;3(7):514–25. 10.1016/S2213-8587(15)00040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van der Velde M, Matsushita K, Coresh J, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with all-cause and cardiovascular mortality. A collaborative meta-analysis of high-risk population cohorts. Kidney Int. 2011;79(12):1341–52. 10.1038/ki.2010.536. [DOI] [PubMed] [Google Scholar]
- 51.Naderi N, Kleine CE, Park C, et al. Obesity paradox in advanced kidney disease: from bedside to the bench. Prog Cardiovasc Dis. 2018;61(2):168–81. 10.1016/j.pcad.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharp CG. Study of Heart and Renal Protection (SHARP): randomized trial to assess the effects of lowering low-density lipoprotein cholesterol among 9,438 patients with chronic kidney disease. Am Heart J. 2010;160(5):785–794.e10. 10.1016/)’.ahj.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 53.Alonso A, Lopez FL, Matsushita K, et al. Chronic kidney disease is associated with the incidence of atrial fibrillation: the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 2011;123(25):2946–53. 10.1161/CIRCULATIONAHA.111.020982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liakopoulos V, Roumeliotis S, Gorny X, Dounousi E, Mertens PR. Oxidative stress in hemodialysis patients: a review of the literature. Oxid Med Cell Longev. 2017;2017:3081856. 10.1155/2017/3081856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang WH, Kitai T, Hazen SL. Gut microbiota in cardiovascular health and disease. Circ Res. 2017;120(7):1183–96. 10.1161/CIRCRESAHA.117.309715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ridker PM, MacFadyen JG, Glynn RJ, et al. Inhibition of interleukin-1beta by canakinumab and cardiovascular outcomes in patients with chronic kidney disease. J Am Coll Cardiol. 2018;71(21):2405–14. 10.1016/jacc.2018.03.490. [DOI] [PubMed] [Google Scholar]
- 57.Hung AM, Tsuchida Y, Nowak KL, et al. IL-1 inhibition and function of the hdl-containing fraction of plasma in patients with stages 3 to 5 CKD. Clinical journal of the American Society of Nephrology : CJASN. 2019;14(5):702–11. 10.2215/CJN.04360418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andersen K, Kesper MS, Marschner JA, et al. Intestinal dysbiosis, barrier dysfunction, and bacterial translocation account for ckd-related systemic inflammation. J Am Soc Nephrol. 2017;28(1):76–83. 10.1681/ASN.2015111285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhong J, Yang HC, Yermalitsky V, et al. Kidney injury-mediated disruption of intestinal lymphatics involves dicarbonyl-modified lipoproteins. Kidney Int. 2021;100(3):585–96. 10.1016/j.kint.2021.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gobert AP, Boutaud O, Asim M, et al. Dicarbonyl electrophiles mediate inflammation-induced gastrointestinal carcinogenesis. Gastroenterology. 2021;160(4):1256–1268.e9. 10.1053/j.gastro.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferguson JF, Aden LA, Barbaro NR, et al. High dietary salt-induced dendritic cell activation underlies microbial dysbiosis-associated hypertension. JCI Insight. 2019. 10.1172/jci.insight.126241. [DOI] [PMC free article] [PubMed] [Google Scholar]