

Abstract
The three small-conductance calcium-activated KCa2 channels and the related intermediate-conductance KCa3.1 channel are voltage-independent K+ channels that mediate calcium-induced membrane hyperpolarization. When intracellular calcium increases in the channel vicinity it calcifies the flexible N-lobe of the channel-bound calmodulin, which then “swings over” to the S4-S5 linker and opens the channel. KCa2 and KCa3.1 channels are highly druggable and offer multiple binding sites for venom peptides and small molecule blockers as well as for positive and negative gating modulators. In this review we will first briefly summarize the physiological role of KCa channels and then discuss the pharmacophores and the mechanism of action of the most commonly used peptidic and small molecule KCa2 and KCa3.1 modulators. Finally, we will describe the progress that has been made in advancing KCa3.1 blockers, and KCa2.2 negative and positive gating modulators towards the clinic for neurological and cardiovascular diseases, and discuss the remaining challenges.
Keywords: Calcium-activated potassium channel, KCa2.2, KCa2.3, KCa3.1, gating modulation
INTRODUCTION
Potassium (K+) channels are critically involved in regulating fundamental physiological processes such as cellular volume, membrane potential, hormone secretion, calcium signaling, and action potential firing (1). To allow for fine tuning of these processes the human genome contains 78 K+ channels. According to the IUPHAR (International Union of Basic and Clinical Pharmacology) Guide to Pharmacology (2) these channels have been grouped based on sequence similarity and the number of their transmembrane domains (TM), which can be 2, 4, 6 or 7. The small- and intermediate-conductance Ca2+-activated K+ channels belong to the 6TM family and thus resemble the voltage-gated K+ channels, with a 4TM voltage sensor domain (VSD) and a 2TM pore domain (3). Like KV channels, functional KCa channels are tetramers and, at least in expression systems, the different family members are able to form heteromultimers in addition to the more common homotetramers (4). However, unlike KV channels, KCa channels have fewer positively charged residues in the S4 segment of their VSD, and are therefore unresponsive to changes in transmembrane voltage and have essentially linear current voltage-relationships at physiological ion gradients (3; 5). Before their cloning, KCa2 and KCa3.1 channels were referred to as small-conductance (SK) or intermediate-conductance (IK) Ca2+-activated K+ channels based on their unitary conductance of ~10 pS or ~40 pS in symmetrical K+ solutions to differentiate them from the large-conductance BK channel (~200 pS), which is now called KCa1.1 (3). This phenomenological nomenclature is still widely used together with the IUPHAR and HUGO nomenclatures, which is why we wanted to introduce it upfront to avoid confusion.
The three KCa2 channels, KCa2.1 (SK1, KCNN1), KCa2.2 (SK2, KCNN2) and KCa2.3 (SK3, KCNN3) were cloned in 1996 by the group of John Adelman and are highly homologous across their transmembrane cores (80–90%) but diverge in sequence and length in their N- and C-termini (5). Two years later, the same group demonstrated that the calcium-sensor of the SK channels is calmodulin, which is constitutively bound to the calmodulin binding domain (CaM-BD) in the C-terminus and thus functions as a β-subunit that endows these channels with Ca2+ sensitivity (6). The KCa3 family only contains a single member, KCa3.1 (IK, SK4, KCNN4), which was cloned in 1997 and delegated to its own subfamily because it only is ~40% identical to the three KCa2 channels (7; 8). Similar to the KCa2 channels, the Ca2+-dependent activation of KCa3.1 is mediated by calmodulin (9). The reported EC50 values for Ca2+ range from 100 to 400 nM for KCa3.1 and from 300 to 750 nM for the KCa2 channels. This submicromolar Ca2+-sensitivity together with their lack of voltage-dependence enables KCa channels to be open at relatively negative membrane potentials when intracellular Ca2+ is raised in their immediate vicinity and to hyperpolarize towards the K+ equilibrium potential of −90 mV. KCa channels are accordingly expressed in cells that need to be able to prevent premature action potential generation or sustain Ca2+ influx through inward-rectifier Ca2+ channels.
EXPRESSION AND PHYSIOLOGICAL FUNCTION OF KCa2 CHANNELS
KCa2 channels are widely expressed on neurons of the central and peripheral nervous system (10). KCa2 currents underlie the so-called medium afterhyperpolarization (mAHP), the second phase of neuronal hyperpolarization following an action potential, and thus regulate intrinsic excitability and spike firing rates. KCa2 currents are activated by calcium entering neurons via CaV channels activated during the action potential, but may also functionally couple to post-synaptic calcium sources such as NMDA receptors and nicotinic acetylcholine receptors as well as calcium released from intracellular ryanodine or IP3 receptors (10).
The mAHP is absent in KCa2.2 knock-out mice, but not mice lacking KCa2.1 or KCa2.3, revealing that KCa2.2 is the dominant subtype responsible for this current (11). Reversely, 10-fold overexpression of KCa2.2 increases the mAHP and dampens excitatory postsynaptic potentials (EPSP) making postsynaptic neurons less likely to fire, resulting in mice with impairments in hippocampal learning and memory (12). KCa2.3 channels are strongly expressed on dopaminergic substantia nigra neurons, where they maintain regular firing and pace-making control (13) and serotonergic raphe neurons, where they regulate burst firing (14). Pharmacological modulation confirms the role KCa2 channels in neurons (Figure 1). The selective KCa2 blocker apamin increased intrinsic excitability and firing frequency, while KCa2 activators enhance the magnitude of the mAHP and slow down firing rates (10; 15). These altered responses have consequences for long-term potentiation (LTP), the increase in synaptic strength following high-frequency stimulation, which underlies some forms of learning. Inhibiting KCa2 activity with apamin improves learning and memory encoding in rodents (16; 17), while increasing KCa2 activity with an activator impairs associative learning (18). In the peripheral nervous system, KCa2 channels are expressed in DRG primary sensory neurons, where they play a role in nociception (19; 20).
Figure 1.
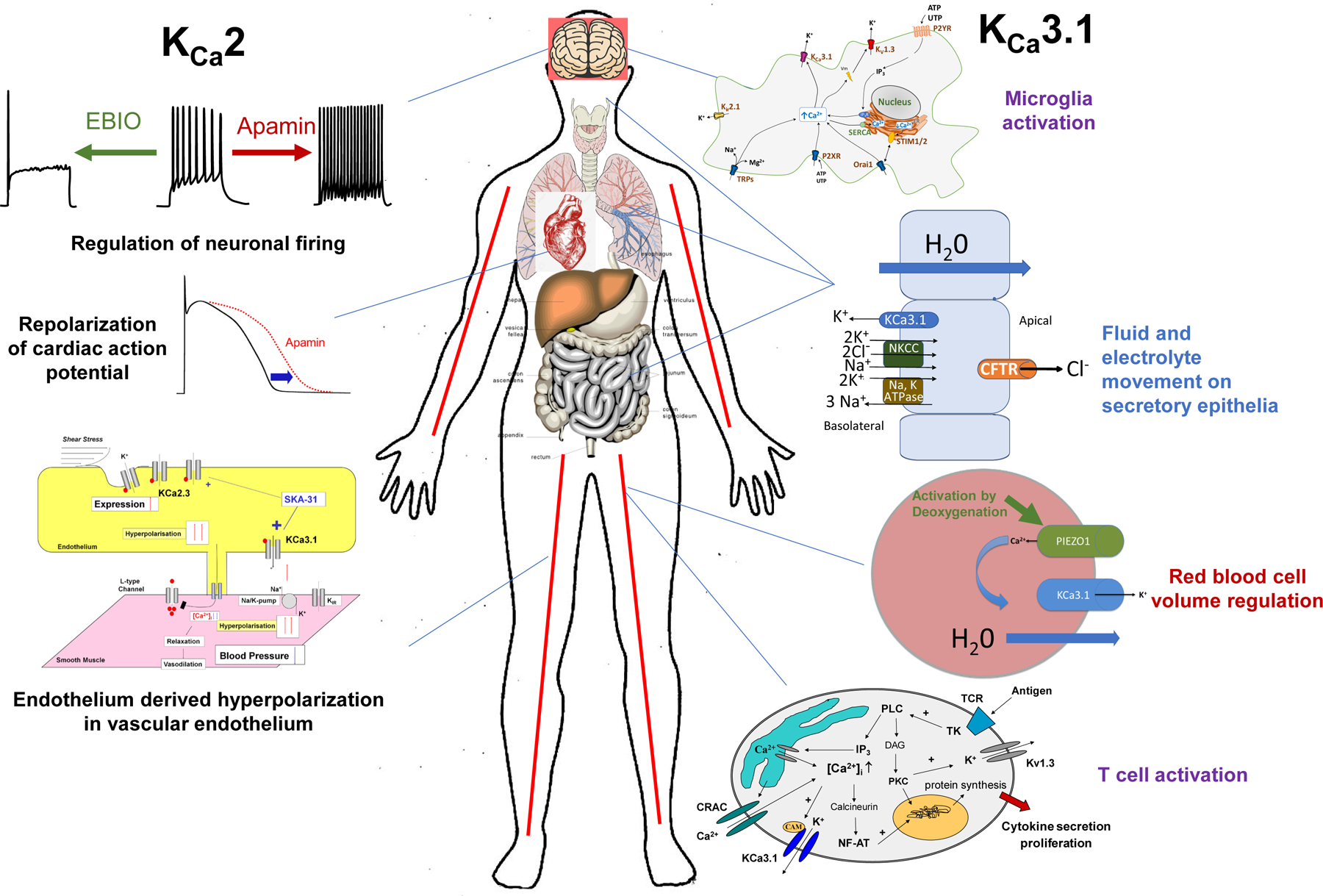
Physiological role of KCa2 and KCa3.1 channels.
While all three KCa2 channels are found in the nervous system, KCa2.3 is the only member found in the endothelial cells of blood vessels, where, together with KCa3.1 (21), KCa2.3 underlies the endothelium derived hyperpolarization (EDH), a phenomenon that, together with prostacyclin and nitric oxide, controls vessel tone (22). Activation of KCa currents on the endothelium leads to hyperpolarization and relaxation of the underlying smooth muscle cells, ultimately reducing blood pressure (Figure 1). KCa2 channels are further expressed in the heart (Figure 1) and have been shown to play an important a role in the repolarization of the cardiac action potential, specifically in atrial myocyte and atrioventricular nodes (23; 24). Multiple single nucleotide polymorphisms (SNPs) in KCa2.3 have been found to be associated with lone atrial fibrillation (AF) (25; 26). Increasing KCa2.3 activity significantly shortens cardiac action potentials resulting in increased susceptibility to AF (27). A KCa2.3 splice variant is expressed in liver hepatocytes, where KCa2.3 plays a role in cellular responses to metabolic stress (28).
EXPRESSION AND PHYSIOLOGICAL FUNCTION OF KCa3.1 CHANNELS
A calcium-activated K+ efflux, which was later demonstrated to be mediated by KCa3.1 and to contribute to volume regulation and hydration state (29; 30), was fist described in erythrocytes in 1958 (31), which is why KCa3.1 is also called the Gárdos channel after the scientist who first described the phenomenon. Heterozygous gain of function mutations in KCa3.1 are responsible for erythrocyte dehydration in a subset of patients with hereditary xerocytosis, a disease characterized by hemolytic anemia associated with erythrocyte dehydration (32; 33). In addition to red blood cells (Figure 1), KCa3.1 is also widely expressed in cells of the immune system such as T cells (34), B cell (35), mast cells (36), macrophages (37), and microglia (38). The primary role of KCa3.1 in the immune cells is to hyperpolarize the cell membrane and create the driving force for the calcium entry that is necessary for activation, proliferation and cytokine production (39). Most, if not all, KCa3.1 expression in the brain seems to be localized to microglia, which upregulate KCa3.1 after activation in vitro and in vivo (40; 41). Although two studies recently reported that KCa3.1 may also be expressed in neurons and contribute to the slow afterhyperpolarization (42; 43), another study presented data that KCa3.1 does not contribute to the slow AHP (44). A potential role for KCa3.1 in neurons therefore currently remains uncertain. The phenotype of the KCa3.1−/− mouse reinforces the channel’s role in the immune system. T cells from KCa3.1−/− mice show reduced T-cell receptor mediated calcium influx and inflammatory cytokine production and the mice develop less severe colitis (45) and arthritis (46). KCa3.1−/− mice further display blunted IgE-mediated anaphylactic reactions and reduced infarction and neuroinflammation after ischemic stroke (40).
KCa3.1 deletion in mice also reduces the EDH response, raises mean arterial blood pressure by 7–9 mm Hg (47) and causes subtle erythrocyte macrocytosis and progressive splenomegaly (48). In secretory epithelia of the lung and gastrointestinal tract KCa3.1 works in concert with the Na-K-2Cl cotransporter to facilitate chloride and fluid secretion (49). While KCa3.1 channels are not expressed in normal vascular smooth muscle cells, expression is turned on in dedifferentiated vascular smooth muscle cells where KCa3.1 activity promotes proliferation and migration, while KCa3.1 inhibition reduces atherosclerosis in mice (37) and restenosis in rats and pigs (50; 51). Likewise, KCa3.1 drives proliferation and migration in many cancers such as glioblastoma (52; 53), breast or prostate cancer (54; 55), which is why KCa3.1 blockers have been proposed to treat diseases that have a proliferative component.
CHANNEL STRUCTURE
The KCa channel field recently obtained some tremendous structural insights when the group of Roderick MacKinnon solved the full-length cryo-EM structures of KCa3.1 in the absence and presence of calcium (56). Unlike KV1.2, KCa3.1 is non-domain swapped and the structure showed four CaMs per channel tetramer, with the CaM C-lobe of each CaM tightly bound to the CaM-BD of each subunit. The CaM N-lobes were only visible in the two open, Ca2+-bound states and poorly resolved in the closed, Ca2+-free structure suggesting that they are flexible in the absence of Ca2+. When Ca2+ binds to the N-lobe, it “swings over” to the S4-S5 linker of another subunit and pulls part of the S4-S5 linker, namely the S45A helix downward, thus expanding the S6 helices and opening the pore (56). This structure solved the long-standing conundrum of the KCa channel gating symmetry. In 2001, Schumacher et al. had crystallized the KCa2.2 channel C-terminal CaM-BD in complex with CaM. This 1.6 Å resolution structure had shown an elongated, anti-parallel dimer of two KCa2.2 C-terminal fragments with CaM tightly bound with its C-lobe to two alpha helices connected by a turn from the same channel subunit (57). The CaM N-lobe was “grabbing” the free end of CaM-BD from the other subunit in the dimer suggesting that CaM-BD dimerization might gate KCa2 channels (57). However, the 2-fold symmetry suggested by this dimer-of-dimers was difficult to reconcile with the 4-fold symmetry of the pore (58) and the new full-length structure now makes it clear that KCa3.1 in fact gates with a more common 4-fold symmetry.
Interestingly, the dimeric crystal has proven very resilient in that it was repeatedly observed in subsequent crystallographic studies addressing the mechanism of action of small molecule KCa channel activators and of PIP2 on KCa2.2 channel function (59–62). Several KCa channel activators were shown to bind in the interface between the CaM N-lobe and the CaM-BD in this dimeric crystal and the interaction was even confirmed by solution state NMR experiments (62). However, the full-length KCa3.1 structure demonstrated that the dimeric crystal is an artefact and suggested that the existing ideas about the binding site of KCa2 and KCa3.1 activators need to be revised (56). We therefore here show a Rosetta refined (63) KCa3.1 model, which is based on open state-1 of the KCa3.1 cryo-EM structure, and a KCa2.2 homology model to illustrate and discuss the binding sites of the commonly used pharmacological tool compounds in context of this structure.
KCa2 AND KCa3.1 CHANNEL PHARMACOLOGY
In 1982 the neurotoxic peptide apamin was shown to inhibit some Ca2+-activated K+ channels (64) and KCa channels were therefore typically differentiated into “apamin-sensitive” and “apamin-insensitive” in the 1980s and 1990s. Following their cloning, KCa channel pharmacology developed relatively rapidly and the field now has quite a range of peptidic and small-molecule inhibitors as well as positive and negative gating modulators available. Since KCa channel pharmacology has been reviewed by us and others a decade ago in great detail (65–67), we here concentrate on the most commonly used modulators and their mechanisms of action.
Venom peptides
The most widely used KCa2 channel blocker is the 18-amino acid honey bee venom peptide apamin (64), which is remarkably selective for KCa2 channels. Apamin is most potent on KCa2.2 (IC50 ~200 pM) and blocks KCa2.1 and KCa2.3 with 10–50-fold lower affinity (5; 10), while it has no effect on KCa3.1. In vitro application of apamin to neurons or brain slices has been instrumental for demonstrating the crucial role of KCa2 channels in neuronal excitability (10). While low concentrations of apamin improve cognitive performance in rodents, higher concentrations induce seizures (67; 68). Apamin was initially assumed to be a simple pore blocker but was later found to inhibit KCa2 channels through an allosteric mechanism involving an outer pore histidine (69) and residues in the S3-S4 extracellular loop (70), a binding configuration that is recapitulated in our docking pose in the KCa2.2 homology model (Figure 2). The larger scorpion toxins scyllatoxin, which is also called leiurotoxin I (71; 72), and tamapin (73) have roughly the same potency as apamin and show comparable preference for KCa2.2. A less potent, but highly KCa2.2 selective blocker, is the scyllatoxin derivative Lei-Dab7 (74), in which one residue is replaced by the unnatural amino acid diaminobutonoic acid (Dab). While Lei-Dab7 thus constitutes an even more selective tool to block KCa2.2 channels in physiological studies (75), there currently are no natural toxins or analogs that selectively inhibit KCa2.1 or KCa2.3.
Figure 2.

Rosetta KCa2.2 homology model based on the KCa3.1 cryo-EM structure (open state 1, pdb: 6cnn). The longer N- and C-terminus of KCa2.2 was not modeled. For clarity only two of the four channel subunits are shown (dark gray). Calmodulin is shown in yellow. Potassium ions in the selectivity filter are colored dark purple. The bee venom apamin with its N-atoms colored dark blue and several small molecule modulators are docked where they have been shown to bind by mutagenesis: Apamin in the outer pore, NS8593 in the inner pore (dark green), CM-TMPF (sky blue) in the inner vestibule, and SKA-111 in the interface between the CaM N-lobe and the S45A helix in the S4-S5 linker. The chemical structures of other KCa2 blockers, negative gating modulators and activators are colored according to where they have either been shown to bind by mutagenesis or are suspected to bind.
Potencies (IC50s for blockers and negative gating modulators; EC50s for activators): Apamin 60–400 pM, d-tubocurarine 5 μM, dequalinium 200 nM, UCL1684 200 pM, UCL1884 110 pM, BBP 400 nM, NS8593 600 nM, AP14145 1 μM, (–)-B-TMPF 31 nM for KCa2.1 and 1 μM for KCa2.2, CM-TMPF 24 nM for KCa2.1 and 290 nM for KCa2.2, SKA-111 8 μM, RA-2 ~100 nM, SKA-31 2 μM, NS309 620 nM, CyPPA 14 μM, NS13001 2 μM.
The best known peptidic blocker of KCa3.1 is the 37-amino acid scorpion toxin charybdotoxin, which anchors itself in the outer vestibule of KCa3.1 by two salt-bridges while inserting its central lysine residue into the selectivity filter (76) as shown in Figure 3. However, charybdotoxin never was an ideal KCa3.1 blocker because it also inhibits KCa1.1 (BK) and the voltage-gated Kv1.3 channels, both cross-reactivities, which initially caused confusion concerning the role of KCa3.1 in the cardiovascular system and in T cells. Another, somewhat more potent scorpion toxin is maurotoxin (77), which unfortunately cross-reacts to KV1.2. While these toxins are sometimes used in vitro to obtain a complete biophysical and pharmacological signature of KCa3.1, they have not been used as KCa3.1 blockers in vivo.
Figure 3.
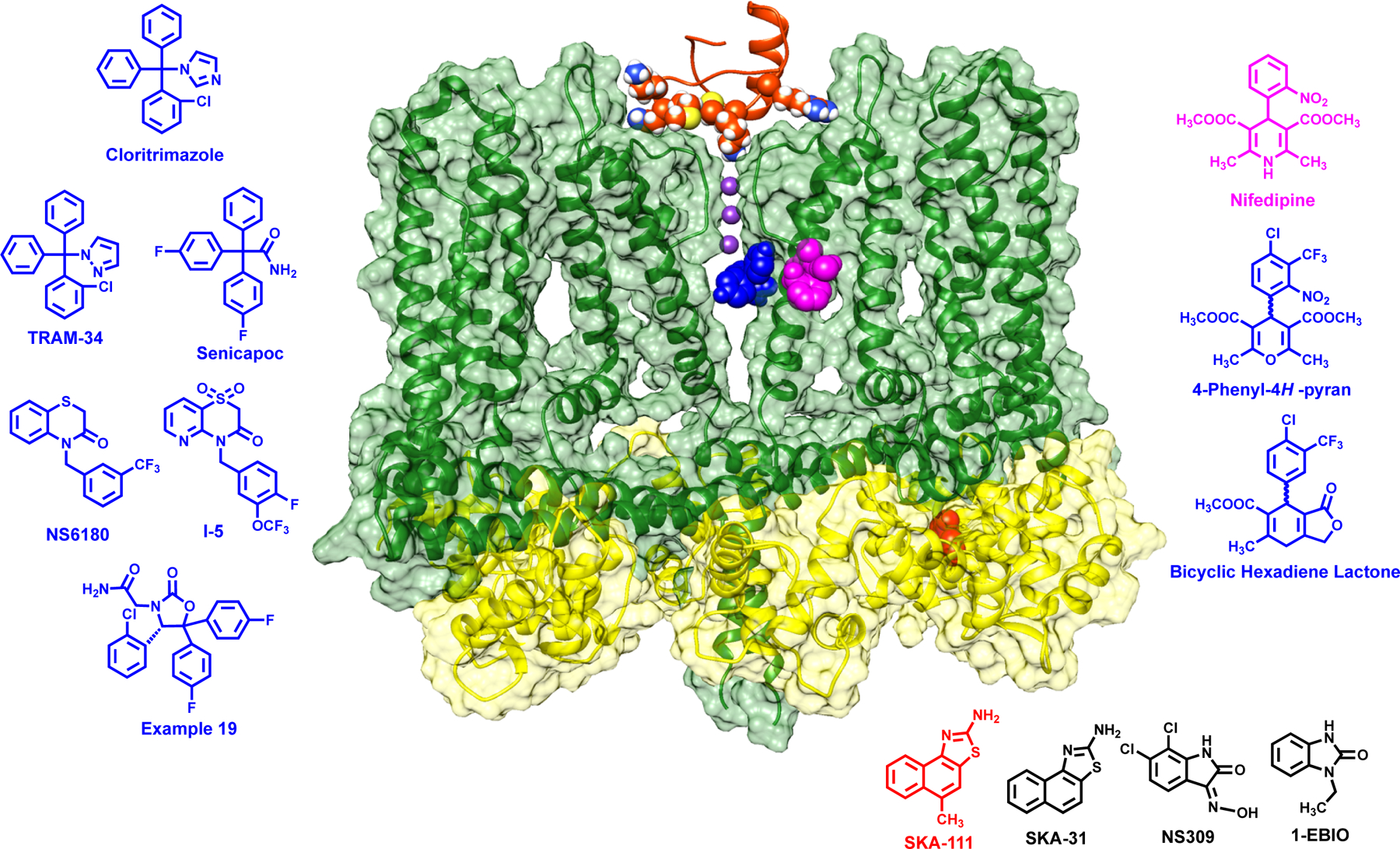
Rosetta refined model of the KCa3.1 cryo-EM structure (open state 1, pdb: 6cnn). For clarity only two of the four channel subunits are shown (dark green). Calmodulin is shown in yellow. Potassium ions in the selectivity filter are colored dark purple. The scorpion toxin charybdotoxin (ChTX) is shown docked into the outer vestibule. Various small molecule modulators are docked were they have been shown to bind by mutagenesis: Senicapoc as a representative triaryl-methane in the inner pore (blue), nifedipine in the fenestration region (pink), and SKA-111 in the interface between the CaM N-lobe and the S45A helix in the S4-S5 linker. The chemical structures of other KCa3.1 blockers and activators are colored according to where they have either been shown to bind by mutagenesis or are suspected to bind.
Potencies (IC50s for blockers; EC50s for activators): ChTX 2–28 nM, clotrimazole 70–250 nM, TRAM-34 10–25 nM, senicapoc 11 nM, NS6180 11 nM, nifedipine 0.8–4 μM, 4-phenyl-4H-pyran 8 nM, SKA-111 150 nM, SKA-31 250 nM, NS309 10–30 nM, 1-EBIO 24–80 μM.
Small molecule KCa2 Channel Blockers
The key structural feature of selective KCa2 blockers is that they carry one or two positive centers, either permanently charged or strongly basic nitrogens, or in some cases acquire a charge by complexing divalent cations. All known KCa2 blockers work from the extracellular side and competitively displace radioactively labeled apamin in binding assays. The positive charges are reminiscences of the essential arginines of apamin (dark blue in the apamin structure in Figure 2). Intriguingly, this requirement for positive charges in the pharmacophore parallels classical anti-cholinergic drugs, many of which like d-tubocurarine and dequalinium (Figure 2), also block KCa2 channel (78; 79). Dequalinium was used as a starting point for several structure-activity-relationship studies focusing on both the nature of the permanently charged ring and the distance between the positive charges (80; 81), efforts which ultimately lead to the discovery of the high-affinity bis-quinolinium cyclophane KCa2 blockers UCL1684 and UCL1848 (Figure 2), which are as potent as apamin in blocking KCa2.2 (82; 83). Interestingly, the commonly used permanently charged derivative of the GABAA receptor antagonist bicucculine, bicucculine methiodide, also blocks KCa2 channels as potently as GABAA receptors (84).
These highly selective but permanently charged KCa2 blockers have been mostly used in academia, while there have only been modest activities in the pharmaceutical industry, probably due to the expected complications of permanently charged molecules, such as low permeability across biological membranes. Icagen published a patent on bis-benzimidazoles (USOO7482373B2), which block KCa2 channels and displace apamin. These molecules are not permanently charged, but form charged complexes with divalent cations, which probably constitute their active form. Bristol Myers Squibb published a series of 2-aminothiazoles, which also chelate divalent cations (85). Recently, the KCa2 channel blocking effect of 2,6-bis(2-benzimidazolyl)pyridine (BBP) (Figure 2), a molecule belonging to this class (86), has been shown to depend on H491 in the extracellular S5-P linker, the same histidine which is also a determinant for apamin binding (69). Despite these quite intriguing approaches, no KCa2 blockers have yet entered clinical development. However, due to their potency and good selectivity several of these molecules have been extremely valuable tools for elucidating the role of KCa2 channels in cardiac arrhythmias (87; 88) or the endothelium derived hyperpolarization response (89).
Small molecule KCa3.1 Blockers and their Preclinical and Clinical Applications
Apart from low affinity inhibitors like the β-blocker cetiedil (90), a compound that been reported to affect erythrocyte K+ fluxes in 1981 (90) and therefore inspired some early medicinal chemistry (67; 91), two compound classes spurred a real interest in finding therapeutically useful small molecule KCa3.1 inhibitors in the late 1990s: Dihydropyridines (92; 93) and triaryl-methanes (94). Both pharmacophores had previously been successfully developed as L-type Ca2+ channel antagonists for hypertension and as P450 inhibitors for topical antifungals. Following the cloning of KCa3.1, scientist at Bayer worked on dihydropyridines (95; 96), a lead optimization resulting in a series of very potent and selective phenyl-pyrans and cyclohexadienes (Figure 3) that showed in vivo efficacy in animal models of traumatic brain injury (97), but never entered clinical development for undisclosed reasons. Following up on work performed by the group of Carlo Brugnara at Harvard showing that clotrimazole reduced erythrocyte dehydration and exerted anti-sickling effects in transgenic mice and in patients with sickle cell disease (98; 99), Icagen pursued the triaryl-methanes and developed the clotrimazole derivative senicapoc. In parallel, one of us used clotrimazole as a template for the design of TRAM-34 (100). In both TRAM-34 and senicapoc (Figure 3) clotrimazole’s toxic effect on the human P450 system have been avoided by substitution of the imidazole ring with either a pyrazole ring or an amide group. While TRAM-34 was not suitable for development, it has become a widely used academic tool compound based on its selectivity and its acceptable pharmacokinetic properties when administered intraperitoneally. For example, TRAM-34 has been utilized to validate KCa3.1 as a potential target for vascular restenosis (50), atherosclerosis (37), asthma (101), allograft vasculopathy (102), inflammatory bowel disease (45), ischemic stroke (40), as well as renal (103; 104) and cardiac fibrosis (105). In many of these disease models the pathophysiological relevance of KCa3.1 was confirmed by parallel experiments in KCa3.1−/− mice.
Senicapoc, which is orally available and has a long 12-day half-live in humans, entered clinical trials for sickle cell anemia but despite showing good effects on several haematological parameters, it unfortunately failed to meet the predefined primary endpoint, which was reduction in the number of painful crisis, in phase-III clinical studies (106). Based on senicapoc’s efficacy in an asthma model in sheep (107), Icagen subsequently tested senicapoc in two small Phase-II trials for asthma (108) and demonstrated encouraging results in allergic asthma. However, senicapoc did not improve lung function in exercise induced asthma. Following these failures Icagen was purchased by Pfizer and senicapoc deposited in the 2012/13 NIH National Center for Advancing Translational Research (NCAT) library as PF-05416266 making it theoretically available for investigator initiated clinical trials. Senicapoc is currently being “repurposed” by the Pfizer spin-out SpringWorks Therapeutics for the hemolytic anemia disease hereditary xerocytosis, a rare condition caused by gain of function mutations in KCa3.1 (109). Another “repurposing” Phase-IIa clinical trial with senicapoc will be conducted by the Alzheimer’s Disease Center at the University of California, Davis. This trial, which is anticipated to start in Fall of 2019, is based on findings that the expression of KCa3.1 is increased on microglia in brains from patients with Alzheimer’s disease (AD) and that KCa3.1 inhibition with senicapoc reduces inflammation and amyloid-β deposition in mouse models of AD (110).
More recently, several new classes of KCa3.1 inhibitors were reported by the pharmaceutical industry. Using a so-called “scaffold hopping” approach (e.g. tetrazole derivatives, US9556132B2) and high-throughput thallium-flux (111) NeuroSearch, a biopharmaceutical company in Denmark, identified a completely new series of benzothiazinone-based KCa3.1 blockers in 2013. One of the exemplary compounds, NS6180 (Figure 3), showed efficacy in an animal model of inflammatory bowel disease despite low in vivo exposure (112). Boehringer Ingelheim pursued the closely related fused thiazine-3-ones (US 2015/0232484) and Roche published a patent on 3,4-disubstituted oxazolidinones (WO 2014/067861), which constitute an interesting variation on the triaryl-methane motive (Fig. 3). Nothing specifically was reported on therapeutic indications by the two companies, but a paper co-authored by Boehringer Ingelheim scientists focused on the role of KCa3.1 in the process of multinucleation of macrophages and osteoclasts (113) suggesting a possible focus on chronic inflammation or bone-diseases.
Atomistic Mechanism of Action of KCa3.1 Blockers
Overall, the various compound classes (Figure 3), which have been reported as potent and selective KCa3.1 inhibitors during the last 20 years, are quite remarkable for their chemical diversity. However, on closer inspection a unifying characteristic is the absence of acidic or basic moieties and the presence of two or usually three substituted aryl groups making most of these compounds quite “greasy”, insoluble and prone to suboptimal pharmaceutical properties, such as high plasma protein binding. Remarkably, the majority of the KCa3.1 blockers shown in Figure 3 are binding to the same site in the inner pore of KCa3.1, just below the selectivity filter. While this canonical site is often touted as not suitable for obtaining subtype selective inhibitors, this seems to be possible for KCa3.1, because it is “alone” in its family. Indeed, none of the currently known KCa3.1 blockers, cross-react to KCa2 channels and the compounds typically also exhibit between 200–1000-fold selectivity over other ion channels.
The triaryl-methane type KCa3.1 blockers clotrimzole, TRAM-34 and senicapoc interact with threonine 250 in the pore loop and valine 275 in S6 as demonstrated by the fact that mutations of these residues completely abolish the sensitivity of KCa3.1 to triaryl-methanes (114). Based on a study using the Rosetta molecular modeling suite (115), TRAM-34 anchors itself through hydrophobic interactions with the V275 residues from all four subunits and forms a hydrogen bond to the T250 side chain from one subunit with its pyrazole nitrogen and thus blocks ion conduction by filling the site that would normally be occupied by a K+ ion before it enters the selectivity filter. Senicapoc is assuming a similar binding pose (Figure 3) but instead of acting as a hydrogen-bond acceptor like TRAM-34, its amide group functions as a hydrogen-bond donor and interacts with T250 side chains from two subunits (115). Interestingly, the same two mutations that “knock off” triaryl-methane binding, also drastically reduce the affinity of the benzothiazinone NS6180 (112). However, in contrast to TRAM-34 and senicapoc, which are positioned directly under the selectivity filter and interact with all four subunits, NS6180 interacts with the T250 and V275 side chains from only two adjacent subunits (115). However, although NS6180 is “sitting” differently is still overlaps with the pore lumen potassium site (116) and thus seems to act by preventing ion permeation.
In contrast to the triaryl-methanes, the binding site of the dihydropyridine nifedipine has been localized to the fenestration region of KCa3.1 (Figure 3), where it binds between the side-chains of T212 in S5 and V272 in S6 from adjacent subunits and has been suggested to stabilize the channel in a non-conducting conformation without directly occluding the pore (115). While this fenestration binding site constitutes a very attractive alternative to the pore site for future design efforts directed towards the identification of KCa3.1 inhibitors with improved pharmaceutical properties, a completely unexpected and somewhat “shocking” observation from our group was that the nifedipine isosteric 4-phenyl-pyran (Figure 3), which had been initially described by Bayer (95) and which we resynthesized (115), is binding in the inner pore at the triaryl-methane site and not in the fenestration like its template nifedipine. As explained in detail elsewhere, this finding is consistent with the published structure-activity-relationship of the phenyl-pyrans (95) and the related carba-analogous cyclohexadienes (96). Medicinal chemists generally assume when making isosteric replacements to improve potency and selectivity that the template and the derivatives bind to the same site, which is clearly not the case here and a caution against making assumptions that are not experimentally tested.
Negative Gating Modulators
Negative gating modulation as applied to KCa2 channels means an inhibitor that shifts the calcium-activation curve towards higher Ca2+ concentrations (in contrast to the left-shifting by positive gating modulators), thereby reducing the apparent Ca2+-sensitivity of the channel. The first molecule in this functional class was the aminobenzimidazolone NS8593 (117) and its analogues (118) which show high selectivity for KCa2 channels compared to KCa3.1. In contrast to most KCa2 blockers, this class of molecules is uncharged at physiological pH and therefore more likely to pass biological barriers. NS8593 does not displace radiolabeled apamin and its activity is not reduced by mutations of the extracellular amino acid residues mediating sensitivity to apamin and the small molecule KCa2 blockers (see above). Instead, the effect of NS8593 was shown through site directed mutagenesis to depend on the same amino acid positions in the pore region of KCa2 (Figure 2) that mediate sensitivity of KCa3.1 to TRAM-34 (119) and introduction of just two mutations into KCa3.1 could render this normally insensitive channel highly sensitive to NS8593 (119). A closer inspection of the binding pose, however, suggests, that unlike TRAM-34, NS8593 is not completely obstructing the permeation pathway. The fact that gating modulation is possible at this position was hypothesized to be a pharmacological reflection of the previously suggested deep pore gating in KCa2 channels (120). The basic characteristics of negative modulators including their pore binding site have recently been confirmed by using the drug candidate AP14145 (Figure 2) from Acesion Pharma, which belongs to the same general class of molecules as NS8593 (121). Interestingly, scientists from Bristol Myers Squibb published another series of molecules, 4-(aminomethylaryl)-pyrrazolopyrimidines, which inhibit KCa2 mediated Tl+ -fluxes at a site not involving the apamin site (122). Although, not rigorously shown in the paper, these compounds may also act via a negative gating modulatory mechanism. While the compounds described so far do not differentiate among the three KCa2 subtypes, subtype selective negative gating modulation was demonstrated for the triazolopyrimidine (−)-B-TPMF (Figure 2), which preferentially inhibited KCa2.1 over the other KCa2 members and KCa3.1, by interaction with Ser293 in KCa2.1, a position not previously identified for modulators of KCa2 (123). Notably, as described later, this site on KCa2.1 can also give rise to positive modulation. Negative gating modulation has recently also been shown to account for the effects of certain dibenzoates, such as RA2 (1,3-phenylenebis(methylene)bis(3-fluoro-4-hydroxybenzoate), which inhibit both KCa2.x and KCa3.1 channels with similar potency (124), demonstrating that this mode of action is also possible for KCa3.1. Although its binding site has never been mapped, the inner vestibule of both KCa3.1 and the KCa2 channels is large enough to accommodate the molecule.
Based on proof-of-concept animal studies demonstrating that NS8593 or AP14145 can terminate atrial fibrillation in rats (125; 126) or even large animals such as pigs (127), negative gating modulators have successfully progressed into clinical development for atrial arrhythmia. According to a press release from Acesion Pharma, a putative analogue of NS8593/AP14145 called AP30663 has recently passed Phase-I clinical trials in human volunteers.
Positive Gating Modulators
Most KCa2 and KCa3.1 activators are “clean” positive gating modulators meaning that they shift the calcium-activation curve concentration-dependently towards lower intracellular Ca2+-concentrations, thereby increasing the apparent Ca2+ affinity, but are unable to activate the channels at “0” intracellular Ca2+. However, there seem to be exceptions to this simple mechanism in that the existence to “true” activators (123) and potentially superagonists (128) has been suggested. The prototype activators of KCa3.1 and KCa2 channels are the benzimidazolone 1-EBIO (129) and its more potent derivative dichloro-EBIO (130), which have both played a significant role as pharmacological ex vivo tool compounds in brain slices, endothelia and epithelia, or smooth muscle preparations. However, several drugs that have been on the market for decades, such as the muscle relaxant chlorzoxazone and the ALS drug riluzole are also quite effective KCa activators (67; 131), which may well be their major therapeutic action. Dedicated search for more potent and selective activators led to NS309 (132), one of the most potent “pan”- KCa3.1/KCa2 activators and an important mechanistic tool compound, but not suited for in vivo studies, due to poor pharmacokinetic properties. With the aim of making more selective and potent riluzole-like compounds that could potentially be used in vivo, Sankaranarayanan et al. (133) identified a series of benzothiazoles including SKA-31 and SKA-121, which has improved selectivity for KCa3.1 and which has been used to demonstrate that selective KCa3.1 activation can lower blood pressure in mice (134).
Also pursuing subtype selectivity, NeuroSearch scientists discovered cyclohexyl-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-pyrimidin-4-yl]-amine (CyPPA) and the more potent analogue (4-chloro-phenyl)-[2-(3,5-dimethyl-pyrazol-1-yl)-9-methyl-9H-purin-6-yl]-amine) (NS13001), which activates KCa2.3 and KCa2.2 while being inactive on KCa2.1 and KCa3.1 (75; 135). A different selectivity profile was exemplified by CM-TPMF, which preferentially activates KCa2.1 (123) and like the negative gating modulator (−)-B-TPMF depends on a serine residue in S5 for its activity (Figure 2). These subtype selective compounds formed the basis for a collaboration between Saniona, a company continuing the research assets of NeuroSearch, and Ataxion (now Cadent Therapeutics), which finally resulted in selection of CAD-1883 for preclinical development for cerebellar dysfunction. According to the Cadent Therapeutics homepage, CAD-1883 is now in phase-II for essential tremor and for spinocerebellar ataxia. The idea of using KCa2 activators for these indications is based on the observations that genetic silencing of KCa2 channels in deep cerebellar neurons induces ataxia in mice (136), while treatment of mice with spinocerebellar ataxia type-2 (SCA2) with the KCa2 activator NS13001 alleviates motor symptoms and prevents neurodegeneration of Purkinje cells (75).
Atomistic Mechanism of KCa Channel Positive Gating Modulators
Mutational studies performed by Pedarzani et al. suggested in 2001 that the binding site of the benzimodazolone-type KCa activators is probably located in the C-terminus, close to or within the CaM-BD, since “swapping” the C-terminus of KCa3.1 into KCa2.2 made this channel as 1-EBIO sensitive as KCa3.1 (137). So, when 1-EBIO, NS309 and riluzole were later reported to bind at the interface between the CaM N-lobe and the KCa2.2 CaM-BD after being soaked into the above described C-terminal dimeric crystal using x-ray crystallography or in solution state NMR (59–62), this interface was widely assumed to be the binding site of this type of KCa activators in both KCa2 and KCa3.1 channels (62; 138). However, the recently published full-length cryo-EM structure of KCa3.1 demonstrated that the analogous segment of the KCa3.1 C-terminus, which is designated the C-terminal HC helix, actually forms a coiled coil at the center of the channel (56) and therefore is unlikely to constitute the binding site. In their study the MacKinnon group also proposed that the “real” binding pocket of 1-EBIO is located in the interface between the S45A helix and the CaM N-lobe, in which 1-EBIO is hypothesized to contact L185 in the S45A linker (56) instead of L480 in the C-terminal crystal complex (59), but did not experimentally test this very plausible alternative binding site hypothesis. Our own group recently picked up on this postulate and confirmed through mutagenesis that at least the SKA-type KCa channel activators as exemplified by SKA-111 (5-methylnaphtho[1,2-d]thiazol-2-amine) are binding in the interface between the CaM N-lobe and the S45A helix (139). In this interface pocket Rosetta modeling shows that SKA-111 makes van der Waals contacts with S181 and L185 in the S45A helix of KCa3.1 but interacts with the same CaM N-lobe residues (M51, E54, and M71) that were previously shown to be involved in binding of the aminothiazole riluzole in the C-terminal crystal dimer (62).
We here show SKA-111 docked into the S45A helix/CaM N-lobe interface of both KCa2.2 (Figure 2) and KCa3.1 (Figure 3), fully recognizing that the KCa2.2 binding pose is currently not supported by experimental data. Based on the high sequence similarity in the S45A helix between KCa3.1 and the three KCa2 channels, we would expect the CaM mediated gating and the putative stabilization of the interaction between the CaM N-lobe and the S45A helix by benzothiazole-type KCa activators to be similar, even if there are some sequence differences between KCa3.1 and KCa2 channels. However, in addition to the S45A helix/CaM N-lobe interface, which is present four times in the KCa3.1 channel, there are certainly more sites on the cytoplasmic surface of KCa channels that could accommodate small molecules and it is feasible that NS309 or the KCa2.2/2.3 selective CyPPA and NS13001 are binding at other sites or occupy a different number of sites. Gating modulation is also possible in the transmembrane domain as has been demonstrated by the fact, that a serine residue in S5 is crucial for the action of both the KCa2.1 selective positive gating modulator CM-TMPF and the negative modulator (−)-B-TPMF (123).
OUTLOOK
As described here, KCa2/3 channels have a relatively well-developed pharmacology and their therapeutic targeting for neurological and cardiovascular diseases is supported by ample preclinical data. The most advanced compound, the KCa3.1 blocker senicapoc unfortunately failed in a phase-III clinical trial in sickle cell anemia (106). However, the trial certainly demonstrated that KCa3.1 inhibition is safe in humans, and, as described above, senicapoc is currently in the process of being repurposed for the treatment of hereditary xerocytosis and Alzheimer’s disease. For Alzheimer’s disease the therapeutic hypothesis is that KCa3.1 inhibition would reduce neuroinflammation by suppressing microglia activation (140). The same hypothesis is used to rationalize repurposing senicapoc for stroke (141) and neuropathic pain (142). Other indications for which repurposing of senicapoc might be worthwhile considering is idiopathic pulmonary fibrosis (143) and glioblastoma (144). While repurposing is an attractive short cut, senicapoc due to its high lipophilicity, low solubility, high plasma protein binding and very long half-live in humans, is not necessarily an “ideal” KCa3.1 blocker and it will be interesting to see if the now available full-length KCa3.1 structure (56) will revive the interest of the pharmaceutical industry in developing better KCa3.1 inhibitors.
The other KCa modulators that have recently entered clinical trials, the yet undisclosed negative KCa2 channel gating modulator AP30663 for atrial fibrillation and the positive KCa2 channel modulator CAD-1883 for cerebellar disorders, have passed phase-I and are currently being tested in patients, which demonstrates the feasibility of balancing benefits and side effects of KCa2 channel modulation by optimizing compound properties. AP30663 is intentionally made peripherally restricted presumably by increasing its polarity or by introducing structural elements favoring its extrusion across the blood brain barrier, thereby strongly reducing liability for inducing tremors and seizures that are observed with the brain-penetrant NS8593 in animal studies (121). In contrast, CAD-1883, which is designed for targeting KCa2 channels in pacemaker neurons of the cerebellar cortex and deep cerebellar nuclei (primarily KCa2.2), is optimized for selectivity and good brain exposure. Similarly, for positive gating modulators targeting peripheral diseases, it will be important to achieve subtype and ideally tissue selectivity. Unselective activators like SKA-31 are useful tool compounds but quickly demonstrated that central KCa2 channel mediated sedation and heart rate reduction (145) constitute undesirable side-effects when attempting to target endothelial KCa3.1 channels to lower blood pressure, even if the approach is effective in large animals such as dogs (146) and pigs (147). A useful KCa3.1 activator for improving endothelial function in hypertension and other cardiovascular disease should therefore ideally be KCa3.1 selective and peripherally restricted.
The very significant advances that have recently been made in elucidation the structure of KCa3.1 (56) and the resulting improvements in modeling have so far not been used for drug design. All current drug candidates in the KCa channel field have been identified by screening or classical medicinal chemistry approaches. However, as more high-resolution protein structures will become available, ideally with KCa channel modulators differing in structure, mode of action, and selectivity positioned at their respective pharmacological sites, there is no doubt that this information will be of increasing importance in future drug optimization programs. Immediate questions to solve are, for example, how to explain the already obtained KCa2.3/KCa2.2 versus KCa2.1/KCa3.1 selectivity of compounds in the CyPPA/NS13001 series at the atomistic level. Another item on the “wish-list” of the pharmacologist and the drug developer is to gain insights into how to design selective blockers or negative gating modulators for KCa2.3, an important channel in all monoaminergic neurons, which could be an important step in developing new drugs for psychiatric diseases. While we believe that KCa2.2 or KCa2.2/KCa2.3 selective activators certainly are also promising for the treatment of dependence on alcohol (148) and other habit forming substances (149), it might be challenging to ever safely translate the beneficial effects of KCa2.2 inhibition on learning and memory into the clinic.
ACKNOWLEGEMENT
H.W. was supported by the CounterACT Program, National Institutes of Health Office of the Director [U54NS079202], and the National Institute of Neurological Disorders and Stroke [R21NS101876]. B.M.B. was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through grant number UL1 TR001860 and linked award TL1 TR001861.
Footnotes
DISCLOSURE STATEMENT
P. C. is a full-time employee of Saniona A/S. H.W. is consulting for Sanonia A/S.
Literature Cited
- 1.Hille B. 2001. Ion Channels of Excitable Membranes Sunderland, MA: Sinauer Associates. 426 pp. [Google Scholar]
- 2.Alexander SP, Kelly E, Marrion NV, Peters JA, Faccenda E, et al. 2017. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Overview. Br. J. Pharmacol 174 Suppl 1:S1–S16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaczmarek LK, Aldrich RW, Chandy KG, Grissmer S, Wei AD, Wulff H. 2017. International Union of Basic and Clinical Pharmacology. C. Nomenclature and Properties of Calcium-Activated and Sodium-Activated Potassium Channels. Pharmacol. Rev 69:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monaghan AS, Benton DC, Bahia PK, Hosseini R, Shah YA, et al. 2004. The SK3 subunit of small conductance Ca2+-activated K+ channels interacts with both SK1 and SK2 subunits in a heterologous expression system. J. Biol. Chem 279:1003–9 [DOI] [PubMed] [Google Scholar]
- 5.Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, et al. 1996. Small-conductance, calcium-activated potassium channels from mammalian brain. Science 273:1709–14 [DOI] [PubMed] [Google Scholar]
- 6.Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, et al. 1998. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395:503–7 [DOI] [PubMed] [Google Scholar]
- 7.Ishii TM, Silvia C, Hirschberg B, Bond CT, Adelman JP, Maylie J. 1997. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. USA 94:11651–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joiner WJ, Wang LY, Tang MD, Kaczmarek LK. 1997. hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc. Natl. Acad. Sci. USA 94:11013–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fanger CM, Ghanshani S, Logsdon NJ, Rauer H, Kalman K, et al. 1999. Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. J. Biol. Chem 274:5746–54 [DOI] [PubMed] [Google Scholar]
- 10.Adelman JP, Maylie J, Sah P. 2012. Small-conductance Ca2+-activated K+ channels: form and function. Ann. Rev. Physiol 74:245–69 [DOI] [PubMed] [Google Scholar]
- 11.Bond CT, Herson PS, Strassmaier T, Hammond R, Stackman R, et al. 2004. Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J. Neurosci 24:5301–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hammond RS, Bond CT, Strassmaier T, Ngo-Anh TJ, Adelman JP, et al. 2006. Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J. Neurosci 26:1844–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolfart J, Neuhoff H, Franz O, Roeper J. 2001. Differential expression of the small-conductance, calcium-activated potassium channel SK3 is critical for pacemaker control in dopaminergic midbrain neurons. J. Neurosci 21:3443–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rouchet N, Waroux O, Lamy C, Massotte L, Scuvee-Moreau J, et al. 2008. SK channel blockade promotes burst firing in dorsal raphe serotonergic neurons. Eur. J. Neurosci 28:1108–15 [DOI] [PubMed] [Google Scholar]
- 15.Stocker M. 2004. Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat. Rev. Neurosci 5:758–70 [DOI] [PubMed] [Google Scholar]
- 16.Messier C, Mourre C, Bontempi B, Sif J, Lazdunski M, Destrade C. 1991. Effect of apamin, a toxin that inhibits Ca2+-dependent K+ channels, on learning and memory processes. Brain Res 551:322–6 [DOI] [PubMed] [Google Scholar]
- 17.Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, et al. 2002. Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J. Neurosci 22:10163–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKay BM, Oh MM, Galvez R, Burgdorf J, Kroes RA, et al. 2012. Increasing SK2 channel activity impairs associative learning. J. Neurophysiol 108:863–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bahia PK, Suzuki R, Benton DC, Jowett AJ, Chen MX, et al. 2005. A functional role for small-conductance calcium-activated potassium channels in sensory pathways including nociceptive processes. J. Neurosci 25:3489–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pagadala P, Park CK, Bang S, Xu ZZ, Xie RG, et al. 2013. Loss of NR1 subunit of NMDARs in primary sensory neurons leads to hyperexcitability and pain hypersensitivity: involvement of Ca2+-activated small conductance potassium channels. J. Neurosci 33:13425–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brahler S, Kaistha A, Schmidt VJ, Wolfle SE, Busch C, et al. 2009. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 119:2323–32 [DOI] [PubMed] [Google Scholar]
- 22.Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. 1998. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 396:269–72 [DOI] [PubMed] [Google Scholar]
- 23.Tuteja D, Xu D, Timofeyev V, Lu L, Sharma D, et al. 2005. Differential expression of small-conductance Ca2+-activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol 289:H2714–23 [DOI] [PubMed] [Google Scholar]
- 24.Zhang Q, Timofeyev V, Lu L, Li N, Singapuri A, et al. 2008. Functional roles of a Ca2+-activated K+ channel in atrioventricular nodes. Circ. Res 102:465–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, et al. 2010. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat. Genetics 42:240–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olesen MS, Jabbari J, Holst AG, Nielsen JB, Steinbruchel DA, et al. 2011. Screening of KCNN3 in patients with early-onset lone atrial fibrillation. Europace 13:963–7 [DOI] [PubMed] [Google Scholar]
- 27.Zhang XD, Timofeyev V, Li N, Myers RE, Zhang DM, et al. 2014. Critical roles of a small conductance Ca2+-activated K+ channel (SK3) in the repolarization process of atrial myocytes. Cardiov. Res 101:317–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barfod ET, Moore AL, Lidofsky SD. 2001. Cloning and functional expression of a liver isoform of the small conductance Ca2+-activated K+ channel SK3. Am. J. Physiol. Cell. Physiol 280:C836–42 [DOI] [PubMed] [Google Scholar]
- 29.Vandorpe DH, Shmukler BE, Jiang L, Lim B, Maylie J, et al. 1998. cDNA cloning and functional characterization of the mouse Ca2+-gated K+ channel, mIK1. Roles in regulatory volume decrease and erythroid differentiation. J. Biol. Chem 273:21542–53 [DOI] [PubMed] [Google Scholar]
- 30.Hoffman JF, Joiner W, Nehrke K, Potapova O, Foye K, Wickrema A. 2003. The hSK4 (KCNN4) isoform is the Ca2+-activated K+ channel (Gardos channel) in human red blood cells. Proc. Natl. Acad. Sci. USA 100:7366–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gardos G. 1958. The function of calcium in the potassium permeability of human erythrocytes. Biochem. Biophys. Acta 30:653–4 [DOI] [PubMed] [Google Scholar]
- 32.Glogowska E, Lezon-Geyda K, Maksimova Y, Schulz VP, Gallagher PG. 2015. Mutations in the Gardos channel (KCNN4) are associated with hereditary xerocytosis. Blood 126:1281–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andolfo I, Russo R, Manna F, Shmukler BE, Gambale A, et al. 2015. Novel Gardos channel mutations linked to dehydrated hereditary stomatocytosis (xerocytosis). Am. J. Hematol 90:921–6 [DOI] [PubMed] [Google Scholar]
- 34.Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, et al. 2000. Up-regulation of the IKCa1 potassium channel during T-cell activation: Molecular mechanism and functional consequences. J. Biol. Chem 275:37137–49 [DOI] [PubMed] [Google Scholar]
- 35.Wulff H, Knaus HG, Pennington M, Chandy KG. 2004. K+ channel expression during B-cell differentiation: implications for immunomodulation and autoimmunity. J. Immunol 173:776–86 [DOI] [PubMed] [Google Scholar]
- 36.Shumilina E, Lam RS, Wolbing F, Matzner N, Zemtsova IM, et al. 2008. Blunted IgE-mediated activation of mast cells in mice lacking the Ca2+-activated K+ channel KCa3.1. J. Immunol 180:8040–7 [DOI] [PubMed] [Google Scholar]
- 37.Toyama K, Wulff H, Chandy KG, Azam P, Raman G, et al. 2008. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J. Clin. Invest 118:3025–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaushal V, Koeberle PD, Wang Y, Schlichter LC. 2007. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci 27:234–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feske S, Wulff H, Skolnik EY. 2015. Ion channels in innate and adaptive immunity. Ann. rev. Immunol 33:291–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen YJ, Nguyen HM, Maezawa I, Grossinger EM, Garing AL, et al. 2016. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J. Cereb. Blood Flow. Metab 36:2146–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen HM, Grossinger EM, Horiuchi M, Davis KW, Jin LW, et al. 2017. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 65:106–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.King B, Rizwan AP, Asmara H, Heath NC, Engbers JD, et al. 2015. IKCa channels are a critical determinant of the slow AHP in CA1 pyramidal neurons. Cell Rep 11:175–82 [DOI] [PubMed] [Google Scholar]
- 43.Sahu G, Asmara H, Zhang FX, Zamponi GW, Turner RW. 2017. Activity-dependent facilitation of Cav1.3 calcium channels promotes KCa3.1 activation in hippocampal neurons. J. Neurosci 37:11255–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang K, Mateos-Aparicio P, Honigsperger C, Raghuram V, Wu WW, et al. 2016. IK1 channels do not contribute to the slow afterhyperpolarization in pyramidal neurons. Elife 5:e11206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di L, Srivastava S, Zhdanova O, Ding Y, Li Z, et al. 2010. Inhibition of the K+ channel KCa3.1 ameliorates T cell-mediated colitis. Proc. Natl. Acad. Sci. USA 107:1541–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raychaudhuri SK, Wulff H, Raychaudhuri SP. 2016. KCa3.1(−/−) Mice do not develop CIA: Regulatory role for KCa3.1 in autoimmune arthritis. J. Cell. Physiol 231:2313–4 [DOI] [PubMed] [Google Scholar]
- 47.Si H, Heyken WT, Wolfle SE, Tysiac M, Schubert R, et al. 2006. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ. Res 99:537–44 [DOI] [PubMed] [Google Scholar]
- 48.Grgic I, Kaistha BP, Paschen S, Kaistha A, Busch C, et al. 2009. Disruption of the Gardos channel (KCa3.1) in mice causes subtle erythrocyte macrocytosis and progressive splenomegaly. Pflüg. Arch 458:291–302 [DOI] [PubMed] [Google Scholar]
- 49.Heitzmann D, Warth R. 2008. Physiology and pathophysiology of potassium channels in gastrointestinal epithelia. Physiol. Rev 88:1119–82 [DOI] [PubMed] [Google Scholar]
- 50.Kohler R, Wulff H, Eichler I, Kneifel M, Neumann D, et al. 2003. Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation 108:1119–25 [DOI] [PubMed] [Google Scholar]
- 51.Tharp DL, Wamhoff BR, Wulff H, Raman G, Cheong A, Bowles DK. 2008. Local Delivery of the KCa3.1 Blocker, TRAM-34, Prevents Acute Angioplasty-Induced Coronary Smooth Muscle Phenotypic Modulation and Limits Stenosis. Arterioscler. Thromb. Vasc. Biol 28:1084–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.D’Alessandro G, Catalano M, Sciaccaluga M, Chece G, Cipriani R, et al. 2013. KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis 4:e773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turner KL, Honasoge A, Robert SM, McFerrin MM, Sontheimer H. 2014. A proinvasive role for the Ca2+ -activated K+ channel KCa3.1 in malignant glioma. Glia 62:971–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steudel FA, Mohr CJ, Stegen B, Nguyen HY, Barnert A, et al. 2017. SK4 channels modulate Ca2+ signalling and cell cycle progression in murine breast cancer. Mol. Oncol 11:1172–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lallet-Daher H, Roudbaraki M, Bavencoffe A, Mariot P, Gackiere F, et al. 2009. Intermediate-conductance Ca2+-activated K+ channels (IKCa1) regulate human prostate cancer cell proliferation through a close control of calcium entry. Oncogene 28:1792–806 [DOI] [PubMed] [Google Scholar]
- 56.Lee CH, MacKinnon R. 2018. Activation mechanism of a human SK-calmodulin channel complex elucidated by cryo-EM structures. Science 360:508–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schumacher MA, Rivard AF, Bachinger HP, Adelman JP. 2001. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature 410:1120–4 [DOI] [PubMed] [Google Scholar]
- 58.Halling DB, Kenrick SA, Riggs AF, Aldrich RW. 2014. Calcium-dependent stoichiometries of the KCa2.2 (SK) intracellular domain/calmodulin complex in solution. J. Gen. Physiol 143:231–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang M, Pascal JM, Schumann M, Armen RS, Zhang JF. 2012. Identification of the functional binding pocket for compounds targeting small-conductance Ca2+-activated potassium channels. Nat. Commun 3:1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang M, Pascal JM, Zhang JF. 2013. Unstructured to structured transition of an intrinsically disordered protein peptide in coupling Ca2+-sensing and SK channel activation. Proc. Natl. Acad. Sci. USA 110:4828–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang M, Meng XY, Cui M, Pascal JM, Logothetis DE, Zhang JF. 2014. Selective phosphorylation modulates the PIP2 sensitivity of the CaM-SK channel complex. Nat. Chem. Biol 10:753–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cho LT, Alexandrou AJ, Torella R, Knafels J, Hobbs J, et al. 2018. An intracellular aAllosteric modulator binding pocket in SK2 ion channels is shared by multiple chemotypes. Structure 26:533–44 e3 [DOI] [PubMed] [Google Scholar]
- 63.Wang RY, Song Y, Barad BA, Cheng Y, Fraser JS, DiMaio F. 2016. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. Elife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hugues M, Romey G, Duval D, Vincent JP, Lazdunski M. 1982. Apamin as a selective blocker of the calcium-dependent potassium channel in neuroblastoma cells: voltage-clamp and biochemical characterization of the toxin receptor. Proc. Natl. Acad. Sci. USA 79:1308–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pedarzani P, Stocker M. 2008. Molecular and cellular basis of small- and intermediate-conductance, calcium-activated potassium channel function in the brain. Cell. Mol. Life Sci 65:3196–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wulff H, Zhorov BS. 2008. K+ channel modulators for the treatment of neurological disorders and autoimmune diseases. Chem. Rev 108:1744–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wulff H, Kolski-Andreaco A, Sankaranarayanan A, Sabatier JM, Shakkottai V. 2007. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr. Med. Chem 14:1437–57 [DOI] [PubMed] [Google Scholar]
- 68.Mourre C, Fournier C, Soumireu-Mourat B. 1997. Apamin, a blocker of the calcium-activated potassium channel, induces neurodegeneration of Purkinje cells exclusively. Brain Res 778:405–8 [DOI] [PubMed] [Google Scholar]
- 69.Lamy C, Goodchild SJ, Weatherall KL, Jane DE, Liegeois JF, et al. 2010. Allosteric block of KCa2 channels by apamin. J. Biol. Chem 285:27067–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nolting A, Ferraro T, D’Hoedt D, Stocker M. 2006. An amino acid outside the pore region influences apamin sensitivity in small conductance Ca2+-activated K+ channels. J. Biol. Chem 82:3478–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Castle NA, Strong PN. 1986. Identification of two toxins from scorpion (Leiurus quinquestriatus) venom which block distinct classes of calcium-activated potassium channel. FEBS Lett 209:117–21 [DOI] [PubMed] [Google Scholar]
- 72.Auguste P, Hugues M, Grave B, Gesquiere JC, Maes P, et al. 1990. Leiurotoxin I (scyllatoxin), a peptide ligand for Ca2+-activated K+ channels. Chemical synthesis, radiolabeling, and receptor characterization. J Biol Chem 265:4753–9 [PubMed] [Google Scholar]
- 73.Pedarzani P, D’Hoedt D, Doorty KB, Wadsworth JD, Joseph JS, et al. 2002. Tamapin, a venom peptide from the Indian red scorpion (Mesobuthus tamulus) that targets small conductance Ca2+-activated K+ channels and afterhyperpolarization currents in central neurons. J. Biol. Chem 277:46101–9 [DOI] [PubMed] [Google Scholar]
- 74.Shakkottai VG, Regaya I, Wulff H, Fajloun Z, Tomita H, et al. 2001. Design and characterization of a highly selective peptide inhibitor of the small conductance calcium-activated K+ channel, SKCa2. J. Biol. Chem 276:43145–51 [DOI] [PubMed] [Google Scholar]
- 75.Kasumu AW, Hougaard C, Rode F, Jacobsen TA, Sabatier JM, et al. 2012. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem. Biol 19:1340–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rauer H, Lanigan MD, Pennington MW, Aiyar J, Ghanshani S, et al. 2000. Structure-guided transformation of charybdotoxin yields an analog that selectively targets Ca2+-activated over voltage-gated K+ channels. J. Biol. Chem 275:1201–8 [DOI] [PubMed] [Google Scholar]
- 77.Castle NA, Lodon DO, Creech C, Fajloun Z, Stocker JW, Sabatier J-M. 2003. Maurotoxin - a potent inhibitor of the intermediate conductance Ca2+-activated potassium channel. Mol. Pharmacol 63:409–18 [DOI] [PubMed] [Google Scholar]
- 78.Ishii TM, Maylie J, Adelman JP. 1997. Determinants of apamin and d-tubocurarine block in SK potassium channels. J. Biol. Chem 272:23195–200 [DOI] [PubMed] [Google Scholar]
- 79.Castle NA, Haylett DG, Morgan JM, Jenkinson DH. 1993. Dequalinium: a potent inhibitor of apamin-sensitive K+ channels in hepatocytes and of nicotinic responses in skeletal muscle. Eur. J. Pharmacol 236:201–7 [DOI] [PubMed] [Google Scholar]
- 80.Galanakis D, Davis CA, Del Rey Herrero B, Ganellin CR, Dunn PM, Jenkinson DH. 1995. Synthesis and structure-activity relationships of dequalinium analogues as K+ channel blockers. Investigations on the role of the charged heterocycle. J. Med. Chem 38:595–606 [DOI] [PubMed] [Google Scholar]
- 81.Galanakis D, Ganellin CR, Malik S, Dunn PM. 1996. Synthesis and pharmacological testing of dequalinium analogues as blockers of the apamin-sensitive Ca2+-activated K+ channel: variation of the length of the alkylene chain. J. Med. Chem 39:3592–5 [DOI] [PubMed] [Google Scholar]
- 82.Campos Rosa J, Galanakis D, Piergentili A, Bhandari K, Ganellin CR, et al. 2000. Synthesis, molecular modeling, and pharmacological testing of bis-quinolinium cyclophanes: potent, non-peptidic blockers of the apamin-sensitive Ca2+-activated K+ channel. J. Med. Chem 43:420–31 [DOI] [PubMed] [Google Scholar]
- 83.Chen JQ, Galanakis D, Ganellin CR, Dunn PM, Jenkinson DH. 2000. bis-Quinolinium cyclophanes: 8,14-diaza-1,7(1, 4)-diquinolinacyclotetradecaphane (UCL 1848), a highly potent and selective, nonpeptidic blocker of the apamin-sensitive Ca2+-activated K+ channel. J. Med. Chem 43:3478–81 [DOI] [PubMed] [Google Scholar]
- 84.Johnson SW, Seutin V. 1997. Bicuculline methiodide potentiates NMDA-dependent burst firing in rat dopamine neurons by blocking apamin-sensitive Ca2+-activated K+ currents. Neurosci. Lett 231:13–6 [DOI] [PubMed] [Google Scholar]
- 85.Gentles RG, Grant-Young K, Hu S, Huang Y, Poss MA, et al. 2008. Initial SAR studies on apamin-displacing 2-aminothiazole blockers of calcium-activated small conductance potassium channels. Bioorg. & Med. Chem. Lett 18:5316–9 [DOI] [PubMed] [Google Scholar]
- 86.Simo-Vicens R, Bomholtz SH, Sørensen US, Bentzen BH. 2018. 2,6-Bis(2-benzimidazolyl)pyridine (BBP) is a potent and selective inhibitor of small conductance calcium-activated potassium (SK) channels. Front. Pharmacol 9:1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Skibsbye L, Poulet C, Diness JG, Bentzen BH, Yuan L, et al. 2014. Small-conductance calcium-activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovas. Res 103:156–67 [DOI] [PubMed] [Google Scholar]
- 88.Kirchhoff JE, Diness JG, Abildgaard L, Sheykhzade M, Grunnet M, Jespersen T. 2016. Antiarrhythmic effect of the Ca2+-activated K+ (SK) channel inhibitor ICA combined with either amiodarone or dofetilide in an isolated heart model of atrial fibrillation. Pflüg. Arch 468:1853–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gluais P, Edwards G, Weston AH, Falck JR, Vanhoutte PM, Feletou M. 2005. Role of SK(Ca) and IK(Ca) in endothelium-dependent hyperpolarizations of the guinea-pig isolated carotid artery. Br. J. Pharmacol 144:477–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Berkowitz LR, Orringer EP. 1981. Effect of cetiedil, an in vitro antisickling agent, on erythrocyte membrane cation permeability. J Clin Invest 68:1215–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roxburgh CJ, Ganellin CR, Shiner MA, Benton DC, Dunn PM, et al. 1996. The synthesis and some pharmacological actions of the enantiomers of the K(+)-channel blocker cetiedil. J Pharm Pharmacol 48:851–7 [DOI] [PubMed] [Google Scholar]
- 92.Ellory JC, Nash GB, Stone PC, Culliford SJ, Horwitz E, Stuart J. 1992. Mode of action and comparative efficacy of pharmacological agents that inhibit calcium-dependent dehydration of sickle cells. Br. J. Pharmacol 106:972–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ellory JC, Culliford SJ, Smith PA, Wolowyk MW, Knaus EE. 1994. Specific inhibition of Ca-activated K channels in red cells by selected dihydropyridine derivatives. Br. J. Pharmacol 111:903–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alvarez J, Montero M, Garcia-Sancho J. 1992. High affinity inhibition of Ca2+-dependent K+ channels by cytochrome P-450 inhibitors. J. Biol. Chem 267:11789–93 [PubMed] [Google Scholar]
- 95.Urbahns K, Horvath E, Stasch JP, Mauler F. 2003. 4-Phenyl-4H-pyrans as IK(Ca) channel blockers. Bioorg. & Med. Chem. Lett 13:2637–9 [DOI] [PubMed] [Google Scholar]
- 96.Urbahns K, Goldmann S, Kruger J, Horvath E, Schuhmacher J, et al. 2005. IKCa-channel blockers. Part 2: discovery of cyclohexadienes. Bioorg. & Med. Chem. Lett 15:401–4 [DOI] [PubMed] [Google Scholar]
- 97.Mauler F, Hinz V, Horvath E, Schuhmacher J, Hofmann HA, et al. 2004. Selective intermediate-/small-conductance calcium-activated potassium channel (KCNN4) blockers are potent and effective therapeutics in experimental brain oedema and traumatic brain injury caused by acute subdural haematoma. Eur. J. Neurosci 20:1761–8 [DOI] [PubMed] [Google Scholar]
- 98.De Franceschi L, Saadane N, Trudel M, Alper SL, Brugnara C, Beuzard Y. 1994. Treatment with oral clotrimazole blocks Ca2+-activated K+ transport and reverses erythrocyte dehydration in transgenic SAD mice. A model for therapy of sickle cell disease. J. Clin. Invest 93:1670–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brugnara C, Armsby CC, Sakamoto M, Rifai N, Alper SL, Platt O. 1995. Oral administration of clotrimazole and blockade of human erythrocyte Ca++-activated K+ channel: the imidazole ring is not required for inhibitory activity. J. Pharmacol. Exp. Ther 273:266–72 [PubMed] [Google Scholar]
- 100.Wulff H, Miller MJ, Haensel W, Grissmer S, Cahalan MD, Chandy KG. 2000. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: A potential immunosuppressant. Proc. Natl. Acad. Sci. USA 97:8151–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Girodet PO, Ozier A, Carvalho G, Ilina O, Ousova O, et al. 2013. Ca2+-activated K+ channel-3.1 blocker TRAM-34 attenuates airway remodeling and eosinophilia in a murine asthma model. Am. J. Respir. Cell. Mol. Biol 48:212–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen YJ, Lam J, Gregory CR, Schrepfer S, Wulff H. 2013. The Ca2+-activated K+ channel KCa3.1 as a potential new target for the prevention of allograft vasculopathy. PLoS ONE 8:e81006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grgic I, Kiss E, Kaistha BP, Busch C, Kloss M, et al. 2009. Renal fibrosis is attenuated by targeted disruption of KCa3.1 potassium channels. Proc. Natl. Acad. Sci. USA 106:14518–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huang C, Shen S, Ma Q, Chen J, Gill A, et al. 2013. Blockade of KCa3.1 ameliorates renal fibrosis through the TGF-beta1/Smad pathway in diabetic mice. Diabetes 62:2923–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ju CH, Wang XP, Gao CY, Zhang SX, Ma XH, Liu C. 2015. Blockade of KCa3.1 Attenuates Left Ventricular Remodeling after Experimental Myocardial Infarction. Cell. Physiol. Biochem 36:1305–15 [DOI] [PubMed] [Google Scholar]
- 106.Ataga KI, Reid M, Ballas SK, Yasin Z, Bigelow C, et al. 2011. Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: a phase III randomized, placebo-controlled, double-blind study of the Gardos channel blocker senicapoc (ICA-17043). Br. J. Haematol 153:92–104 [DOI] [PubMed] [Google Scholar]
- 107.Van Der Velden J, Sum G, Barker D, Koumoundouros E, Barcham G, et al. 2013. KCa3.1 channel-blockade attenuates airway pathophysiology in a sheep model of chronic asthma. PLoS ONE 8:e66886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wulff H, Castle NA. 2010. Therapeutic potential of KCa3.1 blockers: recent advances and promissing trends. Expert. Rev. Clin. Pharmacol 3:385–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rivera A, Vandorpe DH, Shmukler BE, Gallagher DR, Fikry CC, et al. 2017. Erythrocytes from Hereditary Xerocytosis patients heterozygous for KCNN4 V282M exhibit increased spontaneous Gardos channel-like activity inhibited by Senicapoc. Am. J. Hematol 92:E108–E110. [DOI] [PubMed] [Google Scholar]
- 110.Jin LW, Di Lucente J, Nguyen HM, Singh V, Singh L, et al. 2019. Repurposing the KCa3.1 inhibitor senicapoc for Alzheimer’s disease. Ann. Clin. Transl. Neurol [In press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jørgensen S, Dyhring T, Brown DT, Strøbæk D, Christophersen P, Demnitz J. 2013. A high-throughput screening campaign for detection of Ca2+-activated K+ channel activators and inhibitors using a fluorometric imaging plate reader-based Tl+-influx assay. Assay Drug Dev. Technol 11:163–72 [DOI] [PubMed] [Google Scholar]
- 112.Strøbæk D, Brown DT, Jenkins DP, Chen YJ, Coleman N, et al. 2013. NS6180, a new K(Ca) 3.1 channel inhibitor prevents T-cell activation and inflammation in a rat model of inflammatory bowel disease. Br. J. Pharmacol 168:432–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kang H, Kerloc’h A, Rotival M, Xu X, Zhang Q, et al. 2014. KCNN4 is a regulator of macrophage multinucleation in bone homeostasis and inflammatory disease. Cell Rep 8:1210–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wulff H, Gutman GA, Cahalan MD, Chandy KG. 2001. Delination of the clotrimazole/TRAM-34 binding site on the intermediate conductance calcium-activated potassium channel IKCa1. J. Biol. Chem 276:32040–5 [DOI] [PubMed] [Google Scholar]
- 115.Nguyen HM, Singh V, Pressly B, Jenkins DP, Wulff H, Yarov-Yarovoy V. 2017. Structural insights into the atomistic mechanisms of action of small molecule inhibitors targeting the KCa3.1 channel pore. Mol. Pharmacol 91:392–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature 414:43–8 [DOI] [PubMed] [Google Scholar]
- 117.Strøbæk D, Hougaard C, Johansen TH, Sørensen US, Nielsen EO, et al. 2006. Inhibitory gating modulation of small conductance Ca2+-activated K+ channels by the synthetic compound (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphtylamine (NS8593) reduces afterhyperpolarizing current in hippocampal CA1 neurons. Mol. Pharmacol 70:1771–82 [DOI] [PubMed] [Google Scholar]
- 118.Sørensen US, Strøbæk D, Christophersen P, Hougaard C, Jensen ML, et al. 2008. Synthesis and structure-activity relationship studies of 2-(N-substituted)-aminobenzimidazoles as potent negative gating modulators ofsmall conductance Ca2+-activated K+ channels. J. Med. Chem 51:7625–34 [DOI] [PubMed] [Google Scholar]
- 119.Jenkins DP, Strøbæk D, Hougaard C, Jensen ML, Hummel R, et al. 2011. Negative gating modulation by (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphthylamine (NS8593) depends on residues in the inner pore vestibule: pharmacological evidence of deep-pore gating of KCa2 channels. Mol. Pharmacol 79:899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bruening-Wright A, Schumacher MA, Adelman JP, Maylie J. 2002. Localization of the activation gate for small conductance Ca2+-activated K+ channels. J. Neurosci 22:6499–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Simo-Vicens R, Kirchhoff JE, Dolce B, Abildgaard L, Speerschneider T, et al. 2017. A new negative allosteric modulator, AP14145, for the study of small conductance calcium-activated potassium (KCa2) channels. Br. J. Pharmacol 174:4396–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gentles RG, Hu S, Huang Y, Grant-Young K, Poss MA, et al. 2008. Preliminary SAR studies on non-apamin-displacing 4-(aminomethylaryl)pyrrazolopyrimidine KCa channel blockers. Bioorg. & Med. Chem Lett 18:5694–7 [DOI] [PubMed] [Google Scholar]
- 123.Hougaard C, Hammami S, Eriksen BL, Sorensen US, Jensen ML, et al. 2012. Evidence for a common pharmacological interaction site on KCa2 channels providing both selective activation and selective inhibition of the human KCa2.1 subtype. Mol. Pharmacol 81:210–9 [DOI] [PubMed] [Google Scholar]
- 124.Olivan-Viguera A, Valero MS, Coleman N, Brown BM, Laria C, et al. 2015. A novel pan-negative-gating modulator of KCa2/3 channels, fluoro-di-benzoate, RA-2, inhibits endothelium-derived hyperpolarization-type relaxation in coronary artery and produces bradycardia in vivo. Mol. Pharmacol 87:338–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Diness JG, Sørensen US, Nissen JD, Al-Shahib B, Jespersen T, et al. 2010. Inhibition of small-conductance Ca2+-activated K+ channels terminates and protects against atrial fibrillation. Circulation. Arrhyth. Electrophys 3:380–90 [DOI] [PubMed] [Google Scholar]
- 126.Diness JG, Skibsbye L, Jespersen T, Bartels ED, Sørensen US, et al. 2011. Effects on atrial fibrillation in aged hypertensive rats by Ca2+-activated K+ channel inhibition. Hypertension 57:1129–35 [DOI] [PubMed] [Google Scholar]
- 127.Diness JG, Skibsbye L, Simo-Vicens R, Santos JL, Lundegaard P, et al. 2017. Termination of vernakalant-resistant atrial fibrillation by inhibition of small-conductance Ca2+-activated K+ channels in pigs. Circ. Arrhythm. Electrophys 10:e005125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brown BM, Shim H, Wulff H. 2017. Are there superagonists for calcium-activated potassium channels? Channels (Austin) 11:504–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Devor DC, Singh AK, Frizzell RA, Bridges RJ. 1996. Modulation of Cl-secretion by benzimidazolones. I. Direct activation of a Ca2+-dependent K+ channel. Am. J. Physiol 271:L775–84 [DOI] [PubMed] [Google Scholar]
- 130.Singh S, Syme CA, Singh AK, Devor DC, Bridges RJ. 2001. Benzimidazolone activators of chloride secretion: potential therapeutics for cystic fibrosis and chronic obstructive pulmonary disease. J. Pharmacol. Exp. Ther 296:600–11 [PubMed] [Google Scholar]
- 131.Cao Y, Dreixler JC, Roizen JD, Roberts MT, Houamed KM. 2001. Modulation of recombinant small-conductance Ca2+-activated K+ channels by the muscle relaxant chlorzoxazone and structurally related compounds. J. Pharmacol. Exp. Ther 296:683–9 [PubMed] [Google Scholar]
- 132.Strøbæk D, Teuber L, Jørgensen TD, Ahring PK, Kjaer K, et al. 2004. Activation of human IK and SK Ca2+-activated K+ channels by NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime). Biochim. Biophys. Acta 1665:1–5 [DOI] [PubMed] [Google Scholar]
- 133.Sankaranarayanan A, Raman G, Busch C, Schultz T, Zimin PI, et al. 2009. Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol. Pharmacol 75:281–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Coleman N, Brown BM, Olivan-Viguera A, Singh V, Olmstead MM, et al. 2014. New positive Ca2+-activated K+ channel gating modulators with selectivity for KCa3.1. Mol. Pharmacol 86:342–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hougaard C, Eriksen BL, Jorgensen S, Johansen TH, Dyhring T, et al. 2007. Selective positive modulation of the SK3 and SK2 subtypes of small conductance Ca2+-activated K+ channels. Br. J. Pharmacol 151:655–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shakkottai VG, Chou CH, Oddo S, Sailer CA, Knaus HG, et al. 2004. Enhanced neuronal excitability in the absence of neurodegeneration induces cerebellar ataxia. J. Clin. Invest 113:582–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pedarzani P, Mosbacher J, Rivard A, Cingolani LA, Oliver D, et al. 2001. Control of electrical activity in central neurons by modulating the gating of small conductance Ca2+-activated K+ channels. J. Biol. Chem 276:9762–9 [DOI] [PubMed] [Google Scholar]
- 138.Brown BM, Shim H, Zhang M, Yarov-Yarovoy V, Wulff H. 2017. Structural determinants for the selectivity of the positive KCa3.1 gating modulator 5-methylnaphtho[2,1-d]oxazol-2-amine (SKA-121). Mol. Pharmacol 92:469–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shim H, Brown BM, Singh L, Singh V, Fettinger JC, et al. 2019. The trials and tribulations of structure assisted design of KCa channel activators. Mol. Pharmacol submitted [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Maezawa I, Jenkins DP, Jin BE, Wulff H. 2012. Microglial KCa3.1 channels as a potential therapeutic target for Alzheimer’s disease. Int. J. Alzheimers Dis 2012:868972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dale E, Staal RG, Eder C, Moller T. 2016. KCa3.1-a microglial target ready for drug repurposing? Glia 64:1733–41 [DOI] [PubMed] [Google Scholar]
- 142.Staal RG, Khayrullina T, Zhang H, Davis S, Fallon SM, et al. 2017. Inhibition of the potassium channel KCa3.1 by senicapoc reverses tactile allodynia in rats with peripheral nerve injury. Eur. J. Pharmacol 795:1–7 [DOI] [PubMed] [Google Scholar]
- 143.Organ L, Bacci B, Koumoundouros E, Kimpton WG, Samuel CS, et al. 2017. Inhibition of the KCa3.1 channel alleviates established pulmonary fibrosis in a large animal model. Am. J. Respir. Cell. Mol. Biol 56:539–50 [DOI] [PubMed] [Google Scholar]
- 144.D’Alessandro G, Grimaldi A, Chece G, Porzia A, Esposito V, et al. 2016. KCa3.1 channel inhibition sensitizes malignant gliomas to temozolomide treatment. Oncotarget 7:30781–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Radtke J, Schmidt K, Wulff H, Kohler R, de Wit C. 2013. Activation of KCa3.1 by SKA-31 induces arteriolar dilation and lowers blood pressure in normo- and hypertensive connexin40-deficient mice. Br. J. Pharmacol 170:293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Damkjaer M, Nielsen G, Bodendiek S, Staehr M, Gramsbergen JB, et al. 2012. Pharmacological activation of KCa3.1/KCa2.3 channels produces endothelial hyperpolarization and lowers blood pressure in conscious dogs. Br. J. Pharmacol 165:223–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mishra RC, Mitchell JR, Gibbons-Kroeker C, Wulff H, Belenkie I, et al. 2015. A pharmacologic activator of endothelial KCa channels increases systemic conductance and reduces arterial pressure in an anesthetized pig model. Vascul. Pharmacol 79:24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hopf FW, Bowers MS, Chang SJ, Chen BT, Martin M, et al. Reduced nucleus accumbens SK channel activity enhances alcohol seeking during abstinence. Neuron 65:682–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cannady R, McGonigal JT, Newsom RJ, Woodward JJ, Mulholland PJ, Gass JT. 2017. Prefrontal cortex KCa2 channels regulate mGlu5-dependent plasticity and extinction of alcohol-seeking behavior. J. Neurosci 37:4359–69 [DOI] [PMC free article] [PubMed] [Google Scholar]