

Abstract
Rotenone is a naturally occurring insecticide that inhibits mitochondrial complex I and leads to neurochemical and neuropathological deficits closely resembling those in Parkinson’s disease (PD). Deficits include loss of dopaminergic neurons (DAn) in the substantia nigra pars compacta (SNpc), decreased dopamine levels and aggregation of misfolded alpha-synuclein (p129). In rat models of rotenone-induced parkinsonism, the progression of neuronal injury has been associated with activation of microglia and astrocytes. However, these neuroinflammatory changes have been challenging to study in mice, in part because the systemic rotenone exposure model utilized in rats is more toxic to mice. To establish a reproducible murine model of rotenone-induced PD, we therefore investigated the progression of neuroinflammation, protein aggregation and DAn loss in C57Bl/6 mice by exposing animals to 2.5mg/kg/day rotenone for 14 days, followed by a two-week period where neuroinflammation is allowed to progress. Our results indicate that initial cellular dysfunction leads to increased formation of proteinase K-resistant p129 aggregates in the caudate-putamen and SNpc. Clearance of these aggregates was region- and cell type-specific, with the early appearance of reactive astrocytes coinciding with accumulation of p129 in the SNpc. Phagocytic microglial cells containing p129 aggregates were observed proximal to p129+ DAn in the SNpc. The majority of neuronal loss in the SNpc occurred during the two-week period after rotenone exposure, subsequent to the peak of microglia and astrocyte activation, as well as the peak of p129 aggregation. A secondary peak of p129 coincided with neurodegeneration at later timepoints. These data indicate that systemic exposure to rotenone in C57Bl/6 mice causes progressive accumulation and regional spread of p129 aggregates that precede maximal loss of DAn. Thus, activation of glial cells and aggregation of p129 appear to drive neuronal loss following neurotoxic stress imposed by exposure to rotenone.
Keywords: rotenone, microglia, astrocyte, neurodegeneration, Parkinson’s disease, pathology, protein aggregation, alpha-synuclein
Graphical Abstract
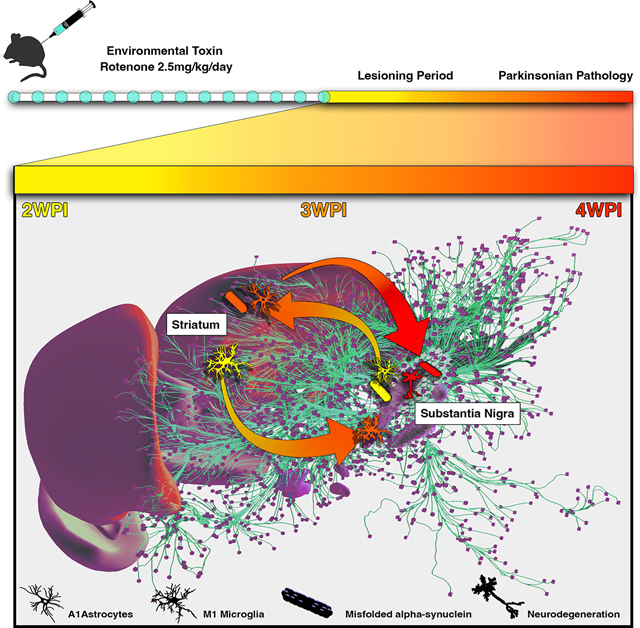
1. Introduction
Parkinson’s disease (PD) is a debilitating movement disorder affecting the central nervous system (CNS) and the second most common neurodegenerative disease worldwide (Halliday et al., 2011; Pringsheim et al., 2014). PD is characterized by loss of dopaminergic neurons (DAn) in the substantia nigra pars compacta (SNpc), loss of striatal dopamine (DA), neuroinflammatory activation of glial cells and aggregation of the phosphorylated form of α-synuclein (phosphor-serine 129/p129) (Domingues et al., 2020; Grayson, 2016; Rocha et al., 2018; Samii et al., 2004). There are no disease modifying therapies for PD in part because of limitations in many of the standard animal models used to study PD including a lack of critical features relevant to the idiopathic disease (Radhakrishnan and Goyal, 2018; Schober, 2004). Two widely used models that induce loss of DAn involve administration of the neurotoxins 1-methyl-4-phenyl-1,2,3,6-tetrahydropyuridine (MPTP) and 6-hydroxydopamine (6-OHDA). However, there are aspects of these models that differ significantly from idiopathic PD, including the absence of p129-α-Syn aggregates. The progressive development and spread of p129-α-Syn aggregates leading to Lewy body formation in the SNpc is the central neuropathological feature of PD but is notoriously difficult to recapitulate in mice. Because the etiology and progression of PD is complex, involving genetic, environmental and biological factors, as well as early and progressive neuroinflammatory changes in glial cells, additional models that more closely recapitulate the neurological and pathological deficits observed in PD are necessary to study disease mechanisms.
It has been shown that systemic administration of pesticides which inhibit mitochondrial complex I, such as paraquat, maneb and rotenone, can cause selective loss of DAn in the SNpc and might better recapitulate certain clinical and pathological features of PD than other neurotoxin-based models (Giasson and Lee, 2000; Johnson and Bobrovskaya, 2015; Kamel et al., 2007; Tanner et al., 2011). Rotenone, in particular, is a potent systemic inhibitor of mitochondrial complex I that results in high levels of reactive oxygen species (ROS) and peroxynitrate, as well as neuroinflammation and progressive accumulation of p129 aggregates within neuronal dendrites and soma (Betarbet et al., 2000; Cannon et al., 2009). Rotenone is distributed and used worldwide as a pesticide and piscicide to reduce crop destruction and invasive fish species (Radad et al., 2019). Despite evidence that rotenone is associated with increased risk for PD, it is still used within the USA, Canada and 30 other countries worldwide (Betarbet et al., 2000; EPA., 2007; Guenther, 2011). Amongst pesticides that inhibit mitochondrial complex I, rotenone closely models pathological features of PD in both mammalian and non-mammalian species. Non-mammalian models of rotenone-induced dopaminergic neurotoxicity utilizing Caenorhabditis elegans, drosophila, zebrafish and Lymnaea stagnalis have been successful in achieving DAn cell loss and motor deficits following exposure (Fontana et al., 2018; Harrington et al., 2010; Hirth, 2010; Vehovszky et al., 2007). These models allow for rapid, high-throughput screening methodologies to investigate potential therapeutics and environmental exposures but are limited with respect to the array of motor and non-motor neurological symptoms that can be modeled, as well as the spread of p129 aggregates across teleologically relevant brain regions (Dung and Thao, 2018; Lim, 2010). In contrast, systemic rotenone exposure in Lewis rats induces behavioral deficits, loss of DAn and formation of Lewy bodies, closely mirroring the pathological changes seen in PD (Betarbet et al., 2000; Cannon et al., 2009; Heikkila et al., 1985). However, it has proven challenging to adapt this model to mice due to variations in lesion profiles and high mortality rates (Johnson and Bobrovskaya, 2015). This precludes the advantages offered by a greater array of transgenic mouse lines to study signaling pathways and molecular mechanisms. Attempts to orally deliver rotenone in mice take at least two months (Inden et al., 2011; Liu et al., 2017), thus decreasing throughput for analyzing the therapeutic efficacy of pharmacologic and genetic interventions in the disease process.
We set out to characterize the efficacy of a 14 day systemic rotenone model used in Lewis rats by Greenamyre and colleagues (Betarbet et al., 2000; Cannon et al., 2009; Sherer et al., 2003) in eliciting PD-like neuropathological changes in mice, with particular emphasis on the capacity of this route of administration to promote progressive neuroinflammation and the formation of p129 aggregates. The key neuropathological features examined within lesioned brain regions included vacuolation of nerve cells, neuronal loss within the basal midbrain spatially progressing in a latero-temporal manor, glial cell activation and the accumulation of p129 aggregates within neurons. These changes were examined weekly across entire serial brain sections using scanning microscopy starting at the conclusion of the rotenone dosing period at 2 weeks through the end of the study at 4 weeks. This approach permitted the quantification of both regional and temporal patterns of gliosis, neuronal loss and p129 across multiple brain regions throughout the entire dosing and lesioning period. The results of this analysis revealed distinct patterns of neuropathology with maximal loss of DAn occurring nearly two weeks after the end of rotenone administration (4 WPI), after the peak of glial activation and coinciding with p129 aggregation. These data suggest that induction of p129 is a stress response to rotenone-induced mitochondrial dysfunction and that glial inflammatory responses are a key potentiator of both p129 aggregation and neuronal injury in this rotenone model of PD in mice.
2. Materials and Methods
2.1. Animals and In vivo Imaging
All animal protocols were approved by the Institutional Animal Care and Use Committee at Colorado State University (IACUC), mice were handled in compliance with the PHS Policy and Guide for the Care and Use of Laboratory Animals and procedures were performed in accordance with National Institutes of Health guidelines. Mice were housed in microisolator cages (3–4 animals per cage), kept on a 12-h light/dark cycle and had access to both food and water ad libitum. Male and female C57Bl/6 background mice were used in studies at 2 months of age (n=12/control, n=21/rotenone). Male and female homozygous Tg(HIV-EGFP,luc)8Tsb/J mice were obtained from The Jackson Laboratory (Stock No. 027529, Bar Harbor ME) and bred to homozygosity onto a C57Bl/6 background for use in this study (n=8/control, n=9/rotenone). For imaging luciferase reporter activity in situ, Tg(HIV-EGFP,luc)8Tsb/J mice (2 WPI: n=8 control, n=9 rotenone; 3 WPI: n=7 control, n=7 rotenone; 4 WPI: n=6 control, n=5 rotenone) were anesthetized at weekly timepoints following exposure with rotenone (2 WPI) and then given a subcutaneous dose of luciferin substrate (150mg/kg) to the dorsal cervical spine 17 minutes prior to imaging. Exposure time was kept consistent for all animals. Animals from. Living Image Software was used to obtain and analyze images on the IVIS Spectrum in vivo imaging system utilizing epi-illumination 3D diffuse fluorescence tomography.
2.2. Rotenone Preparation and Dosing
Rotenone was first prepared in a 50X stock solution that was diluted in 100% dimethyl sulfoxide (DMSO). The rotenone solution was then diluted in medium-chain triglyceride, Miglyol 812 (Cannon et al., 2009) to obtain final working concentrations of 2.5mg/kg at a dosage of 2μL/gram body weight in 98% miglyol and 2% DMSO. The rotenone was prepared fresh every other day. Rotenone powder was pipetted until thoroughly dissolved into DMSO. The stock solution was aliquoted into amber septa vials within a −20 °C freezer to avoid contact with light. The head space of the vial was purged with nitrogen to prevent oxidation of the compound. The optimal concentration of rotenone was determined from a dosing regimen consisting of intraperitoneal dosing daily for 14 days. Male and female C57Bl/6 mice (n=4/control group/time-point, n=7/rotenone group/time-point) were injected daily for 14 days with a dose of 2μL/g weight. Mice were weighed daily before injection to determine the appropriate dosing amount. The rotenone/miglyol was carefully measured using a 50μL Hamilton syringe and then transferred to an insulin syringe with permanently attached needle which was used to administer the volume of solution. Control animals received injections of miglyol only. Hamilton syringes were cleaned every-day after use to prevent precipitation buildup within the needle. The syringe and plunger were submersed in ten percent bleach for a duration of 10 minutes, then 70% ethanol was aspirated into the syringe followed by sterile water. The plunger was removed from the syringe and the instrument was allowed to air dry for 24 hours.
To identify the optimal dose of rotenone to induce loss of DAn, pilot studies were conducted in which mice were administered several concentrations of rotenone at different dosing schedules. Rotenone was dissolved in 100% miglyol 812 as described above. The final dosage used in subsequent studies was determined by quantitative analysis of DAn numbers within the SNpc using design-based stereology and scanning fluorescence microscopy. Regions of interest (ROI) were mapped for the ST and the SNpc. Male and female C57Bl/6 mice (n=4/group) were injected intraperitoneally with 2.0 mg/kg/day, 2.5 mg/Kg/day, 3.0 mg/kg/day and 1.4mg/kg twice daily. Twice daily dosing at 1.4 mg/Kg was highly toxic and omitted from further evaluation. Mice were terminated 14 days after the initial rotenone dose and neurodegeneration was assessed. Based upon initial results and minimal mortalities, a dose of 2.5 mg/Kg/day was selected for use in the 4-week study (n=4 animals/control groups, n=7 animals/rotenone groups).
2.3. Open Field Behavioral Analysis
Open field activity was assessed using the Versamax behavioral system with an infrared beam grid detection array (Accuscan Instruments, Inc., Columbus, OH). Mice were monitored for 5 minutes under low ambient light in the presence of white noise. Animals were pre-conditioned to the chambers the day before exposure started. The animals were recorded 1 hour prior to initial injection (0 DPI) to establish a baseline. Several behavioral assessments were performed at weekly timepoints (1 WPI, 2 WPI, 3 WPI and 4 WPI). Multiple behavioral parameters were obtained and analyzed using Versadat Software (Accuscan Instruments, Inc.) including rearing number, center time, margin time and rest time. We previously reported that these locomotor and behavioral parameters are accurate determinants of basal ganglia function associated with loss of striatal DA (Liu et al., 2006; Moreno et al., 2009). Data is reported as change from baseline (0 DPI) behavioral assessment. (n=10/control, n=14/rotenone).
2.4. Tissue Extraction and Fixation
Based upon initial pilot studies of rotenone-induced neurodegeneration in the SNpc, it was determined that a timepoint of 14 days or 2 weeks post initial injection (2 WPI) directly following the conclusion of the rotenone exposure would represent the first time-point for neuropathological analysis. Groups of mice were also evaluated each week for an additional two weeks (3 and 4 WPI, respectively) to identify neuropathological changes associated with progressive neuroinflammation following rotenone exposure. Mice (n=4/control group, n=7/rotenone group) were euthanized by isoflurane anesthesia followed by cardiac puncture. Perfusion-fixation was immediately performed using 20 mL of 0.1M phosphate buffered-saline sodium cacodylate buffer supplemented with heparin followed by 20 mL of 4% paraformaldehyde. Brain tissue was carefully removed once fully perfused and post-fixed in 10% neutral buffered formalin for two weeks at room temperature. The tissue was then dissected for cassette preparation, paraffin embedding, sectioning and histological examination.
2.5. Tissue Preparation and Automated High-throughput Immunofluorescent Staining
Paraffin embedded brain tissue was sectioned at 6μm thickness and mounted onto polyionic slides (Histotox Labs, Boulder, CO). Slides were deparaffinized and immunofluorescently labeled using a Leica Bond RXm automated robotic staining system. Antigen retrieval was performed by using Bond Epitope Retrieval Solution 1 and 2 for 20 minutes in conjunction with heat application. Sections were then incubated with primary antibodies diluted in 0.1% triton-X containing phosphate buffered saline (PBS): rabbit tyrosine hydroxylase (TH; Millipore; 1:500), mouse neuronal nuclei (NeuN; Millipore, 1:100), mouse glial acidic fibrillary protein (GFAP, Cell Signaling, 1:100), rabbit S100β (Abcam; 1:750), rat complement C3 (Abcam; 1:250), goat ionized calcium binding adaptor molecule 1 (IBA1; Abcam; 1:50), and mouse anti-alpha synuclein phosphorylation at serine position 129 (p129; Wako; 1:100). Sections were stained for DAPI (Sigma) and mounted on glass coverslips in ProLong Gold Antifade hard set mounting medium and stored at 4°C until time of imaging.
2.6. Histological Staining and Examination
Paraffin embedded brain tissue was sectioned at 6μm thickness and mounted to polyionic slides (Histotox Labs, Boulder, CO). Sections were deparaffinized and stained with hematoxylin and eosin. Each section was analyzed at a 10x and 40x magnification using an Olympus IX71 microscope (Center Valley, PA) with Retiga 2000R (Qimaging, Surrey, BC, Canada) and Qcolor3 (Olympus) camera and Slidebook software (v6.0, Intelligent Imaging Innovations, Inc., Denver, CO) for image acquisition and analysis. Stained SN and ST sections were investigated and read by a board-certified veterinary pathologist who was blinded to all groupings.
2.7. Unbiased Stereological Neuronal Counting and Striatal Terminal Analysis
Quantification of neurons was adapted from those previously reported (Bantle et al., 2019; Sadasivan et al., 2015; Tapias and Greenamyre, 2014). In brief, every 20th tissue section was selected for staining and counted, resulting in six sections per animal being counted. The studies described here were conducted blindly by a single investigator. Images were captured using an automated Olympus BX63 fluorescence microscope equipped with a Hamamatsu ORCA-flash 4.0 LT CCD camera and collected using Olympus CellSens software (v1.15). Quantitative analysis was performed on dually labeled fluorescent images generated by montage imaging of the entire coronal brain section by compiling single 100X magnification images acquired by using an Olympus X-Apochromat 10X air objective (0.40 N.A.). One hemisphere of the section was quantified by creating anatomically specific ROIs based on TH immunolabeling and reference to a coronal atlas of the mouse brain (Allen Brain Atlas). All images were obtained and analyzed under the same conditions for magnification, exposure time, LED intensity, camera gain and filter settings.
For quantitative assessment, TH+ and NeuN+ soma from the selected areas determined by the ROI parameters were semi-automatically counted by adaptive thresholding in the Count and Measure feature on the Olympus CellSens platform. Object filters for cellular perimeter size, minimum and maximum area, and shape factor thresholding were applied. Quantitative stereology analysis using the motorized stage method was performed as descried (Tapias and Greenamyre, 2014).
Caudal striatal sections containing the ST and the globus pallidus (GP) were stained dually or triply including TH as the main marker of interest. All slides were stained and imaged simultaneously to reduce variability in intensity measurements. Whole brain montaging was performed using a 10X Olympus X-Apochromat air objective (0.04 N.A.). ROIs specific to the individualized striatal anatomical brain region were applied and total average fluorescence intensity was determined by manual threshold masking within the CellSens platform. Representative whole brain montage images were generated using the 10X air objective and high magnification inserts were acquired using the Olympus X-Apochromat 100X oil objective (1.4 N.A.).
2.8. Skeletonization and Determination of Microglial Cell Phenotype
Skeletonization analysis was performed as previously described (Morrison et al., 2017). In brief, five randomized 400X images spanning the entirety of the substantia nigra pars reticulata (SNpr) were taken using a Olympus X-Apochromat 40X air objective (0.95 N.A). Image software (National Institutes of Health, Version 1.5a) and appropriate plug-ins (FFT bandpass filter, unsharp mask, and close) were downloaded and applied to each of the images prior to converting to binary and skeletonized images. Cell soma quantification was obtained by counting DAPI positive nuclei that co-localized with IBA1 cytoplasmic staining. The Analyze Skeleton Plugin (developed and maintained by http://imagej.net/AnalyzeSkeleton3) was applied to all the skeletonized images. DAPI-positive nuclei were measured using the straight-line tool in order to determine a minimum positive threshold length that a respective generated data point had to meet to be considered in the data set allowing only for the counting of cells within the representative Z-plane counting frame. Data was then compiled and filtered further by endpoint number and threshold sizing. Microglia soma counts were then used to determine microglial branches/cell, branch length/cell and microglial junctions/cell.
2.9. Semi-Automated Glial Counting and Intensity Measurements within the Substantia Nigra and Striatum
Methods used to quantify values of glial cells throughout brain regions were performed as previously described (Bantle et al., 2019). Two total sections per animal were selected for glial counts based on anatomical region and proximity to slides that were used to quantify DAn values. The studies described herein were performed blinded by a single investigator. Images utilized for quantification were captured using an automated BX63 fluorescence microscope equipped with a Hamamatsu ORCA-flash 4.0 LT CCD camera and collected using Olympus CellSens software. Quantitative analysis was performed on dual or triple-labeled fluorescent images through full brain montage assessment. Each montage consisted of compiled individual 100X images taken using an Olympus X-Apochromat 10X air objective (0.4 N.A.). Dual hemispherical analysis was performed on active ROIs determined by anatomical structures identified by immunofluorescent labeling and referenced to the coronal mouse brain atlas (Allen Brain Atlas). All slides were imaged and scanned using the same conditions of exposure time, binning time, magnification, lamp intensity and camera gain.
For identification of GFAP+ and IBA1+ cells, soma from the ROI labeled areas were detected using semi-automated cell counting software on the CellSens platform. Total area of soma, fluorescent intensity, mean grey intensity and cell shape object filters were used to determine positive cells by manual adjustment to identify thresholding parameters for accurate quantification of glial cells. Colocalization of S100β and complement protein C3 (C3) was determined by using the co-localization function on the CellSens platform, channel minimum and maximum were manually determined to ensure accuracy of dual label identification. Pearson correlation coefficients were determined and the colocalization algorithm was not accepted if the R value was below 0.85. Two sections per animal were used to identify glial specific markers within manually created ROIs, the total number of cells were then divided by the overall area (mm2) of the ROI to allow for normalization across dosing groups.
S100β and C3 colocalization was quantified by utilizing the co-localization feature within the Count and Measure module of CellSens. Minimum and maximum intensity thresholds were manually determined for each staining channel per individual section to account for intensity variability between sections. Area of soma exclusion criterion was applied following completion of the algorithm further selecting for co-localized, dual-labeled cells. The number of infiltrating monocytes were quantified based upon immunohistochemical staining and morphological criteria as reported previously (Bantle et al., 2021).
2.10. Artificial Intelligence-Based Identification of S100β+ Astrocytes in Whole Brain Montage Images
Whole brain montage images acquired at 10X magnification using the same imaging parameters per section were utilized for quantification of S100β+ cellular populations. Anatomical ROIs were hand drawn for each section to encompass the SNpc and SNpr, per standard anatomical landmarks (Allen Brain Atlas). Olympus CellSens software was used for analysis and identification of cells using the Deep Learning module (v3.1). Training labels were created on a total of 8 sections across 4 control animals to train the artificial intelligence (AI) algorithm what a ‘positive event’ is (e.g., a cell), based on area, diameter, circumference, fluorescence intensity, multiple channel intensity ratios and cellular elongation factor. After 250 training labels were created spanning all animals and ROIs, neural networking generation was performed on standard basis with a total of 25,000 – 50,000 iterations including cellular identification based on fluorescence intensity of S100β+ cells and DAPI intensity, with exclusion of background tissue architecture and intensity. Therefore, identification of astrocytic cells was made only possible if all criteria were met. The network was deemed adequate at the iteration where the similarity score was ≥0.80. Total objects within each ROI were detected and counted using the Count and Measure feature of Olympus CellSens where manual thresholding was applied to accurately detect the AI identified S100β+ cells. Total cell counts were then normalized to the total area of the ROI.
2.11. Immunohistochemical Staining and Quantification of Protein Aggregates
Sections were processed for histology and immunohistochemistry using a Leica Bond-III RXm automated staining system according to the manufacturer’s protocols. Antigen retrieval was performed in Bond Epitope Retrieval Solution 1 and 2 for 20 minutes at 37 °C. Reactive p129 cell/cell aggregates were stained using mouse monoclonal anti-phospho Ser129 (P129) antibody (1:100, clone pSYN#64, WAKO ) (Jang et al., 2009). Immunoreactions and neuropathological scoring of P129+ protein aggregates were conducted in the midbrain and ST on exposed and unexposed brain sections by a veterinary pathologist blinded to the exposure groups using scoring methodology that was adapted from previous reports (Rey et al., 2016; Rey et al., 2018). We assessed the presence of P129+ inclusions on two coronal sections per animal that were 6μm in thickness with an n=4 control group/timepoint, n=7 rotenone group/timepoint for each exposure group. Each section was analyzed at a 10x and 40x magnification using an Olympus IX71 microscope (Center Valley, PA) with Retiga 2000R (Qimaging, Surrey, BC, Canada) and Qcolor3 (Olympus) camera and slidebook software (v6.0, Intelligent Imaging Innovations, Inc., Denver, CO) for image acquisition and analysis. A score of 0 to 5 was assigned to each brain region from a single coronal brain section and scored as follows: 0=no aggregation, 1= very sparse/few (1–2) p129+ intra-cellular aggregates per cell per high magnification field, 2=mild (<10) p129+ intra-cellular aggregates per cell per high magnification field, 3= moderate (<15) p129+ intra-cellular aggregates per cell per high magnification field, 4=marked (<20) p129+ intra-cellular aggregates per cell per high magnification field, 5= severe (>20) p129+ intra and extra cellular aggregates per cell/surrounding cellular space per high magnification field.
2.12. Proteinase K Digestion of Paraffin Embedded Tissue Sections
Paraffin embedded tissue sections (6um) were ran on a Leica Bond RXm automated robotic staining system. Sections were dewaxed and epitopes were exposed using Leica Bond Epitope retrieval buffer 1. Sections were then permeabilized and treated with 100ug/mL proteinase K (pK, Roche) at 37 degrees Celsius for 30 minutes. This was followed by serum blocking and primary antibody application. Sections that did not receive pK received 1X tris-buffered saline incubations at 37 °C for 30 minutes. Appropriate secondary antibodies were applied to the sections and all images were obtained on an automated BX63 fluorescence microscope equipped with a Hamamatsu ORCA-flash 4.0 LT CCD camera and collected using Olympus CellSens software. Sections that were previously quantified for being high p129 aggregate yield through IF and IHC pathological scoring were used as pK controls, to determine pK effectivity, along with absence of IBA1 and TH staining positivity.
2.13. Generation of Representative Normalized Pathological Overlays
To generate the representative overlays at each time point, the total number of DAn, microglia, macrophages, astrocytes cellular counts and P129 pathological scores from infected mice were normalized to controls according to the following equation: ((x-y)/(y-z))*100=normalized percentage value (Gopal Krishna Patro and Kumar Sahu, 2015). The control values obtained for each pathological parameter were averaged and then subtracted from the individual experimentally infected animal values yielding a pathological representative of activation (x). The minimum (y) and maximum (z) values were determined for each activation parameter dataset. Normalization was then performed by determining the difference between the control subtracted experimental infected values and the minimum overall value. The total obtained from this calculation serves as the numerator. The range of the data set was then determined by obtaining the difference from the minimum (y) and maximum (z) values, this would serve as the denominator. The value obtained from this overall calculation was then multiplied by 100 to represent total percentage activation of each parameter. The respective percentages at each time point (2, 3 and 4 WPI) were averaged and plotted using spline curve fitting within GraphPad software (version 9.1.0; Graph Pad Software, San Diego, CA).
2.14. Nigrostriatal Tract Mapping
Online access of Allen Brain Atlas mouse brain connectivity atlas (https://connectivity.brain-map.org/) was utilized to identify source search of substantia nigra (SNpc, SNpr) and target source of ST within C57BL/6J mice. Beta 3D browser-based atlas was then used to visualize mouse anatomy and axial projection from database AAV injection experiment number 100141993. Structural mapping was added with visualization tools and view planes of the tract were downloaded.
2.15. Statistical Analysis
All data was presented as mean +/− SEM, unless otherwise noted. Experimental values from each mean were analyzed with a ROUT (alpha=0.05) test to identify significant outliers and validate exclusions. Differences between time-exposure group was identified using a two-way ANOVA, where individual timepoint comparisons between exposure groupings were carried out using Sidak’s post hoc multiple comparison test. Significance was identified as *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. All statistical analysis was performed using Prism (version 9.1.0; Graph Pad Software; San Diego, CA).
3. Results
3.1. Behavioral and locomotor deficits resulting from chronic rotenone exposure in mice
Following optimization of rotenone dosing based on survival curve analysis (Fig. 1A), the effects of subacute systemic rotenone exposure on neurobehavioral function in C57Bl/6 mice were determined by open-field activity analysis. Heat maps (Fig. 1B,C) and linear pattern tracking graphs (Fig. 1D,E) of behavioral function revealed anxiety-like and depressive behavior and an overall reduction in exploratory movement, with increased time spent towards the margins of the chamber. Relative to individual baseline measurements, there were increases in rest time in mice exposed to rotenone compared to miglyol-exposed control animals (Fig. 1F). There were also trens of decreased rearing events from the rotenone animals at all timepoints, where the parameter of time accounted for the most variance. There were also decreases observed with the amount of time spent in the center of the chamber at 1 WPI in the rotenone-exposed group (Fig. 1G,I), with a corresponding increase in margin time (Fig. 1H) where the combination thereof rotenone exposure and time accounted for significant variance within regards to center time.
Figure 1. Behavioral and locomotor changes within rotenone treated animals occur prior to Nf-kappaB activation.
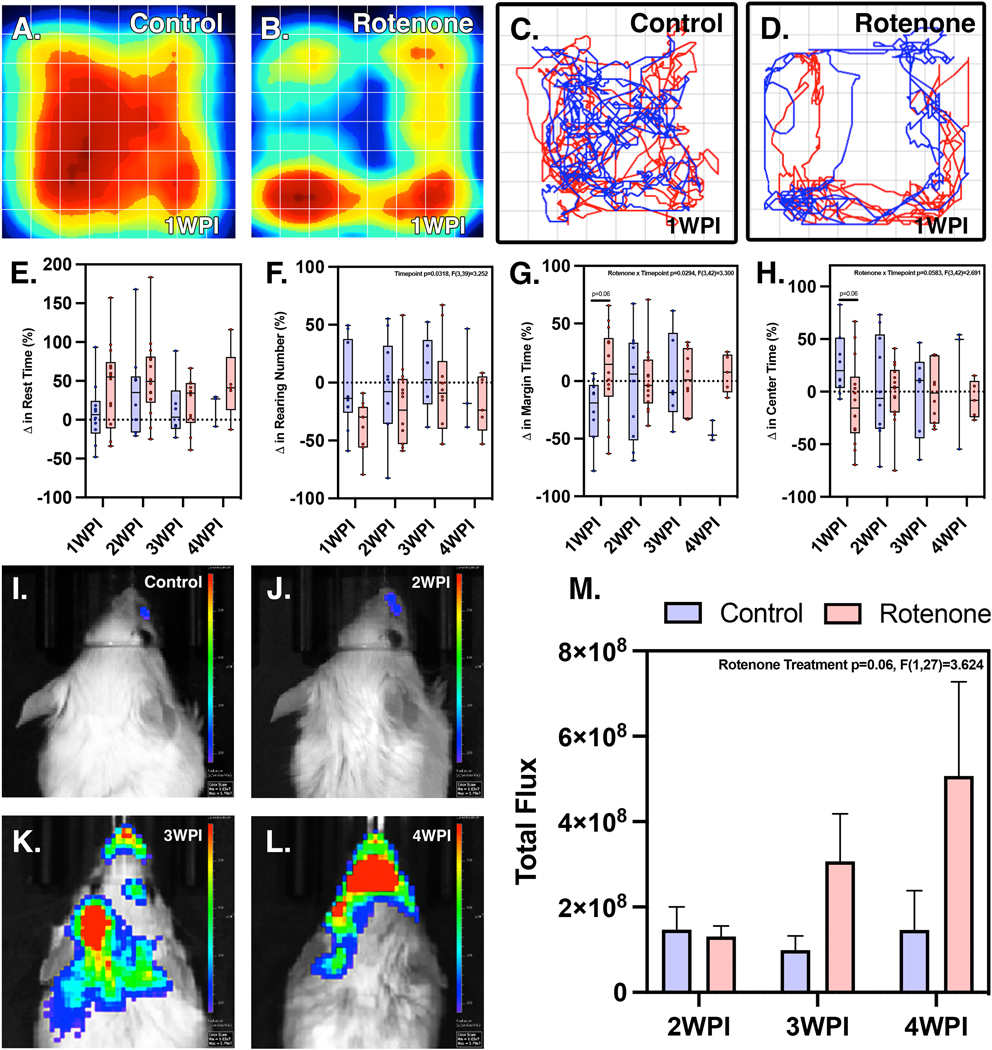
Open field tracking of C57/Bl6 behavioral patterns through heat mapping and linear movement recognition, respectively, in control (A,B) and rotenone treated animals (C,D) show behavioral differences. Quantitative analysis of changes in rest time (E), rearing number (F), margin time (G), and center time (H) are shown. (N=4 control mice/group, N=7 rotenone mice/group) NF-kappaB-GFP-Luc dual reporter animals show increased levels of Nf-kappB luciferase photon flux beginning at 3 WPI (K) and increasing at 4 WPI (L). Whereas 2 WPI (J) reflects that of control (I) levels. Rotenone exposure within these animals is sufficient enough to induce inflammatory activation and successive progression to the 4 WPI time-point (M). (2 WPI: n=8 control, n=9 rotenone; 3 WPI: n=7 control, n=7 rotenone; 4 WPI: n=6 control, n=5 rotenone)
To determine the extent of neuroinflammation associated with rotenone exposure, transgenic NF-κB-GFP-luciferase dual reporter mice were administered rotenone for 14 days followed by two weeks of recovery during which the bioluminescence signal of the NF-κB-luciferase reporter was measured weekly. Following systemic administration of rotenone, expression of the NF-κB reporter as determined by whole-body bioluminescence imaging revealed an increase in reporter signal from 3 – 4 WPI (Fig. 1J–M). Total flux of photons per second was determined for an equivalent region of interest (ROI) for each timepoint (Fig. 1N), indicating an increasing linear trend in the rotenone exposed group (Two-way ANOVA, p<0.06, F(1,27)=3.624) from 2 WPI to 4 WPI.
These findings were consistent with histopathological examination of nigro-striatal nuclei performed by a veterinary pathologist at 2, 3 and 4 WPI with rotenone (Supplementary Figure 1). This analysis revealed focal neuronal apoptosis in the substantia nigra and caudate-putamen in rotenone-treated animals compared to the control group, where affected neurons appeared more angular and contained hyper-eosinophilic cytoplasm. Pyknotic and karyolitic nuclei were present with focal gliosis manifesting in occasional satellitosis and scattered Alzheimer Type II astrocytosis. This was accompanied by multifocal gliosis within the ST, especially within the striosomes, which exhibited axonal profile interruptions. By 4 WPI, the SN showed an overall decrease in neuronal density that was more pronounced in the left hemisphere. Apoptotic neurons were pyknotic and angular with decreased overall size, as well as separated from the neuropil, leaving prominent pericellular clear spaces.
3.2. Microglial activation and phenotypic profiling reveals dynamic M2-M1 conversion in response to rotenone
Phenotypic conversion and pathological activation of microglia was assessed by quantification of IBA1+ cell number and morphology within the SNpc, SNpr, ST and GP (Figure 2). Dual immunolabeling with IBA1 and TH was used to identify microglial cell populations within DA brain regions. Montage images of entire coronal brain sections were scanned using a 10X objective and high-resolution images were acquired using a 100X objective. Representative baseline levels of microglia were present in the SN, ST and GP of control animals (Fig. 2 A,B). Exposure to rotenone resulted in increased microglial cell populations in the SNpc and SNpr by 2 WPI (Fig. 2C), whereas the ST had minimal levels of microglial hypercellularity at this timepoint (Fig. 2D). Microglial phenotyping conducted at 2 WPI by skeletonization and filament tracing analysis revealed conversion of resting/ramified microglia (M2) within the SN to activated ameboid microglia (M1), characterized by reduction in branch length, number of branches per cell and the total number of junctions per cell (Fig. 2J, Q–S). At 3 WPI there was reduced activation and recruitment of microglia in all brain regions examined, except for the SNpr where increases in microglial cell population and activation states were still present (Fig. 2E,F). Individual microglial cells within the SN were seen retaining a M1 phenotypic state (Fig. 2K, Q–S), similar to the activated amoeboid state observed at 2 WPI. By 4 WPI microglia showed decreased activation and proliferation within the SNpc, SNpr, ST, and GP (Fig. 2G, H), with a phenotypic return to a more ramified state characterized by increased branch length, number of branches and number of junctions per cell (Fig. 2L, Q – S). Quantification of the number of microglia within each region over time was calculated by semi-automated analysis of cells in designated ROIs encompassing anatomically consistent sections within the SNpc (Fig. 2M), SNpr (Fig. 2N), ST (Fig. 2O) and GP (Fig. 2P). Microglia were discriminated from infiltrating peripheral macrophages by a previously established protocol that differentiates based on size and morphological differences and slides were read blinded by a pathologist (Smeyne et al., 2016). The number of infiltrating macrophages was determined within the entire SN and showed an increasing trend starting at 2 WPI and peaking at 4 WPI (Fig. 2T). Inter-regional normalization of IBA1 positive cells was performed in each brain region relative to the start of rotenone exposure to model the temporal changes in cellular number over time with respect to the progression of rotenone-induced pathological changes. Regions are denoted as follows (Fig. 2U): SNpc (pink), SNpr (cyan), ST (red), SN - infiltrating macrophages (dashed green).Normalization of cellular responses indicated that microglial reactivity peaked first in the ST at 2 WPI followed by the SNpr at 3 WPI, with a secondary increase in microglia in the ST at 4 WPI. In contrast, the number of microglia in the SNpc peaked between 2 and 3 WPI. Infiltrating macrophage numbers within the SN increased steadily from 2 WPI to 4 WPI, coinciding with the decrease of IBA1+ microglia within the SNpc and SNpr.
Figure 2. Systemic administration of rotenone causes temporally and regionally distinct patterns of microglia activation.

Microglial activation from an M2 phenotypic state to an active ameboid M1 phenotype was investigated in control (A,B) and rotenone-treated animals at 2 WPI (C,D), 3 WPI (E,F) and 4 WPI (G,H) within the substantia nigra and the ST, respectively (TH, green). Analysis of microglia phenotype by skeletonization and filament tracing was conducted in the SNpr in control (I), 2 WPI (J), 3 WPI (K) and 4 WPI (L) animals. Quantification of IBA1+ cells (cyan) associated with microgliosis/microcytosis was performed within the SNpc (M), SNpr (N), ST (O) and GP (P) at all timepoints. (Q-S) Changes in length of branches per cell (Q), number of branches per cell (R) and number of junctions per cell (S) was also quantified at each timepoint. (T) The number of invading monocytes was quantified in the substantia nigra. (U) Normalized counts of IBA1+ cells in the SNpc (pink), SNpr (cyan), ST (red) and invading macrophages within the SNpc (green dashed line) were modeled over the 4-week time course of the study. (N=4 mice/control group, N=7 mice/rotenone group) *p<0.05, **p<0.01, ***p<0.001
3.3. Rotenone toxicity induces a reactive A1 phenotype in astrocytes in multiple brain regions
The changes in the inflammatory phenotype of astrocytes in response to rotenone exposure was determined by quantifying the number of S100β+ (Supplemental Figure 2) and GFAP+ cells in concert with overall fluorescence intensity of GFAP, as well as the number of cells co-expressing S100β and C3, a marker for A1 reactive astrocytes (Figure 3). In control animals, astrocytes displayed a resting stellate morphology in all brain regions examined, without evidence of hypertrophy or other features of inflammatory reactivity (Fig. 3A,B). By 2 WPI there were increased numbers of hypertrophic GFAP+ cells in rotenone-treated animals, primarily in the SNpc, with no increase detected in the SNpr, ST or GP (Fig. 3C,D). This hypertrophy in GFAP+ cells was less evident by 3 WPI in the SNpc and decreased further by 4 WPI (Fig. 3E,F and G,H). Quantification of the number of GFAP+ cells and GFAP fluorescence intensity in each brain region supported these observations (Fig. 3I,J); two-way ANOVA analysis indicated a significant change in the number of GFAP+ cells where rotenone exposure and time were parameters investigated within the SNpc (p=0.026, F(2,51)=3.923) and GFAP+ intensity in the SNpr (p=0.038, F(2,43)=4.583). Expression of GFAP in the SNpr increased from 3 WPI to 4 WPI, later than that in the SNpc. The ST, interestingly, showed decreases in the number of reactive GFAP+ astrocytes at 2 WPI (Fig. 3D), followed by levels similar to control at 3 WPI and 4 WPI (Fig. 3F,H,K). In rotenone-exposed mice, the GP did not show any differences from that of control animals at any timepoint investigated (Fig. 3L). The number of A1 astrocytes was determined in each region by co-immunolabeling to identify cells positive for expression of S100β and C3. The number of S100β+ cells co-expressing C3 did not change in the SNpc at any timepoint (Fig. 3M) but increased in the SNpr at 3 WPI (Fig. 3N). Modeling normalized changes in GFAP-expressing cells over time in the SN and ST indicated peaks in A1 astrocytes in the SNpc occurring early at 2 WPI, followed by the SNpr and ST at 3 WPI (Fig. 3O).
Figure 3. Rotenone exposure induces activation of astrocytes in the substantia nigra prior to the appearance of reactive A1 astrocytes in the striatum.
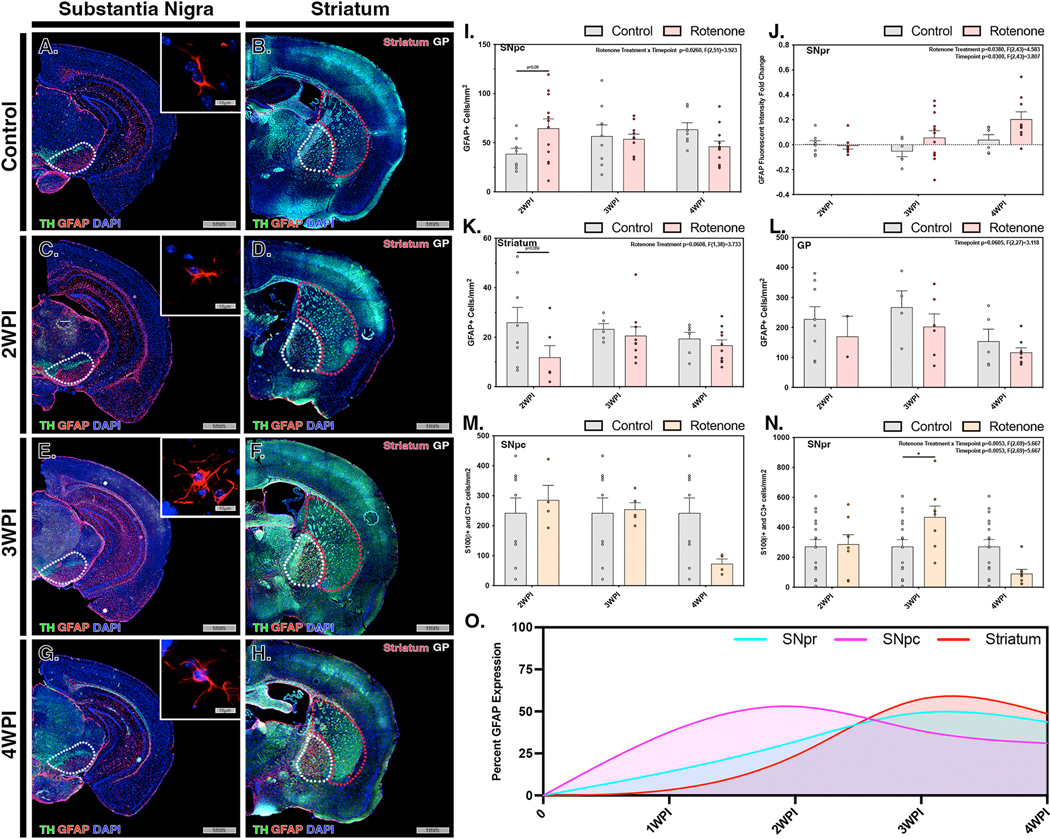
Activation of astrocytes (GFAP, red) within the substantia nigra (TH, green) and ST were investigated in control (A,B) and rotenone-treated mice at 2 WPI (C,D), 3 WPI (E,F) and 4 WPI (G,H). Astrocytosis/astrogliosis was quantified in the SNpc (I), SNpr (J), ST (K) and GP (L) at 2 WPI, 3 WPI, and 4 WPI. Cell number and intensity was normalized to the respective control for each region to determine overall activation patterns throughout multiple brain regions at each timepoint. The number of A1 reactive astrocytes was determined by immunolabeling and co-localization of S100β + C3 in the SNpc (M) and SNpr (N) for all timepoints. (O) Normalized GFAP intensity for the SNpr (cyan), SNpc (pink) and ST (red) was modeling for all regions at each timepoint. (N=4 mice/control group, N=7 mice/rotenone group) *p<0.05
3.4. Rotenone exposure causes progressive loss of dopaminergic neurons and projecting striatal fibers
To determine the extent of neuronal loss in response to systemic administration of rotenone in C57Bl/6 mice, the number of neurons in multiple brain regions were assessed by quantitative stereology using scanning fluorescence microscopy of whole-brain sections coupled to semi-automated image analysis as described previously (Tapias et al., 2013) (Figure 4). Imaging of DAn within the SNpc of control animals showed healthy neuronal cells forming a densely populated layer projecting to an intact neuropil, as well as intact DA terminals in the ST (Fig. 4A,B). By the end of rotenone exposure (2 WPI), there were evident deprecations in neuronal integrity and prevalence in the SNpc, without clear changes in TH staining in the ST (Fig. 4C,D). At 3 WPI, neurons began to decrease in radial size and lose morphological integrity, including loss of dendritic branching, decreased axon size and the appearance of cells with pyknotic nuclei. (Fig. 4E). Increases in the intensity of TH+ DA terminals in the ST also became evident by 3 WPI (Fig. 4F). By the second week following the end of rotenone exposure (4 WPI), there was clear loss of DA neurons within the SNpc with progressive neurodegeneration characterized by decreases in the diameter of neuronal soma, loss of morphological integrity and an increase in the number of remaining cells that had pyknotic and fragmented nuclei (Fig. 4G). Striatal terminal integrity largely decreased at 4 WPI, with progressive loss of terminal intensity compared to the end of rotenone exposure at 2 WPI (Fig. 4H). Quantification of the number of TH+ neurons in the SNpc and the intensity of TH staining in the ST confirmed these observations (Fig. 4I,J), indicating an approximate 35% loss in DA neurons, most of which occurred in the two week lesioning period following the end of rotenone administration.
Figure 4. Rotenone induces selective loss of dopaminergic neurons in the substantia nigra pars compacta occurs that follows maximal inflammatory activation of glial cells.
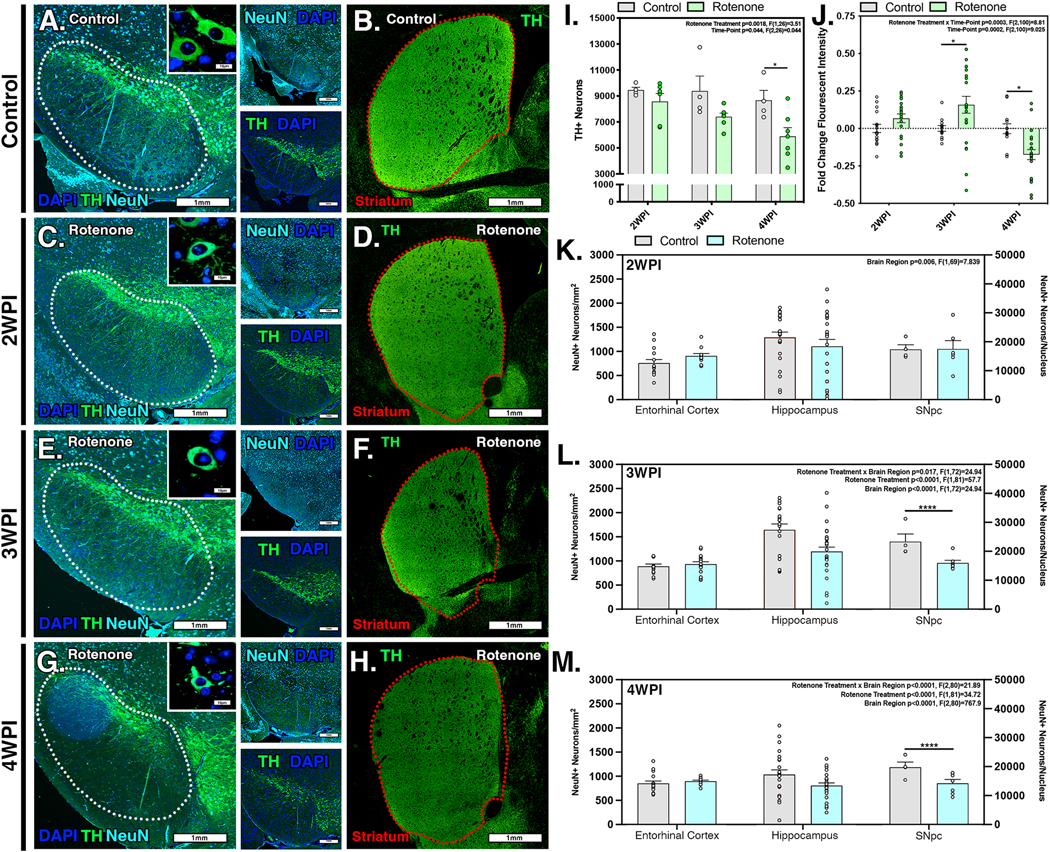
The number of DAn (TH, green) and total neurons (NeuN, cyan) in the substantia nigra was examined by quantitative stereology in control (A) and rotenone-treated mice at 2 WPI (C), 3 WPI (E), and 4 WPI (G). The intensity of TH+ DAn terminals within the ST was also determined at each timepoint (B,D,F,H). (I) Quantification of the number of DAn and (J) dopaminergic terminals in the ST. The number of NeuN+ neurons was quantified in the hippocampus, entorhinal cortex and SNpc at 2 WPI (K), 3 WPI (L), and 4 WPI (M). (N=4 mice/control group, N=7 mice/rotenone group) *p<0.05, ****p<0.0001
To determine if neurodegeneration was specific to TH+ neurons in the SNpc or generalized throughout the brain, NeuN staining was used to examine the total number of neurons in multiple brain regions. Total neuronal counts within the SNpc were unchanged at 2 WPI, indicating that no neurodegeneration had yet occurred after two weeks of rotenone exposure (Fig. 4K). Likewise, no loss of NeuN+ cells was detected in either the hippocampus or entorhinal cortex at this timepoint. However, during the lesioning period from 3 to 4 WPI with rotenone, there was a significant and progressive decrease in the total number of NeuN+ cells within the nucleus of the SNpc (p<0.001 for rotenone exposure, F(1,81)=34.72). No change in the number of NeuN+ cells was detected in the hippocampus or entorhinal cortex during the same period (Fig. 4L,M).
3.5. Rotenone exposure induces the formation of proteinase K-resistant α-synuclein aggregates
Alpha-synuclein aggregation and vacuolization is a hallmark of PD and is an important determinant of disease progression (Mehra et al., 2019). To characterize the formation and spread of p129 aggregates in response to rotenone exposure, immunohistochemical staining and pathological scoring of p129 was conducted in the SNpc and ST. Pathological scoring of the extent of aggregate formation was conducted by a veterinary pathologist using a ranking scale ranging from 0–5 according to the following metrics: 0=no aggregation, 1= very sparse/few (1–2) p129+ intracellular aggregates per cell per high magnification field, 2=mild (<10) p129+ intracellular aggregates per cell per high magnification field, 3= moderate (<15) p129+ intracellular aggregates per cell per high magnification field, 4=marked (<20) p129+ intracellular aggregates per cell per high magnification field, 5= severe (>20) p129+ intra and extra cellular aggregates per cell/surrounding cellular space per high magnification field. Three dimensional maps of the nigro-striatal tract showing neuronal connectivity (highlighted in white) were generated using the pathway mapping feature of the Allan Brain Atlas (Fig. 5A,B). Region-specific analysis did not show significant increases in p129 accumulation at the 2 WPI timepoint within the SN (Fig. 5E, K) but did show increases in the p129 staining within the ST, as noted by change in pathological score (Fig. 5F, L) compared to control animals (Fig. 5C,D). Interestingly, at 3 WPI there was increased p129 aggregation in the SN (Fig. 5G, K) and a corresponding decrease within the ST (Fig. 5H, L). By the 4 WPI timepoint there was further accumulation of p129 aggregates in the SN and elevation in the pathological score within the ST above that of controls (Fig. 5I–L). The most prominent difference in misfolded protein occurred two weeks after the end of rotenone exposure (4 WPI), with the appearance of intensely staining intracellular aggregates of p129-Syn in neuronal perikariya, with additional staining evident in glial cells surrounding p129+ neurons.
Figure 5. Pathological scoring of alpha-synuclein aggregates in the substantia nigra and striatum following systemic administration of rotenone.

(A,B) Three dimensional models of neuronal connectivity (white) in the nigro-striatal pathway with spatial orientation to the olfactory bulb (OB) were generated from the Allan Brain Atlas. Paraffin-embedded sections were immunolabeled for phospho-(Ser129)−α−synuclein (p129) evaluated by blinded pathological scoring for the severity of aggregate formation in the SN and ST in control (C,D) and rotenone-treated animals at 2 WPI (E,F), 3 WPI (G,H) and 4 WPI (I,J). Quantitative comparison of pathological scores for the SN (K) and ST (L) for control and rotenone-treated animals was performed at each timepoint. (N=4 mice/control group, N=7 mice/rotenone group) *p<0.05, ****p<0.0001
To further investigate cellular responses to accumulation of p129 aggregates within individual brain regions, we used immunofluorescence imaging to detect the presence of p129 aggregates within both DAn and microglia in the SN and ST (Figure 6). Overall accumulation of p129 was measured within the SNpc, SNpr and ST at each timepoint (Fig. 6A–H). In control animals, little to no staining for p129 was detected in neuronal soma or in surrounding microglia, which were few in number and displayed a reticulated resting phenotype (Fig. 6A,E). From 2 WPI at the conclusion of rotenone exposure to 4 WPI, progressive increases in intracellular p129 were evident as intracellular puncta within both TH+ neurons and microglia in the SN (Fig. 6B–D), as well as in TH+ fibers and microglia in the ST (Fig. 6F–H). Quantification of total p129 fluorescence intensity by brain region indicated that p129+ aggregates increased in the ST by 3 WPI, before the increase observed in the SN at 4 WPI (Fig. 6I–K). Levels of p129 in microglia increased at 4 WPI in the SN but were unchanged in the ST at the timepoints evaluated (Fig. 6L–N). In TH+ neurons, p129 aggregates detected as fluorescence puncta in peri-nuclear regions were elevated over control at all timepoints (Fig. 6O). Normalized expression of overall p129 expression was modeled within the brain regions, indicating peak accumulation within the SNpr at 2 WPI followed by the ST at 3 WPI and a bimodal increase within the SNpc at 2 and 4 WPI (Fig. 6P).
Figure 6. Formation and trafficking of alpha-synuclein protein aggregates in neurons and microglia following rotenone exposure.
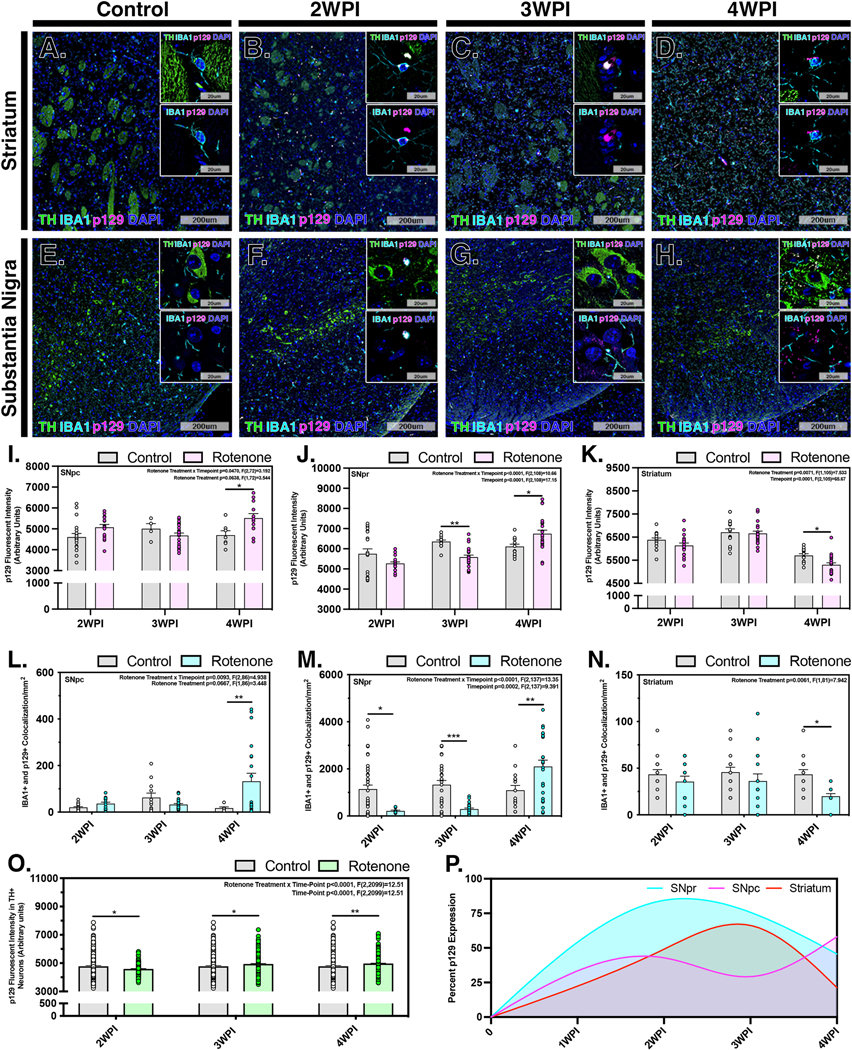
Accumulation of p129 was determined in the ST and SNpc in control (A,E) and rotenone-treated animals at 2 WPI (B,F), 3 WPI (C,G) and 4 WPI (D,H). Representative images of brain sections immunnolabeled for p129 aggregates (p129, pink), microglia (IBA1, cyan) and dopaminergic neurons and terminals (TH, green) are depicted for respective brain regions at each timepoint. Overall intensity measurements of p129 accumulation were determined within the SNpc, SNpr and ST at 2 WPI, 3 WPI and 4 WPI in the SNpc (I), SNpr (J) and ST (K). Co-localization of p129 aggregates in microglia was determined at each timepoint in the SNpc (L), SNpr (M) and ST (N). The intensity of p129 aggregates in DAn was also determined at each timepoint (O). (P) The overall fluorescence intensity of p129 was plotted for each brain region over time to established a normalized pathological model of disease progression. (N=4 mice/control group, N=7 mice/rotenone group) *p<0.05, **p<0.01, ***p<0.001
Proteinase K (pK) digestion studies were performed to determine the stability of protein aggregates detected in the SN in rotenone-exposed animals to proteolytic degradation (Figure 7). In brain sections from control animals very little aggregated p129 was detected in the SN by 4 WPI, seen as a lack of staining intensity following digestion with pK, which fully degraded the intact TH epitope (Fig. 7A). In brain sections from rotenone-exposed animals subjected to pK digestion, intense staining for p129 was still evident (Fig. 7B) that co-localized with TH+ DAn, as seen in companion sections not incubated with pK (Fig. 7C). High resolution microscopic analysis revealed the presence of largely intracellular puncta within DAn in the SNpc that gradually accumulated from 2 – 4 WPI, with the highest levels occurring at latest timepoint evaluated. At this timepoint, p129+ puncta were present throughout the neuronal perikaryon as well as in neighboring microglia (Fig. 7D–H). A similar pattern of accumulation was detected in TH+ fibers in the ST, which peaked at 3 WPI and were similarly present within surrounding microglia (Fig. 7I–M). Microglial trafficking and dissemination of p129+ aggregates was also investigated, revealing IBA1+ cells located within the corpus callosum (Fig. 7S), peri-vascular areas (Fig. 7T), choroid plexus (Fig. 7U), third ventricle (Fig. 7U) and migrating along TH+ projecting neuronal fibers within the nigro-striatal pathway (Fig. 7V), as well as localized to peri-nuclear regions of the cell. There was a relative paucity of microglia in control animals in these brain regions and none that contained P129+ aggregates (Fig. 7N–R).
Figure 7. Microglia clear proteinase K-resistant α-synuclein aggregates in the brains of rotenone-treated mice.
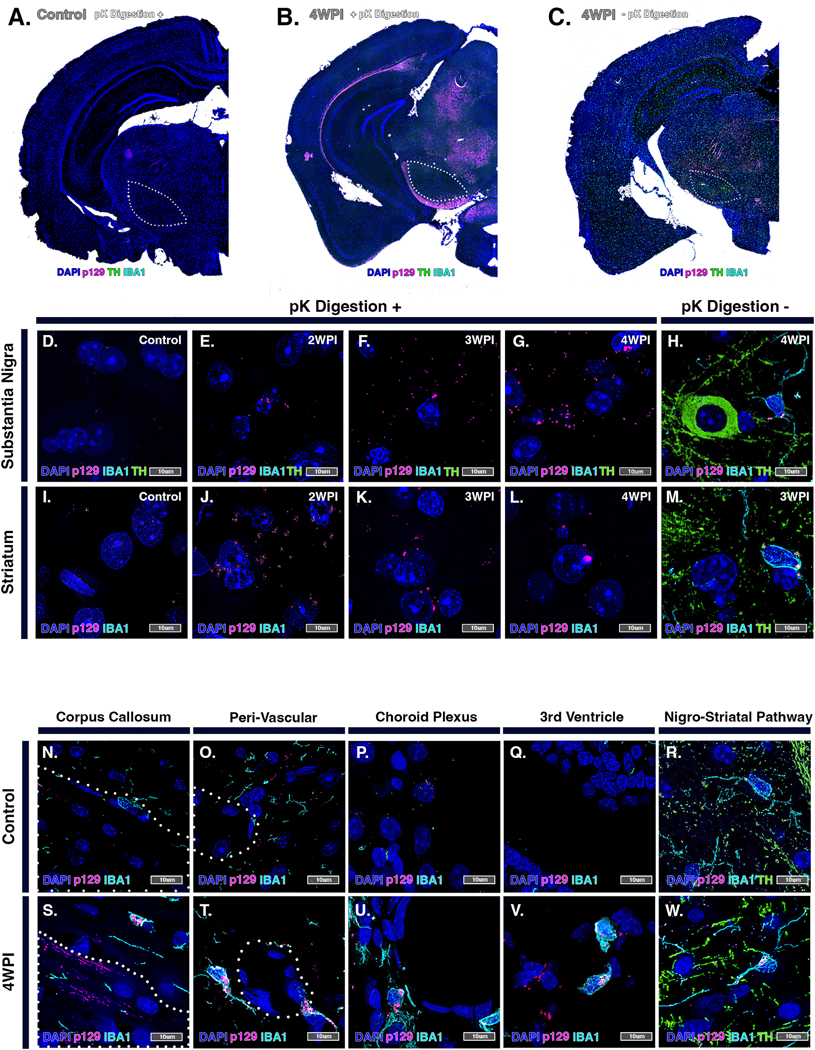
(A-C) High resolution hemispheric montage images of pK-digested brain sections from control (A) and rotenone-treated (B) animals, as well as a representative section from a rotenone-treated animal (−)pK digestion at 4 WPI (C). TH (green), p129 (pink) and IBA1 (cyan). High magnification images of pK-digested brain sections from control (D,I) and rotenone-treated groups at 2 WPI (E,J), 3 WPI (F,K) and 4 WPI (G,L). (H,M) 4 WPI rotenone-treated tissue without pK digestion. By comparing control (N-R) and rotenone-treated (S-W) brain tissue immunolabeled for IBA1, p129 and TH, IBA1+ cells were identified trafficking intracellular p129+ aggregates in rotenone-treated brains within the corpus callosum (S), peri-vascular areas (T), choroid plexus (U), third ventricle (V) and nigro-striatal pathway (W).
3.6. Glial-glial and glial-neuronal interactions mediate the progression of protein misfolding and neuronal injury following exposure to rotenone
The overall progression of pathological changes in the nigro-striatal pathway following systemic administration of rotenone for two weeks was modeled in Figure 8. Three dimensional pathways were overlayed with region-specific changes in microglial activation, p129 accumulation, astrocyte activation and loss of DA neurons, presented as a longitudinal model to depict connectivity of these pathological alterations between brain regions (Fig. 8A). This mapping approach indicates temporal, regional and cellular specificity with respect to the pathology induced by systemic exposure to rotenone. Within the SNpc, GFAP expression and initial accumulation of p129 aggregates were seen by the end of rotenone exposure at 2 WPI. This was followed by a peak of microglial cell increase at 3 WPI that coincided with a decrease in p129+ aggregates and loss of DAn in this brain region (Fig. 8B). In the SNpr there was a similar early increase in p129 by 2 WPI, followed by the peak of microglia activation and A1 astrocyte reactivity at 3 WPI (Fig. 8C). The ST displayed an initial peak of microglia activation at 2 WPI followed by p129 aggregation at 3 WPI and then A1 astrocyte activation and degradation of DA terminals at 4 WPI (Fig. 8D). Schematic representation of all brain regions spanning the postrotenone lesioning periods reveals specific cellular responses and activation in association with movement and accumulation of p129 leading to neurodegeneration (Fig. 8E).
Figure 8. Modeling the progression of cytopathological changes in the nigro-striatal pathway following systemic exposure to rotenone.

(A) Three dimensional pathways depicting neuronal connectivity were overlayed with region-specific changes at each timepoint for microglial activation, p129 accumulation, astrocyte activation and loss of DA neurons. Following two weeks of rotenone exposure, microglia activate initially in the ST, whereas activation of astrocytes and formation of p129 aggregates occurs first in in the SN, spreading thereafter to the ST. By 4 WPI these changes in glial phenotype and protein aggregation lead to loss of DAn in the SNpc. Changes in individual parameters were modeled to accurately depict normalized temporal changes in pathological features for the SNpc (B), SNpr (C) and ST (D). (E) Summary schematic representing cell-cell interactions, aggregation and trafficking of p129 and neurodegeneration for all brain regions over time.
4. Discussion
Rotenone has been used successfully to model multiple pathological features of PD, most notably in Lewis rats, where intraperitoneal administration induces behavioral deficits, loss of DA neurons, degeneration of striatal terminals and p129 aggregation (Betarbet et al., 2000; Cannon et al., 2009; Zeng et al., 2018). Studies attempting to adapt this model to mice have suffered from a number of limitations, including high mortality rates and a necessity for increased sample size due to systemic toxicity (Duty and Jenner, 2011). Additionally, translating doses of rotenone from rats to mice has proven challenging due to the two- to three-fold higher metabolic expenditure on a per gram basis in mice compared to rats (Radermacher and Haouzi, 2013). In mammals, the parent molecule, (2R, 6aS, 12aS)-1,2,6,61,12,12a-hexahydro-2-isopropenyl-8,9-dimethoxychromeno[3,4-b]furo(2,3-h)chromen-6-one, is a potent mitochondrial complex I inhibitor, whereas the rotenone metabolites are 4-fold less toxic, underscoring the importance of species differences in expression of drug metabolizing enzymes that influence dosing with rotenone (Fukami et al., 1969). In addition, typical routes of administration such as oral gavage, inhalation, environmental-contact, and unilateral stereotactic injection are laborious and prone to variation in dosage between individual subjects (Inden et al., 2007; Inden et al., 2011; Liu et al., 2015; Pan-Montojo et al., 2010; Rojo et al., 2007; Wang et al., 2020). In the present study, we show that daily administration of rotenone to mice by intraperitoneal injection for 14 days results in low mortality, marked activation of glial cells, formation of p129+ aggregates and progressive neurodegeneration, all of which reaches a maximum during the two-week period following rotenone exposure. The progression of neuropathology during this postrotenone lesioning period was characterized by behavioral changes, DAn loss, increased glial cell activation, recruitment of peripheral monocytes and the appearance of p129 inclusions similar to that seen in idiopathic PD (Cheng et al., 2010).
Behavioral changes such as depression and anxiety are common non-motor symptoms of PD and precede the onset of motor symptoms. These early neuropsychiatric changes can manifest from alterations in dopaminergic, serotonergic, cholinergic and adrenergic neurotransmission (Belovicova et al., 2017; Gallagher and Schrag, 2012; Shiba et al., 2000). Interestingly, mice that were exposed to rotenone presented with anxiety and depressive-like behaviors in open field assays at 1 WPI, evidenced by an increase in margin time and rest time and decreased in center time and rearing number (Fig. 1). Following this timepoint, the increase in luciferase signal in NF-κB-GFP-luciferase reporter mice indicated an overall progressive increase in neuroinflammation, similar to the spread of dystrophic, inflammatory microglia observed in idiopathic PD (Braak et al., 2007; Yao et al., 2021) and in rat models of rotenone-induced PD (Betarbet et al., 2000; Cannon et al., 2009).
Key cytopathological features of PD, such as mitochondrial dysfunction, p129 synuclein accumulation, neuroinflammation and oxidative stress (Schapira and Jenner, 2011), are not all reproduced in many mouse models of the disease. Mitochondrial dysfunction and aggregation of p129 are thought to drive activation of glia and progressive neurodegeneration, but many toxin-based and genetic models lack both of these important phenotypic features of PD in the SN (Lucking and Brice, 2000; Robinson, 2008; Xie et al., 2010). In this murine model of rotenone-induced PD, histopathological examination of treated mice revealed progressive glial cell activation within the nigro-striatal system. This activation was associated with neuronal resection from the neuropil, overall cellular loss, gliosis, ventricular edema and multifocal myelinic edema advancing from 3 – 4 WPI (Supplementary Figure 1). Importantly, changes in astrocyte reactivity and phenotype in response to systemic exposure with rotenone occurred prior to neuronal loss in the brain regions evaluated, consistent with the appearance of reactive, p129+ astrocytes in idiopathic PD (Braak et al., 2007).
Reactive, dystrophic microglia appear prior to the majority of neurodegeneration in both PD and Lewy Body disease (Doorn et al., 2014). Microglial cells act as resident innate immune cells in the CNS, whereupon encountering pathogens or toxins, they quickly convert from a resting M2 state to a pro-inflammatory M1 state. M1 microglia are characterized by production of ROS and inflammatory cytokines such as TNF-α, IL-6, IL-12, IL-1beta and CCL2 (Subramaniam and Federoff, 2017). These early innate inflammatory immune responses have been detected in PD patients as increased cerebrospinal fluid levels of IL-1beta, IL-6 and human leukocyte antigen-DR isotype reactive microglia (Blum-Degen et al., 1995; McGeer et al., 1988). Notably, a study employing chronic exposure of C547Bl/6 mice to low levels of rotenone in diet reported increases in the astrocyte-derived cytokine, CXCL1, as well as IL-1beta, that were diminished in mice deficient in NLR Family Pyrin Domain Containing 3 (Nlrp3), a key mediator of innate immunity in microglia (Martinez et al., 2017). In the present study, increases in both the number of microglia and the fraction of microglia displaying an M1 phenotype was seen within the SNpr and SNpc at 2 WPI in response to rotenone exposure (Fig. 2). Microglia remained activated at the 3 WPI timepoint, with a gradual return to a more resting, M2-like phenotype by 4 WPI (Fig. 2). Following rotenone exposure, there was an apparent increase in peripheral macrophages in the SN that peaked at 4 WPI, although this trend will require a greater number of animals to validate appropriately. This has also been reported in post-mortem PD tissue (Fuzzati-Armentero et al., 2019). Infiltration of peripheral monocytes is likely mediated by disruption of blood-brain-barrier (BBB) integrity due to cytokines and chemokines released from M1 microglia (Salvi et al., 2017). This infiltration and recruitment of macrophages aids in the clearance of p129 aggregates but also increases inflammatory activity and worsens the overall disease state.
Similar to the phenotypic responses observed in microglia, there was an increase in the number and reactivity of astrocytes in the nigro-striatal system that followed the peak of microglia activation in each brain region and coincided closely with the maximum extent of p129 aggregate formation. We observed an initial peak in hypertrophic, GFAP+ cells expressing C3 at 2 WPI in the SNpc and overall regional decreases in total S100β + cell populations in the SNpc and SNpr, followed by an increase in C3+ astrocytes within the striatum and increased populational distribution of astrocytes within the SNpr at 3 WPI (Fig. 3, Supplementary Figure 2). C3 is reported to be a marker for reactive, neurotoxic A1 astrocytes (Liddelow et al., 2017) that is induced in response to C1q and IL-1alpha released by M1 microglia (Wei et al., 2021). Intra-astrocytic C3 processing to C3a is followed by extra-cellular release and recognition by C3aR on microglia, thus reinforcing glial-glial interactions that amplify neuronal injury (Wei et al., 2021). In PD, activated astrocytes are seen in the SN in areas of DA neuron loss (Middeldorp and Hol, 2011), as we also noted in rotenone-treated mice. The initial appearance of reactive astrocytes in the SN at 2 WPI coincided with the peak of p129 in the SNpc (Fig. 6) and was followed by the peak of C3-expressing astrocytes in the striatum at 3 WPI. This suggests that rotenone-induced mitochondrial dysfunction occurs initially in the SN and drives innate immune activation of glial cells and protein aggregation in this brain region.
Stereological analysis revealed DAn loss within the SNpc and striatal terminal loss by 4 WPI (Fig. 4), similar to the pattern of DAn loss seen in idiopathic PD (Michel et al., 2016). Notably, loss of DAn was not observed following rotenone exposure at 2 WPI but occurred two weeks later (4 WPI), after the appearance of reactive microglia and astrocytes in the SNpc. This strongly supports the assertion that innate immune activation of glia is an important mediator of neuronal injury during exposure to rotenone. Loss of TH+ terminal density in the striatum also occurred at 4 WPI but, interestingly, was preceded by an increase in TH intensity within the striatum at 3 WPI (Fig. 4). Tyrosine-hydroxylase is the rate limiting enzyme of catecholamine biosynthesis and its expression is modulated by the redox-sensitive proteins, DJ-1 (PARK7), which is up-regulated by oxidative stress, such as that caused by inhibition of complex I by rotenone (Ariga et al., 2013; Zhong et al., 2006). This may at least partly explain the apparent transient increase in TH intensity in the striatum observed at 3 WPI in rotenone-treated mice. The regional selectivity of rotenone-induced neuronal loss within the basal ganglia was assessed by determining the number of NeuN+ neurons in the hippocampus and entorhinal cortex, which did not show loss of neurons compared to the SNpc (Fig. 4). Because rotenone is a systemic complex I inhibitor, these data indicate that dopaminergic neurons within the SNpc are selectively vulnerable to complex I inhibition in C57Bl/6 mice.
Formation of Lewy bodies in DA neurons in the SNpc is one of the central pathological features of PD but is difficult to replicate in most murine models of the disease. Because previous studies using intraperitoneal administration of rotenone in male Lewis rats reported accumulation of p129 in DAn in the SNpc (Sherer et al., 2003), we examined the formation of synuclein aggregates using immunostaining for the phosphorylated form of the protein (phospho-Serine129-α-synuclein) in rotenone-treated mice (Fig. 3 and 4). There was increased formation of p129+ puncta within TH neurons beginning at 3 WPI and progressing to 4 WPI. The size and intensity of these aggregates within microglia varied amongst brain region and timepoint (Fig. 6), suggesting that microglial clearance via the autophagy pathway is an important modulator of the neuronal burden of p129. Interestingly, rotenone reduces autophagy of p129 aggregates in DAn that is rescued by overexpression of DJ-1 (De Miranda et al., 2018), highlighting the importance of microglial clearance of p129+ aggregates in limiting neuronal injury. The kinetics of p129+ aggregate formation in response to rotenone suggests that this mouse model is relevant to understanding mechanisms underlying the alterations in autophagy and protein processing associated with the progression of PD (Lynch-Day et al., 2012; Pan et al., 2008).
Alpha-synuclein has been investigated as a potential biomarker for PD due to its presence in the blood and CSF of PD patients (Chang et al., 2019; El-Agnaf et al., 2003; Gao et al., 2015). However, the mechanism of clearance from the CNS by glial cells is not fully understood (Choi et al., 2020). Because microglial cells are long-lived and perform critical phagocytic functions in the CNS (Hefendehl et al., 2014), they are likely important modulators of p129 clearance. Data from rotenone-treated mice in these studies supports this mechanism, where IBA1+ cells containing p129+ aggregates were identified in perivascular regions and along projecting fibers of the nigro-striatal pathway, as well as within the corpus callosum at 4 WPI (Fig. 7). Migration of p129+ microglia along these routes in rotenone-treated mice is consistent with glial-lymphatic clearance of p129and elevated circulating levels of p129 seen in PD patients. Moreover, the resistance of p129+ aggregates to digestion by pK (Fig. 7) echoes the results of p129 self-seeding studies reporting that recombinant fibrillar p129 aggregates were resistant to pK digestion (Kushnirov et al., 2020). The presence of highly stable, pK-resistant p129+ aggregates in rotenone-exposed mice is an important pathological feature of this model that reproduces aspects of protein misfolding observed in idiopathic PD (Angot et al., 2012; Kordower et al., 2008; Lee et al., 2010; Li et al., 2008). Notably, the presence of pK-resistant p129+ aggregates resembles the pattern of p129 aggregation we recently reported in virally-induced parkinsonism in mice (Bantle et al., 2019; Bantle et al., 2021). Additional studies will be required to determine more precisely the role of microglia autophagy pathways in modulating levels of p129+ aggregates in rotenone-treated mice.
These findings reveal a complex temporo-spatial progression of neuropathology in rotenone-induced parkinsonism involving numerous cell types, cell-to-cell signaling mechanisms and differential susceptibility amongst nigro-striatal brain regions. In addition, distinct patterns of p129 aggregation, movement of these aggregates along interconnected neural tracts and glial clearance all appear to influence the progression of pathology. The summary model of rotenone-induced neuropathology presented here (Fig. 8) highlights the relationship between both temporal and spatial patterns of glial activation and p129 aggregation that lead to the loss of DAn within the SN. Early peaks of glial activation seen in the ST and SN after 2 WPI with rotenone resulted in rapid accumulation of pK-resistant p129+ aggregates in these brain regions, which were followed by progressive loss of DAn in the SNpc (Fig. 8). Interestingly, p129+ aggregates were seen within projecting TH+ fibers of the nigro-striatal tract and in M1 phagocytic microglia surrounding DAn in the SNpc, suggesting that distribution of p129 in rotenone-treated mice is heavily influenced both by inter-neuronal spread and by glial clearance. The early appearance of reactive, dystrophic glial cells and deposition of p129+ aggregates prior to the majority of neuronal loss are features of this murine model of rotenone-induced parkinsonism that compare favorably to the progression of neuropathology in idiopathic PD (Braak et al., 2007). Moreover, the rapid appearance of p129+ aggregates within weeks of systemic rotenone exposure increases the utility of this model for basic as well as translational research. It will be important in future studies to determine more precisely the role of glial activation in modulating the formation and distribution of p129 following rotenone exposure in the basal ganglia. It will also be important to examine the effects of rotenone on additional brain regions involved with control of motor function to better understand the pathology resulting from deprecations in mitochondrial function in the brain. Studies will also need to delineate cell-specific changes in gene expression that regulate phenotypic switching of glia to a reactive, neurotoxic state that negatively impacts neuronal survival in the basal midbrain.
Supplementary Material
Highlights.
Rotenone induces replicable features of Parkinson’s Disease pathology in mice
Spread of misfolded α-synuclein follows rotenone-induced glial activation
Microglia modulate the distribution of protein aggregation in response to rotenone
Reactive astrocytes initiate the glial response to rotenone toxicity
The progression of rotenone-induced neuropathology is region-specific
Acknowledgements
We thank Lab Animal Resources (Colorado State University) for outstanding care of the animals used in these studies. We would also like to thank Carol Dewbre for administrative support and Tenley French for project coordination and animal management.
Funding Information
This work was supported by the National Institutes of Health grants ES021656 and ES021656 [RBT].
Abbreviations:
- PD
Parkinson’s Disease
- DAn
dopaminergic neurons
- SNpc
substantia nigra pars compacta
- p129
misfolded α-synuclein
- CNS
central nervous system; DA, dopamine
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- 6-OHDA
6-hydroxydopamine
- ROS
reactive oxygen species
- SNpr
substantia nigra pars reticulate
- ST
striatum
- GP
globus pallidus
- C3
complement protein 3
- pK
proteinase K
- OB
olfactory bulb
- M1
activated ameboid pro-inflammatory microglia
- M2
resting/ramified non-inflammatory microglia
- A1
activated pro-inflammatory astrocytes; A2, neurotrophic astrocytes
Footnotes
Competing Interests
Declarations of interest: none
Credit Author Statement
Savannah M. Rocha: Conceptualization, methodology, validation, formal analysis, investigation, data curation, writing- original draft, writing- review and editing, visualization, project administration. Collin M. Bantle: Conceptualization, methodology, formal analysis, writing- review and editing. Tawfik Aboellail: Formal analysis, investigation, writing- review and editing. Debotri Chatterjee: Investigation, formal analysis, writing- review and editing. Richard J. Smeyne: Investigation, formal analysis, data curation, writing- review and editing. Ronald B. Tjalkens: Conceptualization, investigation, resources, data curation, writing- review and editing, visualization, supervision, project administration, funding acquisition.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
References
- Angot E, et al. , 2012. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS One. 7, e39465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariga H, et al. , 2013. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid Med Cell Longev. 2013, 683920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantle CM, et al. , 2019. Infection with mosquito-borne alphavirus induces selective loss of dopaminergic neurons, neuroinflammation and widespread protein aggregation. NPJ Parkinsons Dis. 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantle CM, et al. , 2021. Astrocyte inflammatory signaling mediates alpha-synuclein aggregation and dopaminergic neuronal loss following viral encephalitis. Exp Neurol. 346, 113845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belovicova K, et al. , 2017. Animal tests for anxiety-like and depression-like behavior in rats. Interdiscip Toxicol. 10, 40–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, et al. , 2000. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 3, 1301–6. [DOI] [PubMed] [Google Scholar]
- Blum-Degen D, et al. , 1995. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett. 202, 17–20. [DOI] [PubMed] [Google Scholar]
- Braak H, Sastre M, Del Tredici K, 2007. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 114, 231–41. [DOI] [PubMed] [Google Scholar]
- Cannon JR, et al. , 2009. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol Dis. 34, 279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CW, et al. , 2019. Plasma and Serum Alpha-Synuclein as a Biomarker of Diagnosis in Patients With Parkinson’s Disease. Front Neurol. 10, 1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HC, Ulane CM, Burke RE, 2010. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol. 67, 715–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I, et al. , 2020. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 11, 1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miranda BR, et al. , 2018. Astrocyte-specific DJ-1 overexpression protects against rotenone-induced neurotoxicity in a rat model of Parkinson’s disease. Neurobiol Dis. 115, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingues AV, et al. , 2020. Glial cells in Parkinson s disease: protective or deleterious? Cell Mol Life Sci. 77, 5171–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorn KJ, et al. , 2014. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol Commun. 2, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dung VM, Thao DTP, 2018. Parkinson’s Disease Model. Adv Exp Med Biol. 1076, 41–61. [DOI] [PubMed] [Google Scholar]
- Duty S, Jenner P, 2011. Animal models of Parkinson’s disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol. 164, 1357–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Agnaf OM, et al. , 2003. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 17, 1945–7. [DOI] [PubMed] [Google Scholar]
- EPA., U., 2007. Re-registration eligibility decision for rotenone. US EPA Case #0255. [Google Scholar]
- Fontana BD, et al. , 2018. The developing utility of zebrafish models of neurological and neuropsychiatric disorders: A critical review. Exp Neurol. 299, 157–171. [DOI] [PubMed] [Google Scholar]
- Fukami JI, et al. , 1969. Oxidative Metabolism of Rotenone in Mammals, Fish, and Insects and Its Relation to Selective Toxicity. [Google Scholar]
- Fuzzati-Armentero MT, Cerri S, Blandini F, 2019. Peripheral-Central Neuroimmune Crosstalk in Parkinson’s Disease: What Do Patients and Animal Models Tell Us? Front Neurol. 10, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher DA, Schrag A, 2012. Psychosis, apathy, depression and anxiety in Parkinson’s disease. Neurobiol Dis. 46, 581–9. [DOI] [PubMed] [Google Scholar]
- Gao L, et al. , 2015. Cerebrospinal fluid alpha-synuclein as a biomarker for Parkinson’s disease diagnosis: a systematic review and meta-analysis. Int J Neurosci. 125, 645–54. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Lee VM, 2000. A new link between pesticides and Parkinson’s disease. Nat Neurosci. 3, 1227–8. [DOI] [PubMed] [Google Scholar]
- Gopal Krishna Patro S, Kumar Sahu K, 2015. Noramlization: A Preporcessing Stage [Google Scholar]
- Grayson M, 2016. Parkinson’s disease. Nature. 538, S1. [DOI] [PubMed] [Google Scholar]
- Guenther H, 2011. Rotenone review advisory committee final report and recommendations to the Arizona game and fish department Vol., U.S.D.o. Agriculture, ed.^eds., pp. 1–122. [Google Scholar]
- Halliday G, Lees A, Stern M, 2011. Milestones in Parkinson’s disease--clinical and pathologic features. Mov Disord. 26, 1015–21. [DOI] [PubMed] [Google Scholar]
- Harrington AJ, et al. , 2010. C. elegans as a model organism to investigate molecular pathways involved with Parkinson’s disease. Dev Dyn. 239, 1282–95. [DOI] [PubMed] [Google Scholar]
- Hefendehl JK, et al. , 2014. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell. 13, 60–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkila RE, et al. , 1985. Dopaminergic toxicity of rotenone and the 1-methyl-4-phenylpyridinium ion after their stereotaxic administration to rats: implication for the mechanism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity. Neurosci Lett. 62, 389–94. [DOI] [PubMed] [Google Scholar]
- Hirth F, 2010. Drosophila melanogaster in the study of human neurodegeneration. CNS Neurol Disord Drug Targets. 9, 504–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inden M, et al. , 2007. Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4-phenylbutyrate, a chemical chaperone. J Neurochem. 101, 1491–1504. [DOI] [PubMed] [Google Scholar]
- Inden M, et al. , 2011. Parkinsonian rotenone mouse model: reevaluation of long-term administration of rotenone in C57BL/6 mice. Biol Pharm Bull. 34, 92–6. [DOI] [PubMed] [Google Scholar]
- Jang H, et al. , 2009. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A. 106, 14063–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ME, Bobrovskaya L, 2015. An update on the rotenone models of Parkinson’s disease: their ability to reproduce the features of clinical disease and model gene-environment interactions. Neurotoxicology. 46, 101–16. [DOI] [PubMed] [Google Scholar]
- Kamel F, et al. , 2007. Pesticide exposure and self-reported Parkinson’s disease in the agricultural health study. Am J Epidemiol. 165, 364–74. [DOI] [PubMed] [Google Scholar]
- Kordower JH, et al. , 2008. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 14, 504–6. [DOI] [PubMed] [Google Scholar]
- Kushnirov VV, Dergalev AA, Alexandrov AI, 2020. Proteinase K resistant cores of prions and amyloids. Prion. 14, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, et al. , 2010. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 285, 9262–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, et al. , 2008. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 14, 501–3. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, et al. , 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KL, 2010. Non-mammalian animal models of Parkinson’s disease for drug discovery. Expert Opin Drug Discov. 5, 165–76. [DOI] [PubMed] [Google Scholar]
- Liu HF, et al. , 2017. Combined LRRK2 mutation, aging and chronic low dose oral rotenone as a model of Parkinson’s disease. Sci Rep. 7, 40887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, et al. , 2006. Manganese-induced neurotoxicity: the role of astroglial-derived nitric oxide in striatal interneuron degeneration. Toxicol Sci. 91, 521–31. [DOI] [PubMed] [Google Scholar]
- Liu Y, et al. , 2015. Environment-contact administration of rotenone: A new rodent model of Parkinson’s disease. Behav Brain Res. 294, 149–61. [DOI] [PubMed] [Google Scholar]
- Lucking CB, Brice A, 2000. Alpha-synuclein and Parkinson’s disease. Cell Mol Life Sci. 57, 1894–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch-Day MA, et al. , 2012. The role of autophagy in Parkinson’s disease. Cold Spring Harb Perspect Med. 2, a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez EM, et al. , 2017. Editor’s Highlight: Nlrp3 Is Required for Inflammatory Changes and Nigral Cell Loss Resulting From Chronic Intragastric Rotenone Exposure in Mice. Toxicol Sci. 159, 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, et al. , 1988. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 38, 1285–91. [DOI] [PubMed] [Google Scholar]
- Mehra S, Sahay S, Maji SK, 2019. alpha-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim Biophys Acta Proteins Proteom. 1867, 890–908. [DOI] [PubMed] [Google Scholar]
- Michel PP, Hirsch EC, Hunot S, 2016. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron. 90, 675–91. [DOI] [PubMed] [Google Scholar]
- Middeldorp J, Hol EM, 2011. GFAP in health and disease. Prog Neurobiol. 93, 421–43. [DOI] [PubMed] [Google Scholar]
- Moreno JA, et al. , 2009. Age-dependent susceptibility to manganese-induced neurological dysfunction. Toxicol Sci. 112, 394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison H, et al. , 2017. Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci Rep. 7, 13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan T, et al. , 2008. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 131, 1969–78. [DOI] [PubMed] [Google Scholar]
- Pan-Montojo F, et al. , 2010. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One. 5, e8762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringsheim T, et al. , 2014. The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov Disord. 29, 1583–90. [DOI] [PubMed] [Google Scholar]
- Radad K, et al. , 2019. Rotenone: from modelling to implication in Parkinson’s disease. Folia Neuropathol. 57, 317–326. [DOI] [PubMed] [Google Scholar]
- Radermacher P, Haouzi P, 2013. A mouse is not a rat is not a man: species-specific metabolic responses to sepsis - a nail in the coffin of murine models for critical care research? Intensive Care Med Exp. 1, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan DM, Goyal V, 2018. Parkinson’s disease: A review. Neurol India. 66, S26–S35. [DOI] [PubMed] [Google Scholar]
- Rey NL, et al. , 2016. Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med. 213, 1759–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, et al. , 2018. Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol. 135, 65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PA, 2008. Protein stability and aggregation in Parkinson’s disease. Biochem J. 413, 1–13. [DOI] [PubMed] [Google Scholar]
- Rocha EM, De Miranda B, Sanders LH, 2018. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis. 109, 249–257. [DOI] [PubMed] [Google Scholar]
- Rojo AI, et al. , 2007. Chronic inhalation of rotenone or paraquat does not induce Parkinson’s disease symptoms in mice or rats. Exp Neurol. 208, 120–6. [DOI] [PubMed] [Google Scholar]
- Sadasivan S, et al. , 2015. Induction of microglia activation after infection with the non-neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS One. 10, e0124047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi V, et al. , 2017. Role of Atypical Chemokine Receptors in Microglial Activation and Polarization. Front Aging Neurosci. 9, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samii A, Nutt JG, Ransom BR, 2004. Parkinson’s disease. Lancet. 363, 1783–93. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Jenner P, 2011. Etiology and pathogenesis of Parkinson’s disease. Mov Disord. 26, 1049–55. [DOI] [PubMed] [Google Scholar]
- Schober A, 2004. Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res. 318, 215–24. [DOI] [PubMed] [Google Scholar]
- Sherer TB, et al. , 2003. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 179, 9–16. [DOI] [PubMed] [Google Scholar]
- Shiba M, et al. , 2000. Anxiety disorders and depressive disorders preceding Parkinson’s disease: a case-control study. Mov Disord. 15, 669–77. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, et al. , 2016. Assessment of the Effects of MPTP and Paraquat on Dopaminergic Neurons and Microglia in the Substantia Nigra Pars Compacta of C57BL/6 Mice. PLoS One. 11, e0164094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam SR, Federoff HJ, 2017. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front Aging Neurosci. 9, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner CM, et al. , 2011. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect. 119, 866–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapias V, Greenamyre JT, Watkins SC, 2013. Automated imaging system for fast quantitation of neurons, cell morphology and neurite morphometry in vivo and in vitro. Neurobiol Dis. 54, 158–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapias V, Greenamyre JT, 2014. A rapid and sensitive automated image-based approach for in vitro and in vivo characterization of cell morphology and quantification of cell number and neurite architecture. Curr Protoc Cytom. 68, 12 33 1–22. [DOI] [PubMed] [Google Scholar]
- Vehovszky A, et al. , 2007. Behavioural and neural deficits induced by rotenone in the pond snail Lymnaea stagnalis. A possible model for Parkinson’s disease in an invertebrate. Eur J Neurosci. 25, 2123–30. [DOI] [PubMed] [Google Scholar]
- Wang DX, et al. , 2020. Protective effect of metformin against rotenone-induced parkinsonism in mice. Toxicol Mech Methods. 30, 350–357. [DOI] [PubMed] [Google Scholar]
- Wei Y, et al. , 2021. The complement C3-C3aR pathway mediates microglia-astrocyte interaction following status epilepticus. Glia. 69, 1155–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Wan OW, Chung KK, 2010. New insights into the role of mitochondrial dysfunction and protein aggregation in Parkinson’s disease. Biochim Biophys Acta. 1802, 935–41. [DOI] [PubMed] [Google Scholar]
- Yao L, et al. , 2021. Genetic Imaging of Neuroinflammation in Parkinson’s Disease: Recent Advancements. Front Cell Dev Biol. 9, 655819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng XS, Geng WS, Jia JJ, 2018. Neurotoxin-Induced Animal Models of Parkinson Disease: Pathogenic Mechanism and Assessment. ASN Neuro. 10, 1759091418777438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N, et al. , 2006. DJ-1 transcriptionally up-regulates the human tyrosine hydroxylase by inhibiting the sumoylation of pyrimidine tract-binding protein-associated splicing factor. J Biol Chem. 281, 20940–20948. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.