

Abstract
Rhabdomyosarcoma (RMS) is one of the common malignant soft-tissue sarcomas affecting children. It originates from the embryonic mesenchyme precursor of striated muscle and is frequently seen in the head-and-neck region, genitourinary system and extremities. Occasionally, it arises from the retroperitoneum, biliary tract and abdomen and is rarely seen in the sacrococcygeal area. A 4-month-male child presented with a nodule over the sacrum. Based on histopathology and immunohistochemical marker studies, a final diagnosis of RMS was rendered. There was no evidence of any teratomatous elements.
Keywords: Congenital, desmin, immunohistochemistry, infants, rhabdomyosarcoma, sacrococcygeal tumour
INTRODUCTION
Rhabdomyosarcoma (RMS) arises from immature mesenchymal cells committed to skeletal muscle differentiation.[1] It is the most frequent soft tissue sarcoma in children and represents 5% of all paediatric malignant tumours.[2] It accounts for 4.5 cases per 1 million people aged 0–20 years.[3] Embryonal RMS (ERMS) is the most common subtype, with one-third of cases occurring in children aged <5 years with a male preponderance.[4] RMS arising before the age of 1 month is termed as congenital RMS and represents 0.4% of all RMS cases in the Intergroup Rhabdomyosarcoma Study Group (IRSG).[5] It primarily involves the head-and-neck region but may also occur at other sites such as the genitourinary tract, extremities and trunk.[6] A rare presentation of RMS as a sacral mass in a 4-month-old baby with presence for 22 days of age is highlighted in this report.
CASE REPORT
A 4-month-old male child presented with a nodule over the sacral region. The nodule started as a small swelling at the age of 22 days which gradually increased in size. The clinical diagnosis was a sacrococcygeal teratoma. Radiological examination revealed a solid heterogeneous lesion in the sacral region suggestive of a sacrococcygeal teratoma. A wide excision biopsy was done, and we received a skin-covered nodular tissue mass measuring 6 cm × 6 cm × 4 cm size. Cut section of the specimen showed a circumscribed grey white lesion with multiple cystic areas, the largest cyst measuring 3 cm × 3 cm filled with haemorrhagic fluid [Figure 1]. Microscopically, the tumour exhibited round-to-oval cells in hypocellular and cellular patterns [Figure 2a]. Furthermore, seen are alveolar, solid, perivascular and discohesive patterns [Figure 2b]. Individual cells show round nuclei with scant cytoplasm and prominent nucleoli [Figure 3a]. Few scattered rhabdomyoblasts with abundant eosinophilic cytoplasm and tadpole-like appearance were seen. Multinucleated giant cells are prominent with multiple peripherally placed nuclei (wreath-like nuclei) [Figure 3b]. There are areas of necrosis, and the tumour is seen infiltrating into surrounding adipose tissue and muscle. The overlying skin is uninvolved by the tumour. A diagnosis of RMS with alveolar and embryonal patterns was rendered as immunohistochemical markers; desmin and vimentin showed diffuse strong cytoplasmic positivity [Figure 4a and b].
Figure 1.
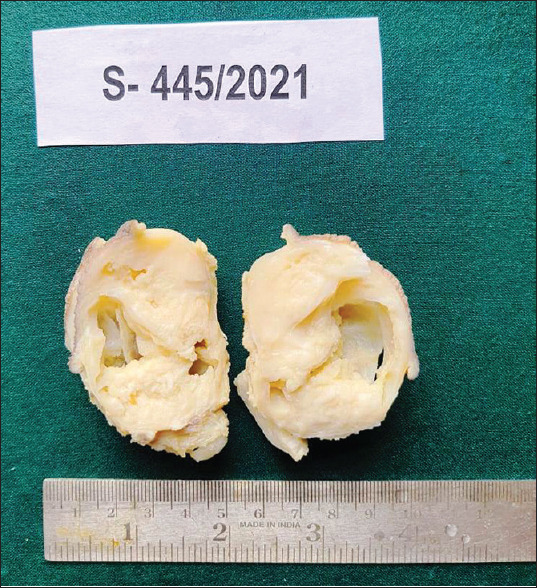
Gross: Circumscribed grey white lesion with multiple cystic areas
Figure 2.
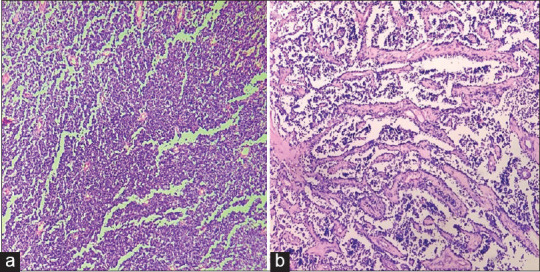
(a) Tumour shows round to oval cells in cellular solid patterns. (H and E × 100). (b) Tumour with cells in alveolar patterns and discohesive patterns. (H and E × 100)
Figure 3.
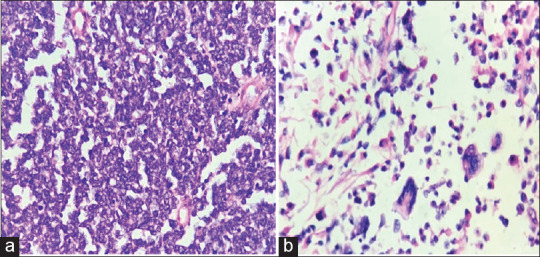
(a) Cells showing uniform round nuclei with scant cytoplasm. (H and E × 400). (b) Rhabdomyoblasts with abundant eosinophilic cytoplasm and tadpole-like appearance and multinucleated giant cells with multiple peripherally placed nuclei (wreath-like nuclei). (H and E × 400)
Figure 4.
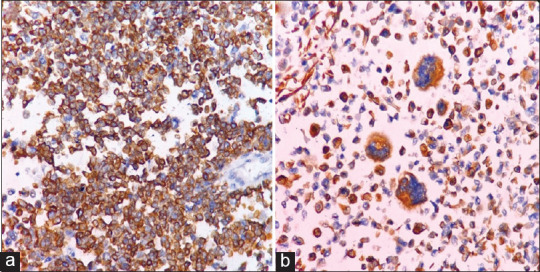
(a) Tumour cells showing cytoplasmic positivity for desmin (immunohistochemistry × 100). (b) Cytoplasm of Rhabdomyoblasts and tumour giant cells positive for desmin (immunohistochemistry × 400)
DISCUSSION
RMS is a malignant mesenchymal tumour of children first described by Zenker in 1856.[7] It is the most common soft-tissue sarcoma in children. Furthermore, it affects adults, but the incidence is much less compared to children, accounting for <1% of all malignancies.[8] RMS shows a bimodal distribution. It usually involves children below the age of 10 years, with a peak incidence reported by 4 years. Ferrari et al., in an Italian study, reported on 50 infants with RMS over 20 years with 15 cases of congenital RMS.[9] Our patient was a 4-month-old, and as his nodule was present since he was 22 days, it is considered congenital RMS.
RMS occurs predominantly in the head-and-neck region with orbit, paranasal sinuses and nasopharynx being the frequently involved sites. Other sites of occurrence are the genitourinary tract, extremities and retroperitoneum.[6] Presentation as a sacrococcygeal RMS is infrequent with scant literature availability. In a 10 years study by Agarwal A et al.,[10] of the 23 paediatric patients with sacrococcygeal masses, only one was a RMS. Our patient is a 4-month boy who presented with a swelling near the sacrum, which was present since the baby was 22 days old, which gradually increased in size. A primary diagnosis of RMS was made on histopathological features, which was confirmed by immunohistochemical examination. Based on the International Classification of RMS created by the IRSG, four histological subtypes are identified. These are embryonal, alveolar, botryoid, spindle cell and undifferentiated.[11] ERMS is the most common and usually affects neonates and younger children. Alveolar RMS affects older individuals. Spindle cell RMS occurs primarily in the paratesticular region in children. It carries a good prognosis, whereas, in adults, the spindle cell variant involves the head-and-neck region and is more aggressive. Spindle cell and botryoids have a good prognosis, ERMS carries intermediate prognosis and alveolar and undifferentiated RMS carries a poor prognosis. Apart from histology, other prognostic factors include the site of the lesion, size of tumour and extent, lymph node status and metastasis. Lobe et al. showed that tumour histology, size and type of surgery did not predict clinical outcome, while the presence of necrosis and small round cell configuration coincides with a poor prognosis regardless of histological diagnosis.[12] Our report shows a case of RMS at a rare sacrococcygeal site in an infant. RMS should be considered in the differential diagnosis of a sacral tumour in a neonate. Histopathology and immunohistochemical studies are needed to confirm the diagnosis and to distinguish it from other small-round-blue-cell tumours such as lymphoma, neuroblastoma and primitive neuroectodermal tumour. Treatment modalities include surgical removal of the tumour followed by chemotherapy and radiotherapy. Complete resection of the tumour with safe margins is mandatory, which was done in this case. ERMS responds well to chemotherapy with a vincristine, adriamycin and cyclophosphamide (VAC) regimen.[13] RMS is moderately radiation-sensitive, and hence higher doses of radiation are required.[12] Our patient was treated with the VAC regimen and is doing well.
To conclude congenital, RMS at the sacral region is rare, and diagnosis is a challenge. Clinicians should be aware of its occurrence in the sacrococcygeal site. Histopathological examination with immunohistochemical studies is required to confirm the diagnosis. Imaging provides an assessment about the extension and for follow-up. The management would be complete excision followed by chemotherapy with the VAC regimen.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Burrows NP, Ratnavel RC, Grant JW, Cormack GC, Pye RJ. Auricular embryonal rhabdomyosarcoma. Dermatology. 2014;189:301–3. doi: 10.1159/000246867. [DOI] [PubMed] [Google Scholar]
- 2.Chirat M, Dainese L, Fasola S, Couloigner V, Denoyelle F, Garabedian EN, et al. Unusual outer ear swelling: Childhood auricular rhabdomyosarcoma. Eur Ann Otorhinolaryngol Head Neck Dis. 2016;133:23–6. doi: 10.1016/j.anorl.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Skapek SX, Ferrari A, Gupta AA, Lupo PJ, Butler E, Shipley J, et al. Rhabdomyosarcoma. Nat Rev Dis Primers. 2019;5:1. doi: 10.1038/s41572-018-0051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer. 2009;115:4218–26. doi: 10.1002/cncr.24465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brecher AR, Reyes-Mugica M, Kamino H, Chang MW. Congenital primary cutaneous rhabdomyosarcoma in a neonate. Pediatr Dermatol. 2003;20:335–8. doi: 10.1046/j.1525-1470.2003.20413.x. [DOI] [PubMed] [Google Scholar]
- 6.Singh O, Gupta SS, Upadhyaya V, Sharma SS, Lahoti BK, Mathur KR. Rhabdomyosarcoma of the posterior chest wall in a newborn: A case report. Cases J. 2009;2:6818. doi: 10.4076/1757-1626-2-6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stuart A, Radhakrishnan J. Rhabdomyosarcoma. Indian J Pediatr. 2004;71:331–7. doi: 10.1007/BF02724100. [DOI] [PubMed] [Google Scholar]
- 8.Arul AS, Verma S, Arul AS, Verma R. Oral rhabdomyosarcoma-embryonal subtype in an adult: A rarity. J Nat Sci Biol Med. 2014;5:222–5. doi: 10.4103/0976-9668.127347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrari A, Casanova M, Bisogno G, Zanetti I, Cecchetto G, De Bernardi B, et al. Rhabdomyosarcoma in infants younger than one year old: A report from the Italian Cooperative Group. Cancer. 2003;97:2597–604. doi: 10.1002/cncr.11357. [DOI] [PubMed] [Google Scholar]
- 10.Agarwal A, Das S, Ghosh D, Agarwal A. Sacrococcygeal masses other than meningomyelocele. Indian J Surg. 2011;73:206–9. doi: 10.1007/s12262-010-0014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tewari V, Mehta R, Tewari K. Congenital embryonal rhabdomyosarcoma presenting as a cutaneous nodule in a neonate. Int J Clin Pediatr. 2016;5:3–4. [Google Scholar]
- 12.Lobe TE, Wiener ES, Hays DM, Lawrence WH, Andrassy RJ, Johnston J, et al. Neonatal rhabdomyosarcoma: The IRS experience. J Pediatr Surg. 1994;29:1167–70. doi: 10.1016/0022-3468(94)90302-6. [DOI] [PubMed] [Google Scholar]
- 13.Baker KS, Anderson JR, Link MP, Grier HE, Qualman SJ, Maurer HM, et al. Benefit of intensified therapy for patients with local or regional embryonal rhabdomyosarcoma: Results from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol. 2000;18:2427–34. doi: 10.1200/JCO.2000.18.12.2427. [DOI] [PubMed] [Google Scholar]