

Abstract
Since the discovery of the caspase-2 (Casp2)-mediated Δtau314 cleavage product and its associated impact on tauopathies such as Alzheimer's disease, the design of selective Casp2 inhibitors has become a focus in medicinal chemistry research. In the search for new lead structures with respect to Casp2 selectivity and druglikeness, we have taken an approach by looking more closely at the specific sites of Casp2-mediated proteolysis. Using seven selected protein cleavage sequences, we synthesized a peptide series of 53 novel molecules and studied them using in vitro pharmacology, molecular modeling, and crystallography. Regarding Casp2 selectivity, AcITV(Dab)D-CHO (23) and AcITV(Dap)D-CHO (26) demonstrated the best selectivity (1-6-fold), although these trends were only moderate. However, some analogous tetrapeptides, most notably AcDKVD-CHO (45), showed significantly increased Casp3 selectivities (>100-fold). Tetra- and tripeptides display decreased or no Casp2 activity which support the assumption that a motif of five amino acids is required for efficient Casp2 inhibition. Overall, the results provide a reasonable basis for the development of both selective caspase-2 and caspase-3 inhibitors.
Keywords: Caspase-2, Alzheimer’s Disease, tauopathies, Caspase-2 inhibitors, protein cleavage
Graphical Abstract
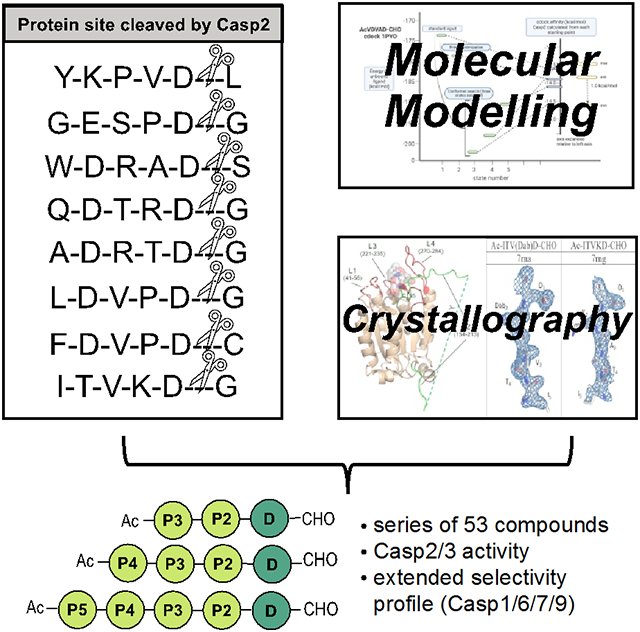
In search of new lead compounds in terms of caspase-2 (Casp2) inhibition and drug suitability we examined specific sites of Casp2-mediated proteolysis. Selected protein cleavage sequences yielded deduced inhibitors such as AcGESPD-CHO, AcLDVPD-CHO, AcFDVPD-CHO, and AcITVKD-CHO which form the basis for our series of 53 peptides. Supported by data from crystallography (Casp3) and molecular modelin, important insights for the further development of selective caspase-2 inhibitors emerged.
Introduction
In recent years, the caspase-2 (Casp2) enzyme has been increasingly associated with neurodegenerative diseases, specifically tauopathies like Alzheimer's disease (AD), Huntington's disease (HD), frontotemporal dementia (FTD), and Lewy body dementia (LBD).[1-6] Casp2 cleavage of tau at Asp314 generates the cleavage product Δtau314 and leads to synaptic dysfunction and impaired cognition by enabling tau to mislocalize to dendritic spines and reduce AMPA receptors in the post-synaptic membrane.[1,7] The Δtau314 fragment may be a biomarker of impaired cognition, as Δtau314 was increased approximately threefold in brain samples from patients with mild cognitive impairment (MCI) and Alzheimer's disease (AD) compared to cognitively intact subjects.[1,4] Making tau resistant to Casp2 cleavage preserves memory function and lowering Casp2 restores memory function in mice expressing mutant human tau associated with frontotemporal dementia.[1]
Casp2 is one of the 12 human caspases, a family of cysteine-aspartic proteases, that perform both apoptotic and non-apoptotic effects which include tau cleavage described above.[8-10] In contrast to the other caspase inhibitors, the so far described Casp2 inhibitors have a pentapeptide instead of a tetrapeptide sequence.[11,12] The aspartic acid motif at P1 is also thought to be essential at P4 for binding to the Casp2 enzyme.[13,14] In order to improve these structures with respect to selectivity and pharmacokinetic properties toward peptidomimetic structures, it is of immense importance to find new peptide sequences that might offer clues to the structural elements that could be used in the design of Casp2 enzyme inhibitors. The cleavage site of tau (GSVQIVYKPVD∣LSKVTSKCGSLGNI-; D∣L cleavage site), which creates the Δtau314 fragment, and the canonical inhibitor AcVDVAD-CHO were the starting points for the development of recently synthesized Casp2 pentapeptide inhibitors.[15] The appearance of Casp2 within the nucleus, Golgi, and mitochondria enables access to further cellular substrates located in these organelles.[16] Many of these cleavage events can also be mediated by Casp3 or other caspases but some of these peptide sequences have also been reported to be cleaved under physiological conditions only or preferentially by Casp2.[9]
We have selected specific sites of Golgin-160[17], Protein transport protein Sec16A[18], Transcriptional-regulating factor 1[18], Ral GTPase-activating protein subunit alpha-1[18], Holliday junction recognition protein[18], and MDM2[19] for three reasons: (1) specificity of Casp2 cleavage (except for Holliday junction recognition protein which will serve as a control and SAR point), (2) our interest in characterizing arginine-containing sequences since we have modeling evidence that this basic amino acid will interact with Glu52 in the Casp2 binding site, and (3) non-canonical sequences (sequence ≠ -DXXD-) containing lysine at P2 (Table 1). Our strategy is to start with peptides that represent the cleavage sequences at these sites (Figure 1) and to modify them to exploit specific features in the Casp2 binding pocket, guided by SAR and structural considerations of peptide binding We expect that the pentapeptides based on these sequences would be an excellent starting point for probe discovery. New insights into selectivity and druglikeness are needed to develop effective in vivo Casp2 probes. In this context, novel active peptide sequences could provide important information on where modifications can be made in the molecule, compared to the canonical inhibitors, in order to achieve the attributes addressed above. Shortening of the pentapeptides to tetra- and tripeptides should give us more information about the importance of the P5/P4 positions and the possibility of decreasing molecular weight for small molecules.
Table 1.
Cleavage sequences of selected substrates specifically and/or efficiently cleaved by Caspase-2 and initial synthetic targets.
| Protein Name, UniProt designation |
Sub-Sequence/Sequence D∣X indicates cleavage site |
Initial Synthetic Target |
Casp2* |
kcat/Km (M−1 s−1)** |
Ref. |
|---|---|---|---|---|---|
| Tau A0A024RA17 (A0A024RA17_HUMAN) |
YKPVD∣L GSVQIVYKPVD∣LSKVTSKCGSLGNI |
AcYKPVD-CHO (1) | YES | ND | [1] |
| Golgin subfamily A member 3 Q08378 (GOGA3_HUMAN) |
GESPD∣G NRASTEGESPD∣GPGQGGLCQNGPTP |
AcGESPD-CHO (3) | YES | 3.3×104 | [17] |
| Protein Transport Protein Sec16A O15027 (SC16A_HUMAN) |
WDRAD∣S SEAPPGWDRAD∣SGPTQPPLSLSPAP |
AcWDRAD-CHO (4) | YES | 5.4×103 | [18] |
| Transcriptional-regulating factor 1 Q96PN7 (TREF1_HUMAN) |
QDTRD∣G ISPHFPQDTRD∣GLGLPVGSKNLGQM |
AcQDTRD-CHO (5) | YES | 1.3×103 | [18] |
| Holliday junction recognition protein Q8NCD3 (HJURP_HUMAN) |
ADRTD∣G DSSMKPADRTD∣GSVQAAAWGPELPS |
AcADRTD-CHO (6) | Casp3 8.3× | 1.2×103 | [18] |
| E3 ubiquitin-protein ligase Mdm2 P23804 (MDM2_MOUSE) | LDVPD∣G AQAEEGLDVPD∣GKKLTENDAKEPCA |
AcLDVPD-CHO (7) | YES | ND | [19] |
| E3 ubiquitin-protein ligase Mdm2 Q00987 (MDM2_HUMAN) | FDVPD∣C TQAEEGFDVPD∣CKKTIVNDSRESCV |
AcFDVPD-CHO (8) | YES | ND | [19] |
| Ral GTPase-activating protein subunit alpha-1 Q6GYQ0 (RGPA1_HUMAN) |
ITVKD∣G NECLEDITVKD∣GLSLQFKRFRETVP |
AcITVKD-CHO (9) | YES | 1.1×103 | [18] |
Is the protein sequence cleaved specifically by casp2? 6 is cleaved by Casp2 but 8.3× more efficiently by Casp3.
kcat/Km represents the catalytic efficiency of Casp2. ND = not determined.
Figure 1.

Specific Casp2 cleavage sequences of selected natural proteins and our proposed inhibitors: AcYKPVD-CHO (1), AcGESPD-CHO (3), AcWDRAD-CHO (4), AcQDTRD-CHO (5), AcADRTD-CHO (6), AcLDVPD-CHO (7), AcFDVPD-CHO (8), and AcITVKD-CHO (9).
Results and discussion
Chemistry
Preparation of the aspartic acid-loaded amino-Merrifield resin was carried out according to previously described protocols[20-22] with minor modifications[15] in the sequence of the synthetic steps. The peptides were synthesized by manual solid-phase peptide synthesis (SPPS) on an aspartic acid-loaded amino-Merrifield resin in accordance to the standard Fmoc strategy (see Scheme 1). The Fmoc protection group of the N-terminal amino acid was deprotected with piperidine 20% in DMF (a). The amino acids were coupled to the amino-Merrifield resin by using HATU/DIPEA or Oxyma/DIC (b). The deprotecting and coupling steps were repeated until the desired tri-, tetra-, or pentapeptide was built up. Finally, the N-terminus was acetylated by using acetic anhydride (c). Subsequently, the peptides were cleaved from the resin and the side chains of the amino acids were deprotected by treatment with trifluoroacetic acid (90%) in water (d). Chemical stability of pentapeptides 7, 38, 41, 42, 43, 44, and 55 was analyzed at room temperature for 28 days and is shown in the Supporting Information (Figures S203-S209).
Scheme 1.

Preparation of the peptides on the modified amino-Merrifield resin: (a) piperidine 20% in DMF, 35 °C ,15 min; (b) amino acid (5 eq), HATU/DIPEA (5/5 eq) or Oxyma/DIC (5/5 eq), DMF/NMP (8/2 v/v), 35 °C, 45 min (single coupling); (c) acetic anhydride (10 eq), DIPEA (10 eq), DMF, rt, 30 min; (d) trifluoroacetic acid (90% in water), rt, 1 h.
Pharmacology
The penta-, tetra-, and tripeptides were investigated in a fluorescence-based enzyme inhibition assay using Casp1, Casp2, Casp3, Casp6, Casp7, and Casp9 to elucidate their structure-activity relationships and selectivity profile. The results are presented as binding constants (pKi values) in Table 2 and Table 3.
Table 2.
Binding data (pKi values) and selectivity ratios of peptides 1-55 at Casp2 and Casp3.
| Sequence | Comp. | pKi ± SEM |
Selectivity ratios of Ki (Casp2: Casp3) |
|||
|---|---|---|---|---|---|---|
| Casp2 | N | Casp3 | N | |||
| AcYKPVD-CHO | 1 | 4.56 ± 0.43a,c | 3 | 4.73 ± 0.10c | 4 | 1:1 |
| AcVDVAD-CHO | 2 | 7.85 ± 0.08a,c | 5 | 7.73 ± 0.09c | 5 | 1:1 |
| AcGESPD-CHO | 3 | 5.23 ± 0.12a | 4 | 6.73 ± 0.14 | 3 | 32:1 |
| AcWDRAD-CHO | 4 | 7.34 ± 0.04a | 2 | 7.34 ± 0.05 | 4 | 1:1 |
| 6.99 ± 0.05b | 2 | 2:1 | ||||
| AcQDTRD-CHO | 5 | 7.02 ± 0.06a | 3 | 7.14 ± 0.16 | 3 | 1:1 |
| AcADRTD-CHO | 6 | 6.73 ± 0.05a | 3 | 7.00 ± 0.09 | 3 | 2:1 |
| AcLDVPD-CHO | 7 | 7.88 ± 0.01a | 2 | 8.16 ± 0.04 | 4 | 2:1 |
| 7.66 ± 0.02b | 2 | 3:1 | ||||
| AcFDVPD-CHO | 8 | 8.15 ± 0.09a | 2 | 8.52 ± 0.06 | 4 | 2:1 |
| 8.03 ± 0.07b | 2 | 3:1 | ||||
| AcITVKD-CHO | 9 | 6.40 ± 0.16a | 4 | 6.05 ±0.06 | 4 | 1:2 |
| AcDRAD-CHO | 10 | 6.39a | 1 | 6.67 ± 0.08 | 3 | 2:1 |
| 5.99 ± 0.02b | 2 | 5:1 | ||||
| AcRAD-CHO | 11 | < 4a | 1 | < 4 | 2 | - |
| < 4b | 2 | - | ||||
| AcDTRD-CHO | 12 | 6.35 ± 0.03a | 2 | 7.09 ± 0.05 | 3 | 5:1 |
| 6.32 ± 0.16b | 2 | 5:1 | ||||
| AcTRD-CHO | 13 | < 4a | 2 | < 4 | 2 | - |
| AcDRTD-CHO | 14 | 5.80 ± 0.02a | 2 | 7.09 ± 0.05 | 3 | 18:1 |
| 5.49 ± 0.11b | 2 | 39:1 | ||||
| AcRTD-CHO | 15 | < 4b | 2 | 4.96 ± 0.09 | 2 | > 9:1 |
| AcDVPD-CHO | 16 | 6.44 ± 0.01a | 2 | 8.18 ± 0.12 | 4 | 55:1 |
| 6.33 ± 0.03b | 2 | 71:1 | ||||
| AcVPD-CHO | 17 | < 4b | 2 | 5.24 ± 0.04 | 2 | > 17:1 |
| AcTVKD-CHO | 18 | 5.11 ± 0.04a | 2 | 4.95 ± 0.04 | 2 | 1:1 |
| AcVKD-CHO | 19 | < 4a | 2 | < 4 | 2 | - |
| AcITV(Orn)D-CHO | 20 | 6.14 ± 0.12a | 2 | 5.68 ± 0.11 | 2 | 1:3 |
| 6.14 ± 0.04b | 2 | 1:3 | ||||
| AcTV(Orn)D-CHO | 21 | 4.62 ± 0.14a | 2 | 4.77 ± 0.01 | 2 | 1:1 |
| AcV(Orn)D-CHO | 22 | < 4a | 2 | < 4 | 2 | - |
| AcITV(Dab)D-CHO | 23 | 5.85 ± 0.04a | 2 | 5.04 ± 0.11 | 3 | 1:6 |
| 5.38 ± 0.18b | 2 | 1:2 | ||||
| AcTV(Dab)D-CHO | 24 | 4.21 ± 0.11a | 2 | 4.43 ± 0.21 | 2 | 2:1 |
| AcV(Dab)D-CHO | 25 | 4.28 ± 0.18a | 2 | < 4 | 2 | > 1:2 |
| AcITV(Dap)D-CHO | 26 | 5.93 ± 0.58a | 2 | 5.32 ± 0.17 | 3 | 1:4 |
| 5.48 ± 0.01b | 2 | 1:1 | ||||
| AcTV(Dap)D-CHO | 27 | < 4a | 2 | 4.43 ± 0.21 | 2 | 3:1 |
| AcV(Dap)D-CHO | 28 | < 4a | 2 | < 4 | 2 | - |
| AcITV(AcK)D-CHO | 29 | 6.30 ± 0.06a | 2 | 7.34 ± 0.12 | 3 | 11:1 |
| 6.24 ± 0.01b | 2 | 13:1 | ||||
| AcTV(AcK)D-CHO | 30 | 4.70 ± 0.18a | 2 | 6.65 ± 0.10 | 3 | 89:1 |
| 4.68 ± 0.03b | 2 | 93:1 | ||||
| AcV(AcK)D-CHO | 31 | < 4a | 2 | 5.02 ± 0.15 | 2 | > 10:1 |
| AcITGKD-CHO | 32 | 5.57 ± 0.01a | 2 | 5.75 ± 0.22 | 2 | 2:1 |
| AcTGKD-CHO | 33 | 4.96 ± 0.03a | 2 | 5.47 ± 0.02 | 2 | 3:1 |
| AcGKD-CHO | 34 | < 4a | 1 | < 4 | 2 | - |
| < 4b | 2 | - | ||||
| AcITAKD-CHO | 35 | 6.63 ± 0.04a | 3 | 6.27 ± 0.05 | 3 | 1:2 |
| AcTAKD-CHO | 36 | 5.79 ± 0.10a | 2 | 5.92 ± 0.01 | 2 | 1:1 |
| AcAKD-CHO | 37 | < 4a | 1 | < 4 | 2 | - |
| < 4b | 2 | - | ||||
| AcATVKD-CHO | 38 | 5.86 ± 0.03b | 2 | 5.93 ± 0.05 | 2 | 1:1 |
| AcATV(Dab)D-CHO | 39 | 4.72 ± 0.02b | 2 | 4.68 ± 0.09 | 2 | 1:1 |
| AcITVSD-CHO | 40 | 6.01 ± 0.10b | 3 | 6.24 ± 0.04 | 3 | 2:1 |
| AcVDVSD-CHO | 41 | 7.26 ± 0.06b | 3 | 7.29 ± 0.03 | 3 | 1:1 |
| AcDVAD-CHO | 42 | 5.95 ± 0.19b | 3 | 7.37 ± 0.11 | 3 | 26:1 |
| AcVAD-CHO | 43 | < 4b | 2 | < 4 | 2 | - |
| AcDVKD-CHO | 44 | 6.05 ± 0.07b | 3 | 6.61 ± 0.03 | 3 | 4:1 |
| AcDKVD-CHO | 45 | 5.61 ± 0.06a | 2 | 7.79 ± 0.02 | 4 | 151:1 |
| 5.22 ± 0.03b | 2 | 371:1 | ||||
| AcDV(Dab)D-CHO | 46 | 4.93 ± 0.07b | 2 | 5.48 ± 0.03 | 2 | 4:1 |
| AcITV(R-K)D-CHO | 47 | < 4b | 2 | < 4 | 2 | - |
| AcITV(R-Dab)D-CHO | 48 | < 4b | 2 | < 4 | 2 | - |
| AcVDV(R-K)D-CHO | 49 | < 4b | 2 | < 4 | 2 | - |
| AcVDV(R-Dab)D-CHO | 50 | 4.41 ± 0.05b | 2 | < 4 | 2 | > 1:3 |
| AcVD(R-K)VD-CHO | 51 | < 4b | 3 | 6.04 ± 0.02 | 3 | > 110:1 |
| AcVD(R-Dab)VD-CHO | 52 | 4.19 ± 0.14b | 2 | 5.64 ± 0.06 | 2 | 28:1 |
| AcVD(R-K)AD-CHO | 53 | 4.31 ± 0.26b | 2 | < 4 | 2 | > 1:2 |
| AcVD(R-Dab)AD-CHO | 54 | < 4b | 2 | 4.71 ± 0.06 | 2 | > 5:1 |
| AcTDTAD-CHO | 55 | 7.48 ± 0.14a | 2 | 7.22 ± 0.12 | 3 | 1:2 |
| 7.19 ± 0.15b | 2 | 1:1 | ||||
Data shown are mean values ± SEM of N independent experiments, each performed in duplicate or triplicate. Data were analyzed by nonlinear regression and were best fitted to sigmoidal concentration–response curves.
Data represent pKi values determined with Casp2.
Data represent pKi values determined with cpCasp2.
Data (pKi ± SD) described in Bresinsky et al.[15]. Conversion of Table 2 data to Ki values within the 95% confidence interval is shown in Table S2 in the SI.
Table 3.
Binding data (pKi values) of selected peptides (1-9, 16, 23, 26, 29, 35, and 45) at Casp1, Casp6, Casp7, and Casp9.
| Sequence | pKi ± SEM | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Casp1 | N | Casp6 | N | Casp7 | N | Casp9 | N | ||
| AcYKPVD-CHO | 1 | < 4a | 2 | < 4a | 2 | 4.53 ± 0.19a | 2 | < 4.5a | 2 |
| AcVDVAD-CHO | 2 | 7.06 ± 0.06a | 2 | < 4a | 2 | 7.08 ± 0.06a | 2 | 5.57 ± 0.31a | 2 |
| AcGESPD-CHO | 3 | 6.52 ± 0.10 | 2 | < 4 | 2 | 5.72 ± 0.01 | 2 | 5.61 ± 0.11 | 2 |
| AcWDRAD-CHO | 4 | 6.48 ± 0.02 | 2 | 4.64 ± 0.03 | 2 | 6.83 ± 0.01 | 2 | 4.96 ± 0.05 | 2 |
| AcQDTRD-CHO | 5 | 5.51 ± 0.01 | 2 | < 4 | 2 | 6.55 ± 0.03 | 2 | < 4 | 2 |
| AcADRTD-CHO | 6 | 5.83 ± 0.07 | 2 | < 4 | 2 | 6.93 ± 0.07 | 2 | 4.81 ± 0.14 | 2 |
| AcLDVPD-CHO | 7 | 7.30 ± 0.02 | 2 | 4.57 ± 0.08 | 2 | 7.68 ± 0.01 | 2 | 5.66 ± 0.10 | 2 |
| AcFDVPD-CHO | 8 | 7.34 ± 0.07 | 2 | 4.85 ± 0.04 | 2 | 8.07 ± 0.11 | 2 | 5.98 ± 0.57 | 2 |
| AcITVKD-CHO | 9 | 6.35 ± 0.06 | 2 | 5.48 ± 0.07 | 2 | 5.25 ± 0.14 | 2 | 4.46 ± 0.35 | 2 |
| AcDVPD-CHO | 16 | 7.15 ± 0.05 | 2 | < 4 | 2 | 7.74 ± 0.07 | 2 | 6.32 ± 0.08 | 2 |
| AcITV(Dap)D-CHO | 26 | 5.82 ± 0.29 | 2 | 4.42 ± 0.37 | 2 | 4.61 ± 0.47 | 2 | 4.68 ± 0.02 | 2 |
| AcITV(AcK)D-CHO | 29 | 6.74 ± 0.19 | 2 | 6.01 ± 0.39 | 2 | 6.65 ± 0.05 | 2 | 5.33 ± 0.04 | 2 |
| AcITAKD-CHO | 35 | 5.84 ± 0.04 | 2 | 4.69 ± 0.02 | 2 | 5.48 ± 0.02 | 2 | 4.59 ± 0.12 | 2 |
| AcDKVD-CHO | 45 | 6.17 ± 0.06 | 2 | < 4 | 2 | 7.51 ± 0.10 | 2 | 5.41 ± 0.05 | 2 |
Data shown are mean values ± SEM of N independent experiments, each performed in duplicate or triplicate. Data were analyzed by nonlinear regression and were best fitted to sigmoidal concentration–response curves.
Data (pKi ± SD) described in Bresinsky et al.[15].
The pentapeptide inhibitors based on natural proteins (see Figure 1) cleaved by Casp2 resulted in a series of peptides with distinct affinities. While the inhibitors AcLDVPD-CHO (7) and AcFDVPD-CHO (8) derived from the MDM2 protein (mouse/human) show high affinities for both Casp2 (pKi: 7.88/7.66 and 8.15/8.03) and Casp3 (pKi: 8.16 and 8.52), the inhibitors based on the protein Golgin subfamily A member 3 AcGESPD-CHO (3) and Ral GTPase-activating protein subunit alpha-1 AcITVKD-CHO (9) demonstrate lower affinities for Casp2 (pKi: 5.23 and 6.40) and Casp3 (pKi: 6.73 and 6.05) (Table 2). Interestingly 9 is the first Casp2 inhibitor within our series with the absence of aspartate (Asp) at position 4, showing moderate affinity for Casp2. The inhibitors AcWDRAD-CHO (4) (based on Protein Transport Protein Sec16A), AcQDTRD-CHO (5) (based on Transcriptional-regulating factor 1), and AcADRTD-CHO (6) (based on Holliday junction recognition protein) have moderate inhibitory affinity for Casp2 (pKi value of 6.73-7.34) with approximately the same affinity for Casp3 (pKi value of 7.00-7.34) (Table 2). All natural proteins were selected because of their specificity of Casp2 cleavage except for the Holliday junction recognition protein, which serves as a control and SAR point. Apart from this, 3, 4, 5, 6, 7, and 8 show a lack of selectivity for Casp2 (selectivity ranging from 0.03 to 1.00), as already seen for 1[15] (cf. Table 2 and Figure 2). Only AcITVKD-CHO (9) exhibits a marginal tendency for Casp2 with a selectivity factor of 2 (cf. Table 2 and Figure 2).
Figure 2.

Concentration-response curves of AcYKPVD-CHO (1), AcGESPD-CHO (3), AcWDRAD-CHO (4), AcQDTRD-CHO (5), AcADRTD-CHO (6), AcLDVPD-CHO (7), AcFDVPD-CHO (8), and AcITVKD-CHO (9) at Casp 2 (A) and Casp3 (B) in the fluoremetric enzyme assay. Data points represent mean values ± SD from representative experiments, each performed in duplicate.
The inhibitors derived from these various natural cleavage sequences (except AcGESPD-CHO (3)) were truncated to verify the extent to which this affects the affinities for Casp2 and Casp3. The purpose of shortening was to make the molecules more drug-like with respect to Lipinski’s rule of 5[23] and to pave the way for future small molecules. Truncations of 4 resulted in the tetrapeptide AcDRAD-CHO (10) and the tripeptide AcRAD-CHO (11). While 10 continues to show affinity for Casp2 (pKi: 6.39/5.97) and Casp3 (pKi: 6.59), 11 evidenced a sharp loss in affinity for Casp2 (pKi: < 4) and Casp3 (pKi: < 4). The same can be observed for the tetrapeptides AcDTRD-CHO (12), AcDRTD-CHO (14), and AcDVPD-CHO (16) (Casp2 pKi: 6.35/6.16, 5.80/5.60, and 6.44/6.33; Casp3 pKi: 7.09, 7.09, and 8.18) and the respective tripeptides AcTRD-CHO (13), AcRTD-CHO (15), and AcVPD-CHO (17) (Casp2 pKi: <4; Casp3 pKi: < 4, 4.96, and 5.24) (Table 2). The peptide AcTVKD-CHO (18) represents a slightly lower affinity for Casp2 (pKi: 5.11) and Casp3 (pKi: 4.95) compared to the other tetrapeptides, which can be explained by the absence of aspartic acid at position 4. In turn, the tripeptide AcVKD-CHO (19) completely loses its affinity for Casp2 (pKi: < 4) and Casp3 (pKi: < 4) (Table 2). It can be summarized that the truncation of the pentapeptides by one amino acid is well tolerated for all tetrapeptides (10, 12, 14, and 16) containing an aspartic acid at position 4. The loss of affinity for Casp2 and Casp3 is somewhat greater for tetrapeptides having threonine (Thr) at position 4 (e.g. AcTVKD-CHO (18)). However, the truncation by a further amino acid is accompanied by a sharp loss in affinity for all tripeptides (11, 13, 15, 17, and 19). Overall, a more significant decrease in Casp2 affinity (ranging from 0.67-1.70 logarithmic units) than Casp3 (ranging from 0.20-0.75 logarithmic units) can be observed for all tetrapeptides. In case of AcDRTD-CHO (14) and AcDVPD-CHO (16) even a slight increase (ranging from 0.02-0.09 logarithmic units) in Casp3 affinity can be observed compared to AcADRTD-CHO (6) and AcLDVPD-CHO (7) (Table 2). These observations go hand-in-hand with the assumption that a motif of five amino acids is required for efficient Casp2 inhibition. Regarding the tetrapeptide’s selectivity between Casp2 and Casp3, it can be observed that 10, 12, 14, and 16 are showing a trend toward Casp3 selectivity (2-71-fold), whereas 18 demonstrates no trend in selectivity (Table 2).
Due to the fact that AcITVKD-CHO (9) shows the best Casp2 selectivity profile, further inhibitors based on 9 were developed to gain more insight in the structure-activity relationships between our peptides and the Casp2/Casp3 binding site. Moreover, 9 is the only reasonably active Casp2 inhibitor that we prepared that does not contain an aspartic acid at the P4 position, which should be beneficial for its pharmacokinetic properties. The Casp2 inhibitor AcVDV(Dab)D-CHO[15], just recently synthesized in our laboratory, served as a starting point for this series. This peptide inhibitor has affinities of pKi: 7.26 (Casp2) and pKi: 5.82 (Casp3), displaying a 27.7-fold Casp2 selectivity over Casp3[15]. The stepwise shortening of the side chain and the acetylation of lysine at P2 led to the peptides AcITV(Orn)D-CHO (20; Casp2 pKi: 6.14; Casp3 pKi: 5.68), AcITV(Dab)D-CHO (23; Casp2 pKi: 5.85/5.56; Casp3 pKi: 5.04), AcITV(Dap)D-CHO (26; Casp2 pKi: 5.93/5.48; Casp3 pKi: 5.32), and AcITV(AcK)D-CHO (29; Casp2 pKi: 6.30/6.23; Casp3 pKi: 7.34), all of them showing lower affinities for Casp2 (Table 2). While 20, 23, and 26 demonstrate lower affinities for Casp3, AcITV(AcK)D-CHO (29) has a higher affinity for Casp3 than 9. In terms of selectivity, we found a selectivity advantage for Casp2 over Casp3 due to the stepwise shortening of the lysine sidechain. While 9 (containing lysine) has a selectivity factor of 2, 20 (containing ornithine) exhibits a factor of 3, 23 (containing diaminobutyric acid) of 6 or 3, respectively, and 26 (containing diaminopropionic acid) of 4 or 1, respectively (Table 2). The acetylation of lysine (AcITV(AcK)D-CHO, 29) generates a selectivity shift toward Casp3 (Casp3/Casp2 selectivity 11 or 13, respectively). The inhibitors 20, 23, 26, and 30 were also truncated to get a deeper insight into the structure-activity relationship of tetra- and tripeptides and the target enzymes. This resulted in the tetrapeptides AcTV(Orn)D-CHO (21; Casp2 pKi: 4.62; Casp3 pKi: 4.77), AcTV(Dab)D-CHO (24; Casp2 pKi: 4.21; Casp3 pKi: 4.43), and AcTV(Dap)D-CHO (27; Casp2 pKi: < 4; Casp3 pKi: 4.43) which show slightly lower affinities for Casp2 and Casp3 relative to AcTVKD-CHO (18) (Table 2). While AcTV(AcK)D-CHO (30, Casp2 pKi: 4.70; Casp3 pKi: 6.65) represents low affinity for Casp2, the affinity for Casp3 almost remains. Thus, the peptide displays selectivity for Casp3 over Casp2 (Casp3/(cp)Casp2 selectivity 89 or 93) (Table 2). The corresponding tripeptides of this series AcV(Orn)D-CHO (22), AcV(Dab)D-CHO (25), AcV(Dap)D-CHO (28), and AcV(AcK)D-CHO (31) do not show notable affinity for Casp2 (pKi values < 4) nor Casp3 (pKi values < 4 - 5.02) (Table 2).
Starting from AcITVKD-CHO (9) further modifications were made, e.g. valine at position P3 was replaced by glycine and alanine. At the same time we prepared tetra- and tripeptides of these sequences. This resulted in AcITGKD-CHO (32), AcTGKD-CHO (33), AcGKD-CHO (34), AcITAKD-CHO (35), AcTAKD-CHO (36), and AcAKD-CHO (37). The pentapeptide 32 (Casp2 pKi: 5.57 ; Casp3 pKi: 5.75) shows slightly lower affinity for Casp2 and Casp3 than 9 (Casp2 pKi: 6.40 ; Casp3 pKi: 6.05), whereas the affinities maintained for 35 (Casp2 pKi: 6.63 ; Casp3 pKi: 6.27). Tetrapeptides 33 (Casp2 pKi: 4.96; Casp3 pKi: 5.47) and 36 (Casp2 pKi: 5.79; Casp3 pKi: 5.92) show lower affinities for Casp2 and Casp3 than the corresponding pentapeptides (Table 2) but the affinity decrease is more pronounced for Casp2 (0.61 and 0.84) than for Casp3 (0.28 and 0.37). The tripeptides 34 and 37 did not show any affinity for Casp2 (pKi: < 4) and Casp3 (pKi: < 4). We have also replaced isoleucine at position P5 by alanine resulting in AcATVKD-CHO (38, Casp2 pKi: 5.86; Casp3 pKi: 5.93). AcATV(Dab)D-CHO (39, Casp2 pKi: 4.72; Casp3 pKi: 4.68) was an unsuccessful approach to gain selectivity for Casp2 by exchanging lysine at P2 with diaminobutyric acid (Dab). In addition, we have introduced serine at position P2, but the expected selectivity advantage for Casp2 described by Poreba et al.[24] could not be determined for AcITVSD-CHO (40, Casp2 pKi: 6.01; Casp3 pKi: 6.24) (Table 2).
To establish further structure-activity relationships with respect to our recent study by Bresinsky et al.[15], we made further modifications starting from the already described canonical inhibitor AcVDVAD-CHO (2, Casp2 pKi: 7.85; Casp3 pKi: 7.73). First, we truncated the pentapeptide to a tetra- and tripeptide, resulting in AcDVAD-CHO (42) and AcVAD-CHO (43). 42 demonstrates a massive loss of affinity for Casp2 (pKi: 5.95) by 2 logarithmic units, while the affinity for Casp3 (pKi: 7.37) is almost completely preserved (Table 2). Thus, AcDVAD-CHO (42) shows a selectivity factor of 26 for Casp3 over Casp2. The tripeptide 43 completely loses its inhibitory affinity for Casp2 (pKi: < 4) and Casp3 (pKi: < 4). We also introduced serine at position P2 of the canonical inhibitor in an attempt to gain selectivity for Casp2 but again the desired effect could not be detected for AcVDVSD-CHO (41, Casp2 pKi: 7.26; Casp3 pKi: 7.29) (Table 2). AcDVKD-CHO (44), AcDKVD-CHO (45), and AcDV(Dab)D-CHO (46) are the corresponding tetrapeptides of the peptides AcVDVKD-CHO (Casp2 pKi: 7.63; Casp3 pKi: 6.91), AcVDKVD-CHO (Casp2 pKi: 7.40; Casp3 pKi: 7.28), and AcVDV(Dab)D-CHO (Casp2 pKi: 7.26; Casp3 pKi: 5.82) we have recently described[15]. While for AcDVKD-CHO (44) and AcDKVD-CHO(45) a significant decrease in affinity for Casp2 (pKi: 5.99 and 5.61/5.22) can be observed, the affinity for Casp3 (pKi: 6.62 and 7.79) remains. In the case of 45, the affinity for Casp3 even increases, making the tetrapeptide a selective Casp3 inhibitor with a selectivity factor of 151 or 371, respectively. AcDV(Dab)D-CHO (46) shows low affinity for Casp2 (pKi: 4.93) and Casp3 (pKi: 5.48) (Table 2).
Finally, AcITV(R-K)D-CHO (47), AcITV(R-Dab)D-CHO (48), AcVDV(R-K)D-CHO (49), AcVDV(R-Dab)D-CHO (50), AcVD(R-K)VD-CHO (51), AcVD(R-Dab)VD-CHO (52), AcVD(R-K)AD-CHO (53), and AcVD(R-Dab)AD-CHO (54) were synthesized to potentially better address specific amino acids (Glu52) in the Casp2 binding site due to the changed spatial arrangement of the inhibitors. 47, 48, 49, 50, 53, and 54 demonstrate low affinities for Casp2 (pKi values < 4 - 4.41) and Casp3 (pKi values < 4 - 4.71). 51 and 52 show low affinities for Casp2 (pKi values < 4 - 4.19) and moderate affinities for Casp3 (pKi values 6.04 and 5.64). AcVD(R-K)VD-CHO (51) indicates an at least 100-fold selectivity for Casp3 compared to Casp2 (Table 2). AcTDTAD-CHO (55) represents a precursor for future stapled peptides. It displays high affinities for Casp2 (pKi: 7.48/7.04) and Casp3 (pKi: 7.22) with no distinct selectivity trend (Table 2).
In addition, a panel assay at Casp1, Casp6, Casp7, and Casp9 (Table 3) was carried out for selected peptides (3-9, 16, 26, 29, 35, and 45). The pentapeptides AcGESPD-CHO (3) and AcADRTD-CHO (6) show moderate inhibitory affinities for Casp1 (pKi: 6.52 and 5.83), Casp7 (pKi: 5.72 and 6.93), and Casp9 (pKi: 5.61 and 4.81) and no affinity for Casp6 (pKi: < 4). AcWDRAD-CHO (4) and AcQDTRD-CHO (5) exhibit moderate affinities for Casp1 (pKi: 6.48 and 5.51) and Casp7 (pKi: 6.83 and 6.55) and low affinities for Casp6 (pKi: 4.64 and < 4) and Casp9 (pKi: 4.96 and < 4) (Table 3). AcLDVPD-CHO (7, Casp1 pKi: 7.30; Casp6 pKi: 4.57; Casp7 pKi: 7.68; Casp9 pKi: 5.66) and AcFDVPD-CHO (8, Casp1 pKi: 7.34; Casp6 pKi: 4.85; Casp7 pKi: 8.07; Casp9 pKi: 5.98) derived from the MDM2 protein (mouse/human) are highly active for Casp1 and Casp7. The affinity for Casp9 is lower, while for Casp6 no noticable affinity can be observed (Table 3). The peptide AcITVKD-CHO (9) exhibits moderate to low affinity for Casp1 (pKi: 6.35), Casp6 (pKi: 5.48), Casp7 (pKi: 5.25), and Casp9 (pKi: 4.46). In comparison, the peptide AcYKPVD-CHO (1) shows no major affinity for Casp1, Casp6, Casp7, and Casp9, while the canonical inhibitor AcVDVAD-CHO (2) is active at Casp1 (pKi: 7.06), Casp7 (pKi: 7.08), and Casp9 (pKi: 5.57) (Table 3). The tetrapeptide AcDVPD-CHO (16, Casp1 pKi: 7.15; Casp6 pKi: < 4; Casp7 pKi: 7.74; Casp9 pKi: 6.32) shows a similar affinity profile as AcLDVPD-CHO (7) and AcFDVPD-CHO (8) (Table 3). AcITV(Dap)D-CHO (26) indicates slightly lower affinities for Casp1, Casp6, Casp7, and Casp9 compared to lead compound AcITVKD-CHO (9) (Casp1 pKi: 6.35 vs. 5.82; Casp6 pKi: 5.48 vs. 4.42; Casp7 pKi: 5.25 vs. 4.61; Casp9 pKi: 4.46 vs. 4.68), while AcITV(AcK)D-CHO (29) exhibits slightly higher affinities (Casp1 pKi: 6.74; Casp6 pKi: 6.01; Casp7 pKi: 6.65; Casp9 pKi: 5.33). Finally, AcITAKD-CHO (35) demonstrates moderate to low affinities for Casp1 (pKi: 5.84), Casp6 (pKi: 4.69), Casp7 (pKi: 5.48), and Casp9 (pKi: 4.59) (Table 3) and AcDKVD-CHO (45) displays high affinity for Casp7 (pKi: 7.51), moderate affinity for Casp1 (pKi: 6.17), Casp9 (pKi: 5.41), and low affinity for Casp6 (pKi: < 4) (Table 3).
Molecular Modeling
To test whether the relative binding affinities of these inhibitors could be understood computationally we implemented a covalent inhibitor docking protocol employing the CovDock module of the Schrödinger Software Suite (Schrödinger, LLC, New York, NY, Version 2021.3 unless otherwise noted.).[25-33] We recently reported that ligand poses predicted by this method recapitulate crystallographically determined structures of similar pentapepties with good fidelity.[15] Table 4 lists the covdock affinity results from this protocol for three reference compounds and the new inhibitors reported in this study. The docking results give a good indication of good and bad binders to Casp2 and 3, although they do not entirely match the in vitro results from Table 2. However, the consistency of the poor pKi and cdock affinity values for AcYKPVD-CHO (1) (Casp2 and 3), as well as the increased cdock affinities for AcQDTRD-CHO (5), AcITVKD-CHO (9) and AcITV(Dap)D-CHO (26) at Casp2 is noticeable. For compound 9 and 26, preferential binding to Casp2 over Casp3 is observed both in vitro and in silico.
Table 4.
Molecular Modeling of Casp2 and Casp3 and potential ligands.
| Sequence | Comp. | Casp2 cdock affinity |
Casp3 cdock affinity |
|---|---|---|---|
| AcYKPVD-CHO | 1 | −9.561 | −10.126 |
| AcGESPD-CHO | 3 | −13.073 | −12.564 |
| AcWDRAD-CHO | 4 | −13.771 | −12.706 |
| AcQDTRD-CHO | 5 | −15.376 | −13.369 |
| AcADRTD-CHO | 6 | −13.113 | −12.475 |
| AcLDVPD-CHO | 7 | −13.080 | −13.271 |
| AcFDVPD-CHO | 8 | −12.728 | −13.634 |
| AcITVKD-CHO | 9 | −15.328 | −13.100 |
| AcDVPD-CHO | 16 | −11.856 | −11.896 |
| AcITV(Dab)D-CHO | 23 | −12.972 | −12.876 |
| AcITV(Dap)D-CHO | 26 | −14.278 | −12.721 |
| AcDKVD-CHO | 45 | −12.641 | −11.952 |
| Reference compounds | |||
| AcVDVAD-CHO | 2 | −13.614 | −12.148 |
| AcLDESD-CHO | 56 | −13.519 | −13.369 |
| AcDEVD-CHO | 57 | −13.439 | −12.218 |
Calculation using covalent docking in Schrödinger 2021.3. Casp2 = PDBid: 1pyo, Casp3 = PDBid: 3edq. Proteins and ligands prepared as described in the section Molecular Modeling. cdock affinity = covalent docking affinity (kcals/mol).
Additionally, we wondered whether the initial structure of the ligand, since this is generated by methods that are not easily standardized, would affect the final cdock affinity. Cdock affinities were computed starting from five conformations of AcVDVAD-CHO with relative energies spanning 10 kcal/mol (Figure 3). This experiment confirmed that cdock affinities are dependent on the starting conformation of the ligand (at least for these highly flexible ligands) and vary in this simple case over a range of 1.0 kcal/mol with an average value of −13.92 kcals/mol (SD 0.41 kcal/mol, Table S3, SI). For potent ligands like AcVDVAD-CHO the final five structures that were arrived at by cdock optimization were roughly identical (RMSD = 0.103 - 0.067, Table S4, SI) whereas for weak ligands, like AcYKPVD-CHO, the final structures were significantly different (RMSD = 0.153 – 0.977, Table S4, SI).
Figure 3.

Cdock affinities (right axis (kcal/mol)) resulting from the docking of five different conformations of AcVDVAD-CHO (energies on left axis (kcal/mol)) with PDBid: 1pyo. Energies on the left axis calculated with Prime using the OPLS4 forcefield. Energies on the right axis calculated using Glide and the Covalent Docking module in Maestro (Schrödinger, 2021.3).
From these experiments we conclude that cdock optimization for this class of pentapeptide inhibitors are useful for generally determining whether or not a ligand will bind to the target. If the pentapeptide inhibitor has a cdock affinity < - 11.0 kcal/mol, the predicted structure from any starting point will be a good approximation of the crystal structure. Further results along these lines will be reported in due course.
Crystallography
While there has been some exploration of the structural consequences of variation of the P4 amino acid from the preferred aspartic acid in complexes with caspase 3[34] and caspase 7[35,36], these have been limited to tetrapeptide inhibitors, and none have specifically involved the P4 threonine found to be preferred from our work. Motivated by curiosity regarding details of how the threonine substitutes for the well-characterized aspartic acid, crystallographic studies were initiated and complexes of five inhibitors with caspase 3 were obtained: one with compound 9 (AcITVKD-CHO), with 16 (AcDVPD-CHO), with 20 (AcITV(Orn)D-CHO), with compound 23 (AcITV(Dab)D-CHO), and one with 36 (AcITAKD-CHO). A summary of crystallographic data and refinement statisitics is provided in Suplemental Table S1. In the 1.9 Å complex with 26 (AcITV(Dab)D-CHO, the position of the N-terminal acetyl group is not resolved, but in other structures, the ligand backbone geometry is otherwise well-defined by electron density across the length of the penta-peptide. As expected, the CHO leaving group has been displaced and a covalent bond joins the P1 Asp to Cys163, in both structures. The P4 Thr sidechain occupies the same position and engages in similar H-bonding in these two structures.
A comparison of the complex of compound 23 to a more typical canonical inhibitor sequence (the AcVDVAD-CHO complex previously described by Bresinsky et al.[15]) is provided as Figure S1 in the Supporting Information (SI). Backbone H-bonds observed in the two complexes are the same, but the lone hydroxyl group of the threonine sidechain (T4) is not capable of accepting hydrogen bonds from both Phe250 and Asn208 as can a D4 carboxylate. Only the hydrogen bond from the T4 OH to Phe 250 NH is observed. We had imagined that this might cause either the inhibitor to slide away from L4 toward Asn208 and L3, or L4 to move in tighten packing against the inhibitor with a smaller footprint, but neither change has occurred. The threonine seems to be an isosteric substitute for an aspartic acid in P4, albeit with a predictable loss in potency due to the loss of one H-bond.
A comparison of the complexes with compounds 16 and 23 is also provided as Figure S1 in the SI. Backbone H-bonds are conserved in both complexes, illustrating that truncation of pentapeptides to tetrapeptides does not affect the backbone binding conformation. Interestingly, a proline in the tetrapeptide sequence does not alter the backbone binding confirmation either (Figure S1 of SI), leading to the hypothesis that the ring in the proline structure locks the peptide into the binding confirmation. Limiting the number of possible binding confirmations creates more favorable enthalpy of binding (Gibbs free energy of binding) and could help explain the increase in potency of compound 16 at both Casp2 and Casp3 as compared to other tetrapeptides in the series (for example compound 42 (AcDVAD-CHO) and compound 44 (AcDVKD-CHO)).
Conclusion
In this study, we report the design and characterization of peptidic caspase-2 inhibitors originating from specific sites of caspase-2-mediated proteolysis. Besides the classic key motif of caspase-2 inhibitors, consisting of five amino acids, we have also truncated our inhibitors to tetrapeptides and tripeptides which should result in a better bioavailability and a more drug-like character of our ligands relative to Lipinski's rule of 5 (RO5). In summary, we have synthesized and characterized a series of 53 peptides for their inhibitory affinity at Casp2 and Casp3. Selected inhibitors 3-9, 16, 23, 26, 29, 35, and 45 were tested in a panel assay screening to examine affinities at Casp1, Casp6, Casp7, and Casp9 to receive more information about the selectivity within the caspase family. Within the new cleavage sequences the inhibitors AcLDVPD-CHO (7) and AcFDVPD-CHO (8) demonstrate the highest affinity for Casp2 (pKi: 7.88/7.66 and 8.15/8.03). Nevertheless, it must be noted that 7 and 8 display little selectivity for Casp2 over Casp3 (Casp2/Casp3 selectivity: 2-3). Truncation of 7 and 8 resulted in the tetrapeptide AcDVPD-CHO (16, Casp2 pKi: 6.44/6.33; Casp3 pKi: 8.18) with 16 showing Casp3 selectivity. Overall the truncation of pentapeptides to tetra- and tripeptides do not lead to Casp2 selectivity. In the case of tripeptides, even a total collapse of Casp2 and Casp3 activities can be observed. These findings support the assumption that a motif of five amino acids is required for efficient Casp2 inhibition in this class of peptidic, reversible inhibitors. The pentapeptide AcITVKD-CHO (9, Casp2 pKi: 6.40) is the only inhibitor derived from the natural cleavage sequences to show marginal selectivity for caspase-2 (2-fold selectivity). Interestingly, it does not contain aspartic acid at position P4. A broader series of AcITVKD-CHO analogs including AcITV(Orn)D-CHO (20), AcITV(Dab)D-CHO (23), AcITV(Dap)D-CHO (26), and AcITVKD-CHO (35) exhibited moderate selectivity for Casp2. Regarding Casp3 selectivity AcDKVD-CHO (45), (Casp2 pKi: 5.61/5.22; Casp3 pKi: 7.79), the corresponding tetrapeptide of the recently described AcVDKVD-CHO (Casp2 pKi: 7.40; Casp3 pKi: 7.28)[15], showed up as the most selective Casp3 inhibitor in this study with a selectivity factor of 151 or 371, respectively. The associated calculations via molecular modeling are predominantly in agreement with the in vitro data. Crystal structures with bound AcITV(Dab)D-CHO in Casp3 confirm that the P4 Thr is a good isostere for the P4 Asp that induces little change in protein conformation: a good prerequisite for future modeling with the series. In summary, the results of this study provide a vast knowledge of the structure–affinity relationships (SAR) of a large number of peptides with various activities and selectivities within the caspase family. These findings provide a reasonable basis for the development of selective Caspase-2 inhibitors.
Experimental section
General
Unless otherwise listed, chemicals and solvents were purchased from commercial suppliers and used as received. All the solvents were of analytical grade or distilled prior to use. Dimethylformamide (DMF) and N-methyl-2-pyrrolidone (NMP) were purchased from Iris Biotech (Marktredwitz, Germany). Sodium borhydride, oxalyl chloride, methanol, dichloromethane, diethyl ether, toluene, ethyl acetate, tetrahydrofuran, and hydrochloric acid 37% were obtained from Fisher Scientific/Acros Organics (Schwerte, Germany). If lower concentrations of hydrochloric acid were required, these were diluted accordingly. Isobutyl chloroformate, tert-butylcarbazat, p-toluenesulfonic acid, trans-4-(aminomethyl)cyclohexanecarboxylic acid, N,N′-diisopropylcarbodiimide (DIC), Oxyma and N-methylmorpholine were purchased from TCI (Eschborn, Germany). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), 1-hydroxybenzotriazole hydrate (HOBt), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), N,N-diisopropylethylamine (DIPEA), and trifluoroacetic acid (TFA) were obtained from ABCR (Karlsruhe, Germany). 1,1'-Carbonyldiimidazole was purchased from Fluorochem (Derbyshire, United Kingdom). Aminomethylated polystyrene HL (100-200 mesh), acetic anhydride, and triethylamine were purchased from Merck (Darmstadt, Germany). Benzyl alcohol, dimethylsulfoxide, acetic acid, and acetonitrile for high-performance liquid chromatography (HPLC) were obtained from Sigma-Aldrich (Taufkirchen, Germany). Protected amino acids Fmoc-Tyr(tBu)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Phe-OH, Fmoc-Pro-OH, Fmoc-Val-OH, Fmoc-Asp(O tBu)-OH, Fmoc-Ala-OH, Fmoc-Orn(Boc)-OH, Fmoc-Dab(Boc)-OH, Fmoc-Dap(Boc)-OH, Fmoc-Lys(Ac)-OH, Fmoc-Thr(tBu-OH, Fmoc-Ile-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Gly-OH, Fmoc-Ser(tBu)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Gln-OH, Fmoc-Leu-OH, Fmoc-D-Lys(Boc)-OH and Fmoc-D-Dab(Boc)-OH were procured from Carbolution Chemicals (St. Ingbert, Germany). Deuterated solvents for nuclear magnetic resonance (NMR) spectroscopy were purchased from Deutero (Kastellaun, Germany). The orbital shaker (Multi Reax) was from Heidolph (Schwabach, Germany). The frits had a pore size of 35 μm and were procured from Roland Vetter Laborbedarf (Ammerbuch, Germany). The infrared lamp was from Medisana (Neuss, Germany), and the thermostat was from PEARL.GmbH (Buggingen, Germany). As syringes were used Injekt Luer Solo from Braun (Melsungen, Germany). For the preparation of stock solutions, buffers, and HPLC eluents Millipore water was used. NMR spectra (1H NMR, 13C NMR, 19F NMR, DEPT, 1H COSY, HSQC, HMBC) were recorded on a Bruker Avance-300 (7.05 T, 1H: 300 MHz, 13C: 75.5 MHz, 19F: 188) or Avance-400 (9.40 T, 1H: 400 MHz, 13C: 100.6 MHz, 19F: 282) NMR spectrometer (Bruker, Karlsruhe, Germany). All chemical shifts are reported in the δ-scale as parts per million (ppm) relative to the solvent's residual peaks as the internal standard. Moreover, the multiplicity, coupling constant (J), and number of protons are stated. Multiplicities are specified with the following abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), m (multiplet), as well as combinations thereof. High-resolution mass spectrometry (HRMS) was performed on a Q-TOF 6540 ultrahigh definition (UHD) LC/MS system (Agilent Technologies) using an electrospray ionization (ESI) source or on an AccuTOF GCX GC/MS system (Jeol, Peabody, MA) using an electron ionization (EI) source. Preparative HPLC was performed with a system from Waters (Eschborn, Germany) consisting of a Waters 2545 binary gradient module, a Waters 2489 UV/vis-detector, a Waters Fraction Collector 3, and the column was a YMC Triart C18 (150 x 10 mm, 5 μm) (YMC, Dinslaken, Germany) at a flow rate of 20 mL/min or a HPLC from Knauer (Berlin, Germany) consisting of two K-1800 pumps, a K-2001 detector, and the column was a Phenomenex Gemini (250 x 21 mm, 5 μm) (Phenomenex, Aschaffenburg, Germany) at a flow rate of 15 mL/min. As a mobile phase, mixtures of MeCN and 0.1% aqueous (aq) TFA were used. The UV detection was carried out at 220 nm. For sample preparation, all compounds were dissolved in a mixture of water/acetonitrile (95/5 v/v) and filtered with PTFE filters (25 mm, 0.2 μm) (Phenomenex, Aschaffenburg, Germany). The purified peptides were lyophilized using a CHRIST Alpha 2-4 LD freeze dryer (Osterode am Harz, Germany) equipped with a RZ 6 rotary vane vacuum pump (Vacuubrand, Wertheim, Germany). Analytical purity control was performed on a 1100 HPLC system from Agilent Technologies equipped with an Instant Pilot controller, a G1312A binary pump, a G1329A ALS autosampler, a G1379A vacuum degasser, a G1316A column compartment, and a G1315B diode array detector (DAD). The column was a Phenomenex Kinetex XB-C18 column (250 x 4.6 mm 2, 5 μm) (Phenomenex, Aschaffenburg, Germany) or a Phenomenex Gemini NX-C18 column (250 x 4.6 mm, 5 μm). The oven temperature during HPLC analysis was 30 °C. As a mobile phase, mixtures of MeCN/aqueous TFA were used. Absorbance was detected at 220 nm. The injection volume was 20-80 μL at compound concentrations of 1 mM. The following linear gradient was applied: MeCN/TFA (0.05%) (v/v) 0 min: 10:90, 25 min: 95:5, 35 min: 95:5; flow rate: 1.0 mL/min, t0(Kinetix XB-C18) = 2.75 min, t0(Gemini NX-C18) = 2.99 min (t0 = dead time). Retention (capacity) factors k were calculated from the retention times tR according to k = (tR − t0)/t0. The purities of the compounds were calculated by the percentage peak area of the chromatograms. The tested compounds have been screened for PAINS and aggregation by publicly available filters (http://zinc15.docking.org/patterns/home, http://advisor.docking.org).[37,38] Compounds have not been previously reported as PAINS or aggregators. None of the data showed abnormalities, e.g. high Hill slopes, that could be a hint for PAINS.[38]
Computational Chemistry
Molecular Modeling
Covalent docking studies were performed using the “Covalent Docking (CovDock)” module within the Schrödinger small-molecule drug discovery software suite (Schrödinger, LLC, New York, NY, Version 2021.3 unless otherwise noted. The crystal structures of Casp2 (PDBid: 1pyo) and Casp3 (PDBid: 3edq) were prepared using the module “Protein Preparation Wizard” in Maestro with the default protein parameters. Hydrogen atoms were added and water molecules that were beyond 5 Å from heterocyclic groups were deleted. The covalently bound ligands AcLDESD-CHO:1pyo, AcLDESD-CHO:3edq where D-CHO represents “aspartic acid aldehyde” were included in the protein structures during their preparation for the covalent docking. In the experiments below, the protein structures prepared in this fashion are denoted by PP:PDBid, e.g. PP:1pyo. Hydrogen bonds were optimized, the partial charges were assigned, and the protein structure was energy-minimized using OPLS3e force field.[39] Following this preparation, the covalent bond connecting the ligand to the protein was broken and the now-separated aldehyde (reactive functional group) and cysteine (nucleophilic reaction group) were reconstituted by adjusting bond orders, adding hydrogens, and minimizing these groups in place. This free ligand (the “Workspace Ligand”) was employed to create the covalent docking grid used in the covalent docking and scoring (vide infra). The individual target receptors were set up using the following reactive cysteine residues (A:155, 1pyo; A:163, 3edq). The “Reaction Type” SMARTS string {[H]C=O} was built as a customized nucleophilic addition to a double bond. The “Box Center” for the docking grid was set using the “Centroid of (the) Workspace Ligand”. Docking was performed in the “Pose Prediction (Thorough)” mode. A “Minimization radius” of 3.0 Å was used, “Perform MM-GBSA scoring” was selected, and three (3) “Output poses per ligand reaction site” were selected (only the lowest energy pose is reported). Ligands for covalent docking experiments were drawn in ChemDraw, imported into Maestro as sdf, and refined into 3D structures using the “Ligand Preparation” module and its default parameters. These 3D structures, with the appropriate tautomers and charges, were directly used in “Covalent Docking” experiments and are designated as PL:ligand name, e.g. PL:AcLDESD-CHO. “Cdock Affinity” is reported in kcal/mol. Five starting points for cdock affinity prediction were created by the following protocol. These are structures 1-5 (state number) in Figure 3. (1) Standard input as described above; (2) Prime (module in the Schrödinger small-molecule drug discovery software suite) minimization of state 1; (3-5) states selected from 433 states generated by searching of the conformational space of e.g. AcVDVAD-CHO with a 10 kcal/mol energy window (3 = lowest energy state, 4 = middle energy state, and 5 = highest energy state).
Compound Characterization
The peptides were characterized using the following methods: HRMS, 1H NMR spectroscopy (for spectra, see Supporting Information), and 13C NMR spectroscopy (for spectra, see Supporting Information). Additionally, two-dimensional (2D) NMR spectra like HSQC (for spectra, see Supporting Information) were made. For purity control, HPLC (RP-HPLC) analysis was performed (for chromatogram, see Supporting Information) with a minimum purity standard of ≥ 95%.
Synthesis and analytical data
General procedure for the synthesis of the peptides
The aspartic acid loaded semicarbazide amino-Merrifield resin (300 mg, 0.188 mmol, 1 eq) was weighed into a fritted 10 mL syringe. Subsequently, 5 mL of a mixture of piperidine 20% in DMF was drawn up to remove the N-terminal Fmoc protecting group. The syringe was shaken on an orbital shaker at 35 °C for 15 min. The orbital shaker was covered with a box, which was insulated from the inside with aluminum foil. An infrared lamp was placed on an aperture on top. In order to keep the temperature constant at 35 °C the lamp was controlled by a thermostat. The liquid was then removed with the aid of a vacuum flask and the residual resin was washed with DMF (3 x 8 mL). The coupling of the amino acids to the N-terminus was performed by two different methods. The corresponding amino acid (5 eq) and HATU (357 mg, 0.94 mmol, 5 eq) (Method A) or Oxyma (134 mg, 0.94 mmol, 5 eq) (Method B) were weighed in two separate Erlenmeyer flasks. Subsequently, both were dissolved in 3-4 mL of a mixture of DMF/NMP (8/2 v/v). Then N,N-diisopropylethylamine (164 μL, 0.94 mmol, 5 eq) (Method A) or N,N'-diisopropylcarbodiimide (151 μL, 0.94 mmol, 5 eq) (Method B) was added to the solution of HATU (procedure A) / Oxyma (procedure B). Subsequently, both solutions were drawn up with the resin-loaded syringe and shaken at 35 °C for 45 min. The liquid was then removed with the aid of a vacuum flask and the residual resin was washed with DMF (3 x 8 mL).
The coupling and deprotection steps were repeated until the desired tri-, tetra-, or pentapeptide was built up. Then the N-terminal Fmoc protecting group was removed using piperidine in DMF (20%). Subsequently, the N-terminus of the peptide was acetylated by dissolving acetic anhydride (178 μL, 1.88 mmol, 10 eq) and N,N-diisopropylethylamine (328 μL, 1.88 mmol, 10 eq) in 6-8 mL DMF. The solution was drawn up with the syringe and shaken for 30 min at room temperature. After completion, the liquid was removed, and the resin was washed with DMF (2 x 8 mL), methanol (2 x 8 mL), dichloromethane (2 x 8 mL), and finally with diethyl ether (2 x 8 mL). The peptide was cleaved off the resin, and the side chains were deprotected by drawing up 6 mL of trifluoroacetic acid 90% in water. The syringe was shaken for 1 h at room temperature. The liquid was then poured into a round-bottomed flask. The step was repeated again, and then the cleavage cocktail was diluted with 50 mL water and freeze-dried. The crude product was purified by HPLC, yielding the corresponding peptide.
(S)-4-(2-Acetamidoacetamido)-5-({(S)-1-[(S)-2-{[(S)-1-carboxy-3-oxopropan-2-yl]carbamoyl}pyrrolidin-1-yl]-3-hydroxy-1-oxopropan-2-yl}amino)-5-oxopentanoic acid (3)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (16.7 mg, 17%): RP-HPLC: >99%, (tR = 3.67, k = 0.22). 1H NMR (400 MHz, D2O) δ 4.95 – 4.85 (m, 1H), 4.66 – 4.60 (m, 1H), 4.42 – 4.04 (m, 3H), 3.80 (s, 2H), 3.77 – 3.54 (m, 4H), 2.71 – 2.38 (m, 2H), 2.35 (t, J = 7.3 Hz, 2H), 2.23 – 1.94 (m, 2H), 1.93 (s, 3H), 1.91 – 1.74 (m, 4H). 13C NMR (101 MHz, D2O) δ 177.05, 175.24, 174.94, 174.27, 173.77, 173.25 (d, J = 3.5 Hz), 171.70, 170.38, 89.67, 60.93, 60.74, 53.51, 52.77, 51.59, 48.09, 42.50, 33.96, 29.89, 29.42, 26.20, 24.46 (d, J = 27.4 Hz), 21.73. m/z [M+H+] calculated for C21H32N5O11+: 530.2093, found 530.2100.; C21H31N5O11 (529.50).
(4S,7S,10S,13S,16S)-4-[(1H-Indol-3-yl)methyl]-7-(carboxymethyl)-16-formyl-10-(3-guanidinopropyl)-13-methyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (4)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (12.0 mg, 8%): RP-HPLC: 98%, (tR = 7.48, k = 1.49). 1H NMR (300 MHz, D2O) δ 7.50 – 7.30 (m, 2H), 7.17 – 6.94 (m, 3H), 4.87 (dd, J = 4.6, 2.7 Hz, 1H), 4.42 (t, J = 6.9 Hz, 1H), 4.34 (t, J = 6.3 Hz, 1H), 4.17 – 3.91 (m, 3H), 3.09 (d, J = 7.0 Hz, 2H), 2.97 (t, J = 6.8 Hz, 2H), 2.65 – 2.28 (m, 4H), 1.83 (s, 3H), 1.71 – 1.46 (m, 2H), 1.45 – 1.30 (m, 2H), 1.25 – 1.16 (m, 3H). m/z [M+H+] calculated for C30H42N9O10+: 688.3049, found 688.3058.; C30H41N9O10 x C2HF3O2 (801.73).
(4S,7S,10S,13S,16S)-4-(3-Amino-3-oxopropyl)-7-(carboxymethyl)-16-formyl-13-(3-guanidinopropyl)-10-[(R)-1-hydroxyethyl]-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (5)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (15.0 mg, 10%): RP-HPLC: >99%, (tR = 3.43, k = 0.14). 1H NMR (300 MHz, D2O) δ 4.88 (d, J = 2.2 Hz, 1H), 4.24 – 4.03 (m, 5H), 3.04 (t, J = 6.9 Hz, 2H), 2.90 – 2.27 (m, 5H), 2.22 (t, J = 7.5 Hz, 2H), 1.98 – 1.77 (m, 5H), 1.72 – 1.55 (m, 2H), 1.52 – 1.39 (m, 2H), 1.04 (d, J = 6.3 Hz, 3H). 13C NMR (75 MHz, D2O) δ 177.77, 175.19, 174.51, 174.30, 173.64, 173.50, 173.01, 172.68, 171.89, 171.62, 156.72, 89.68, 66.84, 59.33, 53.54, 53.46, 51.51, 50.22, 40.48, 35.21, 31.04, 28.08, 26.70, 25.84, 24.34, 21.64, 18.79. m/z [M+H+] calculated for C25H42N9O12+: 660.2947, found 660.2947.; C25H41N9O12 x C2HF3O2 (773.68).
(4S,7S,10S,13S,16S)-7-(Carboxymethyl)-16-formyl-10-(3-guanidinopropyl)-13-[(R)-1-hydroxyethyl]-4-methyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (6)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (12.2 mg, 9%): RP-HPLC: >99%, (tR = 3.21, k = 0.07). 1H NMR (300 MHz, D2O) δ 4.93 – 4.87 (m, 1H), 4.56 – 4.49 (m, 1H), 4.28 – 4.20 (m, 1H), 4.17 – 3.99 (m, 4H), 3.06 (t, J = 6.8 Hz, 2H), 2.84 – 2.35 (m, 4H), 1.87 (s, 3H), 1.83 – 1.59 (m, 2H), 1.54 – 1.41 (m, 2H), 1.21 (d, J = 7.2 Hz, 3H), 1.09 – 1.00 (m, 3H). 13C NMR (75 MHz, D2O) δ 176.37, 175.47, 175.34, 174.61, 174.35, 173.52, 172.54, 171.41, 171.29, 156.72, 89.66, 67.06, 59.47, 53.45, 51.60, 50.23, 50.04, 40.47, 35.44, 34.23, 28.08, 24.30, 21.59, 18.80, 16.30. m/z [M+H+] calculated for C23H39N8O11+: 603.2733, found 603.2741.; C23H38N8O11 x C2HF3O2 (716.63).
(S)-3-[(S)-2-Acetamido-4-methylpentanamido]-4-{[(S)-1-((S)-2-{[(S)-1-carboxy-3-oxopropan-2-yl]carbamoyl}pyrrolidin-1-yl)-3-methyl-1-oxobutan-2-yl]amino}-4-oxobutanoic acid (7)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (17.4 mg, 55%): RP-HPLC: >99%, (tR = 9.35, k = 2.12). 1H NMR (300 MHz, D2O) δ 4.99 – 4.84 (m, 1H), 4.57 (t, J = 6.8 Hz, 1H), 4.40 – 3.90 (m, 4H), 3.78 – 3.31 (m, 2H), 2.90 – 2.27 (m, 4H), 2.44 – 2.01 (m, 1H), 2.00 – 1.63 (m, 7H), 1.54 – 1.32 (m, 3H), 0.91 – 0.61 (m, 12H). 13C NMR (75 MHz, D2O) δ 175.33, 175.21, 174.74, 174.26, 174.01, 173.77, 171.71, 171.66, 171.60, 89.62, 60.77, 57.12, 52.56, 51.51, 51.39, 49.92, 48.25, 39.86, 35.00, 34.05, 33.86, 29.94, 29.66, 29.48, 24.69, 24.54, 24.32, 22.01, 21.59, 20.84, 18.42, 18.34, 17.36. m/z [M+H+] calculated for C26H42N5O10+: 584.2926, found 584.2934.; C26H41N5O10 (583.64).
(S)-3-[(S)-2-Acetamido-3-phenylpropanamido]-4-{[(S)-1-((S)-2-{[(S)-1-carboxy-3-oxopropan-2-yl]carbamoyl}pyrrolidin-1-yl)-3-methyl-1-oxobutan-2-yl]amino}-4-oxobutanoic acid (8)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (51.6 mg, 45%): RP-HPLC: >99%, (tR = 10.04, k = 2.35). 1H NMR (300 MHz, D2O) δ 7.31 – 6.93 (m, 5H), 4.91 (dd, J = 5.3, 4.4 Hz, 1H), 4.52 – 4.35 (m, 2H), 4.27 – 4.17 (m, 2H), 4.17 – 3.99 (m, 1H), 3.79 – 3.24 (m, 2H), 3.06 – 2.27 (m, 6H), 2.17 – 2.02 (m, 1H), 2.01 – 1.62 (m, 7H), 0.90 – 0.64 (m, 6H). 13C NMR (75 MHz, D2O) δ 175.32, 175.20, 174.00, 173.88, 173.73, 172.98, 171.79, 171.71, 171.65, 171.43, 136.18, 129.12, 128.78, 127.22, 89.61, 60.79, 57.20, 55.05, 51.51, 51.38, 49.86, 48.27, 36.97, 35.04, 34.06, 33.85, 29.89, 29.64, 29.47, 24.54, 21.60, 18.39, 18.29, 17.51. m/z [M+H+] calculated for C29H40N5O10+: 618.2770, found 618.2779.; C29H39N5O10 (617.66).
(4S,7S,10S,13S,16S)-13-(4-Aminobutyl)-4-[(S)-sec-butyl]-16-formyl-7-[(R)-1-hydroxyethyl]-10-isopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (9)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (30.0 mg, 22%): RP-HPLC: >99%, (tR = 6.86, k = 1.29). 1H NMR (400 MHz, D2O) δ 4.94 (t, J = 4.8 Hz, 1H), 4.43 – 3.94 (m, 6H), 2.88 (t, J = 7.6 Hz, 2H), 2.69 – 2.32 (m, 2H), 1.94 (s, 4H), 1.80 – 1.51 (m, 5H), 1.43 – 1.23 (m, 3H), 1.16 – 1.03 (m, 4H), 0.90 – 0.72 (m, 12H). 13C NMR (101 MHz, D2O) δ 176.01, 175.64, 174.46, 174.24, 173.15, 172.91, 171.64, 171.57, 89.74, 67.14, 59.46, 58.98, 58.71, 53.59, 51.66, 39.20, 36.18, 34.62, 30.68, 30.12, 26.27, 24.64, 21.95, 21.66, 18.86, 18.43, 17.84, 14.88, 10.32. m/z [M+H+] calculated for C27H49N6O9+: 601.3556, found 601.2558.; C27H48N6O9 x C2HF3O2 (714.74).
(4S,7S,10S,13S)-4-(Carboxymethyl)-13-formyl-7-(3-guanidinopropyl)-10-methyl-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (10)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (17.6 mg, 15%): RP-HPLC: >99%, (tR = 3.66, k = 0.22). 1H NMR (400 MHz, D2O) δ 4.93 (t, J = 3.8 Hz, 1H), 4.56 (d, J = 6.7 Hz, 1H), 4.28 – 4.07 (m, 3H), 3.10 (t, J = 6.9 Hz, 2H), 2.87 – 2.37 (m, 4H), 1.92 (s, 3H), 1.84 – 1.44 (m, 4H), 1.30 – 1.20 (m, 3H). 13C NMR (101 MHz, D2O) δ 175.30, 175.23, 174.66, 174.51, 174.25, 174.16, 173.03, 172.98, 172.90, 172.80, 156.79, 89.72, 53.35, 53.20, 51.49, 50.24, 49.89, 49.83, 40.51, 35.38, 34.12, 34.07, 28.08, 27.98, 24.31, 21.72, 16.81, 16.64. m/z [M+H+] calculated for C19H32N7O9+: 502.2257, found 502.2256.; C19H31N7O9 x C2HF3O2 (615.52).
(S)-3-{(S)-2-[(S)-2-Acetamido-5-guanidinopentanamido]propanamido}-4-oxobutanoic acid (11)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (9.6 mg, 10%): RP-HPLC: >99%, (tR = 3.74, k = 0.25). 1H NMR (400 MHz, D2O) δ 4.94 (dd, J = 4.5, 3.0 Hz, 1H), 4.28 – 4.05 (m, 3H), 3.10 (t, J = 6.8 Hz, 2H), 2.72 – 2.36 (m, 2H), 1.94 – 1.88 (m, 3H), 1.78 – 1.46 (m, 4H), 1.35 – 1.19 (m, 3H). 13C NMR (101 MHz, D2O) δ 175.34, 175.30, 174.66, 174.50, 174.42, 174.36, 173.56, 156.80, 89.72, 53.44, 53.33, 51.51, 49.77, 49.68, 40.57, 34.18, 28.22, 24.27, 21.65, 16.83, 16.67. m/z [M+H+] calculated for C15H27N6O6+: 387.1987, found 387.1985.; C15H26N6O6 x C2HF3O2 (500.43).
(4S,7S,10S,13S)-4-(Carboxymethyl)-13-formyl-10-(3-guanidinopropyl)-7-[(R)-1-hydroxyethyl]-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (12)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (26.2 mg, 22%): RP-HPLC: >99%, (tR = 3.75, k = 0.25). 1H NMR (400 MHz, D2O) δ 4.92 (dd, J = 4.6, 2.3 Hz, 1H), 4.35 – 4.04 (m, 4H), 3.15 – 3.04 (m, 2H), 2.89 – 2.37 (m, 4H), 1.94 (s, 3H), 1.84 – 1.39 (m, 4H), 1.14 – 1.05 (m, 3H). 13C NMR (101 MHz, D2O) δ 175.24, 175.17, 174.40, 174.24, 173.21, 173.13, 173.05, 171.66, 171.57, 156.77, 89.71, 89.66, 66.89, 59.23, 59.13, 53.54, 53.48, 51.54, 51.51, 50.24, 40.50, 35.41, 34.21, 33.99, 28.33, 28.12, 24.35, 24.31, 21.76, 18.78. m/z [M+H+] calculated for C20H34N7O10+: 532.2362, found 532.2366.; C20H33N7O10 x C2HF3O2 (645.55).
(S)-3-{(S)-2-[(2S,3R)-2-Acetamido-3-hydroxybutanamido]-5-guanidinopentanamido}-4-oxobutanoic acid (13)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (16.0 mg, 16%): RP-HPLC: >99%, (tR = 3.36, k = 0.22). 1H NMR (400 MHz, D2O) δ 4.98 – 4.90 (m, 1H), 4.32 – 4.21 (m, 1H), 4.19 – 4.00 (m, 2H), 3.17 – 3.02 (m, 2H), 2.73 – 2.34 (m, 2H), 2.05 – 1.88 (m, 3H), 1.83 – 1.43 (m, 4H), 1.32 – 1.04 (m, 3H). m/z [M+H+] calculated for C16H29N6O7+: 417.2092, found 417.2093.; C16H28N6O7 x C2HF3O2 (530.46).
(4S,7S,10S,13S)-4-(Carboxymethyl)-13-formyl-7-(3-guanidinopropyl)-10-[(R)-1-hydroxyethyl]-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (14)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (17.4 mg, 14%): RP-HPLC: 97%, (tR = 3.62, k = 0.21). 1H NMR (300 MHz, D2O) δ 4.90 (dd, J = 4.6, 1.6 Hz, 1H), 4.53 (t, J = 6.2 Hz, 1H), 4.36 – 3.93 (m, 4H), 3.06 (t, J = 6.9 Hz, 2H), 2.84 – 2.35 (m, 4H), 1.87 (s, 3H), 1.82 – 1.38 (m, 5H), 1.11 – 0.97 (m, 3H). m/z [M+H+] calculated for C20H34N7O10+: 532.2362, found 532.2364.; C20H33N7O10 x C2HF3O2 (645.55).
(S)-3-{(2S,3R)-2-[(S)-2-Acetamido-5-guanidinopentanamido]-3-hydroxybutanamido}-4-oxobutanoic acid (15)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (5.3 mg, 5%): RP-HPLC: >99%, (tR = 3.60, k = 0.20). 1H NMR (400 MHz, D2O) δ 4.97 – 4.91 (m, 1H), 4.30 – 4.01 (m, 4H), 3.11 (t, J = 6.8 Hz, 2H), 2.73 – 2.39 (m, 2H), 1.93 (s, 3H), 1.77 – 1.61 (m, 2H), 1.60 – 1.48 (m, 2H), 1.16 – 1.01 (m, 3H). m/z [M+H+] calculated for C16H29N6O7+: 417.2092, found 417.2096.; C16H28N6O7 x C2HF3O2 (530.46).
(S)-3-Acetamido-4-{[(S)-1-((S)-2-{[(S)-1-carboxy-3-oxopropan-2-yl]carbamoyl}pyrrolidin-1-yl)-3-methyl-1-oxobutan-2-yl]amino}-4-oxobutanoic acid (16)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (41.7 mg, 47%): RP-HPLC: 99%, (tR = 6.17, k = 1.06). 1H NMR (400 MHz, D2O) δ 5.01 – 4.89 (m, 1H), 4.61 – 4.55 (m, 1H), 4.37 – 4.31 (m, 1H), 4.30 – 4.19 (m, 1H), 4.18 – 4.04 (m, 1H), 3.83 – 3.29 (m, 2H), 2.87 – 2.34 (m, 4H), 2.28 – 2.06 (m, 1H), 2.05 – 1.69 (m, 7H), 0.90 – 0.73 (m, 6H). 13C NMR (101 MHz, D2O) δ 175.36, 175.24, 174.18, 173.96, 173.79, 172.38, 171.84, 171.77, 171.69, 89.65, 60.83, 60.80, 57.08, 57.01, 51.55, 51.43, 50.12, 48.34, 35.39, 34.08, 33.92, 30.08, 30.03, 29.69, 29.52, 24.68, 24.56, 21.69, 18.42, 18.35, 17.30, 17.28. m/z [M+H+] calculated for C20H31N4O9+: 471.2086, found 417.2093.; C20H30N4O9 (470.48).
(S)-3-[(S)-1-(Acetyl-L-valyl)pyrrolidine-2-carboxamido]-4-oxobutanoic acid (17)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (26.2 mg, 39%): RP-HPLC: >99%, (tR = 6.66, k = 1.22). 1H NMR (400 MHz, D2O) δ 4.98 – 4.92 (m, 1H), 4.35 – 4.19 (m, 2H), 4.17 – 4.03 (m, 1H), 3.85 – 3.72 (m, 1H), 3.64 – 3.34 (m, 1H), 2.71 – 2.34 (m, 2H), 2.24 – 2.08 (m, 1H), 2.04 – 1.68 (m, 7H), 0.94 – 0.75 (m, 6H). 13C NMR (101 MHz, D2O) δ 175.37, 175.26, 174.21, 173.87, 172.51, 172.44, 172.36, 89.65, 60.86, 60.82, 57.30, 57.24, 51.56, 51.43, 48.31, 34.10, 33.92, 29.76, 29.69, 29.65, 29.48, 24.75, 24.61, 21.51, 18.35, 18.28, 17.36. m/z [M+H+] calculated for C16H26N3O5+: 356.1816, found 356.1821.; C16H25N3O5 (355.39).
(4S,7S,10S,13S)-10-(4-Aminobutyl)-13-formyl-4-[(R)-1-hydroxyethyl]-7-isopropyl-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (18)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (18.8 mg, 25%): RP-HPLC: >99%, (tR = 3.45, k = 0.15). 1H NMR (300 MHz, D2O) δ 4.89 (dd, J = 4.6, 2.0 Hz, 1H), 4.21 – 3.91 (m, 5H), 2.83 (t, J = 7.6 Hz, 2H), 2.72 – 2.33 (m, 2H), 2.00 – 1.81 (m, 4H), 1.73 – 1.47 (m, 4H), 1.38 – 1.17 (m, 2H), 1.05 (d, J = 6.4 Hz, 3H), 0.79 (t, J = 6.5 Hz, 6H). m/z [M+H+] calculated for C21H38N5O8+: 488.2715, found 488.2724.; C21H37N5O8 x C2HF3O2 (601.58).
(S)-3-{(S)-2-[(S)-2-Acetamido-3-methylbutanamido]-6-aminohexanamido}-4-oxobutanoic acid (19)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (20.0 mg, 32%): RP-HPLC: >99%, (tR = 3.50, k = 0.17).1H NMR (300 MHz, D2O) δ 4.88 (dd, J = 4.6, 2.3 Hz, 1H), 4.25 – 3.82 (m, 3H), 2.83 (t, J = 7.6 Hz, 2H), 2.70 – 2.30 (m, 2H), 1.96 – 1.80 (m, 4H), 1.75 – 1.46 (m, 4H), 1.39 – 1.15 (m, 2H), 0.80 (t, J = 5.5 Hz, 6H). 13C NMR (75 MHz, D2O) δ 206.60, 175.42, 174.44, 173.77, 173.27, 173.16, 89.66, 59.77, 53.50, 51.51, 39.17, 34.28, 30.51, 29.91, 26.19, 21.96, 21.56, 18.33, 17.75. m/z [M+H+] calculated for C17H31N4O6+: 387.2238, found 387.2243.; C17H30N4O6 x C2HF3O2 (500.47).
(4S,7S,10S,13S,16S)-13-(3-Aminopropyl)-4-[(S)-sec-butyl]-16-formyl-7-[(R)-1-hydroxyethyl]-10-isopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (20)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (42.2 mg, 48%): RP-HPLC: >99%, (tR = 7.07, k = 1.36). 1H NMR (300 MHz, D2O) δ 4.92 – 4.84 (m, 1H), 4.40 – 3.85 (m, 6H), 2.85 (t, J = 7.2 Hz, 2H), 2.68 – 2.28 (m, 2H), 2.00 – 1.79 (m, 4H), 1.77 – 1.23 (m, 6H), 1.13 – 0.95 (m, 3H), 0.86 – 0.64 (m, 12H). 13C NMR (75 MHz, D2O) δ 206.59, 175.94, 175.48, 174.45, 174.23, 172.96, 172.56, 171.65, 89.67, 67.12, 59.60, 58.92, 58.69, 53.15, 51.59, 38.85, 36.13, 34.56, 30.07, 28.16, 24.62, 23.20, 21.63, 18.83, 18.40, 17.86, 14.84, 10.29. m/z [M+H+] calculated for C26H47N6O9+: 587.3399, found 587.3410.; C26H46N6O9 x C2HF3O2 (700.71).
(4S,7S,10S,13S)-10-(3-Aminopropyl)-13-formyl-4-[(R)-1-hydroxyethyl]-7-isopropyl-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (21)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (21.9 mg, 30%): RP-HPLC: >99%, (tR = 3.46, k = 0.15). 1H NMR (300 MHz, D2O) δ 4.88 (dd, J = 4.6, 3.1 Hz, 1H), 4.40 – 3.88 (m, 5H), 2.85 (t, J = 6.1 Hz, 2H), 2.69 – 2.32 (m, 2H), 1.98 – 1.81 (m, 4H), 1.75 – 1.47 (m, 4H), 1.05 (d, J = 6.4 Hz, 3H), 0.78 (t, J = 6.7 Hz, 6H). m/z [M+H+] calculated for C20H36N5O8+: 474.2558, found 474.2561.; C20H35N5O8 x C2HF3O2 (587.55).
(S)-3-{(S)-2-[(S)-2-Acetamido-3-methylbutanamido]-5-aminopentanamido}-4-oxobutanoic acid (22)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (36.3 mg, 60%): RP-HPLC: >99%, (tR = 3.43, k = 0.14). 1H NMR (300 MHz, D2O) δ 4.92 – 4.82 (m, 1H), 4.27 – 4.18 (m, 1H), 4.15 – 4.03 (m, 1H), 3.92 – 3.79 (m, 1H), 2.85 (t, J = 7.0 Hz, 2H), 2.69 – 2.27 (m, 2H), 1.96 – 1.80 (m, 4H), 1.78 – 1.44 (m, 4H), 0.78 (t, J = 6.8 Hz, 6H). 13C NMR (75 MHz, D2O) δ 206.60, 175.17, 174.52, 173.82, 172.65, 89.66, 59.93, 53.05, 51.51, 38.85, 34.32, 29.89, 28.12, 23.22, 21.55, 18.37, 17.81. m/z [M+H+] calculated for C16H29N4O6+: 373.2082, found 373.2084.; C16H28N4O6 x C2HF3O2 (486.45).
(4S,7S,10S,13S,16S)-13-(2-Aminoethyl)-4-[(S)-sec-butyl]-16-formyl-7-[(R)-1-hydroxyethyl]-10-isopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (23)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (41.0 mg, 48 %): RP-HPLC: >99 %, (tR = 7.11, k = 1.37). 1H NMR (300 MHz, D2O) δ 4.89 (t, J = 4.9 Hz, 1H), 4.38 – 3.88 (m, 6H), 2.97 – 2.82 (m, 2H), 2.70 – 2.30 (m, 2H), 2.10 – 1.84 (m, 6H), 1.77 – 1.25 (m, 2H), 1.04 (d, J = 6.4 Hz, 3H), 0.84 – 0.68 (m, 12H). 13C NMR (75 MHz, D2O) δ 175.69, 175.26, 174.47, 174.26, 173.13, 172.10, 171.69, 171.55, 89.64, 67.13, 59.61, 58.84, 58.72, 51.57, 51.10, 36.18, 36.12, 34.52, 30.07, 29.04, 24.62, 21.63, 18.83, 18.37, 17.88, 14.84, 10.30. m/z [M+H+] calculated for C25H45N6O9+: 573.3243, found 573.3246.; C25H44N6O9 x C2HF3O2 (686.68).
(4S,7S,10S,13S)-10-(2-Aminoethyl)-13-formyl-4-[(R)-1-hydroxyethyl]-7-isopropyl-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (24)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (18.2 mg, 25%): RP-HPLC: >99%, (tR = 3.44, k = 0.15). 1H NMR (300 MHz, D2O) δ 4.89 (t, J = 4.7 Hz, 1H), 4.40 – 3.90 (m, 5H), 2.99 – 2.80 (m, 2H), 2.70 – 2.30 (m, 2H), 2.06 – 1.84 (m, 6H), 1.05 (d, J = 6.4 Hz, 3H), 0.83 – 0.75 (m, 6H). m/z [M+H+] calculated for C19H34N5O8+: 460.2402, found 460.2412.; C19H33N5O8 x C2HF3O2 (573.52).
(S)-3-{(S)-2-[(S)-2-Acetamido-3-methylbutanamido]-4-aminobutanamido}-4-oxobutanoic acid (25)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (8.9 mg, 15%): RP-HPLC: 96%, (tR = 3.53, k = 0.18). 1H NMR (400 MHz, D2O) δ 4.95 (d, J = 4.8 Hz, 1H), 4.44 – 4.29 (m, 1H), 4.18 – 4.10 (m, 1H), 3.92 (d, J = 7.3 Hz, 1H), 3.04 – 2.86 (m, 2H), 2.72 – 2.37 (m, 2H), 2.16 – 2.01 (m, 1H), 2.02 – 1.86 (m, 5H), 0.89 – 0.75 (m, 6H). 13C NMR (101 MHz, D2O) δ 175.06, 174.65, 174.20, 174.01, 172.20, 171.66, 89.68, 59.99, 51.54, 51.07, 36.27, 34.37, 29.86, 28.99, 21.57, 18.37, 17.84. m/z [M+H+] calculated for C15H27N4O6+: 359.1925, found 359.1930.; C15H26N4O6 x C2HF3O2 (472.42).
(4S,7S,10S,13S,16S)-13-(Aminomethyl)-4-[(S)-sec-butyl]-16-formyl-7-[(R)-1-hydroxyethyl]-10-isopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (26)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (24.2 mg, 29%): RP-HPLC: >99%, (tR = 7.01, k = 1.34). 1H NMR (300 MHz, D2O) δ 4.91 (dd, J = 8.1, 4.6 Hz, 1H), 4.64 – 4.57 (m, 1H), 4.50 – 3.92 (m, 5H), 3.41 – 3.05 (m, 2H), 2.77 – 2.27 (m, 2H), 2.02 – 1.85 (m, 4H), 1.76 – 1.23 (m, 2H), 1.05 (d, J = 6.4 Hz, 3H), 0.86 – 0.68 (m, 12H). 13C NMR (75 MHz, D2O) δ 175.48, 174.95, 174.92, 174.52, 174.36, 173.56, 173.39, 171.91, 171.87, 169.73, 169.26, 89.56, 67.16, 67.01, 59.53, 58.81, 58.71, 58.12, 51.64, 51.52, 50.80, 50.57, 40.02, 36.08, 35.93, 34.40, 30.16, 29.89, 24.65, 24.52, 21.62, 21.51, 18.81, 18.37, 18.35, 17.73, 17.71, 17.69, 14.84, 14.68, 10.29, 10.12. m/z [M+H+] calculated for C24H43N6O9+: 559.3086, found 559.3093.; C24H42N6O9 x C2HF3O2 (672.66).
(4S,7S,10S,13S)-10-(Aminomethyl)-13-formyl-4-[(R)-1-hydroxyethyl]-7-isopropyl-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (27)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (14.6 mg, 21%): RP-HPLC: >99%, (tR = 3.44, k = 0.15). 1H NMR (300 MHz, D2O) δ 4.91 (dd, J = 8.4, 4.6 Hz, 1H), 4.63 – 4.51 (m, 1H), 4.27 – 3.86 (m, 4H), 3.41 – 3.04 (m, 4H), 2.73 – 2.29 (m, 1H), 2.02 – 1.82 (m, 4H), 1.06 (d, J = 6.4 Hz, 3H), 0.89 – 0.74 (m, 6H). m/z [M+H+] calculated for C18H32N5O8+: 446.2245, found 446.2253.; C18H31N5O8 x C2HF3O2 (559.50).
(S)-3-{(S)-2-[(S)-2-Acetamido-3-methylbutanamido]-3-aminopropanamido}-4-oxobutanoic acid (28)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (20.2 mg, 35%): RP-HPLC: >99%, (tR = 3.45, k = 0.15). 1H NMR (300 MHz, D2O) δ 4.94 – 4.86 (m, 1H), 4.64 – 4.54 (m, 1H), 4.20 – 3.82 (m, 2H), 3.40 – 3.06 (m, 2H), 2.73 – 2.32 (m, 2H), 2.02 – 1.84 (m, 4H), 0.87 – 0.75 (m, 6H). 13C NMR (75 MHz, D2O) δ 206.61, 175.36, 174.90, 174.79, 174.22, 169.41, 89.58, 59.88, 51.47, 50.45, 40.02, 34.31, 29.89, 21.58, 18.31, 17.66. m/z [M+H+] calculated for C14H25N4O6+: 345.1769, found 345.1776.; C14H24N4O6 x C2HF3O2 (458.39).
(4S,7S,10S,13S,16S)-13-(4-Acetamidobutyl)-4-[(S)-sec-butyl]-16-formyl-7-[(R)-1-hydroxyethyl]-10-isopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (29)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (26.0 mg, 39%): RP-HPLC: >99%, (tR = 8.20, k = 1.73). 1H NMR (400 MHz, D2O) δ 4.93 (d, J = 4.7 Hz, 1H), 4.32 – 4.24 (m, 1H), 4.22 – 3.95 (m, 5H), 3.08 – 3.00 (m, 2H), 2.73 – 2.36 (m, 3H), 2.04 – 1.90 (m, 4H), 1.86 (s, 3H), 1.78 – 1.52 (m, 2H), 1.45 – 1.32 (m, 2H), 1.30 – 1.17 (m, 2H), 1.07 (d, J = 6.5 Hz, 3H), 0.89 – 0.69 (m, 12H). 13C NMR (75 MHz, DMSO-d6) δ 206.43, 175.40, 175.40, 172.16, 171.93, 171.92, 171.88, 171.04, 169.47, 169.47, 102.87, 66.89, 58.57, 58.01, 57.50, 52.83, 38.75, 36.85, 33.48, 29.22, 24.84, 23.30, 23.05, 22.89, 20.05, 19.54, 18.28, 15.84, 11.44. m/z [M+H+] calculated for C29H51N6O10+: 643.3661, found 643.3670.; C29H50N6O10 (642.75).
(4S,7S,10S,13S)-10-(4-Acetamidobutyl)-13-formyl-4-[(R)-1-hydroxyethyl]-7-isopropyl-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (30)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (25.8 mg, 39%): RP-HPLC: >99%, (tR = 5.74, k = 0.91). 1H NMR (300 MHz, D2O) δ 4.88 (d, J = 4.7 Hz, 1H), 4.20 – 3.87 (m, 5H), 2.99 (t, J = 2.0 Hz, 2H), 2.79 – 2.30 (m, 2H), 1.95 – 1.85 (m, 4H), 1.81 (s, 3H), 1.69 – 1.48 (m, 2H), 1.40 – 1.29 (m, 2H), 1.25 – 1.12 (m, 2H), 1.03 (d, J = 6.4 Hz, 3H), 0.78 (dd, J = 6.8, 4.6 Hz, 6H). 13C NMR (75 MHz, D2O) δ 175.20, 175.11, 174.52, 173.93, 173.41, 173.29, 173.01, 172.09, 89.66, 67.10, 59.55, 59.42, 56.09, 53.69, 51.48, 39.15, 34.07, 30.74, 30.11, 27.70, 22.36, 21.86, 21.69, 18.86, 18.39, 17.77. m/z [M+H+] calculated for C23H40N4O9+: 530.2821, found 530.2825.; C23H39N4O9 (529.59).
(S)-3-{(S)-6-Acetamido-2-[(S)-2-acetamido-3-methylbutanamido]hexanamido}-4-oxobutanoic acid (31)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (32.6 mg, 61%): RP-HPLC: >99%, (tR = 5.50, k = 0.83). 1H NMR (300 MHz, D2O) δ 4.88 (d, J = 5.0 Hz, 1H), 4.24 – 4.01 (m, 2H), 3.95 – 3.86 (m, 1H), 3.00 (t, J = 6.7 Hz, 2H), 2.72 – 2.28 (m, 2H), 1.96 – 1.85 (m, 4H), 1.81 (s, 3H), 1.71 – 1.47 (m, 2H), 1.43 – 1.29 (m, 2H), 1.27 – 1.10 (m, 2H), 0.79 (dd, J = 6.8, 3.2 Hz, 6H). 13C NMR (75 MHz, D2O) δ 206.61, 175.14, 175.07, 174.43, 173.93, 173.66, 173.37, 89.66, 59.82, 53.60, 51.47, 39.14, 34.05, 30.67, 29.96, 27.70, 22.34, 21.84, 21.61, 18.39, 18.32, 17.68, 17.66. m/z [M+H+] calculated for C19H31N4O7+: 427.2198, found 427.2202.; C19H30N4O7 (428.49).
(4S,7S,13S,16S)-13-(4-Aminobutyl)-4-[(S)-sec-butyl]-16-formyl-7-[(R)-1-hydroxyethyl]-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (32)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (25.4 mg, 30%): RP-HPLC: 98%, (tR = 5.81, k = 0.94). 1H NMR (300 MHz, D2O) δ 4.86 (d, J = 4.2 Hz, 1H), 4.44 – 3.99 (m, 5H), 3.90 – 3.72 (m, 2H), 2.83 (t, J = 7.4 Hz, 2H), 2.47 – 2.16 (m, 1H), 1.89 (s, 3H), 1.78 – 1.17 (m, 9H), 1.07 (d, J = 6.4 Hz, 4H), 0.80 – 0.68 (m, 6H). 13C NMR (75 MHz, D2O) δ 206.60, 174.62, 174.40, 173.18, 172.39, 172.30, 170.80, 170.34, 90.02, 67.08, 59.17, 58.73, 58.48, 53.58, 52.39, 42.57, 42.40, 39.19, 36.02, 30.84, 26.25, 24.58, 21.81, 21.66, 18.71, 14.86, 10.31. m/z [M+H+] calculated for C24H43N6O9+: 559.3086, found 559.3088.; C24H42N6O9 x C2HF3O2 (672.66).
(4S,10S,13S)-10-(4-Aminobutyl)-13-formyl-4-[(R)-1-hydroxyethyl]-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (33)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (27.5 mg, 39%): RP-HPLC: >99%, (tR = 3.08, k = 0.12). 1H NMR (300 MHz, D2O) δ 4.92 – 4.85 (m, 1H), 4.31 – 4.02 (m, 4H), 3.89 – 3.72 (m, 2H), 2.83 (t, J = 7.5 Hz, 2H), 2.73 – 2.29 (m, 2H), 1.95 (s, 3H), 1.73 – 1.47 (m, 4H), 1.32 – 1.04 (m, 5H). m/z [M+H+] calculated for C18H32N5O8+: 446.2245, found 446.2251.; C18H31N5O8 x C2HF3O2 (559.50).
(S)-3-{(S)-2-[2-Acetamidoacetamido]-6-aminohexanamido}-4-oxobutanoic acid (34)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (35.1 mg, 41%): RP-HPLC: >99%, (tR = 2.58, k = −0.14). 1H NMR (400 MHz, D2O) δ 4.92 (t, J = 4.6 Hz, 1H), 4.35 – 4.01 (m, 2H), 3.89 – 3.71 (m, 2H), 2.91 – 2.81 (m, 2H), 2.74 – 2.56 (m, 1H), 2.54 – 2.30 (m, 1H), 1.93 (s, 3H), 1.88 – 1.47 (m, 4H), 1.44 – 1.17 (m, 2H). 13C NMR (101 MHz, D2O) δ 175.51, 175.25, 175.23, 175.01, 174.92, 173.60, 171.68, 89.67, 53.61, 53.57, 52.33, 51.57, 51.52, 42.55, 42.46, 39.22, 34.22, 33.99, 29.97, 26.22, 26.13, 22.02, 21.69. m/z [M+H+] calculated for C14H25N4O6+: 345.1769, found 345.1769.; C14H24N4O6 x C2HF3O2 (458.39).
(4S,7S,10S,13S,16S)-13-(4-Aminobutyl)-4-[(S)-sec-butyl]-16-formyl-7-[(R)-1-hydroxyethyl]-10-methyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (35)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (26.9 mg, 31%): RP-HPLC: 96%, (tR = 5.98, k = 0.99). 1H NMR (300 MHz, D2O) δ 4.90 – 4.81 (m, 1H), 4.32 – 3.83 (m, 6H), 2.84 (t, J = 7.4 Hz, 2H), 2.73 – 1.98 (m, 2H), 1.88 (s, 3H), 1.75 – 1.45 (m, 5H), 1.37 – 1.21 (m, 5H), 1.05 (d, J = 6.4 Hz, 4H), 0.80 – 0.66 (m, 6H). 13C NMR (75 MHz, D2O) δ 206.60, 178.81, 174.71, 174.51, 174.37, 174.31, 173.24, 173.13, 171.48, 171.29, 90.08, 67.26, 67.11, 58.83, 58.72, 53.84, 53.70, 52.48, 49.76, 49.57, 39.18, 36.08, 30.75, 30.49, 26.27, 24.62, 21.83, 21.64, 18.81, 16.56, 14.84, 10.27. m/z [M+H+] calculated for C25H45N6O9+: 573.3243, found 573.3246.; C25H44N6O9 x C2HF3O2 (686.68).
(4S,7S,10S,13S)-10-(4-Aminobutyl)-13-formyl-4-[(R)-1-hydroxyethyl]-7-methyl-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (36)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (17.6 mg, 24%): RP-HPLC: >99%, (tR = 3.09, k = 0.11). 1H NMR (300 MHz, D2O) δ 4.88 (dd, J = 4.6, 1.9 Hz, 1H), 4.33 – 3.94 (m, 5H), 2.83 (t, J = 7.5 Hz, 2H), 2.73 – 2.25 (m, 2H), 2.01 – 1.88 (m, 3H), 1.75 – 1.45 (m, 4H), 1.36 – 1.03 (m, 8H). m/z [M+H+] calculated for C19H34N5O8+: 460.2402, found 460.2408.; C19H33N5O8 x C2HF3O2 (573.52).
(S)-3-{(S)-2-[(S)-2-Acetamidopropanamido]-6-aminohexanamido}-4-oxobutanoic acid (37)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (46.2 mg, 52%): RP-HPLC: >99%, (tR = 3.25, k = 0.19). 1H NMR (400 MHz, D2O) δ 4.92 (dd, J = 4.6, 2.5 Hz, 1H), 4.33 – 4.02 (m, 3H), 2.87 (t, J = 6.0 Hz, 2H), 2.73 – 2.58 (m, 1H), 2.51 – 2.33 (m, 1H), 1.89 (s, 3H), 1.86 – 1.48 (m, 4H), 1.43 – 1.18 (m, 5H). 13C NMR (101 MHz, D2O) δ 175.51, 175.44, 175.36, 175.25, 175.16, 174.22, 174.09, 173.52, 173.45, 89.65, 53.50, 52.27, 51.49, 49.79, 49.72, 39.23, 34.14, 33.95, 29.95, 26.20, 26.11, 22.01, 21.54, 16.45. m/z [M+H+] calculated for C15H27N4O6+: 359.1925, found 359.1929.; C15H26N4O6 x C2HF3O2 (472.42).
(4S,7S,10S,13S,16S)-13-(4-Aminobutyl)-16-formyl-7-[(R)-1-hydroxyethyl]-10-isopropyl-4-methyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (38)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (35.8 mg, 28%): RP-HPLC: >99%, (tR = 3.98, k = 0.33). 1H NMR (400 MHz, D2O) δ 4.93 (dd, J = 4.6, 2.3 Hz, 1H), 4.33 – 3.90 (m, 6H), 2.87 (t, J = 7.6 Hz, 2H), 2.75 – 2.35 (m, 2H), 2.01 – 1.86 (m, 4H), 1.79 – 1.46 (m, 4H), 1.33 – 1.18 (m, 5H), 1.08 (d, J = 6.4 Hz, 3H), 0.82 (t, J = 6.3 Hz, 6H). 13C NMR (101 MHz, D2O) δ 175.73, 175.27, 175.08, 174.28, 173.73, 173.23, 173.13, 173.02, 171.87, 171.81, 89.70, 67.11, 59.63, 59.49, 58.98, 53.63, 53.54, 51.51, 49.92, 49.89, 39.20, 34.19, 33.89, 30.63, 30.57, 30.17, 30.11, 26.24, 22.00, 21.96, 21.61, 18.86, 18.41, 18.36, 17.85, 17.81, 16.55. m/z [M+H+] calculated for C24H43N6O9+: 559.3086, found 559.3093.; C24H42N6O9 x C2HF3O2 (672.66).
(4S,7S,10S,13S,16S)-13-(2-Aminoethyl)-16-formyl-7-[(R)-1-hydroxyethyl]-10-isopropyl-4-methyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (39)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (32.5 mg, 27%): RP-HPLC: >99%, (tR = 3.68, k = 0.23). 1H NMR (400 MHz, D2O) δ 4.97 – 4.91 (m, 1H), 4.44 – 4.30 (m, 1H), 4.30 – 4.04 (m, 4H), 4.11 – 3.94 (m, 1H), 3.03 – 2.86 (m, 2H), 2.75 – 2.36 (m, 2H), 2.22 – 1.84 (m, 6H), 1.32 – 1.23 (m, 3H), 1.09 (d, J = 6.4 Hz, 3H), 0.89 – 0.78 (m, 6H). 13C NMR (101 MHz, D2O) δ 175.80, 175.34, 175.02, 174.33, 173.29, 171.97, 171.76, 89.59, 67.13, 59.60, 58.90, 51.58, 51.23, 49.94, 36.18, 33.90, 30.09, 29.12, 21.61, 18.86, 18.34, 17.86, 16.54. m/z [M+H+] calculated for C22H39N6O9+: 531.2773, found 531.2782.; C22H38N6O9 x C2HF3O2 (644.60).
(4S,7S,10S,13S,16S)-4-[(S)-sec-Butyl]-16-formyl-7-[(R)-1-hydroxyethyl]-13-(hydroxymethyl)-10-isopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (40)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (36.7 mg, 36%): RP-HPLC: 99%, (tR = 8.30, k = 1.77). 1H NMR (400 MHz, D2O) δ 4.95 (d, J = 4.4 Hz, 1H), 4.41 – 4.23 (m, 2H), 4.22 – 3.98 (m, 4H), 3.81 – 3.62 (m, 2H), 2.73 – 2.35 (m, 2H), 2.07 – 1.85 (m, 4H), 1.81 – 1.67 (m, 2H), 1.47 – 1.27 (m, 1H), 1.19 – 0.99 (m, 4H), 0.88 – 0.69 (m, 12H). m/z [M+H+] calculated for C24H42N5O10+: 560.2926, found 560.2932.; C24H41N5O10 (559.62).
(4S,7S,10S,13S,16S)-7-(Carboxymethyl)-16-formyl-13-(hydroxymethyl)-4,10-diisopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (41)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (45.2 mg, 43%): RP-HPLC: >99%, (tR = 7.17, k = 1.39). 1H NMR (400 MHz, D2O) δ 4.94 (d, J = 4.5 Hz, 1H), 4.65 – 4.64 (m, 1H), 4.39 – 4.28 (m, 1H), 4.19 – 3.89 (m, 3H), 3.87 – 3.64 (m, 2H), 3.07 – 2.38 (m, 4H), 2.11 – 1.87 (m, 5H), 0.90 – 0.76 (m, 12H). 13C NMR (101 MHz, D2O) δ 175.25, 175.17, 174.51, 174.18, 173.62, 173.36, 173.32, 172.35, 172.31, 171.18, 171.10, 89.65, 61.18, 61.09, 59.78, 59.66, 55.81, 55.62, 51.59, 50.08, 35.19, 34.09, 33.95, 29.94, 21.65, 18.45, 18.34, 17.61, 17.44. m/z [M+H+] calculated for C23H38N5O11+: 560.2562, found 560.2572.; C23H37N5O11 (559.57).
(4S,7S,10S,13S)-4-(Carboxymethyl)-13-formyl-7-isopropyl-10-methyl-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (42)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (45.5 mg, 55%): RP-HPLC: >99%, (tR = 5.36, k = 0.79). 1H NMR (400 MHz, D2O) δ 4.93 (dd, J = 4.5, 3.0 Hz, 1H), 4.61 (t, J = 6.8 Hz, 1H), 4.27 – 4.07 (m, 2H), 4.11 – 3.94 (m, 1H), 2.87 – 2.35 (m, 4H), 2.05 – 1.92 (m, 1H), 1.91 (s, 3H), 1.32 – 1.19 (m, 3H), 0.87 – 0.75 (m, 6H). 13C NMR (101 MHz, D2O) δ 175.27, 175.15, 174.97, 174.56, 174.40, 174.18, 174.08, 172.89, 172.80, 172.70, 89.69, 59.48, 59.30, 51.46, 50.13, 49.79, 35.32, 34.06, 33.89, 30.23, 30.13, 21.69, 18.42, 18.36, 17.43, 16.82, 16.70. m/z [M+H+] calculated for C18H29N4O9+: 445.1929, found 445.1933.; C18H28N4O9 (444.44).
(S)-3-{(S)-2-[(S)-2-Acetamido-3-methylbutanamido]propanamido}-4-oxobutanoic acid (43)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (35.8 mg, 58%): RP-HPLC: >99%, (tR = 5.37, k = 0.79). 1H NMR (400 MHz, D2O) δ 4.93 (dd, J = 4.6, 2.2 Hz, 1H), 4.30 – 4.05 (m, 2H), 4.00 – 3.93 (m, 1H), 2.71 – 2.37 (m, 2H), 2.02 – 1.88 (m, 4H), 1.33 – 1.21 (m, 3H), 0.83 (dd, J = 6.8, 3.0 Hz, 6H). 13C NMR (101 MHz, D2O) δ 175.28, 175.18, 174.59, 174.52, 174.42, 173.65, 173.49, 89.70, 89.67, 59.67, 59.52, 51.48, 51.45, 49.74, 49.65, 34.09, 33.95, 30.08, 30.02, 21.66, 18.38, 17.53, 16.85, 16.73. m/z [M+H+] calculated for C14H24N3O6+: 330.1660, found 330.1666.; C14H23N3O6 (329.35).
(4S,7S,10S,13S)-10-(4-Aminobutyl)-4-(carboxymethyl)-13-formyl-7-isopropyl-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (44)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (42.4 mg, 37%): RP-HPLC: >99%, (tR = 3.77, k = 0.26). 1H NMR (400 MHz, D2O) δ 4.92 (dd, J = 4.6, 1.5 Hz, 1H), 4.59 (t, J = 6.4 Hz, 1H), 4.27 – 4.06 (m, 2H), 4.04 – 3.94 (m, 1H), 2.86 (t, J = 7.6 Hz, 2H), 2.85 – 2.35 (m, 4H), 2.02 – 1.85 (m, 4H), 1.77 – 1.48 (m, 4H), 1.40 – 1.18 (m, 2H), 0.86 – 0.75 (m, 6H). 13C NMR (101 MHz, D2O) δ 175.24, 175.07, 174.18, 174.06, 173.77, 173.38, 173.28, 173.18, 173.15, 172.71, 172.63, 89.69, 59.65, 59.51, 53.62, 53.56, 51.49, 50.15, 39.22, 35.33, 34.18, 33.88, 30.57, 30.45, 30.14, 30.07, 26.21, 26.18, 22.00, 21.95, 21.70, 18.43, 18.40, 17.62. m/z [M+H+] calculated for C21H36N5O9+: 502.2508, found 502.2513.; C21H35N5O9 x C2HF3O2 (615.56).
(4S,7S,10S,13S)-7-(4-Aminobutyl)-4-(carboxymethyl)-13-formyl-10-isopropyl-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (45)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (47.6 mg, 41%): RP-HPLC: >99%, (tR = 3.67, k = 0.22). 1H NMR (400 MHz, D2O) δ 4.91 (dd, J = 6.4, 4.8 Hz, 1H), 4.55 (t, J = 6.8 Hz, 1H), 4.28 – 4.20 (m, 1H), 4.20 – 4.08 (m, 1H), 3.95 (d, J = 7.9 Hz, 1H), 2.87 (t, J = 7.6 Hz, 2H), 2.83 – 2.36 (m, 4H), 1.99 – 1.84 (m, 4H), 1.78 – 1.47 (m, 4H), 1.39 – 1.19 (m, 2H), 0.88 – 0.72 (m, 6H). 13C NMR (101 MHz, D2O) δ 175.25, 175.15, 174.22, 173.98, 173.66, 173.45, 173.38, 172.99, 172.88, 172.71, 172.68, 172.63, 172.57, 89.81, 89.69, 59.73, 53.50, 53.44, 51.58, 51.50, 50.21, 39.22, 35.41, 34.08, 33.92, 30.26, 30.15, 30.11, 26.21, 21.98, 21.93, 21.72, 18.39, 18.33, 18.29, 17.71, 17.65. m/z [M+H+] calculated for C21H36N5O9+: 502.2508, found 502.2514.; C21H35N5O9 x C2HF3O2 (615.56).
(4S,7S,10S,13S)-10-(2-Aminoethyl)-4-(carboxymethyl)-13-formyl-7-isopropyl-2,5,8,11-tetraoxo-3,6,9,12-tetraazapentadecan-15-oic acid (46)
The title compound was synthesized according to the general procedure A, yielding a fluffy white solid (76.1 mg, 69%): RP-HPLC: 99%, (tR = 3.68, k = 0.23). 1H NMR (400 MHz, D2O) δ 4.93 (t, J = 4.7 Hz, 1H), 4.59 (t, J = 6.4 Hz, 1H), 4.39 – 4.30 (m, 1H), 4.17 – 4.09 (m, 1H), 4.04 – 3.94 (m, 1H), 3.00 – 2.86 (m, 2H), 2.84 – 2.37 (m, 4H), 2.13 – 1.87 (m, 6H), 0.87 – 0.77 (m, 6H). 13C NMR (101 MHz, D2O) δ 175.26, 174.98, 174.21, 174.12, 173.41, 173.36, 172.81, 172.78, 171.77, 171.63, 89.66, 89.59, 59.69, 59.62, 51.55, 51.51, 51.24, 51.14, 50.13, 36.24, 35.34, 34.29, 33.87, 30.00, 29.04, 28.92, 21.69, 18.40, 18.38, 17.69, 17.65. m/z [M+H+] calculated for C19H32N5O9+: 474.2195, found 474.2200.; C19H31N5O9 x C2HF3O2 (587.51).
(4S,7S,10S,13R,16S)-13-(4-Aminobutyl)-4-[(S)-sec-butyl]-16-formyl-7-[(R)-1-hydroxyethyl]-10-isopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (47)
The title compound was synthesized according to the general procedure A (using 150 mg resin), yielding a fluffy white solid (27.3 mg, 41%): RP-HPLC: 99%, (tR = 6.85, k = 1.28). 1H NMR (400 MHz, D2O) δ 4.91 (t, J = 4.5 Hz, 1H), 4.32 – 4.25 (m, 1H), 4.22 – 3.82 (m, 5H), 2.87 (t, J = 7.7 Hz, 2H), 2.75 – 2.32 (m, 2H), 2.04 – 1.78 (m, 4H), 1.84 – 1.50 (m, 5H), 1.46 – 1.22 (m, 3H), 1.20 – 1.00 (m, 4H), 0.92 – 0.70 (m, 12H). 13C NMR (101 MHz, D2O) δ 175.57, 174.58, 174.36, 174.28, 173.39, 173.35, 173.19, 171.76, 171.60, 89.80, 67.07, 59.94, 59.80, 58.93, 58.86, 58.78, 53.68, 53.61, 51.70, 51.62, 39.17, 36.16, 30.67, 30.28, 30.06, 29.93, 26.25, 26.20, 24.66, 22.15, 22.11, 21.68, 18.86, 18.82, 18.42, 17.88, 17.84, 14.88, 10.34. m/z [M+H+] calculated for C27H49N6O9+: 601.3556, found 601.3564.; C27H48N6O9 x C2HF3O2 (714.74).
(4S,7S,10S,13R,16S)-13-(2-Aminoethyl)-4-[(S)-sec-butyl]-16-formyl-7-[(R)-1-hydroxyethyl]-10-isopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (48)
The title compound was synthesized according to the general procedure A (using 150 mg resin), yielding a fluffy white solid (27.0 mg, 42%): RP-HPLC: >99%, (tR = 6.63, k = 1.21). 1H NMR (400 MHz, D2O) δ 4.95 – 4.91 (m, 1H), 4.41 – 4.23 (m, 2H), 4.22 – 3.83 (m, 4H), 3.07 – 2.87 (m, 2H), 2.77 – 2.34 (m, 2H), 2.22 – 2.03 (m, 1H), 2.03 – 1.86 (m, 5H), 1.82 – 1.68 (m, 1H), 1.45 – 1.31 (m, 1H), 1.22 – 0.98 (m, 4H), 0.92 – 0.73 (m, 12H). 13C NMR (101 MHz, D2O) δ 175.46, 175.36, 174.59, 174.42, 174.35, 173.47, 173.39, 171.85, 171.73, 89.76, 67.10, 59.97, 58.89, 58.83, 51.65, 51.34, 36.38, 36.14, 34.43, 29.88, 29.80, 29.03, 24.68, 21.68, 18.82, 18.40, 17.89, 14.88, 10.33. m/z [M+H+] calculated for C25H45N6O9+: 573.3243, found 573.3250.; C25H44N6O9 x C2HF3O2 (686.68).
(4S,7S,10S,13R,16S)-13-(4-Aminobutyl)-7-(carboxymethyl)-16-formyl-4,10-diisopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (49)
The title compound was synthesized according to the general procedure A (using 150 mg resin), yielding a fluffy white solid (23.2 mg, 35%): RP-HPLC: >99%, (tR = 5.89, k = 0.96). 1H NMR (400 MHz, D2O) δ 4.91 (dd, J = 8.1, 4.6 Hz, 1H), 4.67 – 4.59 (m, 1H), 4.24 – 4.08 (m, 2H), 3.99 – 3.86 (m, 2H), 2.95 – 2.34 (m, 6H), 2.06 – 1.75 (m, 5H), 1.83 – 1.48 (m, 4H), 1.41 – 1.17 (m, 2H), 0.91 – 0.76 (m, 12H). 13C NMR (101 MHz, D2O) δ 175.66, 175.52, 175.50, 174.76, 174.73, 174.65, 174.62, 173.78, 173.69, 173.46, 172.62, 172.59, 172.48, 89.78, 60.10, 60.02, 59.93, 59.91, 53.73, 53.66, 51.66, 51.60, 50.35, 50.28, 39.19, 35.62, 34.33, 34.23, 30.55, 30.34, 29.86, 26.18, 22.14, 21.68, 18.46, 18.41, 18.32, 17.68, 17.64, 17.60. m/z [M+H+] calculated for C26H45N6O10+: 601.3192, found 601.3203.; C26H44N6O10 x C2HF3O2 (714.69).
(4S,7S,10S,13R,16S)-13-(2-Aminoethyl)-7-(carboxymethyl)-16-formyl-4,10-diisopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (50)
The title compound was synthesized according to the general procedure A (using 150 mg resin), yielding a fluffy white solid (25.7 mg, 40%): RP-HPLC: > 99%, (tR = 5.79, k = 0.93). 1H NMR (400 MHz, D2O) δ 4.93 (t, J = 4.7 Hz, 1H), 4.65 – 4.61 (m, 1H), 4.38 – 4.30 (m, 1H), 4.20 – 4.11 (m, 1H), 4.00 – 3.86 (m, 2H), 2.99 – 2.38 (m, 6H), 2.20 – 1.86 (m, 7H), 0.88 – 0.77 (m, 12H). 13C NMR (101 MHz, D2O) δ 175.24, 175.19, 174.36, 174.26, 173.81, 173.74, 173.65, 173.61, 172.52, 172.33, 171.97, 171.90, 89.73, 60.15, 60.03, 59.95, 51.63, 51.60, 51.40, 50.13, 50.08, 36.39, 35.17, 34.22, 34.03, 29.87, 29.69, 29.65, 28.89, 28.72, 21.67, 18.45, 18.31, 17.67. m/z [M+H+] calculated for C24H41N6O10+: 573.2879, found 573.2888.; C24H40N6O10 x C2HF3O2 (686.64).
(4S,7S,10R,13S,16S)-10-(4-Aminobutyl)-7-(carboxymethyl)-16-formyl-4,13-diisopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (51)
The title compound was synthesized according to the general procedure A (using 150 mg resin), yielding a fluffy white solid (32.1 mg, 48%): RP-HPLC: >99%, (tR = 6.11, k = 1.04). 1H NMR (400 MHz, D2O) δ 4.92 (d, J = 4.7 Hz, 1H), 4.67 – 4.59 (m, 1H), 4.30 – 3.87 (m, 4H), 3.02 – 2.37 (m, 6H), 2.06 – 1.87 (m, 5H), 1.83 – 1.48 (m, 4H), 1.40 – 1.23 (m, 2H), 0.89 – 0.71 (m, 12H). 13C NMR (101 MHz, D2O) δ 175.28, 175.22, 174.95, 174.10, 173.93, 173.57, 172.91, 172.87, 172.34, 172.28, 89.79, 60.18, 59.73, 59.68, 53.82, 51.60, 50.34, 50.22, 39.18, 35.58, 35.49, 34.09, 34.02, 30.54, 30.50, 30.18, 30.01, 29.76, 26.15, 22.07, 21.75, 18.43, 18.40, 18.36, 18.31, 17.66. m/z [M+H+] calculated for C26H45N6O9+: 601.3192, found 601.3203.; C26H44N6O9 x C2HF3O2 (714.69).
(4S,7S,10R,13S,16S)-10-(2-Aminoethyl)-7-(carboxymethyl)-16-formyl-4,13-diisopropyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (52)
The title compound was synthesized according to the general procedure A (using 150 mg resin), yielding a fluffy white solid (29.8 mg, 46%): RP-HPLC: >99%, (tR = 6.07, k = 1.02). 1H NMR (400 MHz, D2O) δ 4.95 – 4.91 (m, 1H), 4.60 (t, J = 6.9 Hz, 1H), 4.40 – 4.31 (m, 1H), 4.23 – 4.11 (m, 1H), 4.03 – 3.87 (m, 2H), 3.03 – 2.91 (m, 2H), 2.90 – 2.39 (m, 4H), 2.22 – 2.06 (m, 1H), 2.06 – 1.88 (m, 6H), 0.87 – 0.75 (m, 12H). 13C NMR (101 MHz, D2O) δ 175.33, 175.27, 174.99, 174.27, 174.05, 172.80, 172.76, 172.66, 172.63, 172.04, 172.01, 89.75, 60.21, 59.96, 59.86, 51.63, 51.59, 51.57, 51.52, 50.51, 36.37, 35.50, 34.10, 29.99, 29.92, 29.76, 28.86, 21.76, 18.28, 17.71. m/z [M+H+] calculated for C24H41N6O10+: 573.2879, found 573.2888.; C24H40N6O10 x C2HF3O2 (686.64).
(4S,7S,10R,13S,16S)-10-(4-Aminobutyl)-7-(carboxymethyl)-16-formyl-4-isopropyl-13-methyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (53)
The title compound was synthesized according to the general procedure A (using 150 mg resin), yielding a fluffy white solid (24.6 mg, 38%): RP-HPLC: 99%, (tR = 4.80, k = 0.60). 1H NMR (400 MHz, D2O) δ 4.93 (dd, J = 7.6, 4.6 Hz, 1H), 4.64 – 4.58 (m, 1H), 4.24 – 4.07 (m, 3H), 3.93 (d, J = 7.0 Hz, 1H), 2.93 – 2.37 (m, 6H), 2.02 – 1.90 (m, 4H), 1.83 – 1.48 (m, 4H), 1.42 – 1.20 (m, 5H), 0.88 – 0.79 (m, 6H). 13C NMR (101 MHz, D2O) δ 175.25, 174.93, 174.56, 174.50, 174.10, 174.01, 173.53, 173.34, 172.47, 89.76, 60.12, 53.81, 53.76, 51.55, 51.50, 50.41, 50.38, 49.88, 39.18, 35.46, 35.38, 34.20, 34.13, 30.28, 29.76, 26.15, 22.02, 21.73, 18.28, 17.63, 16.95, 16.78. m/z [M+H+] calculated for C24H41N6O10+: 573.2879, found 573.2887.; C24H40N6O10 x C2HF3O2 (686.64).
(4S,7S,10R,13S,16S)-10-(2-Aminoethyl)-7-(carboxymethyl)-16-formyl-4-isopropyl-13-methyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (54)
The title compound was synthesized according to the general procedure A (using 150 mg resin), yielding a fluffy white solid (18.0 mg, 29%): RP-HPLC: 99%, (tR = 4.71, k = 0.57). 1H NMR (400 MHz, D2O) δ 4.96 – 4.90 (m, 1H), 4.61 – 4.54 (m, 1H), 4.36 – 4.24 (m, 1H), 4.24 – 4.08 (m, 2H), 3.93 (d, J = 7.0 Hz, 1H), 3.01 – 2.90 (m, 2H), 2.89 – 2.37 (m, 4H), 2.20 – 2.07 (m, 1H), 2.05 – 1.88 (m, 5H), 1.32 – 1.22 (m, 3H), 0.88 – 0.77 (m, 6H). 13C NMR (101 MHz, D2O) δ 175.27, 175.23, 174.96, 174.48, 174.45, 174.13, 172.71, 172.69, 171.95, 171.82, 171.80, 89.75, 60.14, 51.52, 51.46, 50.51, 50.06, 36.37, 35.21, 34.10, 29.77, 28.65, 21.72, 18.26, 17.68, 16.90, 16.73. m/z [M+H+] calculated for C22H37N6O10+: 545.2566, found 545.2576.; C22H36N6O10 x C2HF3O2 (658.59).
(4S,7S,10S,13S,16S)-7-(Carboxymethyl)-16-formyl-4,10-bis[(R)-1-hydroxyethyl]-13-methyl-2,5,8,11,14-pentaoxo-3,6,9,12,15-pentaazaoctadecan-18-oic acid (55)
The title compound was synthesized according to the general procedure B, yielding a fluffy white solid (9.3 mg, 9%): RP-HPLC: >99%, (tR = 3.75, k = 0.25). 1H NMR (400 MHz, D2O) δ 4.92 (t, J = 4.7 Hz, 1H), 4.78 – 4.68 (m, 1H), 4.33 – 3.94 (m, 6H), 3.24 – 2.29 (m, 4H), 2.03 – 1.92 (m, 3H), 1.40 – 1.00 (m, 9H). 13C NMR (101 MHz, D2O) δ 175.24, 175.21, 174.78, 174.57, 174.50, 174.24, 172.68, 172.57, 172.16, 171.41, 171.31, 89.71, 67.00, 59.40, 59.25, 56.19, 51.51, 50.15, 49.91, 35.17, 34.08, 21.75, 18.82, 16.83, 16.65. m/z [M+H+] calculated for C21H39N5O12+: 548.2198, found 548.2198.; C21H38N5O12 (547.52).
Fluorometric Enzyme Assay
The expression and purification of recombinant caspase-2, circularly permutated caspase-2 (cpCasp2) and recombinant caspase-3 was performed as previously described by Bresinsky et al.[15]
384-well protocol:
Compound affinity for caspases were measured in fluorometric assays. Casp2, cpCasp2, and Casp3 were produced in house as described below. Human recombinant Casp1, 6, 7, and 9 were purchased from BioVision (Milpitas/CA, USA). AFC fluorogenic substrates and control peptides (AcYVAD-CHO, AcVDVAD-CHO, AcDEVD-CHO, AcVEID-CHO, and AcLEHD-CHO) were purchased from Bachem (Torrance/CA, USA). Km values were determined experimentally to be the following: Casp1: 5.9 μM; Casp2: 37.1 μM; cpCasp2: 89.2 μM; Casp3: 7.6 μM; Casp6: 43.1 μM; Casp7: 13.8 μM; Casp9: 149.1 μM. Enzymes were diluted in buffer: 100 mM MES (pH 6.5) for Casp2 and cpCasp2 or 100 mM HEPES (pH 7.0) for all other caspases, plus 150 mM NaCl, 0.1% CHAPS, 1.5% sucrose, 10 mM DTT. Enzyme concentrations were 0.05 U/well for Casp1, 6, and 7; 0.5 U/well for Casp9, 20 nM/well for Casp2 and cpCasp2; and 2 nM/well for Casp3. Enzyme in buffer (19 μL) was added per well in a black 384-well Corning 4514 assay plate. Test compounds were serially diluted in dimethyl sulfoxide (DMSO) and plated in duplicate into a Corning 3656 transfer plate. Test compound was added to assay plates in 0.5 μL aliquots per well and mixed 10 times using a BiomekFX (Beckman Coulter). Compound and enzyme mixture was incubated at 37 °C for 5 min for reversible inhibitors. The BiomekFX was then used to add and mix 0.5 μL of the AFC substrate in DMSO from a Corning 3656 transfer plate (final assay concentrations: 5 μM AcYVAD-AFC for Casp1, 10 μM Z-VDVAD-AFC for Casp2 and cpCasp2, 5 μM AcDEVD-AFC for Casp3, 5 μM Z-VEID-AFC for Casp6, 5 μM AcDEVD-AFC for Casp7, and 34 μM AcLEHD-AFC for Casp9) to the assay plate for a total assay volume of 20 μL. Fluorescence from free AFC was read at 37 °C every 5 min over an hour using a CLARIOstar (BMG Labtech) plate reader (λex = 400 nm, λem = 505 nm). The 40 minute time point was reported, consistent with reported literature.[40-43]
96-well protocol (Casp2/3):
Compound affinity for caspases were measured in fluorometric assays. Casp2, cpCasp2, and Casp3 were produced in house as described below. AFC fluorogenic substrates Z-VDVAD-AFC and AcDEVD-AFC and control peptides AcVDVAD-CHO and AcDEVD-CHO were purchased from Bachem (Torrance/CA, USA). Enzyme was diluted in buffer: 100 mM MES (pH 6.5) for Casp2 and cpCasp2 or 100 mM HEPES (pH 7.0) for Casp3, plus 150 mM NaCl, 0.1% CHAPS, 1.5% sucrose, and 10 mM DTT. Enzyme concentrations were 5 nM/well for Casp2 and cpCasp2; and 2 nM/well for Casp3. Enzyme in buffer (96.5 μL) was added per well in a black Corning 3356 96-well assay plate. Test compounds were serially diluted in dimethyl sulfoxide (DMSO) and plated in triplicate in a Corning 3357 transfer plate. Test compound was added to assay plates in 1 μL aliquots per well and mixed 10 times using a BiomekFX (Beckman Coulter). Compound and enzyme mixture was incubated at 37 °C for 5 min. The BiomekFX was then used to add and mix 2.5 μL of the AFC substrate in DMSO from a transfer plate (final assay concentrations: 25 μM Z-VDVAD-AFC for Casp2 and cpCasp2, 10 μM AcDEVD-AFC for Casp3) to the assay plate for a total assay volume of 100 μL in the assay plate. Fluorescence from free AFC was read at 37 °C every 5 min over an hour using a CLARIOstar (BMG Labtech) plate reader (λex = 400 nm, λem = 505 nm). The 40 minute time point was reported, consistent with reported literature.[40-43]
Crystallography
Casp3 crystallography
The preparation of Casp3 and co-crystals with covalently bound inhibitors followed protocols recently described.[15] Diffraction data were collected at IMCA-CAT beamline 17-ID at the Advanced Photon Source (APS), Argonne, Illinois, USA. Diffraction data processing were completed as previously described.[15] The Casp3 structure bound with AcVDVAD-CHO (PDBid: 2h65) served as the search model. Model building and refinement were conducted iteratively using Phenix and Coot. Summary data collection and refinement statistics for both structures are given in Table S1 (Supporting Information).
Data analysis
GraphPad Prism version 9 (GraphPad Software, La Jolla, CA) was used to calculate the IC50 by fitting the dose-response data with four parameter variable slope nonlinear regression. These were transformed into pKi values using the Cheng–Prusoff equation.[44] Since our compounds are covalent reversible inhibitors, they were characterized using pKi values and not “kinact / Ki”, as would be necessary for covalent irreversible inhibitors.
Supplementary Material
Acknowledgement
We thank Vivien Czipper for excellent technical assistance and Prof. Dr. Sigurd Elz for providing infrastructure. We thank Prof. Dr. Michelle Arkin for Casp2 and Casp3 plasmids and scientific discussion. Steffen Pockes was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, Forschungsstipendium 436921318). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the IMCA-CAT beamline 17-ID (or 17-BM) at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. This work was supported from NIH grant R01-AG62199, Caspase-2 Probe Compounds (awarded to KHA and MAW). The authors thank the Lucas Brothers Foundation for financial support.
Abbreviations
- AcK
Acetylated lysine
- AD
Alzheimers desease
- AFC
7-amino-4- trifluoromethyl coumarin
- APS
Advanced Photon Source
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- AU
absorption units
- BCA
bicinchoninic acid
- Casp1
caspase-1
- Casp2
caspase-2
- Casp3
caspase-3
- Casp4
caspase-4
- Casp5
caspase-5
- Casp6
caspase-6
- Casp7
caspase-7
- Casp8
caspase-8
- Casp9
caspase-9
- Casp10
caspase-10
- Casp11
caspase-11
- cdock
covalent docking
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- Comp.
compound
- cpCasp2
circularly permuted caspase-2
- D2O
deuterium oxide
- Dab
diaminobutyric acid
- Dap
diaminopropionic acid
- DIC
N,N′-diisopropylcarbodiimide
- DIPEA
N,N-diisopropylethylamine
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- FTD
frontotemporal dementia
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HCl
hydrochloric acid
- HD
Huntington's disease
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HOBt
1-hydroxybenzotriazole hydrate
- HRMS
high resolution mass spectrometry
- k
retention/capacity factor
- LBD
Lewy body dementia
- MDM2
Mouse double minute 2 homolog
- MES
2-(N-morpholino)ethanesulfonic acid
- MCI
mild cognitive impairment
- Orn
ornithine
- OXYMA
Ethyl cyanohydroxyiminoacetate
- PTFE
polytetrafluoroethylene
- R-Dab
D-isomer of diaminobutyric acid RFU, relative fluorescence units
- R-K
D-isomer of lysine
- RMSD
Root Mean Square Deviation
- RO5
Lipinsky´s rule of five
- RP-HPLC
reversed phase high performance liquid chromatography
- SAR
structure affinity relationship
- SD
standard deviation
- SEM
standard error of the mean
- t0
dead time
- SPPS
solid-phase peptide synthesis
- UHD
ultrahigh definition
Footnotes
Accession Codes
Atomic coordinates of crystallographic complexes with Casp3 have been deposited with the RCSB protein Data bank with accession code 7rna (Ac-ITV(Dab)D-CHO) and 7rng (Ac-ITAKD-CHO).
The authors declare no competing financial interest.
Supporting Information
Crystallography (Table S1 and Figure S1), Structures of synthesized peptides 3-55 (Figure S2-S54), NMR spectra of peptides 3-55 (Figure S55-S149), RP-HPLC chromatograms of peptides 3-55 (Figure S150-S202), Chemical stability of peptides 7, 38, 41, 42, 43, 44, and 55 (Figure S203-S209), Pharmacology (Table S2) and Molecular Modeling (Table S3-S4) are shown in the Supporting Information.
References
- [1].Zhao X, Kotilinek LA, Smith B, Hlynialuk C, Zahs K, Ramsden M, Cleary J, Ashe KH, Nat. Med 2016, 22, 1268. [DOI] [PubMed] [Google Scholar]
- [2].Carroll JB, Southwell AL, Graham RK, Lerch JP, Ehrnhoefer DE, Cao L-P, Zhang W-N, Deng Y, Bissada N, Henkelman RM, Mol. Neurodegener 2011, 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pozueta J, Lefort R, Ribe EM, Troy CM, Arancio O, Shelanski M, Nat. Commun 2013, 4, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu P, Smith BR, Montonye ML, Kemper LJ, Leinonen-Wright K, Nelson KM, Higgins L, Guerrero CR, Markowski TW, Zhao X, Sci. Rep 2020, 10, 1–18.31913322 [Google Scholar]
- [5].Smith BR, Nelson KM, Kemper LJ, Leinonen-Wright K, Petersen A, Keene CD, Ashe KH, Acta Neuropathol. Commun 2019, 7, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liu P, Smith BR, Huang ES, Mahesh A, Vonsattel JPG, Petersen AJ, Gomez-Pastor R, Ashe KH, Acta Neuropathol. Commun 2019, 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan L-L, Ashe KH, Liao D, Neuron 2010, 68, 1067–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Troy CM, Shelanski ML, Nat. Med 2016, 22, 1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Miles MA, Kitevska-Ilioski T, Hawkins CJ, Int. Rev. Cell Mol. Biol 2017, 332, 155–212. [DOI] [PubMed] [Google Scholar]
- [10].Kurokawa M, Kornbluth S, Cell 2009, 138, 838–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fuentes-Prior P, Salvesen GS, Biochem. J 2004, 384, 201–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schweizer A, Briand C, Grütter MG, J. Biol. Chem 2003, 278, 42441–42447. [DOI] [PubMed] [Google Scholar]
- [13].Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW, J. Biol. Chem 1997, 272, 9677–9682. [DOI] [PubMed] [Google Scholar]
- [14].Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, J. Biol. Chem 1997, 272, 17907–17911. [DOI] [PubMed] [Google Scholar]
- [15].Bresinsky M, Strasser JM, Vallaster B, Liu P, McCue WM, Fuller J, Hubmann A, Singh G, Nelson KM, Cuellar ME, Wilmot CM, Finzel BC, Ashe KH, Walters MA, Pockes S, ACS Pharmacol. Transl. Sci 2022, 5, 20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kitevska T, Spencer DM, Hawkins CJ, Apoptosis 2009, 14, 829–848. [DOI] [PubMed] [Google Scholar]
- [17].Mancini M, Machamer CE, Roy S, Nicholson DW, Thornberry NA, Casciola-Rosen LA, Rosen A, J. Cell Biol 2000, 149, 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Julien O, Zhuang M, Wiita AP, O’Donoghue AJ, Knudsen GM, Craik CS, Wells JA, Proc. Natl. Acad. Sci 2016, 113, E2001–E2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ, Hubbard D, Bhutkar A, Jacks T, Mol. Cell 2011, 43, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Murphy AM, Dagnino R Jr, Vallar PL, Trippe AJ, Sherman SL, Lumpkin RH, Tamura SY, Webb TR, J. Am. Chem. Soc 1992, 114, 3156–3157. [Google Scholar]
- [21].Marlowe CK, Scarborough RM, Laibelman AM, Sinha U, Zhu B-Y, Inhibitors of Factor XA, 2000, US006046169A. [Google Scholar]
- [22].Black C, Grimm EL, Isabel E, Renaud J, Nicotinyl Aspartyl Ketones as Inhibitors of Caspase-3, 2001, WO01/27085A1. [DOI] [PubMed] [Google Scholar]
- [23].Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Adv. Drug Deliv. Rev 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- [24].Poreba M, Rut W, Groborz K, Snipas SJ, Salvesen GS, Drag M, Cell Death Differ. 2019, 26, 2695–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sun L-Y, Chen C, Su J, Li J-Q, Jiang Z, Gao H, Chigan J-Z, Ding H-H, Zhai L, Yang K-W, Bioorganic Chem. 2021, 112, 104889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kumalo HM, Bhakat S, Soliman ME, Molecules 2015, 20, 1984–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Schröder J, Klinger A, Oellien F, Marhöfer RJ, Duszenko M, Selzer PM, J. Med. Chem 2013, 56, 1478–1490. [DOI] [PubMed] [Google Scholar]
- [28].Carlesso A, Chintha C, Gorman AM, Samali A, Eriksson LA, Sci. Rep 2019, 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chowdhury SR, Kennedy S, Zhu K, Mishra R, Chuong P, Nguyen A, Kathman SG, Statsyuk AV, Bioorg. Med. Chem. Lett 2019, 29, 36–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].London N, Miller RM, Krishnan S, Uchida K, Irwin JJ, Eidam O, Gibold L, Cimermančič P, Bonnet R, Shoichet BK, Nat. Chem. Biol 2014, 10, 1066–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Toledo Warshaviak D, Golan G, Borrelli KW, Zhu K, Kalid O, J. Chem. Inf. Model 2014, 54, 1941–1950. [DOI] [PubMed] [Google Scholar]
- [32].Zhu K, Borrelli KW, Greenwood JR, Day T, Abel R, Farid RS, Harder E, J. Chem. Inf. Model 2014, 54, 1932–1940. [DOI] [PubMed] [Google Scholar]
- [33].Xue S, Liu H, Zheng Z, Int. J. Mol. Sci 2019, 20, 4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fang B, Fu G, Agniswamy J, Harrison RW, Weber IT, Apoptosis 2009, 14, 741–752. [DOI] [PubMed] [Google Scholar]
- [35].Agniswamy J, Fang B, Weber IT, FEBS J. 2007, 274, 4752–4765. [DOI] [PubMed] [Google Scholar]
- [36].Agniswamy J, Fang B, Weber IT, Apoptosis 2009, 14, 1135–1144. [DOI] [PubMed] [Google Scholar]
- [37].Baell JB, Holloway GA, J. Med. Chem 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- [38].Aldrich C, Bertozzi C, Georg GI, Kiessling L, Lindsley C, Liotta D, Merz KM, Schepartz A, Wang S, ACS Cent. Sci 2017, 3, 143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Roos K, Wu C, Damm W, Reboul M, Stevenson JM, Lu C, Dahlgren MK, Mondal S, Chen W, Wang L, J. Chem. Theory Comput 2019, 15, 1863–1874. [DOI] [PubMed] [Google Scholar]
- [40].Maillard MC, Brookfield FA, Courtney SM, Eustache FM, Gemkow MJ, Handel RK, Johnson LC, Johnson PD, Kerry MA, Krieger F, Meniconi M, Muñoz-Sanjuán I, Palfrey JJ, Park H, Schaertl S, Taylor MG, Weddell D, Dominguez C, Bioorg. Med. Chem 2011, 19, 5833–5851. [DOI] [PubMed] [Google Scholar]
- [41].Chauvier D, Renolleau S, Holifanjaniaina S, Ankri S, Bezault M, Schwendimann L, Rousset C, Casimir R, Hoebeke J, Smirnova M, Debret G, Trichet A-P, Carlsson Y, Wang X, Bernard E, Hébert M, Rauzier J-M, Matecki S, Lacampagne A, Rustin P, Mariani J, Hagberg H, Gressens P, Charriaut-Marlangue C, Jacotot E, Cell Death Dis. 2011, 2, e203–e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Choong IC, Lew W, Lee D, Pham P, Burdett MT, Lam JW, Wiesmann C, Luong TN, Fahr B, DeLano WL, McDowell RS, Allen DA, Erlanson DA, Gordon EM, O’Brien T, J. Med. Chem 2002, 45, 5005–5022. [DOI] [PubMed] [Google Scholar]
- [43].Boxer MB, Quinn AM, Shen M, Jadhav A, Leister W, Simeonov A, Auld DS, Thomas CJ, ChemMedChem 2010, 5, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cheng Y-C, Prusoff WH, Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.