

Abstract
We studied small vessel disease (SVD) pathology in Familial Alzheimer's disease (FAD) subjects carrying the presenilin 1 (PSEN1) p.Glu280Ala mutation in comparison to those with sporadic Alzheimer's disease (SAD) as a positive control for Alzheimer's pathology and Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) bearing different NOTCH3 mutations, as positive controls for SVD pathology. Upon magnetic resonance imaging (MRI) in life, some FAD showed mild white matter hyperintensities and no further radiologic evidence of SVD. In post‐mortem studies, total SVD pathology in cortical areas and basal ganglia was similar in PSEN1 FAD and CADASIL subjects, except for the feature of arteriosclerosis which was higher in CADASIL subjects than in PSEN1 FAD subjects. Further only a few SAD subjects showed a similar degree of SVD pathology as observed in CADASIL. Furthermore, we found significantly enlarged perivascular spaces in vessels devoid of cerebral amyloid angiopathy in FAD compared with SAD and CADASIL subjects. As expected, there was greater fibrinogen‐positive perivascular reactivity in CADASIL but similar reactivity in PSEN1 FAD and SAD groups. Fibrinogen immunoreactivity correlated with onset age in the PSEN1 FAD cases, suggesting increased vascular permeability may contribute to cognitive decline. Additionally, we found reduced perivascular expression of PDGFRβ AQP4 in microvessels with enlarged PVS in PSEN1 FAD cases. We demonstrate that there is Aβ‐independent SVD pathology in PSEN1 FAD, that was marginally lower than that in CADASIL subjects although not evident by MRI. These observations suggest presence of covert SVD even in PSEN1, contributing to disease progression. As is the case in SAD, these consequences may be preventable by early recognition and actively controlling vascular disease risk, even in familial forms of dementia.
Keywords: Alzheimer's disease, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, dementia, FAD, presenilin, small vessel disease, vascular disease
Abbreviations
- AQP4
Aquaporine‐4
- BBB
blood–brain‐barrier
- BG
basal ganglia
- CAA
cerebral amyloid angiopathy
- CADASIL
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- FC
frontal cortex
- HIP
hippocampus
- OC
occipital cortex
- PC
parietal cortex
- PDGFRβ
Platelet‐derived growth factor receptor beta
- PVS
perivascular spacing
- SVD
small vessel disease
- TC
temporal cortex
- WMH
white matter hyperintensities
1. INTRODUCTION
Alzheimer's disease (AD) is the most prevalent neurodegenerative dementia affecting approximately 46 million people worldwide [1, 2]. Familial Alzheimer's disease (FAD) accounts for up to 5% of all AD cases [3], caused by mutations in the amyloid precursor protein (APP) or presenilins 1 and 2 (PSEN1/2) genes. The p.Glu280Ala mutation in PSEN1 is the most common known cause of FAD with more than 600 symptomatic carriers in the Colombian kindred alone and presents with neuropsychological symptoms such as memory and language impairment and behavioural changes [3]. We have recently shown multiple factors are responsible in the age of onset heterogeneity in FAD caused by mutations in PSEN1 gene [4]. Other factors including pre‐existing vascular disease should be considered whether they modify severity or disease progression. Accumulating evidence suggests that vascular factors modify disease onset proportional to severity and the threshold for dementia [5, 6, 7].
We have also detected individuals in the same region of Colombia with Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL), caused by two different point mutations, Cys455Arg and Arg1031Cys, in the NOTCH3 gene. Notch3 is a substrate of the γ‐secretase of which presenilin 1 is the catalytic unit. CADASIL is a cerebral small vessel disease (SVD) characterized by the presence of subcortical strokes, microbleeds and white matter hyperintensities (WMHs) on magnetic resonance imaging (MRI) that leads to vascular dementia [8]. CADASIL show several key features of SVD involving arteriopathy in small, perforating vessels leading to subcortical infarcts, microinfarcts, WM changes as well as perivascular spacing (PVS) and cerebral atrophy [9]. PVS been suggested as a significant feature of radiologically defined SVD [10] and are thought to result from periarteriolar obstruction of fluid transport and clearance of waste products by accumulation of proteins or cell debris [11, 12]. Lacunar infarcts and arterial disease were previously reported in late‐onset AD patients that showed no overt evidence of vascular involvement [7]. The neurovascular unit (NVU) is comprised by different cell types, for example, endothelial cells, pericytes, astrocytic end‐feet. These control blood–brain barrier (BBB) function and they were shown to be disrupted in late‐onset AD [13]. In apolipoprotein E (APOE) ε4 carriers, BBB breakdown contributes to cognitive decline independent of Aβ‐pathology [14]. Therefore, the study of SVD in FAD patients provides a unique opportunity to assess vascular pathology without confounding factors associated with ageing and common in the late‐onset variants with usually several comorbidities.
WMH of vascular origin, frequently used as the clinical signature for early detection and progression of SVD [15] are common in late‐onset AD [16, 17] just as they are a key diagnostic feature of CADASIL. Current observations from the Dominantly Inherited Alzheimer Network (DIAN) have described that FAD mutation carriers have greater total WMH volumes and white matter (WM) degeneration, which appear to increase several years prior to expected symptom onset [18, 19]. WMHs have propensity for a posterior distribution and are largely attributed to cerebral amyloid angiopathy (CAA) which tends to be similarly distributed [20]. These findings collectively suggest WMHs are a core feature of SVD in AD and a target for potential therapeutic strategy.
Here, we assessed the extent of SVD pathology in a large FAD cohort of PSEN1 FAD. We hypothesise that clinical FAD may be modified by covert cerebral microvascular pathology. We compared previously established features of SVD pathology [21, 22] in FAD with that in SAD and in a large cohort of CADASIL subjects from two centres. CADASIL is an ideal model for SVD and microvascular pathology, largely free of AD or Aβ pathology. Further with Notch3 being cleaved by the γ‐secretase, the rational to compare FAD to CADASIL is given because the one pathway is affected by the mutations. Quantitative histopathological and immunohistochemical methods were undertaken to evaluate SVD in a total 88 subjects including brain tissues from normal ageing old and young control subjects.
2. MATERIALS AND METHODS
2.1. Human subjects
Table 1 shows the demographics and relevant clinical and pathological features of the 88 subjects. The PSEN1 p.Glu280Ala genealogy was identified 30 years ago and carriers were regularly followed up using the CERAD protocol, NINCDS‐ADRA and DSM‐IV criteria until end‐stage dementia and death [23, 24]. We searched the Neuroscience Group of Antioquia (GNA) database for subjects with confirmed positive genetic status for p.Glu280Ala in PSEN1 or any mutation in the NOTCH3 gene that would have been evaluated using imaging during their follow up and would have donated their brain for research. A total of 31 cases fulfilling these criteria, 21 PSEN1 FAD and 10 CADASIL subjects, were selected for the study (Table 1). The PSEN1 FAD subjects were chosen to be representative for the p.Glu280Ala family. We also assessed 12 CADASIL subjects from the Newcastle cohort [25] and 28 SAD cases from both the Columbian and Newcastle Centres [26]. There are no AD cases in the family history of the SAD patients. Unless otherwise stated, for further analysis we considered the CADASIL and SAD groups from the two centres as two combined respective disease groups since there were no significant differences in age, gender, brain weights or vascular risk factor distribution (Table 1). In addition, we assessed brain tissues from 17 old and 10 young controls to match all the AD and CADASIL subjects (Table 1). For the purpose of this study we considered the SAD cases to be the positive control for AD related vascular pathology and CADASIL cases as the positive control for brain SVD. Brain donation and procedures were preformed following the ethical approval from the respective institutions, Universidad de Antioquia, Medellin, Colombia and Newcastle Health Hospitals NHS Trust, Newcastle, and informed consent from the patients or family members.
TABLE 1.
Demographic details and clinical features of the PSEN1 p.Glu280Ala FAD, late‐onset AD and CADASIL cohorts
| Group | N | Genotypes (n) a | Gender (% female) | Age (years) b Mean ± SEM (range) | Age of onset (years) Mean ± SEM (range) | Duration (years) c Mean (range) | Brain weight (g) mean ± SEM | Braak staging, mean (range) | CERAD score mean (range) | APOE ε4 allele (%) | Vascular risk factors (%) d | Stroke (%) | CVA e (mean no.) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HTN | DM | BMI > 25 | Smoking | Dyslipidaemia | IHD | |||||||||||||
|
FAD PSEN1 |
21 | p.Glu280Ala (21) | 62 | 56.7 ± 1.1 (48–64) | 49.8 ± 0.9 (45–58) | 6.9 (3–12) | 945 ± 31 | 6.0 (6–6) | 3.0 (3–3) | 9.5 | 33 | 5 | 17 | 52 | 19 | 0 | 0 | 0 |
|
CADASIL f NOTCH3 |
22 | Cys445Arg (2), Arg1031Cys (8), Arg133Cys (2), Arg141Cys (3), Arg153Cys (2), Arg169Cys (2), Arg558Cys (1), Arg985Cys (1), D239_D253del (1) | 55 | 59.9 ± 1.8 (44–76) | 49.7 ± 1.6 (36–65) | 10.1 (0–28) | 1181 ± 17 | NA | NA | NA | 32 | 32 | 27 | 32 | 14 | 0 | 91 | 3.8 |
| SAD f | 28 | NA | 57 | 83.8 ± 1.6 (61–96) | 75.6 ± 1.9 (50–95) | 8.6 (1–15) | 1168 ± 40 | 5.6 (4–6) | 2.9 (2–3) | 57 | 32 | 11 | 0 | 18 | 0 | 7 | 0 | 0.1 |
| Old Controls | 17 | NA | 71 | 86.1 ± 2.7 (72–99) | NA | NA | 1207 ± 27 | 0.3 (0–4) | 0.5 (0–2) | 12 | NA | NA | NA | NA | NA | 6 | 6 | 0 |
| Young Controls | 10 | NA | 70 | 60.9 ± 2.6 (52–74) | NA | NA | 1229 ± 33 | 0.0 (0–0) | 0.0 (0–0) | 10 | NA | NA | NA | NA | NA | 10 | 0 | 0 |
Abbreviations: BMI, body mass index; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; DM, Type 2 diabetes mellitus; FAD, familial Alzheimer's disease; IHD, ischaemic heart disease.
Genotypes of presenilin 1 (PSEN1) or NOTCH3 mutations. Amongst the Columbian cases, there were either Cys445Arg (n = 2) or Arg1031Cys (n = 8) genotypes.
Age, age at death.
Duration, duration of disease from the onset of disease and death.
Vascular risk factors including hypertension (HTN).
CVA, cerebrovascular accident including transient ischaemic attack.
Individual values for cases by each institution are available in Supplementary files. Postmortem interval (PM) were between 5 and31 h.
2.2. Histological and immuno‐histochemical analysis
We evaluated brain tissues from Brodmann areas 10 (frontal cortex), 20/21 (temporal), seven (parietal), 17 (occipital), the basal ganglia (caudate‐putamen at level of anterior commissure and the anterior hippocampal formation. The number of samples used for the different immunohistochemical experiments ranged 10 to 22 per group (Table 1). 4 μm thick sections were stained with haematoxylin and eosin (H&E) and luxol fast blue and further processed for immunohistochemical (IHC) staining for amyloid beta (Aβ, 1:100; BAM‐10, Mob410; DBS Emergo Europe, The Hague, The Netherlands), platelet derived growth factor receptor beta (PDGFRβ, 1:50, AF385, R&D systems, MN, USA), aquaporin‐4 (AQP4, 1:2000, HPA014784, Merck KGaA, Darmstadt, Germany), fibrinogen (1:2000, DAKO A0080, DAKO GmbH, Jena, DE) and glial fibrillary acidic protein (GFAP, 1:100; M761, DAKO GmbH, Jena, DE). Automatic immunostaining was performed with a Ventana Benchmark XT system (Roche AG, Basel, Switzerland) according to manufacturer instructions. Briefly, after dewaxing and inactivation of endogenous peroxidases (PBS/3% hydrogen peroxide), antibody specific antigen retrieval was performed, sections were blocked and afterwards incubated with the primary antibody. For detection of specific binding, the Ultra View Universal 3,3´‐Diaminobenzidine (DAB) Detection Kit (Ventana, Roche) was used which contains secondary antibodies, DAB stain and counter staining reagent. For detection of PDGFRβ, sections were blocked with rabbit serum and the anti‐goat Histofine Simlpe Stain MAX PO immune‐enzyme polymer (medac GmbH, 414161F) was used as secondary antibody. Sections were scanned using a Hamamatsu NanoZoomer automatic digital slide scanner (Hamamatsu Photonics, Hamamatsu, Japan) and obtained images of whole stained sections at a resolution of at least 1 pixel per μm. Signal of total area was assessed using ImageJ Software (version 1.52p, NIH, Bethesda, MA).
2.3. Assessment of vascular pathology
Total vascular pathology scores were adapted by the methods of Deramecourt et al. [21] consistent with type of lesion as described in Skrobot et al. [22]. Pathological features evaluated included arteriosclerosis, CAA, perivascular hemosiderin leakage, PVS dilatation and myelin loss. All features evaluated in the intracerebral compartment unless otherwise specified. These features were evaluated visually, and scores were given based on severity. The presence of cortical microinfarcts, large or lacunar infarcts and haemorrhages were considered in the final scoring. In order to derive average scores, we added the scores for all areas per case together and divided by the number of areas. White matter pathology was further assessed by quantifying luxol fast blue signal intensity in white matter relative to its intensity gray matter of occipital cortices of evaluated cases, using region of interest (ROI) and signal intensity features of ImageJ Software (version 1.52p, NIH, Bethesda, MA, USA).
2.4. Quantification of CAA
CAA was quantified using the scale described by Love et al. [27] in which leptomeningeal vessels, cortical microvessels and capillaries were graded for Aβ reactivity. Scores were in range between zero (no CAA present) to three (severe CAA present). Capillary CAA was evaluated as present (one) or not present (zero).
2.5. PVS quantification
We quantified perivascular space dilatation for 15 CAA and non‐CAA vessels in the occipital cortex of each case. Sections stained with Aβ were used for PSEN1 FAD cases while HE stained sections were used for CADASIL, since latter were not stained for Aβ. Images at 5X magnification were exported from the whole‐image scan file, using the NDP.view2 software (Hamamatsu Photonics, Hamamatsu, Japan). Then, ImageJ Software (version 1.52p, NIH, Bethesda, MA, USA) was used to measure the longest distance between the parenchyma and the vessel to determine the exact size of the perivascular spaces. PVS ratio was calculated by dividing this distance by the diameter of the measured vessel. To exclude vessel size as the determinant for larger perivascular spaces, the caliber of the vessels <50 and 50–90 μm diameters were measured in the same manner as the perivascular space dilatation.
2.6. Fibrinogen immunoreactivity
For BBB leakage assessment, whole stained sections were used to quantify fibrinogen immunoreactivity around 100 cortical vessels per case. A vessel was counted as leaking once there was fibrinogen staining around the vessel. For PSEN1 FAD this quantification was additionally performed taking the PVS into account. A vessel was defined as dilated when the ratio was PVS ≥1.
2.7. PDGFRβ immunohistochemistry
PDGFRβ immunoreactivity was determined to assess perivascular pericyte coverage for assessing mural cell integrity. Using ImageJ Software (version 1.52p, NIH, Bethesda, MA, USA), we determined periarteriolar PDGFRβ reactivity in 100 cortical vessels per case. Colour deconvolution [28], using the H&E vectors, was used to separate the channels. The brown channel with the following RGB values: R: 0.26814753, G: 0.57031375, B: 0.77642715 was used. After applying the automatic threshold, particles defined as any continuous PDGFRβ signal positive object were measured. Particles smaller than 100 μm [2] were excluded from this analysis. To account for measurements of large artefacts, the top 1% of each dataset was subtracted. In addition, PDGFRβ total signal restricted to vessels was also assessed, discriminating between those with thickened walls and PVS dilation.
2.8. Ultrastructural analysis
Three PSEN1 FAD formalin‐fixed temporal cortices were fixed in glutaraldehyde and chrome‐osmium, dehydrated in ethanol and embedded in Epon 812 (Serva Electrophoresis GmbH, Heidelberg, Germany). After polymerization, 1‐mm‐thick sections were cut, stained with toluidine blue and checked for presence of arterioles. Relevant specimens were further processed for electron microscopy by cutting 100 nm‐thick sections which were contrasted with uranyl replacement stain (22405, Electron Microscopy Sciences, Hatfield, PA) and lead citrate solution. Sections were viewed and representative pictures taken using a LEO EM 912AB electron microscope (Zeiss, Oberkochen, Germany).
2.9. Aquaporin‐4 (AQP4) immunoreactivity
AQP4 immunoreactivity was determined to assess perivascular end‐feet. Using ImageJ Software we assessed 15 vessels with dilated PVS and 15 PVS undilated vessels per case. Immunostained vessel profiles were exported at 80X magnification from the whole‐image scan, resulting in images with 1280 × 1032 pixels. Again, colour deconvolution was used to obtained separated channels and automated threshold was applied and measured, resulting in total AQP4 signal.
2.10. Statistical analyses
Statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, La Jolla California USA, www.graphpad.com) and IBM SPSS Statistics for Windows, Version 21.0. For cognitive assessment all scores were standardized as Z scores and averaged for each cognitive domain assessed (memory, attention, executive function, reasoning, praxis) as previously described [23]. We used a crosstab with Chi2 test to analyse frequencies on categorical variables. We performed a linear mixed model analysis to longitudinally assess and compare the cognitive evolution at each cognitive domain over time between CADASIL and FAD patients. We also calculated the change (delta) between the first and last evaluation, adjusted to baseline values and the speed (slope) of cognitive decline, for each cognitive domain. For neuropathological analysis, Unpaired t‐tests, one‐way and two‐way ANOVAs, Kruskal–Wallis H test and Mann–Whitney U tests were used according to data distribution. When necessary for analysis, outlier values were removed by using the robust regression outlier removal method (ROUT). p ≤ 0.05 was determined significant.
3. RESULTS
3.1. Characterisation of PSEN1 p.Glu280Ala FAD, SAD and CADASIL patients
There were no age of onset or gender differences between the PSEN1 FAD and the comparison group of CADASIL subjects. As expected, SAD patients showed statistically significant older age of onset but there were no gender differences (Figure 1A and Table 1). Disease duration showed no significant differences between the three groups (Figure 1B). Both PSEN1 FAD and CADASIL groups tended to have high frequencies of vascular risk factors including that acquired by behaviour such as tobacco smoking compared with SAD subjects (Tables 1, S1 and S2). There tended to be greater frequency of tobacco smokers in the Colombian subjects. While there were no overt stroke events recorded in the PSEN1 subjects, >90% of the CADASIL subjects had at least one stroke (Table S2). Where longitudinal follow up data were available, we found that both the PSEN1 FAD and CADASIL groups showed significant decline in cognitive performance over time (Figure S1A and Table S3 and S4). On comparing the trajectories longitudinally between both the groups, controlling for age and education level, the PSEN1 FAD group presented with significantly lower performances in the cognitive domains of memory and reasoning (Figure S1B). The severity of decline in cognitive impairment in general, and in specific cognitive domains, was significantly higher in PSEN1 FAD than in CADASIL subjects, with the sole exception of decline in executive function that was not significantly different between groups (Figures 1C and S2A). On the other hand, the rate of cognitive decline was similar in both groups with no significant differences (Figures 1D and 2B).
FIGURE 1.
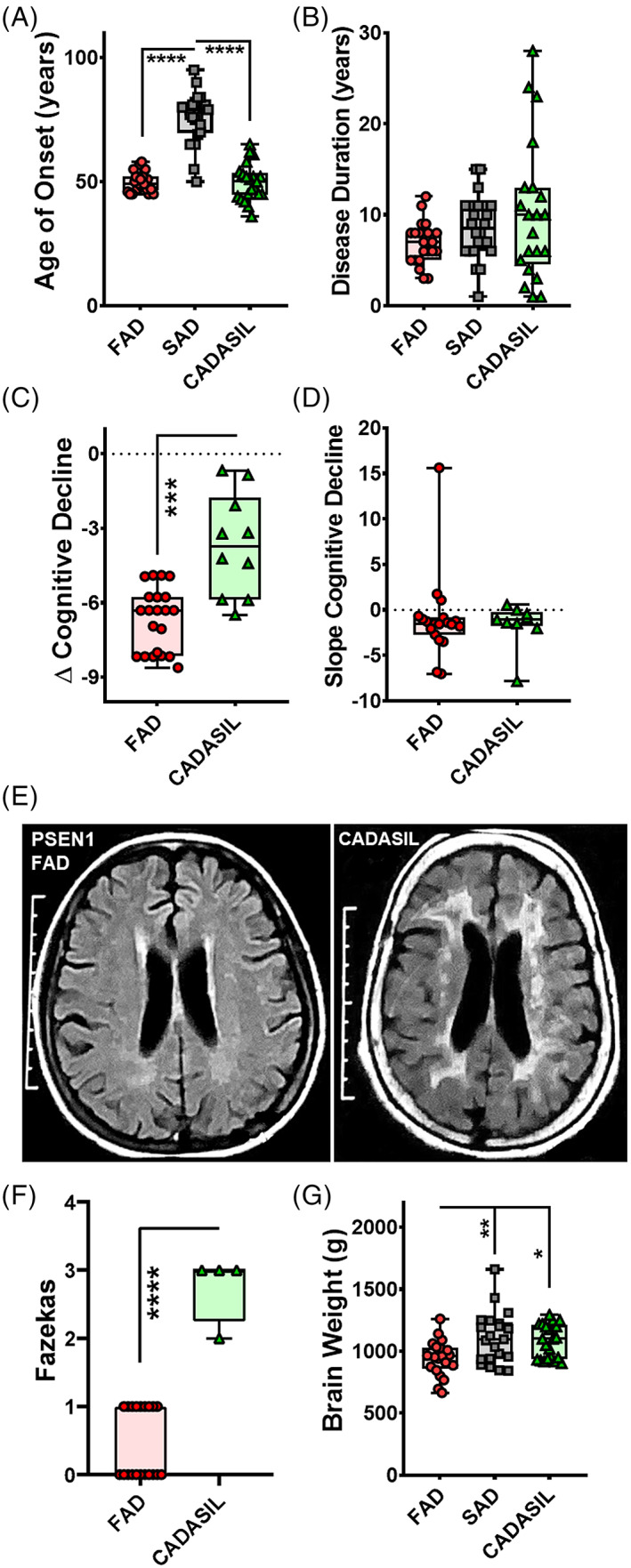
Characterisation of PSEN1 p.Glu280Ala FAD and CADASIL. An overview of demographical, clinical and imaging data for the Colombian and Newcastle cohort. The FAD and CADASIL cases presented with a significantly lower age of onset in comparison with SAD cases (A). There were no significant differences in disease duration (B) while the rate of cognitive decline was significantly faster in PSEN1 FAD than in CADASIL cases (C). The speed of cognitive decline showed no significant differences (D). Representative MRI images are shown for PSEN1 FAD and CADASIL patients (E). PSEN1 FAD patients presented with significantly lower Fazekas scores compared with CADASIL (F). The brain weight was significantly lower for PSEN1 FAD compared with SAD and CADASIL cases (G).
FIGURE 2.
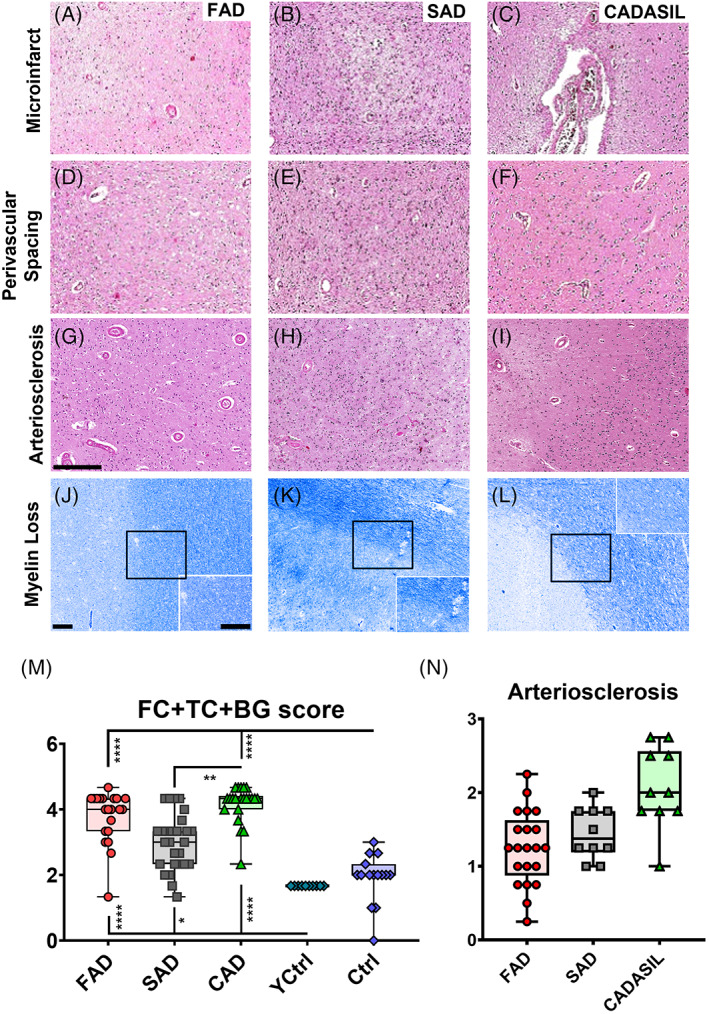
SVD pathology in PSEN1 p.Glu280Ala FAD, SAD and CADASIL. Representative images of vascular pathology are given for PSEN1 FAD, SAD and CADASIL (A–L), scale bar for all panels = 250 μm. The vascular features such as microinfarcts (A–C), perivascular spacing (D–F), arteriosclerosis (G–I) are shown in H&E staining and myelin loss (J–L) is shown in Luxol Fast Blue staining, all features shown in occipital cortex. The average score for frontal (FC) and temporal (TC) cortices and BG (L) was calculated to compare PSEN1 FAD cases with the SAD cohort (Colombian, SAD, n = 10, Newcastle, N‐SAD, n = 17) and different CADASIL mutations (Colombian CADASIL, n = 10, Newcastle CADASIL, n = 12), together with younger (Y‐Ctrl, n = 10) and older healthy (O‐Ctrl, n = 15) controls. Even though PSEN1 FAD tends to show wider variability of vascular pathology, average values are not significantly different to those from pure vascular dementia such as CADASIL. All groups show significant more vascular pathologies than both control groups (p values: **** ≤ 0.0001, *** ≤ 0.001, ** ≤ 0.01, * ≤ 0.05). SAD cases presented with significantly less vascular pathology than the CADASIL cases (p value: ** < 0.01) (M). The arteriosclerosis scores of the evaluated cortices are shown for PSEN1 FAD, SAD and CADASIL (N).
T2W or FLAIR MRI scans of the PSEN1 FAD and CADASIL patients from Colombia were graded for WM changes using Fazekas scoring. Nine out of twenty‐one (42.86%) PSEN1 FAD subjects showed mild WMHs in contrast to the Colombian CADASIL cases who showed significantly higher scores, as expected (Figure 1E,F). Available CADASIL cases (n = 6) from Newcastle also all showed high scores, that is, three with presence of both periventricular as well as deep WMHs on T2W or FLAIR MRI. Further, we assessed imaging patterns in both groups (Table S5), PSEN1 FAD subjects showed evidence of mild to moderate brain and cortical atrophy according to different grading systems (Table S6), while CADASIL cases showed higher frequency of cerebral microbleeds than PSEN1 FAD cases (Tables S4 and S7).
3.2. Assessment of vascular pathology in PSEN1 p.Glu280Ala FAD, SAD and CADASIL
Mean brain weights of PSEN1 FAD cases were significantly lower than those from SAD and CADASIL cases (Tables 1, S1, and Figure 1G). Upon neuropathological examination, the entire spectrum of vascular changes including small infarcts, microinfarcts, hemosiderin leakage, arteriolosclerosis and WM attenuation was evident in PSEN1 FAD subjects (Figure 2A–L). We observed that every PSEN1 FAD subject had at least one lesion attributable to vascular disease. Although cerebral microbleeds were not seen upon MRI in PSEN1 FAD we noted the presence of some perivascular hemosiderin leakage in these subjects as well as in CADASIL subjects.
To determine the degree of SVD in FAD, we quantified cortical and subcortical vascular pathology in PSEN1 FAD compared with SAD, as a control for the possible role of CAA in SVD, and with CADASIL, as a positive control for SVD and dementia [21]. The average scores showed that PSEN1 FAD was similarly affected to CADASIL in frontal and temporal cortices together with basal ganglia. Both PSEN1 FAD and CADASIL scored significantly higher than young or older controls. SAD cases on the other hand, scored significantly lower than CADASIL cases, and significantly higher than young healthy controls but not from controls of similar age (Figures 2M and S3A–C). Lack of differences between PSEN1 FAD and CADASIL remained when all cortices and basal ganglia where evaluated in PSEN1 FAD cases and a subset sample of CADASIL. We also noted that WM attenuation was variable between FAD, SAD and CADASIL subjects and presented no statistically significant features when measured (Figure S3D). We further found that although the degree of arteriolosclerosis was not different in FAD and SAD, CADASIL as expected had greater numbers of arteriosclerotic vessels (Figure 2N). These results suggest that SVD is common in PSEN1 FAD pathology.
3.3. Lack of association between CAA and dilated perivascular spaces in PSEN1 FAD
CAA in AD cases was evaluated as another feature of SVD. We found that both AD groups exhibited similar numbers of CAA affected vessels in all evaluated cortices for all categories of vessels (Figure 3A,B). Next, we quantified the size of enlarged PVS in PSEN1 familial, SAD and CADASIL, as enlargement of PVS is another feature of SVD. The ratio of PVS according to the diameter of microvessels with and without CAA in Aβ stained occipital cortices were quantified in both, FAD and SAD cases. Likewise, PVS ratio was assessed in occipital cortices from CADASIL subjects using H&E staining, given that CAA is not a pathological feature of this disease (Figure 3C). In general, PVS was significantly enlarged in non‐CAA vessels in FAD cases (Figure 3D). This difference was still evident in vessels of both <50 μm and 50–90 μm diameters. This could not be confirmed for vessels with a diameter > 90 μm because the tissue sections did not contain sufficient large numbers of vessels of this size (Figure S4A). Finally, we found a positive correlation between enlarged PVS in non‐CAA vessels and the severity of decline in MMSE scores in FAD cases although these variables were unrelated in CADASIL (Figure S4B). The lack of differences in CAA pathology between AD groups and the presence of significantly dilated PVS without CAA pathology in FAD support the notion that SVD type of arteriosclerotic changes independent of Aβ pathology occur in PSEN1 FAD.
FIGURE 3.
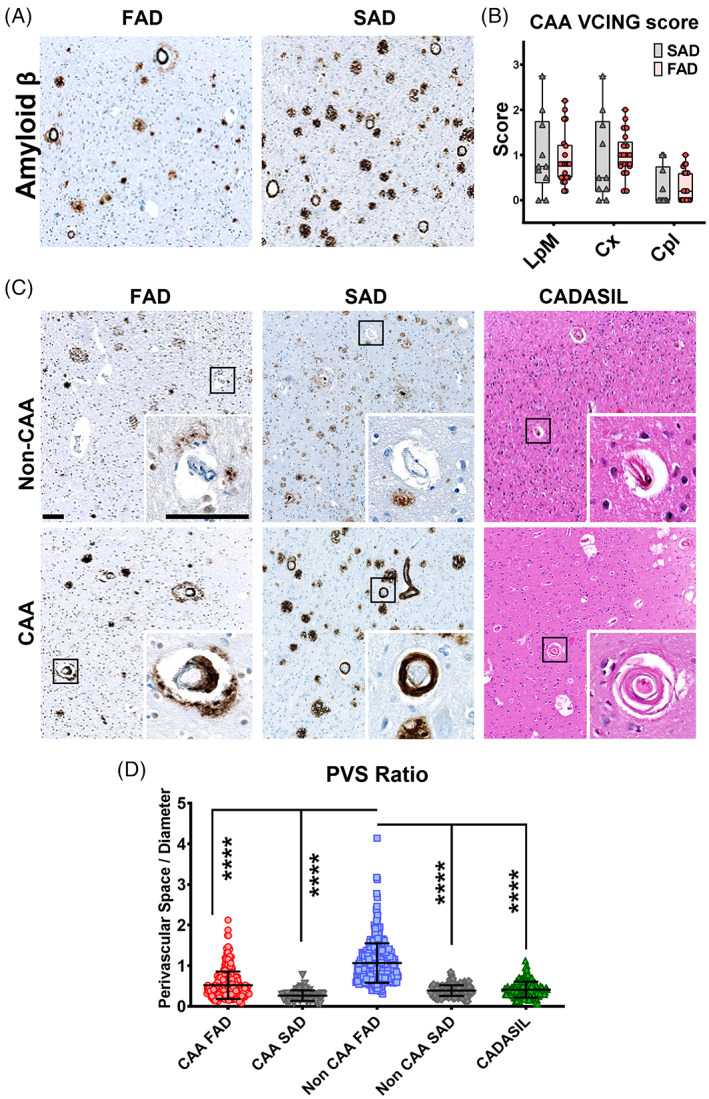
Perivascular spacing in PSEN1 p.Glu280Ala FAD, SAD and CADASIL. Representative images of Aβ pathology are shown for PSEN1 FAD and SAD (A). There are no significant differences in Aβ CAA pathology based on CAA VCING score (B). CAA VCING scores were similar for all vessel types in both groups. Representative images of perivascular spacing for Non CAA and CAA vessels are shown for PSEN1 FAD (n = 21), SAD (n = 10) and CADASIL (n = 10) (C), scale bar all panels = 100 μm. The perivascular space in relation to the diameter of the vessel was calculated to compare perivascular spacing in PSEN1 FAD, SAD and CADASIL. Non CAA FAD vessels showed significant enlargement of the perivascular space compared with all other vessel types measured (D) (p value: **** ≤ 0.0001).
3.4. Blood–brain‐barrier integrity (BBB) and mural cell coverage in PSEN1 FAD, SAD and CADASIL
To further identify pathological events leading to SVD in PSEN1 FAD, we evaluated features of BBB damage as a possible pathological process. Tissue sections from the occipital cortex were immunostained for fibrinogen immunoreactivity in PSEN1 FAD, SAD and CADASIL subjects to identify leaking vessels (Figure 4A). While the majority of vessels in the three disease groups showed to be non‐leaking, we found that the numbers of leaking vessels were similar in PSEN1 FAD and SAD, while being significantly higher in CADASIL (Figure 4B). To assess if our previous finding of dilated PVS in PSEN1 FAD had any impact in BBB integrity, we evaluated the percentage of leaking vessels among those presenting with dilated PVS. The majority of dilated PVS vessels showed to be non‐leaking (Figure 4C), pointing to a lack of association between enlarged PVS and perturbed BBB integrity. Interestingly, as a clinical correlate, we also found that there was a significant relationship with the percentage of fibrinogen leaking vessels with the age of onset in PSEN1 FAD subjects although such association was not significant in CADASIL (Figure 4D,E).
FIGURE 4.
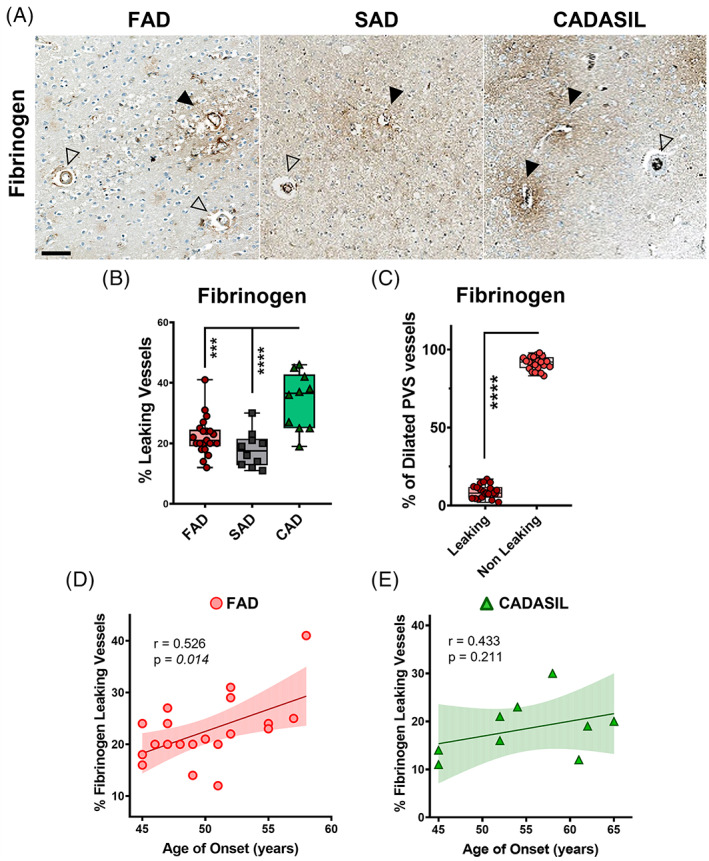
Role of the blood–brain‐barrier in PVS in PSEN1 p.Glu280Ala FAD, SAD and CADASIL. Representative images of leaking (full arrow‐head) and non‐leaking (empty arrow‐head) vessels in PSEN1 FAD (n = 21), SAD (n = 10) and CADASIL (n = 10) are shown (A). PSEN1 FAD and SAD cases presented with significantly less leaking vessels than CADASIL cases (B) (p values: **** ≤ 0.0001, *** ≤ 0.001). Further in PSEN1 FAD significantly less dilated vessels were leaking (C) (p value: **** ≤ 0.0001). The % of fibrinogen leaking vessels is significantly correlated with age of onset in PSEN1 familial Alzheimer's disease but not in CADASIL (D) (p value: 0.014 vs. 0.211, r = 0.526, 0.433).
We then used PDGFRβ, a marker for pericytes, to determine arteriolar pericyte coverage in all the groups (Figure 5A). The percentage of area covered by PDGFRβ‐positive reactivity (Figure 5B) and the PDGFRβ‐positive perivascular signal (Figure 5C) were both significantly lower in PSEN1 FAD cases compared with the SAD and CADASIL groups. However, PDGFRβ‐positive signal intensity was significantly larger in SAD compared with PSEN1 FAD and CADASIL (Figure S5A). We also analysed perivascular PDGFRβ signal in non‐dilated vessels with thickened and normal walls in PSEN1 FAD, SAD and CADASIL. As with total PDGFRβ‐positive perivascular area, the perivascular PDGFRβ‐positive area was significantly lower in PSEN1 FAD compared with SAD and CADASIL but there were no significant differences between PSEN1 FAD vessels regardless of PVS dilation or wall thickness (Figure S5B,C). PDGFRβ‐positive area correlated negatively with the rate of MMSE decline in PSEN1 FAD cases while it showed no correlation in CADASIL subjects (Figure 5D,E). Finally, ultrastructural analysis of PSEN1 FAD capillaries showed thickened basement membranes, without evidencing disruption of tight junctions in EC. Furthermore, there was evidence of apoptotic bodies on pericytes (Figure 5F). These results indicate that even though there is more evidence of altered integrity of BBB in CADASIL, there is also evidence of clinically relevant altered mural cell integrity in PSEN1 FAD.
FIGURE 5.
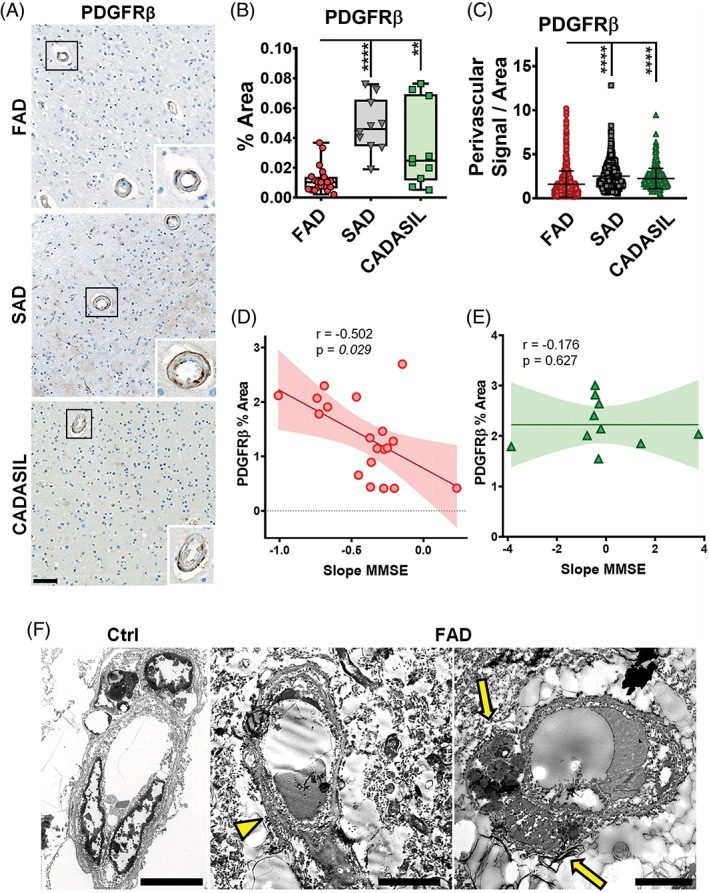
PDGFRβ and the BBB in PSEN1 p.Glu280Ala FAD, SAD and CADASIL. Representative images of PDGFRβ staining are shown for PSEN1 FAD, SAD and CADASIL (A), alongside enlargements of one vessel each, scale bar all panels = 50 μM. The PDGFRβ staining was quantified for PSEN1 FAD (n = 21), SAD (n = 10) and CADASIL (n = 10). The percentage of area covered by PDGFRβ‐positive signal is significantly lower in PSEN1 FAD versus SAD and CADASIL (B) (p values: **** ≤ 0.00001, ** ≤ 0.01). The perivascular PDGFRβ signal was measured for 30 vessels per each group. PSEN1 FAD cases presented with significantly less perivascular PDGFRβ than SAD and CADASIL (C) (p values: **** ≤ 0.0001). The %Area of PDGFRβ is significantly negative correlated with slope of MMSE (r = −0.502, p value: 0.029) while there is no significant correlation observed in CADASIL (r = −0.176, p value: 0.627) (D). Representative EM images are shown for a control (Ctrl) case and a PSEN1 FAD case (F). Thickening of the basement membrane (yellow arrowhead) and a pericyte undergoing apoptosis (yellow arrows) can be observed in FAD (scale bar = 5 μm).
3.5. Astrocytosis and astrocyte end feet pathology in PSEN1 FAD
We observed star‐shaped fibrinogen‐positive astrocytes as well as clasmatodendrotic astrocytes with swollen and vacuolated appearance (Figure 6A), which were previously described in the WM of CADASIL and post‐stroke dementia patients [29, 30]. In prior experiments, we used GFAP + Aβ co‐staining in PSEN1 FAD and SAD, and GFAP in CADASIL cases. Although we observed GFAP+ astrocytic end‐feet surrounding vessels, the intense astrogliosis observed in both groups did not allow for their accurate assessment (Figure S6A). We then used AQP4, an astrocyte marker with preferential localisation of water channel protein in the end feet [31], to determine the distribution of astrocytic end‐feet in PVS dilated and PVS non‐dilated vessels in PSEN1 FAD, SAD and CADASIL cases (Figure 6B). We confirmed the degree of astrocytosis was similar in FAD and SAD with significantly more signal per area in comparison to CADASIL (Figure S6B). Moreover, there were no differences in perivascular AQP4 reactivity between PSEN1 FAD, SAD and CADASIL groups regardless of PVS enlargement. However, AQP4 perivascular reactivity was significantly decreased in dilated PVS vessels when compared with their non‐dilated PVS counterparts in PSEN1 FAD cases (Figure 6C). These results indicate that even though astrocytosis is a common feature in both, PSEN1 FAD and SAD, there are astrocyte‐specific changes in vessels associated with enlarged PVS in PSEN1 FAD.
FIGURE 6.
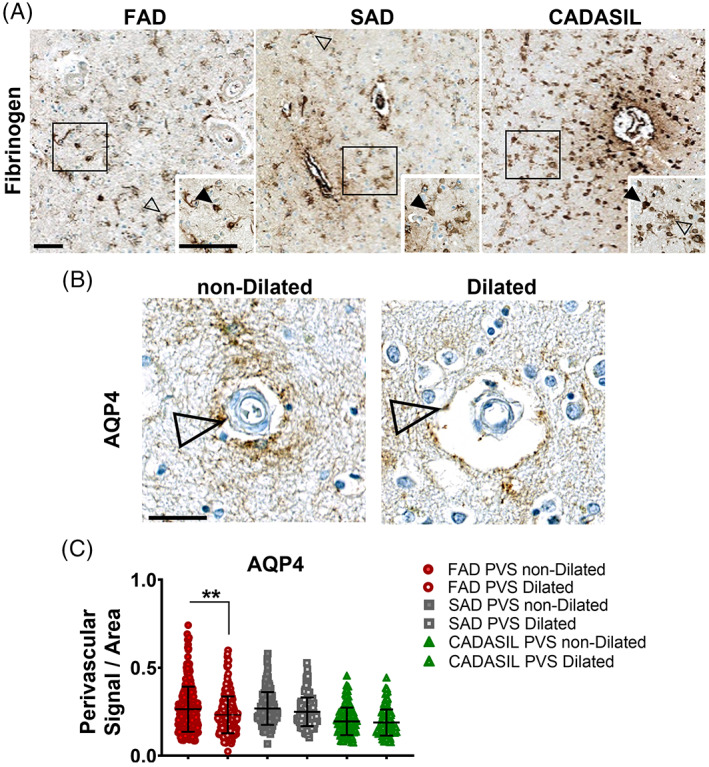
Influence of perivascular astrocytes in PSEN1 p.Glu280Ala FAD, SAD and CADASIL vessel pathology. Representative images of Fibrinogen+ astrocytes in PS1 FAD, SAD and CADASIL are shown, scale bar all panels = 50 μm (A). Clasmatodendrotic (full arrow‐head) cells can be observed in PSEN1 FAD, SAD and CADASIL as well as star‐shaped astrocytes (empty arrow‐head). Additionally, representative images of AQP4 staining of a dilated and a non‐dilated vessel in PSEN1 FAD (n = 21) are shown. AQP4‐positive astrocyte podocytes (empty arrow‐head) are present for dilated and non‐dilated vessels (B). Signal per area was quantified for 15 dilated and non‐dilated vessels in PSEN1 FAD (n = 21), SAD (n = 10) and CADASIL (n = 10). The signal per area for non‐dilated and dilated vessels in PSEN1 FAD (C) is significantly higher for non‐dilated vessels (p value: * ≤ 0.05).
4. DISCUSSION
Our observations represent the first comprehensive study on cerebrovascular pathology utilising a large sample of PSEN1 FAD cases. There was a substantial burden of vascular pathology incorporating all the features of SVD in FAD, comparable in severity to SAD and to a certain extent in CADASIL with known SVD pathology. Despite similar degrees of CAA pathology to SAD, PSEN1 FAD showed (1) an unexpectedly high degree of amyloid‐β independent total SVD pathology, (2) similar extent of WM attenuation, (3) increased PVS, (4) increased tendency of fibrinogen leakage, (5) decreased periarteriolar pericyte coverage and (6) reduced astrocytic end feet reactivity. PSEN1 FAD, SAD and CADASIL subjects showed similar degrees of SVD. We used established vascular pathology scoring systems to identify types of lesions and demonstrate the presence of vascular pathology as a starting point for a more detailed analysis of SVD in PSEN1 FAD. It should be noted that even though we found abundant pathological evidence of SVD in all PSEN1 FAD cases, premortem standard MRI evaluation of WMHs as a radiological marker of SVD did not reveal such in majority of the subjects. It is plausible that the presence of SVD in PSEN1 FAD can be detected using functional MRI approaches such as cerebrovascular reactivity and BBB permeability [32]. It is not unlikely that the cerebrovascular lesions modify severity of presentation or lead to earlier development of cognitive impairment or dementia also in FAD [33].
Previous studies showed that age‐related vascular pathology in PSEN1 FAD mutant transgenic mice [34], and vascular pathology in AD [18, 21, 35, 36] was often concomitant with CAA [37, 38]. It has also been reported that Aβ deposits were significantly less spatially related to blood vessels in FAD when compared with SAD. However, only four clearly genotyped FAD cases were included in that study [39]. While the methods used did not discriminate if FAD had more or less CAA affected vessels [26], our study, with 21 FAD cases from a single PSEN1 mutation, showed no differences in CAA pathology between PSEN1 FAD and SAD, further supporting that the observed vascular pathology in our study was unrelated to Aβ‐pathology.
We observed that non‐CAA vessels exhibited larger PVS than all other vessels making PVS a distinctive feature between PSEN1 FAD and SAD. Enlarged PVS in SVD are gaining relevance due to the association with AD status and SVD, in which enlarged PVS represent the earliest and most consistent neuroimaging finding, predominantly in BG, associated with WM lesions and cerebral microbleeds [40]. Enlarged PVS could also result from loss of brain parenchyma, particularly WM or decreased intramural perivascular drainage. Cerebral atrophy could be excluded as a main cause of enlarged PVS because there was no correlation between brain weight (Figure S7), as an indirect measurement for atrophy, and enlarged PVS in PSEN1 FAD and CADASIL cases. Enlarged PVS involving large vessels has been reported before in SAD and mixed dementia [41] and an association of PVS in centrum semi‐ovale in both AD and CAA was also described [42].
As enlarged PVS has been previously associated with disrupted vessel wall integrity [43], we assessed pericyte cell coverage by assessing various markers of the NVU and their association with PVS and the arterial wall. We previously shown periarteriolar PDGFRβ reactivity in CADASIL as a quantifiable marker in SVD [44]. We found reduced periarteriolar PDGFRβ reactivity indicating lower coverage of cell processes in PSEN1 FAD compared with SAD and CADASIL subjects, regardless of PVS or wall thickening. We showed that vessels with dilated PVS had more PDGFRβ‐expressing pericytes than non‐dilated PVS vessels in FAD, suggesting that enlargement of PVS is not directly associated with loss of PDGFRβ‐expressing processes. We suggest these are novel findings. While pericyte type of mural cells have not been assessed in large microvessels, Sengillo et al. previously identified decreased PDGFRβ‐positive cells and coverage in capillaries of the frontal cortex in a small sample of late‐onset AD [45]. In contrast, a recent stereological study showed preserved PDGFRβ‐positive pericyte cells against increased capillary density in the frontal cortex in late‐onset AD [46]. Our ultrastructural findings indicate mural cell pathology in PSEN1 FAD cases, including evidence of basement membrane alterations and pericyte degeneration, and confirming previous results from Szpak et al., that showed degeneration of pericytes and vascular smooth muscle cells in two FAD cases with changes of endothelial morphology in the presence and absence of amyloid fibrils in the vessel wall [47]. Finally, faster rate of cognitive impairment was shown to correlate with PDGFRβ signal coverage in PSEN1 FAD cases, indicating a possible clinical correlate between cognition and SVD in these patients. Taken together, our findings indicate that PDGFRβ is differentially affected in FAD compared with the other groups and could be related to mural cells pathology. Up to which point this is a reflection of pericyte dysfunction and its possible impact on BBB integrity, cannot be identified without further mechanistic studies, including various marker for pericytes in order to identify pericytes of different origins [48, 49]. Furthermore, a decrease of coverage of pericytes in FAD cases could affect intramural periarterial drainage as a consequence of decreased vasomotor function.
Regarding BBB integrity, CADASIL subjects showed greater leaking vessels than both the AD groups. This can be explained by the presence of higher burden of SVD pathology, more microbleeds and WMHs of vascular origin in the CADASIL compared with FAD. Also, decreased BBB integrity seems to be associated with age of onset in PSEN1 FAD cases, perhaps reflecting the role of ageing in the pathology. However, leaking vessels determined here only represented active leaking at time of death which is supported by distribution of fibrinogen‐positive astrocytes. The astrocytes showed structural changes in form of retracted processes and swollen round cell bodies which had been previously described as clasmatodendrocytes in the WM of CADASIL patients [30] as well as post‐stroke survivors who developed dementia [29]. Higher GFAP coverage of vessels in FAD has been recently reported [50]. However, this can be attributed to the abundant astrogliosis which was apparent in our study. The differences in astrogliosis and astrocyte morphology between PSEN1 FAD and SAD suggest disease specific changes.
We selected AQP4 as a marker for evaluating astrocytic involvement because it plays an important role in water movement, neuronal activity, astrocyte migration as well as the glymphatic system. It has been shown that the perivascular spaces around microvessels were completely covered in astrocytic end‐feet [51, 52]. However, analyses of AQP4 expression in AD are contradictory. Moftakhar et al. showed higher AQP4 expression in a small cohort of AD patients with varying degrees of CAA [53], whilst other studies demonstrated no apparent differences between AD patients and healthy controls [54]. Boespflug et al. showed increased global AQP4 expression alongside qualitative reduction in perivascular AQP4 [41] reactivity, while their previous study demonstrated reduced perivascular AQP4 expression in small vessels of AD patients [55]. The small sample sizes hinder direct comparison with our findings. We showed more AQP4 signal per area in FAD and SAD in comparison to CADASIL, consistent with the findings of increased global expression of AQP4 in the study by Boespflug et al. [41]. However, vessels with dilated PVS in FAD show significantly less AQP4 signal/area compared with vessels with non‐dilated PVS, indicating less astrocytic end‐feet in the PVS of dilated vessels in FAD. This is consistent with the findings of Zeppenfeld et al. [55]. Both studies [41, 55] assessed frontal cortical regions in late‐onset AD patients. Additionally, reduced pericyte coverage increases BBB permeability and pericytes have been shown to induce polarisation of astrocyte end‐feet. Reduced pericyte coverage results in reduced polarisation of AQP4. PDGFβ signalling was shown to be crucial for the development of the glymphatic system as well as recruitment of pericytes to the brain vasculature early in development [56]. The molecular interactions between pericytes and astrocytes in PSEN1 FAD progression remain to be identified in further studies. Although the status of the glymphatic system in AD remains to be explored, it has been suggested that accumulation of Aβ is a product of reduced glymphatic flow [57], caused by constricted capillaries and reduced AQP4 positive astrocytes. Reduced blood flow due to constricting capillaries contributes to the aggregation of amyloid resulting possibly in reduced clearance. Therefore, enlarged PVS with overall reduced pericyte coverage and reduced AQP4‐expressing astrocytic end‐feet in PSEN1 FAD suggest impaired glymphatic flow.
Limitations of this study include restricted assessment of a larger battery of vascular markers for SVD pathology within the whole microvascular structure, such as specific endothelial cell function markers [58] or laminin. In addition, we assessed subjects of a single PSEN1 causative mutation. It remains to be seen whether similar extent of SVD pathology is apparent in other FAD types with mutations in PSEN1 or PSEN2 or APP. However, we previously did note high burden of SVD pathology including WM attenuation in a family with the Ile143Met PSEN1 mutation [59]. Furthermore, this is a descriptive study and additional structural and mechanistic studies are required to identify the mechanism of SVD pathophysiology in FAD. The relevance of our findings for SAD remains to be identified given the wide heterogeneity of pathological presentation found in these patients.
In summary, we provide evidence of amyloid‐independent moderate to severe SVD in PSEN1 FAD, characterized by several features of SVD pathology including enlarged PVS, WM attenuation and arteriolosclerosis with likely involvement of dysfunctional astrocytes and pericytes. Our observations indicate that SVD is a relevant pathological feature in PSEN1 FAD, supporting a multifactorial pathology model for these patients, beyond Aβ or Tau aggregation. Regardless of inheritance and other pathological events, the presence of SVD in PSEN1 FAD cases clearly calls for need to control vascular disease risk factors, encourage healthier lifestyle including cessation of tobacco smoking in these patients. Also, it should lead to efforts to develop future therapeutic strategies for SVD to impact on progression to dementia.
AUTHOR CONTRIBUTIONS
Joseph F. Arboleda‐Velasquez, Rajesh Kalaria and Diego Sepulveda‐Falla designed the study. Jessica Lisa Littau, Lina Velilla, Nelson David Villalba‐Moreno, Christian Hagel, Dagmar Drexler, Santiago Osorio Restrepo and Andres Villegas collected data or performed experiments. Lina Velilla, Andres Villegas, Francisco Lopera and Yakeel T. Quiroz performed neurological and neuropsychological evaluations. Santiago Osorio Restrepo and Sergio Vargas performed imaging evaluations and scale assessments. Andres Villegas collected and sampled donated brains. Yoshiki Hase, Christian Hagel, Susanne Krasemann, Markus Glatzel, Rajesh Kalaria and Diego Sepulveda‐Falla performed structural or neuropathological assessments. Jessica Lisa Littau performed histological quantitative measurements. Jessica Lisa Littau, Lina Velilla, Yakeel T. Quiroz and Diego Sepulveda‐Falla performed data analysis. Jessica Lisa Littau, Lina Velilla, Yoshiki Hase, Markus Glatzel, Susanne Krasemann, Joseph F. Arboleda‐Velasquez, Rajesh Kalaria and Diego Sepulveda‐Falla drafted the manuscript, all authors read it and approved it for submission.
FUNDING INFORMATION
Jessica Lisa Littau, Lina Velilla, Nelson David Villalba‐Moreno, Andres Villegas, Francisco Lopera, Yakeel T. Quiroz, Joseph F. Arboleda‐Velasquez, and Diego Sepulveda‐Falla were supported by US National Institute of Neurological Disorders and Stroke and National Institute on aging co‐funded grant RF1NS110048. Markus Glatzel was supported by the BMBF with the “Understanding disease modifiers and heterogeneity in Alzheimer's disease‐UndoAD” grant.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
Brain donation and procedures, including signature of informed consent by next of kin, were preformed following the ethical approval from the respective institutional boards at Universidad de Antioquia, Medellin, Colombia and at Newcastle Health Hospitals, NHS Trust, Newcastle. Informed consent for brain donations were obtained from the patients or family members following national and international standards.
Supporting information
APPENDIX S1: Supporting information
ACKNOWLEDGEMENT
The authors would like to thank patients and families that generously donated their brains for this study, in particular those belonging to the Colombian PSEN1 FAD and CADASIL families. Open Access funding enabled and organized by Projekt DEAL.
Littau JL, Velilla L, Hase Y, Villalba‐Moreno ND, Hagel C, Drexler D, et al. Evidence of beta amyloid independent small vessel disease in familial Alzheimer's disease. Brain Pathology. 2022;32(6):e13097. 10.1111/bpa.13097
Joseph F. Arboleda‐Velasquez, Rajesh Kalaria and Diego Sepulveda‐Falla have contributed equally to this study.
Funding information Bundesministerium für Bildung und Forschung; National Institute of Neurological Disorders and Stroke, Grant/Award Number: RF1NS110048
Contributor Information
Joseph F. Arboleda‐Velasquez, Email: joseph_arboleda@meei.harvard.edu.
Rajesh Kalaria, Email: raj.kalaria@newcastle.ac.uk.
Diego Sepulveda‐Falla, Email: dsepulve@uke.de.
DATA AVAILABILITY STATEMENT
Additional data supporting the findings of the study are available on request from the corresponding author. The data are not publicly available due to privacy of the patients and ethical restrictions.
REFERENCES
- 1. Prince M, Ali GC, Guerchet M, Wu YT, Prina M, Wimo A. World Alzheimer Report 2015: The Global Impact of Dementia. 1st ed. London, UK: Alzheimer's Disease International; 2015. [Google Scholar]
- 2. Scheltens P, de Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer's disease. Lancet. 2021;397(10284):1577–90. 10.1016/S0140-6736(20)32205-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sepulveda‐Falla D, Glatzel M, Lopera F. Phenotypic profile of early‐onset familial Alzheimer's disease caused by presenilin‐1 E280A mutation. J Alzheimers Dis. 2012;32(1):1–12. 10.3233/JAD-2012-120907 [DOI] [PubMed] [Google Scholar]
- 4. Sepulveda‐Falla D, Chavez‐Gutierrez L, Portelius E, Vélez JI, Dujardin S, Barrera‐Ocampo A, et al. A multifactorial model of pathology for age of onset heterogeneity in familial Alzheimer's disease. Acta Neuropathol. 2021;141(2):217–33. 10.1007/s00401-020-02249-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de la Torre JC, Mussivand T. Can disturbed brain microcirculation cause Alzheimer's disease? Neurol Res. 1993;15(3):146–53. 10.1080/01616412.1993.11740127 [DOI] [PubMed] [Google Scholar]
- 6. Kalaria RN. Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutr Rev. 2010;68(Suppl 2):S74–87. 10.1111/j.1753-4887.2010.00352.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, et al. Vascular dysfunction‐the disregarded partner of Alzheimer's disease. Alzheimers Dement. 2019;15(1):158–67. 10.1016/j.jalz.2018.07.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schoemaker D, Arboleda‐Velasquez JF. NOTCH3 signaling and aggregation as targets for the treatment of CADASIL and other NOTCH3‐associated small‐vessel disease. Am J Pathol. 2021;22:1856–70. 10.1016/j.ajpath.2021.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arboleda‐Velasquez JF, Manent J, Lee JH, Tikka S, Ospina C, Vanderburg CR, et al. Hypomorphic notch 3 alleles link notch signaling to ischemic cerebral small‐vessel disease. Proc Natl Acad Sci U S A. 2011;108(21):E128–35. 10.1073/pnas.1101964108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12(8):822–38. 10.1016/S1474-4422(13)70124-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brown R, Benveniste H, Black SE, Charpak S, Dichgans M, Joutel A, et al. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc Res. 2018;114(11):1462–73. 10.1093/cvr/cvy113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carare RO, Aldea R, Agarwal N, Bacskai BJ, Bechman I, Boche D, et al. Clearance of interstitial fluid (ISF) and CSF (CLIC) group‐part of vascular professional interest area (PIA): cerebrovascular disease and the failure of elimination of amyloid‐β from the brain and retina with age and Alzheimer's disease‐opportunities for therapy. Alzheimers Dement (Amst). 2020;12(1):e12053. 10.1002/dad2.12053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kirabali T, Rust R, Rigotti S, Siccoli A, Nitsch RM, Kulic L. Distinct changes in all major components of the neurovascular unit across different neuropathological stages of Alzheimer's disease. Brain Pathol. 2020;30(6):1056–70. 10.1111/bpa.12895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71–6. 10.1038/s41586-020-2247-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kalaria RN. The pathology and pathophysiology of vascular dementia. Neuropharmacology. 2018;134:226–39. 10.1016/j.neuropharm.2017.12.030 [DOI] [PubMed] [Google Scholar]
- 16. Frings L, Yew B, Flanagan E, Lam BYK, Hüll M, Huppertz HJ, et al. Longitudinal grey and white matter changes in frontotemporal dementia and Alzheimer's disease. PLoS One. 2014;9(3):e90814. 10.1371/journal.pone.0090814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Prins ND, Scheltens P. White matter hyperintensities, cognitive impairment and dementia: an update. Nat Rev Neurol. 2015;11(3):157–65. 10.1038/nrneurol.2015.10 [DOI] [PubMed] [Google Scholar]
- 18. Lee S, Viqar F, Zimmerman ME, Narkhede A, Tosto G, Benzinger TLS, et al. White matter hyperintensities are a core feature of Alzheimer's disease: evidence from the dominantly inherited Alzheimer network. Ann Neurol. 2016;79(6):929–39. 10.1002/ana.24647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Araque Caballero MÁ, Suárez‐Calvet M, Duering M, Franzmeier N, Benzinger T, Fagan AM, et al. White matter diffusion alterations precede symptom onset in autosomal dominant Alzheimer's disease. Brain. 2018;141(10):3065–80. 10.1093/brain/awy229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer's coordinating Centre. Brain. 2013;136(Pt 9):2697–706. 10.1093/brain/awt188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deramecourt V, Slade JY, Oakley AE, Perry RH, Ince PG, Maurage CA, et al. Staging and natural history of cerebrovascular pathology in dementia. Neurology. 2012;78(14):1043–50. 10.1212/WNL.0b013e31824e8e7f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Skrobot OA, Attems J, Esiri M, Hortobágyi T, Ironside JW, Kalaria RN, et al. Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain. 2016;139(11):2957–69. 10.1093/brain/aww214 [DOI] [PubMed] [Google Scholar]
- 23. Acosta‐Baena N, Sepulveda‐Falla D, Lopera‐Gómez CM, Jaramillo‐Elorza MC, Moreno S, Aguirre‐Acevedo DC, et al. Pre‐dementia clinical stages in presenilin 1 E280A familial early‐onset Alzheimer's disease: a retrospective cohort study. Lancet Neurol. 2011;10(3):213–20. 10.1016/S1474-4422(10)70323-9 [DOI] [PubMed] [Google Scholar]
- 24. Sepulveda‐Falla D, Matschke J, Bernreuther C, Hagel C, Puig B, Villegas A, et al. Deposition of hyperphosphorylated tau in cerebellum of PS1 E280A Alzheimer's disease. Brain Pathol. 2011;21(4):452–63. 10.1111/j.1750-3639.2010.00469.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamamoto Y, Hase Y, Ihara M, Khundakar A, Roeber S, Duering M, et al. Neuronal densities and vascular pathology in the hippocampal formation in CADASIL. Neurobiol Aging. 2021;97:33–40. 10.1016/j.neurobiolaging.2020.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dinkel F, Trujillo‐Rodriguez D, Villegas A, Streffer J, Mercken M, Lopera F, et al. Decreased deposition of Beta‐amyloid 1‐38 and increased deposition of Beta‐amyloid 1‐42 in brain tissue of Presenilin‐1 E280A familial Alzheimer's disease patients. Front Aging Neurosci. 2020;12:220. 10.3389/fnagi.2020.00220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Love S, Chalmers K, Ince P, Esiri M, Attems J, Kalaria R, et al. Erratum: development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post‐mortem brain tissue. Am J Neurodegener Dis. 2015;4(2):49. [PMC free article] [PubMed] [Google Scholar]
- 28. Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol. 2001;23(4):291–9. [PubMed] [Google Scholar]
- 29. Chen A, Akinyemi RO, Hase Y, Firbank MJ, Ndung'u MN, Foster V, et al. Frontal white matter hyperintensities, clasmatodendrosis and gliovascular abnormalities in ageing and post‐stroke dementia. Brain. 2016;139(Pt 1):242–58. 10.1093/brain/awv328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hase Y, Chen A, Bates LL, Craggs LJL, Yamamoto Y, Gemmell E, et al. Severe white matter astrocytopathy in CADASIL. Brain Pathol. 2018;28(6):832–43. 10.1111/bpa.12621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ikeshima‐Kataoka H. Neuroimmunological implications of AQP4 in astrocytes. Int J Mol Sci. 2016;17(8):1306. 10.3390/ijms17081306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smith EE, Beaudin AE. New insights into cerebral small vessel disease and vascular cognitive impairment from MRI. Curr Opin Neurol. 2018;31(1):36–43. 10.1097/WCO.0000000000000513 [DOI] [PubMed] [Google Scholar]
- 33. Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA. Relation of cerebral vessel disease to Alzheimer's disease dementia and cognitive function in elderly people: a cross‐sectional study. Lancet Neurol. 2016;15(9):934–43. 10.1016/S1474-4422(16)30029-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gama Sosa MA, Gasperi RD, Rocher AB, Wang ACJ, Janssen WGM, Flores T, et al. Age‐related vascular pathology in transgenic mice expressing presenilin 1‐associated familial Alzheimer's disease mutations. Am J Pathol. 2010;176(1):353–68. 10.2353/ajpath.2010.090482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yamazaki Y, Kanekiyo T. Blood‐brain barrier dysfunction and the pathogenesis of Alzheimer's disease. Int J Mol Sci. 2017;18(9):1965. 10.3390/ijms18091965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu Y, Braidy N, Poljak A, Chan DKY, Sachdev P. Cerebral small vessel disease and the risk of Alzheimer's disease: a systematic review. Ageing Res Rev. 2018;47:41–8. 10.1016/j.arr.2018.06.002 [DOI] [PubMed] [Google Scholar]
- 37. Yamada M. Cerebral amyloid angiopathy: emerging concepts. J Stroke. 2015;17(1):17–30. 10.5853/jos.2015.17.1.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kalaria RN, Sepulveda‐Falla D. Cerebral small vessel disease in sporadic and familial Alzheimer disease. Am J Pathol. 2021;191(11):1888–905. 10.1016/j.ajpath.2021.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Armstrong RA. Spatial correlations between beta‐amyloid (Abeta) deposits and blood vessels in familial Alzheimer's disease. Folia Neuropathol. 2008;46(4):241–8. [PubMed] [Google Scholar]
- 40. Mestre H, Kostrikov S, Mehta RI, Nedergaard M. Perivascular spaces, glymphatic dysfunction, and small vessel disease. Clin Sci (Lond). 2017;131(17):2257–74. 10.1042/CS20160381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boespflug EL, Simon MJ, Leonard E, Grafe M, Woltjer R, Silbert LC, et al. Targeted assessment of enlargement of the perivascular space in Alzheimer's disease and vascular dementia subtypes implicates Astroglial involvement specific to Alzheimer's disease. J Alzheimers Dis. 2018;66(4):1587–97. 10.3233/JAD-180367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Banerjee G, Kim HJ, Fox Z, Jäger HR, Wilson D, Charidimou A, et al. MRI‐visible perivascular space location is associated with Alzheimer's disease independently of amyloid burden. Brain. 2017;140(4):1107–16. 10.1093/brain/awx003 [DOI] [PubMed] [Google Scholar]
- 43. Wardlaw JM, Benveniste H, Nedergaard M, Zlokovic BV, Mestre H, Lee H, et al. Perivascular spaces in the brain: anatomy, physiology and pathology. Nat Rev Neurol. 2020;16(3):137–53. 10.1038/s41582-020-0312-z [DOI] [PubMed] [Google Scholar]
- 44. Craggs LJL, Fenwick R, Oakley AE, Ihara M, Kalaria RN. Immunolocalization of platelet‐derived growth factor receptor‐β (PDGFR‐β) and pericytes in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Neuropathol Appl Neurobiol. 2015;41(4):557–70. 10.1111/nan.12188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, Zlokovic BV. Deficiency in mural vascular cells coincides with blood‐brain barrier disruption in Alzheimer's disease. Brain Pathol. 2013;23(3):303–10. 10.1111/bpa.12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fernandez‐Klett F, Brandt L, Fernández‐Zapata C, Abuelnor B, Middeldorp J, Sluijs JA, et al. Denser brain capillary network with preserved pericytes in Alzheimer's disease. Brain Pathol. 2020;30(6):1071–86. 10.1111/bpa.12897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Szpak GM, Lewandowska E, Wierzba‐Bobrowicz T, Bertrand E, Pasennik E, Mendel T, et al. Small cerebral vessel disease in familial amyloid and non‐amyloid angiopathies: FAD‐PS‐1 (P117L) mutation and CADASIL. Immunohistochemical and ultrastructural studies. Folia Neuropathol. 2007;45(4):192–204. [PubMed] [Google Scholar]
- 48. Cheng J, Korte N, Nortley R, Sethi H, Tang Y, Attwell D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018;136(4):507–23. 10.1007/s00401-018-1893-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Santos GSP, Magno LAV, Romano‐Silva MA, Mintz A, Birbrair A. Pericyte plasticity in the brain. Neurosci Bull. 2019;35(3):551–60. 10.1007/s12264-018-0296-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. González‐Molina LA, Villar‐Vesga J, Henao‐Restrepo J, Villegas A, Lopera F, Cardona‐Gómez GP, et al. Extracellular vesicles from 3xTg‐AD mouse and Alzheimer's disease patient astrocytes impair neuroglial and vascular components. Front Aging Neurosci. 2021;13:593927. 10.3389/fnagi.2021.593927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia. 2010;58(9):1094–103. 10.1002/glia.20990 [DOI] [PubMed] [Google Scholar]
- 52. Tarasoff‐Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain‐implications for Alzheimer disease. Nat Rev Neurol. 2015;11(8):457–70. 10.1038/nrneurol.2015.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moftakhar P, Lynch MD, Pomakian JL, Vinters HV. Aquaporin expression in the brains of patients with or without cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2010;69(12):1201–9. 10.1097/NEN.0b013e3181fd252c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Davis DG, Schmitt FA, Wekstein DR, Markesbery WR. Alzheimer neuropathologic alterations in aged cognitively normal subjects. J Neuropathol Exp Neurol. 1999;58(4):376–88. 10.1097/00005072-199904000-00008 [DOI] [PubMed] [Google Scholar]
- 55. Zeppenfeld DM, Simon M, Haswell JD, D'Abreo D, Murchison C, Quinn JF, et al. Association of perivascular localization of aquaporin‐4 with cognition and Alzheimer disease in aging brains. JAMA Neurol. 2017;74(1):91–9. 10.1001/jamaneurol.2016.4370 [DOI] [PubMed] [Google Scholar]
- 56. Munk AS, Wang W, Bèchet NB, Eltanahy AM, Cheng AX, Sigurdsson B, et al. PDGF‐B is required for development of the Glymphatic system. Cell Rep. 2019;26(11):2955–2969.e3. 10.1016/j.celrep.2019.02.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17(11):1016–24. 10.1016/S1474-4422(18)30318-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kalaria RN. Cerebral vessels in ageing and Alzheimer's disease. Pharmacol Ther. 1996;72(3):193–214. 10.1016/s0163-7258(96)00116-7 [DOI] [PubMed] [Google Scholar]
- 59. Heckmann JM, Low WC, de Villiers C, Rutherfoord S, Vorster A, Rao H, et al. Novel presenilin 1 mutation with profound neurofibrillary pathology in an indigenous southern African family with early‐onset Alzheimer's disease. Brain. 2004;127(Pt 1):133–42. 10.1093/brain/awh009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1: Supporting information
Data Availability Statement
Additional data supporting the findings of the study are available on request from the corresponding author. The data are not publicly available due to privacy of the patients and ethical restrictions.