

Abstract
Methods to functionalize arenes and heteroarenes in a site-selective manner are highly sought after for rapidly constructing value-added molecules of medicinal, agrochemical, and materials interest. One effective approach is the site-selective cross-coupling of polyhalogenated arenes bearing multiple, but identical, halogen groups. Such cross-coupling reactions have proven to be incredibly effective for site-selective functionalization. However, they also present formidable challenges due to the inherent similarities in the reactivities of the halogen substituents. In this Review, we discuss strategies for site-selective cross-couplings of polyhalogenated arenes and heteroarenes bearing identical halogens, beginning first with an overview of the reaction types that are more traditional in nature, such as electronically, sterically, and directing-group-controlled processes. Following these examples is a description of emerging strategies, which includes ligand- and additive/solvent-controlled reactions as well as photochemically initiated processes.
Graphical Abstract
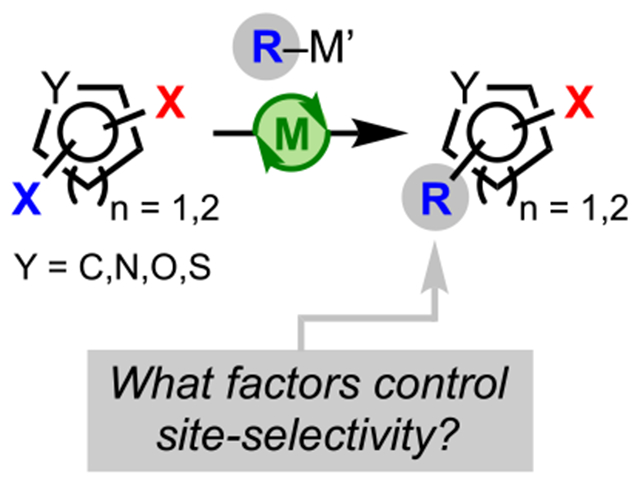
1. INTRODUCTION
Functionalized arenes and heteroarenes (collectively, (hetero)arenes) occur in a diverse array of secondary metabolites (i.e., natural products), drug candidates, ligands (e.g., those employed in transition-metal-catalyzed reactions), polymers, and electronic materials.1–3 Given the ubiquity of these structural motifs, synthetic chemists continue to seek efficient, generalizable syntheses to prepare these scaffolds.4–8 Two approaches have primarily been pursued. In the first approach, the core (hetero)arene is assembled from acyclic precursors already bearing key substituents that will reside on the periphery of the (hetero)arene.9–11 A second approach focuses on functionalizing the periphery of a preformed (hetero)arene core through sequential functionalizations.12 Of the two approaches, the latter has become more commonly employed due to the widespread availability of the unfunctionalized (hetero)arene precursors and the broad range of functionalization strategies that currently exist, including aromatic substitution reactions,13–15 metal-mediated halogen-exchange reactions,16,17 C–H functionalization processes,18 cross-coupling reactions,19 and the more recently developed photoredox catalysis.6
In particular, cross-coupling reactions (selected examples shown in Figure 1),20,21 have been shown to be tremendously powerful tools for functionalizing halogenated/pseudohalogenated arenes by selectively replacing a halogen/pseudohalogen with a desired substituent. Since their inception over 40 years ago, cross-coupling reactions have continued to offer a highly convergent and streamlined approach for generating value-added complexity and are therefore invaluable in all areas of synthetic chemistry, in particular, the fine chemical industry.22 Indeed, it is no surprise that this powerful class of reactions was duly recognized with the 2010 Nobel Prize in Chemistry to Richard F. Heck, Ei-ichi Negishi, and Akira Suzuki.20 The field of cross-coupling has continued to evolve since the pioneering work of Suzuki,23–25 Negishi,26,27 Stille,28,29 and Heck.30 Recent decades have seen the development of myriad new coupling strategies,22 many of which focus on addressing and improving several important aspects of cross-coupling chemistry, such as the site selectivity.31
Figure 1.
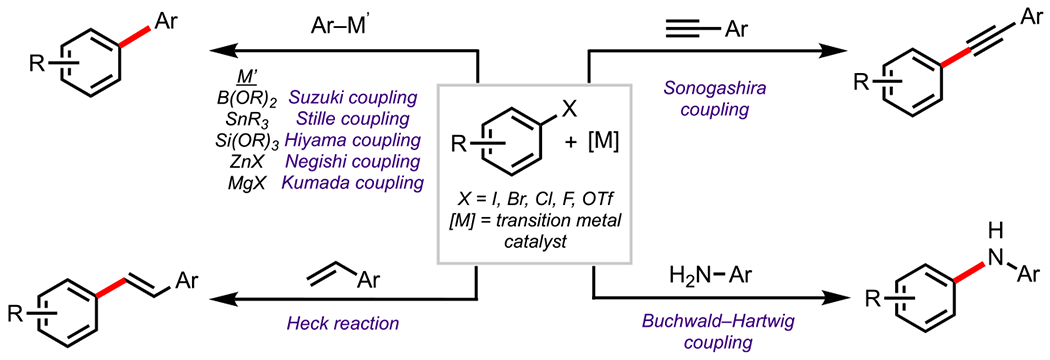
Selected cross-coupling examples.
The appeal of site-selective32,33 coupling reactions stems from the ability to functionalize one position of a multiply halogenated (hetero)arene over all other positions, thus providing a streamlined synthetic approach for structural diversification. Traditionally, site-selective couplings have been achieved by employing (hetero)arene electrophiles bearing multiple, nonidentical halogens or pseudohalogens, such as the triflyloxy (−OTf) group (Scheme 1A).12,34,35 In these cases, the site selectivity relates to the relative bond dissociation energies (BDEs) of carbon–halogen (C–X) bonds (lower C–X BDE = preferred site of oxidative addition) and therefore follows the general reactivity trend: C–I > C–Br > C–Cl > C–F.36–38 This difference in reactivity ultimately facilitates the chemoselective installation of a substituent to a desired position.12 This strategy, however, becomes challenging when the parent (hetero)arene bears identical halogen atoms, as there is little difference in the nature of the aryl–halide bonds (Scheme 1B). In these systems, site-selective coupling at the carbon bearing one halogen atom over another depends on other factors such as the electronic nature of the atom to which the halogen or pseudohalide is attached and the steric environment, which determines the preferred site of reactivity.31 Overall, achieving site-selective couplings is of great interest to the synthetic community not only because of the use of easily accessible polyhalogented aryl precursors but also due to the effectiveness of these processes in generating complexity both rapidly and efficiently. Strategies for achieving site-selective couplings in polyhalogenated (hetero)arenes containing identical halogen atoms continue to emerge.39–44
Scheme 1.
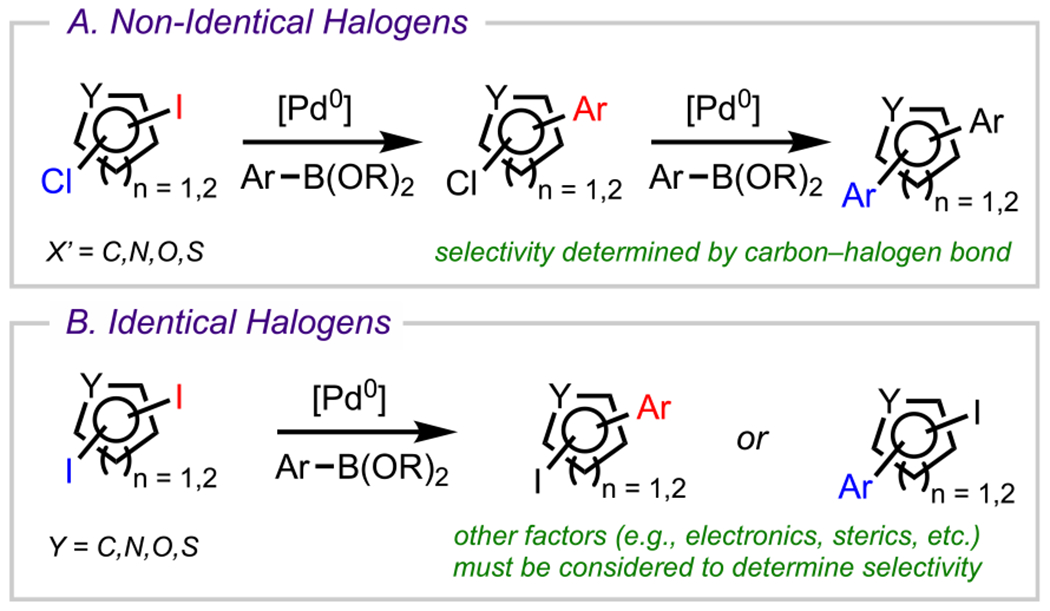
Cross-Couplings of Polyhalogenated Arenes Substituted with (A) Nonidentical Halogens or (B) Identical Halogens
In this Review, we present a comprehensive overview of the site-selective coupling of polyhalogenated (hetero)arenes bearing identical halogen groups by summarizing selected examples in this field, which highlight key factors that enable site selectivity. These examples are further categorized on the basis of factors that control the selectivity in each system, including electronic control, steric control, directing group (DG) control, ligand control, and additive/solvent control. The examples range from traditional approaches to controlling site selectivity, such as electronic and steric control, to more contemporary strategies, such as those that leverage additive/solvent control, to highlight the evolution of site-selective coupling. Additionally, the evolution in methods for achieving selective coupling is further supported with recent examples that deviate mechanistically from the norm, such as photochemical and C–F activation processes. It is important to note that aspects of classical approaches to site-selective cross-couplings (e.g., those influenced by DGs and electronic/steric factors) discussed herein have also been discussed in previous, insightful reviews;31,39–42 however, more modern approaches, such as the aforementioned C–F activation, additive/solvent-controlled, and photochemically initiated processes, have not been previously discussed. By focusing on a large array of examples, including those previously covered, we hope to place the methods in context, illustrate the growth of this field, and provide insight into the potential future directions of this highly valuable but rapidly evolving area of cross-coupling chemistry.
2. OVERVIEW OF MECHANISTIC CONSIDERATIONS AND EXISTING METHODS FOR PREDICTING SITE SELECTIVITY
To understand how selectivity arises in the cross-coupling of polyhalogenated (hetero)arenes, one must first consider the mechanism of cross-coupling. A simplistic depiction of the catalytic cycle for a transition-metal catalyzed cross-coupling, such as the Suzuki–Miyaura reaction, reveals three key elementary steps: oxidative addition, transmetalation, and reductive elimination (Scheme 2).34,35,45–48 The cycle begins with oxidative addition of the ligand-bound metal catalyst (MnLn) into one of the carbon–halogen bonds to generate a Mn+2 intermediate. This intermediate then transmetalates with a suitable organometallic reagent (RM′) to generate a new Mn+2 complex, where the halogen on the metal is displaced. This newly generated intermediate, when oriented in the cis geometry (with respect to the metal center), is primed to undergo reductive elimination to furnish the coupled product and regenerate the MnLn complex.45–48 In most instances, the turnover-limiting (or selectivity-determining) step is oxidative addition, where selectivity arises due to differences in the relative electrophilicities of the carbons bound to halogen groups in the parent (hetero)arene.40–42 However, it should be noted that there are instances where transmetalation or reductive elimination are turnover-limiting, and such examples will be discussed on a case-by-case basis.48,49 Because oxidative addition formally involves the addition of electrons to a carbon–halogen bond, the most electrophilic carbon would be expected to be the preferred site of attack, and, in general, the selectivity of cross-coupling follows the same trend observed for nucleophilic aromatic substitution (SNAr) reactions.13,50 Overall, the selectivity-determining step is influenced by several factors, including the electronics of the (hetero)arene, specifically, the relative electrophilicities of the carbon positions; the steric environment around the (hetero)arene, usually favoring positions that minimize steric interactions in the transition state; the reactivity of the ligand-bound catalyst from both an electronic and a steric standpoint; and the transmetalating reagent, in cases where transmetalation is turnover-limiting.51 Therefore, a site-selective coupling could be achieved by controlling any of these factors, depending on which step is turnover-limiting and irreversible.
Scheme 2.
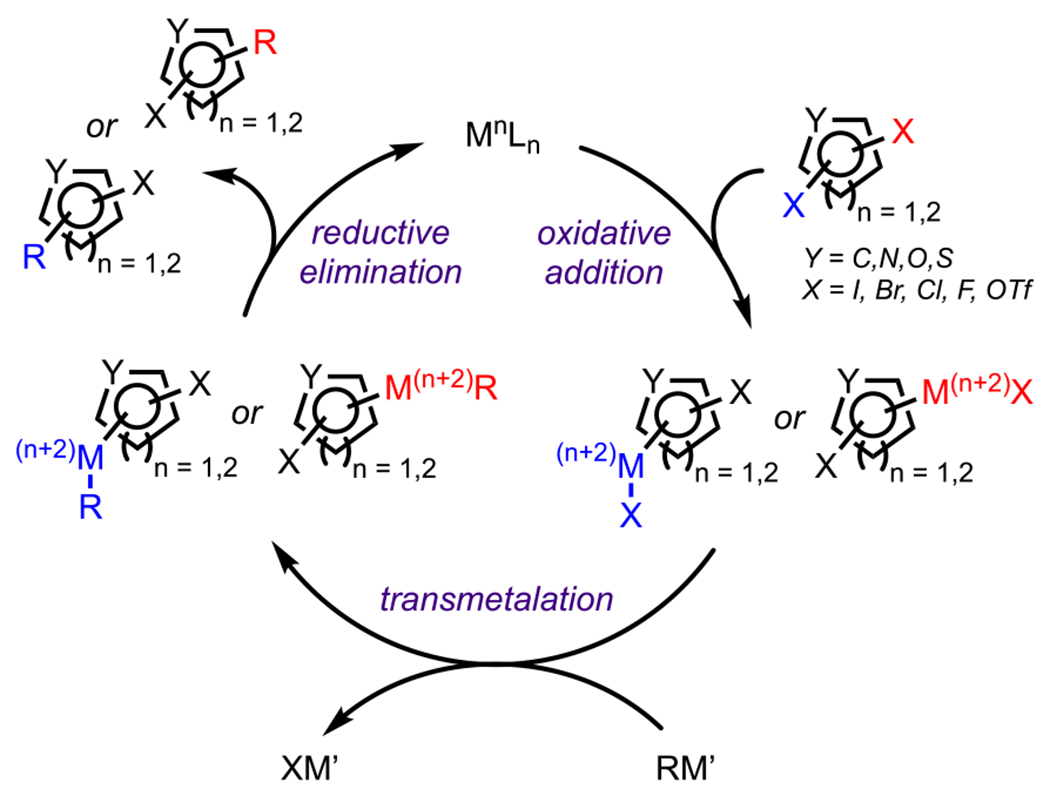
General Reaction Mechanism for Transition Metal-Catalyzed Cross-Coupling
In their highly informative review on site-selective Suzuki–Miyaura couplings of heteroaryl halides, Spivey and coworkers highlighted several key paradigms for predicting site selectivity.31 Because many of these factors will be invoked throughout our Review and to ensure comprehensive and consistent coverage, we will briefly summarize these key paradigms as well. Several methods have been employed in predicting the selectivity outcome in cross-couplings involving polyhalogenated (hetero)arenes. Often, empirically determined SNAr data correlate with site-selective cross-coupling outcomes.42,52,53 In some cases, the reactivity trend observed with lithium–halogen exchange to generate a more stabilized aryllithium intermediate can also correlate with the preferred site for transition-metal-mediated coupling.54,55 Furthermore, 13C NMR chemical shifts (δC) can provide insight into the most electrophilic carbon positions on a given system.41,56 Similarly, 1H NMR chemical shifts (δH) of the C–H bonds in the corresponding nonhalogenated aromatic compounds can guide the prediction of site-selective transformations as well.57 However, this method is not reliable for systems where ΔδH is <0.3 ppm because the site selectivity for such (hetero)arenes can be reversed depending on the reaction conditions.31,41,58 Another potential limitation in this NMR-guided approach to predicting reactivity order is that an initial substitution at one of the carbon–halogen bonds could affect the electronics of the remaining carbon positions, thus making subsequent substitutions more challenging to predict by NMR.
Recently, computational strategies have also been applied in predicting the selectivity outcome in cross-coupling for various heterocyclic systems.38 Advantageously, computations evaluate several related factors, such as steric influences and neighboring group participation, in addition to the intrinsic electronic properties of the parent heterocycle, to predict selectivity outcomes. Overall, this represents a more comprehensive and practical approach to predicting site selectivity compared with previous methods like NMR, which rely solely on intrinsic electronic differences to rationalize preferred sites of coupling. For example, Houk, Merlic, and coworkers have recently determined that the site selectivity of Pd-catalyzed cross-couplings cannot be predicted/rationalized by analyzing only the BDEs of respective carbon–halogen bonds.38 They have instead employed a density functional theory (DFT)-based “distortion–interaction” model to understand the factors that control oxidative addition (Figure 2A). This model considers both the energy required to distort the reactants (metal and heterocycle) to the oxidative addition transition-state geometry (known as the distortion energy, ΔE‡dist) and the energy from major orbital interactions between the distorted reactants (known as the interaction energy, ΔE‡int) to determine the activation energy for oxidative addition (ΔE‡). In general, the distortion energy is related to the BDE of the C–X bond, and the interaction energy is related to the extent of stabilization that results from the overlap between the heterocycle π* lowest unoccupied molecular orbitals (LUMOs) and the Pd dxy highest occupied molecular orbitals (HOMOs) (Figure 2B).38 For polyhalogenated arenes, both of these factors (ΔE‡dist and ΔE‡int) determine the relative stabilities of oxidative addition transition states at different halogen positions (ultimately conveyed by relative ΔE‡ barriers). These data are, in turn, used to predict and rationalize the preferred site of oxidative addition.
Figure 2.
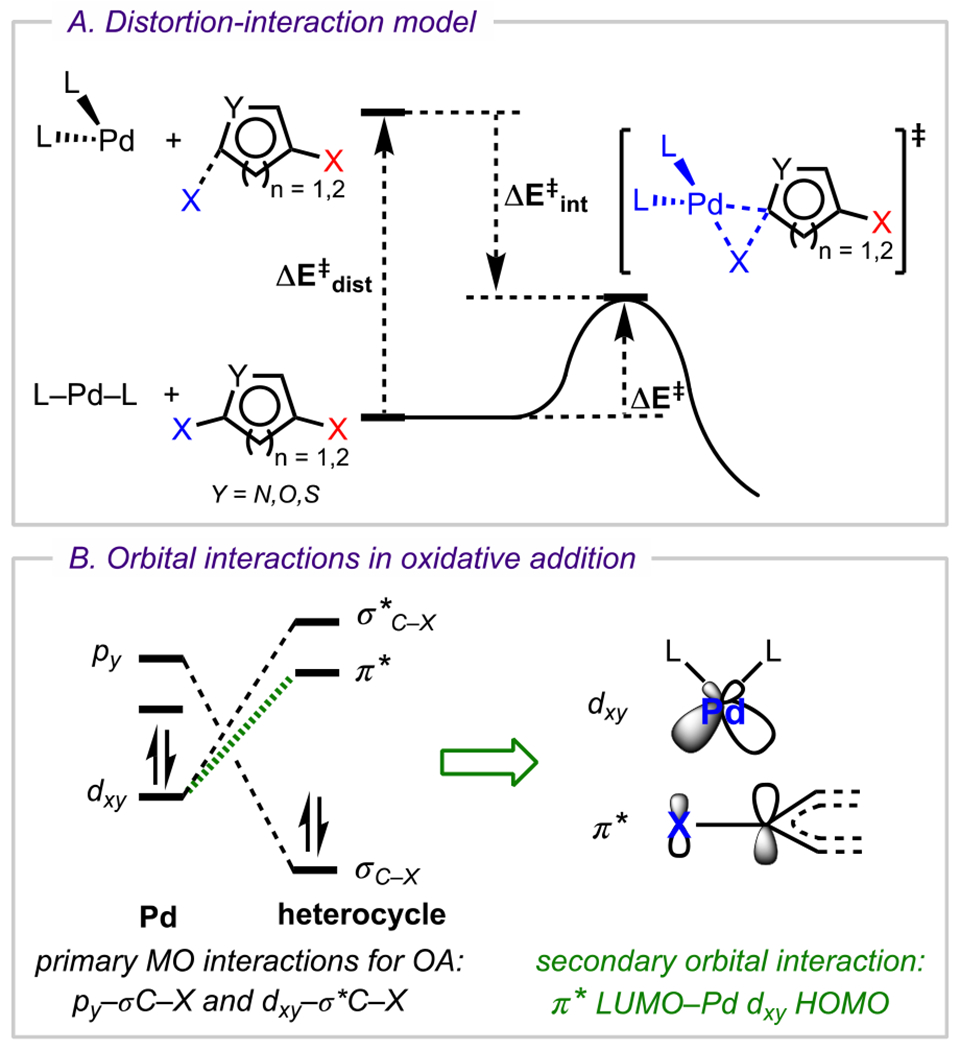
(A) Distortion–interaction model and (B) major orbital interactions between PdL2 and ArX in oxidative addition.
Overall, these methods highlight the importance of considering multiple factors when predicting selectivity outcomes in cross-coupling. On this basis, we will highlight several strategies and tools for controlling site selectivity, beginning with a discussion of how innate electronic properties can guide selectivity outcomes in myriad cross-coupling processes (section 3). After establishing a fundamental understanding of electronic considerations and how they guide selectivity, we will continue our discussion of this expansive field by considering other strategies that can be employed to either reinforce or override innate electronics to control selectivity outcomes, such as leveraging steric, DG, ligand, and additive/solvent effects (sections 4–7). Finally, we will discuss modern photochemical approaches to execute site-selective cross-couplings (section 8).
3. ELECTRONIC CONTROL
The electronics of the (hetero)arene can provide insight into the site that is expected to undergo oxidative addition to predict selectivity outcomes for cross-couplings of polyhalogenated (hetero)arenes bearing the same type of halogen. In fact, unless there are other factors that override electronics and perturb innate reactivity, such as strong steric influences or DGs, electronic effects will usually guide the reaction selectivity. Along these lines, there are two major factors to consider when assessing the electronics of a system: the (hetero)arene type and the substituent effects. For arenes (all-carbon framework), electronic effects from substituents (i.e., electron-withdrawing groups (EWGs) or electron-donating groups (EDGs)), beyond the two C–X bonds likely to undergo oxidative addition, often guide the selectivity outcomes (Scheme 3A). On the contrary, heteroarenes have intrinsic electronic properties by virtue of the embedded heteroatom, which can guide site selectivity even in the absence of substituents; in these cases, however, substituent effects can still play a role in guiding site selectivity (Scheme 3B).59 In this section, we discuss representative examples that illustrate how the (hetero)arene type and the substituent effects influence the electronic properties and therefore control selectivity outcomes. We begin with a discussion of all-carbon systems followed by examples of various six- and five-membered heterocyclic systems. Whereas this section is categorized on the basis of (hetero)arene type, the discussion will focus on outlining the fundamental concepts surrounding the electronic effects and properties of each system. Overall, this section is meant to provide a general guide for understanding electronic properties of arenes and heteroarenes in the context of site-selective cross-coupling.
Scheme 3.
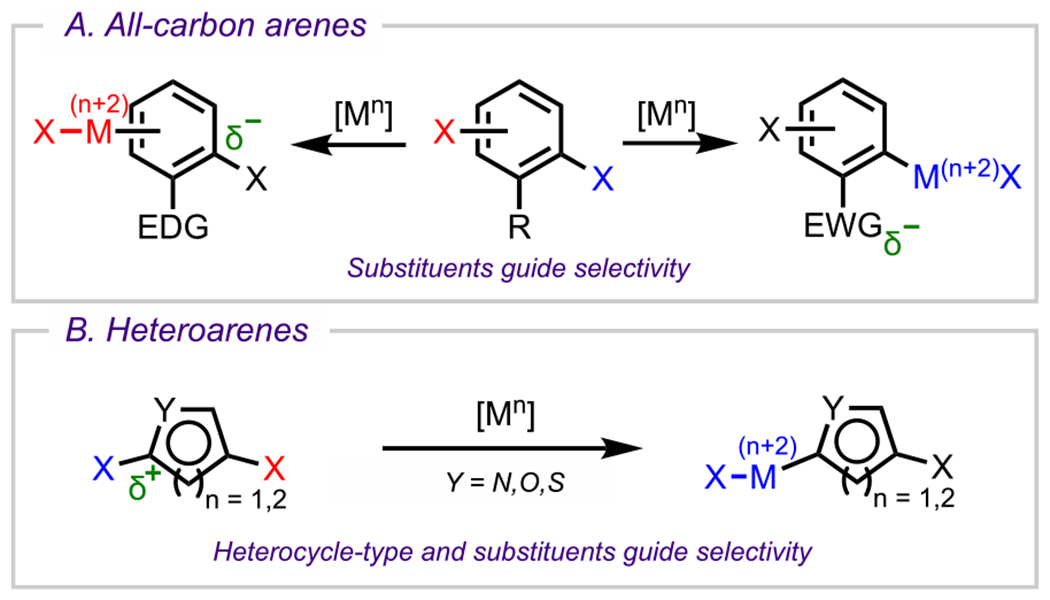
General Electronic Considerations for Both (A) Arenes (All-Carbon) and (B) Heteroarenes
3.1. Selected Examples of Arenes (All-Carbon Framework)
Substituents play a major role in defining the electronic environment of arenes and therefore heavily influence the site of oxidative addition. One example of substituent effects on electronics and, by extension, guiding site selectivity was described by Hearn and coworkers in their syntheses of unsymmetrical para-terphenyl moieties, which have been shown to possess medicinally relevant biological properties (4, Scheme 4).60 This transformation was achieved through a sequential ligand-free Suzuki–Miyaura coupling of 1,4-dibromo-nitrobenzene (1) and diverse arylboronic acids (2 and 3) with a Pd(OAc)2 precatalyst (Scheme 4). By virtue of a nitro (−NO2) substituent, cross-coupling to an electron-deficient polyhalogenated arene gave selectivities that mirrored that of an SNAr reaction, where the first oxidative addition occurred preferentially at the more electrophilic C–Br bond ortho to the nitro group rather than the meta C–Br bond; this reaction resulted in the ortho-substituted (with respect to the NO2 group) biaryl intermediate, which subsequently underwent a second coupling at the meta position upon the addition of the second arylboronic acid (3) to provide the desired terphenyl scaffold (4).
Scheme 4.
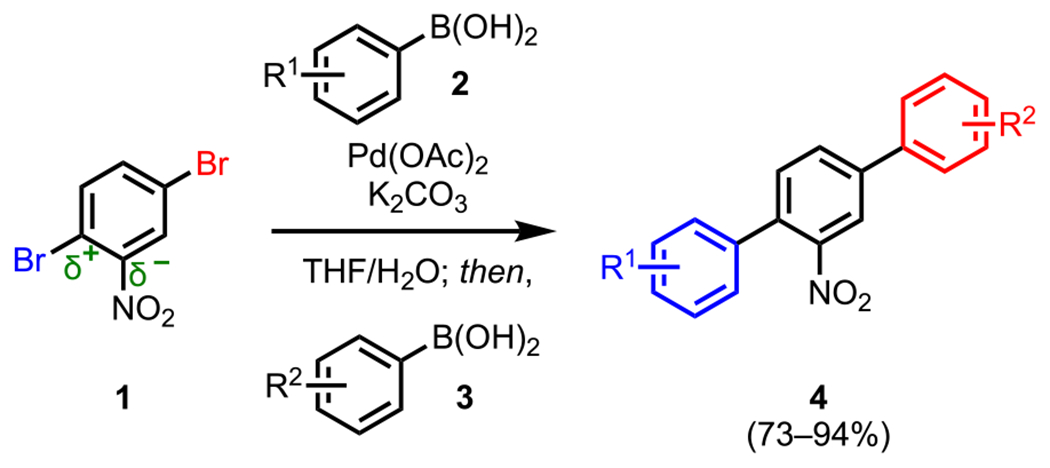
Site-Selective, One-Pot Suzuki–Miyaura Coupling to Form Unsymmetrical Terphenyl Cores
In addition to the work by Hearn and coworkers, site-selective cross-coupling has also been achieved in site-selective C–F bond cross-coupling reactions. Organofluorine compounds have proven valuable in pharmaceuticals because C–F bonds are bioisosteres of highly ubiquitous C–H bonds but are not susceptible to oxidation.61 For this reason, a considerable amount of research has been dedicated toward developing new C–F bond-forming processes.62 On the contrary, the functionalization of C–F bonds by transition-metal activation has been relatively less studied. Nonetheless, this alternative approach has proven to be an attractive strategy for diversifying organofluorine compounds. The main challenge associated with oxidative addition into C–F bonds arises from the strength of the C–F bond, which is not only the strongest bond in the aryl carbon–halide series (Figure 3) but also the strongest carbon–heteroatom bond in nature.61 This bond strength is attributed to the strong electronegativity of fluorine, which polarizes the C–F bond and creates a strong electrostatic attraction between Cδ+ and Fδ−.63
Figure 3.
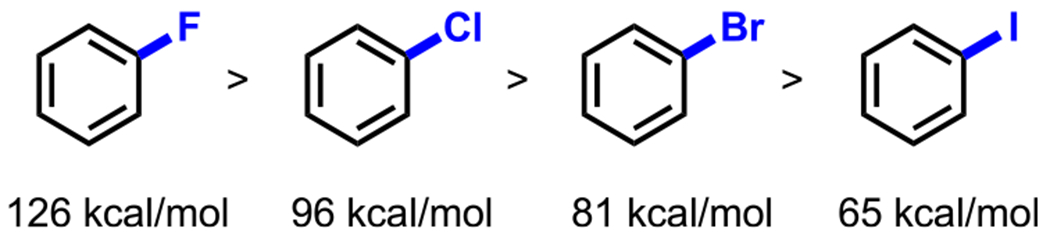
Relative BDEs of aryl C–X bonds.
Nonetheless, considerable progress has been made in the field of C–F bond activation,64 in particular, for the site-selective cross-coupling of polyfluorinated arenes. In general, selectivity outcomes for cross-couplings of these systems can be understood by examining the analogous SNAr reactivity (Scheme 5). In SNAr reactions of these systems, nucleophilic addition is preferred at sites that result in a Meisenheimer65 intermediate, with the anionic charge at a carbon bearing a nonfluorine substituent (usually R = H, EWG, or EDG) instead of one bearing a fluorine atom, which would otherwise result in destabilizing electron–electron repulsive interactions. To satisfy this preference, nucleophilic additions usually occur at a position that is not para to a fluorine atom.66 Using pentafluorobenzene derivative 5 as a representative example, addition is favored para to the nonfluorine R group, which results in intermediate 6, whereas, addition ortho to the R group (para to a fluorine substituent) is disfavored due to the formation of intermediate 7, which is destabilized by electron–electron repulsion. Analogously, 8 would also favor oxidative addition para to R, providing intermediate 9.
Scheme 5.
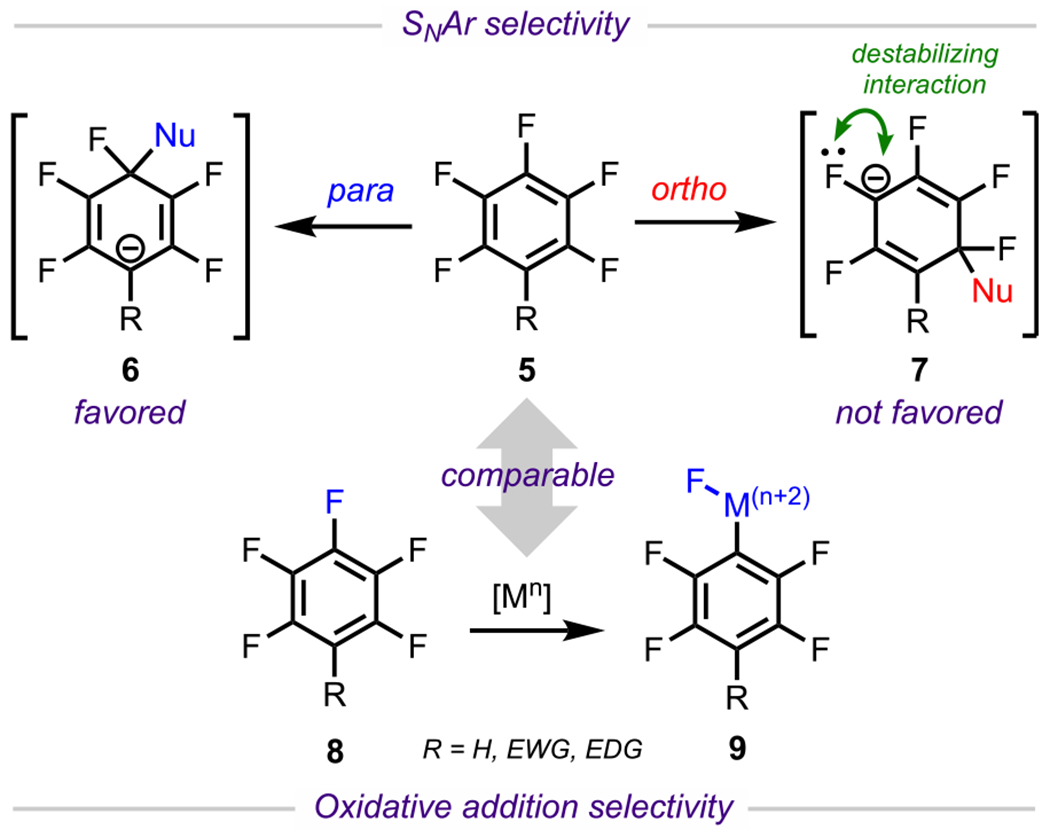
Electronic Considerations of Polyfluorinated Arenes and General Reactivity toward SNAr and Cross-Coupling
A prime example of this selectivity is the site-selective Pd-catalyzed C–F arylation of polyfluoroarenes that was demonstrated by Zhang and coworkers (Scheme 6).67 This methodology involved the treatment of various polyfluoroarenes (10) with a diverse array of arylboronic acids and an in situ generated Pd(0) catalyst to synthesize monoarylated cross-coupled products. These reactions proceeded in accordance with their known SNAr selectivity, where cross-coupling occurred para to a nonfluorine R substituent to provide monoarylated products 11a–d. In addition to para-arylated products, the authors also reported ortho-arylated products, such as 11d, where C–F activation selectivity was achieved between the two ortho sites by the strategic placement of the fluorine substituents. Similar to SNAr, the site that was ortho to the ester but also para to a hydrogen atom (instead of a fluorine atom) was the preferred site of coupling.
Scheme 6.
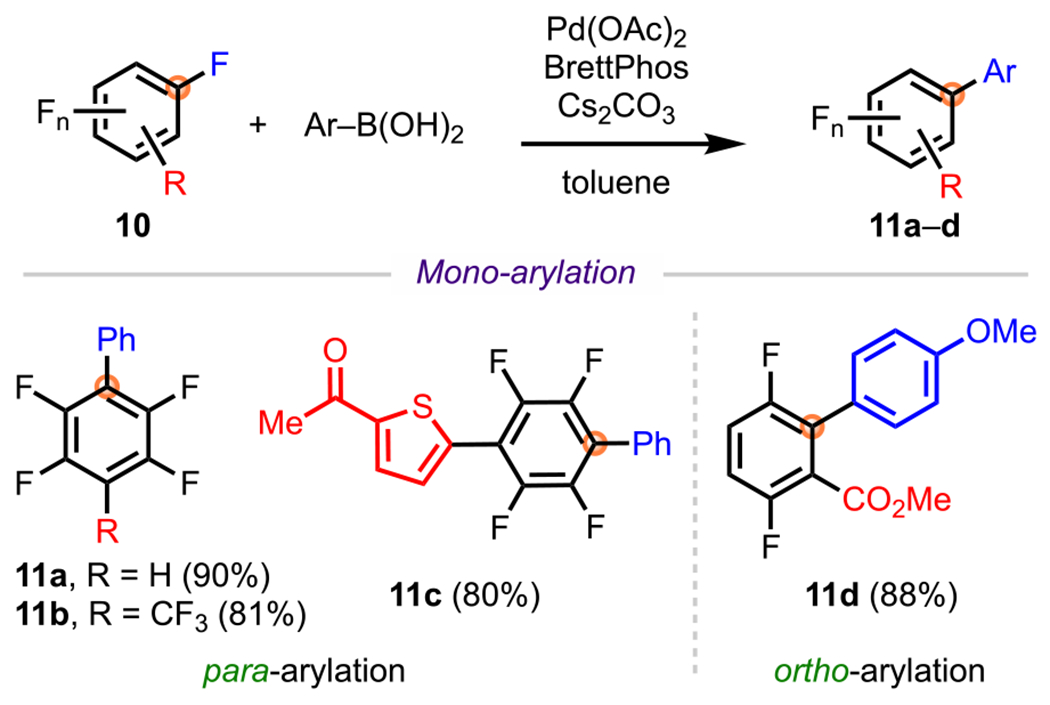
Site-Selective Cross-Coupling of Polyfluorinated Arenes
Similarly, the base-free Ni(0)-catalyzed Hiyama coupling reported by Ogoshi and coworkers in 2014 (Scheme 7) proceeded with a similar selectivity outcome.68 Using [Ni2(iPr2Im)4(cod)], the authors effectively coupled perfluorotoluene (12) with various arylsiloxanes (13) to furnish the corresponding para-arylated products (14a–d). One key feature of this work is that cross-coupling was achieved between arylsiloxanes and electron-rich (14a and 14b) or electron-deficient (14c and 14d) arenes. Of note, the site selectivity of this reaction could be rationalized similarly to the previous example, where selectivity analogous to an SNAr reaction was observed.
Scheme 7.
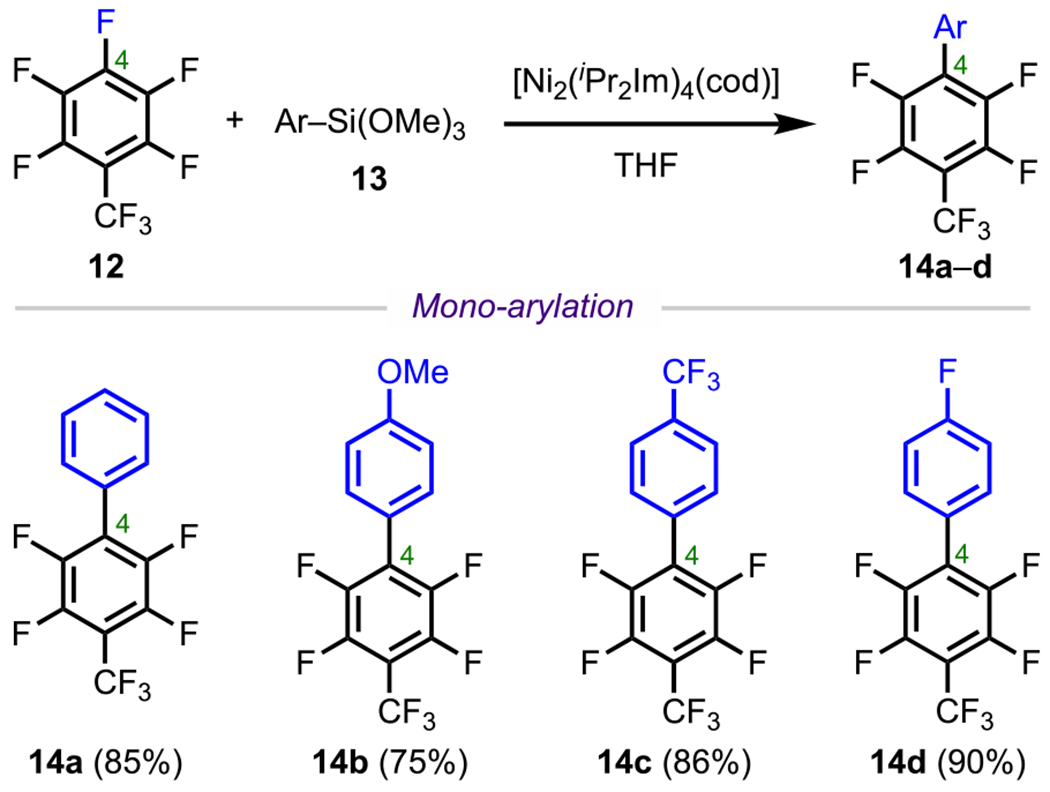
Site-Selective Monoarylation of Perfluorotoluene
3.2. Selected Examples of Six-Membered Heteroarenes
Heteroarenes, many of which are considered privileged scaffolds69 in medicinal chemistry, have also gained significant traction in site-selective cross-coupling chemistry, which provides an effective and modular strategy for structural diversification. In fact, the majority of small-molecule drugs employed in human medicine are composed of heterocyclic subunits, with >85% of all bioactive molecules containing some type of heterocycle.70,71 Unlike arenes, such as benzene and derivatives thereof, heteroarenes are inherently electronically biased due to inductive effects imparted by the embedded heteroatom(s).72 The selected examples of heteroarenes that will be discussed in this section (pyridine (15), diazines (pyridazine (16), pyrimidine (17), and pyrazine (18)), and 2-pyrone (19)) are shown in Figure 4.
Figure 4.
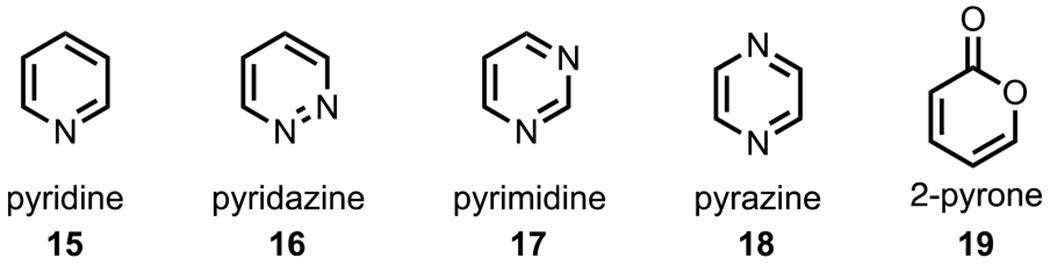
Selected six-membered heterocycles employed in site-selective cross-coupling.
3.2.1. Pyridine.
Pyridines (e.g., 20 and 20′, Scheme 8) are inherently π-deficient by virtue of the embedded electronegative nitrogen atom, which inductively withdraws electron density from C2 (ortho) and C4 (para). The increased electrophilicity at these positions activates them toward nucleophilic attack in SNAr (Scheme 8).72 In an analogous sense, for cross-couplings of polyhalogenated pyridines, oxidative addition is also favored at the C2 and C4 positions, but this selectivity also greatly depends on specific reaction conditions, other substituents, and substitution patterns.73 In general, for 2,3-, 2,4-, and 2,5-dihalopyridines, oxidative addition is favored α to the nitrogen (i.e., at C2). Recently, Houk, Merlic, and coworkers calculated the BDEs of the C–Cl bonds in various dichloropyridines (Figure 5) and found that the C2–Cl bond in each of these systems has the lowest calculated BDE, thus accounting for the observed selectivity in cross-coupling.73 The lowest BDE associated with the C2 position can be rationalized by the α-nitrogen effect in which there is a stabilizing interaction between the nitrogen lone pair orbital and the C2 singly occupied molecular orbital (SOMO) resulting from homolysis.74,75 It is important to note, however, that whereas there is an overall preference for C2 oxidative addition in 2,4-dihalopyridines, site selectivity for the C4-position can be achieved under certain reaction conditions (see 29d, Scheme 9B).76
Scheme 8.
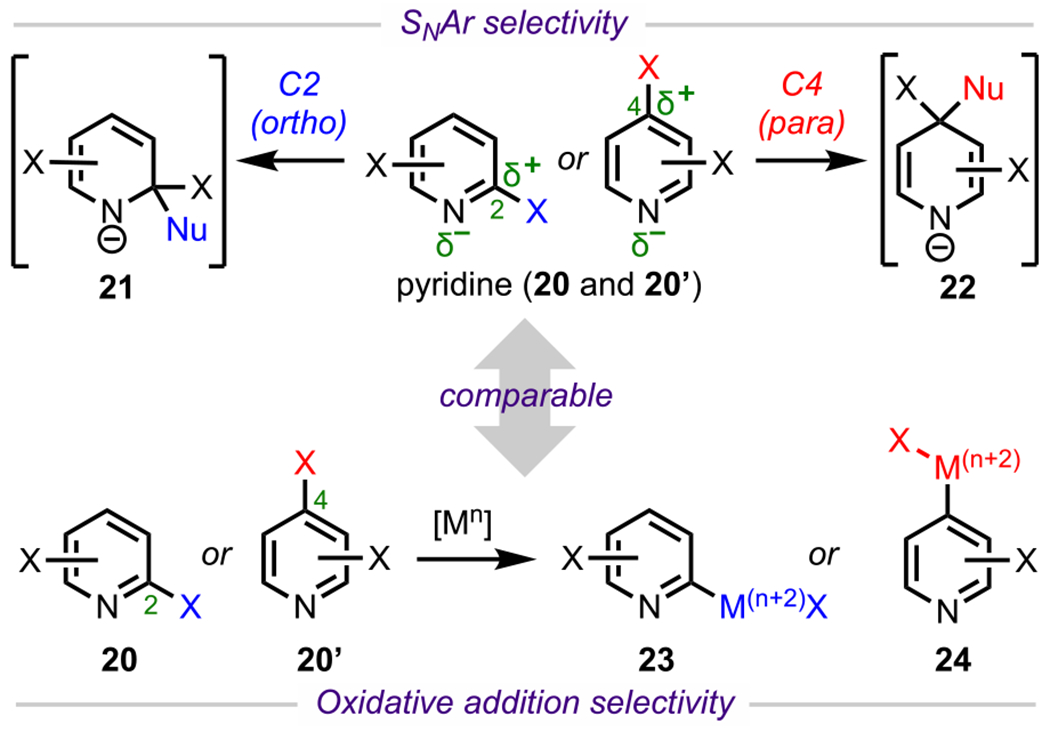
Electronic Considerations of Pyridine and General Reactivity toward SNAr and Cross-Coupling
Figure 5.
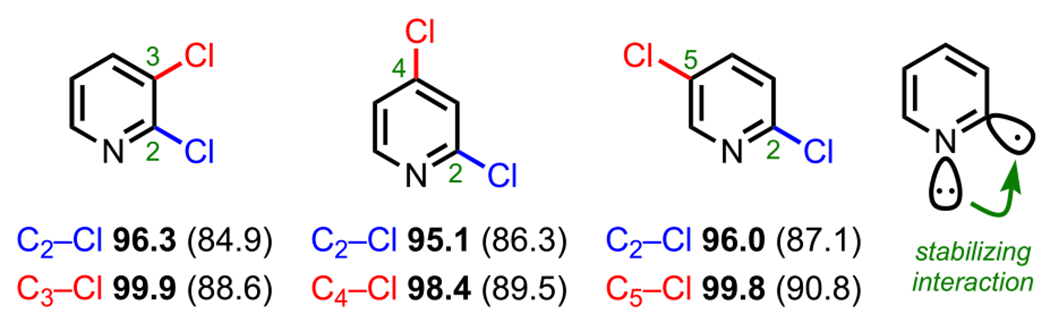
C–Cl BDEs (kcal/mol) of dichloropyridines using G3B3 (bold) and B3LYP (in parentheses).
Scheme 9.
Site-Selective (A) Allenylation and (B) Arylation of Dibrominated Pyridines
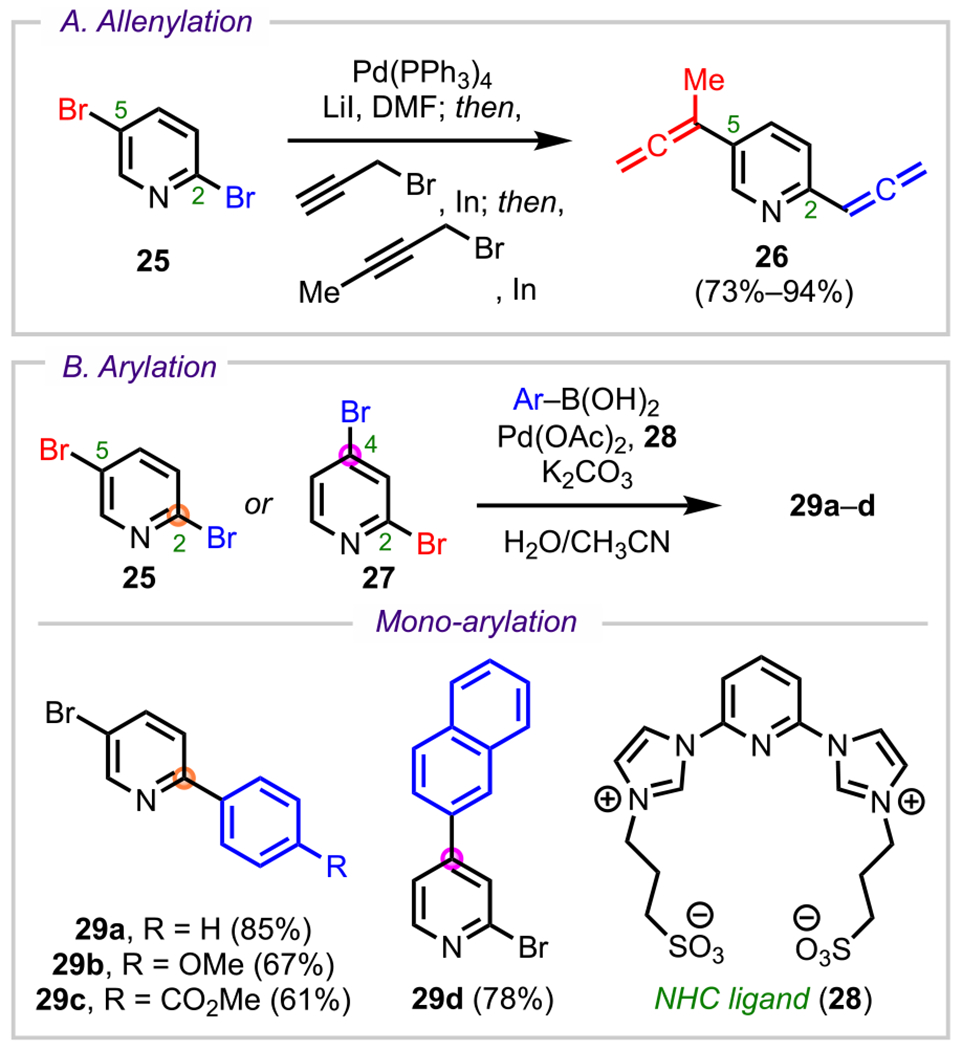
Preferential reactivity at the C2 position has been observed in many cross-coupling examples involving dihalogented pyridines, such as the Pd-catalyzed allenylation of 2,5-dibromopyridine (25) described by Lee and coworkers (Scheme 9A).77 In this work, the authors demonstrated C2-selective coupling with an in situ generated allenylindium reagent derived from the corresponding propargyl bromide. After installation of the C2 allene, the addition of a different allenylindium reagent resulted in C5 coupling, furnishing the C2/C5-allenylated product (26). Similarly, Kapdi and coworkers (Scheme 9B) demonstrated the C2- and C4-selective arylation of 2,5- and 2,4-dibromopyridines (25 and 27) with arylboronic acids using Pd(OAc)2 and an N-heterocyclic carbene (NHC) ligand (28).76 As expected, 2,5-dibromopyridines underwent monoarylation at the more electrophilic C2 position (29a–c). Interestingly, 2,4-dibromopyridine 27 underwent preferential arylation at C4 over C2 to furnish 29d, thus demonstrating how under certain reaction conditions, selectivity can be achieved between two similarly electrophilic positions.
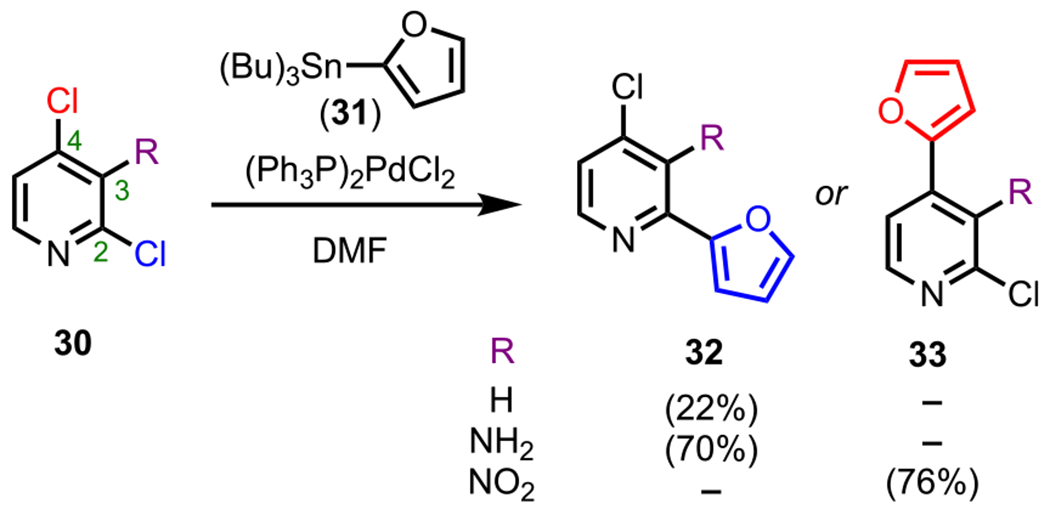
Along these lines, Gundersen and coworkers demonstrated how selectivity can be achieved between the electronically similar C2 and C4 positions of 2,4-dichloropyridines (30) by altering the substituent at the C3 position (Scheme 10).78 The intrinsic reactivity (when R = H) of this system upon subjection to Stille coupling conditions with 2-furyl(tributyl) tin (31) favored cross-coupling at the C2 position to furnish 32, albeit in low yields. Adding an EDG (R = NH2) to the three-position also favored coupling at the C2 position, presumably due to the deactivation of the C4 position. However, adding an EWG (R = NO2) to C3 activated C4 toward oxidative addition, thus completely reversing the selectivity to favor C4 coupling and providing 33. Overall, this example showcases how substituents can be used to alter intrinsic heteroarene electronics to control selectivity outcomes.
Scheme 10.
Substituent Effects in C2 versus C4 Stille Coupling
Complementary to Gundersen’s work is the report by Langer and coworkers, who investigated site-selective cross-couplings of 2,6-dichloro-3-(trifluoromethyl)pyridines (34) to access myriad medicinally relevant trifluoromethyl-substituted pyridine derivatives (35, Scheme 11).79 Focusing specifically on the monoarylation of these systems, the authors showcased how 34 in the presence of one equivalent of various arylboronic acids and a Pd(0) catalyst underwent selective arylation at C2 over C6. Whereas these two positions on unsubstituted pyridine systems are electronically equivalent, the presence of an adjacent trifluoromethyl (CF3) group further activated the C2 position toward oxidative addition, thus rendering this reaction selective for C2 coupling. This preference for C2 selectivity has also been observed by Bourissou and coworkers in their studies of Suzuki–Miyaura couplings involving the similar substrate, 2,6-dichloro-3-nitropyridine.80
Scheme 11.
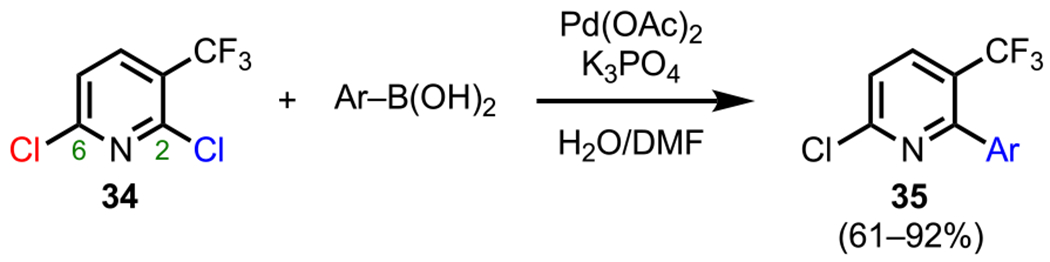
Site-Selective Monoarylation of 2,6-Dichloro-4-(trifluoromethyl)pyridines
In addition to investigating site-selective cross-coupling in dihalogenated pyridines, Langer and coworkers have also investigated these processes in more heavily substituted systems, such as perchloropyridine (36, Scheme 12).81 In this work, the authors investigated the selectivity of coupling pyridine 36 with various arylboronic acids through a Pd-catalyzed Suzuki–Miyaura coupling. This system underwent arylation only at the C2 and C6 positions, with no arylation observed at the electronically similar C4 position. It should also be noted that to achieve the second arylation, more forcing reaction conditions were required (i.e., higher temperatures around 90–100 °C). This is presumably because the aryl group is less electron-withdrawing than the chlorine atom that was substituted.
Scheme 12.
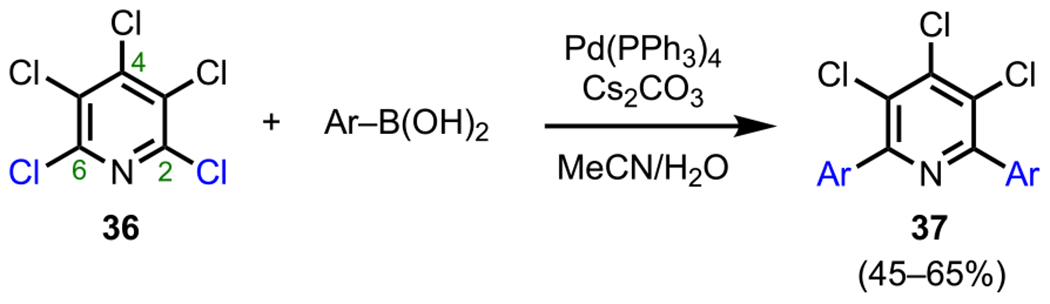
Site-Selective Bisarylation of Perchloropyridine
In contrast with the C2/C6 selectivity observed by Langer and coworkers, Zhang and coworkers observed completely different selectivity while investigating a Pd-catalyzed cross-coupling of perfluoropyridine (38, Scheme 13) with phenyl boronic acid. This cross-coupling was selective for the C4 position to provide 4-phenylpyridine 39.67 This selectivity can be explained through the same analysis that was applied to the polyfluorinated arenes discussed previously (Schemes 5 and 7), where coupling occurs preferentially at a position para to a nonfluorine substituent.
Scheme 13.
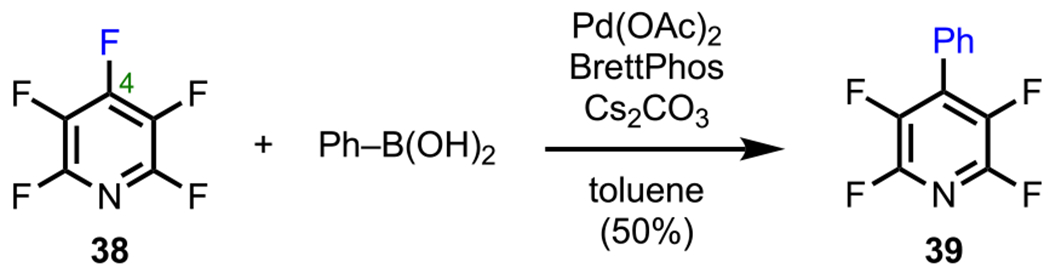
Site-Selective Monoarylation of Perfluoropyridine
In addition to serving as a robust strategy for functionalizing the periphery of simple arenes, site-selective cross-coupling has also served as an effective approach for constructing even more complex polycyclic heteroarenes, such as the carbazole (42, Scheme 14A) and α-carboline (45, Scheme 14B) structural motifs.82 Ackermann and coworkers were one of the first to demonstrate this approach to construct these heterocycles using a Pd-catalyzed amination/C–H functionalization domino reaction between anilines (40 and 43) and dichlorinated arene electrophiles (41 and 44). These reactions generally first proceed through a Buchwald–Hartwig-type amination between the aniline and the more electrophilic C–Cl bond, which in 41 is the para-C–Cl bond (with respect to the ketone) and in 44 is the ortho-C–Cl bond (with respect to the nitrogen), both in accordance with predicted selectivity. This initial amination was then followed by a second oxidative addition/intramolecular C–H functionalization event to provide either the carbazole (42) or the α-carboline (45) product. Following this work, a number of similar strategies for synthesizing α-carbolines were demonstrated by Maes and coworkers83 and Cuny and coworkers.84
Scheme 14.
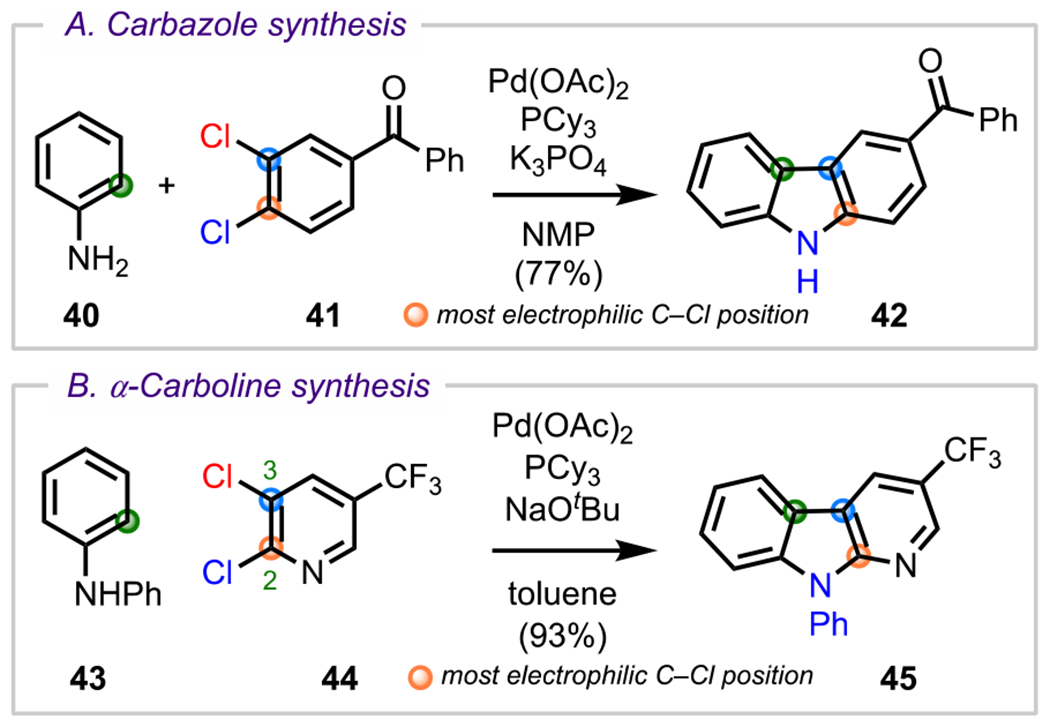
(A) Carbazole and (B) α-Carboline Synthesis Using a Site-Selective Domino Cross-Coupling Reaction
3.2.2. Diazines: Pyrimidine, Pyridazine, and Pyrazine.
In addition to pyridines, polyhalogenated diazines (Figure 6), including pyrimidine (46), pyridazine (47), and pyrazine (48), have also been widely used as substrates in site-selective cross-couplings. Similar to pyridine, in general, oxidative addition in diazines occurs at the site with the lowest C–X BDE.73
Figure 6.
Selected diazines: pyrimidine, pyridazine, and pyrazine.
For polyhalogenated pyrimidines, the general order of reactivity is C4 > C2 > C5.73 Analogous to the case for dichloropyridines, Houk, Merlic, and coworkers also calculated the relative BDEs for both 2,4- and 2,5-dichloropyrimidines and found that the C4–Cl bond has the lowest BDE relative to the C2 and C5 positions (Figure 7). On the basis of the α-nitrogen effect observed in pyridine systems, one might have expected the C2–X bond in pyrimidines, which is “doubly” α (situated next to two nitrogen atoms), to have a lower BDE compared with the C4–X bond (due to added stabilization of the C2-pyrimidyl radical from an additional N(nonbonding orbital)–C2(SOMO) interaction). However, through computational studies, Hadad and coworkers found that the C4-pyrimidyl radical is actually more stable than the C2-pyrimidyl radical, thus accounting for the lower BDE at the C4 position.85 Specifically, the authors reported that following C–H homolysis at C4, C2, and C5, the bond angles at these positions widen by 2.5, 3.8, and 5.0°, respectively. Overall, these data convey that less deviation from the ideal sp2-hybridized 120° bond angle provides greater radical stabilization and thus a lower BDE for the parent C–X bond.
Figure 7.
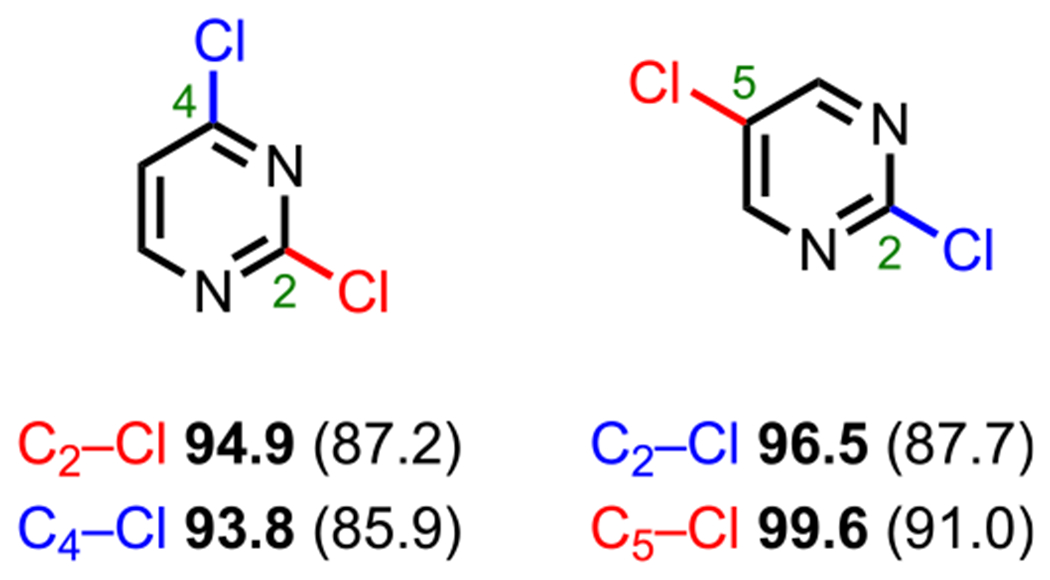
C–Cl BDEs (kcal/mol) of dichloropyrimidines using G3B3 (bold) and B3LYP (in parentheses).
The order of reactivity that was predicted computationally (C4 > C2 > C5) is supported by empirical observations by Undheim and Solberg, who reported identical trends from their studies of Stille couplings between various polychlorinated pyrimidines (49, 52, and 54, Scheme 15) and tributyl-styryl-stannane (50).86 Ultimately, these reactions afforded the expected C4- and C2-vinylated adducts (C4: 51 and 55; C2: 53) in accordance with the selectivity trend predicted by relative BDE values.
Scheme 15.
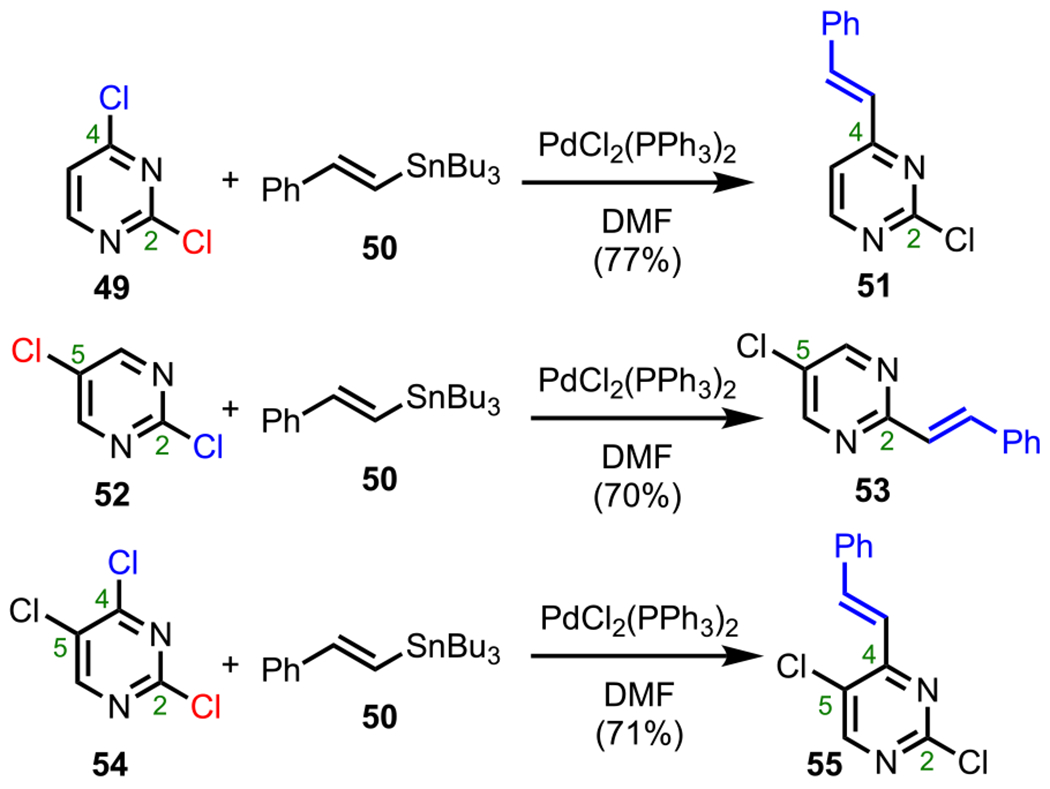
Site-Selective Stille Coupling of Polyhalogenated Pyrimidines
Polyhalogenated pyridazines generally favor cross-coupling at C3, which has the lowest C–X BDE relative to the C5 position due to C3 being α to a nitrogen (Scheme 16).73 Young and coworkers observed this selectivity trend in their Pd-catalyzed amidation reaction between 3,5-dichloropyridazine (56) and 2-pyrrolidinone (57) to give C3-amidated adduct 58.87
Scheme 16.
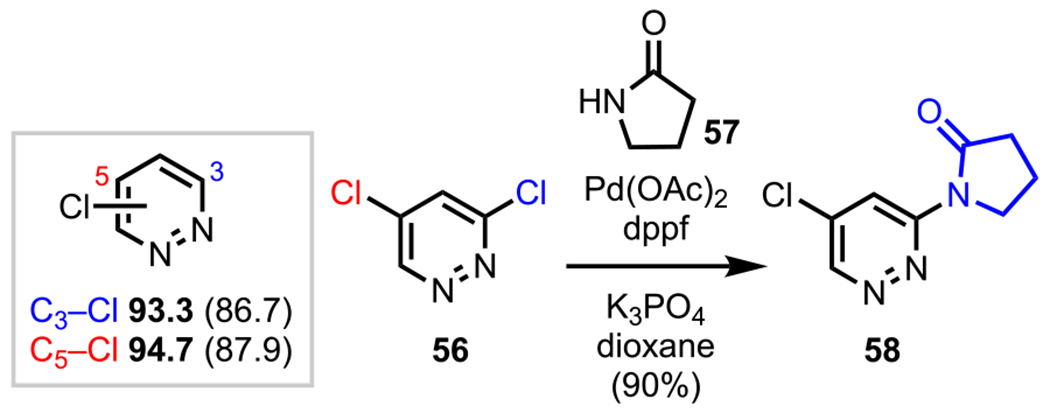
Pyridazine C–Cl BDEs in kcal/mol (in Box) Using G3B3 (Bold) and B3LYP (in Parentheses) and Site-Selective Amidation of 3,5-Dichloropyridazine
Unlike pyridine and pyrimidine derivatives, dihalogenated pyrazines are symmetrical for all substitution patterns, with both halogens residing α to a nitrogen in every case (Figure 6).31 These C–X bonds are electronically identical and, in the case of a C–Cl bond, have a calculated BDE of 92.9 kcal/mol (Figure 8).73 Because these positions cannot be differentiated electronically, other strategies, such as employing a DG, are required to achieve site selectivity; such an example is described in section 5. (See Scheme 31.)
Figure 8.
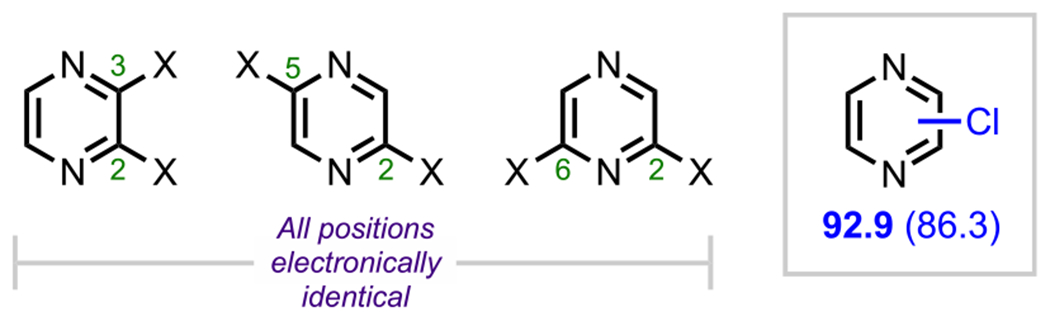
Electronically identical dihalogenated pyrazines and the pyrazine C–Cl BDE in kcal/mol (in box) using G3B3 (bold) and B3LYP (in parentheses).
Scheme 31.
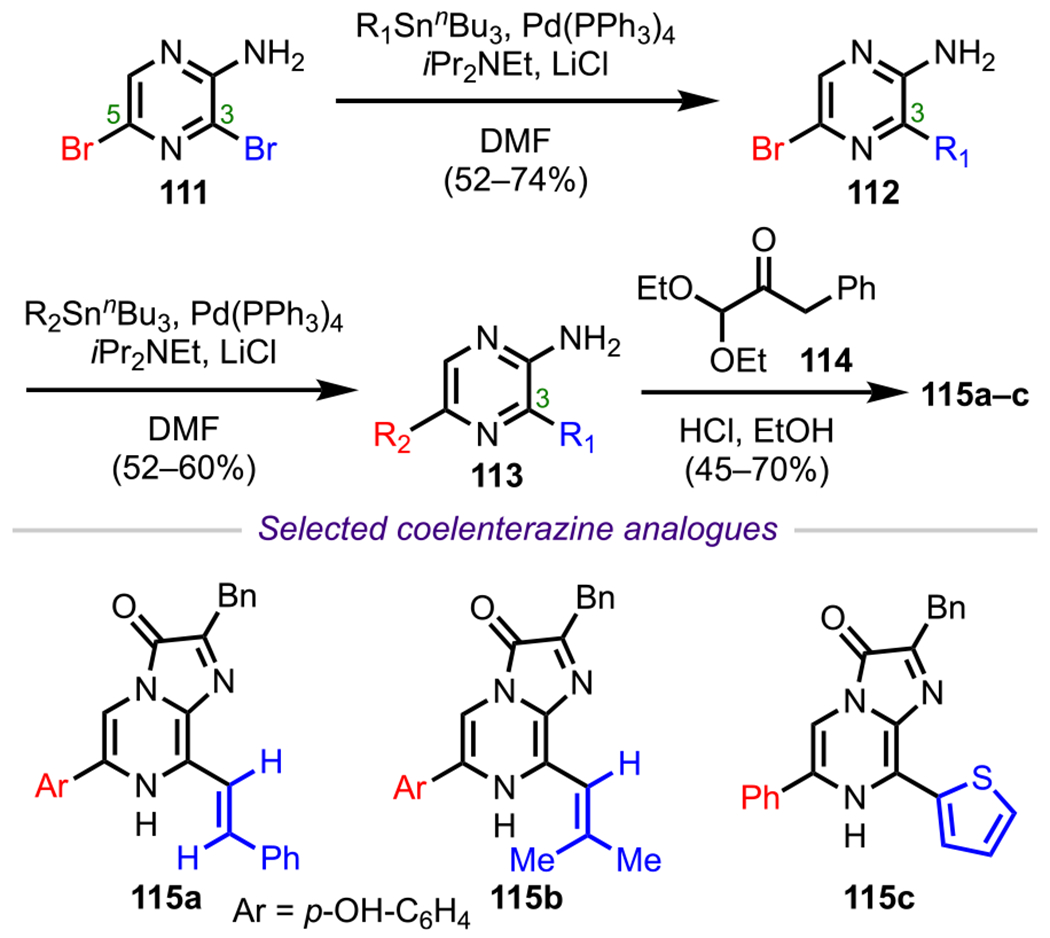
Synthesis of Coelenterazine Analogues Starting from 2-Amino-3,5-dibromo Pyrazine (111)
3.2.3. 2-Pyrone.
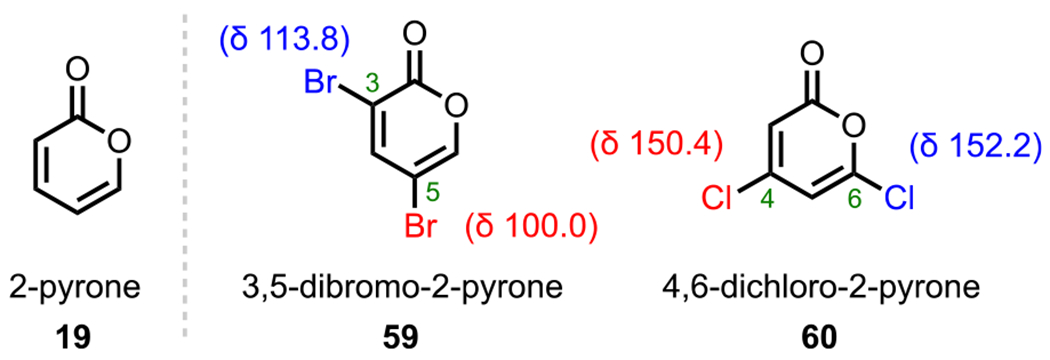
The 2-pyrone motif (19, Figure 9) is found in a number of biologically active secondary metabolites and has also become a useful synthetic precursor to access numerous biologically important molecules.88–91 Specifically, various cross-coupling adducts originating from 2-pyrone derivatives exhibit promising anticancer activity. Given the promising bioactivity of 2-pyrone derivatives, 3,5-dibromo-2-pyrone (59) and 4,6-dichloro-2-pyrone (60) have been the subject of various studies on site-selective cross-coupling. The 3,5-dibromo-2-pyrone motif (59, Scheme 17A) has been shown to undergo C3-selective Sonogashira coupling with various terminal alkynes to afford the corresponding C3-alkynylated products (61).92 The selectivity observed under these conditions is in accordance with what would be predicted by 13C NMR shifts (Figure 9), where C3 is more deshielded (electron-deficient) than C5 (Δδ = 3.8).93–95 With a decrease in electron density at C3, oxidative addition is more favorable at this position over the more shielded C5 position. Whereas reactivity at C3 of 3,5-dibromo-2-pyrone is favored by virtue of the innate electronics, the selectivity can be completely reversed to instead favor the C5 position under certain conditions.43,44 An example of employing additive/solvent control to reverse the intrinsic site selectivity of 3,5-dibromo-2-pyrone is discussed in section 7.
Figure 9.
2-Pyrone and 13C NMR shifts for 3,5-dibromo-2-pyrone and 4,6-dichloro-2-pyrone.
Scheme 17.
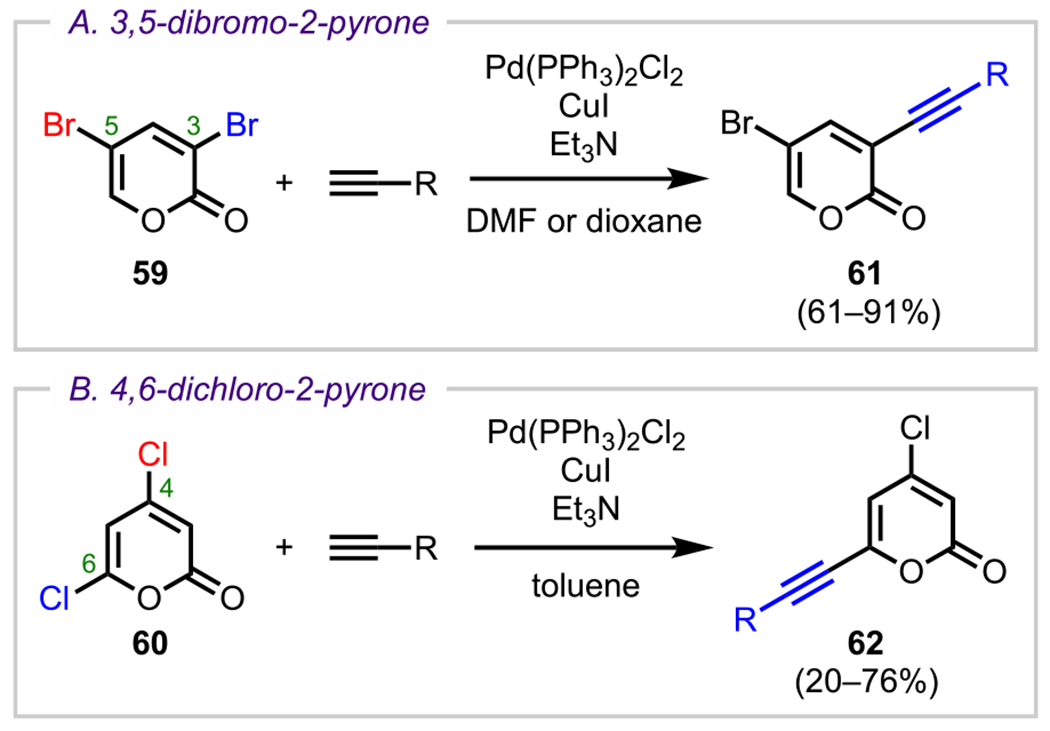
Site-Selective Sonogashira Coupling of (A) 3,5-Dibromo-2-pyrone and (B) 4,6-Dichloro-2-pyrone
For 4,6-dichloro-2-pyrone (60), Sonogashira coupling with terminal alkynes preferentially gives C6-alkynylated products (62, Scheme 17B).95 Similar to 3,5-dibromo-2-pyrone, the 13C NMR shifts for 4,6-dichloro-2-pyrone (C6: δ = 152.2 vs C4: δ = 150.4) also correlate with this observed selectivity (Figure 9). Upon analysis of the energy profile for both C4 and C6 oxidative addition (using PMe3 model ligands), it can be seen that oxidative addition leading to intermediate I via TS-I is both kinetically and thermodynamically favored over the formation of intermediate II via TS-II (Figure 10). This favorability for C6 oxidative addition can be rationalized by the α-oxygen, which presumably stabilizes the developing positive charge in the transition state by serving as a π-donor.95
Figure 10.
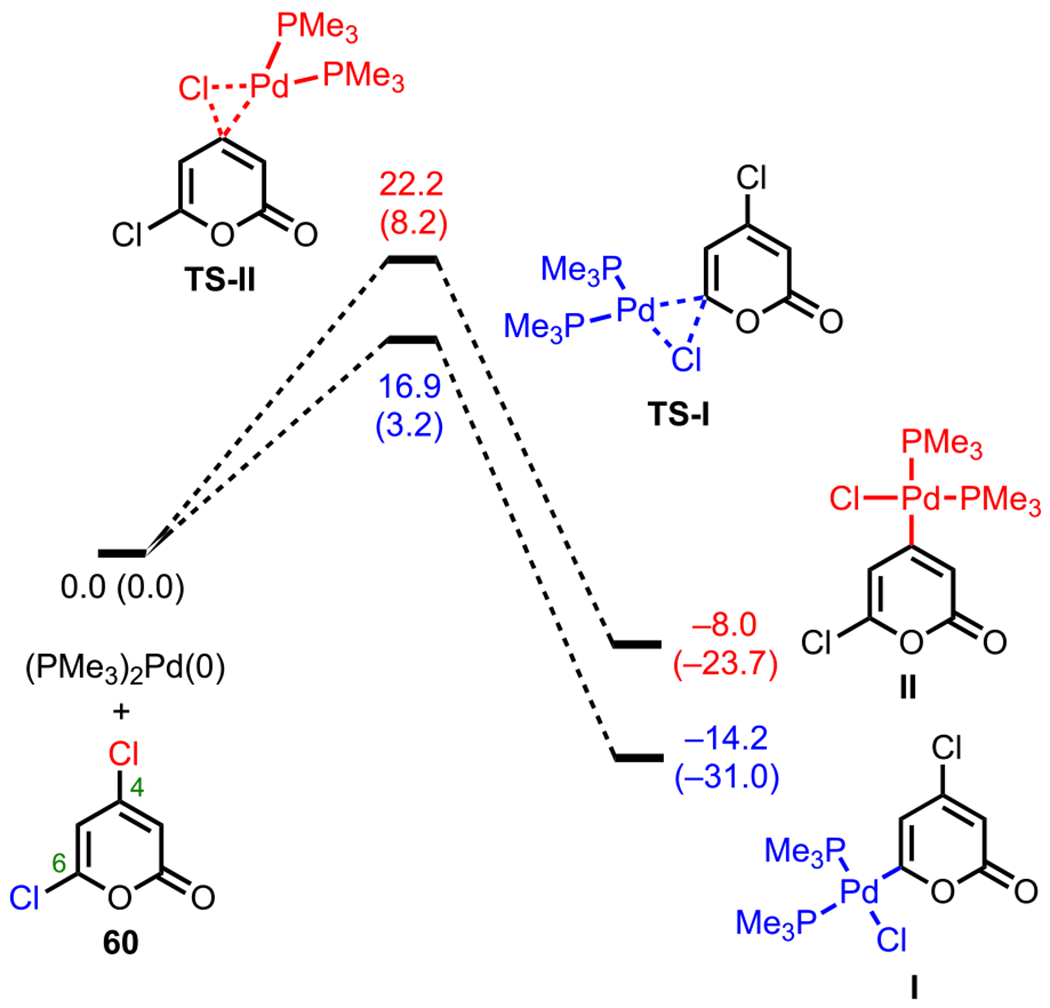
Energy profiles for C4 versus C6 oxidative addition of 4,6-dichloro-2-pyrone. Relative free energies and reaction energies (in parentheses) listed in kcal/mol (ref 95).
3.3. Selected Examples of Five-Membered Heteroarenes
In addition to six-membered heteroarenes, five-membered heteroarenes (Figure 11) are also prevalent in many pharmaceuticals and therefore constitute an important class of medicinally relevant compounds.70 In this section, we discuss selected examples of site-selective cross-couplings of five-membered polyhalogenated heteroarenes along with electronic considerations that help guide the prediction of site selectivity. As with the previous sections, these examples are further categorized based on the subclass of five-membered heterocycles they belong to, which in this section will include five-membered heterocycles with either one heteroatom or two heteroatoms (i.e., 1,3-azoles and 1,2-azoles).
Figure 11.
Selected five-membered heterocycles.
3.3.1. Thiophene, Pyrrole, and Furan.
Polyhalogenated pyrroles, thiophenes, and furans are known, in general, to preferentially undergo oxidative addition α to the heteroatom (C2 or C5) in cross-coupling reactions. Unlike most of the six-membered aza-aromatics previously discussed (with the exception of pyrimidines), the site selectivity for oxidative addition involving polyhalogented five-membered heteroarenes with one heteroatom (i.e., thiophene, pyrrole, and furan) does not necessarily correlate with relative BDEs.73 In fact, for both 2,4- and 2,3-dichlorinated thiophene, pyrrole, and furan, the BDEs for both C–Cl bonds (Figure 12) are very similar, if not nearly identical, which means that the experimentally observed C2/C5 selectivity is controlled by other factors that go beyond the relative bond strength.
Figure 12.
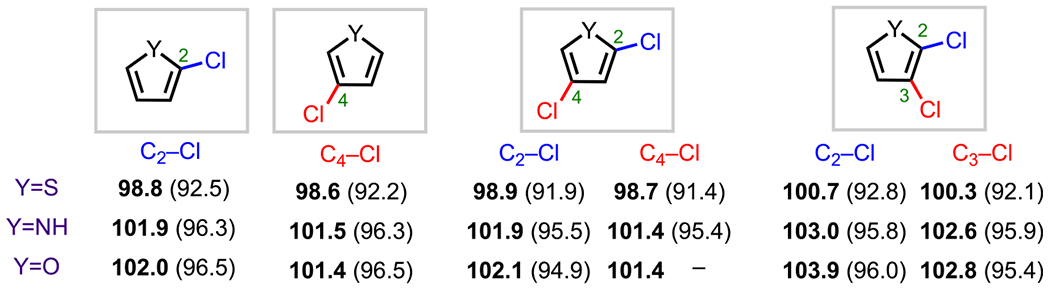
2,4- and 2,3-Thiophene, pyrrole, and furan C–Cl BDEs in kcal/mol using G3B3 (bold) and B3LYP (in parentheses).
A simple analysis of these systems using the Handy–Zhang57 method of assessing electronics based on 1H NMR chemical shifts shows that the C2/C5 positions for these systems are the most deshielded positions (i.e., most electron-deficient positions) relative to other positions, thus providing one rationalization as to why oxidative addition occurs preferentially at C2/C5 (Figure 13)41,96 Of course, this deshielding effect is more pronounced for thiophene and furan, as conveyed by the larger differences in 1H NMR chemical shifts relative to pyrrole, due to sulfur and oxygen being more electronegative relative to nitrogen; this greater electronegativity ultimately causes the inductive effect imparted by the heteroatom to be the dominant effect, which results in an overall dipole moment directed toward the heteroatom. For pyrrole, the nitrogen is still somewhat inductively electron-withdrawing from the C2 position; however, in contrast with thiophene and furan, the mesomeric effect in pyrrole dominates over the inductive effect, thus accounting for the π-excessive character of pyrrole and producing an overall dipole directed away from the nitrogen.96
Figure 13.
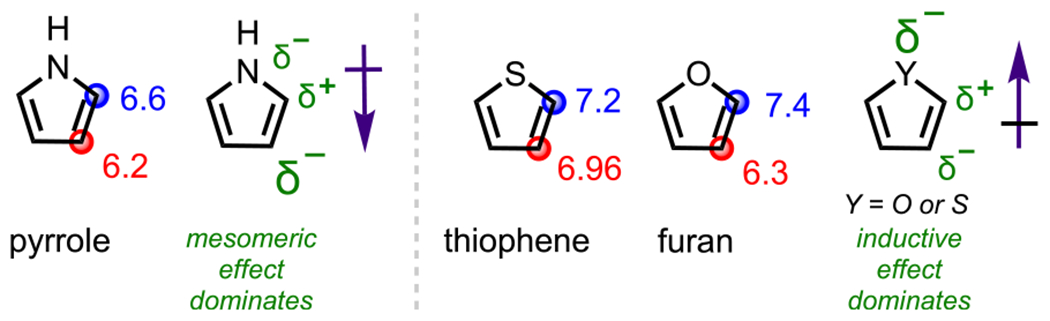
1H NMR chemical shifts (highlighted in blue and red) and major electronic effects for pyrrole, thiophene, and furan.
In addition to considering inductive effects, a second, and more comprehensive, rationalization for C2 selectivity can be ascertained using Houk’s distortion–interaction model. (See Figure 2.) In the case of 2,3-dibromofuran (63, Scheme 18A), the C2 and C3 positions have identical C–Br BDEs (88.9 kcal/mol). Because BDEs are related to the distortion energy, one can conclude, based on these identical BDEs, that the distortion energies for C2 and C3 oxidative addition do not play a prominent role in controlling the site selectivity. Instead, it is the interaction energy (ΔE‡int), the extent of stabilization from the interaction between the heterocycle π* LUMOs and the Pd dxy, HOMOs, that controls the site selectivity for oxidative addition; for the C2 position of 63, there is greater stabilization in the oxidative addition transition state and a lower activation barrier (lower ΔE‡) for oxidative addition, thus leading to C2 selectivity.73 Overall, the selectivity predicted by this model is in complete agreement with experimental results by Tang and coworkers, who showcased a Suzuki–Miyaura coupling between 63 and 4-methoxyphenylboronic acid, which provided the predicted C2-arylated product (64, Scheme 18B).97
Scheme 18.
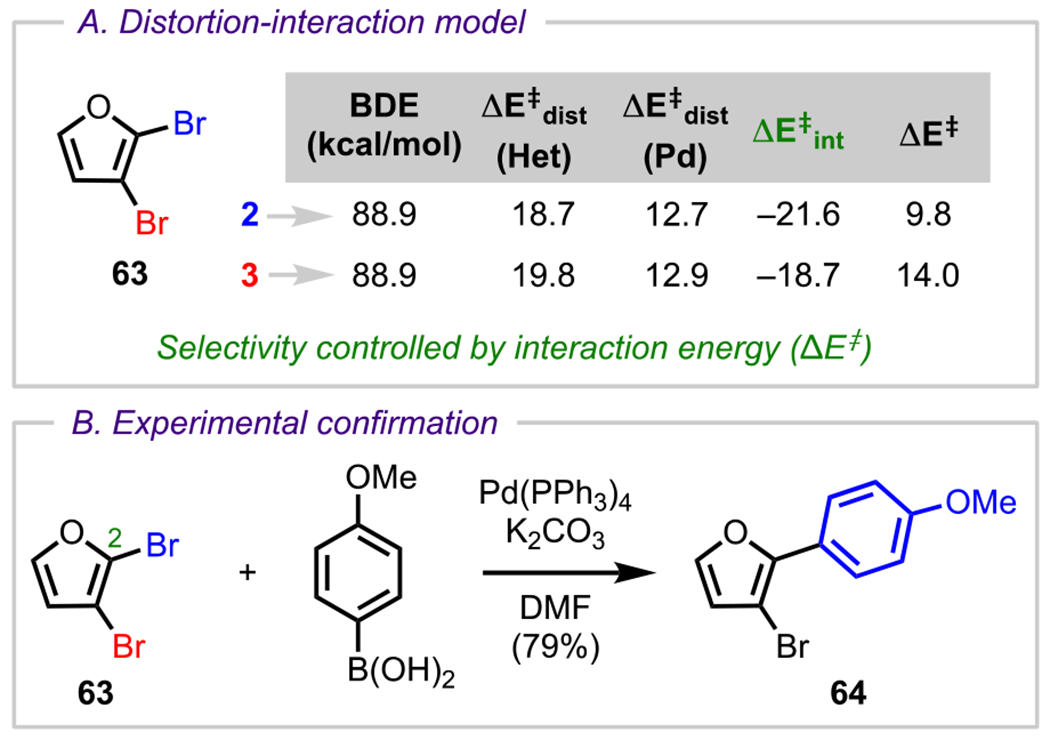
(A) Distortion–Interaction Analysis for the Oxidative Addition of a Model Pd(PH3)2 Catalyst into the C2 and C3 C–Br Bonds of 2,3-Dibromofuran and (B) Experimental Confirmation of Predicted Selectivity
This preference for C2/C5 has also been observed for polyhalogenated pyrroles. One such example is 3,4,5-tribromopyrrole-2-carboxylate (65, Scheme 19A), which has been shown to undergo C5-selective arylation to give various cross-coupled adducts (66a–g).98 Complementary to this study was additional work by Handy and Zhang, who demonstrated how electronic effects from substituents at the C2 and C5 positions can control cross-coupling selectivity at the C3 and C4 positions (Scheme 19B). Specifically, they showed how an electron-withdrawing ester moiety at the C2 position could facilitate C3 selectivity over C4 (due to the increased electrophilicity of C3 compared with C4).99
Scheme 19.
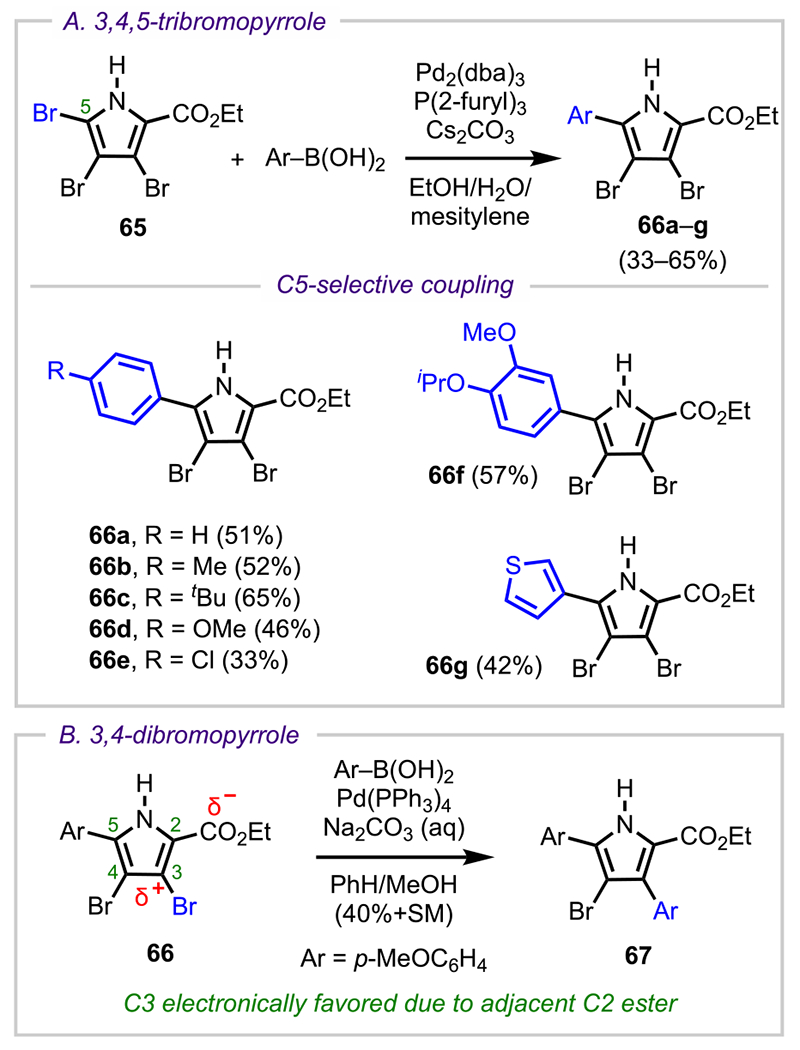
(A) C5- and (B) C3-Selective Arylation of Polybrominated Pyrroles
Finally, similar C2/C5-favored selectivity outcomes have also been reported for thiophene derivatives, such as 2,3,5,6-tetrabromothieno[3,2-b]thiophene (68, Scheme 20). For 68, Dang and coworkers reported a C2-selective Suzuki–Miyaura coupling with either one or two equivalents of various arylboronic acids to afford the mono- or bisarylated products (69 and 70).100
Scheme 20.
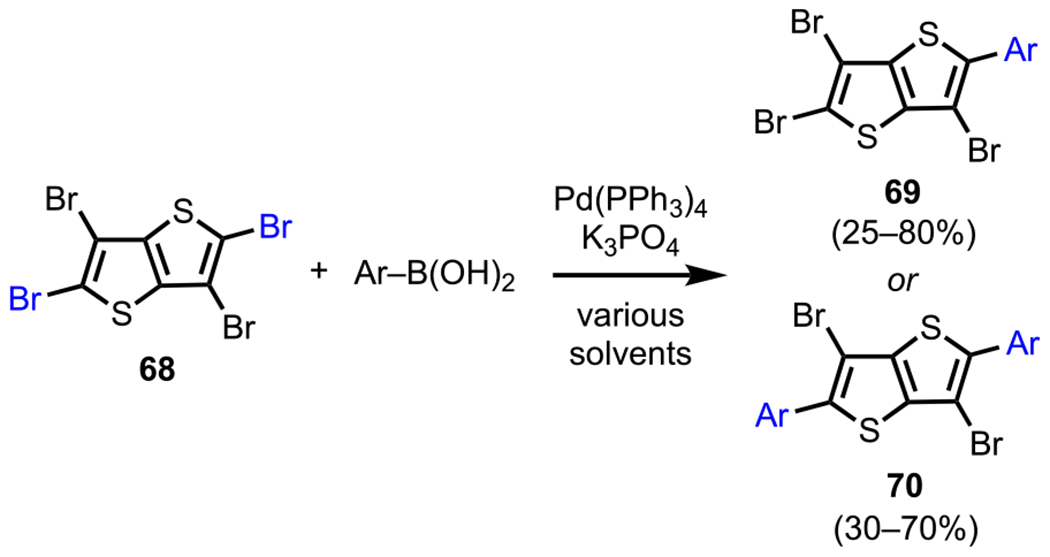
Site-Selective Mono/Bis-Arylation of 2,3,5,6-Tetrabromothieno[3,2-b]thiophene
3.3.2. 1,3-Azoles: Thiazole, Imidazole, and Oxazole.
Unlike the five-membered heteroarenes that contain one heteroatom (i.e., thiophene, pyrrole, and furan), 1,3-azoles (i.e., thiazole, imidazole, and oxazole) have BDEs that differ significantly among the various positions, especially the C2 position, which has the lowest relative BDE in both mono- and dichloro-1,3-azole systems (Figure 14) by virtue of being α to two heteroatoms.73
Figure 14.
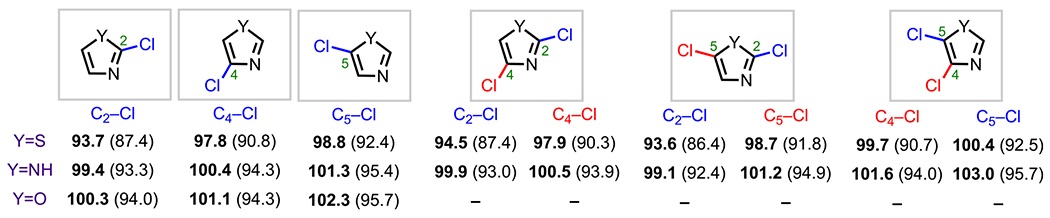
C–Cl BDEs in kcal/mol of mono- and dichloro-1,3-azoles using G3B3 (bold) and B3LYP (in parentheses).
For 2,4- and 2,5-dibromothiazoles (71 and 72, Scheme 21A), cross-coupling selectivity has been observed to parallel these BDE trends, with coupling occurring preferentially at the C2 position with various arylboronic acids to give both the 2-aryl-4-bromothiazole (74) and the 2-aryl-5-bromothiazole (75) products.101 Similar to dibromothiazoles, 2,4-dibromoimidazoles (76, Scheme 21B) also exhibit a preference for the doubly α C2-position and undergo monoarylation with diverse arylboronic acids to give the corresponding C2-arylated products (78).101
Scheme 21.
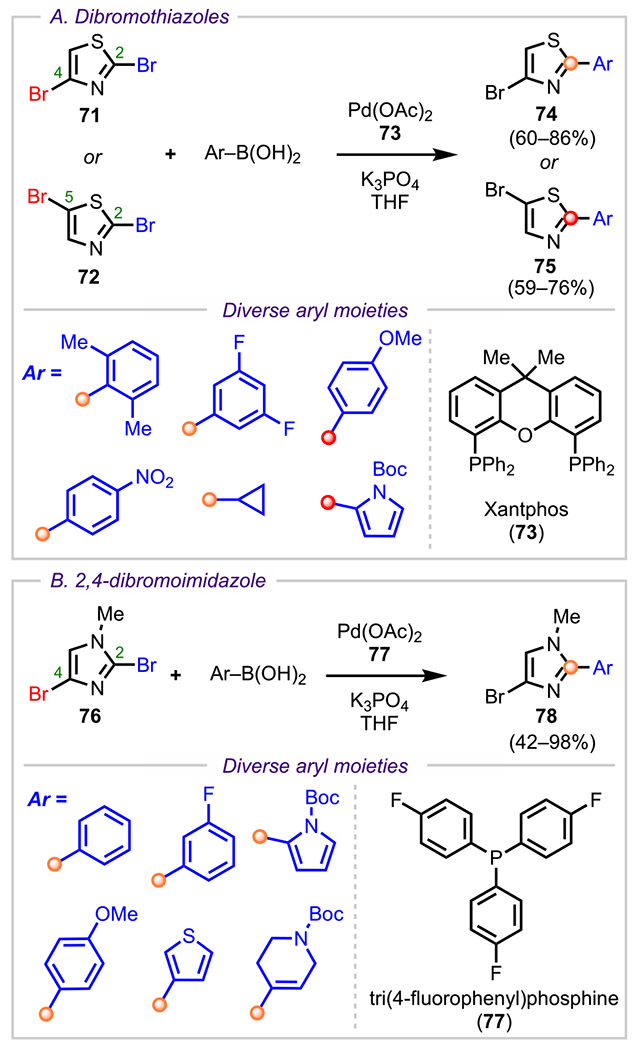
Selectivity Outcomes for (A) 2,4- and 2,5-Dibromothiazole and (B) N-Methyl-2,4-dibromoimidazole
Site-selective sequential Suzuki–Miyaura couplings of N-protected 2,4,5-tribromoimidazoles (79, Scheme 22A) have become a popular, effective strategy to access trisubstituted imidazoles (80), a common structural motif found in natural products with diverse biological activities ranging from anticancer to cell differentiation properties.102,103 In one study by Springer and coworkers, which focused on the synthesis of BRAF104 inhibitors, the 2,4,5-tribromoimidazole scaffold underwent sequential Suzuki–Miyaura couplings in the order C2 > C5 > C4, demonstrating the modularity of this strategy in accessing diverse trisubstituted imidazoles (Scheme 22A).102 Along similar lines, in their total synthesis of topsentin (Scheme 22B), Ohta and coworkers demonstrated a C5-selective Suzuki–Miyaura coupling between an N-SEM-protected-4,5-diiodoimidazole (81) and an indole boronic acid (82) to access the corresponding C5 coupled adduct (83).105 The reactivity pattern C2 > C5 > C4 is the same as that observed in metal–halogen exchange reactions,106,107 and whereas the preference for oxidative addition at C2 over C5/C4 can be explained just as before (α to two heteroatoms), the preference for oxidative addition at C5 over C4 that is observed in both of these studies can potentially be understood by comparing the stereoelectronic environments of both positions. Analogous to the work of Sames and coworkers on the site-selective palladation/arylation of N-protected imidazoles,108 the C4–Pd(II)L2X complex in this case may be less favored (relative to the alternative C5–Pd(II)L2X complex) due to destabilization by electron repulsion from the adjacent nitrogen lone pair at the three-position of the imidazole (highlighted in red; Scheme 22B).
Scheme 22.
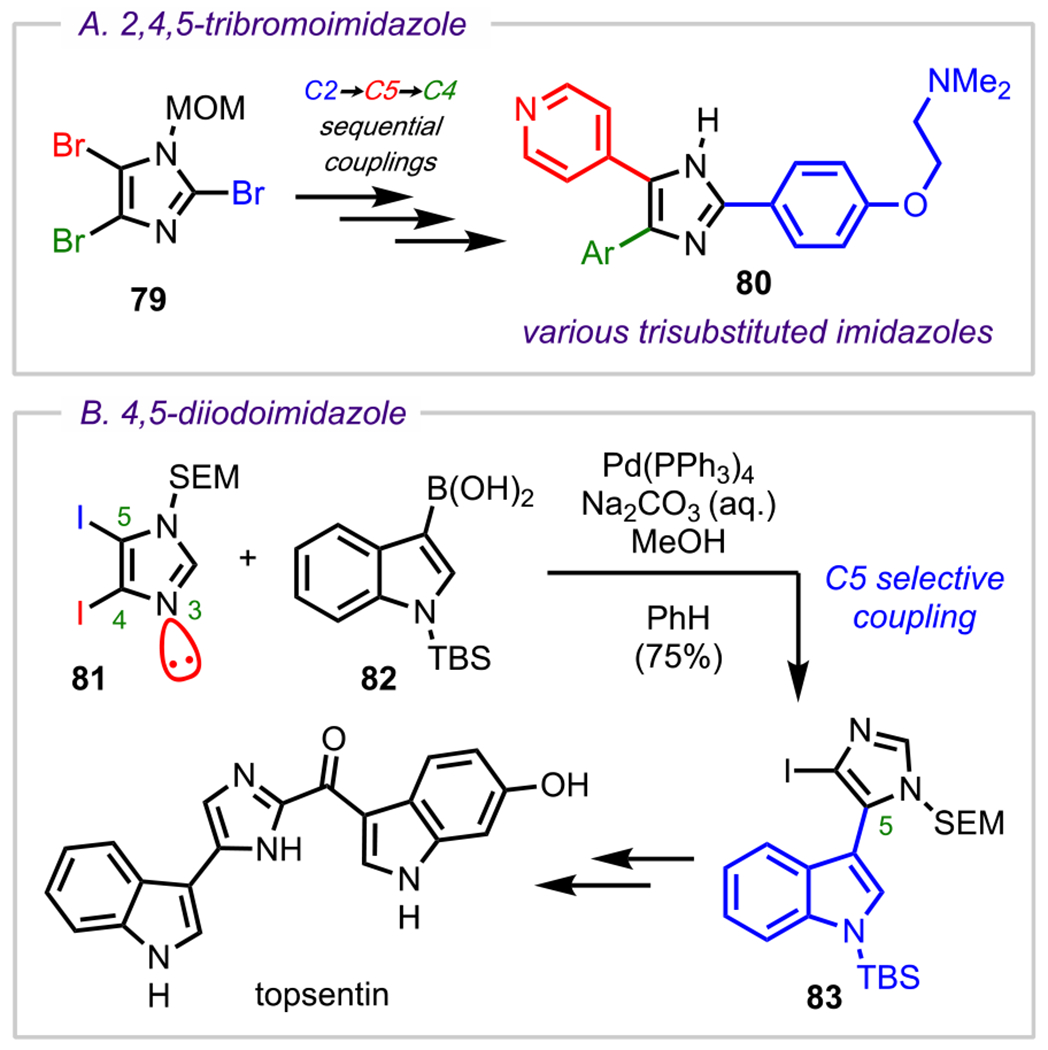
Selectivity Outcomes for (A) N-MOM-2,4,5-Tribromoimidazole and (B) N-SEM-4,5-Diiodoimidazole
It is important to note that the preference for C2 oxidative addition observed for many 1,3-azoles does not apply to all cases, as it has also been shown, namely, for N-methyl-2,5-dibromoimidazole (84, Figure 15) and 2,4-diiodooxazole (85), that selectivity can be reversed to favor C5 oxidative addition (for N-methyl-2,5-dibromoimidazole) or C4 oxidative addition (for 2,4-diiodooxazole) over C2 by simply altering the reaction conditions, specifically the metal/ligand complex (discussed in more detail in section 6; see Scheme 64).101 Finally, for ethyl 2,5-dibromooxazole-4-carboxylate (86), Hodgetts and coworkers have reported that no selectivity could be achieved using standard Suzuki–Miyaura conditions, with a complex mixture of products resulting instead.109
Figure 15.
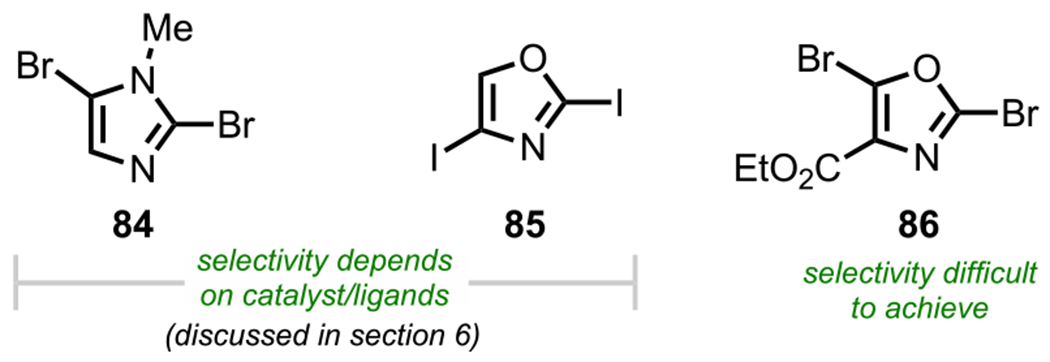
Selectivity outcomes for N-methyl-2,5-dibromoimidazole, 2,4-diiodooxazole, and ethyl 2,5-dibromooxazole-4-carboxylate.
Scheme 64.
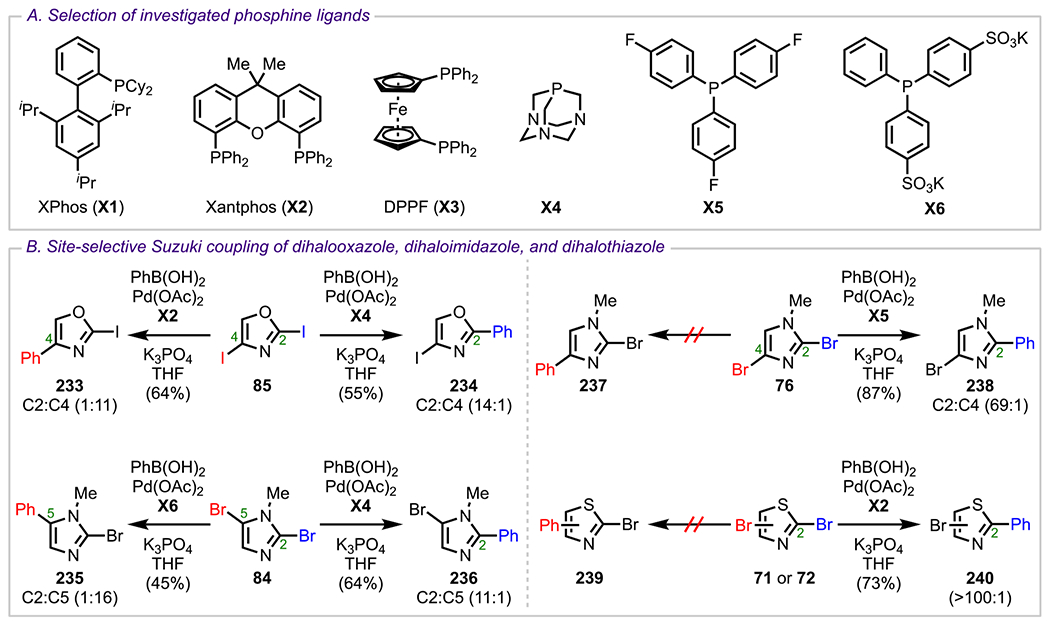
(A) Investigated Ligands and (B) Ligand-Controlled Site-Selective Suzuki–Miyaura Couplings of Various Dihaloazoles
3.3.3. 1,2-Azoles: Pyrazole and Isothiazole.
The last series of heteroarenes to be discussed in this section are the 1,2-azoles, specifically pyrazole and isothiazole. To the best of our knowledge, there are currently no reports of site-selective cross-couplings of polyhalogenated isoxazoles substituted with the same type of halogen. For pyrazoles and isothiazoles, recent studies convey that there is no direct correlation between BDE trends and cross-coupling site selectivity.110,111 On the basis of relative BDE values (Figure 16), one would expect that oxidative addition would occur first at the C3 position because C3 has the lowest relative BDE.73 However, for polyhalogenated pyrazoles and isothiazoles, cross-coupling has been shown to occur preferentially at the C5 position.110,111.
Figure 16.
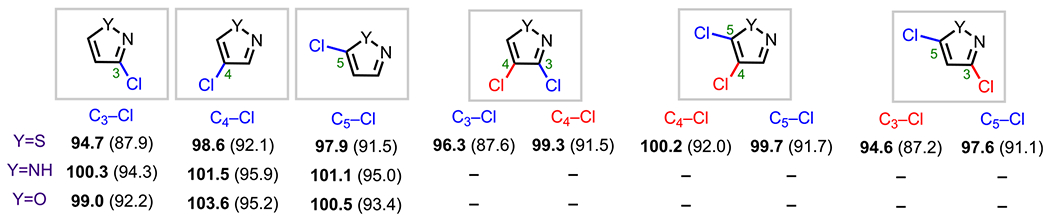
C–Cl BDEs in kcal/mol of mono- and dichloro-1,2-azoles using G3B3 (bold) and B3LYP (in parentheses).
N-Protected 3,4,5-tribromopyrazoles (87, Scheme 23) have been observed to undergo oxidative addition (and subsequent cross-coupling) in the order C5 > C3 > C4.31 This preference for C5 coupling has been demonstrated before by Langer and coworkers, who synthesized a variety of C5-arylated pyrazoles (88a–e) from the corresponding arylboronic acids.110 Cross-coupling selectivity for N-protected 3,4,5-tribromopyrazoles follows the same selectivity pattern observed in the lithium–halogen exchange reaction for the N-vinyl system, where lithiation occurs first at the C5 position.112 The preference for reactivity at C5/C3 over C4 arises from the inductive effect of the adjacent heteroatoms; as for the preference for C5 oxidative addition over C3, this may result from destabilizing electron repulsion at C3 by the adjacent nitrogen lone pair at C2, similar to the observed selectivity in imidazole systems.108 (See Scheme 22B.)
Scheme 23.
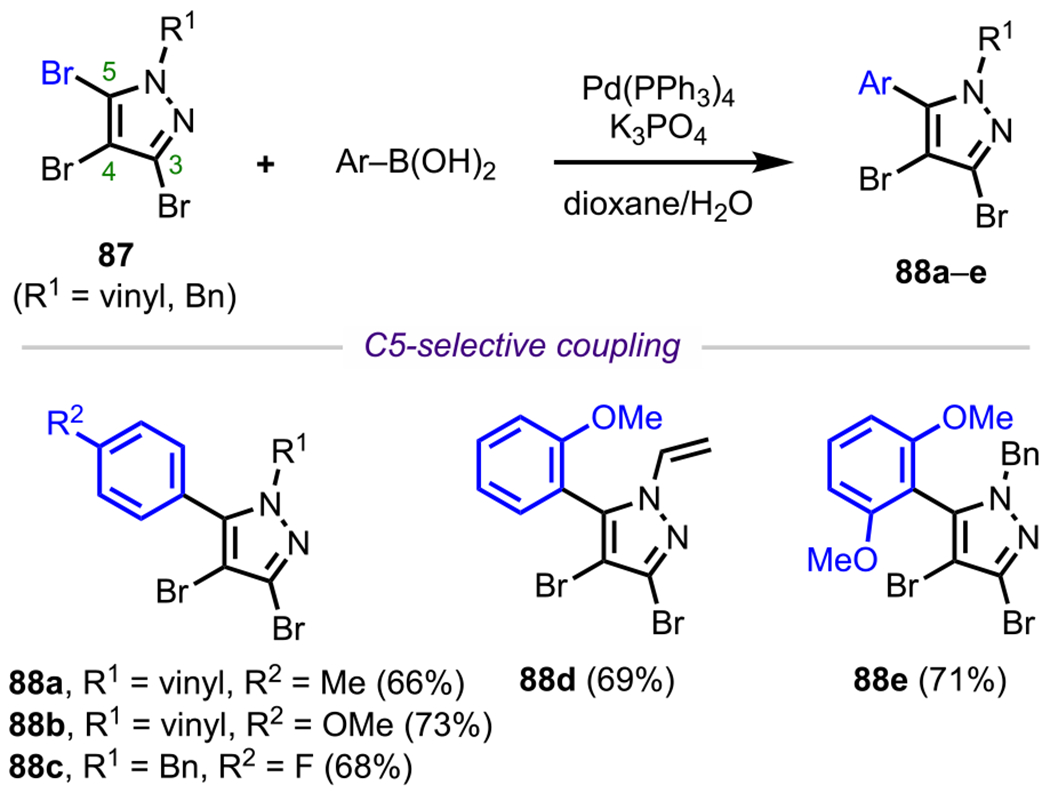
Site-Selective Coupling of N-Protected 3,4,5-Tribromoimidazole
In addition to pyrazole systems, the site-selective cross-coupling of isothiazoles such as 3,5-dichloroisothiazole-4-carbonitrile (89, Scheme 24) has been studied. These heterocycles are the basis for the observed antiviral properties of various derivatives.113,114 Therefore, the modular synthesis of these derivatives have garnered significant attention. Polyhalogenated isothiazole systems tend to undergo coupling at C5 followed by reaction at C3 and then C4, similar to pyrazoles.31 From their studies on site-selective Suzuki–Miyaura couplings between 3,5-dichloroisothiazole-4-carbonitrile (89) and one equivalent of various boronic acids, Koutentis and coworkers demonstrated that a series of diverse isothiazole products (90a–l) could be obtained following selective coupling at C5.111
Scheme 24.
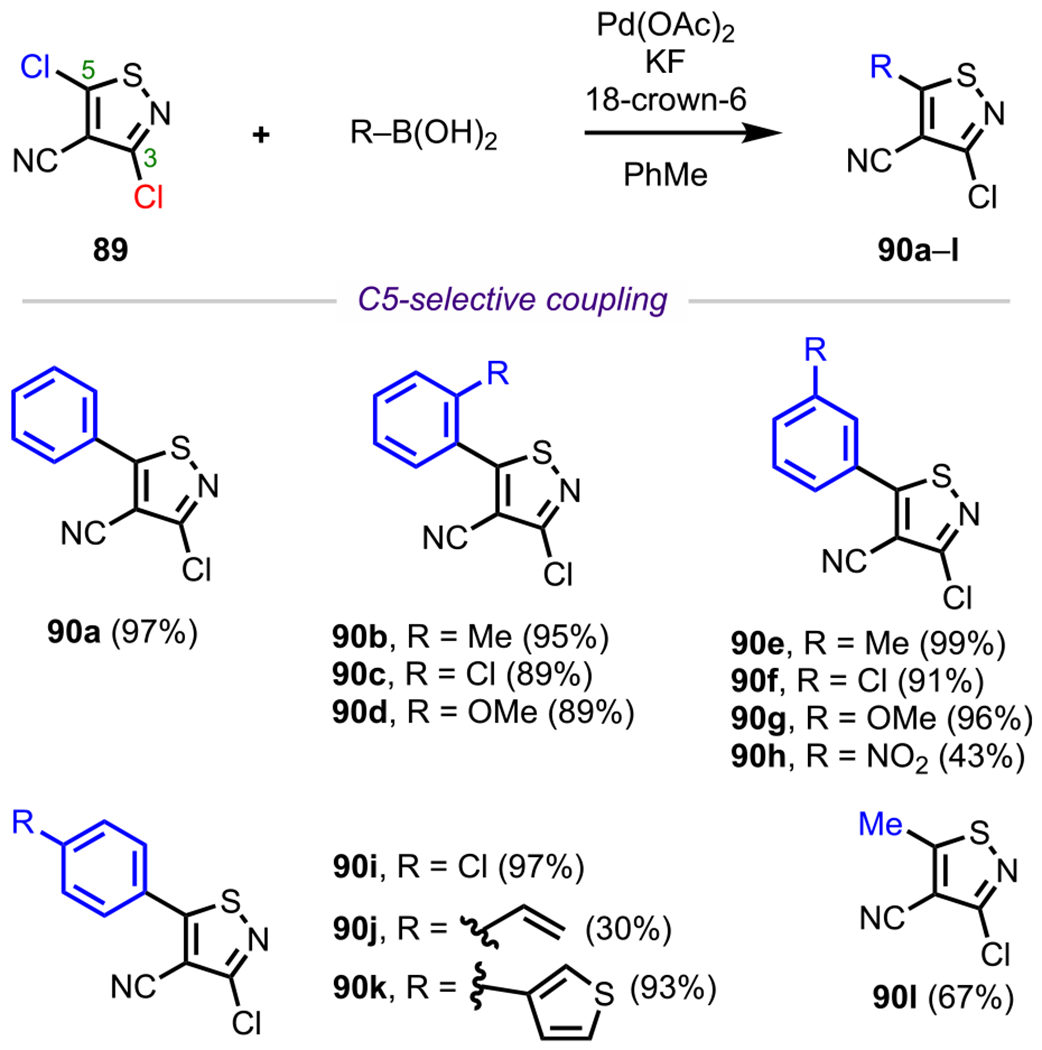
Site-Selective Coupling of 3,5-Dichlorothiazole-4-carbonitrile
4. STERIC CONTROL
Whereas the relative carbon electrophilicities in a (hetero)aromatic ring play a prominent role in oxidative addition selectivity, as conveyed in the previous section, one must also consider the steric environment and its effect on site selectivity (Scheme 25). Overall, selectivity is often governed by the interplay between electronics and sterics,31 with the most electrophilic and most sterically accessible position being primed for oxidative addition.115 However, it is not always the case that these two factors will be additive (or multiplicative). Examples discussed herein illustrate instances where the steric environment of the aryl halide guides selectivity in a number of possible scenarios, including: steric effects overriding innate electronics, steric effects differentiating between two electronically similar positions, and steric effects reinforcing the selectivity dictated by innate electronics. (Representative examples are shown in Scheme 26.) It should be noted that whereas the following examples focus on the intrinsic steric environment of the (hetero)arene, steric effects introduced by a metal complex can also play a primary role in governing selectivity;116 specific examples of this class of reactions are discussed in section 6 (Ligand Control).
Scheme 25.
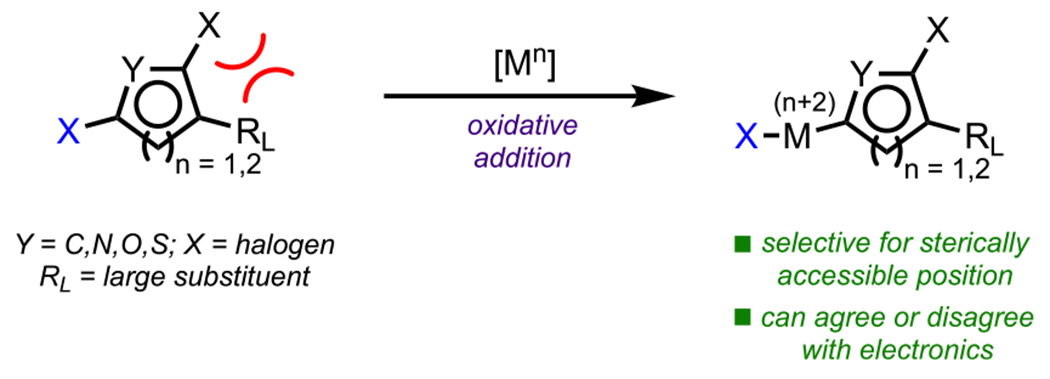
Steric Effects Guiding Cross-Coupling Selectivity
Scheme 26.
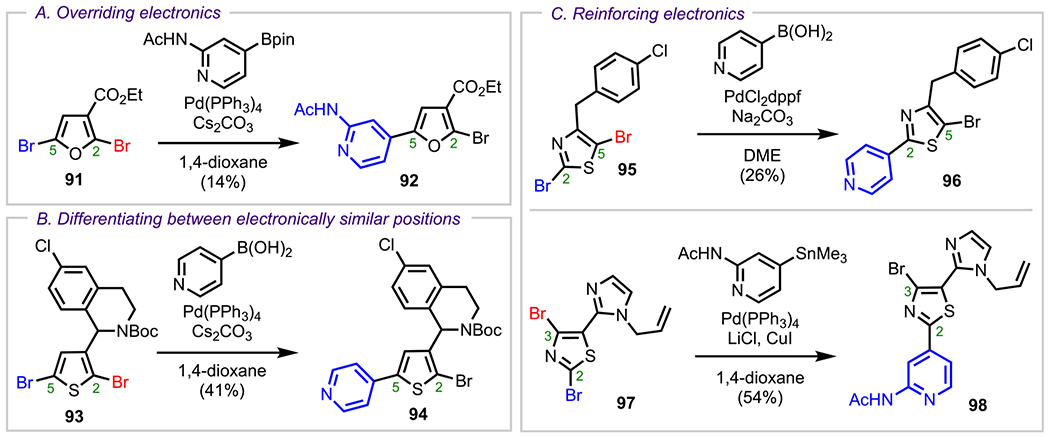
Representative Examples of Steric Effects: (A) Overriding Electronic Effects, (B) Differentiating between Electronically Similar Positions, and (C) Reinforcing Electronic Effects
One example of steric effects overriding electronic effects is shown in Scheme 26A, where coupling takes place at the more sterically accessible C5 position of 2,5-dibromo-3-furoate (91), despite the C2 position being electronically favored (i.e., most electron-deficient position) by virtue of being adjacent to both an oxygen atom and an electron-withdrawing ester moiety.117 In the case of 2,5-dibromothiophene 93 (Scheme 26B), there are no pronounced electronic effects from the pendant tetrahydroisoquinoline derivative.118 Therefore, the C2 and C5 positions are electronically similar, and in this case, the difference in the steric environments between these two positions facilitates reactivity at the more sterically accessible C5 position to give adduct 94. For both 95 → 96 and 97 → 98 (Scheme 26C), the C2 positions of thiazoles 95 and 97 are both electronically preferred over the C5 and C3 positions and are more sterically accessible as well. Therefore, sterics reinforce the innate electronics of these systems, and coupling occurs at the C2 position in both cases.118
Sterically controlled cross-coupling has also proven to be an effective strategy in total synthesis, as exemplified by the synthesis of the thiazolyl peptide antibiotic GE2270 A (102)119,120 by Bach and coworkers in 2007 (Scheme 27).121 Here an 11-step sequence (longest linear sequence) was established to access peptide 99, which features a key northern 2,6-dibromopyridine moiety poised to undergo sequential cross-couplings. The first coupling between peptide 99 and organozinc reagent 100 took place at the less sterically hindered C6 position (relative to the more sterically hindered C2 position) to install the northern C6 thiazole subunit. The target macrocycle was then forged downstream in the synthesis using an intramolecular Stille coupling at the remaining pyrido-C2 bromide.
Scheme 27.
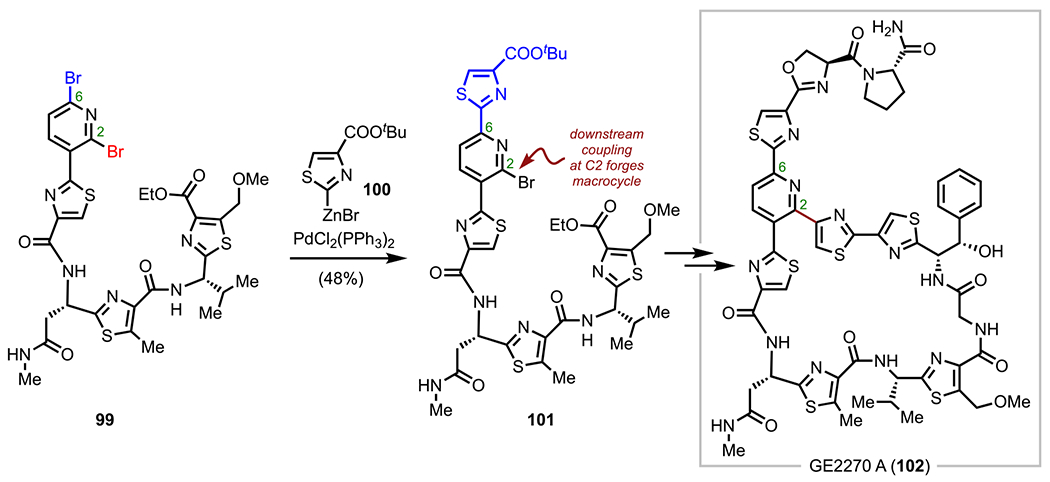
Sterically Controlled Coupling in the Total Synthesis of GE2270 A
Similar to Bach and coworkers’ synthesis of GE2270 A, Miller and coworkers also leveraged steric control in their syntheses of atropisomerically defined biaryl scaffolds through a sequential cross-coupling strategy (Scheme 28).122 In 2010, Miller and coworkers described a peptide-catalyzed enantioselective bromination of atropisomeric compounds to access enantioenriched biaryls, such as the acid and free-hydroxy variant of 103.123 In 2011, they developed a sequential coupling approach to advance these tribrominated biaryls to diverse, medicinally relevant polyaryl systems without a loss of enantiopurity.122 One representative example of this is shown in Scheme 28, where tribromide 103a first underwent selective coupling with a N-methyl-iminodiacetic acid (MIDA) boronate124 at the more sterically accessible C1 position to give 105. Coupling with a different boronate then occurred at the next sterically more accessible C5 position to give 106, which was followed by a final coupling at the least sterically accessible C3 position to give the final polyaryl scaffold (104a) in an enantioenriched fashion.
Scheme 28.
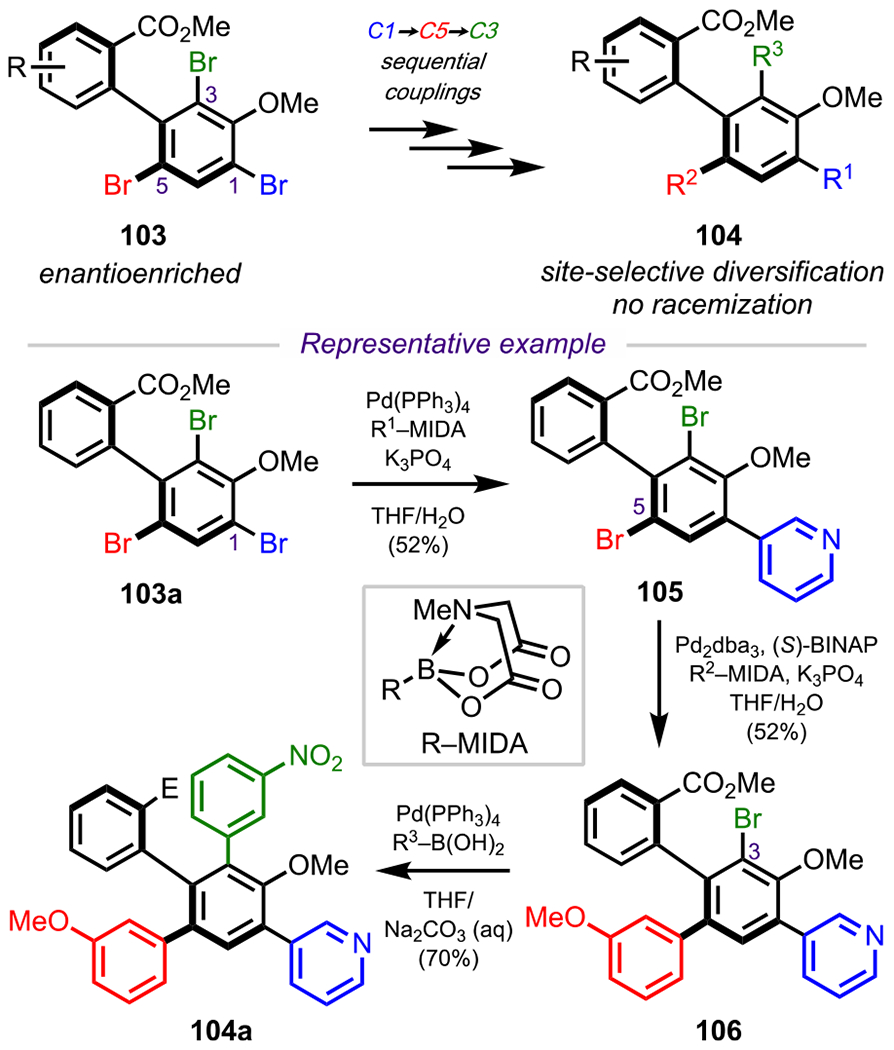
Synthesis of Highly Substituted, Enantioenriched Biaryls through Sequential Site-Selective Cross-Couplings
Finally, it has also been shown that halogen atoms can also have a significant steric influence in guiding site-selective couplings (Scheme 29).115,125 In a 2005 study by Nakamura and coworkers on Ni-catalyzed couplings of polyhalogenated arenes, it was shown that 1,2,3-trichlorobenzene (107) underwent coupling at the more sterically accessible C1 and C3 positions over the electronically favored but more sterically hindered C2 position.125 Likewise, a similar trend was observed by Al-Zoubi and coworkers in 2015 when a variety of 1,2,3-triiodobenzene derivatives (109) underwent coupling with arylboronic acids at the more sterically accessible C1/C3 position over the C2 position to give myriad C1-arylated adducts (110).115
Scheme 29.
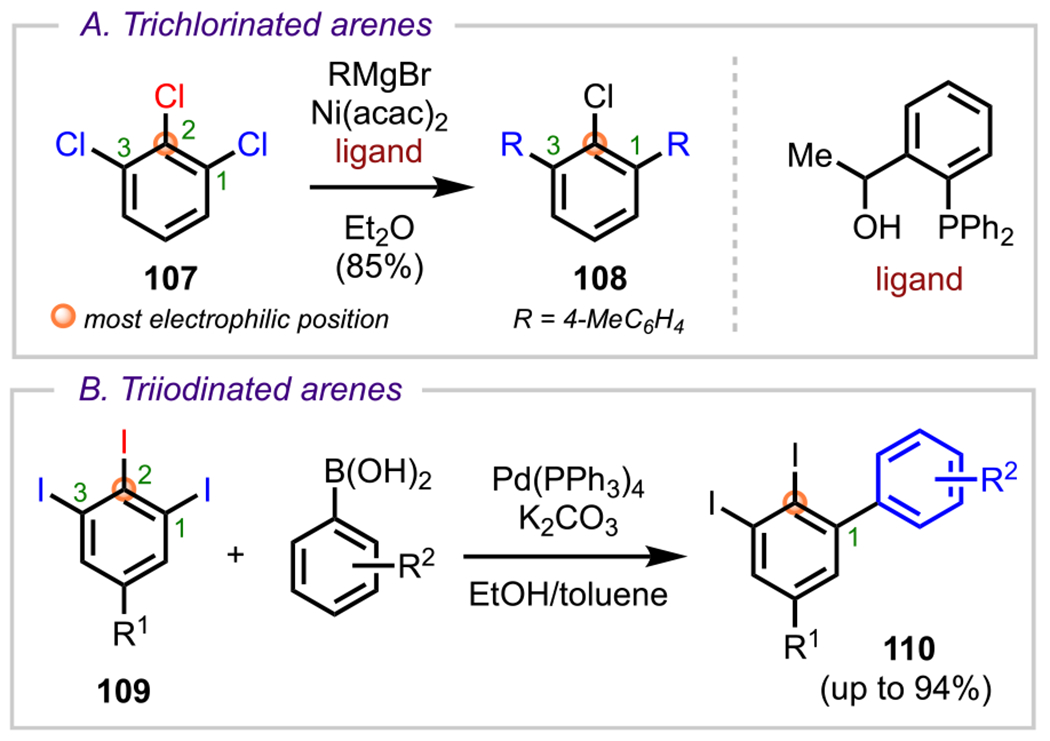
Sterically Controlled Cross-Coupling of (A) 1,2,3-Trichlorinated Benzene and (B) 1,2,3-Triiodinated Benzene
5. DIRECTING GROUP CONTROL
As discussed in the previous section, the steric influence of various groups can result in site selectivity, sometimes contrary to the outcome based solely on the difference in electron density at different carbons bearing an X group. However, for polyhalogenated arenes with electronically comparable C–X bonds and sterically less-pronounced substituents, a substrate-driven strategy becomes more challenging to execute. To obviate this challenge and achieve site-selective cross-couplings in such systems, synthetic chemists have often relied on a strategy that leverages the coordinating ability of a DG, a pendant functional group preinstalled on a molecule that can coordinate to a metal catalyst or ligand, to direct a metal catalyst to a proximal (or sometimes distal) C–X bond, thus enabling site-selective oxidative addition and subsequent functionalization.126 Historically, there have been two major classes of DGs employed in such processes: (1) transition-metal-binding DGs, functional groups on the substrate that coordinate directly to a metal (Scheme 30A), and (2) ligand-binding DGs, functional groups on the substrate that coordinate to a ligand that is, in turn, bound to a transition-metal complex, thus linking the substrate and catalyst (Scheme 30B). Both classes of DGs have proven effective in facilitating site-selective oxidative addition, with transition-metal-binding DGs being optimal for proximal C–X bond cleavage, whereas ligand-binding DGs are applicable in both proximal and distal C–X bond cleavage.
Scheme 30.
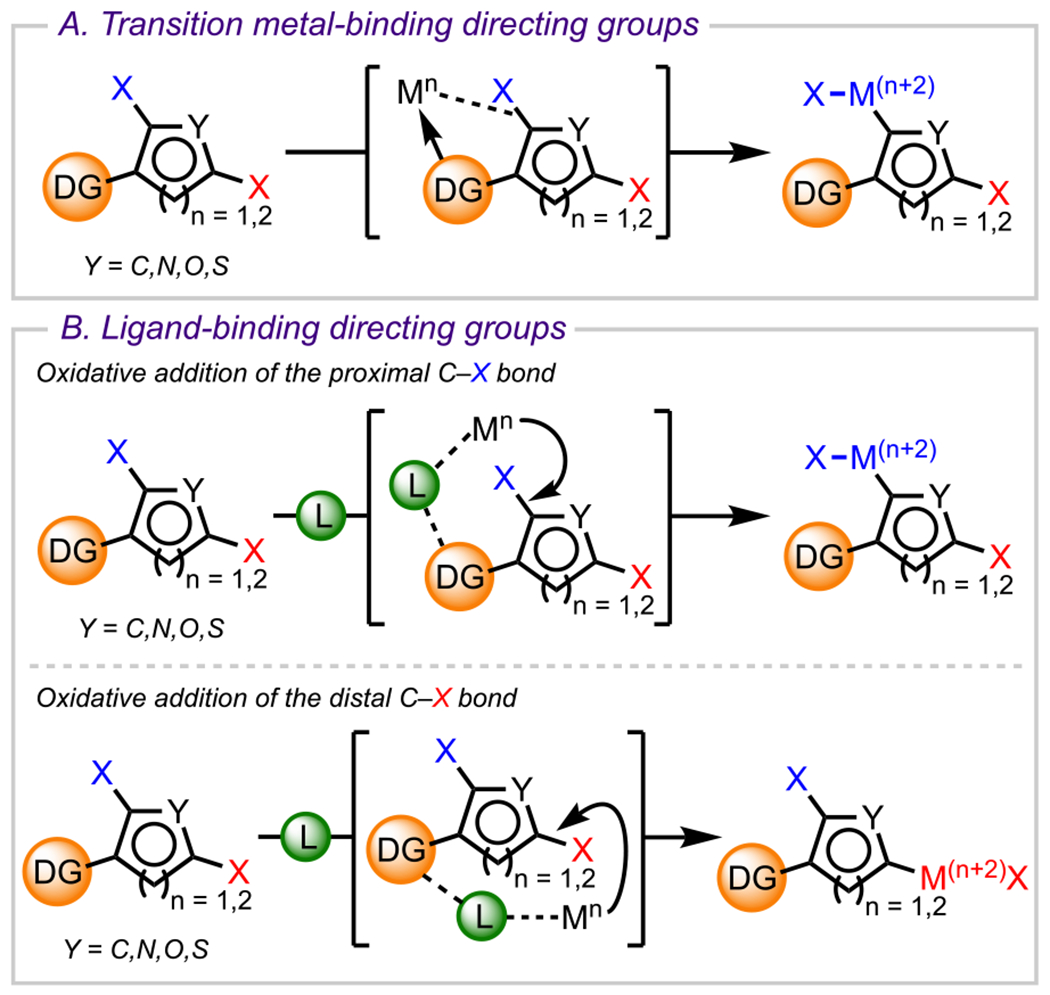
General Strategy of Achieving Selective Coupling by Using Directing Groups That Either (A) Bind Directly to a Metal Center (Metal-Binding Directing Groups) or (B) Bind to a Ligand (Ligand-Binding Directing Groups)
Whereas DGs can be incredibly effective for enabling site selectivity, employing this strategy is often reserved for functional groups that either are ubiquitous in natural or commercially available compounds or do not negatively impact the step count and atom economy in a synthesis. Along these lines, one must be mindful of DGs that need to be removed in a later stage in a synthesis, which can ultimately inflate the step count and reduce the synthetic efficiency. Overall, these considerations have resulted in a collection of diverse functional groups, which have proven to be suitable DGs.126 DG-controlled site-selective cross-coupling in polyhalogenated arenes first gained momentum in the early 2000s and has since experienced steady growth, as exemplified by several recent publications, many of which are discussed in this Review. In this section, we have categorized a wide array of examples based on the specific DG facilitating site-selective cross-coupling. Here the discussion focuses first on examples employing substrates with DGs that bind directly to a transition metal (section 5.1), which is then followed by examples employing DGs that interact with a ligand that links the substrate to a transition-metal complex (section 5.2).
5.1. Transition-Metal-Binding Directing Groups
The first category of DGs that we discuss here are those that reside on the substrate and have been shown to coordinate directly to the metal, thus enabling site-selective oxidative addition at a proximal C–X bond. This section is organized based on the specific DG employed. These are often ubiquitous functional groups with coordinating capabilities (e.g., amines, hydroxy groups, carboxylic acids, ketones, amides, etc.).
5.1.1. Amine.
An early example of using a DG to facilitate site-selective cross-coupling was reported by Nakamura and coworkers in 1995.127 Their work entailed synthesizing 2-amino-3,5-disubstituted pyrazines en route to bioluminescent and chemiluminescent compounds such as coelenterazine128 and analogues thereof. To develop a concise and convergent synthetic route, they chose to commence their synthesis from 2-amino-3,5-dibromo pyrazine (111). They initially adopted a sequential Pd-mediated Stille coupling strategy to access disubstituted pyrazines by using excess stannane. Interestingly, when an equimolar mixture of stannane and 111 was treated under the same reaction conditions, they mostly observed the formation of monocoupled pyrazines (112) substituted at the three-position, presumably directed by the nearby primary amino group (Scheme 31). This exceptional selectivity enabled them to introduce two different substituents on the 2-aminopyrazines. From these monosubstituted pyrazines (112), a second Stille coupling at C5 was effected with a different stannane to furnish disubstituted pyrazines (113). Finally, condensation with ketoaldehyde derivative 114 enabled access to a diverse range of coelenterazine analogues (115a–c, Scheme 31).129
Following this initial report, several groups attempted to utilize the coordination ability of amines to selectively cleave a proximal C–X bond. Toward this end, Hikawa and Yokoyama presented an interesting example where they investigated the possibility of achieving the site-selective Suzuki–Miyaura coupling of N-(3,5-dibromo-2-pyridyl)piperazine 116 by coordinating the Pd complex to the tertiary piperazine nitrogen atom and directing it to the nearby C–Br bond (Scheme 32).130 In their study, they analyzed the influence of several palladium catalyst precursors and solvents on the selectivity outcome. After extensive screening, they found that employing PdCl2(dppf) as the precatalyst, KF as the base, and tetrahydrofuran (THF) as the solvent facilitated the site-selective Suzuki–Miyaura coupling between 116 and methylboronic acid to furnish the monomethylated derivative 117 in 71% yield. Eventually, they extended the scope of this reaction to other boronic acids to access a wide variety of monosubstituted products. In addition to primary (Scheme 31) and tertiary (Scheme 32) amines, secondary amines (not shown) were also identified as suitable DGs.131
Scheme 32.
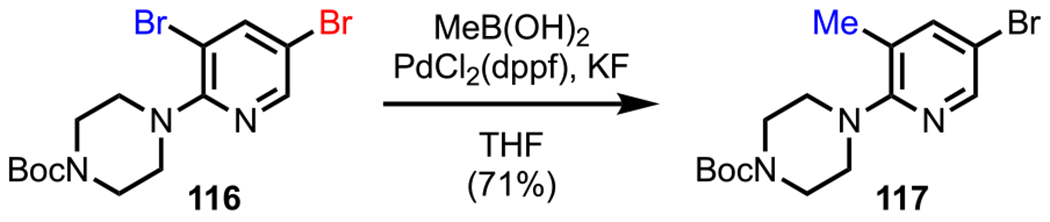
Site-Selective Suzuki–Miyaura Coupling of N-(3,5-Dibromo-2-pyridyl)piperazine 116
More recently, amines have also been used as DGs to achieve site-selective C–F bond activation in difluorinated systems. As discussed in previous examples (section 3.1, Schemes 6 and 7), the selective functionalization of C–F bonds through direct C–F bond activation is of high significance. However, known methods for achieving this transformation are mainly limited to palladium and nickel catalysis. Whereas great effort has been devoted to the development and identification of highly effective phosphine and NHC ligands for Pd- and Ni-catalyzed C–F bond activation, there is a necessity to develop novel, alternative transition-metal-catalyzed approaches to enable the activation of these highly stable bonds.132–134,64 Toward this end, Duan and coworkers reported a cobalt-catalyzed biaryl coupling reaction between aryl fluorides and aryl Grignard reagents in the presence of Ti(OEt)4 (Scheme 33).135 In the case of difluorinated systems, such as 118, the authors identified the free amino group to be an effective ortho DG, exclusively furnishing the monoarylated product (120) in high yield (Scheme 33). Overall, these examples validate the versatility of using amino groups as a DG to facilitate site-selective cross-couplings under various transition-metal-catalyzed conditions.
Scheme 33.
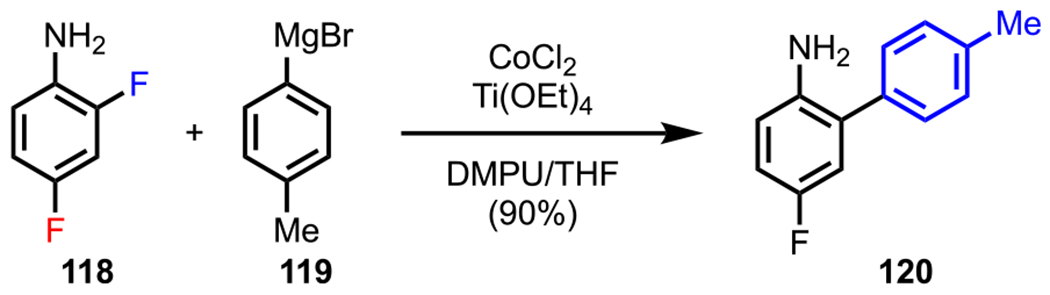
Amine-Directed C–F Bond Activation under Cobalt-Catalyzed Conditions
5.1.2. Hydroxy Group.
Synthetic chemists have also explored the potential of other ubiquitous functional groups as DGs. Hydroxy groups have attracted particular attention. Much effort has been invested to effect the ortho functionalizations of polyhalogenated phenol and benzylic alcohol derivatives by tuning the coordinating ability of the hydroxy group. It is important to note that whereas hydroxy groups may serve as DGs (similar to amines in aniline derivatives), the electron-donating properties of hydroxy groups may result in the deactivation of the ortho position from both an electronic and a steric standpoint. To circumvent this inherent challenge, Manabe and coworkers developed a novel methodology to enable ortho-selective Kumada coupling in difluorinated phenol derivatives by using the nearby hydroxy group as a DG.136 After screening several catalyst precursors, PdCl2(PCy3)2 was identified as the most suitable precatalyst for this transformation. As an example, difluorinated phenol 121 and Grignard reagent 122 underwent ortho-selective Kumada coupling to provide the biaryl product (123) in good yield (Scheme 34). In addition to hydroxy groups, hydroxymethyl groups also worked well to give the ortho-substituted product, where difluorinated benzylic alcohol 124 and the corresponding Grignard reagent (125) gave the coupled adduct 126. Importantly, Manabe and coworkers found that a protic DG was necessary to effect the desired Kumada coupling, as the use of a methyl ether led to unsatisfactory selectivities.136
Scheme 34.
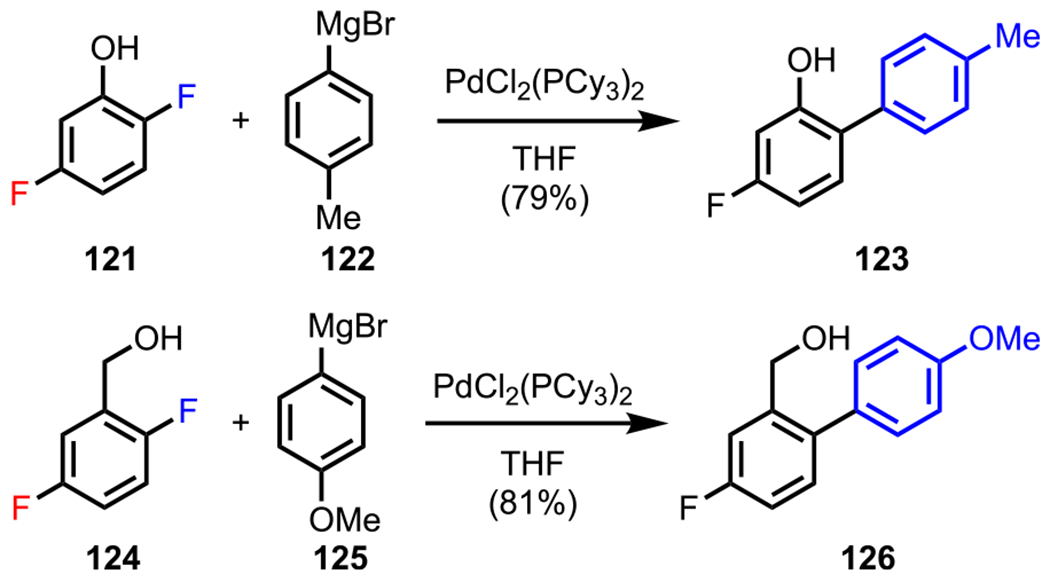
ortho-Selective Kumada Coupling of Difluorinated Phenol and Benzylic Alcohol Derivatives
In most examples of DG-assisted site-selective cross-coupling, the identification of the most suitable catalyst precursors is of utmost importance. In this regard, Ackermann and coworkers observed that palladium complexes generated from air-stable heteroatom-substituted137,138 secondary phosphine oxide (HASPO) preligands139–141 could be used to achieve highly ortho-selective Kumada coupling involving dichloroarenes.142 Although originally developed for Kumada couplings with aryl and alkenyl tosylates,143 the scope of this methodology was later extended to aryl chlorides144–146 as well. Specifically, they prepared a well-defined palladium complex (128) upon treating 4 equiv of an inexpensive HASPO preligand (127) with PdCl2(PhCN)2 (Scheme 35). The generated HASPO complex (128), which presumably interacts with the phenolic group by hydrogen bonding, was then used to accomplish the hydroxy-group-directed site-selective Kumada coupling of 2,4-dichlorophenol with a range of aryl Grignard reagents.142
Scheme 35.
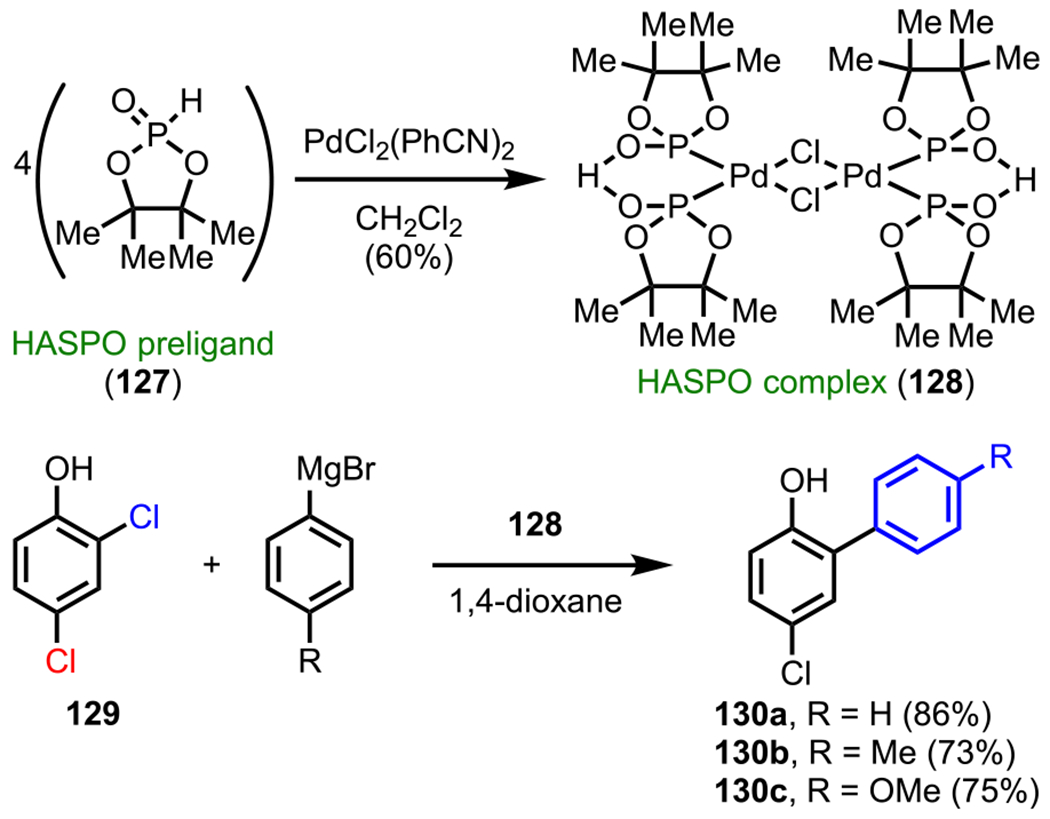
Site-Selective Kumada Coupling of 2,4-Dichlorophenol with the HASPO Complex
5.1.3. Carboxylic Acid.
Although carboxylic acids are ubiquitous functional groups, exploration into their potential as DGs only began in the late 2000s.147–149 In addition to their abundance, an added advantage of employing carboxylic acids as DGs is their versatility; they can be easily removed150 or further diversified.151,152 However, carboxylates are known to coordinate weakly to catalysts and are therefore often disfavored as DGs in the presence of other strongly coordinating DGs (e.g., pyridines, amides, etc.). In addition, under transition-metal catalysis, carboxylates can also undergo undesired side reactions (e.g., hydrodecarboxylation),153 which are sometimes challenging to avoid. Despite these challenges, several groups have devised innovative strategies to leverage carboxylic acids as effective DGs in myriad cross-coupling processes.
Contemporaneous with Yu and coworkers’ report on using sodium carboxylate as a DG in C–H activation methodology,148 Houpis and coworkers devised a strategy to use lithium carboxylate as a DG in coupling reactions of brominated benzoic acid derivatives (Scheme 36).154 During this study, initial attempts focused on coupling 2,5-dibromobenzoic acid (131b) to a dioxolane Grignard reagent. However, under almost all of the conditions that were investigated, they predominantly observed the formation of the undesired styrene derivative (133). Following extensive experimentation, the authors postulated that after an initial oxidative addition to furnish the magnesium salt of trans-Pd(II) species (133a), an undesired β-alkoxide elimination occurred to give enol ether complex 133b. This was followed by an intramolecular migratory insertion to provide 133c, which then underwent a second β-alkoxide elimination to form the styrene derivative (133). To outcompete this undesired pathway, they envisioned promoting the formation of the cis-Pd(II) intermediate after oxidative addition, which was then expected to undergo facile reductive elimination to furnish 132. Toward this end, they anticipated beginning with a more nucleophilic carboxylate salt, which would favor the formation of a cis-Pd(II) intermediate through stronger coordination of the carboxylate group to the Pd center. This was ultimately realized by commencing with lithium salt 131a, where the desired product (132) was isolated in 78% yield, and the undesired styrene derivative was limited to trace quantities.154
Scheme 36.
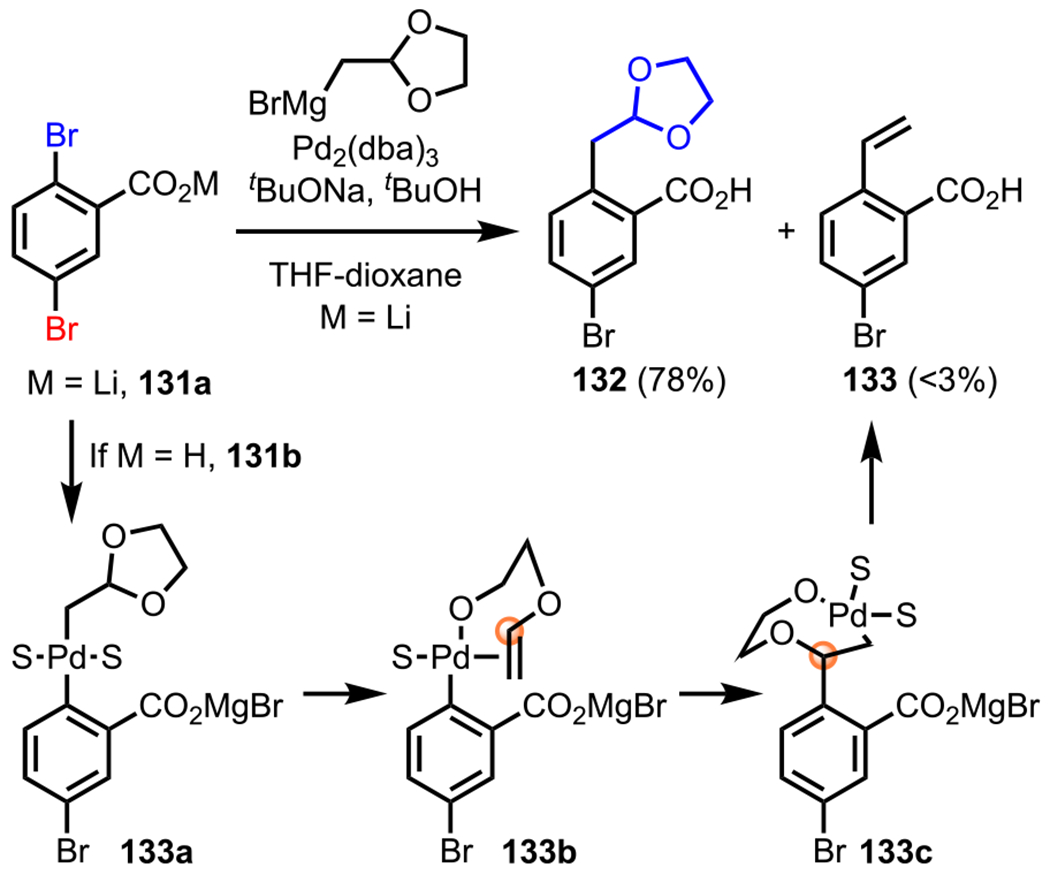
ortho-Selective Kumada Coupling with the Lithium Salt of 2,5-Dibromobenzoic Acid
Following this novel finding, the authors investigated the possibility of applying this concept to the widely used Suzuki–Miyaura coupling reaction.155 They commenced their study with 2,4-dibromobenzoic acid (134) and p-tolylboronic acid (Scheme 37). After a thorough investigation, they concluded that the choice of the Pd precatalyst and the equivalents of an appropriate base were crucial for the carboxylate-assisted ortho-selective Suzuki–Miyaura coupling of 134. Eventually, they found that using Pd2(dba)3 as the precatalyst in the presence of 2.2 equiv of LiOH furnished the desired product (135) with high selectivity, presumably via intermediate 135a. Furthermore, carbonates were also identified as appropriate bases when the equivalents remained unchanged. In an attempt to reverse the selectivity, the authors chose to add an external phosphine ligand: bis[(2-diphenylphosphino)phenyl] ether (DPEPhos). They hypothesized that in the presence of this phosphine ligand, the coordination between the carboxylate and the Pd metal center would be disrupted, and consequently, the bulky Pd(0)Ln species would be oxidatively added into the less sterically hindered para C–Br bond to generate intermediate 136a. Upon adding DPEPhos to the reaction mixture, the para-coupled product (136) was isolated in 68% yield.
Scheme 37.
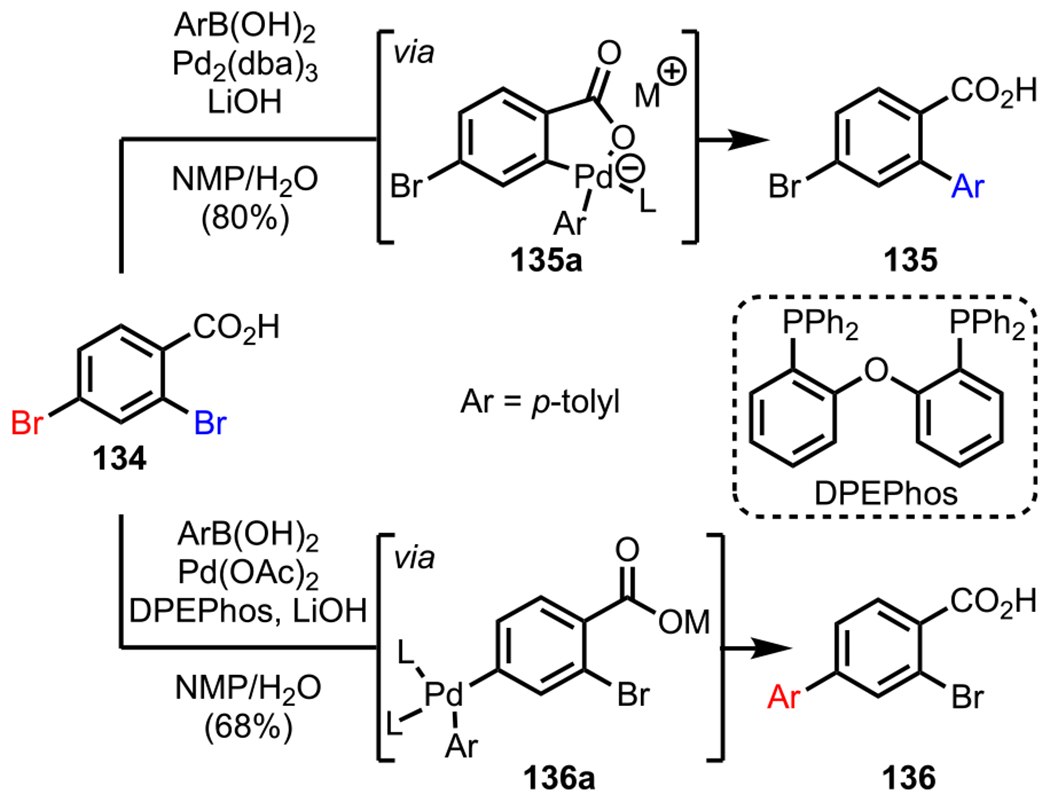
Carboxylate-Directed Site-Selective Suzuki–Miyaura Coupling of 2,4-Dibromobenzoic Acid
Houpis and coworkers then attempted to extend the scope of the coordinating ability of the carboxylate group to other transformations such as Pd-catalyzed amination.156 Their study included coupling reactions between 2,4-dibromobenzoic acid 134 and various electronically and sterically diverse amines (Scheme 38A). They observed that under the previously disclosed conditions to obtain para selectivity, they isolated the expected coupled product (137) when benzylamine was treated with 134. However, they were unable to reverse the selectivity under all of the conditions that they investigated, even when the phosphine ligand was excluded. They then examined other transition-metal complexes to achieve ortho selectivity. Taking inspiration from Ullmann and Goldberg’s pioneering work157 on Cu-catalyzed amination reactions with halobenzoic acids, Houpis and coworkers turned their attention toward investigating Cu-catalyzed conditions. They were also inspired by very similar work by Wolf and coworkers where an ortho-selective amination was achieved with both dichlorobenzoic acid 139 and dibromobenzoic acid 131b when treated with phenethylamine in the presence of Cu/Cu2O to furnish 2-aminobenzoic acid derivatives, 140 and 141, respectively (Scheme 38B).158 Eventually, Houpis and coworkers were also able to couple benzylamine to the ortho position of 134 in the presence of Cu2O to give the desired 2-aminobenzoic derivative 138 in 91% yield (Scheme 38A).
Scheme 38.
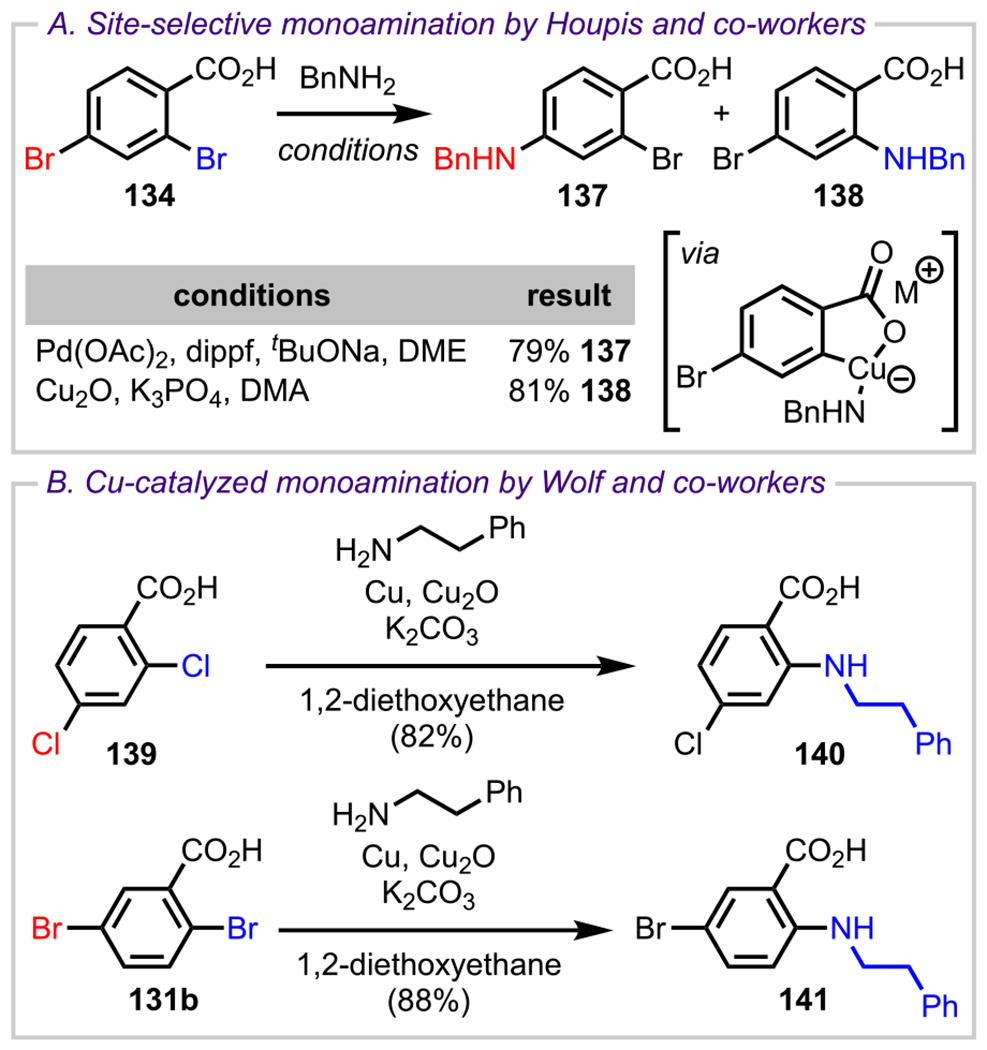
(A) Complementary Selectivity Observed under Pd- and Cu-Catalyzed Conditions and (B) Site-Selective Monoamination under Cu-Catalyzed Conditions
The versatility of using a carboxylate as a DG was also extended to difluorobenzoic acid derivatives. In addition to exploring an amine as a DG in difluorinated systems (Scheme 33), Duan and coworkers also investigated the possibility of using a carboxylate as a DG in difluorobenzoic acids under Co-catalyzed conditions.135 Gratifyingly, they observed that coupling between 142 and Grignard reagent 143 in the presence of CoCl2 furnished the ortho-coupled product (144) in 81% yield (Scheme 39).
Scheme 39.
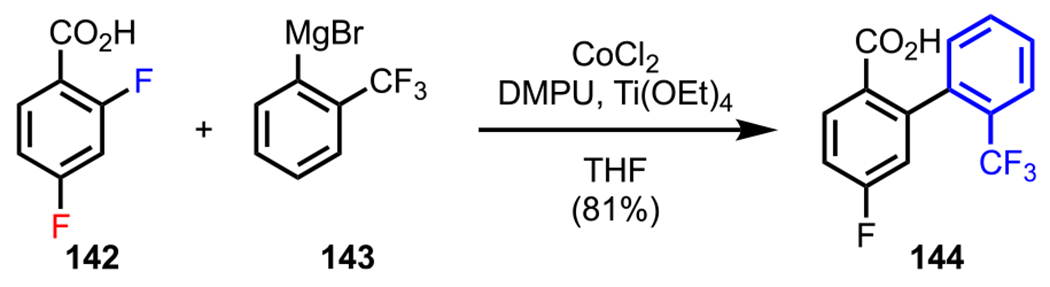
ortho-Selective Kumada Coupling of Difluorobenzoic Acid
5.1.4. Ketone and Imine.
The ketone carbonyl group was one of the first DGs to be used in site-selective coupling chemistry.159 New contributions continue to be made to expanding the scope of the coordination ability of the carbonyl group. In 2014, Wang and coworkers reported a coupling between aryl fluorides and organozinc reagents under Ni-catalyzed conditions.160 When polyfluoroarenes were used as substrates, they introduced a suitable DG to avoid multiple substitutions as a result of ring-walking.161 As shown in Scheme 40, they showed that the installation of a benzoyl group [PhC(O)] enabled the selective functionalization of difluoroarene 145 in the cross-coupling with various organozinc reagents to favor the formation of the ortho-substituted product (146). In addition to this study, Kakiuchi and coworkers also showed that ketones can be employed as DGs to achieve the ortho-selective arylation of polyfluoroarenes with several arylboron reagents under Ru-catalyzed conditions (not shown).162
Scheme 40.
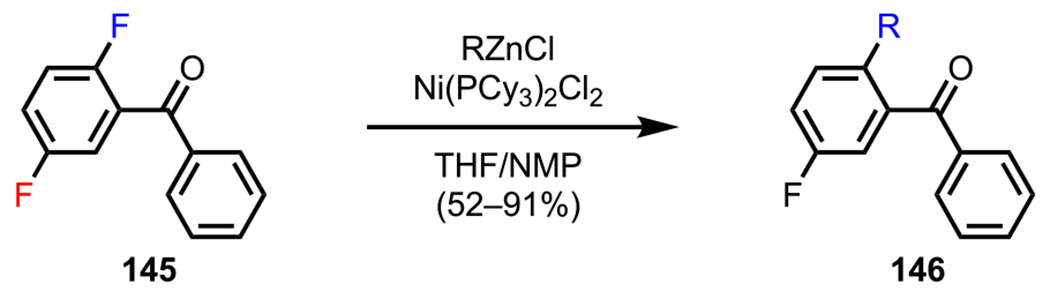
ortho-Selective Functionalization of Difluoroarene 145
Similar to ketones, imines have also been widely employed as DGs in recent years. One of the main approaches has been to leverage the “transient” directing ability of imines, that is, to install an imine for a directed site-selective functionalization with subsequent cleavage of the imine group, which ideally would all occur in a single pot. Along these lines, Love and coworkers devised several novel methodologies to achieve the ortho functionalization of polyfluoroarenes. Taking inspiration from work by Crespo and Martinez,163 who achieved stoichiometric C–F activation in a series of polyfluoroaryl imines in the presence of [Me2Pt(μ-SMe2)]2,164 the Love group investigated the possibility of performing this transformation in a catalytic process. Toward this end, they found that by using [Me2Pt(μ-SMe2)]2 as the catalyst with substoichiometric loading of Me2Zn as the methylating agent, the ortho-methylation of a variety of polyfluoroaryl imines (147) could be achieved to furnish 148 (Scheme 41).165–168 Additionally, they extended the scope of this methodology to achieve monomethoxylation in polyfluoroaryl imines by using tetramethoxysilane as the transmetalating reagent.169
Scheme 41.
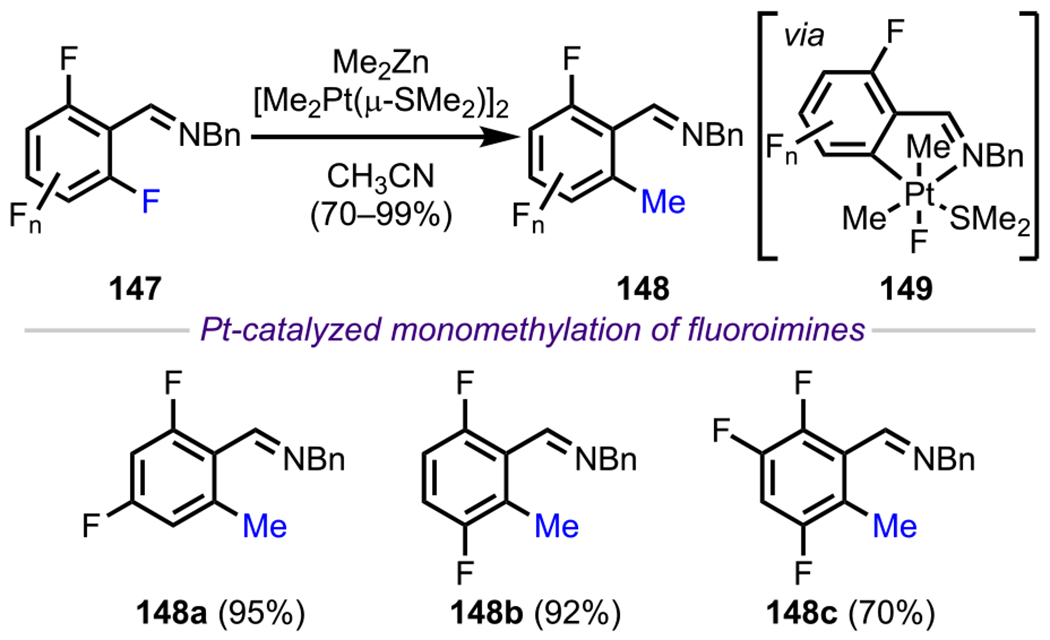
Platinum(II)-Catalyzed ortho-Methylation of Polyfluoroaryl Imines
After their initial work exploiting the coordinating ability of mines to achieve the site-selective functionalization of the ortho C–F bond in various polyfluoroaryl mines, Love and coworkers extended this methodology to include other coupling processes as well. For example, they demonstrated a Ni-catalyzed cross-coupling reaction between several polyfluoroaryl mines (150) and 4-methoxyphenylboronic acid to access a wide variety of biaryl aldehydes (151) upon hydrolysis (Scheme 42).170 This study showcases the “transient” nature of imines as DGs to achieve site-selective functionalization with the subsequent, in situ hydrolysis of the mine to unveil the corresponding aldehyde. Following these reports by the Love group, Li and coworkers extended the scope of employing imines as DGs to include the ortho-selective alkylative cross-coupling of polyfluoroarenes with various organozinc reagents under Ni-catalyzed conditions.171
Scheme 42.
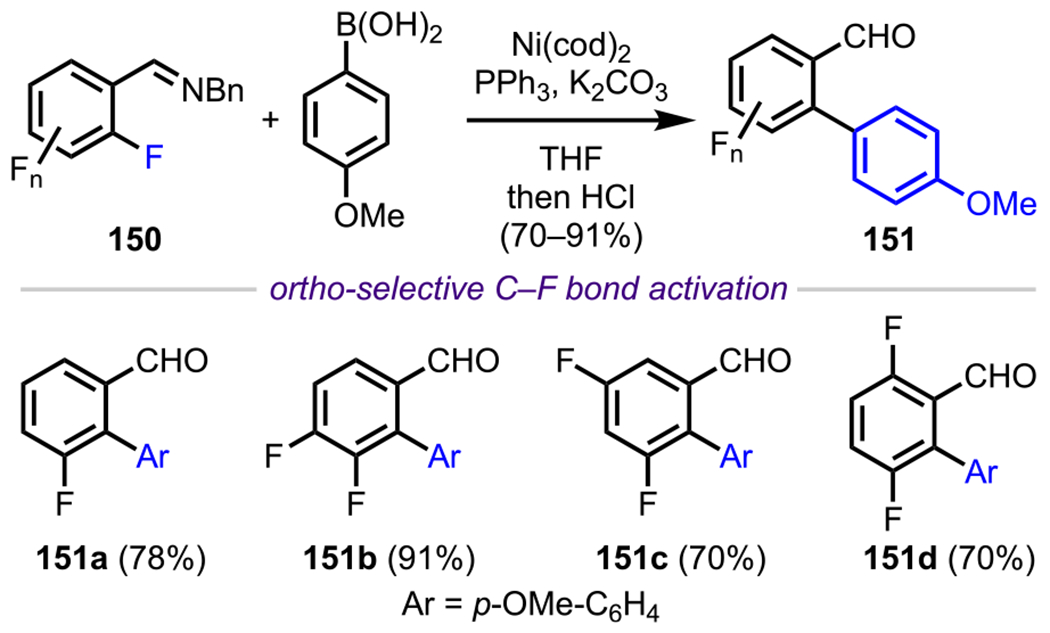
Nickel-Catalyzed ortho-Arylation of Polyfluoroaryl Imines
5.1.5. Ester and Amide.
Esters are versatile functional groups because they can be either introduced or interconverted easily through a variety of methods; therefore, the development of strategies for ester-directed site-selective functionalization has become highly desirable. Although the directing potential of ester groups was first explored in the mid-1990s,172,173 very little progress has been made in this area. This is mainly attributed to the weak coordination of the carbonyl group of esters to metal catalysts. One of the first successful methodologies to enable the site-selective functionalization of polyhalogenated heterocycles directed by an ester group was reported by Yang and coworkers in 2003.174 In their study, they observed that Suzuki–Miyaura coupling with the methyl ester of 2,6-dichloronicotinic acid (152) predominantly gave the six-substituted product (154) presumably due to the steric influence of the C3 ester (entry 1, Scheme 43A). To reverse the selectivity, they sought to coordinate the Pd(0) metal center to the ester group to promote oxidative addition into the C2 C–Cl bond. To enable this coordination, they envisioned reducing the ligand to palladium ratio to provide an open coordination site on the palladium. To test their hypothesis, they conducted the Suzuki–Miyaura coupling with a Pd/PtBu3 (1:1) catalyst system,175 which had previously been employed by Hartwig and coworkers in their amination studies,176,177 where they proposed the generation of a monophosphine Pd(0) complex as the catalytically active species. As predicted, Yang and coworkers achieved the desired complementary selectivity, with the C2-coupled product (153) forming as the major product in a ratio of 1.7:1 (entry 2).
Scheme 43.
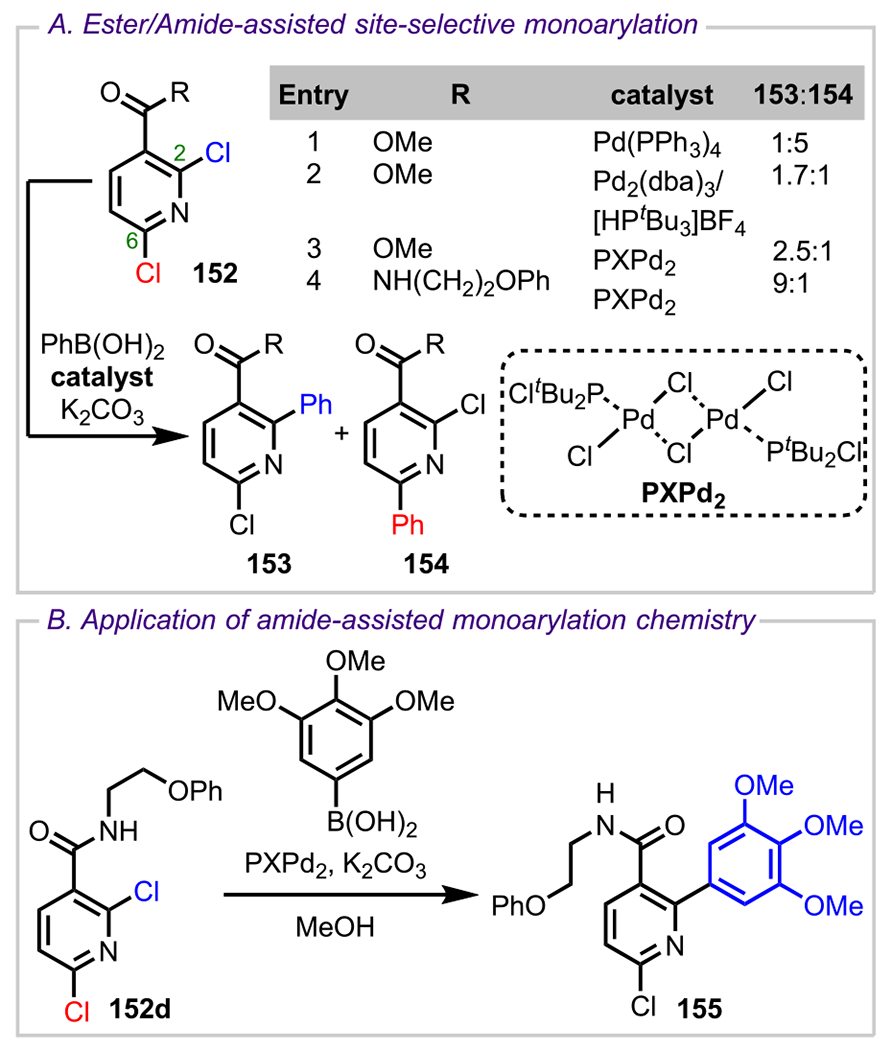
(A) Ester- and Amide-Directed Suzuki–Miyaura Coupling in the Presence of PXPd2 and (B) Application toward the Syntheses of Negative Allosteric Modulator Analogues
This promising initial result was followed by a thorough catalyst screening, where the best result was observed with an air-stable PXPd2 system (a 1:1 Pd/chlorophosphine complex),178–181 which furnished 153/154 in a ratio of 2.5:1 (entry 3, Scheme 43A). With this newly identified catalyst, they anticipated being able to further improve the selectivity by employing a more strongly coordinating amide generated from 2,6-dichloronicotinic acid (152d). Indeed, Suzuki–Miyaura coupling with the nicotinamide substrate in the presence of PXPd2 resulted in a 9:1 mixture of 153/154 (entry 4), thereby significantly enhancing selectivity. This chemistry was applied to the synthesis of novel negative allosteric modulator analogues,182 which included a C2-selective arylation of nicotinamide 152d to access myriad biaryl compounds, such as 155 (Scheme 43B).183,184 The directing ability of the amide group was also explored by Love and coworkers in their selective Ni-catalyzed trifluoromethylthiolation chemistry.185 Over the course of their studies, they developed a novel protocol to access monotrifluoromethylthiolated adducts (157) starting from dichloronicotinamide 156 using AgSCF3 as a nucleophilic trifluoromethylthiolating reagent in the absence of ligands and additives (Scheme 44).
Scheme 44.
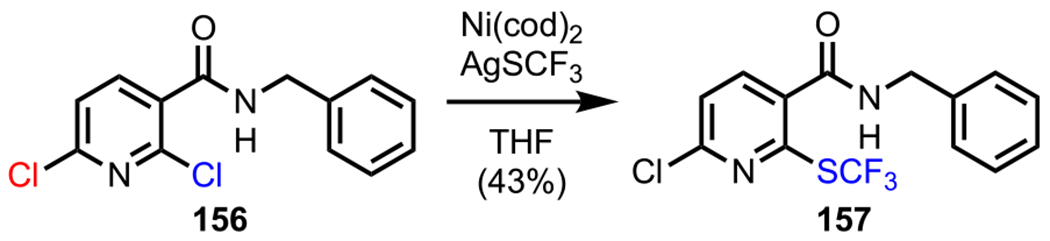
Amide-Directed Mono-Trifluoromethylthiolation of Dichloronicotinamide 156
5.1.6. N-Heterocycle.
N-Heterocycles are a very common class of DGs for the selective functionalization of polyhaloarenes. Several factors have contributed to their popularity as DGs, including: (1) N-heterocycles are found in various pharmaceutically relevant compounds, which helps facilitate late-stage modifications and prevents the need for additional DG installation/removal steps; (2) there are many strategies for introducing N-heterocycles; and (3) there is flexibility in introducing different substituents or heteroatoms to alter the coordinating ability for a specific transformation.186 Consequently, several heterocycles have been exploited as DGs to facilitate the selective cleavage of a specific C–X bond in polyhaloarenes (Scheme 45).
Scheme 45.
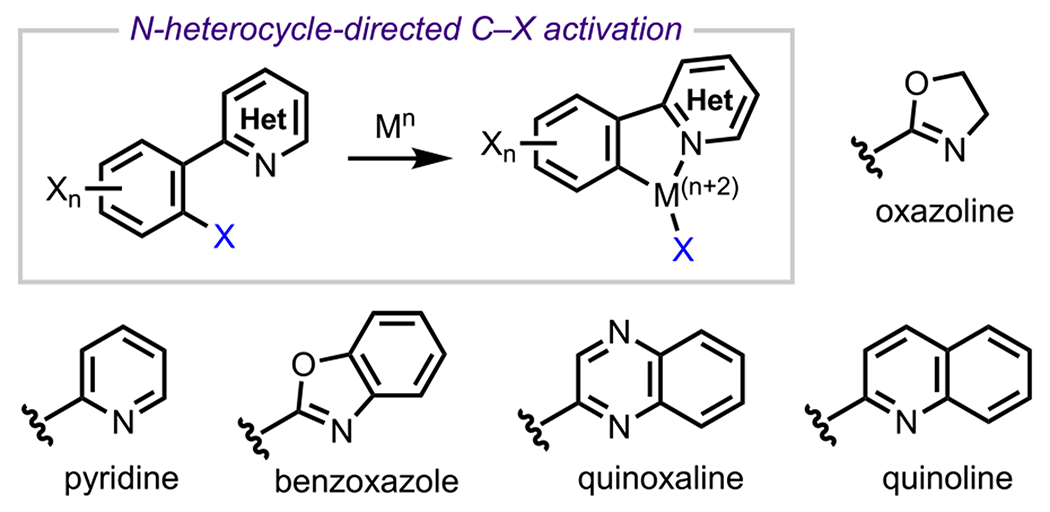
Selected Examples of N-Heterocycles Employed for the Selective Functionalization of Polyhaloarenes
Inspired by pioneering work in applying an oxazoline motif as a DG in C–H activation chemistry,187–189 Shen and Lu investigated the possibility of extending this strategy toward selective C–F bond activation in polyfluorinated arenes. They developed a Suzuki–Miyaura coupling between polyfluoroaryl oxazolines (158) and arylboronic acids to furnish ortho-arylated products (159) in the presence of Pd(CH3CN)2Cl2 as the precatalyst and Cs2CO3 as the base (Scheme 46A).190 It was concluded that 1,1′-ferrocenediyl-bis(diphenylphosphine) (DPPF) was the most effective ligand for this process because reactions using DPPF were much faster than reactions employing other phosphine ligands.
Scheme 46.
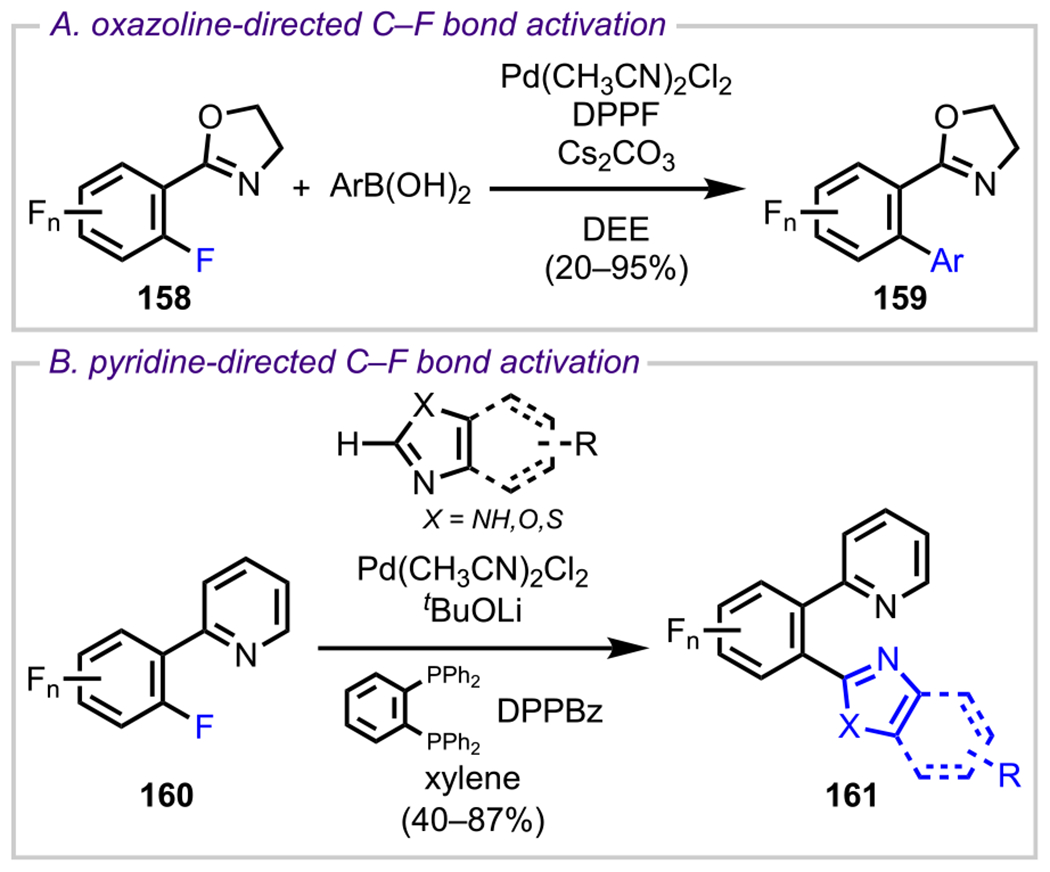
(A) Oxazoline-Directed ortho-Arylation and (B) Pyridine-Directed Concurrent C–F/C–H Bond Activation
Following this report, the authors envisioned performing a C–F bond activation of a polyfluoroarene with a tandem C–H bond activation event191–197 occurring with a second coupling partner to achieve a more efficient synthesis of partially fluorinated arenes.198,199 This strategy came with its own synthetic challenge of suppressing the undesired, but kinetically favorable, activation of a C–H bond in the polyfluoroarene substrate. After a careful investigation, they ultimately accomplished this tandem C–F/C–H bond activation process between polyfluoroarenes 160 and benzothiazole by employing a pyridyl group as a DG to effect selective C–F bond cleavage. The authors found that when oxazoline was installed as the DG, the desired product could be obtained only in poor yields with the predominant formation of undesired side products. The highest yield was observed using lithium tert-butoxide as the base and a catalyst generated in situ from Pd(MeCN)2Cl2 and 1,2-bis(diphenylphosphino)benzene (DPPBz). Impressively, the scope of this methodology was extended to other heterocycles such as thiazole, benzothiazole, benzoimidazole, oxazole, and oxadiazole to access a wide variety of partially fluorinated arenes (161) (Scheme 46B).200
Having developed Pd-catalyzed conditions to leverage the directing ability of N-heterocycles, Shen and Lu began investigating other transition-metal-catalyzed conditions to effect other selective transformations. They developed the first Ni-catalyzed α-arylation using Reformatsky reagents (163) and polyfluorinated arenes (162) via an N-heterocycle-directed C–F bond activation process (Scheme 47).201 Following optimization studies, they identified DIOP to be the most suitable ligand for this process. Furthermore, they also showed that in addition to pyridine, quinoxaline and oxazoline groups were effective DGs to access a wide variety of α-arylated amides (164).
Scheme 47.
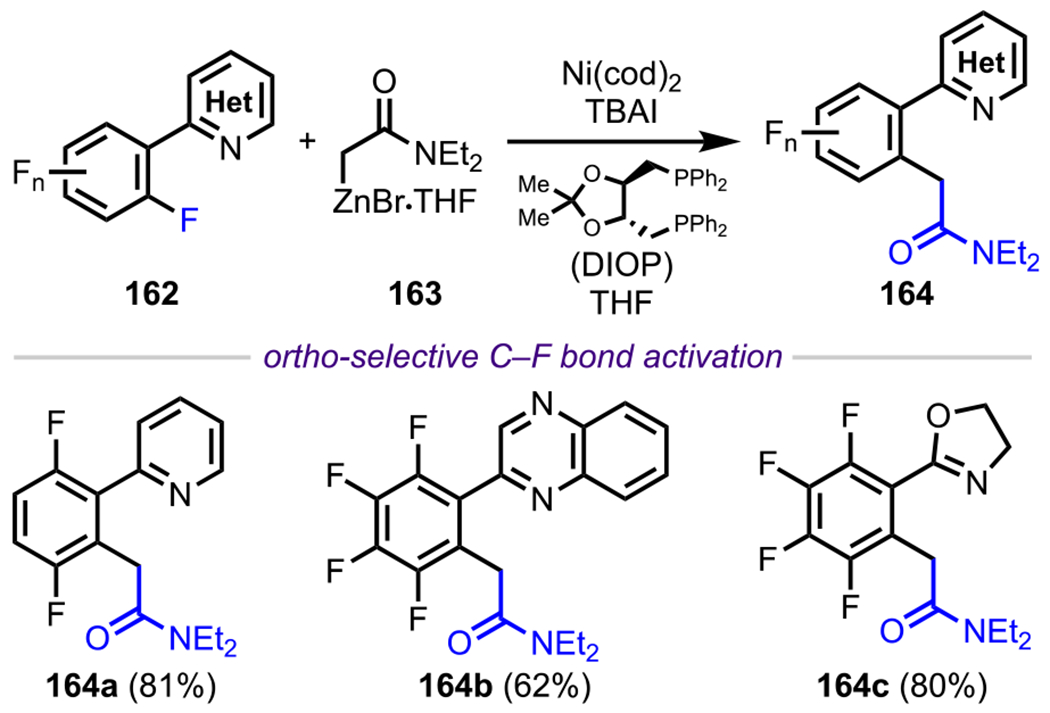
ortho-Selective α-Arylation of Polyfluoroarenes with Reformatsky Reagents
In a related study, Cao and coworkers demonstrated that organozinc reagents, such as Me2Zn, Et2Zn, and PhCH2ZnBr, can also be selectively coupled to polyfluoroarenes using N-heterocycle DGs.202 Additionally, in the methodology developed by Love and coworkers detailing amide-assisted ortho-selective trifluoromethylthiolation under Ni-catalyzed conditions (156 → 157, Scheme 44), the authors demonstrated the same transformation by employing a pyridine DG to provide biaryl trifluoromethyl sulfide 166 (Scheme 48).185
Scheme 48.
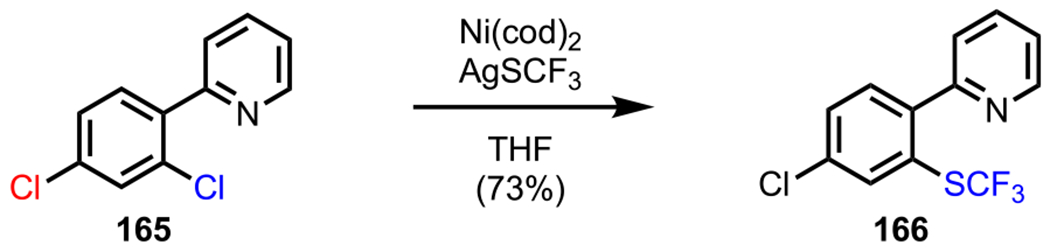
Pyridine-Directed Mono-Trifluoromethylthiolation of Dichloroarene 165
So far, we have mainly discussed strategies focused on the construction of C–C bonds through the transition-metal-catalyzed, site-selective C–F bond activation of polyfluoroarenes. Other types of couplings to install noncarbon substituents by the C–F bond activation of polyfluoroarenes enable the diversification of fluorinated molecules. Toward this end, Zhang and coworkers disclosed a novel strategy for achieving the N-heterocycle-directed ortho-borylation of polyfluoroarenes (167) to furnish boronate esters such as 168 (Scheme 49).203 Rh(I) catalysts were investigated, and after a comprehensive screen, [Rh(cod)2]BF4 was identified as the most suitable catalyst for this process. Several N-heterocycles including pyridine, quinoline, and benzoxazole emerged as viable DGs in this methodology.
Scheme 49.
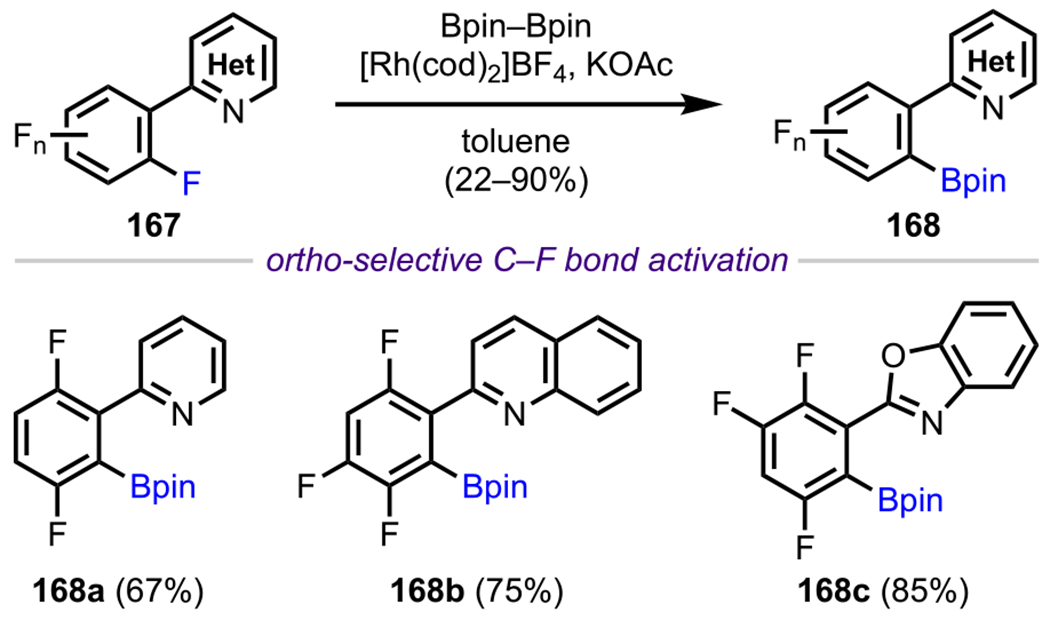
N-Heterocycle-Directed ortho-Borylation of Polyfluoroarenes
5.1.7. Other Miscellaneous Directing Groups.
5.1.7.1. Nitro.
In the early 2000s, strategies for the Pd-catalyzed C–F bond activation and functionalization of monofluorinated nitrobenzene derivatives were reported by the groups of Yu204 and Widdowson.205 In their studies, the authors proposed that in addition to its selectivity-enhancing electron-withdrawing properties, a nitro group also has the potential to direct a Pd catalyst to an adjacent C–F bond for its site-selective cleavage. Taking inspiration from these initial reports, Sandford and coworkers envisioned that a polyfluorinated nitroarene could also undergo a nitro-directed ortho functionalization via a Pd-catalyzed C–F bond activation event. When a Suzuki–Miyaura coupling reaction between perfluorinated nitroarene 169 and phenylboronate ester 170 was conducted in the presence of Pd(PPh3)4, the desired ortho-coupled product (171) was isolated in 80% yield (Scheme 50).206 Of note, the absence of the electronically favored para-coupled product (see Scheme 5) further supports transition state 172, which depicts the directing ability of the nitro group. Additionally, the scope of the reaction was then expanded to include a variety of polyfluoro nitroarenes and arylboronic acids with equally satisfying results.
Scheme 50.
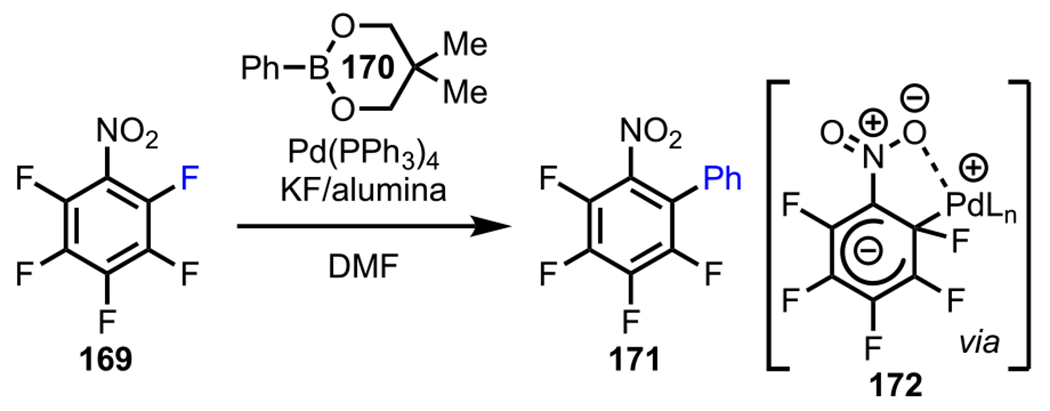
ortho-Selective C–F Bond Activation in Perfluorinated Nitroarene 169
5.1.7.2. Aminide.
Pyridinium N-(3′,5′-dibromopyrid-2′-yl)aminides (e.g., 173a)207–210 have been employed as building blocks in the synthesis of various azine derivatives.211–214 When they possess two C–Br bonds, these compounds are primed to undergo late-stage modifications via cross-coupling reactions. Accordingly, Alvarez-Builla and coworkers observed that upon subjecting these compounds to Suzuki–Miyaura coupling conditions with one equivalent of a boronic acid, substitution occurred exclusively at C3 to afford the monocoupled product (174) as the major product (Scheme 51). The authors attributed the observed site selectivity to the coordination ability of the aminide nitrogen to direct the Pd catalyst to the C3 position. On the basis of this result, a series of 3-(hetero)arylated derivatives were synthesized beginning from both 173a and the analogous pyrazine derivative, 173b.215,216
Scheme 51.
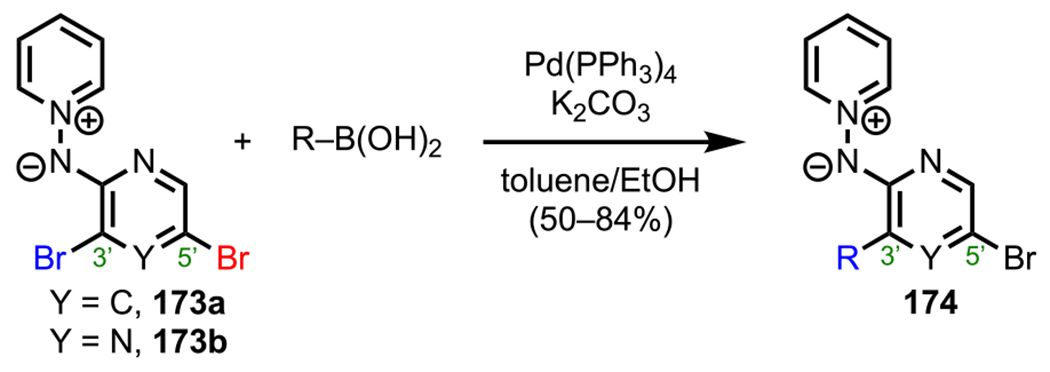
Aminide-Directed Selective Suzuki–Miyaura Coupling
5.1.7.3. Olefin.
The DGs that have been discussed thus far have mainly served to direct oxidative addition to a proximal C–X bond. Beaudry and coworkers reported the first example of using a coordinative functional group, such as an olefin, to achieve a site-selective coupling at a distal C–X bond. In their study, they observed that a Suzuki–Miyaura coupling between dibromostyrene 175 and boronate ester 176 predominantly furnished the monocoupled product (177) arylated at C4 (Scheme 52A).217 From their subsequent mechanistic studies, they ruled out the possibility of the observed selectivity arising from either the sterics or the inherent electronics of 175. They attributed the selectivity outcome to the relative rates of oxidative addition of the two bromides (Scheme 52B). Specifically, they proposed a reversible coordination of the Pd(0) species with the C–Br bond flanked by the alkene to generate the coordinately saturated complex 178. This coordination decreases the rate of oxidative addition at the C1 position, which leads to the observed site selectivity. The effects of olefins218 have been well studied, and it is reported that the rate of oxidative addition of Pd(0) into iodobenzene is significantly decreased in the presence of olefin additives;219–221 overall, this rationale is in agreement with the reactivity observed with olefin 175.
Scheme 52.
(A) Olefin-Mediated Site-Selective Suzuki–Miyaura Coupling of Dibromostyrene 175; (B) Mechanistic Considerations for the Observed Selectivity; (C) One-Pot Three-Component Suzuki–Miyaura Reaction for the Synthesis of Terphenyl 184; and (D) One-Pot, Three-Component Suzuki–Miyaura Reaction for the Synthesis of (+)-Cavicularin (189)
Beaudry and coworkers then leveraged this observed selectivity in developing an elegant synthesis of target compound 184, which was identified as a proteomimetic compound by Hamilton and coworkers.222 A one-pot, three-component Suzuki–Miyaura reaction was developed to achieve the synthesis of terphenyl 183 in 54% overall yield as a single isomer, starting from 175 and boronate esters 181 and 182. A subsequent hydrogenation furnished the target compound (184) in high yield (Scheme 52C). Overall, the authors accomplished an efficient synthesis of 184 compared with a previously established synthetic route involving seven steps.217 Prior to this work, an analogous one-pot, three component Suzuki–Miyaura reaction was employed in the synthesis of the bisbibenzyl natural product (+)-cavicularin (189). Beaudry and coworkers found that dibromomethoxystyrene 185 and boronate esters 186 and 187 could react sequentially in a one-pot Suzuki–Miyaura coupling reaction to furnish triaryl compound 188 in 63% overall yield (Scheme 52D). The development of this reaction proved to be pivotal in achieving an efficient synthesis of 189.223,224 In summary, these examples emphasize the significance of leveraging the coordinating ability of functional groups that are inherent in a target to achieve site-selective cross-couplings in various polyhalogenated (hetero)arenes. Furthermore, these tactics ultimately enabled the efficient syntheses of several target compounds.
5.2. Ligand-Binding Directing Groups
The examples that we have discussed thus far have employed DGs on substrates that coordinate directly to and guide the metal center to a proximal carbon position. However, some functional groups, depending on the reaction conditions, may retard the reaction rate by altering the electronics of the substrate. Furthermore, for reactions where the functionalization of a distal C–X bond is desired, a transition-metal-binding DG may not be suitable if the metal center is too distant from the desired site of reactivity. To circumvent this challenge and achieve distal functionalization, certain ligands that are of adequate length and capable of binding to a DG on a substrate can serve as a linker between the substrate and the transition-metal complex, enabling the transition metal to get in reasonable proximity to the remote C–X bond for activation (Scheme 30B). In the following section, we discuss examples where this “ligand-binding directing group strategy” is employed to either enhance the rate of oxidative addition or achieve the selective cleavage of a distal C–X bond. It is important to note that whereas the following examples do illustrate a form of ligand control, we have chosen to include them in this Directing Group Control section, as opposed to the Ligand Control section (section 6), because their primary role is to interact with a directing functional group on the substrate. Ligands that do not interact with a DG but instead coordinate exclusively to a transition-metal complex will be discussed in section 6.
Kumada coupling reactions with hydroxylated aryl substrates generally suffer from reduced reaction rates because the Grignard reagent deprotonates the hydroxy group to generate a highly electron-donating oxido group, which electronically retards oxidative addition. A general strategy to overcome this challenge would involve binding the generated oxido group to a ligand (that is, in turn, bound to a transition metal), which would place the transition-metal center proximal to the C–X bond, thus accelerating the reaction. In this regard, Manabe and coworkers synthesized a series of hydroxylated oligoarene-type phosphine (HOP)225–228 ligands for transition-metal catalysis, which are based on the Buchwald-type biphenylphosphine ligands.229–232 Specifically, they synthesized mono-hydroxyterphenylphosphine (HTP) and dihydroxyterphenylphosphine (DHTP) ligands (Scheme 53A), which were expected to perform as bifunctional ligands.233,234 Mechanistically, the authors anticipated that a linkage (via an electrostatic interaction) between the hydroxy group of the HOP ligand and the metal-oxido group of the substrate would increase the reaction rate by bringing the phosphino-coordinated transition-metal center closer to the desired C–X bond for site-selective oxidative addition (Scheme 53B).235,236
Scheme 53.
(A) Different HTP and DHTP Ligands and (B) Anticipated Heteroaggregate Formation
To test this hypothesis, Manabe and coworkers conducted a Kumada coupling between 2,4-dibromophenol (192) and Grignard reagent 125 in the presence of Pd2dba3 and various ligands. Interestingly, employing Ph-HTP as the ligand promoted coupling ortho to the hydroxy group to furnish 193 with excellent selectivity. On the contrary, using DPPF as the ligand completely reversed the selectivity to provide the para-coupled product 194, presumably due to steric effects (Scheme 54).237 Notably, the reaction was drastically accelerated with Ph-HTP and proceeded to completion in only 2 h; in contrast, the reaction with DPPF took 24 h to reach completion. Following mechanistic studies, the authors determined that HOP ligands favor ortho coupling, with poor selectivity occurring at both the meta and para positions.235,236 Altogether, these studies supported the mechanistic hypothesis of forming a bimetallic, bridged intermediate capable of facilitating site-selective ortho coupling.
Scheme 54.
Site-Selective Kumada Coupling with 2,4-Dibromophenol 192
In addition to the Ph-HTP ligand, the use of the HBF4 salt of Cy-HTP was also found to be effective for Kumada coupling reactions; however, both of these ligands were limited in their substrate scope. Subsequently, Manabe and coworkers found that the DHTP ligands were more effective than the HTP ligands and significantly broadened the substrate scope with enhanced selectivity. The higher reactivity of the DHTP ligands is attributed to the higher probability of the palladium and magnesium phenoxide to be on the same face of the para-terphenyl system due to the presence of two hydroxy groups.238 Eventually, the scope of the Kumada coupling reaction in the presence of HTP/DHTP ligands was also extended to include dibromoaniline and dibromoindole derivatives (e.g., 195) to give highly selective ortho-coupled products, such as 196 (Scheme 55).239
Scheme 55.
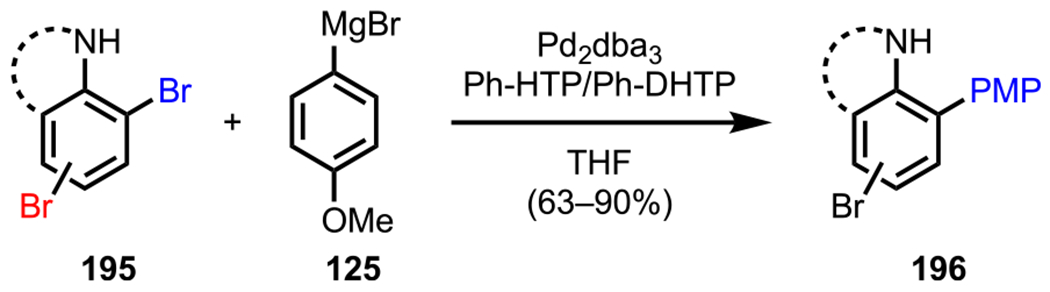
ortho-Selective Kumada Coupling with Dibromoaniline Derivatives
Manabe and coworkers then investigated the possibility of achieving HTP/DHTP-mediated ortho-selective Sonogashira coupling with dihalophenol and aniline derivatives. Toward this end, they devised a Sonogashira/cyclization sequence with dichlorophenols and terminal alkynes. Additionally, they also aimed to conduct a sequential Suzuki–Miyaura coupling at the unreacted C–X bond. Eventually, they achieved a one-pot synthesis for the preparation of myriad disubstituted benzo-[b]furans (198) from a series of dichlorophenols (197), terminal alkynes, and boronic acids (Scheme 56A).240–242 They identified the HBF4 salt of Cy-DHTP as the most suitable ligand and tBuOLi as the ideal base to effect ortho-selective Sonogashira coupling to arrive at alkynyl phenoxide 198b, presumably through the formation of the heteroaggregate, 198a. Mechanistically, a subsequent intramolecular cyclization and a sequential Suzuki–Miyaura coupling reaction afforded 198. This development encouraged them to explore the Sonogashira/cyclization sequence with dihaloanilines as well. Analogous to the sequence with dichlorophenols, N-tosyldichloroanilines 199 were also transformed to disubstituted indoles (200) using various terminal alkynes and boronic acids (Scheme 56B).240,243
Scheme 56.
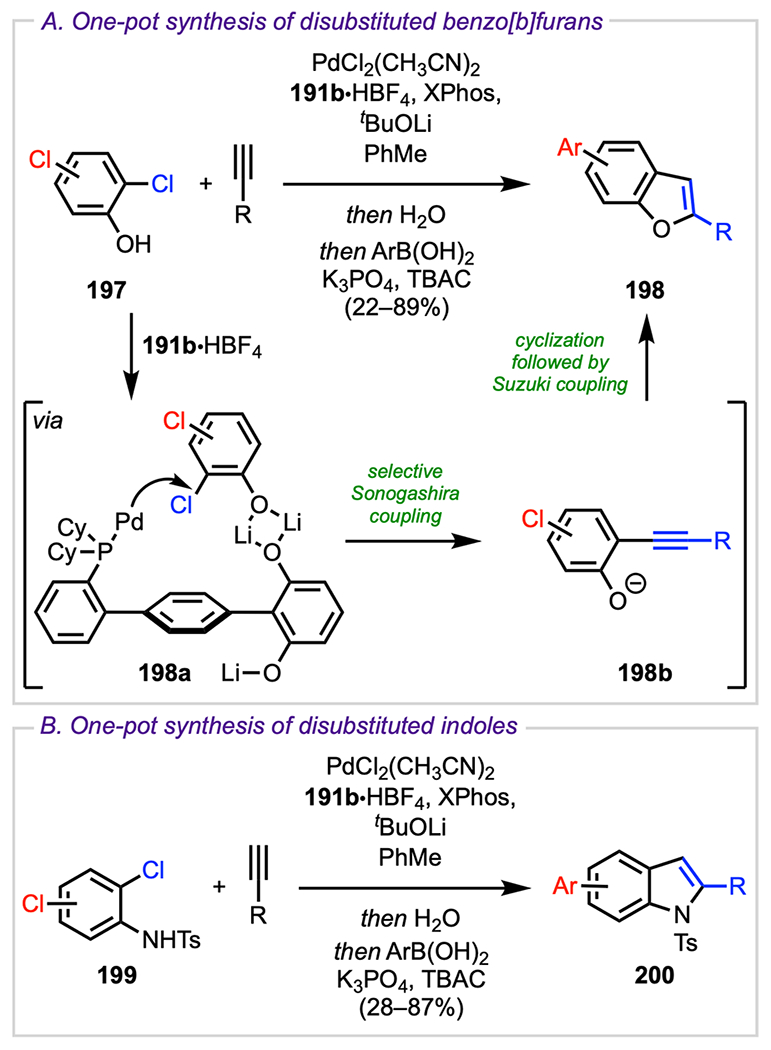
(A) Reaction Pathway to Access Disubstituted Benzo[b]furans and (B) Synthesis of Disubstituted Indoles from N-Tosyldichloroaniline Derivatives
In addition to tosylated aniline derivatives, acetylated aniline derivatives were also suitable substrates for this transformation. Specifically, N-acetyl-2,4,6-trichloroaniline 201 reacted with various terminal alkynes to furnish a series of 2-substituted 5,7-dichloroindoles (202). Mechanistically, similar to the previous examples, the Sonogashira coupling proceeds via the heteroaggregate intermediate 202a, which installs the alkyne moiety ortho to the acetamido group. However, in contrast with the previously observed indole synthesis, 202b cyclizes followed by a deacetylation of the resulting N-acetylated indole to furnish 1H-indole 202 (Scheme 57A). Importantly, this compound could be directly utilized in a subsequent C7-selective Kumada coupling with several aryl Grignard reagents under DHTP ligand conditions to furnish disubstituted indoles, such as 203 (Scheme 57B). This strategy allows for the stepwise substitution of each of the chloro groups to install different substituents in a well-controlled manner, thus proving to be effective for preparing a series of multisubstituted indoles.244,245
Scheme 57.
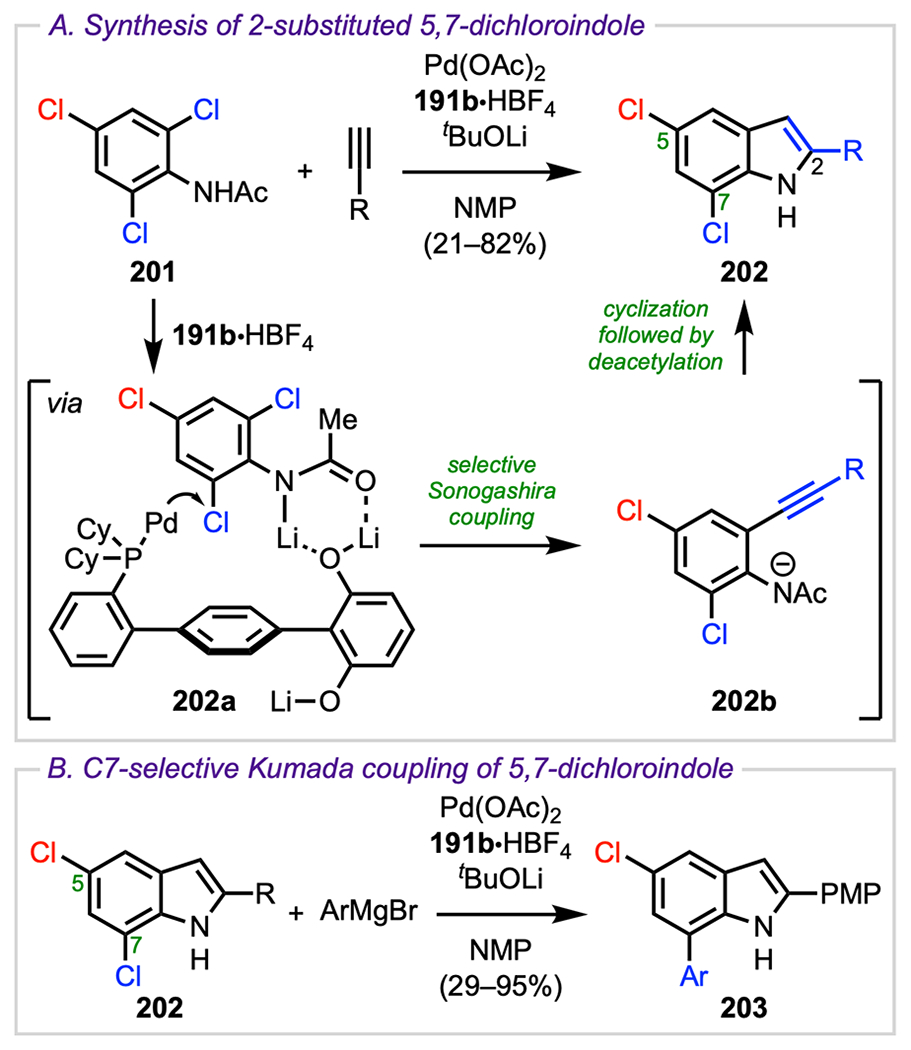
(A) Reaction Pathway to Access Two-Substituted 5,7-Dichloroindoles and (B) Site-Selective Kumada Coupling of 5,7-Dichloroindole
Whereas the aforementioned reports by Manabe and coworkers exemplify the utility of bifunctional ligands (HTP and DHTP) in palladium catalysis to accelerate the rate of oxidation addition at a nearby ortho C–X bond, there currently exists a dearth in methodologies that address site-selective coupling at more distal positions with minimal steric or electronic bias. To address this challenge, Phipps and coworkers focused on leveraging bifunctional phosphine ligands to direct oxidative addition at a distal position. They postulated a pseudointramolecular transition state stabilized by some noncovalent interactions between the substrate and the ligand,246–249 which would bring the phosphino-coordinated metal center in close proximity to a specific C–X bond. Toward this end, they considered sulfonylated phosphine ligands to serve this bifunctional purpose, where the sulfonate group was anticipated to engage the substrate via an electrostatic interaction. Accordingly, they chose to employ sSPhos and sXPhos, the sodium salts of sulfonylated SPhos and XPhos, which have been previously utilized by Anderson and Buchwald for the cross-coupling of aryl chlorides in water (Scheme 58A).250–255
Scheme 58.
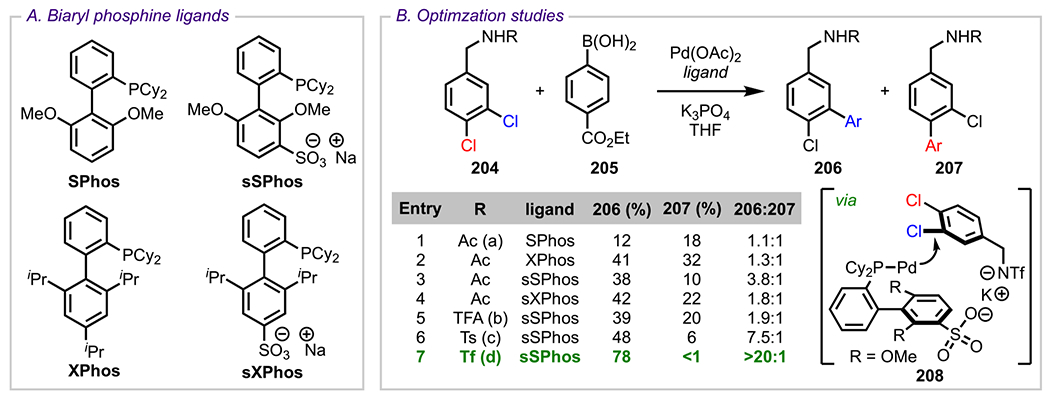
(A) Selection of Investigated Biarylphosphine Ligands and (B) Optimization Studies to Achieve meta-Selective Coupling
They probed the coordination ability of these ligands in the Suzuki–Miyaura coupling between N-acetyl-3,4-dichlorobenzylamine 204a and boronic acid 205 (Scheme 58B).256 They hypothesized that the interaction between the ligand and the amine group of the substrate would result in site selectivity. Consistent with this notion, they observed no selectivity with either SPhos or XPhos (entries 1 and 2). In contrast, the addition of either sSPhos or sXPhos favored meta coupling to predominantly give 206 over 207 (entries 3 and 4). Subsequently, the N-group was varied to study the selectivity outcome. Interestingly, as the N-protecting groups became more electron-withdrawing, the selectivity was significantly enhanced. Whereas trifluoroacetyl gave poor selectivity (entry 5), tosyl increased the ratio to 7.5:1 (entry 6). Eventually, triflyl was identified to provide both excellent selectivity and conversion (entry 7). Overall, the authors rationalized the observed results by invoking an electrostatic interaction of the potassium salt of 204d with the sulfonate group of the ligand to generate intermediate 208. In this intermediate, the phosphine-bound Pd is proximal to the meta-C–Cl bond, thus enabling subsequent oxidative addition in a selective manner.
Next, the scope of the Suzuki–Miyaura coupling of 204d was expanded to include various aromatic boronic acids, which led to the formation of biaryl products (206d) with excellent selectivity (Scheme 59A).256 Additionally, Sonogashira coupling to form Csp2–Csp bonds was investigated with several alkyne derivatives, and the corresponding aryl alkyne products (209) were isolated with superior selectivity. Furthermore, Buchwald–Hartwig amination was also explored with 204d to access a variety of meta-substituted aryl amine products, such as 210 (Scheme 59B). Gratifyingly, aryl amines proved to be suitable substrates for this transformation, and after the optimization, s(tBuSPhos), the P(tBu)2 variant of sSPhos, was found to give the best selectivity. Applying these optimized conditions enabled a range of aryl amines to be successfully coupled.
Scheme 59.
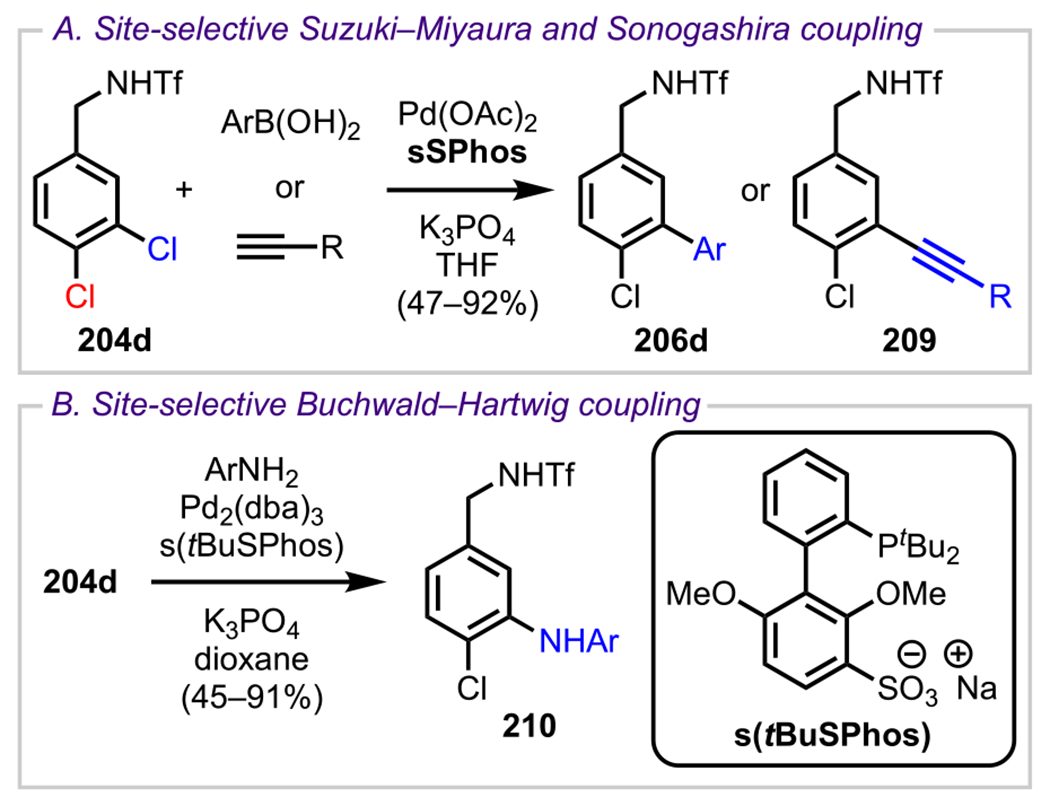
(A) Site-Selective Suzuki–Miyaura and Sonogashira Couplings with 204d and (B) Site-Selective Buchwald–Hartwig Amination with 204d
Following this work, the authors then investigated the possibility of coupling 204d to a suitable coupling partner via a C–H bond activation process (Scheme 60).257 This approach would be highly beneficial because it avoids the necessity of prefunctionalizing the second coupling partner; furthermore, this strategy would demonstrate the viability of concerted metalation–deprotonation (CMD) in this putative electrostatically bound transition state (208, Scheme 58B). Toward this end, fluoroarenes and fluoroheteroarenes (211) were chosen as the second coupling partner.258,259 Surprisingly, no product was observed under the previously disclosed conditions (Scheme 58B). However, on the basis of previous literature data, adding pivalic acid260 and switching the solvent to iPrOAc261,262 proved to be crucial for executing the desired C–H activation/C–C bond forming event. The yield was substantially improved by using the more soluble tetrabutylammonium salt of sSPhos, sSPhos(NBu4), instead of the typical sodium salt. In addition, following an evaluation of Pd precatalysts, [(cinnamyl)PdCl]2 was identified to give the product in an enhanced yield. Next, several fluoroarenes were coupled to 204d to produce myriad biaryl products (212) under this established set of optimized conditions (Scheme 60).
Scheme 60.
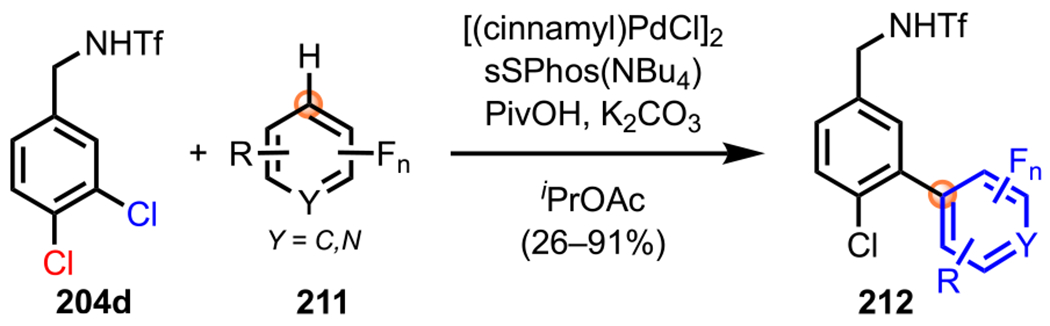
meta-Selective C–H Activation/C–C Bond Formation with Fluoroarenes and Fluoroheteroarenes
Phipps and coworkers next explored the dichloroarene component by varying the amino functional handle (Scheme 61). They established that extended triflamide derivatives were tolerated, albeit with reduced selectivity. Additionally, α-substitution with both methyl and phenyl groups did not interfere with the catalytic process. Interestingly, sulfamate, sulfonate, and phosphonate derivatives were all found to be suitable substrates for this site-selective cross-coupling process. Furthermore, carboxylic acid derivatives with an extended chain length resulted in excellent site selectivity, and the presence of an alkene in the chain also did not disrupt the observed selectivity.
Scheme 61.
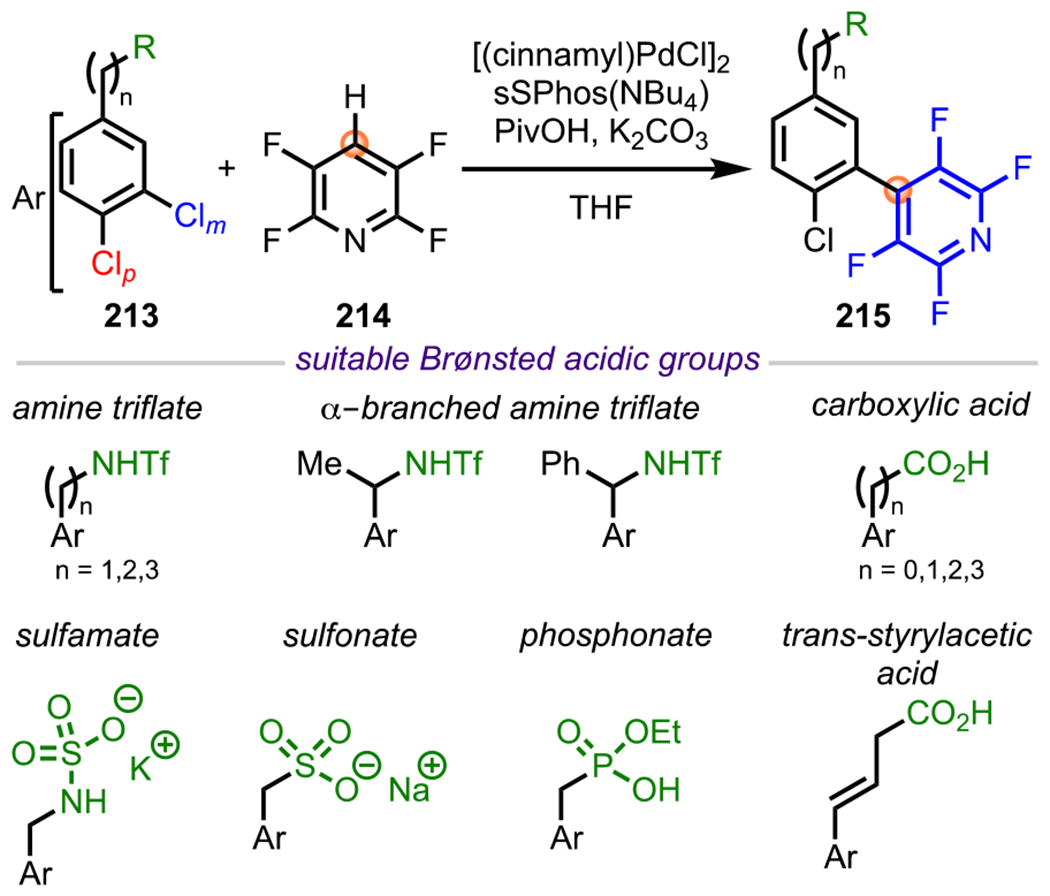
Evaluation of Various Brønsted Acidic Functional Groups
Adding to their repertoire of site-selective cross-couplings, Phipps and coworkers next established a comprehensive “toolkit” where various ligand/base combinations were identified for site-selective Suzuki–Miyaura and Buchwald–Hartwig couplings via the electrostatic model proposed for intermediate 208.263 (See Scheme 58.) Through a systematic screening process, the authors identified various ligand/base combinations for transforming dichlorinated arenes with diverse substitution patterns (216–218) into their corresponding monocoupled products (219–221) under both Suzuki–Miyaura and Buchwald–Hartwig conditions (Scheme 62A). In the same publication, the authors reported ligand/base combinations suitable for facilitating site-selective Suzuki–Miyaura and Buchwald–Hartwig couplings of various trichlorinated arenes (222–224) to monocoupled products (225–227) as well (Scheme 62B). Using the ligand/base combinations shown in Scheme 62, the authors successfully executed sequential Suzuki–Miyaura cross-couplings on both trichlorinated and tetrachlorinated arenes to obtain polyarylated products in a site-selective manner (not shown). In summary, Phipps and coworkers demonstrated that electrostatically directed palladium catalysis employing bifunctional sulfonylated biarylphosphine ligands is compatible with various cross-coupling and CMD-type C–H activation processes on several functionalized dichloroarene derivatives.
Scheme 62.
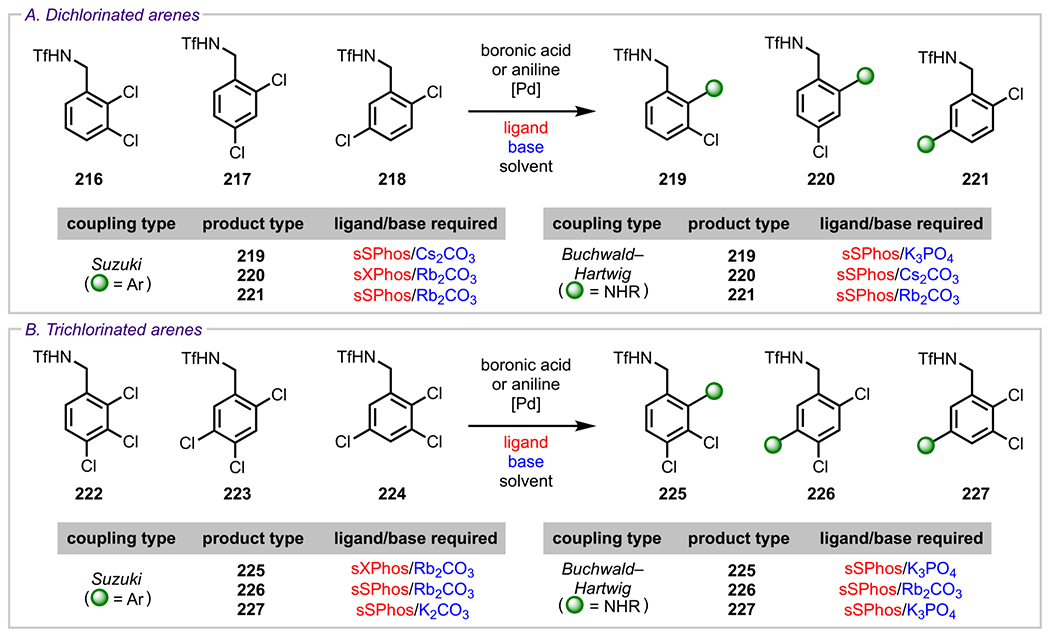
Site-Selective Suzuki–Miyaura and Buchwald–Hartwig Couplings of (A) Dichlorinated and (B) Trichlorinated Arenes
Overall, the contributions of Manabe and Phipps clearly mark the advent of employing bifunctional phosphine ligands to promote site-selective cross-coupling in polyhalogenated (hetero)arenes. This series of thought-provoking examples demonstrates how a ligand can be finely tuned to interact with halogenated precursors to facilitate site-selective oxidative addition. In this stage, one can anticipate the development of several new reactions that employ bifunctional ligands along with transition-metal catalysts to effect cross-couplings at positions that are otherwise challenging to activate under the previously disclosed strategies.
6. LIGAND CONTROL
The majority of examples for the site-selective cross-coupling of polyhalogenated arenes discussed so far have been based on a “substrate-controlled” strategy, which focuses on leveraging key differences in reactivity between various C–X bonds and DG abilities of functional groups present on the substrate. Thus far, the preference for a reaction to occur at one C–X bond over another has been ascribed to three main factors: (1) the electronics of the carbon position, (2) the steric environment, and (3) the influence of nearby DGs. Examples relevant to these categories have been discussed in detail in sections 3–5. This section focuses on examples related to a “ligand-controlled” strategy (i.e., a non-substrate-controlled strategy), wherein ligands that bind exclusively to the transition metal (and not the substrate) can be used to alter/tune the electronic and steric properties of the transition-metal complex, thus controlling the selectivity outcome. In other words, depending on the choice of ligands, different products can be obtained selectively from the same polyhaloarene precursor. Employing this strategy would enable one to obtain a product selectively, regardless of the steric and electronic nature of the starting material. This provides an advantage over the previously described strategies in instances where the desired selectivity and electronically preferred outcomes are mismatched. Substrate control requires the preinstallation of either a sterically bulky substituent or a coordinating functional group to bias oxidative addition toward a specific C–X bond. However, this can decrease the atom and step economy if the added substituent must be removed later in the synthesis. Hence, strategies that enable site-selective reactivity without modification of the substrate are highly desirable. Recent reports of novel ligand platforms to guide site-selective couplings represent a significant advancement in this field.
In 2005, Chung and coworkers reported a Suzuki–Miyaura coupling between 5,7-dichloro-1-(2,6-dichlorophenyl)-1,6-naphthyridin-2-(1H)-one (228) and electron-deficient arylboronic acid 229 (Scheme 63).264–266 During their initial investigation, the authors observed selectivity between monocoupled products, with 230 being favored over 231. However, in the case of both monocoupled products, the electron-deficient difluorophenyl group activated the second C–Cl bond toward a second coupling, which formed the bis-coupled product (232) in almost equal ratio to 230 (entry 1). Eventually, Chung and coworkers rationalized that the site selectivity observed with 228 was completely substrate-dependent, and they anticipated minimizing the second coupling by altering the ligand employed in the reaction. Consequently, they screened ~80 commercially available ligands with various bases and solvents,266 with selected results shown in Scheme 62 (see table). Furthermore, some improvements were also observed by either lowering the temperature or using a monodentate ligand, such as PPh3 (entries 2 and 3). Interestingly, using SPhos, a highly activating ligand, enabled the reaction to occur at a lower temperature and gave a slightly improved selectivity for the monocoupled product 230 (entry 4). Further improvement in selectivity was observed using IMes●Cl, the NHC derived from 1,3-bis(2,4,6-trimethylphenyl)imidazolium chloride. A combination of this carbene along with Pd(OAc)2 or Pd2(dba)3●CHCl3 was identified as a suitable catalytic system for this transformation (entries 5 and 6). Additionally, for a large-scale manufacturing process, the precatalyst/ligand system of Pd2(dba)3●CHCl3/P(2-MeOC6H4)3 was revealed to be more robust and cost-effective, providing >99% conversion and 230 in 92% yield (entry 7).
Scheme 63.
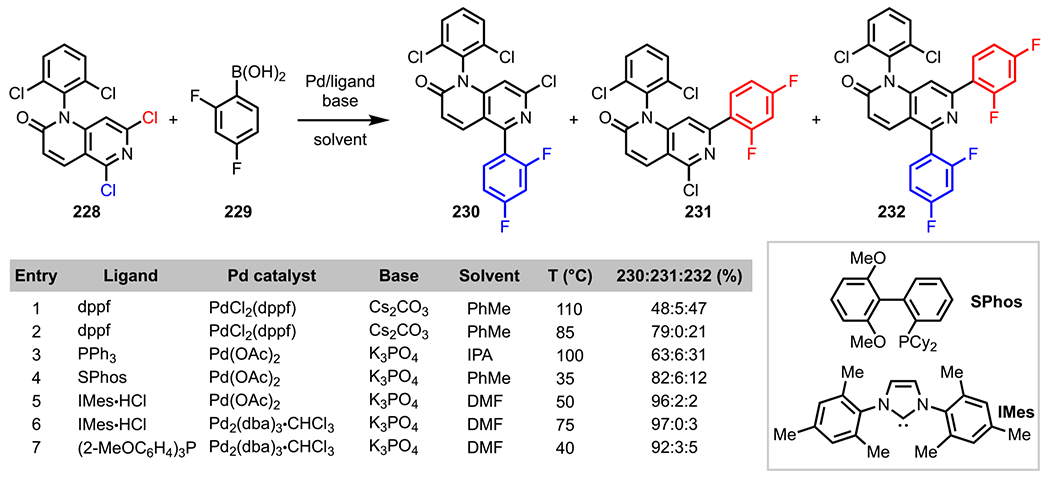
Site-Selective Suzuki–Miyaura Coupling of 228
These studies were followed by a report by Strotman, Chobanian, and coworkers, where various ligands (Scheme 64A) were identified to facilitate complementary selectivity outcomes in the cross-coupling of several dihaloazoles.267 Because oxazole, imidazole, and thiazole frameworks are present in several bioactive and pharmaceutically relevant molecules,268,269 devising efficient strategies to access their derivatives is highly valuable. Toward this end, the authors investigated site-selective Suzuki–Miyaura couplings of dihaloimidazoles, dihalooxazoles, and dihalothiazoles as a strategy to rapidly diversify diarylazoles in a modular fashion (Scheme 64B). Taking inspiration from the work of Greaney and coworkers,270 the authors commenced their study with the Suzuki–Miyaura coupling of 2,4-diiodooxazole (85) and phenylboronic acid. The more reactive C2 position271,272 (as predicted by Handy’s method273) was expected to be the preferred site of coupling; however, contrary to this expectation, the majority of ligands screened favored coupling at the C4 position. The highest yielding result was obtained by employing Xantphos (X2) as the ligand, which furnished the C4-phenylated product 233 in 64% yield. To explore C2-selective coupling, the authors next screened ~200 achiral phosphine ligands and identified 1,3,5-triaza-7-phosphaadamantane (X4) as a ligand that produced the desired C2-phenylated product (234) in 55% yield.
With these complementary conditions established, the authors next investigated a Suzuki–Miyaura coupling of other commercially available dihaloazoles. As with 85, high C2 selectivity was observed with 2,5-dibromo-1-methylimidazole (84) when X4 was employed as the ligand; these conditions provided 236 in 64% yield. Concurrently, C5 selectivity was also obtained with a wide variety of ligands, with bis(p-sulfonatophenyl)phenylphosphine dihydrate dipotassium salt (X6) furnishing 235 in 45% yield. While exploring C2 versus C4 site selectivity with 2,4-dibromo-1-methylimidazole (76), the previously described ligand choices were investigated. Surprisingly, the major site of reaction for all the explored ligands, including X4 and X2, was at the C2 position. The best selectivity and highest yield of 238 (produced almost exclusively) were obtained using tri(4-fluorophenyl)phosphine ligand X5. Furthermore, no ligands in their collection provided any appreciable reaction at C4, which was the more reactive position observed for 2,4-diiodooxazole (85). A similar outcome was also observed with dibromothiazoles (71 and 72). Out of all of the investigated ligands, X2 proved to be highly selective for C2 coupling in both 2,4- and 2,5-dibromothiazoles; notably, no ligand was identified that achieved selective coupling at either C4 or C5. These results stand in stark contrast with what was observed with both 84 and 85, where coupling could take place at the C4 and C5 positions in a selective manner. The scope of the Suzuki–Miyaura coupling with these heterocycles was extended to include a wide variety of electron-rich, electron-poor, and sterically hindered arylboronic acids. Selectivity with heteroaryl and cyclopropylboronic acids followed similar trends to that observed with phenylboronic acid. The dependence of the relative rates of C2 versus C4 (or C5) coupling on the identity of the ligand is not fully understood but is believed to be a cumulative effect of both the steric and the electronic nature of the ligand.
The potential of ligand-controlled site-selective couplings has also been investigated by Dai and coworkers,274 who were interested in constructing a wide array of pyridazine derivatives from 3,5-dichloropyridazine (56) as part of a drug discovery program. The authors envisioned catalytic systems that could selectively favor coupling at either the C3 or the C5 positions of 56, thus enabling a modular approach to accessing diverse diarylpyridazines. On the basis of the findings by Houk and Merlic,38,73 3,5-dichloropyridazine is expected to react more readily at C3 than C5 due to the lower BDE of the C–Cl bond α to the nitrogen atom. Dai and coworkers focused on a Suzuki–Miyaura coupling between 56 and phenylboronic acid under different ligand conditions (Scheme 65). They observed that electron-deficient bidentate ligands and PPh3 favored the formation of the C3-coupled product (241), and DPPF was found to give the best yield and selectivity (entries 1–3). Notably, DTBPF, an electron-rich bidentate ligand, gave the opposite selectivity, albeit in a moderate ratio between mono- and bis-coupled products (entry 4). Alternatively, C5 selectivity was also achieved with electron-rich monodentate ligands (entries 5–7). Q-Phos gave the best selectivity and afforded the C5-coupled product (242) in 80% yield. Furthermore, Buchwald’s biaryl-based monophosphine ligands (e.g., RuPhos, DavePhos, and XPhos) predominantly favored the formation of the bis-coupled product (243), even when only 1 equiv of phenylboronic acid was used (entries 8–10).
Scheme 65.
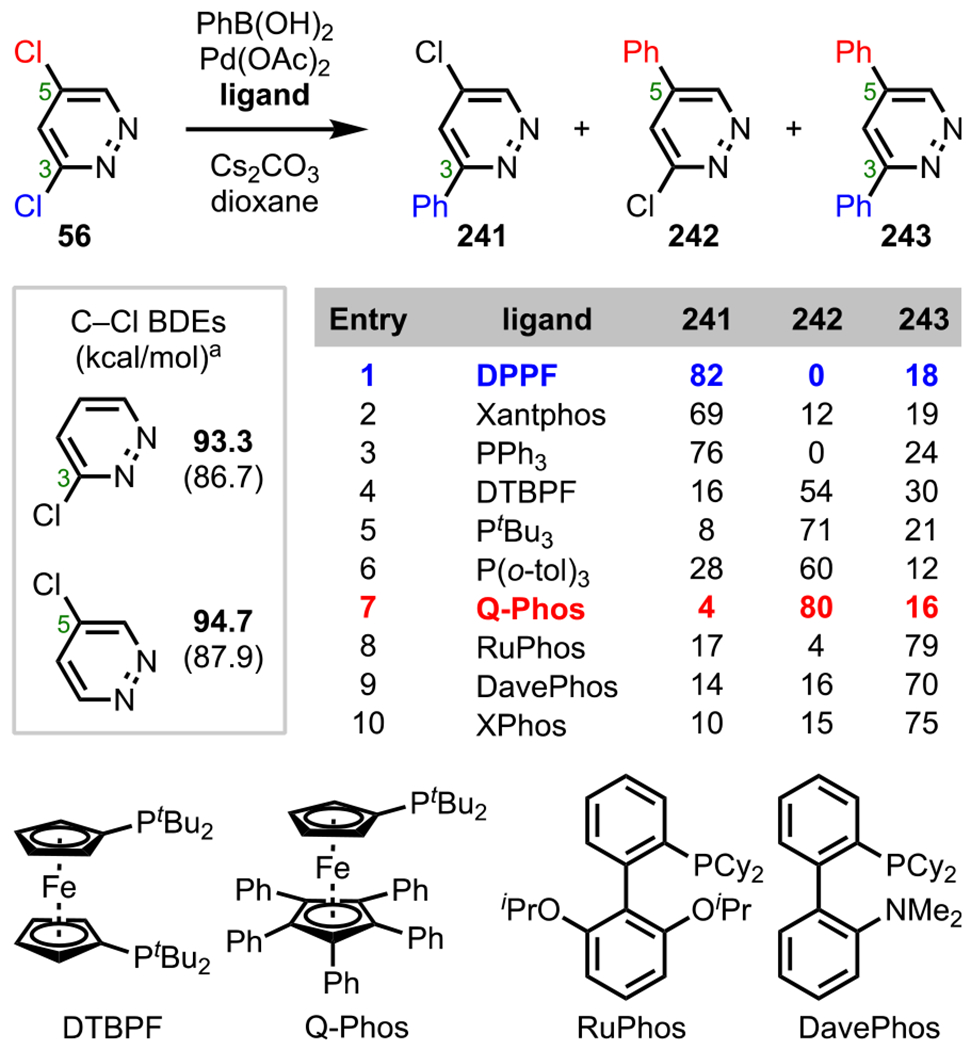
C3 versus C5 Complementary Selectivity Observed in 3,5-Dichloropyridazine (56)
Although the exact nature of how substrate–ligand interactions facilitate complementary selectivity remains unclear, one could envision selectivity arising from an effect of the steric and electronic influences of both the ligand and the substrate. For instance, electron-deficient ligands favor slower oxidative addition; therefore, the insertion of the metal center at the more reactive C3 position of 56 (Scheme 65) occurs, as predicted by the corresponding C–Cl BDE values reported by Houk and Merlic.38,73 On the contrary, electron-rich ligands bias the metal center to insert into the more sterically accessible C5 position of 56. These principles guided Dai and coworkers in their investigation of a complementary C2 versus C4 coupling strategy for 2,4-dichloropyridine (244, Scheme 66). C2 coupling was achieved with DPPF, which furnished the desired product (245) in 90% yield. On the basis of the report by Houk and Merlic, coupling at C2 over C4 is favored by nearly 1000-fold;38,73 however, Dai and coworkers were able to overcome this preference by implementing their previously discovered conditions employing Q-Phos (see entry 7, Scheme 65) to give the C4-coupled product (246) in 36% yield. Despite the modest 36% yield of 246, this is the first example of 244 undergoing a C4 coupling in the absence of a DG.
Scheme 66.
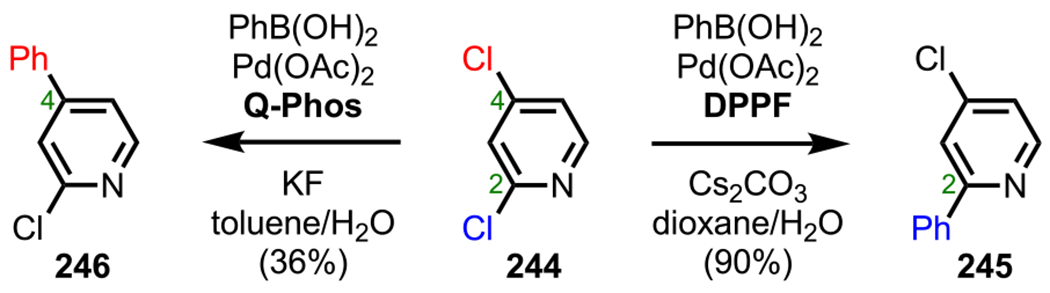
Complementary Selectivity Observed in 2,4-Dichloropyridine (244)
Recently, Yang and coworkers also investigated this challenging C4 coupling of 2,4-dichloropyridine (244).275–280 The authors began their exploration by using 244 and 4,4,5,5-tetramethyl-2-phenyl-1,3,2,-dioxaborolane (PhBpin) (Scheme 67). They observed that using Pd(PPh3)4 as the catalyst afforded the C2-coupled product (245) in high yield (entry 1). Next, they conducted an extensive screening with Buchwald Pd-G3 precatalysts. Over the past decade, the Buchwald group has developed a series of Pd precatalysts from the corresponding family of biarylphosphine ligands.281–287 The Pd-G3 precatalysts are, in general, bulky, electron-rich ligands, which rapidly convert to the active L–Pd(0) species under mild reaction conditions; these catalysts have therefore been prominently used in various cross-coupling processes.284,288–293 Buchwald Pd-G3 precatalysts improved the ratio of C4:C2-coupled products, albeit with a moderate overall selectivity (entries 2–6). A plausible mechanistic rationale is that the precatalysts containing bulky electron-rich ligands favor oxidative addition at the more sterically accessible C4 position; this logic would be in agreement with what Dai and coworkers observed during their studies.274 (See Schemes 65 and 66.)
Scheme 67.
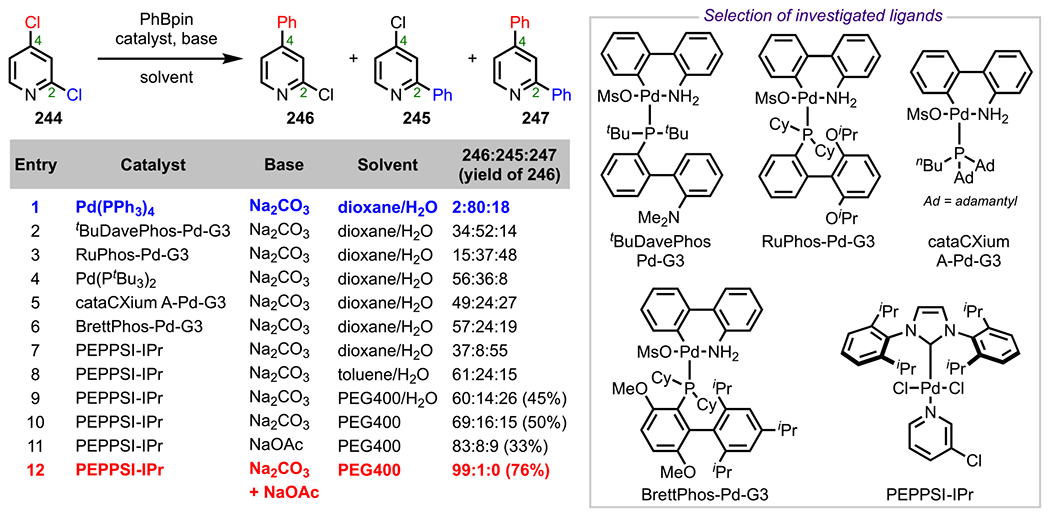
Optimization for the Site-Selective Suzuki–Miyaura Coupling of 2,4-Dichloropyridine (244)
In an effort to further improve the yield of this C4 coupling, Yang and coworkers turned their attention toward “pyridine-enhanced precatalyst preparation stabilization and initiation” (PEPPSI) catalysts. These catalysts form highly stable organopalladium complexes containing bulky NHC ligands. PEPPSI catalysts were originally developed by Organ and coworkers and have since been found to accelerate various aminations and cross-coupling reactions.294–298 Gratifyingly, when the Suzuki–Miyaura coupling was conducted in the presence of a PEPPSI-IPr precatalyst, a large proportion of 246:245 (~4.5:1) was observed (entry 7, Scheme 67). This selectivity was attributed to the steric bulk of the PEPPSI-IPr precatalyst, which inserted into the more sterically accessible C4 position of 244. However, a significant amount of the bis-coupled product (247) was also formed along with the desired C4-coupled product (246). Further optimization with PEPPSI-IPr was conducted to minimize the formation of 247. Among the explored solvents, PEG400 was found to give the highest yield (entries 8–10). In varying the base, NaOAc emerged as beneficial for achieving a greater ratio of 246 to 245; however, it drastically affected the conversion of 244 (entry 11). Eventually, using a combination of both Na2CO3 and NaOAc, along with employing KI as an additive, provided an optimal ratio of 246/245/247 (99:1:0), where 246 was isolated in 76% yield (entry 12). This optimized set of conditions was then utilized to couple 244 to both electron-donating and electron-withdrawing boronate esters to construct a variety of pyridine-based dyes.
In summary, the examples illustrated here demonstrate the role that ligands play in achieving unprecedented selectivity in cross-couplings. Notably, there are few reported cases where the careful tuning and choice of ligands were leveraged to achieve complementary selectivity outcomes. Whereas the origin of the site selectivity in many cases remains underexplored, further studies making use of Merlic and Houk’s DFT-based “distortion–interaction” model38,73 could prove useful in uncovering the underlying mechanisms governing observed selectivity patterns. As a deeper understanding of these mechanisms is obtained, “ligand-controlled” approaches will undoubtedly continue to move to the forefront of the field of site-selective cross-coupling due to the tunable nature of ligands in facilitating unique selectivity outcomes without the need for substrate modification or neighboring group effects.
7. SOLVENT/ADDITIVE CONTROL
The previous section discussed how ligands can be carefully tuned to favor coupling at a particular position with minimal substrate modification. In many of those examples, once the optimal ligand was identified, other factors such as the solvent, base, additives, and temperature were also tuned to further improve the selectivity and isolated yield of the desired coupling product. In one previous example (Scheme 67), Yang and coworkers showed that employing PEPPSI-IPr as a precatalyst, using a combination of two bases (Na2CO3 and NaOAc), along with KI as an additive and running the reaction in PEG400 proved to be essential for obtaining high selectivity for the C4 coupling of 244.275 On the basis of these results, one could postulate that the selectivity, in both this and other examples, could be altered by manipulating variables other than the identity of the ligand (i.e., solvent, temperature, additives, etc.) in the reaction mixture. In principle, this practice of modifying other variables besides metal salt/ligand mixtures, would present the added advantage of being more cost-effective as compared with screening catalysts and ligands, which can often be expensive. The following examples provide insight into the types of substrates that have proven competent in cross-couplings where selectivity is guided by modifying solvents and additives.
In 2008, while investigating the site selectivity of the Suzuki–Miyaura coupling of dihalopyrrole esters,99,299–301 the Handy group disclosed the first example of altering the site selectivity by modifying the reaction solvent (Scheme 68).302 Specifically, they observed that Suzuki–Miyaura coupling between dibromopyrrole 248 and 4-methoxyphenylboronic acid using Pd(PPh3)4 as the catalyst with aqueous sodium carbonate as the base in a 3:1 mixture of toluene and ethanol afforded a mixture of 249 and 250 in a ratio of 22 to 1, where the C5-coupled product (249) was isolated in 70% yield. Alternatively, when the reaction solvent was changed to dimethylformamide (DMF), the C3-coupled product 250 was obtained in 53% yield. Intrigued by this reversal of selectivity upon changing the solvent, the authors investigated the reaction in other solvents as well. In doing so, they found that more polar solvents, such as DMF and dimethylsulfoxide (DMSO), favored C3 coupling, whereas relatively less polar solvents favored C5 coupling.
Scheme 68.
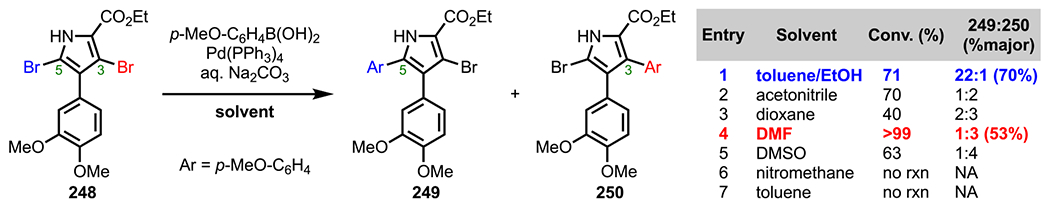
Reversal of Selectivity in 248 upon Changing Reaction Solvent
To explain this differential reactivity, the authors proposed two hypotheses. First, they envisioned that the nitrogen atom could direct C5 coupling303–305 in less polar solvents. This coordination could, however, be disrupted in solvents like DMF or DMSO, thus resulting in C3 coupling. Second, they proposed that the reversal of selectivity could also be attributed to changes in the electronics of C3 versus C5 caused by the differential solvation of 248 on the basis of solvent choice. To investigate this second hypothesis, they compared 1H NMR chemical shifts of the nonhalogenated precursor of 248 in different solvents. In CDCl3, the difference in 1H NMR chemical shifts (ΔδH) between the C3 and C5 protons was barely detectable (ΔδH = 0.08 ppm), and in DMSO-d6, the difference was more pronounced (ΔδH = 0.15 ppm), with the C3 proton remaining more deshielded. However, in a 3:1 mixture of benzene-d6 and methanol-d4, the electronics were reversed, with the C5 proton being more deshielded (ΔδH = −0.25 ppm).
Given that oxidative addition is generally favored at the more electron-deficient positions,40–42 these NMR studies were in good agreement with this experimentally observed selectivity trend. Specifically, C3 coupling was favored in polar solvents (e.g., DMSO or DMF), whereas C5 coupling was preferred in nonpolar solvents (e.g., toluene).306 The authors ultimately proceeded to study the scope of this reaction, where both electron-rich and electron-deficient boronic acids were competent for obtaining complementary selectivity upon solvent changes. Finally, this novel protocol was applied in a second-generation synthesis of the lamellarins.307 In this context, Handy and coworkers performed a sequential, one-pot Suzuki–Miyaura coupling between 248 and boronic acids 251 and 252 to obtain the bis-coupled product (253) in 36% yield (Scheme 69). In summary, this report provides fascinating insight into the reversal in site selectivity that can result by merely changing the reaction solvent. Furthermore, this study clearly set the stage for future investigations into this exciting subfield of site-selective cross-coupling chemistry.
Scheme 69.
One-Pot Sequential Suzuki–Miyaura Coupling of 248
In 2019, Sarpong and coworkers studied the cross-coupling of 3,5-dibromo-2-pyrone (59), an α-pyrone-derived dihalogenated heterocycle bearing two chemically inequivalent C–Br bonds.43 Building on the precedent for the site-selective cross-coupling of 59 established by Cho and coworkers,92,94,308 the authors sought to understand the underlying factors that guide selectivity, which was not easily explained using existing models.41 Consistent with the results of Cho and coworkers,94 it was observed that Suzuki–Miyaura coupling between 59 and phenylboronic acid in nonpolar solvents,309,310 such as 1,2-DCE, THF, and toluene, and in both the presence and the absence of CuI predominantly delivered the C3-coupled product (254) (Scheme 70A). On the contrary, in more polar solvents, such as DMSO and DMF, and in the presence of CuI, the C5-coupled product 255 was generally formed as the major product. Notably, the C5-coupled product (255) was observed only in the presence of CuI, and conducting the reaction in the absence of CuI exclusively generated the C3-coupled product (254).
Scheme 70.
(A) Suzuki–Miyaura Coupling with 3,5-Dibromo-2-pyrone (59) and (B) Oxidation Addition Experiments with 3,5-Dibromo-2-pyrone (59) (Adapted from ref 43. Copyright 2019 American Chemical Society)
Additionally, experiments were conducted to study the oxidative addition of 59 in the presence of Pd(PPh3)4, which results in the formation of both the corresponding C3 and C5 oxidative adducts, 257 and 258 (Scheme 70B).311 Consistent with the Suzuki–Miyaura coupling results, C3 Pd complex 257 was formed in nonpolar solvents (toluene), irrespective of whether CuI was added. In polar solvents (DMF), the formation of 257 was favored in the absence of CuI, whereas C5 Pd complex 258 was formed as the major product in the presence of CuI. The C3 site selectivity was attributed to the lower BDE of the C3–Br bond and the larger LUMO coefficient at the C3 position, which were both supported by computational findings on the basis of the conditions that favored C3 coupling. However, even more intriguing was the observed C5 site selectivity under a specified set of conditions. From both the Suzuki–Miyaura coupling and the oxidative addition results, it is clearly evident that C5 coupling was favored only in polar solvents and in the presence of CuI. To gain further insight into the effect of CuI and solvent on C5 selectivity, the authors chose to analyze the relative stabilities of the C3 and C5 Pd complexes (257 and 258) (Scheme 71).
Scheme 71.
Curtin–Hammett Scenario to Favor C5 Coupling over C3 Coupling
When the oxidative addition was conducted in DMF and in the presence of CuI, it was determined that C5 Pd complex 258 is kinetically favored and gives rise to 255 following a relatively low barrier for transmetalation. The C3 Pd complex 257 was identified to be thermodynamically favored and possesses a relatively higher transmetalation barrier. In 257-cis, the coordination of Cu to both the pyrone carbonyl oxygen and the bromide attached to the Pd center results in greater stabilization compared with 258, and this coordination is favored in polar solvents. Subsequently, the corresponding kinetic and thermodynamic stabilities of 257 and 258 were both experimentally and computationally established to arise from an initial CuI-assisted PPh3 dissociation312–314 to furnish the monophosphine ligated Pd complex 259315–317 as the active catalyst for the oxidative addition. Additionally, under the conditions that favor C5 coupling, it was established that the isomeric Pd complexes are interconvertible. Overall, this describes a Curtin–Hammett scenario,318 wherein rapid interconversion of the Pd complexes (257 and 258) occurs, and the ratio of the resulting cross-coupled products (i.e., 254/255) is solely dependent on the energy difference between the two respective turnover-limiting transition states for transmetalation.
This enhanced mechanistic understanding of the site-selective cross-coupling of 3,5-dibromo-2-pyrone (59) facilitated a unified retrosynthetic disconnection of two structurally related natural products, delavatine A and incarviatone A (Scheme 72).43,44 Consequently, a synthetic route was developed commencing with a sequential Stille–Stille coupling of 59 using stannanes 260a and 260b followed by the selective ring-opening of pyrone 261a, which furnished methyl ester 261 in a single pot. The sequential Stille–Stille coupling involves a site-selective C5 coupling between 59 and stannane 260a in N-methylpyrrolidone (NMP) (polar solvent) and in the presence of copper(I) thiophene-2-carboxylate (CuTC) (copper source), which is then followed by a second Stille coupling with stannane 260b at the remaining C3–Br bond. Next, 261 was converted to intermediate 262 via an electrocyclization cascade sequence, which was then elaborated to delavatine A and incarviatone A in two and eight steps, respectively.
Scheme 72.
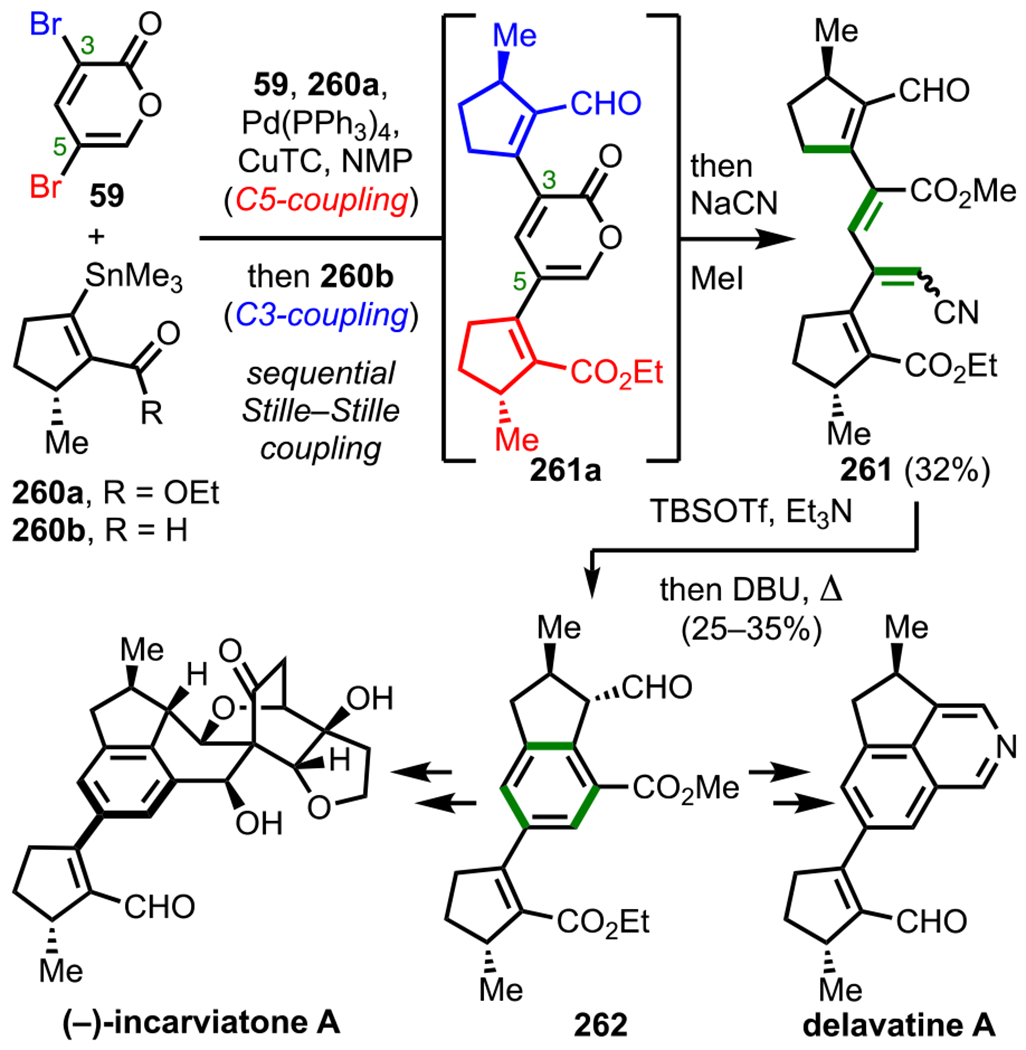
Application of 3,5-Dibromo-2-pyrone (59) Site-Selective Cross-Coupling to the Synthesis of Delavatine A and (–)-Incarviatone A
Recently, Sarpong and coworkers also studied a method to access pyrido[1,2-a]indole scaffolds starting from indole–pyrone adducts (Scheme 73).319 For one of the substrates, they conducted a site-selective C3 coupling between indole boronate ester 263 and 3,4-dibromo-2-pyrone (59) under the previously disclosed C3 coupling conditions92,94,308 to give the indole–pyrone adduct 264. Upon the ring-opening of 264 with sodium methoxide to reveal intermediate 264a, a spontaneous intramolecular cyclization proceeded to furnish pyrido[1,2-a]indole 264b in 29% yield.
Scheme 73.
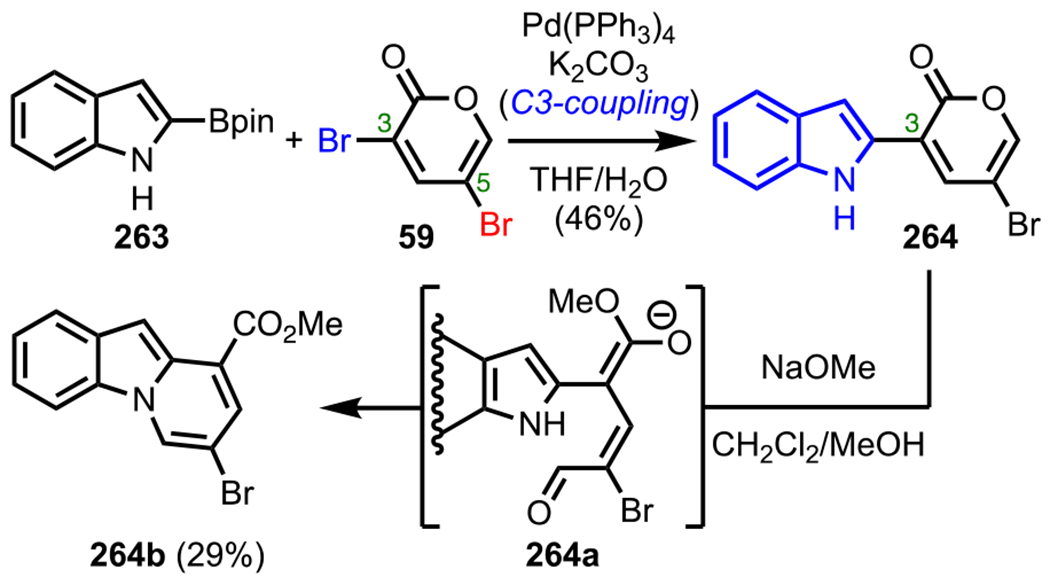
Application of 3,5-Dibromo-2-pyrone (59) Site-Selective Cross-Coupling to the Synthesis of Pyrido[1,2-α]indole 264b
In summary, the studies by Sarpong and coworkers provide new insight into how solvents and additives can be tuned to achieve complementary selectivity. Many of the previous examples involving site-selective cross-coupling considered oxidative addition to be the rate-determining step (RDS) or the turnover-limiting step (TLS). However, this more recent study from Sarpong and coworkers provides an example of transmetalation as the TLS, thus underscoring the notion that other elementary steps in the catalytic cycle can be turnover-limiting as well.38,73 Furthermore, it is illustrated that for unbiased systems (i.e., those that contain C–X bonds with relatively little intrinsic electronic differences), solvents and additives can be leveraged to control site-selectivity outcomes. Currently, there are limited examples where this paradigm for selectivity has been applied; however, the two examples discussed in this section showcase the potential of this approach to serve as an effective strategy in chemical synthesis.
8. PHOTOCHEMICALLY INITIATED
The last set of site-selective cross-coupling reactions to be discussed are those we classify as photochemically initiated. Recently, there have been significant developments in photocatalytic strategies for chemical synthesis, including photoredox catalysis,320,321 which have drastically expanded the repertoire of radical-based couplings. In the context of the site-selective cross-coupling of polyhalogenated arenes bearing identical halogen groups, these processes are fairly new (only a handful of reported examples to date) but have nonetheless proven to be effective in accessing products that have remained elusive using the traditional cross-coupling or SNAr chemistry. Photochemically initiated processes do not follow a traditional two-electron nucleophilic addition pathway but instead proceed through photomediated radical generation followed by the addition of this radical to a suitable acceptor (Scheme 74), thus providing complementary reactivity to more traditional methods. Because either coupling partner can serve as the radical precursor or acceptor, there are diverse and tunable reaction manifolds that can be developed for diverse product generation. The examples shown here illustrate, through distinct radical-based mechanisms, general strategies for both C–C and C–heteroatom formation.
Scheme 74.
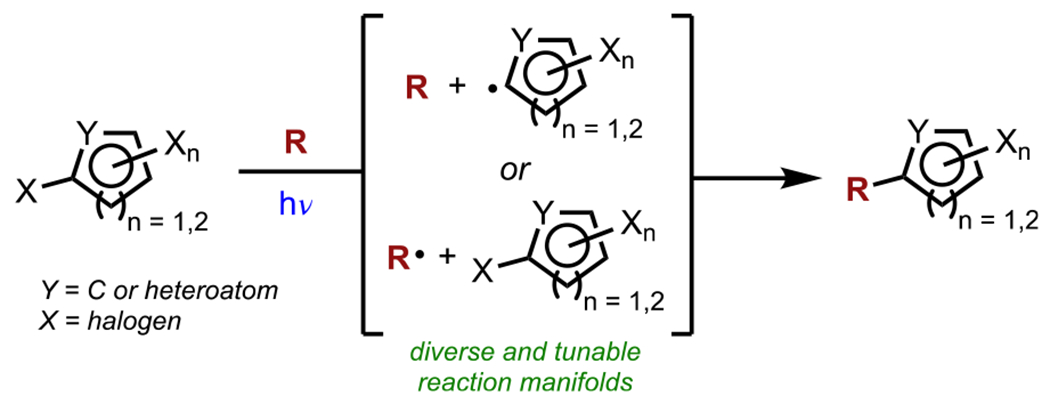
Photochemically Initiated Site-Selective Cross-Coupling
The first examples to be discussed are the photocatalytic C–F alkylation322 and C–F arylation323 of perfluoroarenes developed by Weaver and coworkers. As discussed in section 3 (electronic control), polyfluorinated arenes are of particular interest from a medicinal chemistry perspective; however, accessing functionalized fluorinated aryls from readily available perfluoroarenes through direct C–F activation remains challenging due to the thermodynamic and kinetic robustness of the C–F bond. The studies by Weaver and coworkers circumvents this inherent challenge through a novel, divergent radical-based mechanism to access both alkylated and arylated perfluoroarenes (Scheme 75). The proposed mechanism322,323 for these processes begins with the formation of perfluoroaryl radical 265 following a single electron reduction facilitated by a photoexcited Ir catalyst and diisopropylethylamine (DIPEA) as an electron donor. At this point, depending on the acceptor, 265 can lead to either the alkylated product or the arylated product. For alkylation, radical 265 adds to an alkene to generate alkyl radical 266, which then performs an H-atom abstraction from the DIPEA radical cation to give the final reduced alkylated perfluoroarene (267) and the iminium ion as a byproduct. Similarly, radical 265 can instead add to an arene to give aryl radical 268. The rearomatization of this aryl radical intermediate via either 269a or 269b then leads to the final arylated product (270). Notably, the arylation pathway results in dual C–F and C–H functionalization.
Scheme 75.
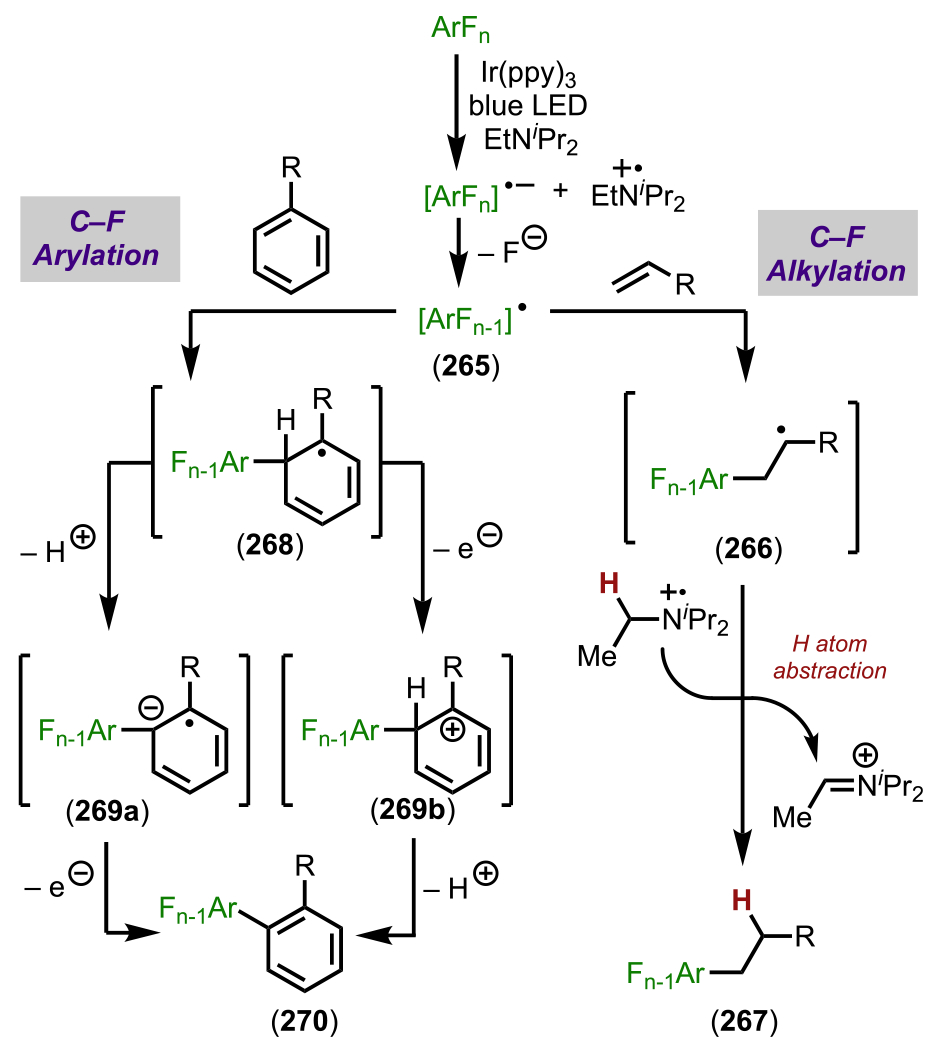
Mechanism of Photocatalytic C–F Alkylation and Arylation
For the reductive C–F alkylation (Scheme 76A), the treatment of various perfluoroarenes (271) with cyclic and acyclic alkenes, an Ir catalyst, and DIPEA under blue LED irradiation afforded a wide array of alkylated perfluoroarenes (272a–g). The C–F arylation (Scheme 76B) proceeded under similar Ir-catalyzed conditions to give fluorinated biaryls (273a–f) from the corresponding perfluoroarenes (271) and arene acceptors, which included both all-carbon and heteroarene acceptors.
Scheme 76.
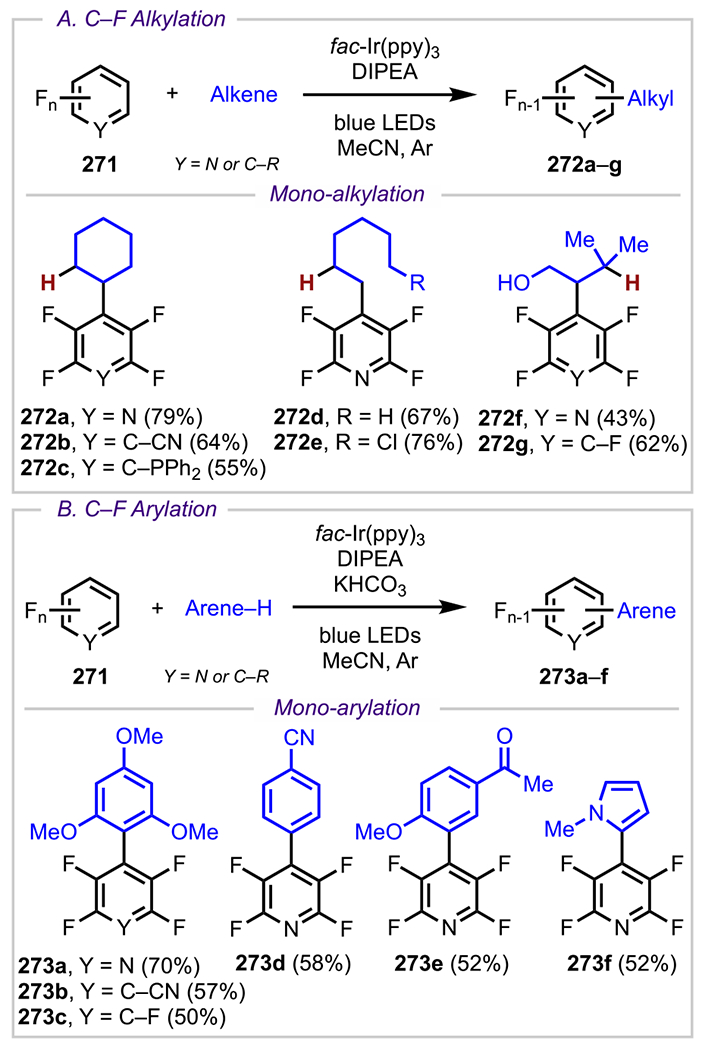
Photocatalyzed (A) C–F Alkylation and (B) C–F Arylation
In addition to the work by Weaver and coworkers, there have also been studies into leveraging photocatalysis for arene–heteroatom couplings, such as thiolation.324 Traditionally, the thiolation of halogenated arenes has been achieved via an SNAr pathway using a thiol nucleophile and an electron-deficient arene. However, comparable thiolations to electron-rich arenes, which are not readily amenable to SNAr, have proven to be challenging. In addition, such thiolations are not favored to proceed via traditional two-electron oxidative addition pathways. To obviate this clear challenge, in 2020, Glorius and coworkers developed a novel photo/Ir-catalyzed homolytic aromatic substitution reaction (Scheme 77A), which provided access to a variety of C5-thiolated heteroarenes (275a–j) from the corresponding polyhalogenated heteroarene precursors (274). This reaction is proposed to follow a radical chain mechanism (Scheme 77B) that initiates with the formation of a thiyl radical from thiol via a photocatalytic oxidation/deprotonation, which may either be stepwise or occur through an oxidative proton-coupled electron transfer (PCET).325 The thiyl radical then undergoes homolytic aromatic substitution at the more electron-rich C5 position of heteroarene 276 to give radical cation intermediate 277 with the concomitant extrusion of a chloride anion. Finally, electron transfer to 277 following a second oxidation/deprotonation of thiol provides product 275a and subsequent chain propagation. Overall, this method, which facilitates the addition of a heteroatom to an electron-rich system, provides complementary reactivity to traditional SNAr processes, which involve the addition of a nucleophile to electron-poor arenes.
Scheme 77.
(A) Photocatalytic Thiolation and (B) Proposed Mechanism
9. CONCLUSIONS AND OUTLOOK
Site-selective cross-couplings of polyhalogenated (hetero)arenes have received widespread attention in recent years due to their importance in building complex molecules, in particular, in medicinal, agrochemical, and materials chemistry. Applications toward developing elegant syntheses of pharmaceutically relevant drugs have been particularly impactful and have been highlighted in this Review. Whereas the selective functionalization of a C–X bond in a molecule bearing multiple, distinct halogens might follow the trend of I > Br > Cl > F, predicting and rationalizing the selectivity outcome in an arene bearing multiple, but identical, halogen types has remained challenging. Therefore, several factors have been identified to enable the differentiation between carbon positions bearing identical halogen atoms. Initially, this distinction was achieved purely by leveraging the inherent electronics of the system, where the most electrophilic C–X bond would undergo faster oxidative addition. In addition to electronically driven site-selective cross-coupling processes, steric effects were also identified to guide oxidative addition to the least sterically encumbered carbon position. Following these advances, the coordinating ability of functional groups was leveraged to direct oxidative addition to a particular position. Subsequently, methods were developed to achieve site selectivity by tuning the electronic and steric influences of an external ligand. More recently, selectivity has been achieved by altering the choice of reaction solvents and additives.
Whereas leveraging the intrinsic electronic and sterics of a substrate remains the traditional approach to achieving site-selective cross-coupling, controlling site-selectivity outcomes by altering other factors, such as the metal precatalyst, external ligands, reaction solvent, or choice of additives, presents novel directions to further advance this highly valuable subfield of cross-coupling chemistry. Whereas current investigations have been mainly steered toward identifying conditions to obtain a desired selectivity, more effort is needed to gain a deeper understanding of the mechanistic rationale behind the selectivity observed in various (hetero)arenes. Better insight into these selectivity-guiding processes will provide a more methodical, and therefore more efficient, approach toward predicting conditions suitable for obtaining a particular selectivity outcome. Furthermore, on the basis of the large data sets that are emerging for site-selective cross-coupling, employing other modern strategies, such as linear regression models and machine-learning algorithms, to identify reaction conditions could also prove fruitful in further advancing this field.326,327 These systematic approaches could also prove to be particularly helpful for effecting site-selective cross-couplings in more complex systems, such as natural-product-like molecules.
Finally, an emerging area of interest includes achieving site-selective cross-couplings under photocatalytic conditions. Whereas many subfields of photocatalysis are still in their early development, the examples discussed in this Review showcase how new, state-of-the-art reaction manifolds can be accessed, thus enabling nonintuitive, site-selective cross-couplings that do not abide by the traditional cross-coupling catalytic cycle (Scheme 2). Overall, the site-selective cross-coupling of (hetero)arenes with identical halogen substitution is a highly diverse and versatile field, which, as exemplified by this Review, presents countless future opportunities to not only advance this specific field but also help advance chemical synthesis as a whole.
Funding
V.P. acknowledges TRDRP for a predoctoral fellowship. M.A.P. acknowledges both the NSF (DGE 1752814) and the NIH (F31GM139368) for predoctoral fellowships. R.S. is grateful to the NSF (CHE-1856228) for funding of the work described here conducted in the Sarpong laboratory.
Biographies
Vignesh Palani received his B.S. in chemistry with honors (summa cum laude) from the University of Minnesota-Twin Cities in 2016. He earned his Ph.D. in chemistry from the University of California, Berkeley in 2020, where he carried out his research in the laboratory of Professor Richmond Sarpong. At UC Berkeley, he worked on the total synthesis of complex natural products and new reaction development.
Melecio Perea received his B.S. in chemistry (graduating summa cum laude) from New Mexico Highlands University in 2016. That same year he began his graduate studies at the University of California, Berkeley, working in the lab of Professor Richmond Sarpong. As an NSF and NIH predoctoral fellow, his research at Berkeley has focused on developing strategies for the synthesis of complex natural products.
Richmond Sarpong is a Professor of Chemistry at the University of California Berkeley, where his research group specializes in synthetic organic chemistry. He completed his undergraduate studies at Macalester College in St. Paul, Minnnesota (with Prof. Rebecca C. Hoye) and his graduate work with Prof. Martin Semmelhack at Princeton. He conducted postdoctoral studies at Caltech with Prof. Brian Stoltz and joined the faculty at Berkeley in 2004.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.chemrev.1c00513
The authors declare no competing financial interest.
Contributor Information
Vignesh Palani, Department of Chemistry, University of California, California 94720, United States.
Melecio A. Perea, Department of Chemistry, University of California, California 94720, United States.
Richmond Sarpong, Department of Chemistry, University of California, California 94720, United States.
REFERENCES
- (1).Carey JS; Laffan D; Thomson C; Williams MT Analysis of the Reactions Used for the Preparation of Drug Candidate Molecules. Org. Biomol Chem 2006, 4, 2337–2347. [DOI] [PubMed] [Google Scholar]
- (2).Pozharskii AF; Soldatenkov AT; Katrizky AR Heterocycles in Life and Society; Wiley: New York, 1997; pp 1–361. [Google Scholar]
- (3).Reis J; Gaspar A; Milhazes N; Borges F Chromone as a Privileged Scaffold in Drug Discovery: Recent Advances. J. Med. Chem 2017, 60, 7941–7957. [DOI] [PubMed] [Google Scholar]
- (4).Corbet J-P; Mignani G Selected Patented Cross-Coupling Reaction Technologies. Chem. Rev 2006, 106, 2651–2710. [DOI] [PubMed] [Google Scholar]
- (5).de Meijere A; Diederich F Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004; pp 1–882. [Google Scholar]
- (6).Boubertakh O; Goddard J-P Construction and Functionalization of Heteroarenes by Use of Photoredox Catalysis. Eur. J. Org. Chem 2017, 2017, 2072–2084. [Google Scholar]
- (7).Li JJ; Gribble GW Palladium in Heterocyclic Chemistry; Pergamon: Oxford, U.K., 2000; pp 1–650. [Google Scholar]
- (8).Bellina F; Rossi R Recent Advances in the Synthesis of (Hetero)Aryl-Substituted Heteroarenes via Transition Metal-Catalysed Direct (Hetero)Arylation of Heteroarene C–H Bonds with Aryl Halides or Pseudohalides, Diaryliodonium Salts, and Potassium Aryltrifluoroborates. Tetrahedron 2009, 65, 10269–10310. [Google Scholar]
- (9).Nair V; Rajesh C; Vinod AU; Bindu S; Sreekanth AR; Mathen JS; Balagopal L Strategies for Heterocyclic Construction via Novel Multicomponent Reactions Based on Isocyanides and Nucleophilic Carbenes. Acc. Chem. Res 2003, 36, 899–907. [DOI] [PubMed] [Google Scholar]
- (10).Andraos J A New Paradigm for Designing Ring Construction Strategies for Green Organic Synthesis: Implications for the Discovery of Multicomponent Reactions to Build Molecules Containing a Single Ring. Beilstein J. Org. Chem 2016, 12, 2420–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Keerthi Krishnan K; Ujwaldev SM; Saranya S; Anilkumar G; Beller M Recent Advances and Perspectives in the Synthesis of Heterocycles viaZinc Catalysis. Adv. Synth. Catal 2019, 361, 382–404. [Google Scholar]
- (12).Dobrounig P; Trobe M; Breinbauer R Sequential and Iterative Pd-Catalyzed Cross-Coupling Reactions in Organic Synthesis. Monatsh. Chem 2017, 148, 3–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rohrbach S; Smith AJ; Pang JH; Poole DL; Tuttle T; Chiba S; Murphy JA Concerted Nucleophilic Aromatic Substitution Reactions. Angew. Chem., Int. Ed 2019, 58, 16368–16388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Galabov B; Nalbantova D; Schleyer P. v. R.; Schaefer HF III Electrophilic Aromatic Substitution: New Insights into an Old Class of Reactions. Acc. Chem. Res 2016, 49, 1191–1199. [DOI] [PubMed] [Google Scholar]
- (15).Bunnett JF; Zahler RE Aromatic Nucleophilic Substitution Reactions. Chem. Rev 1951, 49, 273–412. [Google Scholar]
- (16).Schnürch M; Spina M; Khan AF; Mihovilovic MD; Stanetty P Halogen Dance Reactions–A Review. Chem. Soc. Rev 2007, 36, 1046–1057. [DOI] [PubMed] [Google Scholar]
- (17).Evano G; Nitelet A; Thilmany P; Dewez DF Metal-Mediated Halogen Exchange in Aryl and Vinyl Halides: A Review. Front. Chem 2018, 6, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Davies HML; Morton D Recent Advances in C–H Functionalization. J. Org. Chem 2016, 81, 343–350. [DOI] [PubMed] [Google Scholar]
- (19).Jana R; Pathak TP; Sigman MS Advances in Transition Metal (Pd, Ni, Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners. Chem. Rev 2011, 111, 1417–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wu X-F; Anbarasan P; Neumann H; Beller M From Noble Metal to Nobel Prize: Palladium-Catalyzed Coupling Reactions as Key Methods in Organic Synthesis. Angew. Chem., Int. Ed 2010, 49, 9047–9050. [DOI] [PubMed] [Google Scholar]
- (21).Dorel R; Grugel CP; Haydl AM The Buchwald–Hartwig Amination After 25 Years. Angew. Chem. Int. Ed 2019, 58, 17118–17129. [DOI] [PubMed] [Google Scholar]
- (22).Campeau L-C; Hazari N Cross-Coupling and Related Reactions: Connecting Past Success to the Development of New Reactions for the Future. Organometallics 2019, 38, 3–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Suzuki A Cross-Coupling Reactions of Organoboranes: An Easy Way to Construct C–C Bonds (Nobel Lecture). Angew. Chem., Int. Ed 2011, 50, 6722. [DOI] [PubMed] [Google Scholar]
- (24).Miyaura N; Yamada K; Suzuki A A New Stereospecific Cross-Coupling by the Palladium-Catalyzed Reaction of 1-Alkenylboranes with 1-Alkenyl or 1-Alkynyl Halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar]
- (25).Miyaura N; Suzuki A Stereoselective Synthesis of Arylated (E)-Alkenes by the Reaction of Alk-1-enylboranes with Aryl Halides in the Presence of Palladium Catalyst. J. Chem. Soc., Chem. Commun 1979, 866–867. [Google Scholar]
- (26).Negishi E Magical Power of Transition Metals: Past, Present, and Future (Nobel Lecture). Angew. Chem., Int. Ed 2011, 50, 6738–6764. [DOI] [PubMed] [Google Scholar]
- (27).Negishi E; King AO; Okukado N Selective Carbon–Carbon Bond Formation via Transition Metal Catalysis. 3 A Highly Selective Synthesis of Unsymmetrical Biaryls and Diarylmethanes by the Nickel- or Palladium-Catalyzed Reaction of Aryl- and Benzylzinc Derivatives with Aryl Halides. J. Org. Chem 1977, 42, 1821–1821. [Google Scholar]
- (28).Milstein D; Stille JK A General, Selective, and Facile Method for Ketone Synthesis from Acid Chlorides and Organotin Compounds Catalyzed by Palladium. J. Am. Chem. Soc 1978, 100, 3636–3638. [Google Scholar]
- (29).Milstein D; Stille JK Palladium-Catalyzed Coupling of Tetraorganotin Compounds with Aryl and Benzyl Halides. Synthetic Utility and Mechanism. J. Am. Chem. Soc 1979, 101, 4992–4998. [Google Scholar]
- (30).Heck RF; Nolley JP Jr. Palladium-Catalyzed Vinylic Hydrogen Substitution Reactions with Aryl, Benzyl, and Styryl Halides. J. Org. Chem 1972, 37, 2320–2322. [Google Scholar]
- (31).Almond-Thynne J; Blakemore DC; Pryde DC; Spivey AC Site-Selective Suzuki–Miyaura Coupling of Heteroaryl Halides – Understanding the Trends for Pharmaceutically Important Classes. Chem. Sci 2017, 8, 40–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Whereas the term “regioselective” may be used, we prefer to use the term “site-selective” instead (see refs 32 and 33):; Huang Z; Dong G Site-Selectivity Control in Organic Reactions: AQuest to Differentiate Reactivity Among the Same Kind of Functional Groups. Acc. Chem. Res 2017, 50, 465–471. [DOI] [PubMed] [Google Scholar]
- (33).Seebach D Methods of Reactivity Umpolung. Angew. Chem., Int. Ed. Engl 1979, 18, 239–258. [Google Scholar]
- (34).Shen C; Wei Z; Jiao H; Wu X-F Ligand- and Solvent-Tuned Chemoselective Carbonylation of Bromoaryl Triflates. Chem. - Eur. J 2017, 23, 13369–13378. [DOI] [PubMed] [Google Scholar]
- (35).Espino G; Kurbangalieva A; Brown JM Aryl Bromide/Triflate Selectivities Reveal Mechanistic Divergence in Palladium-Catalysed Couplings; The Suzuki–Miyaura Anomaly. Chem. Commun 2007, 1742–1744. [DOI] [PubMed] [Google Scholar]
- (36).Miyaura N; Suzuki A Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev 1995, 95, 2457–2483. [Google Scholar]
- (37).Hassan Z; Patonay T; Langer P Regioselective Suzuki–Miyaura Reactions of Aromatic Bis-triflates: Electronic versus Steric Effects. Synlett 2013, 24, 412–423. [Google Scholar]
- (38).Legault CY; Garcia Y; Merlic CA; Houk KN Origin of Regioselectivity in Palladium-Catalyzed Cross-Coupling Reactions of Polyhalogenated Heterocycles. J. Am. Chem. Soc 2007, 129, 12664–12665. [DOI] [PubMed] [Google Scholar]
- (39).Wang J-R; Manabe K Transition-Metal-Catalyzed Site-Selective Cross-Coupling of Di- and Polyhalogenated Compounds. Synthesis 2009, 2009, 1405–1427. [Google Scholar]
- (40).Manabe K; Yamaguchi M Catalyst-Controlled Site-Selectivity Switching in Pd-Catalyzed Cross-Coupling of Dihaloarenes. Catalysts 2014, 4, 307–320. [Google Scholar]
- (41).Fairlamb IJS Regioselective (Site-Selective) Functionalisation of Unsaturated Halogenated Nitrogen, Oxygen and Sulfur Heterocycles by Pd-Catalysed Cross-Couplings and Direct Arylation Processes. Chem. Soc. Rev 2007, 36, 1036–1045. [DOI] [PubMed] [Google Scholar]
- (42).Schröter S; Stock C; Bach T Regioselective Cross-Coupling Reactions of Multiple Halogenated Nitrogen-, Oxygen-, and Sulfur-Containing Heterocycles. Tetrahedron 2005, 61, 2245–2267. [Google Scholar]
- (43).Palani V; Hugelshofer CL; Kevlishvili I; Liu P; Sarpong R A Short Synthesis of Delavatine A Unveils New Insights into Site-Selective Cross-Coupling of 3,5-Dibromo-2-pyrone. J. Am. Chem. Soc 2019, 141, 2652–2660. [DOI] [PubMed] [Google Scholar]
- (44).Palani V; Hugelshofer CL; Sarpong R A Unified Strategy for the Enantiospecific Total Synthesis of Delavatine A and Formal Synthesis of Incarviatone A. J. Am. Chem. Soc 2019, 141, 14421–14432. [DOI] [PubMed] [Google Scholar]
- (45).For selected examples of the mechanism of cross-coupling reactions, see refs 45–48:; Biffis A; Centomo P; Del Zotto A; Zecca M Pd Metal Catalysts for Cross-Couplings and Related Reactions in the 21stCentury: A Critical Review. Chem. Rev 2018, 118, 2249–2295. [DOI] [PubMed] [Google Scholar]
- (46).Meconi GM; Vummaleti SVC; Luque-Urrutia JA; Belanzoni P; Nolan SP; Jacobsen H; Cavallo L; Solá M; Poater A Mechanism of the Suzuki–Miyaura Cross-Coupling Reaction Mediated by [Pd(NHC)(allyl)Cl] Precatalysts. Organometallics 2017, 36, 2088–2095. [Google Scholar]
- (47).Diccianni JB; Diao T Mechanisms of Nickel-Catalyzed Cross-Coupling Reactions. Trends Chem. 2019, 1, 830–844. [Google Scholar]
- (48).Espinet P; Echavarren AM The Mechanisms of the Stille Reaction. Angew. Chem., Int. Ed 2004, 43, 4704–4734. [DOI] [PubMed] [Google Scholar]
- (49).Li J; Jin L; Liu C; Lei A Transmetalation of Ar1ZnX with [Ar2–Pd–X] is the Rate-Limiting Step: Kinetic Insights from a Live Pd-Catalyzed Negishi Coupling. Org. Chem. Front 2014, 1, 50–53. [Google Scholar]
- (50).Stenlid JH; Brinck T Nucleophilic Aromatic Substitution Reactions Described by the Local Electron Attachment Energy. J. Org. Chem 2017, 82, 3072–3083. [DOI] [PubMed] [Google Scholar]
- (51).Busch M; Wodrich MD; Corminboeuf C A Generalized Picture of C–C Cross-Coupling. ACS Catal. 2017, 7, 5643–5653. [Google Scholar]
- (52).Fauvarque J-F; Pflüger F; Troupel M Kinetics of Oxidative Addition of Zerovalent Palladium to Aromatic Iodides. J. Organomet. Chem 1981, 208, 419–427. [Google Scholar]
- (53).Mortier J Arene Chemistry: Reaction Mechanism and Methods for Aromatic Compounds; John Wiley & Sons, Inc.: Hoboken, NJ, 2016; pp 131–295. [Google Scholar]
- (54).Gebauer J; Arseniyadis S; Cossy J A Concise Total Synthesis of Melithiazole C. Org. Lett 2007, 9, 3425–3427. [DOI] [PubMed] [Google Scholar]
- (55).Flegeau EF; Popkin ME; Greaney MF Direct Arylation of Oxazoles at C2. A Concise Approach to Consecutively Linked Oxazoles. Org. Lett 2008, 10, 2717–2720. [DOI] [PubMed] [Google Scholar]
- (56).Fairlamb IJS; O’Brien CT; Lin Z; Lam KC Regioselectivity in the Sonogashira Coupling of 4,6-Dichloro-2-pyrone. Org. Biomol. Chem 2006, 4, 1213–1216. [DOI] [PubMed] [Google Scholar]
- (57).Handy ST; Zhang Y A Simple Guide for Predicting Regioselectivity in the Coupling of Polyhaloheteroaromatics. Chem. Commun 2006, 299–301. [DOI] [PubMed] [Google Scholar]
- (58).Ceide SC; Montalban AG Microwave-Assisted, Efficient and Regioselective Pd-Catalyzed C-Phenylation of Halopyrimidines. Tetrahedron Lett. 2006, 47, 4415–4418. [Google Scholar]
- (59).Maes BUW; Verbeeck S; Verhelst T; Ekomié A; von Wolff N; Lefèvre G; Mitchell EA; Jutand A Oxidative Addition of Haloheteroarenes to Palladium(0): Concerted versus SNAr-Type Mechanism. Chem. - Eur. J 2015, 21, 7858–7865. [DOI] [PubMed] [Google Scholar]
- (60).Kazi SA; Campi EM; Hearn MTW A Convenient and Efficient One Pot Synthesis of Unsymmetrically Substituted p-Terphenyls via a Phosphine-Free Double Suzuki Cross-Coupling Protocol Using 1,4-Dibromo-2-nitrobenzene as the Substrate. Tetrahedron 2018, 74, 1731–1741. [Google Scholar]
- (61).Liu X-W; Echavarren J; Zarate C; Martin R Ni-Catalyzed Borylation of Aryl Fluorides via C–F Cleavage. J. Am. Chem. Soc 2015, 137, 12470–12473. [DOI] [PubMed] [Google Scholar]
- (62).Campbell MG; Ritter T Modern Carbon–Fluorine Bond Forming Reactions for Aryl Fluoride Synthesis. Chem. Rev 2015, 115, 612–633. [DOI] [PubMed] [Google Scholar]
- (63).O’Hagan D Understanding Organofluorine Chemistry. An Introduction to the C–F Bond. Chem. Soc. Rev 2008, 37, 308–319. [DOI] [PubMed] [Google Scholar]
- (64).Ahrens T; Kohlmann J; Ahrens M; Braun T Functionalization of Fluorinated Molecules by Transition-Metal-Mediated C–F Bond Activiation to Access Fluorinated Building Blocks. Chem. Rev 2015, 115, 931–972. [DOI] [PubMed] [Google Scholar]
- (65).Lennox AJJ Meisenheimer Complexes in SNAr Reactions: Intermediates of Transisiton States? Angew. Chem. Int. Ed 2018, 57, 14686–14688. [DOI] [PubMed] [Google Scholar]
- (66).Chambers RD; Martin PA; Sandford G; Williams DLH Mechanisms of Reactions of Halogenated Compounds: Part 7. Effects of Fluorine and Other Groups as Substituents on Nucleophilic Aromatic Substitution. J. Fluorine Chem 2008, 129, 998–1002. [Google Scholar]
- (67).Luo Z-J; Zhao H-Y; Zhang X Highly Selective Pd-Catalyzed Direct C–F Bond Arylation of Polyfluoroarenes. Org. Lett 2018, 20, 2543–2546. [DOI] [PubMed] [Google Scholar]
- (68).Saijo H; Sakaguchi H; Ohashi M; Ogoshi S Base-Free Hiyama Coupling Reaction via a Group 10 Metal Fluoride Intermediate Generated by C–F Bond Activation. Organometallics 2014, 33, 3669–3672. [Google Scholar]
- (69).Welsch ME; Snyder SA; Stockwell BR Privileged Scaffolds for Library Design and Drug Discovery. Curr. Opin. Chem. Biol 2010, 14, 347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Jampilek J Heterocycles in Medicinal Chemistry. Molecules 2019, 24, 3839–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Joule JA; Mills K Heterocyclic Chemistry, 5th ed.; Wiley-John Wiley & Sons Ltd.: West Sussex, U.K., 2014; pp 645–664. [Google Scholar]
- (72).Terrier F Modern Nucleophilic Aromatic Substitution; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp 11–15. [Google Scholar]
- (73).Garcia Y; Schoenebeck F; Legault CY; Merlic CA; Houk KN Theoretical Bond Dissociation Energies of Halo-Heterocycles: Trends and Relationships to Regioselectivity in Palladium-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc 2009, 131, 6632–6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Kikuchi O; Hondo Y; Morihashi K; Nakayama M An Ab Initio Molecular Orbital Study of Pyridyl Radicals. Bull. Chem. Soc. Jpn 1988, 61, 291–292. [Google Scholar]
- (75).Jones J; Bacskay GB; Mackie JC; Doughty A Ab InitioStudies of the Thermal Decomposition of Azaaromatics: Free Radical versus Intramolecular Mechanism. J. Chem. Soc., Faraday Trans 1995, 91, 1587–1592. [Google Scholar]
- (76).Prajapati D; Schulzke C; Kindermann MK; Kapdi AR Selective Palladium-Catalysed Arylation of 2,6-Dibromopyridine Using N-Heterocyclic Carbene Ligands. RSC Adv. 2015, 5, 53073–53085. [Google Scholar]
- (77).Lee K; Seomoon D; Lee PH Highly Efficient Catalytic Synthesis of Substituted Allenes Using Indium. Angew. Chem., Int. Ed 2002, 41, 3901–3903. [DOI] [PubMed] [Google Scholar]
- (78).Khoje AD; Gundersen L-L Reactivity and Regioselectivity in Stille Couplings of 3-Substituted 2,4-Dichloropyridines. Tetrahedron Lett. 2011, 52, 523–525. [Google Scholar]
- (79).Ahmed A; Sharif M; Shoaib K; Reimann S; Iqbal J; Patonay T; Spannenberg A; Langer P Synthesis of 2,6-Diaryl-3-(trifluoromethyl)pyridines by Regioselective Suzuki–Miyaura Reactions of 2,6-Dichloro-3-(trifluoromethyl)pyridine. Tetrahedron Lett. 2013, 54, 1669–1672. [Google Scholar]
- (80).Malacea R; Chahdoura F; Devillard M; Saffon N; Gómez M; Bourissou D Ortho-(Dimesitylboryl)phenylphosphines: Positive Boryl Effect in the Palladium-Catalyzed Suzuki–Miyaura Coupling of 2-Chloropyridines. Adv. Synth. Catal 2013, 355, 2274–2284. [Google Scholar]
- (81).Ehlers P; Reimann S; Erfle S; Villinger A; Langer P Synthesis of 2-Aryl-3,4,5,6-tetrachloropyridines and 2,6-Diaryl-3,4,5-trichloropyridines by Site-Selective Suzuki–Miyaura Reactions of Pentachloropyridine. Synlett 2010, 2010, 1528–1532. [Google Scholar]
- (82).Ackermann L; Althammer A Domino N–H/C–H Bond Activation: Palladium-Catalyzed Synthesis of Annulated Heterocycles Using Dichloro(hetero)arenes. Angew. Chem., Int. Ed 2007, 46, 1627–1629. [DOI] [PubMed] [Google Scholar]
- (83).Hostyn S; Van Baelen G; Lemière GLF; Maes BUW Synthesis of r-Carbolines Starting from 2,3-Dichloropyridines and Substituted Anilines. Adv. Synth. Catal 2008, 350, 2653–2660. [Google Scholar]
- (84).Laha JK; Petrou P; Cuny GD One-Pot Synthesis of y-Carbolines via Sequential Palladium-Catalyzed Aryl Amination and Intramolecular Arylation. J. Org. Chem 2009, 74, 3152–3155. [DOI] [PubMed] [Google Scholar]
- (85).Barckholtz C; Barckholtz TA; Hadad CM C–H and N–H Bond Dissociation Energies of Small Aromatic Hydrocarbons. J. Am. Chem. Soc 1999, 121, 491–500. [Google Scholar]
- (86).Solberg J; Undheim K; Pettersson L; Ohman L-O; Ruiz J; Colacio E; Mulichak AM; Alminger T; Erickson M; Grundevik I; Hagin I; Hoffman K-J; Johansson S; Larsson S; Lofberg I; Ohlson K; Persson B; Skanberg I; Tekenbergs-Hjelte L Regiochemistry in Pd-Catalyzed Organotin Reactions with Halopyrimidines. Acta Chem. Scand 1989, 43, 62–68. [Google Scholar]
- (87).Young IS; Glass A-L; Cravillion T; Han C; Zhang H; Gosselin F Palladium-Catalyzed Site-Selective Amidation of Dichloroazines. Org. Lett 2018, 20, 3902–3906. [DOI] [PubMed] [Google Scholar]
- (88).Dickinson JM Microbial Pyran-2-ones and Dihydropyran-2-ones. Nat. Prod. Rep 1993, 10, 71–98. [DOI] [PubMed] [Google Scholar]
- (89).McGlacken GP; Fairlamb IJS 2-Pyrone Natural Products and Mimetics: Isolation, Characterization, and Biological Activity. Nat. Prod. Rep 2005, 22, 369–385. [DOI] [PubMed] [Google Scholar]
- (90).Marrison LR; Dickinson JM; Fairlamb IJS Bioactive 4-Substituted-6-methyl-2-pyrones with Promising Cytotoxicity Against A2780 and K562 Cell Lines. Bioorg. Med. Chem. Lett 2002, 12, 3509–3513. [DOI] [PubMed] [Google Scholar]
- (91).Fairlamb IJS; Marrison LR; Dickinson JM; Lu F-J; Schmidt JP 2-Pyrones Possessing Antimicrobial and Cytotoxic Activities. Bioorg. Med. Chem 2004, 12, 4285–4299. [DOI] [PubMed] [Google Scholar]
- (92).Lee J-H; Park J-S; Cho C-G Regioselective Synthesis of 3-Alkynyl-5-bromo-2-pyrones via Pd-Catalyzed Couplings on 3,5-Dibromo-2-pyrone. Org. Lett 2002, 4, 1171–1173. [DOI] [PubMed] [Google Scholar]
- (93).Cho H-K; Cho C-G Preparation of 3,5-Dibromo-2-pyrone from Coumalic Acid. Org. Synth 2015, 92, 148–155. [Google Scholar]
- (94).Ryu K-M; Gupta AK; Han JW; Oh CH; Cho C-G Regiocontrolled Suzuki–Miyaura Couplings of 3,5-Dibromo-2-pyrone. Synlett 2004, 12, 2197–2199. [Google Scholar]
- (95).Fairlamb IJS; O’Brien CT; Lin Z; Lam KC Regioselectivity in the Sonogashira Coupling of 4,6-Dichloro-2-pyrone. Org. Biomol. Chem 2006, 4, 1213–1216. [DOI] [PubMed] [Google Scholar]
- (96).Joule JA; Mills K Heterocyclic Chemistry, 5th ed.; Wiley-John Wiley & Sons Ltd.: West Sussex, U.K., 2014; pp 9–10. [Google Scholar]
- (97).Liu J.-t.; Simmons CJ; Xie H; Yang F; Zhao X; Tang Y; Tang W Synthesis of Highly Substituted Benzofuran-Containing Natural Products via Rh-Catalyzed Carbonylative Benzannulation. Adv. Synth. Catal 2017, 359, 693–697. [Google Scholar]
- (98).Schröter S; Bach T Di- and Triarylsubstituted Pyrroles by Sequential Regioselective Cross-Coupling Reactions. Heterocycles 2007, 74, 569–594. [Google Scholar]
- (99).Handy ST; Zhang Y Regioselective Couplings of Dibromopyrrole Esters. Synthesis 2006, 2006, 3883–3887. [Google Scholar]
- (100).Nguyen H; Nguyen DX; Tran TQ; Vo BN; Nguyen TH; Vuong TMH; Dang TT Programmed Synthesis of Tetraarylthieno[3,2-b]Thiophene by Site-selective Suzuki Cross-Coupling Reactions of Tetrabromothieno[3,2-b]Thiophene. Synlett 2013, 25, 93–96. [Google Scholar]
- (101).Strotman NA; Chobanian HR; He J; Guo Y; Dormer PG; Jones CM; Steves JE Catalyst-Controlled Regioselective Suzuki Couplings at Both Positions of Dihaloimidazoles, Dihalooxazoles, and Dihalothiazoles. J. Org. Chem 2010, 75, 1733–1739. [DOI] [PubMed] [Google Scholar]
- (102).Niculescu-Duvaz D; Niculescu-Duvaz I; Suijkerbuijk BMJM; Ménard D; Zambon A; Davies L; Pons J-F; Whittaker S; Marais R; Springer CJ Potent BRAF Kinase Inhibitors Based on 2,4,5-Trisubstituted Imidazole with Naphthyl and Benzothiophene 4-Substituents. Bioorg. Med. Chem 2013, 21, 1284–1304. [DOI] [PubMed] [Google Scholar]
- (103).Recnik L-M; Abd El Hameid M; Haider M; Schnurch M; Mihovilovic M Selective Sequential Cross-Coupling Reactions on Imidazole towards Meurodazine and Analogues. Synthesis 2013, 45, 1387–1405. [Google Scholar]
- (104).Ascierto PA; Kirkwood JM; Grob J-J; Simeone E; Grimaldi AM; Maio M; Palmieri G; Testori A; Marincola FM; Mozzillo N The Role of BRAF V600 Mutation in Melanoma. J. Transl. Med 2012, 10, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Kawasaki I; Katsuma H; Nakayama Y; Yamashita M; Ohta S Total Synthesis of Topsentin, Antiviral and Antitumor Bis(indolyl)imidazole. Heterocycles 1998, 48, 1887–1901. [Google Scholar]
- (106).Alford PE Lithiation-Based and Magnesium-Based Strategies for the Functionalization of Imidazole: 2001–2010. In Metalation of Azoles and Related Five-Membered Ring Heterocycles. Topics in Heterocyclic Chemistry; Gribble GW, Ed.; Springer: Berlin, 2012; Vol. 29, pp 77–102. [Google Scholar]
- (107).Yamauchi T; Shibahara F; Murai T Facile Synthetic Method for Diverse Polyfunctionalized Imidazoles by Means of Pd-Catalyzed C–H Bond Arylation of N-Methyl-4,5-dibromoimidazole. J. Org. Chem 2014, 79, 7185–7192. [DOI] [PubMed] [Google Scholar]
- (108).Joo JM; Touré BB; Sames D C–H Bonds as Ubiquitous Functionality: AGeneral Approach to Complex Arylated Imidazoles via Regioselective Sequential Arylation of All Three C–H Bonds and Regioselective N-Alkylation Enabled by SEM-Group Transposition. J. Org. Chem 2010, 75, 4911–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Hodgetts KJ; Kershaw MT Ethyl 2-Chlorooxazole-4-carboxylate: AVersatile Intermediate for the Synthesis of Substituted Oxazoles. Org. Lett 2002, 4, 2905–2907. [DOI] [PubMed] [Google Scholar]
- (110).Khera RA; Ali A; Hussain M; Tatar J; Villinger A; Langer P Synthesis of Arylated Pyrazoles by Site-selective Suzuki–Miyaura Reactions of Tribromopyrazoles. Synlett 2010, 2010, 1923–1926. [Google Scholar]
- (111).Christoforou IC; Koutentis PA; Rees CW Regiospecific Suzuki Coupling of 3,5-Dichloroisothiazole-4-carbonitrile. Org. Biomol. Chem 2003, 1, 2900–2907. [DOI] [PubMed] [Google Scholar]
- (112).Iddon B; Tønder JE; Hosseini M; Begtrup M The N-vinyl Group as a Protection Group of the Preparation of 3(5)-Substituted Pyrazoles via Bromine–Lithium Exchange. Tetrahedron 2007, 63, 56–61. [Google Scholar]
- (113).Cutrí CCC; Garozzo A; Siracusa MA; Sarvá MC; Tempera G; Geremia E; Pinizzotto MR; Guerrera F Synthesis and Antiviral Activity of a New Series of 4-Isothiazolecarbonitriles. Bioorg. Med. Chem 1998, 6, 2271–2280. [DOI] [PubMed] [Google Scholar]
- (114).Cutrí CCC; Garozzo A; Siracusa MA; Sarvá MC; Castro A; Geremia E; Pinizzotto MR; Guerrera F Synthesis of New 3,4,5-Trisubstituted Isothiazoles as Effective Inhibitory Agents of Enteroviruses. Bioorg. Med. Chem 1999, 7, 225–230. [DOI] [PubMed] [Google Scholar]
- (115).Al-Zoubi RM; Al-Jammal WK; El-Khateeb MY; McDonald R Synthesis of Diiodinated Biphenyls and Iodinated Meta-Terphenyls by Regioselective Suzuki–Miyaura Cross-Coupling Reactions of 5-Substituted 1,2,3-Triiodobenzenes. Eur. J. Org. Chem 2015, 2015, 3374–3384. [Google Scholar]
- (116).For further discussions on the effects of catalyst/ligand sterics, see ref 19.
- (117).Cullis CA; Granger KE; Guo J; Hirose M; Li G; Mizutani M; Vos TJ Heteroaryls and Uses Thereof. Patent WO 2012021615 A1, 2012.
- (118).Freeze BS; Hirose M; Hu Y; Hu Z; Lee HM; Sells TB; Shi Z; Vyskocil S; Xu T Heteroaryls and Uses Thereof. Patent WO 2012021696 A1, 2012.
- (119).Selva E; Beretta G; Montanini N; Saddler GS; Gastaldo L; Ferrari P; Lorenzetti R; Landini P; Ripamonti F; Goldstein BP; et al. Antibiotic GE2270 A: ANovel Inhibitor of Bacterial Protein Synthesis. J. Antibiot 1991, 44, 693–701. [DOI] [PubMed] [Google Scholar]
- (120).Tavecchia P; Gentili P; Kurz M; Sottani C; Bonfichi R; Selva E; Lociuro S; Restelli E; Ciabatti R Degradation Studies of Antibiotic MDL 62,879 (GE2270A) and Revision of Structure. Tetrahedron 1995, 51, 4867–4890. [Google Scholar]
- (121).Müller HM; Delgado O; Bach T Total Synthesis of the Thiazolyl Peptide GE2270 A. Angew. Chem., Int. Ed 2007, 46, 4771–4774. [DOI] [PubMed] [Google Scholar]
- (122).Gustafson JL; Lim D; Barrett KT; Miller SJ Synthesis of Atropisomerically Defined, Highly Substituted Biaryl Scaffolds through Catalytic Enantioselective Bromination and Regioselective Cross-Coupling. Angew. Chem., Int. Ed 2011, 50, 5125–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Gustafson JL; Lim D; Miller SJ Dynamic Kinetic Resolution of Biaryl Atropisomers via Peptide-Catalyzed Asymmetric Bromination. Science 2010, 328, 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Knapp DM; Gillis EP; Burke MD A General Solution for Unstable Boronic Acids: Slow-Release Cross-Coupling from Air-Stable MIDA Boronates. J. Am. Chem. Soc 2009, 131, 6961–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Yoshikai N; Mashima H; Nakamura E Nickel-Catalyzed Cross-Coupling Reaction of Aryl Fluorides and Chlorides with Grignard Reagents under Nickel/Magnesium Bimetallic Cooperation. J. Am. Chem. Soc 2005, 127, 17978–17979. [DOI] [PubMed] [Google Scholar]
- (126).Chen Z; Wang B; Zhang J; Yu W; Liu Z; Zhang Y Transition Metal-Catalyzed C–H Bond Functionalizations by the Use of Diverse Directing Groups. Org. Chem. Front 2015, 2, 1107–1295. [Google Scholar]
- (127).Nakamura H; Takeuchi D; Murai A Synthesis of 5- and 3,5-Substituted 2-Aminopyrazines by Pd Mediated Stille Coupling. Synlett 1995, 1995, 1227–1228. [Google Scholar]
- (128).Shimomura O; Musicki B; Kishi Y Semi-synthetic Aequorins with Improved Sensitivity to Ca2+Ions. Biochem. J 1989, 261, 913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Wu C; Nakamura H; Murai A; Shimomura O Chemi- and Bioluminescense of Coelenterazine Analogues with a Conjugated Group at the C-8 Position. Tetrahedron Lett. 2001, 42, 2997–3000. [Google Scholar]
- (130).Hikawa H; Yokoyama Y Cross-Coupling Reaction on N-(3,5-dibromo-2-pyridyl)piperazines: Regioselective Synthesis of 3,5-Disubstituted Pyridylpiperazines. Tetrahedron 2010, 66, 9552–9559. [Google Scholar]
- (131).Hilton S; Naud S; Caldwell JJ; Boxall K; Burns S; Anderson VE; Antoni L; Allen CE; Pearl LH; Oliver AW; et al. Identification and Characterisation of 2-Aminopyridine Inhibitors of Checkpoint Kinase 2. Bioorg. Med. Chem 2010, 18, 707–718. [DOI] [PubMed] [Google Scholar]
- (132).For selected reviews on C–F bond activation, see refs 132–134:; Braun T; Perutz RN Routes to Fluorinated Organic Derivatives by Nickel Mediated C–F Activation of Heteroaromatics. Chem. Commun 2002, 2749–2757. [DOI] [PubMed] [Google Scholar]
- (133).Amii H; Uneyama K C–F Bond Activation in Organic Synthesis. Chem. Rev 2009, 109, 2119–2183. [DOI] [PubMed] [Google Scholar]
- (134).Weaver J; Senaweera S C–F Activation and Functionalization of Perfluoro- and Polyfluoroarenes. Tetrahedron 2014, 70, 7413–7428. [Google Scholar]
- (135).Wei J; Liu K-M; Duan X-F Cobalt-Catalyzed Biaryl Couplings via C–F Bond Activation in the Absence of Phosphine or NHC Ligands. J. Org. Chem 2017, 82, 1291–1300. [DOI] [PubMed] [Google Scholar]
- (136).Manabe K; Ishikawa S Ortho-Selective Cross-Coupling of Fluorobenzenes with Grignard Reagents: Acceleration by Electron-Donating Ortho-Directing Groups. Synthesis 2008, 2008, 2645–2649. [Google Scholar]
- (137).For selected reviews on secondary phosphine oxide (SPO) preligands, see refs 137 and 138:; Ackermann L Air- and Moisture-Stable Secondary Phosphine Oxides as Preligands in Catalysis. Synthesis 2006, 2006, 1557–1571. [Google Scholar]
- (138).Dubrovina NV; Börner A Enantioselective Catalysis with Chiral Phosphine Oxide Preligands. Angew. Chem., Int. Ed 2004, 43, 5883–5886. [DOI] [PubMed] [Google Scholar]
- (139).For selected reviews on the application of HASPO preligands, see refs 139–141:; Ackermann L Catalytic Arylations with Challenging Substrates: From Air-Stable HASPO Preligands to Indole Syntheses and C–H-Bond Functionalizations. Synlett 2007, 2007, 507–526. [Google Scholar]
- (140).Nemoto T; Hamada Y Pd-Catalyzed Asymmetric Allylic Substitution Reactions Using P-Chirogenic Diaminophosphine Oxides: DIAPHOXs. Chem. Rec 2007, 7, 150–158. [DOI] [PubMed] [Google Scholar]
- (141).Ackermann L Transition-Metal-Catalyzed Direct Arylations via C–H Bond Cleavages. Pure Appl. Chem 2010, 82, 1403–1413. [Google Scholar]
- (142).Ackermann L; Kapdi AR; Fenner S; Kornhaaβ C; Schulzke C Well-Defined Air-Stable Palladium HASPO Complexes for Efficient Kumada–Corriu Cross-Couplings of (Hetero)Aryl or Alkenyl Tosylates. Chem. - Eur. J 2011, 17, 2965–2971. [DOI] [PubMed] [Google Scholar]
- (143).Ackermann L; Althammer A Air-Stable Pin P(O)H as Preligand for Palladium-Catalyzed Kumada Couplings of Unactivated Tosylates. Org. Lett 2006, 8, 3457–3460. [DOI] [PubMed] [Google Scholar]
- (144).For selected examples of coupling aryl bromides, chlorides, and fluorides using HASPO preligands, see refs 144–146:; Ackermann L; Potukuchi HK; Althammer A; Born R; Mayer P Tetra-Ortho-Substituted Biaryls through Palladium-Catalyzed Suzuki–Miyaura Couplings with a Diaminochlorophosphine Ligand. Org. Lett 2010, 12, 1004–1007. [DOI] [PubMed] [Google Scholar]
- (145).Ackermann L; Spatz JH; Gschrei CJ; Born R; Althammer A A Diaminochlorophosphine for Palladium-Catalyzed Arylations of Amines and Ketones. Angew. Chem., Int. Ed 2006, 45, 7627–7630. [DOI] [PubMed] [Google Scholar]
- (146).Ackermann L; Born R Modular Diamino- and Dioxophosphine Oxides and Chlorides as Ligands for Transition-Metal-Catalyzed C–C and C–N Couplings with Aryl Chlorides. Angew. Chem., Int. Ed 2005, 44, 2444–2447. [DOI] [PubMed] [Google Scholar]
- (147).For selected initial reports of the carboxylate directing effect, see refs 147–149:; Tanaka D; Romeril SP; Myers AG On the Mechanism of the Palladium(II)-Catalyzed Decarboxylative Olefination of Arene Carboxylic Acids. Crystallographic Characterization of Non-Phosphine Palladium(II) Intermediates and Observation of Their Stepwise Transformation in Heck-like Processes. J. Am. Chem. Soc 2005, 127, 10323–10333. [DOI] [PubMed] [Google Scholar]
- (148).Giri R; Maugel N; Li J-J; Wang D-H; Breazzano SP; Saunders LB; Yu J-Q Palladium-Catalyzed Methylation and Arylation of sp2 and sp3 C–H Bonds in Simple Carboxylic Acids. J. Am. Chem. Soc 2007, 129, 3510–3511. [DOI] [PubMed] [Google Scholar]
- (149).Mei T-S; Giri R; Maugel N; Yu J-Q PdII-Catalyzed Monoselective OrthoHalogenation of C–H Bonds Assisted by Counter Cations: AComplementary Method to Directed OrthoLi-thiation. Angew. Chem., Int. Ed 2008, 47, 5215–5219. [DOI] [PubMed] [Google Scholar]
- (150).Font M; Quibell JM; Perry GJP; Larrosa I The Use of Carboxylic Acids as Traceless Directing Groups for Regioselective C–H Bond Functionalisation. Chem. Commun 2017, 53, 5584–5597. [DOI] [PubMed] [Google Scholar]
- (151).Schwarz J; König B Decarboxylative Reactions with and without Light – AComparison. Green Chem. 2018, 20, 323–361. [Google Scholar]
- (152).Wei Y; Hu P; Zhang M; Su W Metal-Catalyzed Decarboxylative C–H Functionalization. Chem. Rev 2017, 117, 8864–8907. [DOI] [PubMed] [Google Scholar]
- (153).Modak A; Maiti D Metal Catalyzed Defunctionalization Reactions. Org. Biomol. Chem 2016, 14, 21–35. [DOI] [PubMed] [Google Scholar]
- (154).Houpis IN; Van Hoeck J-P; Tilstam U Carboxylate-Directed Kumada Coupling of an Acetaldehyde Synthon with 2-Bromobenzoates Used towards the Synthesis of Isochromanes. Synlett 2007, 2007, 2179–2184. [Google Scholar]
- (155).Houpis IN; Huang C; Nettekoven U; Chen JG; Liu R; Canters M Carboxylate Directed Cross-Coupling Reactions in the Synthesis of Trisubstituted Benzoic Acids. Org. Lett 2008, 10, 5601–5604. [DOI] [PubMed] [Google Scholar]
- (156).Houpis IN; Weerts K; Nettekoven U; Canters M; Tan H; Liu R; Wang Y Palladium- and Copper-Catalyzed Site Selective Monoamination of Dibromobenzoic Acid. Adv. Synth. Catal 2011, 353, 538–544. [Google Scholar]
- (157).For a thorough review, see:; Lindley J Copper Assisted Nucleophilic Substitution of Aryl Halogen. Tetrahedron 1984, 40, 1433–1456. [Google Scholar]
- (158).Liu S; Pestano JPC; Wolf C Regioselective Copper-Catalyzed C–N and C–S Bond Formation Using Amines, Thiols and Halobenzoic Acids. Synthesis 2007, 2007, 3519–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Murai S; Kakiuchi F; Sekine S; Tanaka Y; Kamatani A; Sonoda M; Chatani N Efficient Catalytic Addition of Aromatic Carbon-Hydrogen Bonds to Olefins. Nature 1993, 366, 529–531. [Google Scholar]
- (160).Zhu F; Wang Z-X Nickel-Catalyzed Cross-Coupling of Aryl Fluorides and Organozinc Reagents. J. Org. Chem 2014, 79, 4285–4292. [DOI] [PubMed] [Google Scholar]
- (161).Nakamura Y; Yoshikai N; Ilies L; Nakamura E Nickel-Catalyzed Monosubstitution of Polyfluoroarenes with Organozinc Reagents Using Alkoxydiphosphine Ligand. Org. Lett 2012, 14, 3316–3319. [DOI] [PubMed] [Google Scholar]
- (162).Kawamoto K; Kochi T; Sato M; Mizushima E; Kakiuchi F Ruthenium-Catalyzed Arylation of Fluorinated Aromatic Ketones via Ortho-Selective Carbon–Fluorine Bond Cleavage. Tetrahedron Lett. 2011, 52, 5888–5890. [Google Scholar]
- (163).Crespo M; Martinez M; Sales J Effect of Fluorine Substituents in Intramolecular Activation of C–F and C–H Bonds by Platinum(II). Organometallics 1993, 12, 4297–4304. [Google Scholar]
- (164).Hill GS; Irwin MJ; Levy CJ; Rendina LM; Puddephatt RJ; Andersen RA; McLean L Platinum(II) Complexes of Dimethyl Sulfide. Inorg. Synth 2007, 32, 149–153. [Google Scholar]
- (165).Wang T; Alfonso BJ; Love JA Platinum(II)-Catalyzed Cross-Coupling of Polyfluoroaryl Imines. Org. Lett 2007, 9, 5629–5631. [DOI] [PubMed] [Google Scholar]
- (166).Wang T; Love JA Insight into the Mechanism of Platinum-Catalyzed Cross-Coupling of Polyfluoroaryl Imines. Organometallics 2008, 27, 3290–3296. [Google Scholar]
- (167).Buckley HL; Sun AD; Love JA User-Friendly Precatalyst for the Methylation of Polyfluoroaryl Imines. Organometallics 2009, 28, 6622–6624. [Google Scholar]
- (168).Sun AD; Love JA Pt(II)Cl2(DMSO)2-Catalyzed Cross-Coupling of Polyfluoroaryl Imines. J. Fluorine Chem 2010, 131, 1237–1240. [Google Scholar]
- (169).Buckley HL; Wang T; Tran O; Love JA Selective Platinum-Catalyzed C–F Bond Activation as a Route to Fluorinated Aryl Methyl Ethers. Organometallics 2009, 28, 2356–2359. [Google Scholar]
- (170).Sun AD; Love JA Nickel-Catalyzed Selective Defluorination to Generate Partially Fluorinated Biaryls. Org. Lett 2011, 13, 2750–2753. [DOI] [PubMed] [Google Scholar]
- (171).Yang X; Sun H; Zhang S; Li X Nickel-Catalyzed C–F Bond Activation and Alkylation of Polyfluoroaryl Imines. J. Organomet. Chem 2013, 723, 36–42. [Google Scholar]
- (172).Trost BM; Imi K; Davies IW Elaboration of Conjugated Alkenes Initiated by Insertion into a Vinylic C–H Bond. J. Am. Chem. Soc 1995, 117, 5371–5372. [Google Scholar]
- (173).Sonoda M; Kakiuchi F; Kamatani A; Chatani N; Murai S Ruthenium-Catalyzed Addition of Aromatic Esters at the Ortho C–H Bonds to Olefins. Chem. Lett 1996, 25, 109–110. [Google Scholar]
- (174).Yang W; Wang Y; Corte JR Efficient Synthesis of 2-Aryl-6-chloronicotinamides via PXPd2-Catalyzed Regioselective Suzuki Coupling. Org. Lett 2003, 5, 3131–3134. [DOI] [PubMed] [Google Scholar]
- (175).Netherton MR; Fu GC Air-Stable Trialkylphosphonium Salts: Simple, Practical, and Versatile Replacements for Air-Sensitive Trialkylphosphines. Applications in Stoichiometric and Catalytic Processes. Org. Lett 2001, 3, 4295–4298. [DOI] [PubMed] [Google Scholar]
- (176).Hartwig JF; Kawatsura M; Hauck SI; Shaughnessy KH; Alcazar-Roman LM Room-Temperature Palladium-Catalyzed Amination of Aryl Bromides and Chlorides and Extended Scope of Aromatic C–N Bond Formation with a Commercial Ligand. J. Org. Chem 1999, 64, 5575–5580. [DOI] [PubMed] [Google Scholar]
- (177).Alcazar-Roman LM; Hartwig JF Mechanism of Aryl Chloride Amination: Base-Induced Oxidative Addition. J. Am. Chem. Soc 2001, 123, 12905–12906. [DOI] [PubMed] [Google Scholar]
- (178).Li GY; Zheng G; Noonan AF Highly Active, Air-Stable Versatile Palladium Catalysts for the C–C, C–N, and C–S Bond Formations via Cross-Coupling Reactions of Aryl Chlorides. J. Org. Chem 2001, 66, 8677–8681. [DOI] [PubMed] [Google Scholar]
- (179).Li GY The First Phosphine Oxide Ligand Precursors for Transition Metal Catalyzed Cross-Coupling Reactions: C–C, C–N, and C–S Bond Formation on Unactivated Aryl Chlorides. Angew. Chem., Int. Ed 2001, 40, 1513–1516. [DOI] [PubMed] [Google Scholar]
- (180).Li GY Highly Active, Air-Stable Palladium Catalysts for the C–C and C–S Bond-Forming Reactions of Vinyl and Aryl Chlorides: Use of Commercially Available [(t-Bu)2P(OH)]2PdCl2, [(t-Bu)2P-(OH)PdCl2]2, and [[(t-Bu)2POPPPHHHHOP(t-Bu)2]PdCl]2 as Catalysts. J. Org. Chem 2002, 67, 3643–3650. [DOI] [PubMed] [Google Scholar]
- (181).Li GY Catalysis Using Phosphine Oxide and Phosphine Sulfide Complexes with Pd and Ni for the Synthesis of Biaryls and Arylamines. Int. Patent WO2002000574A2, January 3, 2002.
- (182).Henderson BJ; Carper DJ; González-Cestari TF; Yi B; Mahasenan K; Pavlovicz RE; Dalefield ML; Coleman RS; Li C; McKay DB Structure–Activity Relationship Studies of Sulfonylpiperazine Analogues as Novel Negative Allosteric Modulators of Human Neuronal Nicotinic Receptors. J. Med. Chem 2011, 54, 8681–8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Hu J; Jiang J; Costanzi S; Thomas C; Yang W; Feyen JHM; Jacobson KA; Spiegel AM A Missense Mutation in the Seven-Transmembrane Domain of the Human Ca2+Receptor Converts a Negative Allosteric Modulator into a Positive Allosteric Modulator. J. Biol. Chem 2006, 281, 21558–21565. [DOI] [PubMed] [Google Scholar]
- (184).Yang W; Ruan Z; Wang Y; Van Kirk K; Ma Z; Arey BJ; Cooper CB; Seethala R; Feyen JHM; Dickson JK Discovery and Structure–Activity Relationships of Trisubstituted Pyrimidines/Pyridines as Novel Calcium-Sensing Receptor Antagonists. J. Med. Chem 2009, 52, 1204–1208. [DOI] [PubMed] [Google Scholar]
- (185).Nguyen T; Chiu W; Wang X; Sattler MO; Love JA Ligandless Nickel-Catalyzed Ortho-Selective Directed Trifluoromethylthiolation of Aryl Chlorides and Bromides Using AgSCF3. Org. Lett 2016, 18, 5492–5495. [DOI] [PubMed] [Google Scholar]
- (186).Sambiagio C; Schönbauer D; Blieck R; Dao-Huy T; Pototschnig G; Schaaf P; Wiesinger T; Zia MF; Wencel-Delord J; Besset T; et al. A Comprehensive Overview of Directing Groups Applied in Metal-Catalyzed C–H Functionalization Chemistry. Chem. Soc. Rev 2018, 47, 6603–6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Giri R; Chen X; Yu J-Q Palladium-Catalyzed Asymmetric Iodination of Unactivated C–H Bonds under Mild Conditions. Angew. Chem., Int. Ed 2005, 44, 2112–2115. [DOI] [PubMed] [Google Scholar]
- (188).Giri R; Liang J; Lei J-G; Li J-J; Wang D-H; Chen X; Naggar IC; Guo C; Foxman BM; Yu J-Q Pd-Catalyzed Stereoselective Oxidation of Methyl Groups by Inexpensive Oxidants under Mild Conditions: ADual Role for Carboxylic Anhydrides in Catalytic C–H Bond Oxidation. Angew. Chem., Int. Ed 2005, 44, 7420–7424. [DOI] [PubMed] [Google Scholar]
- (189).Chen X; Li J-J; Hao X-S; Goodhue CE; Yu J-Q Palladium-Catalyzed Alkylation of Aryl C–H Bonds with sp3Organotin Reagents Using Benzoquinone as a Crucial Promoter. J. Am. Chem. Soc 2006, 128, 78–79. [DOI] [PubMed] [Google Scholar]
- (190).Yu D; Shen Q; Lu L Selective Palladium-Catalyzed C–F Activation/Carbon–Carbon Bond Formation of Polyfluoroaryl Oxazolines. J. Org. Chem 2012, 77, 1798–1804. [DOI] [PubMed] [Google Scholar]
- (191).For selected reviews on C–X/C–H activation, see refs 191–194:; Ackermann L; Vicente R; Kapdi AR Transition-Metal-Catalyzed Direct Arylation of (Hetero)Arenes by C–H Bond Cleavage. Angew. Chem., Int. Ed 2009, 48, 9792–9826. [DOI] [PubMed] [Google Scholar]
- (192).Chen X; Engle KM; Wang D-H; Yu J-Q Palladium(II)-Catalyzed C–H Activation/C–C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem., Int. Ed 2009, 48, 5094–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Alberico D; Scott ME; Lautens M Aryl–Aryl Bond Formation by Transition-Metal-Catalyzed Direct Arylation. Chem. Rev 2007, 107, 174–238. [DOI] [PubMed] [Google Scholar]
- (194).Campeau L-C; Fagnou K Applications of and Alternatives to l-Electron-Deficient Azine Organometallics in Metal Catalyzed Cross-Coupling Reactions. Chem. Soc. Rev 2007, 36, 1058–1068. [DOI] [PubMed] [Google Scholar]
- (195).For selected examples on C–X/C–H activation, see refs 195–197:; Ye M; Gao G-L; Edmunds AJF; Worthington PA; Morris JA; Yu J-Q Ligand-Promoted C3-Selective Arylation of Pyridines with Pd Catalysts: Gram-Scale Synthesis of (y)-Preclamol. J. Am. Chem. Soc 2011, 133, 19090–19093. [DOI] [PubMed] [Google Scholar]
- (196).Sun C-L; Li H; Yu D-G; Yu M; Zhou X; Lu X-Y; Huang K; Zheng S-F; Li B-J; Shi Z-J An Efficient Organo-catalytic Method for Constructing Biaryls through Aromatic C–H Activation. Nat. Chem 2010, 2, 1044–1049. [DOI] [PubMed] [Google Scholar]
- (197).Muto K; Yamaguchi J; Itami K Nickel-Catalyzed C–H/C–O Coupling of Azoles with Phenol Derivatives. J. Am. Chem. Soc 2012, 134, 169–172. [DOI] [PubMed] [Google Scholar]
- (198).Müller K; Faeh C; Diederich F Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- (199).Murphy AR; Fréchet JMJ Organic Semiconducting Oligomers for Use in Thin Film Transistors. Chem. Rev 2007, 107, 1066–1096. [DOI] [PubMed] [Google Scholar]
- (200).Yu D; Lu L; Shen Q Palladium-Catalyzed Coupling of Polyfluorinated Arenes with Heteroarenes via C–F/C–H Activation. Org. Lett 2013, 15, 940–943. [DOI] [PubMed] [Google Scholar]
- (201).Yu D; Wang C-S; Yao C; Shen Q; Lu L Nickel-Catalyzed a-Arylation of Zinc Enolates with Polyfluoroarenes via C–F Bond Activation under Neutral Conditions. Org. Lett 2014, 16, 5544–5547. [DOI] [PubMed] [Google Scholar]
- (202).Xiao S-H; Xiong Y; Zhang X-X; Cao S Nickel-Catalyzed N-Heterocycle-Directed Cross-Coupling of Fluorinated Arenes with Organozinc Reagents. Tetrahedron 2014, 70, 4405–4411. [Google Scholar]
- (203).Guo W-H; Min Q-Q; Gu J-W; Zhang X Rhodium-Catalyzed Ortho-Selective C–F Bond Borylation of Polyfluoroarenes with Bpin–Bpin. Angew. Chem., Int. Ed 2015, 54, 9075–9078. [DOI] [PubMed] [Google Scholar]
- (204).Kim YM; Yu S Palladium(0)-Catalyzed Amination, Stille Coupling, and Suzuki Coupling of Electron-Deficient Aryl Fluorides. J. Am. Chem. Soc 2003, 125, 1696–1697. [DOI] [PubMed] [Google Scholar]
- (205).Widdowson DA; Wilhelm R Palladium Catalysed Suzuki Reactions of Fluoroarenes. Chem. Commun 2003, 34, 578–579. [DOI] [PubMed] [Google Scholar]
- (206).Cargill MR; Sandford G; Tadeusiak AJ; Yufit DS; Howard JAK; Kilickiran P; Nelles G Palladium-Catalyzed C–F Activation of Polyfluoronitrobenzene Derivatives in Suzuki–Miyaura Coupling Reactions. J. Org. Chem 2010, 75, 5860–5866. [DOI] [PubMed] [Google Scholar]
- (207).Sucunza D; Cuadro AM; Alvarez-Builla J; Vaquero JJ Recent Advances in the Synthesis of Azonia Aromatic Heterocycles. J. Org. Chem 2016, 81, 10126–10135. [DOI] [PubMed] [Google Scholar]
- (208).José Reyes M; Luisa Izquierdo M; Alvarez-Builla J Suzuki Reaction on Pyridinium N-(5-Bromoheteroar-2-yl)aminides. Tetrahedron Lett. 2004, 45, 8713–8715. [Google Scholar]
- (209).Sliwa W; Bachowska B; Zelichowicz N Chemistry of Viologens. Heterocycles 1991, 32, 2241–2273. [Google Scholar]
- (210).Ollis WD; Stanforth SP; Ramsden CA Heterocyclic Mesomeric Betaines. Tetrahedron 1985, 41, 2239–2329. [Google Scholar]
- (211).Sánchez A; Núñez A; Alvarez-Builla J; Burgos C Pyridinium N-2’-Pyridylaminide: Synthesis of 3-Aryl-2-aminopyridines through an Intramolecular Radical Process. Tetrahedron 2004, 60, 11843–11850. [Google Scholar]
- (212).Nuñez A; de Viedma AG; Martínez-Barrasa V; Burgos C; Alvarez-Builla J N-Azinylpyridinium N-Aminides: An Approach to Pyrazolopyridines via an Intramolecular Radical Pathway. Synlett 2002, 2002, 1093–1096. [Google Scholar]
- (213).José Reyes M; Delgado F; Luisa Izquierdo M; Alvarez-Builla J Pyridinium N-(2’-Azinyl)Aminides: Regioselective Synthesis of N-(2-Pyridyl) Substituted Polyamines. Tetrahedron 2002, 58, 8573–8579. [Google Scholar]
- (214).Martınez-Barrasa V.ın; Delgado F; Burgos C; Luis García-Navío J; Luisa Izquierdo M; Alvarez-Builla J Pyridinium N-(2’-Azinyl)Aminides: Regioselective Synthesis of 2-Alkylaminoazines. Tetrahedron 2000, 56, 2481–2490. [Google Scholar]
- (215).Castillo R; Reyes MJ; Izquierdo ML; Alvarez-Builla J Suzuki Reaction on Pyridinium N-Haloheteroarylaminides: Regioselective Synthesis of 3,5-Disubstituted 2-Aminopyrazines. Tetrahedron 2008, 64, 1351–1370. [Google Scholar]
- (216).Reyes MJ; Castillo R; Izquierdo ML; Alvarez-Builla J Regioselective Suzuki Coupling on Pyridinium N-(3,5-Dibromoheteroar-2-yl)Aminides. Tetrahedron Lett. 2006, 47, 6457–6460. [Google Scholar]
- (217).Zhao P; Young MD; Beaudry CM Regioselective Suzuki Couplings of Non-Symmetric Dibromobenzenes: Alkenes as Regiochemical Control Elements. Org. Biomol. Chem 2015, 13, 6162–6165. [DOI] [PubMed] [Google Scholar]
- (218).Johnson JB; Rovis T More Than Bystanders: The Effect of Olefins on Transition-Metal-Catalyzed Cross-Coupling Reactions. Angew. Chem., Int. Ed 2008, 47, 840–871. [DOI] [PubMed] [Google Scholar]
- (219).Macé Y; Kapdi AR; Fairlamb IJS; Jutand A Influence of the dba Substitution on the Reactivity of Palladium(0) Complexes Generated from Pd02(dba-n, n’-Z)2 and PPh3 in Oxidative Addition with Iodobenzene. Organometallics 2006, 25, 1795–1800. [Google Scholar]
- (220).Amatore C; Broeker G; Jutand A; Khalil F Identification of the Effective Palladium(0) Catalytic Species Generated in situ from Mixtures of Pd(dba)2 and Bidentate Phosphine Ligands. Determination of their Rates and Mechanism in Oxidative Addition. J. Am. Chem. Soc 1997, 119, 5176–5185. [Google Scholar]
- (221).Amatore C; Jutand A; Khalil F; M’Barki MA; Mottier L Rates and Mechanisms of Oxidative Addition to Zerovalent Palladium Complexes Generated in situ from Mixtures of Pd0(dba)2 and Triphenylphosphine. Organometallics 1993, 12, 3168–3178. [Google Scholar]
- (222).Orner BP; Ernst JT; Hamilton AD Toward Proteomimetics: Terphenyl Derivatives as Structural and Functional Mimics of Extended Regions of an e-Helix. J. Am. Chem. Soc 2001, 123, 5382–5383. [DOI] [PubMed] [Google Scholar]
- (223).Zhao P; Beaudry CM Enantioselective and Regioselective Pyrone Diels–Alder Reactions of Vinyl Sulfones: Total Synthesis of (+)-Cavicularin. Angew. Chem. Int. Ed 2014, 53, 10500–10503. [DOI] [PubMed] [Google Scholar]
- (224).Zhao P; Beaudry CM Total Synthesis of (y)-Cavicularin: Control of Pyrone Diels–Alder Regiochemistry Using Isomeric Vinyl Sulfones. Org. Lett 2013, 15, 402–405. [DOI] [PubMed] [Google Scholar]
- (225).For selected examples of oligoarene-type phosphines, see refs 225–228:; Matsui K; Takizawa S; Sasai H A Brønsted Acid and Lewis Base Organocatalyst for the Aza-Morita–Baylis–Hillman reaction. Synlett 2006, 5, 761–765. [Google Scholar]
- (226).Iwasawa T; Komano T; Tajima A; Tokunaga M; Obora Y; Fujihara T; Tsuji Y Phosphines Having a 2,3,4,5-Tetraphenylphenyl Moiety: Effective Ligands in Palladium-Catalyzed Transformations of Aryl Chlorides. Organometallics 2006, 25, 4665–4669. [Google Scholar]
- (227).Matsumoto T; Kasai T; Tatsumi K Chiral Crystallization of a Linear Two-coordinated Palladium(0) Complex Bearing Propeller-shaped Bulky Phosphine Ligands. Chem. Lett 2002, 31, 346–347. [Google Scholar]
- (228).Yu H-B; Hu Q-S; Pu L The First Optically Active BINOL–BINAP Copolymer Catalyst: Highly Stereoselective Tandem Asymmetric Reactions. J. Am. Chem. Soc 2000, 122, 6500–6501. [Google Scholar]
- (229).Martin R; Buchwald SL Pd-Catalyzed Kumada-Corriu Cross-Coupling Reactions at Low Temperatures Allow the Use of Knochel-type Grignard Reagents. J. Am. Chem. Soc 2007, 129, 3844–3845. [DOI] [PubMed] [Google Scholar]
- (230).Barder TE; Walker SD; Martinelli JR; Buchwald SL Catalysts for Suzuki–Miyaura Coupling Processes: Scope and Studies of the Effect of Ligand Structure. J. Am. Chem. Soc 2005, 127, 4685–4696. [DOI] [PubMed] [Google Scholar]
- (231).Huang X; Anderson KW; Zim D; Jiang L; Klapars A; Buchwald SL Expanding Pd-Catalyzed C–N Bond-Forming Processes: The First Amidation of Aryl Sulfonates, Aqueous Amination, and Complementarity with Cu-Catalyzed Reactions. J. Am. Chem. Soc 2003, 125, 6653–6655. [DOI] [PubMed] [Google Scholar]
- (232).Parrish CA; Buchwald SL Use of Polymer-Supported Dialkylphosphinobiphenyl Ligands for Palladium-Catalyzed Amination and Suzuki Reactions. J. Org. Chem 2001, 66, 3820–3827. [DOI] [PubMed] [Google Scholar]
- (233).For selected examples of phosphines with a hydroxy group, see refs 233 and 234:; Yoshikai N; Matsuda H; Nakamura E Hydroxyphosphine Ligand for Nickel-Catalyzed Cross-Coupling through Nickel/Magnesium Bimetallic Cooperation. J. Am. Chem. Soc 2009, 131, 9590–9599. [DOI] [PubMed] [Google Scholar]
- (234).Meng X; Huang Y; Chen R A Novel Selective Aza-Morita–Baylis–Hillman (Aza-MBH) Domino Reaction and Aza-MBH Reaction of N-Sulfonated Imines with Acrolein Catalyzed by a Bifunctional Phosphine Organocatalyst. Chem. - Eur. J 2008, 14, 6852–6856. [DOI] [PubMed] [Google Scholar]
- (235).Ishikawa S; Manabe K Oligoarene Strategy for Catalyst Development. Hydroxylated Oligoarene-type Phosphines for Palladium-Catalyzed Cross-Coupling. Chem. Lett 2007, 36, 1302–1303. [Google Scholar]
- (236).Ishikawa S; Manabe K Synthesis of Hydroxylated Oligoarene-type Phosphines by a Repetitive Two-Step Method. Tetrahedron 2010, 66, 297–303. [Google Scholar]
- (237).Ishikawa S; Manabe K Ortho-Selective Cross Coupling of Dibromophenols and Dibromoanilines with Grignard Reagents in the Presence of Palladium Catalysts Bearing Hydroxylated Oligoarene-type Phosphine. Chem. Lett 2007, 36, 1304–1305. [Google Scholar]
- (238).Ishikawa S; Manabe K DHTP Ligands for the Highly Ortho-Selective, Palladium-Catalyzed Cross-Coupling of Dihaloarenes with Grignard Reagents: AConformational Approach for Catalyst Improvement. Angew. Chem. Int. Ed 2010, 49, 772–775. [DOI] [PubMed] [Google Scholar]
- (239).Ishikawa S; Manabe K Hydroxylated Terphenylphosphine Ligands for Palladium-Catalyzed Ortho-Selective Cross-Coupling of Dibromophenols, Dibromoanilines, and their Congeners with Grignard Reagents. Tetrahedron 2011, 67, 10156–10163. [Google Scholar]
- (240).Yamaguchi M; Akiyama T; Sasou H; Katsumata H; Manabe K One-Pot Synthesis of Substituted Benzo[b]furans and Indoles from Dichlorophenols/Dichloroanilines Using a Palladium–Dihydroxyterphenylphosphine Catalyst. J. Org. Chem 2016, 81, 5450–5463. [DOI] [PubMed] [Google Scholar]
- (241).Yamaguchi M; Katsumata H; Manabe K One-Pot Synthesis of Substituted Benzo[b]furans from Mono- and Dichlorophenols Using Palladium Catalysts Bearing Dihydroxyterphenylphosphine. J. Org. Chem 2013, 78, 9270–9281. [DOI] [PubMed] [Google Scholar]
- (242).Wang J-R; Manabe K Hydroxyterphenylphosphine–Palladium Catalyst for Benzo[b]furan Synthesis from 2-Chlorophenols. Bifunctional Ligand strategy for Cross-Coupling of Chloroarenes. J. Org. Chem 2010, 75, 5340–5342. [DOI] [PubMed] [Google Scholar]
- (243).Yamaguchi M; Manabe K One-Pot Synthesis of 2,4-Disubstituted Indoles from N-Tosyl-2,3-Dichloroaniline Using Palladium–Dihydroxyterphenylphosphine Catalyst. Org. Lett 2014, 16, 2386–2389. [DOI] [PubMed] [Google Scholar]
- (244).Yamaguchi M; Manabe K Three-Step Synthesis of 2,5,7-Trisubstituted Indoles from N-Acetyl-2,4,6-trichloroaniline Using Pd-Catalyzed Site-Selective Cross-Coupling. Org. Biomol. Chem 2017, 15, 6645–6655. [DOI] [PubMed] [Google Scholar]
- (245).Konishi H; Itoh T; Manabe K Site-Selective Cross-Coupling of Dichlorinated Benzo-Fused Nitrogen-Heterocycles with Grignard Reagents. Chem. Pharm. Bull 2010, 58, 1255–1258. [DOI] [PubMed] [Google Scholar]
- (246).Davis HJ; Phipps RJ Harnessing Non-Covalent Interactions to Exert Control Over Regioselectivity and Site-Selectivity in Catalytic Reactions. Chem. Sci 2017, 8, 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Zhang J; Sha S-C; Bellomo A; Trongsiriwat N; Gao F; Tomson NC; Walsh PJ Positional Selectivity in C–H Functionalizations of 2-Benzylfurans with Bimetallic Catalysts. J. Am. Chem. Soc 2016, 138, 4260–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Dydio P; Reek JNH Supramolecular Control of Selectivity in Transition-Metal Catalysis through Substrate Preorganization. Chem. Sci 2014, 5, 2135–2145. [Google Scholar]
- (249).Raynal M; Ballester P; Vidal-Ferran A; van Leeuwen PWNM Supramolecular Catalysis. Part 1: Non-Covalent Interactions as a Tool for Building and Modifying Homogeneous Catalysts. Chem. Soc. Rev 2014, 43, 1660–1733. [DOI] [PubMed] [Google Scholar]
- (250).Anderson KW; Buchwald SL General Catalysts for the Suzuki–Miyaura and Sonagashira Coupling Reactions of Aryl Chlorides and for the Coupling of Challenging Substrate Combinations in Water. Angew. Chem., Int. Ed 2005, 44, 6173–6177. [DOI] [PubMed] [Google Scholar]
- (251).Surry DS; Buchwald SL Biaryl Phosphane Ligands in Palladium-Catalyzed Amination. Angew. Chem., Int. Ed 2008, 47, 6338–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).For selected applications of sulfonylated phosphine ligands, see refs 252–255:; Rojas AJ; Pentelute BL; Buchwald SL Water-Soluble Palladium Reagents for Cysteine S-Arylation under Ambient Aqueous Conditions. Org. Lett 2017, 19, 4263–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Janusson E; Zijlstra HS; Nguyen PPT; MacGillivray L; Martelino J; McIndoe JS Real-Time Analysis of Pd2(dba)3 Activation by Phosphine Ligands. Chem. Commun 2017, 53, 854–856. [DOI] [PubMed] [Google Scholar]
- (254).Corr MJ; Sharma SV; Pubill-Ulldemolins C; Bown RT; Poirot P; Smith DRM; Cartmell C; Abou Fayad A; Goss RJM Sonogashira Diversification of Unprotected Halotryptophans, Halotryptophan Containing Tripeptides; and Generation of a New to Nature Bromo-Natural Product and its Diversification in Water. Chem. Sci 2017, 8, 2039–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Jiang H; Jia T; Zhang M; Walsh PJ Palladium-Catalyzed Arylation of Aryl Sulfenate Anions with Aryl Bromides under Mild Conditions: Synthesis of Diaryl Sulfoxides. Org. Lett 2016, 18, 972–975. [DOI] [PubMed] [Google Scholar]
- (256).Golding WA; Pearce-Higgins R; Phipps RJ Site-Selective Cross-Coupling of Remote Chlorides Enabled by Electrostatically Directed Palladium Catalysis. J. Am. Chem. Soc 2018, 140, 13570–13574. [DOI] [PubMed] [Google Scholar]
- (257).Golding WA; Phipps RJ Electrostatically-Directed Pd-Catalysis in Combination with C–H Activation: Site-Selective Coupling of Remote Chlorides with Fluoroarenes and Fluoroheteroarenes. Chem. Sci 2020, 11, 3022–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).For selected reviews of C–H activation with fluoroarenes and fluoroheteroarenes, see refs 258 and 259:; Eisenstein O; Milani J; Perutz RN Selectivity of C–H Activation and Competition between C–H and C–F Bond Activation at Fluorocarbons. Chem. Rev 2017, 117, 8710–8753. [DOI] [PubMed] [Google Scholar]
- (259).Clot E; Eisenstein O; Jasim N; Macgregor SA; McGrady JE; Perutz RN C–F and C–H Bond Activation of Fluorobenzenes and Fluoropyridines at Transition Metal Centers: How Fluorine Tips the Scales. Acc. Chem. Res 2011, 44, 333–348. [DOI] [PubMed] [Google Scholar]
- (260).Lafrance M; Fagnou K Palladium-Catalyzed Benzene Arylation: Incorporation of Catalytic Pivalic Acid as a Proton Shuttle and a Key Element in Catalyst Design. J. Am. Chem. Soc 2006, 128, 16496–16497. [DOI] [PubMed] [Google Scholar]
- (261).Lafrance M; Rowley CN; Woo TK; Fagnou K Catalytic Intermolecular Direct Arylation of Perfluorobenzenes. J. Am. Chem. Soc 2006, 128, 8754–8756. [DOI] [PubMed] [Google Scholar]
- (262).Lafrance M; Shore D; Fagnou K Mild and General Conditions for the Cross-Coupling of Aryl Halides with Penta-fluorobenzene and Other Perfluoroaromatics. Org. Lett 2006, 8, 5097–5100. [DOI] [PubMed] [Google Scholar]
- (263).Golding WA; Schmitt HL; Phipps RJ Systematic Variation of Ligand and Cation Parameters Enables Site-Selective C–C and C–N Cross-Coupling of Multiply Chlorinated Arenes through Substrate–Ligand Electrostatic Interactions. J. Am. Chem. Soc 2020, 142, 21891–21898. [DOI] [PubMed] [Google Scholar]
- (264).Chung JYL; Cai C; McWilliams JC; Reamer RA; Dormer PG; Cvetovich RJ Efficient Synthesis of a Trisubstituted 1,6-Naphthyridone from Acetonedicarboxylate and Regioselective Suzuki Arylation. J. Org. Chem 2005, 70, 10342–10347. [DOI] [PubMed] [Google Scholar]
- (265).Chung JYL Development of a Practical Synthesis of Naphthyridone p38 MAP Kinase Inhibitor MK-0913; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp 39–56. [Google Scholar]
- (266).Cai C; Chung JYL; McWilliams JC; Sun Y; Shultz CS; Palucki M From High-Throughput Catalyst Screening to Reaction Optimization: Detailed Investigation of Regioselective Suzuki Coupling of 1,6-Naphthyridone Dichloride. Org. Process Res. Dev 2007, 11, 328–335. [Google Scholar]
- (267).For the cross-coupling of dihaloazoles by Strotman, Chobanian, and coworkers, see ref 101.
- (268).Wiglenda T; Gust R Structure–Activity Relationship Study to Understand the Estrogen Receptor-Dependent Gene Activation of Aryl- and Alkyl-Substituted 1H-Imidazoles. J. Med. Chem 2007, 50, 1475–1484. [DOI] [PubMed] [Google Scholar]
- (269).Stangeland EL; Sammakia T Use of Thiazoles in the Halogen Dance Reaction: Application to the Total Synthesis of WS75624 B. J. Org. Chem 2004, 69, 2381–2385. [DOI] [PubMed] [Google Scholar]
- (270).Flegeau EF; Popkin ME; Greaney MF Direct Arylation of Oxazoles at C2. A Concise Approach to Consecutively Linked Oxazoles. Org. Lett 2008, 10, 2717–2720. [DOI] [PubMed] [Google Scholar]
- (271).The preferred site of lithium halogen exchange on 2,4-dibromothiazole is C2:; Martin T; Laguerre C; Hoarau C; Marsais F Highly Efficient Borylation Suzuki Coupling Process for 4-Bromo-2-ketothiazoles: Straightforward Access to Micrococcinate and Saramycetate Esters. Org. Lett 2009, 11, 3690–3693. [DOI] [PubMed] [Google Scholar]
- (272).The preferred site of SNAr on 2,4-dibromothiazoles with thiols is C2:; Nicolaou KC; Sasmal PK; Rassias G; Reddy MV; Altmann K-H; Wartmann M; O’Brate A; Giannakakou P Design, Synthesis, and Biological Properties of Highly Potent Epothilone B Analogues. Angew. Chem., Int. Ed 2003, 42, 3515–3520. [DOI] [PubMed] [Google Scholar]
- (273).Handy’s method for predicting the site of coupling in dihaloheteroaromatics using the 1H NMR shifts of the parent nonhalogenated compound incorrectly predicts reactivity at the C2 position of 2,4-diiodooxazole and 2,5-dibromo-1-methylimidazole; see ref 57.
- (274).Dai X; Chen Y; Garrell S; Liu H; Zhang L-K; Palani A; Hughes G; Nargund R Ligand-Dependent Site-Selective Suzuki Cross-Coupling of 3,5-Dichloropyridazines. J. Org. Chem 2013, 78, 7758–7763. [DOI] [PubMed] [Google Scholar]
- (275).Yang M; Chen J; He C; Hu X; Ding Y; Kuang Y; Liu J; Huang Q Palladium-Catalyzed C-4 Selective Coupling of 2,4-Dichloropyridines and Synthesis of Pyridine-Based Dyes for Live-Cell Imaging. J. Org. Chem 2020, 85, 6498–6508. [DOI] [PubMed] [Google Scholar]
- (276).Wang RX; Lai XJ; Qiu G; Liu JB Research Progress of Reactive Fluorescent Probe Based on ESIPT Principle. Youji Huaxue 2019, 39, 952–960. [Google Scholar]
- (277).Gong J; Li Y-H; Zhang C-J; Huang J; Sun Q A Thiazolo[4,5-b]Pyridine-Based Fluorescent Probe for Detection of Zinc Ions and Application for in vitro and in vivoBioimaging. Talanta 2018, 185, 396–404. [DOI] [PubMed] [Google Scholar]
- (278).Zhang J; Wang H Photophysical Investigations and the Bioimagings of i-, –, –Pyridine-Based Terpyridine Derivatives. J. Mol. Struct 2018, 1157, 457–462. [Google Scholar]
- (279).Golden JH; Facendola JW; Sylvinson MR,D; Baez CQ; Djurovich PI; Thompson ME Boron Dipyridylmethene (DIPYR) Dyes: Shedding light on Pyridine-Based Chromophores. J. Org. Chem 2017, 82, 7215–7222. [DOI] [PubMed] [Google Scholar]
- (280).Chen J; Liu W; Ma J; Xu H; Wu J; Tang X; Fan Z; Wang P Synthesis and Properties of Fluorescence Dyes: Tetracyclic Pyrazolo[3,4-b]Pyridine-Based Coumarin Chromaphores with Intramolecular Charge Transfer Character. J. Org. Chem 2012, 77, 3475–3482. [DOI] [PubMed] [Google Scholar]
- (281).Biscoe MR; Fors BP; Buchwald SL A New Class of Easily Activated Palladium Precatalysts for Facile C–N Cross-Coupling Reactions and the Low Temperature Oxidative Addition of Aryl Chlorides. J. Am. Chem. Soc 2008, 130, 6686–6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Kinzel T; Zhang Y; Buchwald SL A New Palladium Precatlyst Allows for the Fast Suzuki–Miyaura Coupling Reactions of Unstable Polyfluorophenyl and 2-Heteroarylboronic Acids. J. Am. Chem. Soc 2010, 132, 14073–14075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Shu W; Buchwald SL Use of Precatalysts Greatly Facilitate Palladium-Catalyzed Alkynylations in Batch and Continuous-Flow Conditions. Chem. Sci 2011, 2, 2321–2325. [Google Scholar]
- (284).Yang Y; Oldenhuis NJ; Buchwald SL Mild and General Conditions for Negishi Cross-Coupling Enabled by the Use of Palladacycle Precatalysts. Angew. Chem., Int. Ed 2013, 52, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Bruno NC; Niljianskul N; Buchwald SL N-Substituted 2-Aminobiphenylpalladium Methanesulfonate Precatalysts and Their Use in C–C and C–N Cross-Couplings. J. Org. Chem 2014, 79, 4161–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Friis SD; Skrydstrup T; Buchwald SL Mild Pd-Catalyzed Aminocarbonylation of (Hetero)Aryl Bromides with a Palladacycle Precatalyst. Org. Lett 2014, 16, 4296–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Ingoglia BT; Buchwald SL Oxidative Addition Complexes as Precatalysts for Cross-Coupling Reactions Requiring Extremely Bulky Biarylphosphine Ligands. Org. Lett 2017, 19, 2853–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Ruiz-Castillo P; Buchwald SL Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Bruno NC; Tudge MT; Buchwald SL Design and Preparation of New Palladium Precatalysts for C–C and C–N Cross-Coupling Reactions. Chem. Sci 2013, 4, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Bruno NC; Buchwald SL Synthesis and Application of Palladium Precatalysts that Accommodate Extremely Bulky Di-tert-butylphosphino Biaryl Ligands. Org. Lett 2013, 15, 2876–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Cheung CW; Surry DS; Buchwald SL Mild and Highly Selective Palladium-Catalyzed Monoarylation of Ammonia Enabled by the Use of Bulky Biarylphosphine Ligands and Palladacycle Precatalysts. Org. Lett 2013, 15, 3734–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Cheung CW; Buchwald SL Mild and General Palladium-Catalyzed Synthesis of Methyl Aryl Ethers Enabled by the Use of a Palladacycle Precatalyst. Org. Lett 2013, 15, 3998–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Cheung CW; Buchwald SL Palladium-Catalyzed Hydroxylation of Aryl and Heteroaryl Halides Enabled by the Use of a Palladacycle Precatalyst. J. Org. Chem 2014, 79, 5351–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Hadei N; Kantchev EAB; O’Brie CJ; Organ MG The First Negishi Cross-Coupling Reaction of Two Alkyl Centers Utilizing a Pd–N-Heterocycle Carbene (NHC) Catalyst. Org. Lett 2005, 7, 3805–3807. [DOI] [PubMed] [Google Scholar]
- (295).Nasielski J; Hadei N; Achonduh G; Kantchev EAB; O’Brien CJ; Lough A; Organ MG Structure–Activity Relationship Analysis of Pd–PEPPSI Complexes in Cross-Couplings: AClose Inspection of the Catalytic Cycle and the Precatalyst Activation Model. Chem. - Eur. J 2010, 16, 10844–10853. [DOI] [PubMed] [Google Scholar]
- (296).Valente C; Belowich ME; Hadei N; Organ MG Pd-PEPPSI Complexes and the Negishi Reaction. Eur. J. Org. Chem 2010, 2010, 4343–4354. [Google Scholar]
- (297).O’Brien CJ; Kantchev EAB; Valente C; Hadei N; Chass GA; Lough A; Hopkinson AC; Organ MG Easily Prepared Air- and Moisture-Stable Pd-NHC (NHC = N-Heterocyclic Carbene) Complexes: AReliable, User-Friendly, Highly Active Palladium Precatalyst for the Suzuki–Miyaura Reaction. Chem. - Eur. J 2006, 12, 4743–4748. [DOI] [PubMed] [Google Scholar]
- (298).Organ MG; Avola S; Dubovyk I; Hadei N; Kantchev EAB; O’Brien CJ; Valente C A User-Friendly, All-Purpose Pd-NHC (NHC = N-Heterocyclic Carbene) Precatalyst for the Negishi Reaction: AStep Towards a Universal Cross-Coupling Catalyst. Chem. - Eur. J 2006, 12, 4749–4755. [DOI] [PubMed] [Google Scholar]
- (299).For selected examples of the Handy group’s work on the development of one-pot polycoupling approaches to substituted heteroaromatics, see refs 99 and 299–301:; Handy ST; Sabatini JJ Regioselective Dicouplings: Application to Differentially Substituted Pyrroles. Org. Lett 2006, 8, 1537–1539. [DOI] [PubMed] [Google Scholar]
- (300).Handy ST; Wilson T; Muth A Disubstituted Pyridines: The Double-Coupling Approach. J. Org. Chem 2007, 72, 8496–8500. [DOI] [PubMed] [Google Scholar]
- (301).Handy ST; Mayi D Regioselective Double Suzuki Couplings of 4,5-Dibromothiophene-2-carboxylate. Tetrahedron Lett. 2007, 48, 8108–8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Zhang Y; Handy ST A Solvent-Induced Reversal of Regioselectivity in the Suzuki Coupling of Pyrrole Esters. Open Org. Chem. J 2008, 2, 58–64. [Google Scholar]
- (303).For selected examples of the chelation of the nitrogen atom in pyrrole to transition-metal catalysts, see refs 303–305:; Wiest JM; Pöthig A; Bach T Pyrrole as a Directing group: Regioselective Pd(II)-Catalyzed Alkylation and Benzylation at the Benzene Core of 2-Phenylpyrroles. Org. Lett 2016, 18, 852–855. [DOI] [PubMed] [Google Scholar]
- (304).Jiao L; Bach T Palladium-Catalyzed Direct C–H Alkylation of Electron-Deficient Pyrrole Derivatives. Angew. Chem., int. Ed 2013, 52, 6080–6083. [DOI] [PubMed] [Google Scholar]
- (305).Zhang M; Zhang Y; Jie X; Zhao H; Li G; Su W Recent Advances in Directed C–H Functionalizations Using Monodentate Nitrogen-based Directing Groups. Org. Chem. Front 2014, 1, 843–895. [Google Scholar]
- (306).The Suzuki coupling of 248 in a 3:1 mixture of benzene and methanol proceeded to give an outcome indistinguishable from that of the toluene/ethanol mixture.
- (307).Handy ST; Zhang Y; Bregman H A Modular Synthesis of the Lamellarins: Total Synthesis of Lamellarin G Trimethyl Ether. J. Org. Chem 2004, 69, 2362–2366. [DOI] [PubMed] [Google Scholar]
- (308).For the site-selective Stille coupling of 3,5-dibromo-2-pyrone, see:; Kim W-S; Kim H-J; Cho C-G Regioselectivity in the Stille Coupling Reactions of 3,5-Dibromo-2-pyrone. J. Am. Chem. Soc 2003, 125, 14288–14289. [DOI] [PubMed] [Google Scholar]
- (309).The polarity of the solvents is described based on the reported dielectric constants. For context, see refs 309 and 310:; Vogel AI; Furniss BS; Hannaford AJ; Smith PWG; Tatchell AR Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Pearson Education Limited: Essex, U.K., 1989; pp 395–468. [Google Scholar]
- (310).Common Solvents Used in Organic Chemistry: Table of Properties. https://www.organicdivision.org/wp-content/uploads/2016/12/organic_solvents.html (accessed 2020-07-10).
- (311).Interestingly and in contrast with ref 94, when the oxidative addition was carried out with a stoichiometric amount of Pd(PPh3)4, the formation of C5–Pd complex 258 was not observed, and only C3–Pd complex 257 could be isolated.
- (312).Farina V; Kapadia S; Krishnan B; Wang C; Liebeskind LS On the Nature of the “Copper Effect“ in the Stille Cross-Coupling. J. Org. Chem 1994, 59, 5905–5911. [Google Scholar]
- (313).Beaupérin M; Job A; Cattey H; Royer S; Meunier P; Hierso J-C Copper(I) Iodide Polyphosphine Adducts at Low Loading for Sonogashira Alkynylation of Demanding Halide Substrates: Ligand Exchange Study between Copper and Palladium. Organometallics 2010, 29, 2815–2822. [Google Scholar]
- (314).Aufiero M; Proutiere F; Schoenebeck F Redox Reactions in Palladium Catalysis: On the Accelerating and/or Inhibiting Effects of Copper and Silver Salt Additives in Cross-Coupling Chemistry Involving Electron-Rich Phosphine Ligands. Angew. Chem., Int. Ed 2012, 51, 7226–7230. [DOI] [PubMed] [Google Scholar]
- (315).Li Z; Fu Y; Guo Q-X; Liu L Theoretical Study on Monoligated Pd-Catalyzed Cross-Coupling Reactions of Aryl Chlorides and Bromides. Organometallics 2008, 27, 4043–4049. [Google Scholar]
- (316).Lam KC; Marder TB; Lin Z DFT Studies on the Effect of the Nature of the Aryl Halide Y–C6H4–X on the Mechanism of its Oxidative Addition to Pd0L versus Pd0L2. Organometallics 2007, 26, 758–760. [Google Scholar]
- (317).Ahlquist M; Norrby P-O Oxidative Addition of Aryl Chlorides to Monoligated Palladium(0): A DFT-SCRF study. Organometallics 2007, 26, 550–553. [Google Scholar]
- (318).Seeman JI Effect of Conformational Change on Reactivity in Organic Chemistry. Evaluations, Applications, and Extensions of Curtin-Hammett Winstein-Holness Kinetics. Chem. Rev 1983, 83, 83–134. [Google Scholar]
- (319).Palani V; Perea MA; Gardner KE; Sarpong R A Pyrone Remodeling Strategy to Access Diverse Heterocycles: Application to the Synthesis of Fascaplysin Natural Products. Chem. Sci 2021, 12, 1528–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Matsui JK; Lang SB; Heitz DR; Molander GA Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development. ACS Catal. 2017, 7, 2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Singh A; Kubik JJ; Weaver JD Photocatalytic C–F Alkylation; Facile Access to Multifluorinated Arenes. Chem. Sci 2015, 6, 7206–7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Senaweera S; Weaver JD Dual C–F, C–H Functionalization via Photocatalysis: Access to Multifluorinated Biaryls. J. Am. Chem. Soc 2016, 138, 2520–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Sandfort F; Knecht T; Pinkert T; Daniliuc CG; Glorius F Site-Selective Thiolation of (Multi)Halogenated Heteroarenes. J. Am. Chem. Soc 2020, 142, 6913–6919. [DOI] [PubMed] [Google Scholar]
- (325).Weinberg DR; Gagliardi CJ; Hull JF; Murphy CF; Kent CA; Westlake BC; Paul A; Ess DH; McCafferty DG; Meyer TJ Proton-Coupled Electron Transfer. Chem. Rev 2012, 112, 4016–4093. [DOI] [PubMed] [Google Scholar]
- (326).Ahneman DT; Estrada JG; Lin S; Dreher SD; Doyle AG Predicting Reaction Performance in C–N Cross-Coupling Using Machine Learning. Science 2018, 360, 186–190. [DOI] [PubMed] [Google Scholar]
- (327).Shen Y; Borowski JE; Hardy MA; Sarpong R; Doyle AG; Cernak T Automation and Computer-Assisted Planning for Chemical Synthesis. Nat. Rev. Methods Primers 2021, 1, 23 DOI: 10.1038/s43586-021-00022-5. [DOI] [Google Scholar]