

Summary
Diverse host factors drive microbial variation in plant‐associated environments, whereas their genetic mechanisms remain largely unexplored. To address this, we coupled the analyses of plant genetics and microbiomes in this study. Using 100 tea plant (Camellia sinensis) cultivars, the microbiomes of rhizosphere, root endosphere and phyllosphere showed clear compartment‐specific assembly, whereas the subpopulation differentiation of tea cultivars exhibited small effects on microbial variation in each compartment. Through microbiome genome‐wide association studies, we examined the interactions between tea genetic loci and microbial variation. Notably, genes related to the cell wall and carbon catabolism were heavily linked to root endosphere microbial composition, whereas genes related to the metabolism of metal ions and small organic molecules were overrepresented in association with rhizosphere microbial composition. Moreover, a set of tea genetic variants, including the cytoskeleton‐related formin homology interacting protein 1 gene, were strongly associated with the β‐diversity of phyllosphere microbiomes, implying their interactions with the overall structure of microbial communities. Our results create a catalogue of tea genetic determinants interacting with microbiomes and reveal the compartment‐specific microbiome assembly driven by host genetics.
Keywords: plant–microbiome interactions, microbiome, Camellia sinensis (tea), phyllosphere, rhizosphere, root endosphere
Introduction
Harnessing plant microbiomes is of growing appreciation in sustainable agriculture (Arif et al., 2020; Dessaux et al., 2016; Lawson et al., 2019). Despite the increasing identifications of beneficial and pathogenic plant–microbe interactions (Bulgarelli et al., 2013; Pieterse and Dicke, 2007), our understanding of the host‐driven microbiome assembly is still limited. Crop domestication and cultivation narrow down the ranges of plant genotypes and phenotypes to pursue yield and product quality (Cordovez et al., 2019; Doebley et al., 2006). Meanwhile, these artificial selections may also alter the ecological niches for microorganisms residing in plant‐associated environments (Pérez‐Jaramillo et al., 2017; Xiong et al., 2021). Studies have shown that microbial communities differentiated between plant genotypes (Chen et al., 2019; Kim et al., 2020; Zhang et al., 2021), which in return alter plant performance in nutrition, health and fitness (Bodenhausen et al., 2014; Peiffer et al., 2013; Pérez‐Jaramillo et al., 2017). For these reasons, in the plant holobiont comprised of plant and microbiome, it is of both ecological and agricultural interest to tackle the mechanisms of microbiome assembly driven by host plants (Jones et al., 2019; Vandenkoornhuyse et al., 2015).
Compartment‐specific microbiome assembly in plant‐associated environments is shaped by diverse host factors (Jones et al., 2019). The plant maintains interactions with countless microbes simultaneously, which has a genetic basis (Beilsmith et al., 2019). In rhizosphere environments, root exudates dominate the selection of rhizosphere microbes (Harbort et al., 2020; Liu et al., 2021), whereas cell wall and immune systems are argued to play the major roles in filtering endosphere microbes (Malinovsky et al., 2014; van der Burgh and Joosten, 2019). Compared with root‐associated environments, plant aerial parts are confronted with distinct abiotic and biotic environments and thus interact with microorganisms in different mechanisms (Vorholt, 2012). Both polygenic (Chen et al., 2020b) and monogenic (Zhang et al., 2019b) host effects exist in plant–microbe interactions, whereas a genome‐wide perspective is still needed to reveal the contributions of different genes and functional pathways on compartment‐specific microbiome assembly.
Towards a systemic view of host–microbiome interactions, it is necessary to integrate multi‐omic approaches from both host and microbiome, termed holo‐omics (Nyholm et al., 2020). Notably, a few studies successfully identify plant genetic loci that are associated with microbial variation through microbiome genome‐wide association study (mGWAS) in Arabidopsis thaliana and Picea abies (Elfstrand et al., 2020; Horton et al., 2014). In another recent study, rhizosphere microbiomes are significantly associated with a genomic dataset of Sorghum bicolor (Deng et al., 2021). Moreover, transcriptomic and metabolomic approaches also enable untangling the biological pathways in host–microbiome interplays (Nyholm et al., 2020; Xu et al., 2021a), although still limited by the robustness of sampling and analytical methods. However, these studies almost exclusively lack functional comparisons between plant compartments.
Tea is a perennial woody crop and has been domesticated and cultivated as a beverage plant for a long history (Xia et al., 2020b). Tea plants are featured by diverse secondary metabolites and maintain beneficial or pathogenic relationships with diverse microorganisms (Bag et al., 2022; Tang et al., 2021; Xie et al., 2020). Until recently, the high‐quality reference genome of tea (Camellia sinensis) has been reported (Wang et al., 2020; Wei et al., 2018; Xia et al., 2020a; Zhang et al., 2020), which opens up new possibilities for tea studies from a genome‐wide view. Tea species forms a wide range of diversity panel during domestication and cultivation. C. sinensis var. sinensis and C. sinensis var. assamica are two major varieties. However, due to immense hybridization between cultivars as well as between varieties, widespread gene flow exists between tea accessions (Wang et al., 2020; Xia et al., 2020a), which leads to great challenges for tea genetic studies. Considering the heavy interplay between tea plants and microbes, whether and how tea genetic diversity interacts with microbial variation remain to be elucidated. In previous studies, through investigating the microbiome networks of tea plants across China, we have demonstrated that the core phyllosphere microbiota contributes to the resilience to foliar bacterial pathogens in tea plants (Xu et al., 2022). Moreover, we also uncovered that two metabolites in tea leaves, theophylline and epigallocatechin gallate, can regulate the dynamics of phyllosphere microbiota and promote fungal disease suppression (Xu et al., 2021b). However, considering the complexity of plant–microbiome interactions, the genetic determinants of tea plants driving microbial variation are rarely studied. To address this, we collectively analysed the genetics of 100 tea cultivars and their associated microbiomes residing in the rhizosphere, root endosphere (hereafter root) and phyllosphere environments. We observed clear compartment‐specific microbiome assembly in plant‐associated environments and uncovered multiple plant genetic determinants of microbial diversity and composition.
Results
Compartment‐specific microbiome assembly in plant‐associated environments
The microbiomes of 100 tea cultivars (Table S1) were profiled from bulk soil, rhizosphere soil, root and phyllosphere by targeting the V5–V7 regions of 16S rRNA gene. Principal coordination analysis (PCoA) significantly separated the microbiomes inhabiting the four compartments by the analysis of similarity (ANOSIM, R = 0.46, P = 0.001; Figure 1a). The β‐diversity of phyllosphere microbiomes was distinct from that of the other three compartments (ANOSIM, R = 0.91, P = 0.001; Figure 1b). Alpha diversity (Shannon index) of microbiomes decreased significantly in the order of bulk soil, rhizosphere, root and phyllosphere (one‐way ANOVA and Tukey's test, P < 2 × 10−16; Figure 1c).
Figure 1.
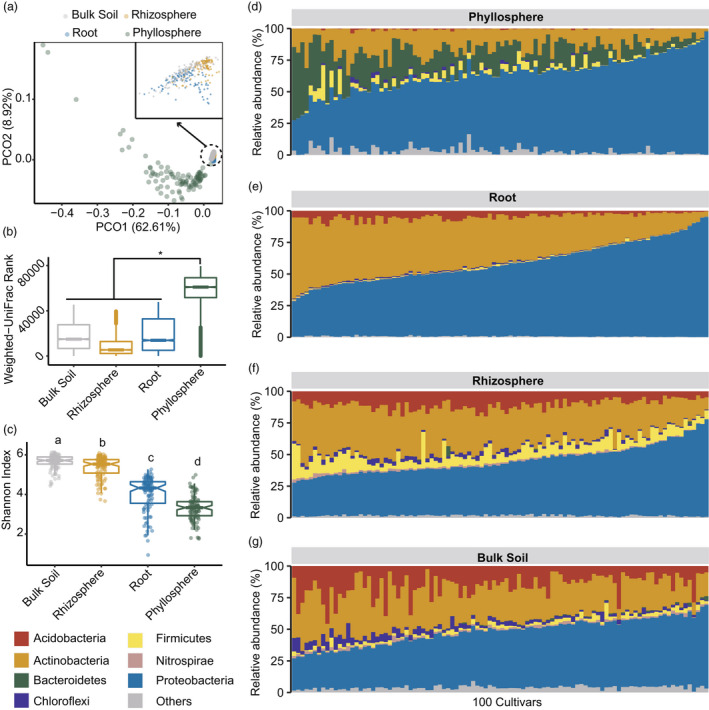
Diversity and composition of microbial communities in 100 tea (Camellia sinensis) cultivars. (a) Principal coordination analysis (PCoA) for the microbiomes in bulk soil, rhizosphere soil, root and phyllosphere compartments (n = 100 for each compartment). Eigenvectors of the top two principal coordinates (PCOs) were used to visualize the microbial β‐diversity. The samples of bulk soil, rhizosphere and root were zoomed‐in for clarity purposes. (b) Comparison of microbial weighted UniFrac dissimilarity between phyllosphere and the other three compartments by the analysis of similarity (ANOSIM, R = 0.91, P = 0.001). (c) Alpha diversity (Shannon index) of the microbiomes in different compartments. The letters above the boxes indicated Tukey's test after one‐way ANOVA (n = 100 for each compartment). (d–g) Microbial composition of 100 tea cultivars in phyllosphere (d), root endosphere (e), rhizosphere (f) and bulk soil (g). The seven most abundant microbial phyla for each compartment were shown and other phyla were aggregated into ‘others’.
Microbial composition at the phylum level significantly differed between compartments (PERMANOVA, R = 0.48, P = 1 × 10−4; Figure 1d–g). Phyllosphere microbiomes were featured by the highest relative abundance of Bacteroidetes and the lowest levels of Acidobacteria and Actinobacteria compared to that of other compartments (P < 2 × 10−16; Figures 1d and S1). Proteobacteria and Actinobacteria dominated root microbiomes and accounted for 93.6% relative abundance (Figure 1e). Rhizosphere microbiomes showed a significantly higher abundance of Firmicutes than that of other compartments (One‐way ANOVA and Tukey's test, P < 2 × 10−16; Figures 1f and S1). Bulk soil microbiomes harboured the highest abundance of Acidobacteria (15.3%) (P < 2 × 10−16) and Chloroflexi (3.6%) (P < 2 × 10−16; Figures 1g and S1).
Enrichment of microbial genera in plant‐associated environments
To further reveal the compartment‐specific compositions of microbiomes, the enrichment of microbial taxa at the genus level was compared between different compartments. Phyllosphere microbiomes were featured by the enrichment of 17 genera belonging to phylum Bacteroidetes versus rhizosphere and root microbiomes (Figures 2a and S2), of which Capnocytophaga (relative abundance = 4.32%) and Chryseobacterium (6.94%) are the most abundant (Figure 2b). Besides, Sphingomonas (22.56%) of phylum Proteobacteria was the most abundant phyllosphere‐enriched taxa (Figure 2b). Most of the genera enriched in root microbiomes belonged to the phyla Actinobacteria and Proteobacteria (Figures 2a and S2), of which the genus Pseudomonas (15.02%) of phylum Proteobacteria showed the greatest relative abundance (Figure 2b,c). Moreover, rhizosphere microbiomes enriched the greatest number of genera belonging to phylum Firmicutes compared to other compartments (Figures 2a and S2), which was highlighted by the genus Bacillus (5.99%; Figure 2b,c).
Figure 2.
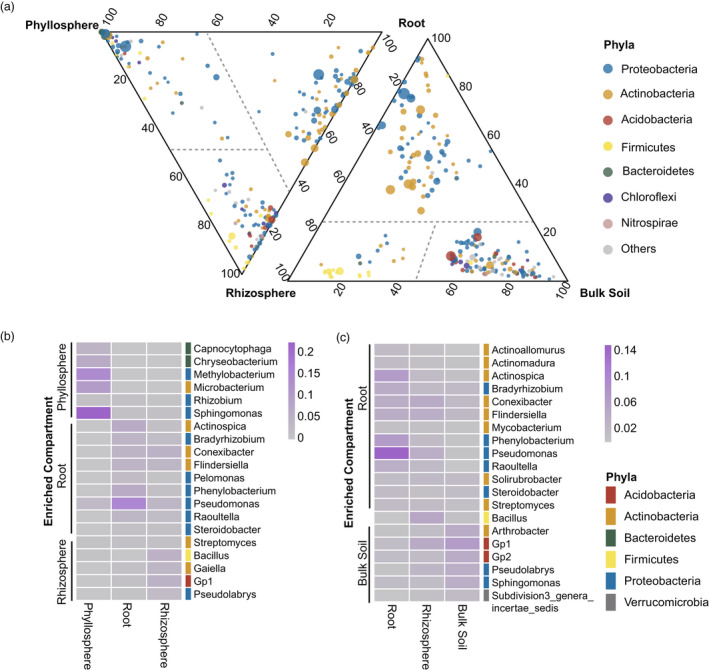
Enrichment of microbial genera in plant‐associated environments. (a) Ternary plots comparing relative abundance of all genera (>0.01%) among phyllosphere, root and rhizosphere microbiomes (left), and among root, rhizosphere and bulk soil microbiomes (right). A negative binomial generalized linear model was performed followed by a likelihood ratio test for comparing the microbial relative abundance between compartments at the genus level. Points represented microbial genera and size represented the average relative abundance across all the three compared compartments. Microbial genera were coloured by their belonged phyla. The seven most abundant microbial phyla for each compartment were shown and other phyla were aggregated into ‘others’. Position of points represented its relative abundance with respect to each compartment. Points were separated by dashed lines, representing their enriched compartment. (b) Heatmap of the 20 most abundant enriched genera compared among phyllosphere, root and rhizosphere microbiomes. (c) Heatmap of the 20 most abundant enriched genera compared between root and rhizosphere and bulk soil microbiomes. Blocks in heatmap were filled with different colour representing the relative abundance of microbial genera. Colour bars on the right of heatmap were labelled by the belonged microbial phyla.
Small effects of tea subpopulation differentiation on microbiomes
We asked whether tea plant genetic variation contributed to the compartment‐specific microbiome assembly. The resequencing of the 100 tea cultivars yielded >13 million high‐quality single nucleotide polymorphisms (SNPs), which formed a dense genetic dataset considering the ~3 Gb genome size of the tea plant (Xia et al., 2020a). According to the ancestry analysis, 100 tea cultivars were divided into three subpopulations, POP1, POP2 and POP3 (Figures 3a and S3). The genetic dissimilarity was significantly differentiated between tea subpopulations (ANOSIM, R = 0.67, P = 0.001; Figure 3b). Forty‐eight cultivars of the POP1 (n = 61) originated from Zhejiang and Fujian provinces of China (Table S1), suggesting the geographical origins contributed to their genetic similarity. In comparison, the cultivars of POP2 (n = 28) originated from diverse regions, including Jiangxi, Hunan and Sichuan provinces of China (Table S1). Interestingly, the POP3 (n = 11) was highlighted by ‘Fuding Dabaicha’ and several cultivars derived from the cross between ‘Fuding Dabaicha’ and ‘Yunnan Dayecha’, including ‘Yingshuang’, ‘Zhenong113’ and ‘Jinfeng’, suggesting their genetic relatedness during tea breeding (Table S1).
Figure 3.
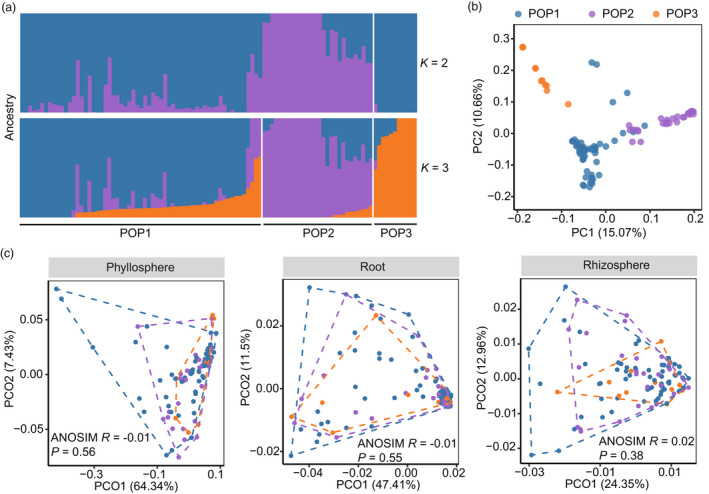
Effects of tea (Camellia sinensis) genetic diversity on plant‐associated microbiomes. (a) Ancestry analysis of 100 tea cultivars. The maximum likelihood estimation of ancestries (K) for tea cultivars (n = 100) was estimated. Five‐fold cross‐validations were performed with K values ranging from 2 to 10. And the most probable number of subpopulations (POP) was estimated by the lowest cross‐validation error, which was K = 3 here (Figure S2). Tea cultivars were clustered into three groups by hierarchical cluster analysis based on their genetic dissimilarity matrix. (b) Principal component analysis of the tea genetic dissimilarity. (c) Comparisons of microbial β‐diversity between tea subpopulations. Principal coordination analysis (PCoA) for microbiomes in phyllosphere, root and rhizosphere was performed. Points represented tea cultivars (n = 100) and were coloured by the predicted subpopulations. The microbial weighted UniFrac dissimilarity between subpopulations was compared by the analysis of similarity (ANOSIM).
The microbiomes in phyllosphere, root and rhizosphere compartments exhibited homogeneous β‐diversity compared between tea subpopulations (Figure 3c), suggesting small effects of tea subpopulation differentiation on microbiomes. In addition, the geographic origins also showed no impact on the microbial β‐diversity in all compartments (ANOSIM, P > 0.05).
Genome‐wide associations of microbial composition
We further examined the effects of individual genetic variants on the relative abundance of microbial taxa through mGWAS. After excluding the rare operational taxonomic units (OTUs), which were present in less than 10% of samples and with the relative abundance lower than 0.01%, 954 rhizosphere OTUs, 596 root OTUs and 403 phyllosphere OTUs were included in the analyses.
By gathering the significant associations from mGWAS results, we observed that 39 OTUs were associated with 44 nonsynonymous exonic variants, lying within 41 genes (Table S2). The gene ontology (GO) of candidate genes was annotated to show the gene functions, and two terms of cellular component, ‘intrinsic component of membrane’ (GO:0031224) and ‘integral component of membrane’ (GO: 0016021, a child term of the former) were over‐represented.
In association with root microbial taxa, the strongest association was observed between the Otu956 (genus Streptomyces) and the SNP Chr1_91139090 in CSS0044082 gene (β‐carotene hydroxylase 2 gene, NR annotation; P = 2.98 × 10−23; Figures 4a and S4). Additionally, in association with rhizosphere microbial taxa, the rhizosphere Otu4877 (genus Conexibacter) was significantly associated with the SNP Chr9_67484586 in CSS0040599 gene (predicted cytochrome P450 81D11‐like, NR annotation; P = 3.09 × 10−17; Figures 4b and S4). In phyllosphere microbiomes, only the Otu2029 (genus Mycoplasma) was linked to the SNP Chr4_146209412 in CSS0043600 (C2H2‐type zinc finger, Pfam annotation; Figures 4c and S4).
Figure 4.
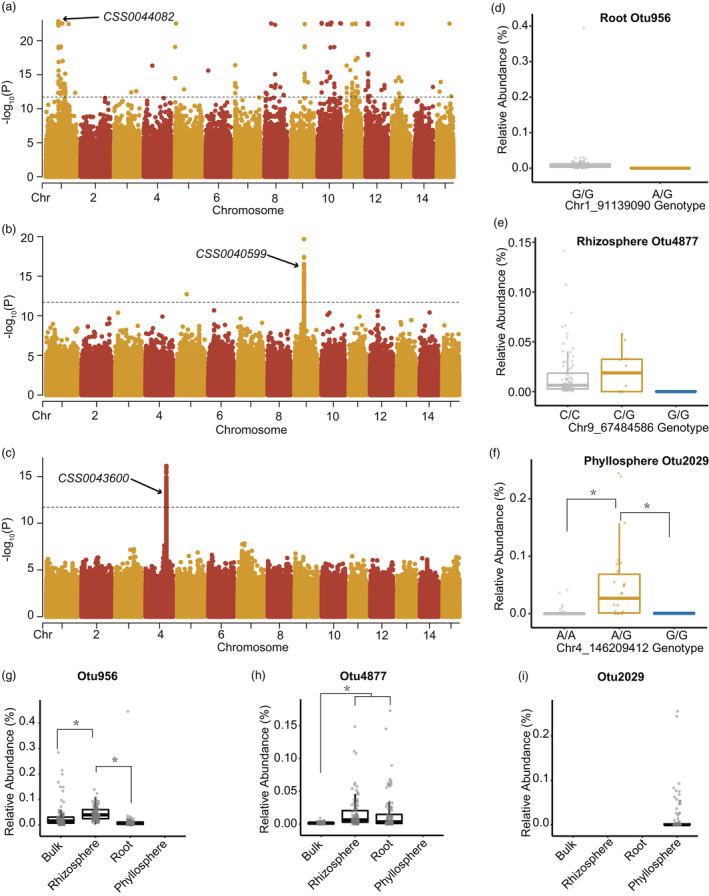
Genome‐wide associations between tea (Camellia sinensis) genetic variants and the abundance of individual microbes. (a–c) Manhattan plots for the genome‐wide associations of root endosphere Otu956 (a), rhizosphere Otu4877 (b) and phyllosphere Otu2029 (c). The dashed line represented the significance level of 1.95 × 10−12 (transformed with –log10; Bonferroni correction). The candidate genes of the nonsynonymous exonic variants were indicated by arrows. (d–f) Relative abundance of the microbial taxa compared between the genotypes of significant genetic variants. (g–i) Relative abundance of the microbial taxa compared between the four compartments.
The genotypes of the associated SNPs showed no effects on the relative abundance of root Otu956 and rhizosphere Otu4877 (Figure 4d,e). In contrast, tea cultivars (29 of 100) with the heterozygote of the SNP Chr4_146209412 showed a significantly higher relative abundance of phyllosphere Otu2029 than other cultivars (Figure 4f). Besides, the Otu956 and Otu4877 were only present in root‐associated environments and showed varied relative abundance compared among bulk soil, rhizosphere and root microbiomes (Figure 4g,h). In comparison, the Otu2029 was uniquely present in phyllosphere microbiomes (Figure 4i).
Compartment‐specific microbiome assembly driven by plant genetics
To reveal the potential functional interactions between microbial composition and tea genetics, we further searched for all genes nearby the significant variants, except for ‘intergenic’ SNPs that were distant from gene coding regions (>21.1 kb) and ‘intronic’ SNPs. The links between microbial taxa and the GO annotation of candidate genes were used to build a bipartite function–taxon network (Figure 5a). Rhizosphere and root microbial OTUs showed heavy overlaps in association with plant biological processes. Twenty‐five GO biological processes were shared between the three compartments, including several terms that were related to cellular vesicle trafficking, plant response to abiotic/biotic factors and signalling mechanisms (Figure 5a, Table S3).
Figure 5.
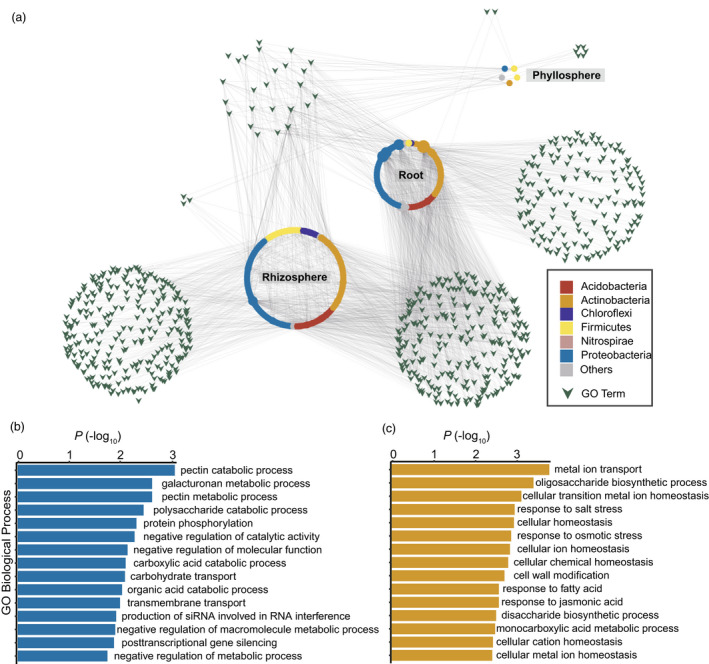
Tea (Camellia sinensis) biological processes in association with microbial composition. (a) Linkages between the gene ontology (GO) biological processes and the microbial taxa. The relationships between plant candidate genes and microbial operational taxonomic units (OTUs) were built by mGWAS. The GO biological processes were annotated to the candidate genes and were then linked to microbial OTUs. Dots represented the microbial OTUs and were coloured by their phylum taxonomy. The triangular arrows represented the GO biological processes for the candidate genes. The size of shapes represented their degrees in the network. (b, c) Enrichment analysis of GO biological processes in association with root (b) and rhizosphere (c) microbial composition. The GO terms were ranked by the –log10 of the P values and the top 15 terms were shown.
GO biological processes exhibited distinct enrichment in association with the microbiomes of different compartments. Associating with root microbial OTUs, cell wall metabolism (pectin and galacturonan) and carbon catabolism (polysaccharide, carboxylic acid and organic acid) were the most enriched biological processes (Figure 5b, Table S4). Associating with rhizosphere microbial OTUs, GO terms related to the metabolism of metal ions (transport and homeostasis) and small organic molecules (oligosaccharide, disaccharide and monocarboxylic acid) were overrepresented (Figure 5c, Table S5). In comparison, only one biological process of ‘response to stimulus’ was enriched in association with phyllosphere microbial OTUs.
Genome‐wide associations of microbial β‐diversity
Followed by the observations of multiple links between individual microbial taxa and tea genetic variants, we also examined whether tea genetics is associated with the overall microbial community structure. Therefore, mGWAS analyses were performed for the top five principal coordinates (PCOs) of rhizosphere, root and phyllosphere microbiomes, which accounted for 83.9%, 50.5% and 47.3% microbial variation respectively (Figure S5).
In association with phyllosphere microbiomes, 73 SNPs passed the significant threshold of P < 3.81 × 10−9, consisting of 1 nonsynonymous exonic, two synonymous exonic, two downstream, one upstream, 59 intergenic and eight intronic variants (Figure S6, Table S6). The genetic dissimilarity of these SNPs was significantly correlated with phyllosphere microbial dissimilarity (Mantel test, R = 0.13, P = 0.01; Figure 6a). In contrast, no significant hits were observed for rhizosphere microbial β‐diversity, and only one noncoding intergenic variant of Chr12_18479397 showed significant association with root microbial PCO2 (P = 2.07 × 10−9; Figure S6).
Figure 6.
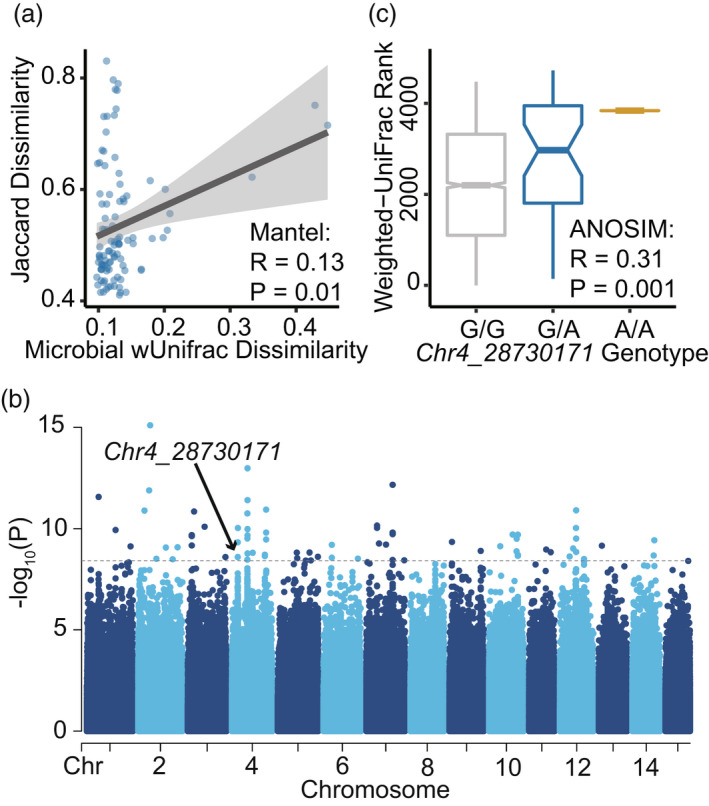
Genome‐wide associations between tea (Camellia sinensis) genetic variants and the β‐diversity of phyllosphere microbiomes. (a) Mantel correlation between the microbial dissimilarity (weighted UniFrac) of phyllosphere microbiome and the genetic variation of 73 significant genetic variants (Jaccard dissimilarity). (b) Manhattan plot for the associations between the first principal coordinate (PCO1) of phyllosphere microbiomes. The dashed line represented the significance level of 3.89 × 10−9 (transformed with –log10) with the P values corrected by the number of SNPs included in the analyses (Bonferroni correction). (c) Comparison of the microbial dissimilarity between the three genotypes of Chr4_28730171 (ANOSIM analysis).
Phyllosphere microbial PCO1 contributed to the most significant associations with tea genetic variants except for one variant associating with PCO2 (Figure 6b). Among the variants, the only nonsynonymous exonic variant Chr4_28730171 (P = 2.55 × 10−9) was located within CSS0046992 gene on chromosome 4 (Figures 6b and S7), which was annotated as formin homology interacting protein 1 (FIP1) against NR database. Notably, the dissimilarities of phyllosphere microbial communities significantly differed between the genotypes of Chr4_28730171 (ANOSIM, R = 0.31, P = 0.001; Figure 6c).
Discussion
Plants provide diverse niches for microbes through physiological and biochemical features (Reinhold‐Hurek et al., 2015). Although being frequently argued, it remains challenging to identify host genetic factors interacting with microbiomes in different compartments (Wagner et al., 2016). In this study, we comprehensively explored the microbiomes residing in plant‐associated environments and their interactions with the genetics of 100 tea cultivars. We uncovered multiple tea genes that hold the potential to modulate microbial diversity and composition. Notably, microbiomes in different compartments were linked to divergent biological processes in tea plants, suggesting obvious compartment‐specific microbiome assembly driven by host genetics. Our results emphasize that plant genetics interact with microbiomes as an intricate genome‐wide network. The associations identified in this study are worthwhile to be further explored aiming at harnessing the roles of microbes in tea breeding.
In our results, compartment‐specific microbiome assembly was observed in plant‐associated environments. Specifically, tea phyllosphere microbiomes were distinct from other compartments in both community structure and composition. These results corroborate our earlier large‐scale study on tea‐associated microbiomes across the main plantation regions of China (Xu et al., 2022). Microbes residing phyllosphere environment are derived from complicated sources including soil, atmosphere and others (Bulgarelli et al., 2013). Compared to root‐associated environments, the phyllosphere is poor in nutrients and heavily affected by extreme abiotic factors of radiation and temperature (Turner et al., 2013). Therefore, the adaption of microorganisms in the phyllosphere includes the ability to scavenge different nutrients at low concentrations (Vorholt, 2012). In our results, the genus Sphingomonas was enriched in phyllosphere and showed a high relative abundance, which is consistent with its predominant roles in the leaf‐associated environments of A. thaliana and soybean (Delmotte et al., 2009; Vorholt, 2012). Besides, the phylum Firmicutes showed a preference for the rhizosphere environment and was nearly absent in the root endosphere. Further examination showed that the Firmicutes genus Bacillus was among the most abundant rhizosphere‐enriched taxa. Soil Bacillus positively responds to the increased levels of epigallocatechin gallate, a bioactive compound from tea plants (Tang et al., 2021). Moreover, the phylum Bacteroidetes exhibited higher abundance in the phyllosphere than other compartments, which was also reported in sugar maple (Acer saccharum) and a seagrass species (Tarquinio et al., 2019; Wallace et al., 2018). These results collectively pointed to the compartment‐specific assembly of microbiomes in plant‐associated environments. It has been well acknowledged that plant factors play varied roles in the selection of microbes in different compartments, including root exudates and immune systems (Malinovsky et al., 2014; Sasse et al., 2018; van der Burgh and Joosten, 2019). However, our understanding of how plants create these selection mechanisms from genetics to metabolism is still in infancy. Therefore, as discussed below, the present study mainly explored the genetic determinants that were associated with microbial diversity and composition.
Through mGWAS, rhizosphere and root microbial compositions were associated with divergent plant biological processes. These results reveal clear compartment‐specific microbiome assembly driven by plant genetics. Specifically, genes related to the metabolism of ions and small organic compounds were overrepresented interacting with rhizosphere microbial abundance. This result points to the vital roles of root organic exudates in filtering and cultivating rhizosphere microbes in exchange for nutrients including mineral ions (Harbort et al., 2020; Sasse et al., 2018; Vieira et al., 2020), which constitute a major proportion of resource trade between plants and microbes in the rhizosphere. In comparison, cell wall metabolism, carbon catabolism and post‐transcriptional regulation were highlighted in plant–microbiome interactions in the root endosphere. Cell walls form dispersion barriers for microbes and carbon catabolic processes provide resources to endophytes (Malinovsky et al., 2014). Additionally, post‐transcriptional regulation is heavily involved in recognizing and responding to microbes (Carpenter et al., 2014). It is hypothesized that microbes experience a ‘two‐step’ selection mechanism when immigrating from the rhizosphere to root endosphere (Bulgarelli et al., 2013). In this hypothesis, rhizodeposition is mainly responsible for filtering microbes from the surrounding soil, and the innate immune system recognizes and responds to microbial endophytes (Bulgarelli et al., 2013). In support of this hypothesis, our observation that root endosphere microbiomes displayed a reduced microbial α‐diversity than that of rhizosphere and bulk soil microbiomes exhibited a clear selection process. More importantly, our results link the microbiomes in the rhizosphere and root endosphere to differential plant biological processes, which reveal the plant genetic basis for the ‘two‐step’ selection hypothesis in root‐associated environments.
The candidate genes interacting with microbial composition were involved in diverse plant biological processes. Notably, the relative abundance of root endosphere Otu956 (genus Streptomyces) was strongly linked to the β‐carotene hydroxylase 2 gene, encoding a key enzyme in the transformation of carotenoids. Besides, the Otu956 showed varied relative abundance in root‐associated environments, suggesting a selection effect from tea plants. Plant carotenoids are involved in different plant primary and secondary metabolisms, including serving as antioxidants and hormone precursors in plant defence (Howitt and Pogson, 2006; Zilber‐Rosenberg and Rosenberg, 2021). Both plants and many Streptomyces species can synthesize carotenoids (Schmidt‐Dannert, 2000), and thus Streptomyces may promote the homeostasis of tea roots via modulating carotenoid metabolism. As shown in our previous study, several metabolites in tea plants, including theophylline and epigallocatechin gallate, are associated with microbial variation (Xu et al., 2021b). Therefore, carotenoid metabolism may also bridge the plant–microbiome interactions in tea plants.
The associations between plant defence‐related genes and the microbial taxa of Mycoplasma and Conexibacter underly the ubiquitous roles of plant defence interacting with microbes. Mycoplasma species are nonculturable prokaryotes and potentially pathogenic to plants (Razin and Hayflick, 2010), which can colonize plants from environments or be transmitted from insects (Hogenhout et al., 2008). Besides, the genus Conexibacter is involved in soil carbon and phosphorus cycling (Ma et al., 2021), and is induced by root‐exuded L‐theanine from tea plants (Xie et al., 2022). Both pathogenic and nonpathogenic microbes can induce plant defence responses (van der Burgh and Joosten, 2019). It has been shown that a cytochrome P450 81D11‐like homologue in A. thaliana is involved in the cis‐jasmone‐induced plant defence against insects (Bruce et al., 2008), and many C2H2‐type zinc finger genes act as transcription factors in plant stress response to pathogens (Kiełbowicz‐Matuk, 2012). Consistently in our results, the C2H2‐type zinc finger gene (CSS0043600) was linked to a potentially pathogenic Mycoplasma OTU, pointing to the role of this gene in response to tea pathogens. In tea plants, it has been reported that several C2H2 transcription factors are involved in the biosynthesis of catechins (Wei et al., 2018), which are important secondary metabolites and are suppressive to tea foliar pathogens (Xu et al., 2021b). However, considering the diverse functions of C2H2 gene family members, the association between the C2H2‐type zinc finger gene (CSS0043600) and Mycoplasma Otu2029 needs further elucidation. Plant defence mechanisms form the primary filters for microbes, including genes involved in the recognition and responses of plants (van der Burgh and Joosten, 2019). Similarly, mGWAS in A. thaliana also identified the associations between microbiomes and plant defence‐related genetic loci (Horton et al., 2014), suggesting these mechanisms are likely conserved across different plant species. It was also noticeable that the SNP Chr_146209412 in association with the Mycoplasma Otu2029 showed a high heterozygosity rate of 29%, reflecting the heavy hybridization between tea genotypes. The self‐incompatibility of tea plants results in high genome heterozygosity (Xia et al., 2020a). Thus, our results linked the hybridization history of tea cultivars to the variation in the Mycoplasma in tea phyllosphere.
Our results also uncovered the genetic determinants of tea plants in association with the phyllosphere β‐diversity. These results are congruent with previous reports of the impacts of host genetic factors on the overall microbial communities (Busby et al., 2017; Chen et al., 2020b; Zhang et al., 2019b). Most interestingly, the predicted FIP1 gene (CSS0046992) harbours the only nonsynonymous exonic SNP that was associated with phyllosphere microbial β‐diversity. This gene is enriched during tea plant varietal improvement (Xia et al., 2020a), indicating the effects of tea breeding on it. In addition, our results also proved that the phyllosphere microbial communities differentiated between the FIP1 genotypes, which further confirmed the role of FIP1 in shaping the phyllosphere microbiomes. FIP1 protein interacts with plant formins (formin homology proteins) and affects the actin cytoskeleton (Banno and Chua, 2000; Doerks et al., 2000). Thus, the interactions between the FIP1 gene and phyllosphere microbial communities may involve the modification of cell structures (Jones and Dangl, 2006; Sassmann et al., 2018). Besides the FIP1 gene, the dissimilarity of phyllosphere microbiomes was significantly correlated with the genetic variation of the 73 associated SNPs. Thus, except for the nonsynonymous variant, other genetic variants may still be functionally interacting with the tea phyllosphere microbiome.
Heavy hybridization between tea cultivars, as well as between varieties, occurs during tea breeding, which raises great challenges for tea genetic studies (Wang et al., 2020). Meanwhile, we identified three subpopulations for the 100 tea cultivars, and considerable overlap of genetic similarity existed between the POP1 and POP2 subpopulations, indicating the homogeneity of these cultivars. The POP3 was relatively distinct from the other two subpopulations, which included ‘Fuding Dabaicha’ and several accessions with the hybridization records between ‘Fuding Dabaicha’ and other tea accessions (Wang et al., 2021). Microbiomes showed no differentiation between the tea subpopulations and between their geographic origins, which is, at least partially, due to the relatedness between the examined tea cultivars. It has been reported that the rhizosphere microbial compositions varied between wild and domesticated common beans (Phaseolus vulgaris; Pérez‐Jaramillo et al., 2017). Thus, the inclusion of wild (or ancient) and genetic distant populations in the future may facilitate the comparisons of microbiomes between tea subpopulations.
Overall, our study reveals clear compartment‐specific microbiome assembly driven by tea plant genetics by incorporating plant genetics and microbiome. We uncovered a set of genetic variants interacting with microbiomes inhabiting different plant‐associated environments. Importantly, the microbial diversity and composition were linked to diverse plant genetic factors, which increased the complexity of plant–microbiome associations. Thus, considering the infancy of holo‐omic studies, further development of suitable methodologies is eagerly needed to reveal the interacting mechanisms in plant holobionts (Goodrich et al., 2016; Nyholm et al., 2020). We used 16S amplicon sequencing to reconstruct the microbial communities in this study, which was effective to capture the microbial taxonomic composition but was limited by the lack of functional information. Therefore, with the increased applications of multi‐omic approaches and the continuously decreased costs, it is worthy to expect more advances in holo‐omic studies incorporating the metabolic and transcriptomic attributes from both host and microbiome (Xu et al., 2021a). Moreover, we note that the causality of mGWAS results needs to be further improved. Promisingly, a few pioneering studies have employed mendelian randomization analysis to estimate the causal links between host genetic loci and microbial taxa (Liu et al., 2022; Xu et al., 2020), whereas the interpretation of results requires caution because of the complexity of mechanisms in host–microbe interactions (Kurilshikov et al., 2021).
Experimental procedures
Plant materials and sampling
The tea cultivars were grown in a common garden of Hangzhou Agriculture Academy (Hangzhou, China, 30.19°N, 120.07°E). Leaves, root tissues, rhizosphere soil and bulk soil (unplanted) of a total of 100 cultivars were sampled for 16S amplicon sequencing (Table S1). The geographic origins of the cultivars include Zhejiang (n = 41), Fujian (n = 24), Jiangxi (n = 8), Hunan (n = 7), Guangxi (n = 6) and other provinces (Table S1), which are main tea production regions in China.
For each tea cultivar, three replicates of healthy plants were randomly sampled. Nine leaves were sampled from the top of three branches and pooled together as one replicate for each plant. Root and soil samples were collected from the four ordinated directions of tea plants and pooled together as one replicate (Xu et al., 2018). The top ~5 cm of soil was removed, and 5–8 g of fine roots from a depth <15 cm were collected. Roots were removed from the soil with a shovel and were manually shaken to remove loosely attached soil. Roots and soil samples were then stored in zipper plastic bags and transferred to the lab on ice on the same day. In the lab, fine roots were washed three times with 25 mL PBS in 50 mL centrifuge tubes. The washed‐off soil was centrifuged at 3000 g for 10 min and used as rhizosphere soil (Xu et al., 2018). After that, roots were placed in centrifuge tubes with 15 mL PBS and sonicated at 50 Hz for 30 s. Three times sonication were performed, which had been proved to effectively remove root surface microbes (Edwards et al., 2015; Lundberg et al., 2012). Three unplanted soil samples (bulk soil) were also collected 1 m away from each tea cultivar with ~20 g for each of the samples. Roots and soil samples were stored at −80 °C for ~2 months until DNA extraction. In total, the 100 tea cultivars yielded 300 bulk soil, 300 rhizosphere soil, 300 root and 300 leaf samples.
Microbial amplicon sequencing and data processing
DNA was extracted by the PowerSoil DNA Isolation kit (MoBio Laboratories, CA) from ~0.5 g sample following the manufacturer's instructions. Leaves and roots were sheared with a scissor to fit the DNA extraction kit. The primers of 799F and 1193R were used in the amplification of the variable regions V5–V7 of bacterial 16S rRNA genes (Horton et al., 2014). An Illumina NovaSeq platform was used for amplicons sequencing and generated 250‐bp paired‐end reads at Personalbio (Shanghai, China). UPARSE pipeline was employed for controlling read quality and clustering OTUs (Edgar, 2013). The OTUs were clustered based on a threshold of 97% similarity. Bacterial taxonomy was classified with the 16S rRNA reference database RDP Version 16 (Cole et al., 2014). Reads counts of OTUs were normalized in the DESeq2 R package (Love et al., 2014). The weighted UniFrac dissimilarities of microbiomes were visualized with PCoA analysis in the phyloseq R package (McMurdie and Holmes, 2013). Enrichment of microbial taxa at genus level was analysed in edgeR and ggtern R packages by fitting the relative abundance of each genus (>0.01%) to a negative binomial generalized linear model (glmFit) followed by a likelihood ratio test (glmLRT; Bonferroni correction, P < 0.05; Zgadzaj et al., 2016).
Genotyping
A short‐size (350 bp) DNA library was constructed and sequenced using an Illumina Novaseq PE150 platform (Illumina, San Diego, CA). Low‐quality bases were removed using Trimmomatic v0.39. The high‐quality reads were aligned to the genome assembly of C. sinensis var. sinensis (cultivar Shuchazao; Xia et al., 2020a) using BWA v0.7.17‐r1188 with default parameters (Li and Durbin, 2009), and sorted with SAMtools v1.9 subsequently (Li et al., 2009). The MarkDuplicates command in GATK v4.1.4.0 (Depristo et al., 2011) was used for the removal of duplicated reads. Genotypes were called using FreeBayes v1.3.1 with the parameters of ‐‐min‐repeat‐entropy 1 ‐‐min‐coverage 10 ‐‐limit‐coverage 100 000 (Garrison and Marth, 2012). The yielded VCF files were finally filtered using the vcffilter command of vcflib software (Garrison et al., 2021) to get high‐quality SNPs.
Principal component analysis was done using PLINK 1.9 (Chang et al., 2015). The genetic similarity of tea cultivars was measured as identity‐by‐sequence (IBS) values using PLINK, and the genetic dissimilarity matrix was constructed with 1–IBS values (Zhao et al., 2011). The population structure of tea cultivars was estimated using ADMIXTURE 1.3.0 (Alexander et al., 2009). The maximum likelihood estimation of ancestries (K) was estimated and fivefold cross‐validations were performed with K values ranging from 2 to 10. The most probable number of subpopulations was estimated by the lowest cross‐validation error, which was K = 3 in this study (Figure S2). The tea cultivars were subsequently classified into three groups by clustering the genetic dissimilarity matrix with hclust and cutree function in R.
Single nucleotide polymorphisms were annotated to the reference genome (Xia et al., 2020a) mentioned above by ANNOVAR (Wang et al., 2010). The files of coding sequences and gene function of the tea reference genome were downloaded from the Tea Plant Information Archive (http://tpdb.shengxin.ren/). The gene function file contained GO terms and the annotations against KEGG, KOG, Pfam, TrEMBL and NCBI NR databases (Xia et al., 2020a). Linkage disequilibrium was estimated by calculating the squared correlation coefficient (r 2) between SNPs and was visualized in PopLDdecay and LDBlockShow (Zhang et al., 2019a).
Microbiome genome‐wide association analysis
The mGWAS was conducted using GEMMA 0.98.1 software (Zhou and Stephens, 2012). A kinship matrix was firstly constructed in GEMMA to control the effects of genetic relatedness between tea cultivars. Wald test using a univariate linear mixed model was performed for each SNP variant and ‘phenotype’ to yield an estimation of the P value (Zhou and Stephens, 2012).
For mGWAS with microbial β‐diversity, the eigenvectors of top five PCOs from PCoA analyses were associated with tea genetic variants separately. Bonferroni correction was performed by dividing the α (=0.05) by the number of SNPs (13 124 663) after quality control in GEMMA, which is α = 3.81 × 10−9. For mGWAS with microbial composition, microbial OTUs present in more than 10% of samples and with a minimum relative abundance of 0.01% in each compartment were included in the analyses, which resulted in 954 rhizosphere OTUs, 596 root OTUs and 403 phyllosphere OTUs. The relative abundance of each OTU was employed as a phenotypic trait in mGWAS and was fitted to a univariate linear mixed model in GEMMA software. Since these analyses yielded a large amount of results, a stricter significance level was set by dividing the α (=0.05) by the total number of OTUs (1953) and the number of SNPs (13 124 663) after quality control in GEMMA, which was α = 1.95 × 10−12.
Quantile–quantile (Q‐Q) plot and Manhattan plot were generated with the rMVP R package to show the distribution of observed and expected P values (−log10 transformed; Yin et al., 2021).
The significant SNPs of mGWAS were annotated to candidate genes and GO functions as stated in genotyping methods. The searches for candidate genes were performed within 21.1 kb distance around SNPs at which linkage disequilibrium dropped to a baseline of 0.02. ‘Intergenic’ SNPs that were distant from gene coding regions (distance > 21.1 kb) and ‘intronic’ SNPs were excluded from the analyses. To build the bipartite network of function–taxon (Ma et al., 2021), the associations between GO terms and OTUs were gathered and the network was constructed in the igraph R package by graph_from_edgelist function (Csardi and Nepusz, 2006) and visualized in Cytoscape 3.7.2 (Shannon et al., 2003). GO enrichment analysis was performed in TBtools (Chen et al., 2020a) to find the over‐represented GO categories for sets of genes.
Statistical analyses
Means were compared by one‐way ANOVA and Tukey's post hoc test in the agricolae R package (De Mendiburu, 2020). In the vegan R package (Oksanen et al., 2020), Shannon index was calculated with diversity function, Mantel test was performed using mantel function with ‘spearman’ method and 9999 times of permutation, ANOSIM was performed to compare dissimilarity matrices using anosim function with 999 times of permutation and PERMANOVA was performed with adonis function with a ‘bray’ method and 9999 times of permutations. For the ANOSIM comparing the microbial dissimilarity between tea geographic origins, only the regions with more than two cultivars were included to control the effects of unbalanced group size on the results (Anderson and Walsh, 2013).
Conflict of interest
The authors declare that they have no conflict of interest.
Author contributions
BM, PX and XT designed the study, XT and HX performed the experiments, XT and JY analysed the data. XT wrote the original manuscript and all authors reviewed and edited the manuscript.
Supporting information
Figure S1 Comparisons of microbial composition on phylum level among the compartments of bulk soil (Bulk), rhizosphere (Rhizo), root endosphere (Root) and phyllosphere.
Figure S2 Counts of enriched microbial genera belonging to different phyla.
Figure S3 Cross‐validation (CV) of ancestry analysis for 100 tea (Camellia sinensis) cultivars.
Figure S4 Local Manhattan plots of the significant genetic loci associated with root endosphere Otu956 (a), rhizosphere Otu4877 (b) and phyllosphere Otu2029 (c).
Figure S5 Variance explained by the top five principal coordinates (PCOs) of phyllosphere, rhizosphere and root endosphere (root) microbiomes.
Figure S6 Associations of microbial principal coordinates (PCOs) and tea (Camellia sinensis) genetic variants through mGWAS.
Figure S7 Local Manhattan plot of the significant genetic variant Chr4_28730171 associated with phyllosphere microbiomes.
Table S1 Tea (Camellia sinensis) cultivars used in this study.
Table S2 Nonsynonymous exonic variants in the genome‐wide associations of the microbial operational taxonomic units (OTUs).
Table S3 Gene ontology (GO) associated with phyllosphere, root endosphere and rhizosphere compartments.
Table S4 Gene ontology (GO) enrichment analysis of the candidate genes associated with root endosphere operational taxonomic units (OTUs).
Table S5 Gene ontology (GO) enrichment analysis of the candidate genes associated with rhizosphere–endosphere operational taxonomic units (OTUs).
Table S6 Significant genetic variants in the genome‐wide associations of the principal coordinates (PCOs) of phyllosphere microbiomes.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (42107314, 42090060 and 4201101246) and the Natural Science Foundation of Zhejiang Province (LD19D060001). The authors are grateful to Prof. Xuxia Zheng of Hangzhou Agriculture Academy for his kind supports on sample collection.
Contributor Information
Ping Xu, Email: zdxp@zju.edu.cn.
Bin Ma, Email: bma@zju.edu.cn.
Data availability statement
Sequencing data of 16S rRNA are available from the Genome Sequence Archive (GSA) under the BioProject PRJCA007800. Sequencing reads of tea plant cultivars reported in this study are available from the National Center for Biotechnology Information (NCBI) under the BioProject PRJNA798546. The Variant Call Format files of the sequencing of tea cultivars can be accessed from the Genome Variation Map (GVM) under the BioProject PRJCA010001. Codes for the full analyses included in this study are available on GitHub repository (https://github.com/hahafengxiang/tea_microbiome).
References
- Alexander, D.H. , Novembre, J. and Lange, K. (2009) Fast model‐based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, M.J. and Walsh, D.C.I. (2013) PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: what null hypothesis are you testing? Ecol. Monogr. 83, 557–574. [Google Scholar]
- Arif, I. , Batool, M. and Schenk, P.M. (2020) Plant microbiome engineering: expected benefits for improved crop growth and resilience. Trends Biotechnol. 38, 1385–1396. [DOI] [PubMed] [Google Scholar]
- Bag, S. , Mondal, A. and Banik, A. (2022) Exploring tea (Camellia sinensis) microbiome: insights into the functional characteristics and their impact on tea growth promotion. Microbiol. Res. 254, 126890. [DOI] [PubMed] [Google Scholar]
- Banno, H. and Chua, N.H. (2000) Characterization of the Arabidopsis formin‐like protein AFH1 and its interacting protein. Plant Cell Physiol. 41, 617–626. [DOI] [PubMed] [Google Scholar]
- Beilsmith, K. , Thoen, M.P.M. , Brachi, B. , Gloss, A.D. , Khan, M.H. and Bergelson, J. (2019) Genome‐wide association studies on the phyllosphere microbiome: embracing complexity in host–microbe interactions. Plant J. 97, 164–181. [DOI] [PubMed] [Google Scholar]
- Bodenhausen, N. , Bortfeld‐Miller, M. , Ackermann, M. and Vorholt, J.A. (2014) A synthetic community approach reveals plant genotypes affecting the phyllosphere microbiota. PLoS Genet. 10, e1004283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce, T.J.A. , Matthes, M.C. , Chamberlain, K. , Woodcock, C.M. , Mohib, A. , Webster, B. , Smart, L.E. et al. (2008) cis‐Jasmone induces Arabidopsis genes that affect the chemical ecology of multitrophic interactions with aphids and their parasitoids. Proc. Natl. Acad. Sci. USA, 105, 4553–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulgarelli, D. , Schlaeppi, K. , Spaepen, S. , van Themaat, E.V.L. and Schulze‐Lefert, P. (2013) Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 64, 807–838. [DOI] [PubMed] [Google Scholar]
- Busby, P.E. , Soman, C. , Wagner, M.R. , Friesen, M.L. , Kremer, J. , Bennett, A. , Morsy, M. et al. (2017) Research priorities for harnessing plant microbiomes in sustainable agriculture. PLoS Biol. 15, e2001793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter, S. , Ricci, E.P. , Mercier, B.C. , Moore, M.J. and Fitzgerald, K.A. (2014) Post‐transcriptional regulation of gene expression in innate immunity. Nat. Rev. Immunol. 14, 361–376. [DOI] [PubMed] [Google Scholar]
- Chang, C.C. , Chow, C.C. , Tellier, L.C.A.M. , Vattikuti, S. , Purcell, S.M. and Lee, J.J. (2015) Second‐generation PLINK: rising to the challenge of larger and richer datasets. GigaScience, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Chen, H. , Zhang, Y. , Thomas, H.R. , Frank, M.H. , He, Y. and Xia, R. (2020a) TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant, 13, 1194–1202. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Li, S. , Zhang, Y. , Li, T. , Ge, H. , Xia, S. , Gu, J. et al. (2019) Rice root morphological and physiological traits interaction with rhizosphere soil and its effect on methane emissions in paddy fields. Soil Biol. Biochem. 129, 191–200. [Google Scholar]
- Chen, T. , Nomura, K. , Wang, X. , Sohrabi, R. , Xu, J. , Yao, L. , Paasch, B.C. et al. (2020b) A plant genetic network for preventing dysbiosis in the phyllosphere. Nature, 580, 653–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, J.R. , Wang, Q. , Fish, J.A. , Chai, B. , McGarrell, D.M. , Sun, Y. , Brown, C.T. et al. (2014) Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordovez, V. , Dini‐Andreote, F. , Carrión, V.J. and Raaijmakers, J.M. (2019) Ecology and evolution of plant microbiomes. Annu. Rev. Microbiol. 73, 69–88. [DOI] [PubMed] [Google Scholar]
- Csardi, G. and Nepusz, T. (2006) The igraph software package for complex network research. Int. J. Complex Syst. 1695, 1–9. [Google Scholar]
- De Mendiburu, F . (2020) agricolae: Statistical procedures for agricultural research. R package version 1.3‐3. https://CRAN.R‐project.org/package=agricolae
- Delmotte, N. , Knief, C. , Chaffron, S. , Innerebner, G. , Roschitzki, B. , Schlapbach, R. , Von Mering, C. et al. (2009) Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proc. Natl. Acad. Sci. USA, 106, 16428–16433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, S. , Caddell, D.F. , Xu, G. , Dahlen, L. , Washington, L. , Yang, J. and Coleman‐Derr, D. (2021) Genome wide association study reveals plant loci controlling heritability of the rhizosphere microbiome. ISME J. 15, 3181–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depristo, M.A. , Banks, E. , Poplin, R. , Garimella, K.V. , Maguire, J.R. , Hartl, C. , Philippakis, A.A. et al. (2011) A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat. Genet. 43, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessaux, Y. , Grandclément, C. and Faure, D. (2016) Engineering the rhizosphere. Trends Plant Sci. 21, 266–278. [DOI] [PubMed] [Google Scholar]
- Doebley, J.F. , Gaut, B.S. and Smith, B.D. (2006) The molecular genetics of crop domestication. Cell, 127, 1309–1321. [DOI] [PubMed] [Google Scholar]
- Doerks, T. , Strauss, M. , Brendel, M. and Bork, P. (2000) GRAM, a novel domain in glucosyltransferases, myotubularins and other putative membrane‐associated proteins. Trends Biochem. Sci. 25, 483–485. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods, 10, 996–998. [DOI] [PubMed] [Google Scholar]
- Edwards, J. , Johnson, C. , Santos‐Medellín, C. , Lurie, E. , Podishetty, N.K. , Bhatnagar, S. , Eisen, J.A. et al. (2015) Structure, variation, and assembly of the root‐associated microbiomes of rice. Proc. Natl. Acad. Sci. USA, 112, E911–E920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfstrand, M. , Zhou, L. , Baison, J. , Olson, Å. , Lundén, K. , Karlsson, B. , Wu, H.X. et al. (2020) Genotypic variation in Norway spruce correlates to fungal communities in vegetative buds. Mol. Ecol. 29, 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison, E. , Kronenberg, Z.N. , Dawson, E.T. , Pedersen, B.S. and Prins, P. (2021) Vcflib and tools for processing the VCF variant call format. bioRxiv . 10.1101/2021.05.21.445151 [DOI] [PMC free article] [PubMed]
- Garrison, E. and Marth, G. (2012) Haplotype‐based variant detection from short‐read sequencing. arXiv . 10.48550/arXiv.1207.3907 [DOI]
- Goodrich, J.K. , Davenport, E.R. , Waters, J.L. , Clark, A.G. and Ley, R.E. (2016) Cross‐species comparisons of host genetic associations with the microbiome. Science, 352, 532–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbort, C.J. , Hashimoto, M. , Inoue, H. , Niu, Y. , Guan, R. , Rombolà, A.D. , Kopriva, S. et al. (2020) Root‐secreted coumarins and the microbiota interact to improve iron nutrition in Arabidopsis . Cell Host Microbe, 28, 825–837.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogenhout, S.A. , Oshima, K. , Ammar, E.D. , Kakizawa, S. , Kingdom, H.N. and Namba, S. (2008) Phytoplasmas: bacteria that manipulate plants and insects. Mol. Plant Pathol. 9, 403–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton, M.W. , Bodenhausen, N. , Beilsmith, K. , Meng, D. , Muegge, B.D. , Subramanian, S. , Vetter, M.M. et al. (2014) Genome‐wide association study of Arabidopsis thaliana leaf microbial community. Nat. Commun. 5, 5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitt, C.A. and Pogson, B.J. (2006) Carotenoid accumulation and function in seeds and non‐green tissues. Plant Cell Environ. 29, 435–445. [DOI] [PubMed] [Google Scholar]
- Jones, J.D.G. and Dangl, J.L. (2006) The plant immune system. Nature, 444, 323–329. [DOI] [PubMed] [Google Scholar]
- Jones, P. , Garcia, B.J. , Furches, A. , Tuskan, G.A. and Jacobson, D. (2019) Plant host‐associated mechanisms for microbial selection. Front. Plant Sci. 10, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiełbowicz‐Matuk, A. (2012) Involvement of plant C2H2‐type zinc finger transcription factors in stress responses. Plant Sci. 185–186, 78–85. [DOI] [PubMed] [Google Scholar]
- Kim, H. , Lee, K.K. , Jeon, J. , Harris, W.A. and Lee, Y.‐H. (2020) Domestication of Oryza species eco‐evolutionarily shapes bacterial and fungal communities in rice seed. Microbiome, 8, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurilshikov, A. , Medina‐Gomez, C. , Bacigalupe, R. , Radjabzadeh, D. , Wang, J. , Demirkan, A. , Le Roy, C.I. et al. (2021) Large‐scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 53, 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson, C.E. , Harcombe, W.R. , Hatzenpichler, R. , Lindemann, S.R. , Löffler, F.E. , O'Malley, M.A. , García Martín, H. et al. (2019) Common principles and best practices for engineering microbiomes. Nat. Rev. Microbiol. 17, 725–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 14, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 16, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Ma, B. , Chen, W. , Schlaeppi, K. , Erb, M. , Stirling, E. , Hu, L. et al. (2021) Rhizobium symbiotic capacity shapes root‐associated microbiomes in soybean. Front. Microbiol. 12, 709012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Tong, X. , Zou, Y. , Lin, X. , Zhao, H. , Tian, L. , Jie, Z. et al. (2022) Mendelian randomization analyses support causal relationships between blood metabolites and the gut microbiome. Nat. Genet. 54, 52–61. [DOI] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg, D.S. , Lebeis, S.L. , Paredes, S.H. , Yourstone, S. , Gehring, J. , Malfatti, S. , Tremblay, J. et al. (2012) Defining the core Arabidopsis thaliana root microbiome. Nature, 488, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, B. , Stirling, E. , Liu, Y. , Zhao, K. , Zhou, J. , Singh, B.K. , Tang, C. et al. (2021) Soil biogeochemical cycle couplings inferred from a function‐taxon network. Research, 2021, 7102769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinovsky, F.G. , Fangel, J.U. and Willats, W.G.T. (2014) The role of the cell wall in plant immunity. Front. Plant Sci. 5, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie, P.J. and Holmes, S. (2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyholm, L. , Koziol, A. , Marcos, S. , Botnen, A.B. , Aizpurua, O. , Gopalakrishnan, S. , Limborg, M.T. et al. (2020) Holo‐Omics: integrated host‐microbiota multi‐omics for basic and applied biological research. iScience 23, 101414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F.G. , Friendly, M. , Kindt, R. , Legendre, P. , Mcglinn, D. , Minchin, P.R. , et al. 2020. vegan: Community ecology package. R package version 2.5‐7. https://CRAN.R‐project.org/package=vegan
- Peiffer, J.A. , Spor, A. , Koren, O. , Jin, Z. , Tringe, S.G. , Dangl, J.L. , Buckler, E.S. et al. (2013) Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA, 110, 6548–6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Jaramillo, J.E. , Carrión, V.J. , Bosse, M. , Ferrão, L.F.V. , De Hollander, M. , Garcia, A.A.F. , Ramírez, C.A. et al. (2017) Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits. ISME J. 11, 2244–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieterse, C.M.J. and Dicke, M. (2007) Plant interactions with microbes and insects: from molecular mechanisms to ecology. Trends Plant Sci. 12, 564–569. [DOI] [PubMed] [Google Scholar]
- Razin, S. and Hayflick, L. (2010) Highlights of mycoplasma research–an historical perspective. Biologicals, 38, 183–190. [DOI] [PubMed] [Google Scholar]
- Reinhold‐Hurek, B. , Bünger, W. , Burbano, C.S. , Sabale, M. and Hurek, T. (2015) Roots shaping their microbiome: global hotspots for microbial activity. Annu. Rev. Phytopathol. 53, 403–424. [DOI] [PubMed] [Google Scholar]
- Sasse, J. , Martinoia, E. and Northen, T. (2018) Feed your friends: do plant exudates shape the root microbiome? Trends Plant Sci. 23, 25–41. [DOI] [PubMed] [Google Scholar]
- Sassmann, S. , Rodrigues, C. , Milne, S.W. , Nenninger, A. , Allwood, E. , Littlejohn, G.R. , Talbot, N.J. et al. (2018) An immune‐responsive cytoskeletal‐plasma membrane feedback loop in plants. Curr. Biol. 28, 2136–2144.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt‐Dannert, C. (2000) Engineering novel carotenoids in microorganisms. Curr. Opin. Biotechnol. 11, 255–261. [DOI] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N.S. , Wang, J.T. and Ramage, D. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, S. , Ma, Q. , Luo, J. , Xie, Y. , MLUR, H. , Pan, W. , Zheng, N. et al. (2021) The inhibition effect of tea polyphenols on soil nitrification is greater than denitrification in tea garden soil. Sci. Total Environ. 778, 146328. [DOI] [PubMed] [Google Scholar]
- Tarquinio, F. , Hyndes, G.A. , Laverock, B. , Koenders, A. and Säwström, C. (2019) The seagrass holobiont: Understanding seagrass‐bacteria interactions and their role in seagrass ecosystem functioning. FEMS Microbiol. Lett. 366, 1–15. [DOI] [PubMed] [Google Scholar]
- Turner, T.R. , James, E.K. and Poole, P.S. (2013) The plant microbiome. Genome Biol. 14, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burgh, A.M. and Joosten, M.H.A.J. (2019) Plant immunity: thinking outside and inside the box. Trends Plant Sci. 24, 587–601. [DOI] [PubMed] [Google Scholar]
- Vandenkoornhuyse, P. , Quaiser, A. , Duhamel, M. , Le Van, A. and Dufresne, A. (2015) The importance of the microbiome of the plant holobiont. New Phytol. 206, 1196–1206. [DOI] [PubMed] [Google Scholar]
- Vieira, S. , Sikorski, J. , Dietz, S. , Herz, K. , Schrumpf, M. , Bruelheide, H. , Scheel, D. et al. (2020) Drivers of the composition of active rhizosphere bacterial communities in temperate grasslands. ISME J. 14, 463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorholt, J.A. (2012) Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10, 828–840. [DOI] [PubMed] [Google Scholar]
- Wagner, M.R. , Lundberg, D.S. , del Rio, T.G. , Tringe, S.G. , Dangl, J.L. and Mitchell‐Olds, T. (2016) Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 7, 12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, J. , Laforest‐Lapointe, I. and Kembel, S.W. (2018) Variation in the leaf and root microbiome of sugar maple (Acer saccharum ) at an elevational range limit. PeerJ 6, e5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Feng, H. , Chang, Y. , Ma, C. , Wang, L. , Hao, X. , Li, A. et al. (2020) Population sequencing enhances understanding of tea plant evolution. Nat. Commun. 11, 4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M. and Hakonarson, H. (2010) ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 38, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Zhuang, H. , Shen, Y. , Wang, Y. and Wang, Z. (2021) The dataset of Camellia cultivars names in the world. Biodivers. Data J. 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, C. , Yang, H. , Wang, S. , Zhao, J. , Liu, C. , Gao, L. , Xia, E. et al. (2018) Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA, 115, E4151–E4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, E. , Tong, W. , Hou, Y. , An, Y. , Chen, L. , Wu, Q. , Liu, Y. et al. (2020a) The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation. Mol. Plant, 13, 1013–1026. [DOI] [PubMed] [Google Scholar]
- Xia, E. , Tong, W. , Wu, Q. , Wei, S. , Zhao, J. , Zhang, Z. , Wei, C. et al. (2020b) Tea plant genomics: achievements, challenges and perspectives. Hortic. Res. 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, H. , Chen, Z. , Feng, X. , Wang, M. , Luo, Y. , Wang, Y. and Xu, P. (2022) L‐theanine exuded from Camellia sinensis roots regulates element cycling in soil by shaping the rhizosphere microbiome assembly. Sci. Total Environ. 837, 155801. [DOI] [PubMed] [Google Scholar]
- Xie, H. , Feng, X. , Wang, M. , Wang, Y. , Kumar Awasthi, M. and Xu, P. (2020) Implications of endophytic microbiota in Camellia sinensis: a review on current understanding and future insights. Bioengineered, 11, 1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, C. , Zhu, Y.G. , Wang, J.T. , Singh, B. , Han, L.L. , Shen, J.P. , Li, P.P. et al. (2021) Host selection shapes crop microbiome assembly and network complexity. New Phytol. 229, 1091–1104. [DOI] [PubMed] [Google Scholar]
- Xu, L. , Dong, Z. , Chiniquy, D. , Pierroz, G. , Deng, S. , Gao, C. , Diamond, S. et al. (2021a) Genome‐resolved metagenomics reveals role of iron metabolism in drought‐induced rhizosphere microbiome dynamics. Nat. Commun. 12, 3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, P. , Fan, X. , Mao, Y. , Cheng, H. , Xu, A. , Lai, W. , Lv, T. et al. (2021b) Temporal metabolite responsiveness of microbiota in the tea plant phyllosphere promotes continuous suppression of fungal pathogens. J. Adv. Res. 39, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, F. , Fu, Y. , Sun, T. , Jiang, Z. , Miao, Z. , Shuai, M. , Gou, W. et al. (2020) The interplay between host genetics and the gut microbiome reveals common and distinct microbiome features for complex human diseases. Microbiome, 8, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, P. , Stirling, E. , Xie, H. , Li, W. , Lv, X. , Matsumoto, H. , Cheng, H. et al. (2022) Continental scale deciphering of microbiome networks in the tea plant untangles the hidden phyllosphere homeostasis. J. Adv. Res. 10.1016/j.jare.2022.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. , Zhang, Y. , Zhang, P. , Trivedi, P. , Riera, N. , Wang, Y. , Liu, X. et al. (2018) The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 9, 4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, L. , Zhang, H. , Tang, Z. , Xu, J. , Yin, D. , Zhang, Z. , Yuan, X. et al. (2021) rMVP: a memory‐efficient, visualization‐enhanced, and parallel‐accelerated tool for genome‐wide association study. Genom. Proteom. Bioinf. 19, 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgadzaj, R. , Garrido‐Oter, R. , Jensen, D.B. , Koprivova, A. , Schulze‐Lefert, P. and Radutoiu, S. (2016) Root nodule symbiosis in Lotus japonicus drives the establishment of distinctive rhizosphere, root, and nodule bacterial communities. Proc. Natl. Acad. Sci. USA, 113, E7996–E8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Dong, S.S. , Xu, J.Y. , He, W.M. and Yang, T.L. (2019a) PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788. [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Liu, Y.X. , Zhang, N. , Hu, B. , Jin, T. , Xu, H. , Qin, Y. et al. (2019b) NRT1.1B is associated with root microbiota composition and nitrogen use in field‐grown rice. Nat. Biotechnol. 37, 676–684. [DOI] [PubMed] [Google Scholar]
- Zhang, W. , Zhang, Y. , Qiu, H. , Guo, Y. , Wan, H. , Zhang, X. , Scossa, F. et al. (2020) Genome assembly of wild tea tree DASZ reveals pedigree and selection history of tea varieties. Nat. Commun. 11, 3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Zhang, J. , Wei, Y. , Hu, W. , Liu, G. , Zeng, H. and Shi, H. (2021) Microbiome‐wide association studies reveal correlations between the structure and metabolism of the rhizosphere microbiome and disease resistance in cassava. Plant Biotechnol. J. 19, 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, K. , Tung, C.‐W. , Eizenga, G.C. , Wright, M.H. , Ali, M.L. , Price, A.H. , Norton, G.J. et al. (2011) Genome‐wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa . Nat. Commun. 2, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. and Stephens, M. (2012) Genome‐wide efficient mixed‐model analysis for association studies. Nat. Genet. 44, 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilber‐Rosenberg, I. and Rosenberg, E. (2021) Microbial‐driven genetic variation in holobionts. FEMS Microbiol. Rev. 45, 1–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Comparisons of microbial composition on phylum level among the compartments of bulk soil (Bulk), rhizosphere (Rhizo), root endosphere (Root) and phyllosphere.
Figure S2 Counts of enriched microbial genera belonging to different phyla.
Figure S3 Cross‐validation (CV) of ancestry analysis for 100 tea (Camellia sinensis) cultivars.
Figure S4 Local Manhattan plots of the significant genetic loci associated with root endosphere Otu956 (a), rhizosphere Otu4877 (b) and phyllosphere Otu2029 (c).
Figure S5 Variance explained by the top five principal coordinates (PCOs) of phyllosphere, rhizosphere and root endosphere (root) microbiomes.
Figure S6 Associations of microbial principal coordinates (PCOs) and tea (Camellia sinensis) genetic variants through mGWAS.
Figure S7 Local Manhattan plot of the significant genetic variant Chr4_28730171 associated with phyllosphere microbiomes.
Table S1 Tea (Camellia sinensis) cultivars used in this study.
Table S2 Nonsynonymous exonic variants in the genome‐wide associations of the microbial operational taxonomic units (OTUs).
Table S3 Gene ontology (GO) associated with phyllosphere, root endosphere and rhizosphere compartments.
Table S4 Gene ontology (GO) enrichment analysis of the candidate genes associated with root endosphere operational taxonomic units (OTUs).
Table S5 Gene ontology (GO) enrichment analysis of the candidate genes associated with rhizosphere–endosphere operational taxonomic units (OTUs).
Table S6 Significant genetic variants in the genome‐wide associations of the principal coordinates (PCOs) of phyllosphere microbiomes.
Data Availability Statement
Sequencing data of 16S rRNA are available from the Genome Sequence Archive (GSA) under the BioProject PRJCA007800. Sequencing reads of tea plant cultivars reported in this study are available from the National Center for Biotechnology Information (NCBI) under the BioProject PRJNA798546. The Variant Call Format files of the sequencing of tea cultivars can be accessed from the Genome Variation Map (GVM) under the BioProject PRJCA010001. Codes for the full analyses included in this study are available on GitHub repository (https://github.com/hahafengxiang/tea_microbiome).