

Summary
Ice plant (Mesembryanthemum crystallinum), a member of the Aizoaceae family, is a typical halophyte crop and a model plant for studying the mechanism of transition from C3 photosynthesis to crassulacean acid metabolism (CAM). Here, we report a high‐quality chromosome‐level ice plant genome sequence. This 98.05% genome sequence is anchored to nine chromosomes, with a total length of 377.97 Mb and an N50 scaffold of 40.45 Mb. Almost half of the genome (48.04%) is composed of repetitive sequences, and 24 234 genes have been annotated. Subsequent to the ancient whole‐genome triplication (WGT) that occurred in eudicots, there has been no recent whole‐genome duplication (WGD) or WGT in ice plants. However, we detected a novel WGT event that occurred in the same order in Simmondsia chinensis, which was previously overlooked. Our findings revealed that ice plants have undergone chromosome rearrangements and gene removal during evolution. Combined with transcriptome and comparative genomic data and expression verification, we identified several key genes involved in the CAM pathway and constructed a comprehensive network. As the first genome of the Aizoaceae family to be released, this report will provide a rich data resource for comparative and functional genomic studies of Aizoaceae, especially for studies on salt tolerance and C3‐to‐CAM transitions to improve crop yield and resistance.
Keywords: ice plant; genome assembly, functional annotation; genome evolution; crassulacean acid metabolism; expression pattern
Introduction
Mesembryanthemum crystallinum (ice plant, 2n = 2× = 18) belongs to the Aizoaceae family, which contains approximately 160 genera and 2500 species. Ice plants are native to southern and eastern Africa and are now grown worldwide. Ice plants are vegetables with high nutritional value and health functions. They are rich in pinitol, which lowers blood sugar and has potential antidiabetic effects (Drira et al., 2016; Zhang et al., 2019a). They also contain myo‐inositol, which prevents fatty liver disease (Drira et al., 2016). Ice plants are inherently more resistant to diseases, insect pests and toxicity compared with most other vegetables. Therefore, these plants are capable of healthy growth without the need of pesticides but only a small amount of fertilizer (Amari et al., 2020; Śliwa‐Cebula et al., 2020). Ice plants can be eaten fresh as a high‐quality green vegetable.
Soil salinization is a global ecological problem that seriously affects the growth and production of most crops (Palansooriya et al., 2019). Clarifying the salt tolerance mechanisms of halophytes is important for preventing soil salinization and increasing crop yields (Mishra and Tanna, 2017). Ice plants are halophytes that convert from C3 photosynthesis to crassulacean acid metabolism (CAM) under high‐salinity stress (Adams et al., 1998). CAM is a special form of photosynthetic carbon assimilation that occurs in an estimated 7% of vascular plants (Crayn et al., 2004; Cushman et al., 2008; Silvera et al., 2005). CAM is characterized by the absorption and assimilation of atmospheric carbon dioxide (CO2) catalysed by phosphoenolpyruvate carboxylase (PEPC) at night, leading to the accumulation of C4 acids (Gilman and Edwards, 2020). By absorbing large amounts of CO2 at night, when the evapotranspiration rate is low, the water use efficiency of CAM plants is three to six times higher than that of C4 and C3 plants (Cushman et al., 2008). The genes related to salt tolerance identified in ice plants are strong candidates for genetic engineering of C3 crops to improve salt tolerance (Tsukagoshi et al., 2015).
Several studies have revealed omics level changes in ice plants in response to salt stress. The first microarray experiment used 5‐week‐old and 14‐day salt‐stressed plants (Cushman et al., 2008). Using second‐generation sequencing, 53 516 cDNAs were detected in the roots of ice plants (Tsukagoshi et al., 2015). A reference transcriptome was constructed that contained 37 341 transcripts from control and salt‐treated ice plant epidermal bladder cells (EBCs) (Oh et al., 2015). One hundred thirty‐five conserved microRNAs (miRNAs) have been identified in the roots of 3‐day‐old ice plant seedlings (Chih‐Pin et al., 2016). The proteomes and metabolomes of ice plant EBCs have also been studied (Barkla et al., 2016; Barkla and Vera‐Estrella, 2015; Li et al., 2021). Recently, the critical transition time for ice plants from C3 to CAM has been determined by measuring several key attributes, including gas exchange, stomatal aperture, titratable acidity, CAM enzyme activity and CAM gene expression (Kong et al., 2020).
Related transcriptome studies have analysed the changes in gene expression of ice plants after salt stress treatment (Kong et al., 2020; Oh et al., 2015; Tsukagoshi et al., 2015). However, the genome of ice plants has not yet been resolved, which has hindered studies of molecular regulation mechanisms related to salt tolerance in ice plants. In the present study, PacBio HiFi, Illumina sequencing and Hi‐C technologies were used to obtain high‐quality ice plant genome sequences. Several key genes for salt tolerance in ice plants were identified by combined genomic and transcriptomic analyses. These data provide an important reference for research on salt‐tolerant genomics and molecular biology of ice plants.
Results
Ice plant genome sequencing, assembly, and assessment
De novo ice plant genome sequencing was performed using PacBio HiFi, Illumina and Hi‐C technologies (Figure 1a, Table 1). First, the ice plant genome was estimated by the K‐mer method using 90.43 Gb of data from Illumina sequencing (Tables 1 and S1). The estimated size of the ice plant genome was 394.89 Mb, and the heterozygosity rate was 0.10% (Figure S1, Table S1). The PacBio HiFi sequencer was adopted to generate 18.77 Gb of data with an average of 47.53× coverage depth (Tables 1 and S3). In total, 109.20 Gb (276.53×) of ice plant DNA sequences generated from the Illumina and PacBio platforms were used to perform the preliminary assembly. The results indicated that the cumulative length of the contig was 377.96 Mb, and that of contig N50 was 6.18 Mb (Tables S4–S6).
Figure 1.
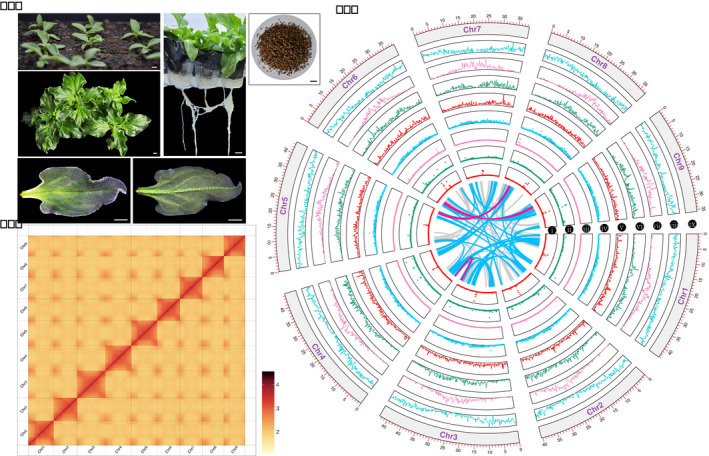
Morphology, Hi‐C contact map, and genome features of the ice plant genome. (a) Morphology of seedlings, leaves and seeds of ice plant. Scale bars, 2 cm. (b) All‐by‐all interactions of all nine chromosomes obtained by Hi‐C in the ice plant genome. (c) The distribution of various repetitive sequences and gene density on each chromosome in ice plants. The window size was set to 200 Kb with non‐overlapping regions. i to iv, distribution of TRF, SSRs, SINE and LINE, respectively; v to viii, density of DNA repeats, Copia transposons, Gypsy transposons and genes, respectively. ix, nine chromosomes of the ice plant. The inner curve lines showed the collinear gene blocks in the ice plant. The grey, blue and red colours indicate 5–10, 10–30 and >30 gene pairs in collinear blocks, respectively.
Table 1.
Summary of ice plant genome sequencing data
| libraries | Insert size (bp) | Total data (G) | Read length (bp) | Sequence coverage (X) |
|---|---|---|---|---|
| Illumina reads | 350 | 90.43 | 150 | 229.00 |
| PacBio reads | – | 18.77 | – | 47.53 |
| Sub‐total | 109.20 | 276.53 | ||
| Hi‐C | – | 61.80 | – | 156.50 |
| Total | – | 171.00 | – | 433.03 |
Hi‐C technology was used to improve the ice plant genome assembly. High‐quality sequences (61.80 Gb, 156.50×) were obtained by Illumina sequencing (Table 1). A Hi‐C contact map was used to divide the distinct regions of each chromosome (Figure 1b). Finally, the assembled genome size was approximately 377.97 Mb, with contig N50 and scaffold N50 reaching 6.18 Mb and 40.45 Mb, respectively (Table 2). In total, 370.61 Mb sequences were anchored to nine chromosomes in the ice plant, accounting for 98.05% of the assembled genome (Figure 1c, Table S7).
Table 2.
Statistics of ice plant genome assembly quality
| Type | Length | Number | ||
|---|---|---|---|---|
| Contig (bp) | Scaffold (bp) | Contig | Scaffold | |
| Total | 377 960 088 | 377 968 988 | 269 | 180 |
| Max | 24 134 815 | 47 886 399 | – | – |
| Number ≥ 2000 | – | – | 269 | 180 |
| N50 | 6 175 963 | 40 449 012 | 18 | 5 |
| N60 | 5 222 265 | 38 645 384 | 25 | 6 |
| N70 | 4 618 777 | 38 543 744 | 33 | 7 |
| N80 | 3 539 318 | 37 272 660 | 42 | 8 |
| N90 | 1 695 433 | 35 725 216 | 57 | 9 |
The reads mapping rate exceeded 99.11%, indicating the assembled ice plant genome was relatively complete (Table S8). The core eukaryotic gene mapping approach (CEGMA) and benchmarking universal single‐copy orthologs (BUSCO) methods were used to assess the quality of assembled genomes and annotations. The CEGMA results showed that 96.77% (241) of core eukaryotic genes were detected in the assembled genome (Table S9). The BUSCO results showed that 98.0% of 1641 genes were found in the ice plant genome (Table S10).
Genome annotation
Repetitive sequences accounted for 48.04% of the estimated ice plant genome (Figure 1c, Table S11). Most repetitive sequences were long‐terminal repeats (LTRs; 121.34 Mb), accounting for 32.11% of the genome (Figure S2, Table S11). Long interspersed nuclear elements (LINE) and DNA transposons only accounted for 6.40% and 7.42% of the ice plant genome, respectively. Most genes were located in the terminal regions of each chromosome, displaying a similar trend to that of DNA transposable elements. However, Copia and Gypsy retrotransposons were almost inversely distributed on each chromosome compared with the genes (Figure 1c).
Among the 24 234 annotated ice plant genes (Figures S3, S4, Tables S12, S13), InterPro, Swiss‐Prot, Kyoto encyclopedia of genes and genomes (KEGG) and non‐redundant protein databases showed functional evidence for 24 064 (99.30%) genes, with 15 512 genes annotated in all databases (Figure S5, Table S14). Concerning RNA, 4269 rRNAs, 2446 miRNAs, 1054 tRNAs and 889 snRNAs were found, in total accounting for 0.91% of the ice plant genome (Figure S6, Table S15).
Gene family expansion analysis and divergence time estimation
We detected gene families in ice plant, nine Caryophyllales species, two other eudicots (Arabidopsis thaliana and Vitis vinifera), and Oryza sativa (Figure 2a, Table S16). In total, 28 686 gene families were identified in the ice plant and the other 12 examined species (Figure 2b, Table S17). There were 14 330 gene families in the ice plant, which was lower than that in Atriplex hortensis (15950), Chenopodium quinoa (15886), Spinacia oleracea (15020), and Chenopodium pallidicaule (14729) (Figure 2b, Table S17). However, only 12 835 and 12 894 gene families were detected in A. thaliana and Beta vulgaris, respectively. Common and specific gene family analyses among the 13 species was performed using a Venn diagram (Figure 2b). A total of 252 single‐copy and 6108 common gene families were found among the 13 species (Figure 2b, Table S17). The 468 ice plant species‐specific gene families exceeded the numbers for C. pallidicaule (103), B. vulgaris (258) and Hylocereus undatus (447) but was less than that of the other nine species (Figure 2b).
Figure 2.
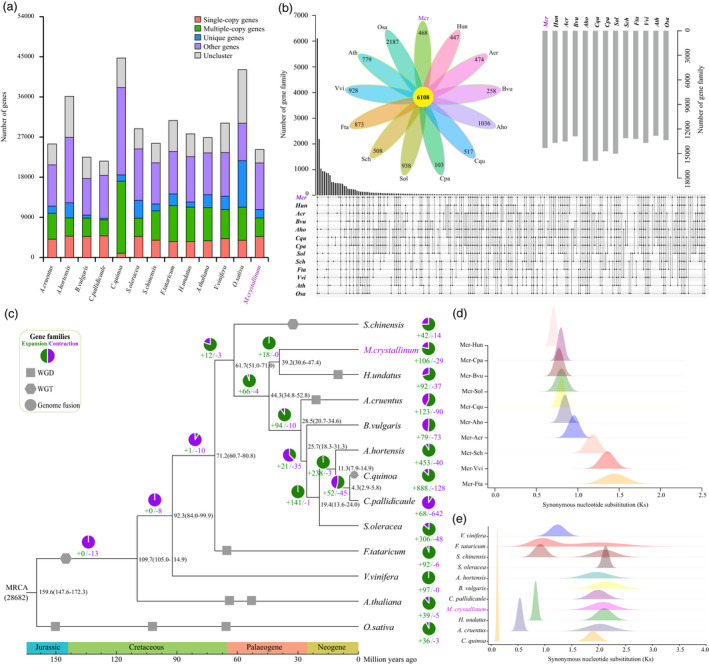
Gene family, phylogenetic and divergence time analyses. (a) The gene numbers of each category in the ice plant and 12 representative species. (b) Common and specific‐specific gene families in ice plant and the other 12 species. (c) Expansion/contraction of gene family and divergence time estimation. The green and purple pies depict the ratios of the numbers of expansion and contraction gene families, respectively. The numbers on the nodes represent the species divergence time, with the confidence range list in brackets. The grey squares, hexagons and circles represent whole‐genome duplication (WGD), whole‐genome triplication (WGT) and genome fusion, respectively. (d) The density of synonymous nucleotide substitutions per synonymous site (Ks) among collinear genes between ice plant (Mcr) and other related species. (e) The density plot of Ks among collinear genes within ice plant (Mcr) and each other related species.
Gene family contraction and expansion were explored in ice plant and the 12 other representative species (Figure 2c). In the ice plant, we detected 106 gene family expansions, which was more than that of the closely related species, H. undatus (92) (Figure 2c). The largest number of gene family expansions was found in C. quinoa (888), followed by A. hortensis (453), and S. oleracea (306). Only 29 gene family contractions were found in ice plants, which was more than in Fagopyrum tataricum (6) and S. chinensis (14), but fewer than that seen in other Caryophyllales species (Figure 2c). We performed a phylogenetic analysis and divergence time estimation using single‐copy gene families among the 13 species (Figure 2c). The ice plant had the closest relationship with H. undatus among all examined species. It diverged from H. undatus 30.6–47.4 million years ago (Mya) (Figure 2c). The divergence time between Caryophyllales and the other species was 84.0–99.9 Mya (Figure 2c).
Evolution and polyploidization of the ice plant genome
We explored the genomic evolution of ice plants using the rate of synonymous nucleotide substitution (Ks) within syntenic blocks among ice plants, grapes, and nine other Caryophyllales species (Figure 2d‐e). In the Ks density plot, only one peak was detected in the ice plant genome. This indicates that only one polyploidization event occurred in the ice plant, which was an ancient whole‐genome triplication (WGT) event shared with grapes and most eudicots (Jaillon et al., 2007). Therefore, there were no recent whole‐genome duplication (WGD) or WGT events in ice plants. Surprisingly, two peaks were detected in the Ks density plot of the S. chinensis genome, indicating that two polyploidization events had occurred in this species (Figure 2e). However, only one polyploidization event was found in a previous report, indicating that one recent WGT event in S. chinensis was overlooked. We also verified that S. chinensis experienced two rounds of WGT events by combining dot plot and syntenic analysis data (Figure 3a‐d). Based on the ancient WGT event times that occurred in most eudicots, the recent WGT event in S. chinensis occurred between 50.60–57.42 Mya (Ks = 0.91). This was later than the time of the recent WGD event in F. tataricum (53.03–59.95 Mya; Ks = 0.95) but earlier than the times of the recent WGD events in H. undatus (46.33–52.38 Mya; Ks = 0.83) and A. cruentus (29.59–33.44 Mya; Ks = 0.53) (Figure 2e, Table S18).
Figure 3.
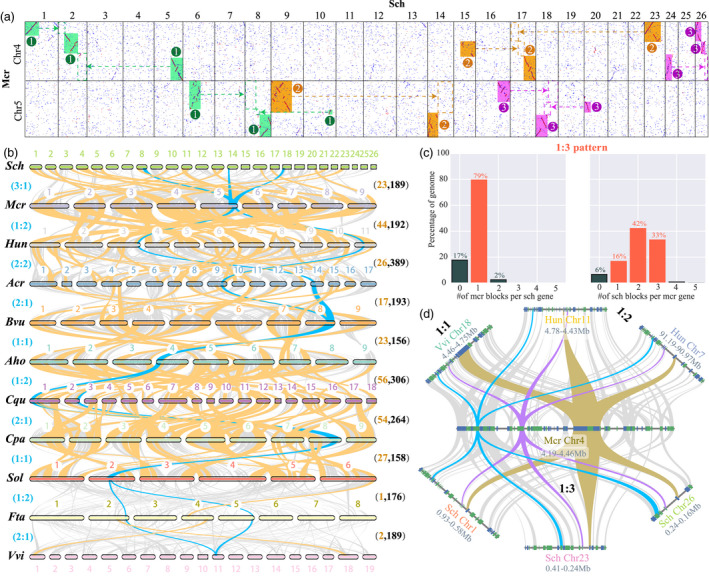
Homologous dot plot and syntenic analyses between ice plant and other representative species. (a) The homologous dot plot between selected ice plant chromosomes (Chr4 and Chr5) and S. chinensis (Sch) chromosomes. Three syntenic regions were indicated by rectangles numbered by 1, 2 and 3 in circles with different colours. An additional novel WGT in S. chinensis was identified via syntenic comparison with ice plant, which was previously overlooked. (b) Syntenic diagram between ice plant and other related species. Syntenic blocks were marked using grey lines. Large syntenic blocks (>200 gene pairs) are highlighted in orange. Blue lines indicate an exemplar syntenic relationship between ice plant and other related species. (c) The syntenic depth analysis between ice plant and S. chinensis also showed the 1:3 ratios between ice plant and S. chinensis. (d) Microsyntenic comparison of genes between ice plant and other related species. A representative syntenic relationship revealed that one ice plant region matched one region in grape, two regions in H. undatus (Hun) and three regions in S. chinensis (Sch). Rectangles showed annotated genes with orientation on the same strand (blue) and the reverse strand (green). The grey lines connected collinear gene pairs, with three regions highlighted in blue, purple and orange colours as example.
Syntenic and gene retention analysis of the ice plant genome
We identified 100 intra‐genomic collinear blocks in an ice plant, containing 2079 collinear genes (Table S19). We then mapped the ice plant gene sequences onto nine other Caryophyllales genomes to infer their inter‐genomic collinearity (Figures 3b, S7–S10). Among the Caryophyllales species, the most collinear blocks were between the ice plant and C. quinoa (305). This might reflect the genomic fusion that occurred in C. quinoa according to a previous report (Zou et al., 2017) (Figure S7). We also found that the number of collinear blocks between ice plants and species that underwent recent WGD or WGT events was greater than that of the other species (Figures 3b, S7–S10). For example, there were more collinear blocks between the ice plant and the four species that underwent recent WGD or WGT events, including Amaranthus cruentus (223 collinear blocks), F. tataricum (217), H. undatus (192) and S. chinensis (189) (Figures 3b, S8, and S9). However, there were fewer collinear blocks between ice plant and the other four species that had not undergone recent WGD or WGT events, including A. hortensis (181), S. oleracea (166), B. vulgaris (142) and C. pallidicaule (131) (Figures S7, S9, S10). Furthermore, the largest number of large collinear blocks (gene pairs >200) were between the ice plant and H. undatus (44), followed by C. quinoa (40) and S. oleracea (32). Syntenic analysis indicated that most species underwent chromosome rearrangement after divergence (Figures 3b, S7–S10). To a certain extent, the syntenic results reflect the genetic relationship between ice plants and other Caryophyllales species.
The ratio of the collinear regions between the ice plant and grape was 1:1 because no recent genome duplication has occurred in the ice plant. However, the ratio between ice plants and the three species (H. undatus, A. cruentus, and F. tataricum) was 1:2 because of the recent WGD event detected in these three species (Figures 3b, 4a, Table S20). The ratio of ice plants to S. chinensis was 1:3 because of the recent WGT event detected in this study. For example, the end of chromosome 5 in the ice plant was collinear with S. chinensis chromosomes 8, 14 and 18, and it was also collinear with H. undatus chromosomes 3 and 11 (Figure 3a, b). Microsynteny analysis was also consistent with the global syntenic analysis, showing a similar ratio between ice plants and other species. For example, the 4.19–4.46 Mb of chromosome 4 in the ice plant was perfectly collinear with grape, H. undatus and S. chinensis (Figure 3d).
Figure 4.
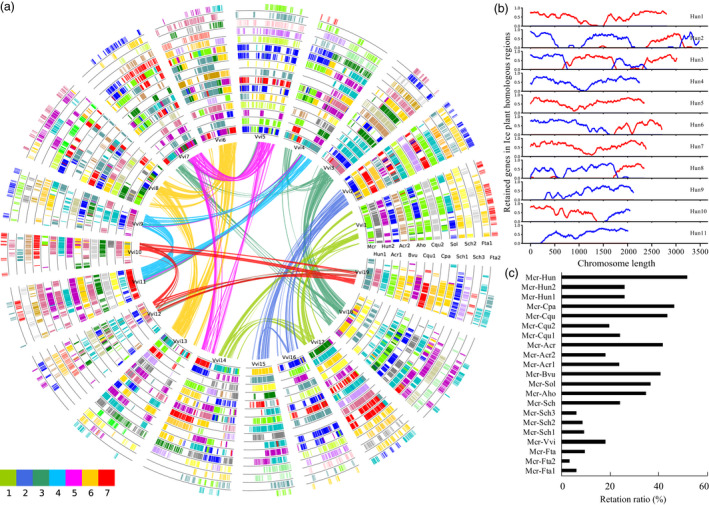
Global alignments and gene retention analyses of the ice plant genome. (a) Global alignment of homologous regions in ice plant and 10 other plant genomes with the grape as a reference. Each genome of H. undatus, A. cruentus, F. tataricum and S. chinensis were further divided into two or three sub‐genomes due to additional WGD or WGT events that occurred in their genomes after divergence from the grape. Collinear genes between each sub‐genome of species and grape were shown in each circle. The curved lines of the inner circle were composed of 19 grape chromosomes corresponding to the seven chromosomes of the ancient core‐eudicot hexaploid ancestor (γ event). (b) The retention of duplicated genes residing in the ice plant genome along with each chromosome of the H. undatus. (c) The summary of gene retention in the ice plant genome compared with other species.
We conducted gene retention analysis of the ice plant genome in homologous regions by comparing it with other species. Different regions on the chromosome showed divergent retention levels (Figures 4a, b, S11–S19, Table S21). Grossly, the highest retention rate of collinear ice plant genes was 51.85% using H. undatus as a reference, followed by C. pallidicaule (46.60%), and C. quinoa (43.66%) (Figure 4c, Table S21). However, the average retention rate of different chromosomes was only 9.66% when F. tataricum was the reference. We counted the number of syntenic gene pairs between the ice plant and the other nine Caryophyllales genomes. The most syntenic gene pairs were found between the ice plant and A. hortensis (5989 gene pairs), followed by C. quinoa (5735), S. chinensis (5656), B. vulgaris (5527), S. oleracea (5350), H. undatus (5177), C. pallidicaule (4975) and A. cruentus (4783). However, only 2426 syntenic gene pairs were identified between the ice plant and F. tataricum. The collective results revealed large‐scale genome fractionation and instability of the ice plant genome after its split from these plants.
Exploring key genes in the CAM pathway
The CAM pathway is highly plastic under salt stress in ice plants, which makes it a good representative model for studying the transition mechanism from C3 to CAM (Cushman et al., 2008; Kong et al., 2020). Here, we attempted to identify the genes involved in regulating the C3 to CAM pathway by combining genomic and transcriptomic analyses (Table S22). Twenty genes encoding six main enzymes involved in the CAM pathway were identified in the ice plant (Figure 5a). Most nodes in the pathway had more gene copies in ice plants, including seven malate dehydrogenase (MDH), five malic enzyme (ME), three phosphoenolpyruvate carboxylase (PEPC) and three carbonic anhydrase (CA) genes.
Figure 5.
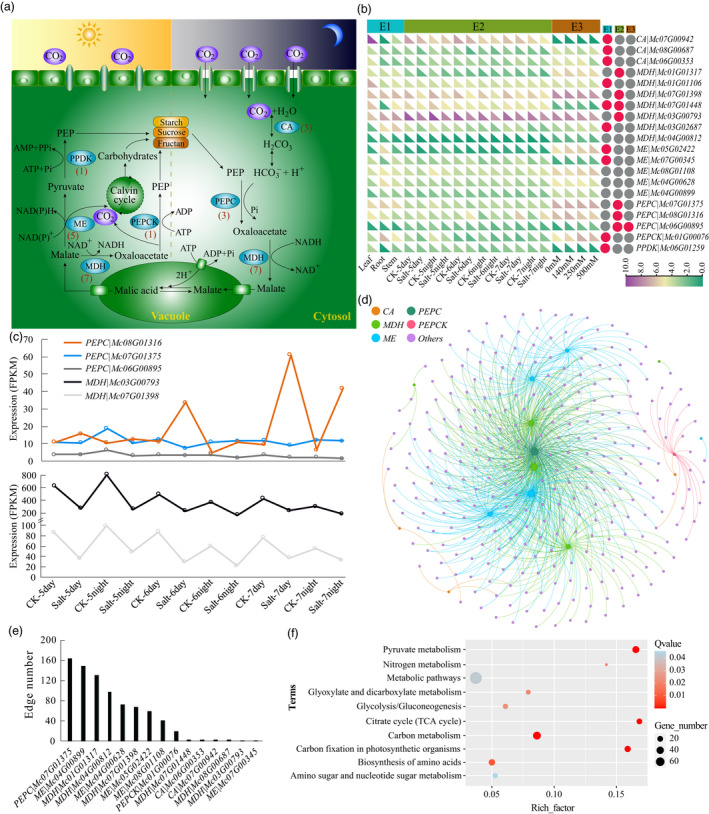
Overview of crassulacean acid metabolism (CAM) pathway in ice plant. (a) The CAM pathway map in the ice plant. Blue colours showed the key enzymes involved in the CAM pathway. The numbers in parenthesis are the number of genes encoding the corresponding enzymes in the ice plant. CA, carbonic anhydrase; PEPC, phosphoenolpyruvate carboxylase; PEPCK, PEPC kinase; MDH, malate dehydrogenase; ME, malic enzyme; PPDK, pyruvate phosphate kinase. (b) Expression profile heatmap of CAM‐related genes in ice plant. The expression values (FPKM) were transformed by log2. The RNA‐seq data set of the experiment (E1) was obtained in this study, and the data sets of E2 and E3 were obtained according to the previous reports. The red circle indicates the DEGs in corresponding experiments (fold‐change >2, q‐value <0.01). (c) Expression level of PEPC and MDH genes under salt treatment from E2 data set in the ice plant. (d) The interaction network between CAM‐related genes and DEGs identified from E2 and E3 data sets in ice plant. (e) The edge number of each CAM‐related gene in the network. (f) Functional enrichment analysis of DEGs involved in the network using KEGG (q‐value <0.05).
To further explore the expression pattern of 20 CAM‐related genes in the ice plant, we used three RNA‐seq data sets from different tissues (E1) and applied salt treatment for different times (E2) or at different concentrations (E3) (Figure 5b, Tables S23–25). The E1 data set was generated in this study, and the E2 and E3 data sets were collected from previous studies (Kong et al., 2020; Tsukagoshi et al., 2015). Among the 20 differentially expressed genes (DEGs), ten, seven, and one were detected in E1, E2 and E3 experiments, respectively. Under salt treatment (E2 and E3), seven DEGs were identified, including four MDH and three PEPC genes. Furthermore, the expression level of the PEPC gene (Mc08G01316) was higher in the salt‐treated plant tissue than in the control, regardless of day or night during days 5 to 7 (Figure 5c). The expression trend of the Mc08G01316 gene was similar to that of the PEPC gene (Contig20312) identified in a previous study (Kong et al., 2020). The expression patterns of the other two PEPC genes (Mc07G01375, Mc06G00895) were the opposite of Mc08G01316. However, these two genes were not detected in a previous study (Kong et al., 2020). Similarly, the expression levels of the two MDH genes (Mc03G00793, Mc07G01398) were lower with salt treatment than in the control, regardless of timing (i.e., day or night).
Interaction network construction for key genes in the ice plant CAM pathway
Based on the identified CAM‐related genes and their expression patterns, we constructed an interaction network for these genes and the genes they regulate in ice plants (Figure 5d). A total of 805 gene pairs formed the network according to the Pearson correlation coefficients (¦PCC¦ >0.95) (Table S26). Among these connections, only 10 represent regulatory interactions between CAM‐related genes. The other 795 connections represent interactions between CAM‐related genes and other DEGs in the ice plant genome (Table S26). This network contained nine CAM‐related genes (five DEGs) and 285 other DEGs (Figure 5d, Table S27). Among these genes, the PEPC gene Mc07G01375 had the most connections (163) with other genes, followed by the ME gene Mc04G00899 (149 connections) and the MDH gene Mc01G01317 (131 connections) (Figure 5d, e, Table S27). These results suggest that genes with more connections might play a core role in the CAM pathway in ice plants.
To explore the functions of the genes involved in the network constructed in the ice plant, we performed enrichment analysis. We identified 10 significantly enriched terms (q‐value <0.05). The most significantly enriched term was pyruvate metabolism (q‐value = 2.08 × 10−11), followed by carbon metabolism, carbon fixation in photosynthetic organisms and citrate cycle (TCA cycle) (Figure 5f, Table S28). Most enriched terms were related to the CAM pathway in plants.
Morphological detection and verification of CAM pathway genes in the ice plant
Ice plants reportedly survive high‐salinity conditions because their EBCs sequester up to 1 M sodium to adjust to osmotic stress (Cushman et al., 1989). The seedlings showed obvious wilt symptoms after 1 day of salt treatment. This phenotype became more pronounced in the cotyledons with increasing treatment time, while the mature leaves examined on days 3 and 6 displayed minor changes, and the whole plants seemed to be more energetic compared to day 1 (Figure 6a). The fresh weight to dry weight ratio after 1, 3 and 6 days of salt stress showed a downward trend compared with that on day 0 (Figure 6b). CAM plants assimilate carbon dioxide at night, accumulate malic acid and transfer it to vacuoles for storage, resulting in an increase in the hydrogen ion concentration in vacuoles. Therefore, the change in titratable acidity in leaves at the start (8:00 pm) and end (8:00 am) was determined as a vital indicator of the presence or absence of CAM activity. On day 0, titratable acidity was very low and did not fluctuate at the end of the day and night (Figure 6c). On day 1 of salt treatment, with obvious phenotypic differences, the nocturnal acid content at dawn was significantly higher than that on day 0. Consistent with this result, on day 0, the stomatal aperture was larger in the day and smaller at night, whereas the plant showed inversible stomatal movement on days 1, 3 and 6 after stress (Figure 6d). Therefore, we inferred that the plant quickly shifted from C3 to CAM photosynthesis after 1 day of salt treatment in our study.
Figure 6.
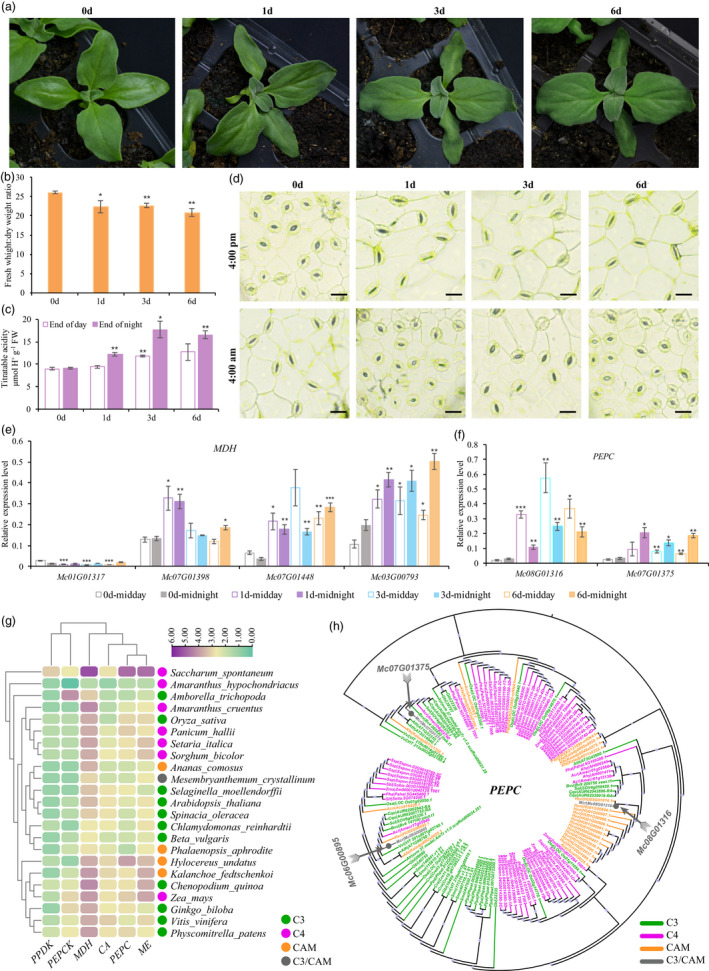
Morphological detection and CAM pathway genes verification in ice plant. (a) Representative images of plants during the course of 0, 1, 3 and 6 days of salt treatments. (b) Fresh weight to dry weight ratio. (c) Levels of titratable acidity of leaves measured at the start (8:00 am) and end (8:00 pm) of the photoperiod. Each value is mean ± standard deviation. The asterisks above the bars indicate Student's t test significance in comparison with day 0 (*P < 0.05; **P < 0.01). (d) The representative images of stomata under different salt treatment time courses. Expression profiles of representative MDH and PEPC genes determined at midday and midnight in leaves of M. crystallinum under different stages of salt stress. (e) The relative expression level of four MDH genes under different stages of salt stress by qRT‐PCR. (f) The relative expression level of two PEPC genes under different stages of salt stress by qRT‐PCR. (g) The heatmap of the CAM pathway genes (PPDK, PEPCK, MDH, CA, PEPC and ME) number in ice plant and other 22 related species, including C3, C4 and CAM plants. The gene number in each species was transformed by log2. (h) Maximum‐likelihood trees of PEPC genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
In ice plants, genes with CAM‐related functions exhibit stress‐induced/putative circadian expression patterns following salt stress (Cushman et al., 2008). The aforementioned analysis showed that the expression of four MDH and three PEPC genes was largely altered during the transition of ice plants from C3 photosynthesis to CAM (Figure 5b). Among these, the expression levels of two MDH genes decreased during the transition phase compared with the control, and two PEPCs showed opposite expression trends: one was time dependently induced, and the other was time dependently repressed with salinity stress (Figure 5c). Several PEPC and MDH genes were selected for qRT‐PCR to validate the expression patterns of the individual genes. Among the four genes encoding MDH, the expression level of Mc01G01317 was relatively low and repressed by salinity stress at midday in all salt treatments (Figure 6e). Mc07G01398 was rapidly induced more than twice, at midday and midnight on day 1. However, compared with day 0, the change in the later stages was not significant. The transcript abundance of the other two MDH genes, Mc07G01448 and Mc03G00793, increased on day 1 both at midday and midnight; the genes maintained a higher expression state on days 3 and 6 after salt treatment (Figure 6e), which correlated with the nocturnal accumulation of acidity (Figure 6c). Remarkably, Mc03G00793 showed a putative circadian expression pattern that peaked at midnight and was more pronounced on day 6 of salinity, suggesting that this gene may be specialized in CAM function. Mc08G01316 and Mc07G01375 encode two PEPC genes (the key enzymes responsible for assimilating CO2 during the night in CAM plants). In our study, two PEPC genes were immediately induced by salt treatment on day 1 and relatively abundant transcripts were maintained on subsequent days (Figure 6f). In addition, they both exhibited an apparent circadian fluctuation in expression level, but in opposite directions. Mc08G01316 had peak expression in the middle of the day, while the expression level of Mc07G01375 peaked at subjective midnight (Figure 6f).
In addition, we detected CAM‐related genes in 22 other species (11 C3, 7 C4 and 4 CAM species) by comparative analysis (Figure 6g). Interestingly, we detected 111 genes in the C4 plant Saccharum spontaneum, which was far more than in any other species (Figure 6g, Table S29). Only eight genes were found in another C4 plant, Amaranthus hypochondriacus. Among the C3 plants examined, the number of CAM genes ranged from 18 to 49, C4 plant genes ranged from 8 to 111 and CAM plant genes ranged from 21 to 46 (Figures S20–S25, Table S29). This phenomenon indicates that the number of genes involved in the CAM pathway varies widely among different species, despite belonging to the same metabolic type. Interestingly, the number of phosphoenolpyruvate carboxylase kinase (PEPCK) genes in the basal angiosperm plant Amborella trichopoda was much higher than that of the other examined species. This result provides abundant data resources and ideas for the evolution of the PEPCK gene in plants. Cluster analysis performed according to the number of CAM‐related genes revealed that ice plants clustered with C3 and CAM plants and were most similar to the CAM plant Ananas comosus (pineapple) (Figure 6g). In addition, the phylogenetic trees of genes encoding the six main enzymes indicated that most genes in the ice plant had a close relationship with CAM or C3 plants. In particular, three PEPC genes in ice plants were distributed in different branches in the phylogenetic tree, indicating their probable function in the C3 and CAM pathways (Figure 6h). To some extent, these results support the position that ice plants are C3 and CAM‐facultative plants.
Discussion
The ice plant is a member of the Aizoaceae family, which belongs to the order Caryophyllales. The genomes of several species in this order have been sequenced and released, including 14 species from Amaranthaceae, four species from Droseraceae, four species from Polygonaceae, two species from Cactaceae and one species each from Simmondsiaceae, Caryophyllaceae and Phytolaccaceae. To date, no species in Aizoaceae has been sequenced and reported. The ice plant genome data in this study will provide a rich resource for conducting comparative and functional genomic studies in Aizoaceae and other families of the order Caryophyllales.
The CAM pathway is highly plastic in ice plants (Cushman, 2001; Cushman et al., 2008; Cushman and Bohnert, 1999). Under non‐stressed conditions, the ice plant shows C3 photosynthesis and can complete its entire life cycle in the C3 mode without exhibiting net CO2 absorption at night (Winter and Holtum, 2007). However, it exhibits all the physiological characteristics of a CAM plant when grown under various stress conditions such as high salinity, water deficit or high light (Matsuoka et al., 2018; Wakamatsu et al., 2021; Winter and Holtum, 2005). The inducibility of CAM and the biochemical properties of C3 and CAM in the same cells of ice plants make it an excellent model for studying the transition mechanism from C3 to CAM (Bohnert and Cushman, 2000; Kong et al., 2020; Kore‐eda et al., 2013; Winter and Holtum, 2014).
The annual succulent ice plant is a typical model halophyte used to explore the basic biochemical, physiological and molecular mechanisms of salt and water stress responses in plants (Barkla et al., 2009; Bohnert and Cushman, 2000). Ice plants can adjust their metabolic pathway from C3 to CAM in response to various stresses (Adams et al., 1998; Oh et al., 2015; Winter and Holtum, 2007).
In vascular plants, PEPC belongs to a multigene family, with each member encoding a specific functional enzyme (O'Leary et al., 2011). In CAM species, at least one form of the gene specializes in CAM function, which catalyses nocturnal CO2 assimilation into C4‐dicarboxylic acids (Gehrig et al., 1998; Lepiniec et al., 1993). The results of phylogenetic analyses of PEPC genes imply a single origin before the divergence of bacterial and plant lineages (Izui et al., 2004; Westhoff and Gowik, 2004). CAM‐specific PEPC genes were thought to have evolved first because of a lack of water supply from the non‐photosynthetic role of PEPC by local gene duplications, which could then allow the subsequent functional specialization of genes divided into different clades (Gehrig et al., 2005; Taybi et al., 2004). In the present study, three PEPC genes were assigned to different clades, indicating functional divergence.
MDH is a ubiquitous enzyme in plants with different genes, with roles in a series of metabolic processes according to its subcellular location (Gietl, 1992). In particular, cytosolic MDH converts oxaloacetate to l‐malate in CAM plants (Holtum and Winter, 1982). In our study, the expression levels of four MDH genes were determined during the transition from C3 to CAM, indicating different expression patterns of the genes. Among these, Mc03G00793 presented a putative circadian fluctuation in mRNA abundance that peaked at subjective midnight, implying its specific function in CAM photosynthesis.
Conclusions
Here, we report a high‐quality and chromosomal‐level ice plant genome. This is the first released genome of an Aizoaceae family member. The total length of the genome is 377.97 Mb comprising 24 234 genes. Although no recent WGD or WGT events occurred in ice plants, we detected a novel WGT event that was overlooked in a previous report on S. chinensis. Several key genes involved in the CAM pathway were identified and a comprehensive network was constructed for CAM‐related genes in ice plants. The ice plant genome sequences, together with the comparative genomic analysis data, will provide rich resources for studies of gene functions and genome evolution in ice plants and other Aizoaceae plants.
Materials and methods
DNA sequencing and genome size estimation
Genomic DNA was extracted from ice plant leaves using a QIAGEN kit, according to the standard procedure. DNA purity was determined using a NanoDrop™ One spectrophotometer. DNA quantification was performed using a Qubit® 3.0 Fluorometer. Sequencing libraries were sequenced using the Illumina (USA) and PacBio (Pacific Biosciences, USA) platforms according to previous reports (Song et al., 2021b; Song et al., 2022). Three sequencing strategies were used. In the first, two paired‐end libraries were constructed with fragments of 350 bp and sequenced on the Illumina platform. In the second strategy, third‐generation sequencing libraries were constructed and sequenced using the PacBio HiFi platform according to the manufacturer's protocol. In the third strategy, Hi‐C technology combined with Illumina sequencing was used to assist genome assembly. The ice plant genome size was estimated according to the 17 nt k‐mers using Illumina sequencing data (Marcais and Kingsford, 2011).
Data quality control and de novo genome assembly
Third‐generation data quality control was performed using SMRT Link (v11.0) software (https://www.pacb.com/support/software‐downloads/). The original data were polymerase reads of dumbbell‐shaped structural sequences containing adapters at both ends. Subreads were obtained after the sequences were interrupted by the adapters and the adapter sequences were filtered out. Subreads were filtered using the minimum length criterion of 50. On the basis of subreads, ccs software (https://github.com/PacificBiosciences/ccs) was used to generate high‐precision HiFi reads. The set parameters were min‐passes = 3 and min‐rq = 0.99; the quality of all read values was above Q20.
Ice plant genome assembly was performed using the Hifiasm program (v0.16.1‐r375), which uses a new haplotype assembly algorithm (Cheng et al., 2021). Hifiasm was developed based on the characteristics of PacBio HiFi reads. Therefore, it is more prominently used than other software in the assembly of HiFi data. Hifiasm assembly is divided into three steps. The first step is error correction, in which Hifiasm uses all HiFi reads for all‐vs.‐all comparisons and error correction. The second is construction of the assembly graph. After correction, a phased string graph was constructed according to the overlap between the sequences. The third, is the generation of assembly sequence, in which Hifiasm selects one side of the bubble to build the primary assembly.
Hi‐C data‐assisted assembly and genome assessment
Hi‐C data quality control includes alignment control and HiCUP quality control (Wingett et al., 2015). A Perl script was used for alignment control for three reasons. First, reads were removed using adapters. Second, reads with a ratio of N (N means that the base could not be determined) > 10% were removed. Third, when the number of low‐quality (<5) bases contained in the single‐end sequencing read exceeded 20% of the read length, the paired reads were removed. HiCUP quality control analysis was performed in three steps: (i) reads were aligned to the reference genome, (ii) the hicup_filter in the HiCUP software was used to filter the sequences in the alignment and the hicup_deduplicator in HiCUP was used to filter duplicate contacts and (iii) the ratio of the number of valid and unique read pairs after deduplication to the number of read pairs after quality control was calculated.
Based on Hi‐C technology, the ALLHiC program was used to assist in ice plant genome assembly (Zhang et al., 2019b). Clustered bam files and genomes from ALLHiC were visualized using the Juicebox program (Durand et al., 2016). Manual correction was performed according to the strength of chromosome interaction. The genome was obtained at the chromosomal level. Finally, the assembled genome was assessed using BUSCO (embryophyta_odb10) and CEGMA software (Manni et al., 2021; Parra et al., 2007). Genomes and second‐generation data were aligned using BWA software (Li and Durbin, 2009). The alignment rate, genome coverage and depth of reads were determined to assess the integrity of the assembly and uniformity of sequencing.
Genome annotation
Repeated sequences were detected using de novo prediction and homologous alignment. First, a repeat sequence database was built using RepeatModeler, LTR_FINDER (Xu and Wang, 2007), RepeatScout (Price et al., 2005) and Piler (Edgar and Myers, 2005) for de novo estimation. Second, repeated sequences were predicted using Repeatmasker. The repeatproteinmask and Repeatmasker programs were used to conduct homologous sequence alignment by searching the RepBase database (Bao et al., 2015; Tarailo‐Graovac and Chen, 2009). Tandem repeat sequences were detected using TRF software (Benson, 1999). snRNAs and miRNAs were identified using INFERNAL (Nawrocki and Eddy, 2013). tRNA and rRNA genes were detected using tRNAscan‐SE and BLAST (E‐value <1e‐5), respectively (Chan and Lowe, 2019). Simple sequence repeat (SSR) was identified according to the previous reports (Song et al., 2021a; Song et al., 2021c).
Protein‐coding gene prediction and functional annotation
De novo prediction was performed using three software programs: GlimmerHMM (Stanke and Morgenstern, 2005), SNAP (Korf, 2004) and Augustus (http://bioinf.uni‐greifswald.de/augustus/). Homologous prediction was conducted using Genewise and BLAST (E‐value <1e‐5) (Birney et al., 2004; Camacho et al., 2009). The predicted results were integrated using the IntegrationModeler (EVM) pipeline (Haas et al., 2008). Finally, the results of EVM gene prediction were corrected by combining them with transcriptomic data using the PASA software (Haas et al., 2003). Several protein databases, including TrEMBL, SwissProt, InterPro and KEGG, were used to conduct gene annotation with an E‐value cut‐off of 1e‐5. The distribution of genes, repeat sequences and non‐coding genes on each chromosome was illustrated using TBtools (Chen et al., 2020).
Detection of gene families and expansion analysis
Gene families were detected using OrthoFinder (Emms and Kelly, 2019). First, alternative splicing was filtered for each species. Only the longest transcript was retained for gene family analysis. Second, genes with amino acid lengths of <50 were removed. Third, all‐vs‐all BLAST was performed using the protein sequences to obtain the similarity relationships of all examined species (E‐value <1e‐5). Finally, single‐copy and multi‐copy gene families were detected by conducting cluster analysis based on the MCL graph clustering algorithm (‐Inflation = 1.5). Gene family contraction and amplification were performed using CAFE software (−p 0.05 ‐t 4 ‐r 10 000; De Bie et al., 2006).
Phylogeny and divergence time analysis
Genes from single‐copy gene families were used to perform multiple sequence alignments using MUSCLE (Edgar, 2004). The RAxML program (−m PROTGAMMAAUTO ‐p 12345 ‐x 12 345 ‐# 100 ‐f ad ‐T 20) was used to construct a phylogenetic tree of 13 species based on the maximum‐likelihood (ML) model (Stamatakis, 2014). Single‐copy gene families, combined with the species trees, were used to estimate the divergence time using the Mcmctree method of the PAML program (burn‐in = 50 000; sample frequency = 50; sample number = 10 000) (Yang, 2007). Time correction points were obtained from the TimeTree database (Kumar et al., 2017).
RNA‐seq and public data set collection
RNA was extracted from the roots, stems and leaves of ice plants. The purity of the RNA(OD260/280) was determined using a NanoDrop spectrophotometer. RNA concentration was quantified using Qubit, and RNA integrity was detected using an Agilent 2100 device. RNA‐seq libraries were constructed using an AMPure XP kit (Beckman, China), according to the manufacturer's instructions. RNA sequencing was performed on an Illumina HiSeq 4000 with 150‐bp paired‐end reads.
Two public RNA‐seq data sets were downloaded from NCBI and DDBJ databases to comprehensively explore the expression patterns of genes under salt stress. The raw reads of the second data set from guard cells of control and salt‐treated ice plants were collected at 12 am and 12 pm from 5 to 7 days after salt treatment (SRX3878746) (Kong et al., 2020). Raw reads of third data sets were from ice plants under salt treatments with 0, 140, 250 and 500 mm NaCl (ADRP002316) (Tsukagoshi et al., 2015).
RNA‐seq analysis
The quality of all raw reads, including our RNA‐seq data and public data sets, was assessed using the FastQC program (https://github.com/s‐andrews/FastQC). The trim‐galore program was used to filter bad quality reads and remove adaptors (https://anaconda.org/bioconda/trim‐galore). Finally, the clean reads were mapped to the ice plant genome using HISAT2 (− p 12 − x Index) (Kim et al., 2015). The expression value of each gene was normalized to fragments per kilobase of exon per million mapped fragments (Trapnell et al., 2010). Analysis of DEGs was performed using DESeq with parameters set as ¦log2(fold‐change)¦ >1 and P‐adj. < 0.05 (Anders and Huber, 2010; Wu et al., 2021).
Genome collinearity and visualization
Genome collinearity was assessed using the WGDI program, which integrates an improved ColinearScan version (‘‐icl’ model) (Sun et al., 2021; Wang et al., 2006). First, homologous genes within or between two species genomes were identified using the Blastp program (E‐value <1e‐5). Then, the ‘–icl’ model was adopted to run the WGDI for collinearity detection. The maximal gap length of collinearity was set to 50, and over 30 gene families were deleted before running ‘–icl.’ A dot plot of collinear genes was generated using WGDI (Sun et al., 2021).
Using the grape genome as a reference, we built collinear alignments for each species. Theoretically, each grape gene has two additional collinear genes because of a WGT event (Jaillon et al., 2007). In grapes, the cell of the column was filled with a gene name if a collinear gene was detected. The cell was marked with a dot if a collinear gene was absent. We also assigned the corresponding number of columns according to the situation of the WGD or WGT events for ice plants and other species. Finally, collinear alignment was visualized using a Circos plot, which was created using the –ci module in WGDI (Sun et al., 2021). Synteny and microsynteny among different species were visualized using MCscan in Python (Tang et al., 2008). The duplicate_gene_classifier program MCScanX was used to predict duplicated gene types (Wang et al., 2012).
Ks calculation and distribution fitting
The MUSCLE program was used to perform alignment using homologous amino acid sequences (−maxiters 1 –diags –sv –distance1 kbit20‐3) (Edgar, 2004). The PAL2NAL program was used to convert the protein alignment into a codon alignment according to the CDS sequence (Suyama et al., 2006). Finally, Ka and Ks were calculated using the yn00 program of PAML with the Nei‐Gojobori method according to the previous reports (Pei et al., 2021a; Pei et al., 2021b; Yang, 2007). In collinear blocks, the median values of Ks between homologous genes were used to classify blocks caused by duplication events. Ks was illustrated on a collinear block of different colours using the WGDI (Sun et al., 2021).
The distribution of Ks density was determined using three modules of WGDI: Kspeaks (−kp), PeaksFit (−pf), and KsFigures (−kf). The curve of the Ks density distribution was drawn using Kspeaks. Multipeak fitting was then performed using PeaksFit. Finally, KsFigures were used to convert multiple fitted density curves into one graph.
CAM pathway genes analyses
Based on the Pfam annotation, CAM‐related genes were identified using accession numbers with E‐values <1e‐5 (Table S22). The chromosomal distribution of CAM‐related genes was determined using TBtools (Chen et al., 2020). The protein sequences of CAM‐related genes were aligned using the Mafft program (−‐maxiterate 1000 ‐‐localpairs) (Nakamura et al., 2018). An ML tree was then constructed using FastTree using the JTT model with a bootstrap set of 1000 (Price et al., 2009; Yu et al., 2022). PCCs between CAM‐related genes and DEGs were calculated based on gene expression using Perl scripts. Based on ¦PCC¦ >0.95, an interaction network was constructed using Gephi software with the algorithm ForceAtlas2 (https://gephi.org) (Jacomy et al., 2014). KEGG functional enrichment analysis was performed using the OmicShare platform (q‐value <0.05) (https://www.omicshare.com/tools).
Morphological detection and CAM pathway gene verification
Plant growth and salt stress
Ice plants were grown in a growth chamber under 200 μmol/m2/s white light with a 12 h (26 °C) day/12 h (18 °C) night cycle. After 1 week, seedlings were transferred to 32‐ounce containers and nourished using 0.5× Hoagland's solution every 4 days. To induce CAM, salt treatment was conducted on day 28 after sowing by irrigation with 0.5 mm NaCl in 0.5× Hoagland's solution once daily. Before salt treatment (day 0, control) and at 1, 3 and 6 days of stress, the second pair of mature leaves was harvested at midday and midnight for three replicate plants.
Stomatal aperture assay
For stomatal movement observation, a part of the abaxial epidermis was peeled with tweezers at 4:00 am and 4:00 pm at each treatment time point. The peeled epidermal strips were immediately placed onto a microscope slide and observed under a Leica DM6000 B microscope.
Titratable acidity measurement
Mature leaves were collected from control and salt‐treated plants at the end of the light and dark periods. After measuring fresh weight, the samples were frozen in liquid nitrogen and ground to a fine powder using a mortar and pestle. For organic acid extraction, the powder was homogenized in 20% ethanol, boiled until volume was reduced by half, suspended in water to the original volume and boiled to half original volume. Finally, the sample was returned to its original volume using distilled water and cooled to room temperature. Titratable acidity was determined by titration with 5 mm NaOH, using phenolphthalein as an indicator. The volume of NaOH was recorded, which could be switched to leaf titratable acidity in the form of μmol H+ g−1 fresh weight.
RNA extraction and RT‐qPCR
Total RNA was isolated using RNAiso Plus (TaKaRa Bio, Japan). Integrity and quality were assessed by electrophoresis and spectrophotometry. First‐strand cDNA was synthesized using the PrimeScript RT Reagent Kit, according to the manufacturer's instructions. The resulting cDNA was diluted 1:5 in double‐distilled water before RT‐qPCR analysis using an Agilent MX3005P qPCR System Cycler. Each sample was run in triplicate and normalized by comparison with FNR1. Primers for the PEPC and MDH genes are listed in Table S30.
Conflicts of interest
The authors declare no competing interests.
Author contributions
X.S. conceived the project and were responsible for the project initiation. X.S. and S.S. supervised and managed the project and research. Experiments and analyses were designed by X.S., N.L. and S.S. Data generation and bioinformatic analyses were led by X.S., S.S., T.Y., Z.L., Y.W., R.Z., P.S., Z.W., X.T., C.Z., S.F. and Y.Z. The manuscript was organized, written and revised by X. S., S.S., N.L., H.L. and W.C. All authors read and revised the manuscript.
Supporting information
Figure S1 K‐mer distribution of the ice plant genome with the K‐mer = 17.
Figure S2 The length and frequency of the main types of repetitive sequences in ice plant genome, including DNA, LINE, LTR, and SINE repeats.
Figure S3 Comparative analysis of CDS length, exon length, exon number, gene length, and intron length in ice plant and other representative species.
Figure S4 The Venn diagram of gene sets evidence in ice plant genome.
Figure S5 The Venn diagram of gene function annotations in ice plant by four databases.
Figure S6 The chromosomal distribution of ice plant ncRNAs, including tRNA, rRNA, miRNA, and snRNA.
Figure S7 Syntenic comparison between ice plant (mcr) and A. hortensis (aho) or C. quinoa (cqu).
Figure S8 Syntenic comparison between ice plant (mcr) and F. tataricum (fta) or V. vinifera (vvi).
Figure S9 Syntenic comparison between ice plant (mcr) and A. cruentus (acr) or B. vulgaris (bvu).
Figure S10 Syntenic comparison between ice plant (mcr) and C. pallidicaule (cpa) or S. oleracea (sol).
Figure S11 Gene retention analysis of ice plant genome comparing with grape.
Figure S12 Gene retention analysis of ice plant genome comparing with A. cruentus.
Figure S13 Gene retention analysis of ice plant genome comparing with A. hortensis.
Figure S14 Gene retention analysis of ice plant genome comparing with B. vulgaris.
Figure S15 Gene retention analysis of ice plant genome comparing with C. pallidicaule.
Figure S16 Gene retention analysis of ice plant genome comparing with C. quinoa.
Figure S17 Gene retention analysis of ice plant genome comparing with F. tataricum.
Figure S18 Gene retention analysis of ice plant genome comparing with S. chinensis.
Figure S19 Gene retention analysis of ice plant genome comparing with S. oleracea.
Figure S20 Maximum‐likelihood trees of CA genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
Figure S21 Maximum‐likelihood trees of MDH genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
Figure S22 Maximum‐likelihood trees of ME genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
Figure S23 Maximum‐likelihood trees of PEPC genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
Figure S24 Maximum‐likelihood trees of PEPCK genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
Figure S25 Maximum‐likelihood trees of PPDK genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
Table S1 Statistics of sequencing data obtained by illumina Hiseq platform for ice plant genome survey.
Table S2 K‐mer statistics of the genomic characteristics of ice plant obtained by genome survey.
Table S3 Statistics of sequencing data of ice plant by Pacbio HiFi platform.
Table S4 The summary of preliminary assembly of the ice plant genome using illumina and Pacbio HiFi technology.
Table S5 Statistics on the base content of the ice plant genome.
Table S6 The SNP statistics of the ice plant genome.
Table S7 The assembled length and cluster number of each chromosome of ice plant genome.
Table S8 The read coverage statistics of the ice plant genome.
Table S9 The summary of CEGMA assessment of the ice plant genome.
Table S10 The summary of BUSCO assessment of the ice plant genome.
Table S11 The statistics of the repeat sequence classification in ice plant genome.
Table S12 The statistical of gene structure prediction in ice plant genome.
Table S13 Statistics of gene structure of ice plant and other related species.
Table S14 Statistics of gene functional annotations in ice plant genome.
Table S15 The statistics of non‐coding RNA in ice plant genome.
Table S16 The statistics of gene family in the ice plant and other 12 plant genomes.
Table S17 The gene number of each family in the ice plant and other 12 plant genomes.
Table S18 Kernel function analysis of Ks distribution related to duplication events within each genome and between two genomes.
Table S19 The duplication type analysis of each gene in the ice plant and other 12 plant genomes.
Table S20 The table listing the homologous gene sets between grape and other related species. A dot (.) is placed where no homolog is identified in the respective genome.
Table S21 Retained genes in ice plant homologous regions comparing with other related species.
Table S22 The main genes encoding enzymes involved in crassulacean acid metabolism (CAM) pathway according to the KEGG.
Table S23 The expression values (FPKM) of CAM pathway genes in different tissues of the ice plant.
Table S24 The expression values (FPKM) of CAM pathway genes under salt treatment with different days in the ice plant.
Table S25 The expression values (FPKM) of CAM pathway genes under salt treatment with different concentrations in the ice plant.
Table S26 The connections of CAM genes and other genes in the network. The regulated connections (¦PCC¦ >0.95) were used in the network.
Table S27 The edge number of each gene in the network of CAM and other DEG genes. The red color indicated the DEG genes.
Table S28 The KEGG enrichment analysis of DEGs involved in the CAM‐related gene network in ice plant (Q‐value < 0.05).
Table S29 The identified genes involved in crassulacean acid metabolism (CAM) pathway in the 23 species using the Pfam database.
Table S30 The primers sequences used in this study for qRT‐PCR.
Acknowledgements
This work was supported by the Natural Science Foundation for Distinguished Young Scholar of Hebei Province (C2022209010), the National Natural Science Foundation of China (32172583), the Natural Science Foundation of Hebei (C2021209005), and the China Postdoctoral Science Foundation (2020M673188, 2021T140097). The genome sequencing and Hi‐C were conducted in the Novogene Corporation.
Data availability statement
The genome sequence and RNA‐seq data sets of ice plant reported in this paper have been deposited in the Genome Sequence Archive (Wang et al., 2017) in BIG Data Center (Members, 2019), Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession numbers CRA005754 and CRA005756 that are publicly accessible at http://bigd.big.ac.cn/gsa. All materials and related data in this study are available upon request.
References
- Adams, P. , Nelson, D.E. , Yamada, S. , Chmara, W. , Jensen, R.G. , Bohnert, H.J. and Griffiths, H. (1998) Growth and development of Mesembryanthemum crystallinum (Aizoaceae). New Phytol. 138, 171–190. [DOI] [PubMed] [Google Scholar]
- Amari, T. , Souid, A. , Ghabriche, R. , Porrini, M. , Lutts, S. , Sacchi, G.A. , Abdelly, C. et al. (2020) Why does the halophyte Mesembryanthemum crystallinum better tolerate Ni toxicity than Brassica juncea: Implication of antioxidant defense systems. Plants (Basel), 9, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders, S. and Huber, W. (2010) Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, W. , Kojima, K.K. and Kohany, O. (2015) Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkla, B.J. and Vera‐Estrella, R. (2015) Single cell‐type comparative metabolomics of epidermal bladder cells from the halophyte Mesembryanthemum crystallinum . Front. Plant Sci. 6, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkla, B.J. , Vera‐Estrella, R. , Hernandez‐Coronado, M. and Pantoja, O. (2009) Quantitative proteomics of the tonoplast reveals a role for glycolytic enzymes in salt tolerance. Plant Cell, 21, 4044–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkla, B.J. , Vera‐Estrella, R. and Raymond, C. (2016) Single‐cell‐type quantitative proteomic and ionomic analysis of epidermal bladder cells from the halophyte model plant Mesembryanthemum crystallinum to identify salt‐responsive proteins. BMC Plant Biol. 16, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, G. (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney, E. , Clamp, M. and Durbin, R. (2004) GeneWise and genomewise. Genome Res. 14, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert, H.J. and Cushman, J.C. (2000) The ice plant cometh: Lessons in abiotic stress tolerance. J. Plant Growth Regulation, 19, 334–346. [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. and Madden, T.L. (2009) BLAST+: architecture and applications. BMC Bioinform. 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, P.P. and Lowe, T.M. (2019) tRNAscan‐SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Chen, H. , Zhang, Y. , Thomas, H.R. , Frank, M.H. , He, Y. and Xia, R. (2020) TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant, 13, 1194–1202. [DOI] [PubMed] [Google Scholar]
- Cheng, H. , Concepcion, G.T. , Feng, X. , Zhang, H. and Li, H. (2021) Haplotype‐resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods, 18, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih‐Pin, C. , Yim, W.C. , Sun, Y.H. , Miwa, O. , Tetsuro, M. , Cushman, J.C. and Yen, H.E. (2016) Identification of ice plant (Mesembryanthemum crystallinum L.) MicroRNAs using RNA‐Seq and their putative roles in high salinity responses in seedlings. Front. Plant Sci., 7, 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crayn, D.M. , Winter, K. and Smith, J.A.C. (2004) Multiple origins of crassulacean acid metabolism and the epiphytic habit in the Neotropical family Bromeliaceae. Proc. Natl. Acad. Sci. USA, 101, 3703–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushman, J.C. (2001) Crassulacean acid metabolism. A plastic photosynthetic adaptation to arid environments. Plant Physiol. 127, 1439–1448. [PMC free article] [PubMed] [Google Scholar]
- Cushman, J.C. and Bohnert, H.J. (1999) CRASSULACEAN ACID METABOLISM: Molecular genetics. Annu. Rev. Plant. Physiol. Plant. Mol. Biol. 50, 305–332. [DOI] [PubMed] [Google Scholar]
- Cushman, J.C. , Meyer, G. , Michalowski, C.B. , Schmitt, J.M. and Bohnert, H.J. (1989) Salt stress leads to differential expression of two isogenes of phosphoenolpyruvate carboxylase during Crassulacean acid metabolism induction in the common ice plant. Plant Cell, 1, 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushman, J.C. , Tillett, R.L. , Wood, J.A. , Branco, J.M. and Schlauch, K.A. (2008) Large‐scale mRNA expression profiling in the common ice plant, Mesembryanthemum crystallinum, performing C3 photosynthesis and Crassulacean acid metabolism (CAM). J. Exp. Bot. 59, 1875–1894. [DOI] [PubMed] [Google Scholar]
- De Bie, T. , Cristianini, N. , Demuth, J.P. and Hahn, M.W. (2006) CAFE: A computational tool for the study of gene family evolution. Bioinformatics, 22, 1269–1271. [DOI] [PubMed] [Google Scholar]
- Drira, R. , Matsumoto, T. , Agawa, M. and Sakamoto, K. (2016) Ice Plant (Mesembryanthemum crystallinum) extract promotes lipolysis in mouse 3T3‐L1 adipocytes through extracellular signal‐regulated kinase activation. J. Med. Food, 19, 274–280. [DOI] [PubMed] [Google Scholar]
- Durand, N.C. , Shamim, M.S. , Machol, I. , Rao, S.S. , Huntley, M.H. , Lander, E.S. and Aiden, E.L. (2016) Juicer provides a one‐click system for analyzing loop‐resolution Hi‐C experiments. Cell Syst. 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. and Myers, E.W. (2005) PILER: Identification and classification of genomic repeats. Bioinformatics 21(Suppl 1), i152–i158. [DOI] [PubMed] [Google Scholar]
- Emms, D.M. and Kelly, S. (2019) OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrig, H.H. , Heute, V. and Kluge, M. (1998) Toward a better knowledge of the molecular evolution of phosphoenolpyruvate carboxylase by comparison of partial cDNA sequences. J. Mol. Evol. 46, 107–114. [DOI] [PubMed] [Google Scholar]
- Gehrig, H.H. , Wood, J.A. , Cushman, M.A. , Virgo, A. , Cushman, J.C. and Winter, K. (2005) Research note: Large gene family of phosphoenolpyruvate carboxylase in the crassulacean acid metabolism plant Kalanchoe pinnata (Crassulaceae) characterised by partial cDNA sequence analysis. Funct. Plant Biol. 32, 467–472. [DOI] [PubMed] [Google Scholar]
- Gietl, C. (1992) Malate dehydrogenase isoenzymes: Cellular locations and role in the flow of metabolites between the cytoplasm and cell organelles. Biochim. Biophys. Acta, 1100, 217–234. [DOI] [PubMed] [Google Scholar]
- Gilman, I.S. and Edwards, E.J. (2020) Crassulacean acid metabolism. Curr. Biol. 30, R57–R62. [DOI] [PubMed] [Google Scholar]
- Haas, B.J. , Delcher, A.L. , Mount, S.M. , Wortman, J.R. , Smith, R.K., Jr. , Hannick, L.I. , Maiti, R. et al. (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J. , Salzberg, S.L. , Zhu, W. , Pertea, M. , Allen, J.E. , Orvis, J. , White, O. et al. (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtum, J.A. and Winter, K. (1982) Activity of enzymes of carbon metabolism during the induction of Crassulacean acid metabolism in Mesembryanthemum crystallinum L. Planta, 155, 8–16. [DOI] [PubMed] [Google Scholar]
- Izui, K. , Matsumura, H. , Furumoto, T. and Kai, Y. (2004) Phosphoenolpyruvate carboxylase: a new era of structural biology. Annu. Rev. Plant Biol. 55, 69–84. [DOI] [PubMed] [Google Scholar]
- Jacomy, M. , Venturini, T. , Heymann, S. and Bastian, M. (2014) ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS One, 9, e98679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon, O. , Aury, J.M. , Noel, B. , Policriti, A. , Clepet, C. , Casagrande, A. , Choisne, N. et al. (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature, 449, 463–467. [DOI] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. and Salzberg, S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nat. Methods, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, W. , Yoo, M.J. , Zhu, D. , Noble, J.D. , Kelley, T.M. , Li, J. , Kirst, M. et al. (2020) Molecular changes in Mesembryanthemum crystallinum guard cells underlying the C3 to CAM transition. Plant Mol. Biol. 103, 653–667. [DOI] [PubMed] [Google Scholar]
- Kore‐eda, S. , Nozawa, A. , Okada, Y. , Takashi, K. , Azad, M.A. , Ohnishi, J. , Nishiyama, Y. et al. (2013) Characterization of the plastidic phosphate translocators in the inducible crassulacean acid metabolism plant Mesembryanthemum crystallinum . Biosci. Biotechnol. Biochem. 77, 1511–1516. [DOI] [PubMed] [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinform. 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Suleski, M. and Hedges, S.B. (2017) TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819. [DOI] [PubMed] [Google Scholar]
- Lepiniec, L. , Keryer, E. , Philippe, H. , Gadal, P. and Crétin, C. (1993) Sorghum phosphoenolpyruvate carboxylase gene family: structure, function and molecular evolution. Plant Mol. Biol. 21, 487–502. [DOI] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C.H. , Tien, H.J. , Wen, M.F. and Yen, H.E. (2021) Myo‐inositol transport and metabolism participate in salt tolerance of halophyte ice plant seedlings. Physiol. Plant. 172, 1619–1629. [DOI] [PubMed] [Google Scholar]
- Manni, M. , Berkeley, M.R. , Seppey, M. , Simao, F.A. and Zdobnov, E.M. (2021) BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcais, G. and Kingsford, C. (2011) A fast, lock‐free approach for efficient parallel counting of occurrences of k‐mers. Bioinformatics, 27, 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka, T. , Onozawa, A. , Sonoike, K. and Kore‐Eda, S. (2018) Crassulacean acid metabolism induction in Mesembryanthemum crystallinum can be estimated by non‐photochemical quenching upon actinic illumination during the dark period. Plant Cell Physiol. 59, 1966–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Members, B.I.G.D.C. (2019) Database resources of the BIG data center in 2019. Nucleic Acids Res. 47, D8–D14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra, A. and Tanna, B. (2017) Halophytes: Potential resources for salt stress tolerance genes and promoters. Front. Plant Sci. 8, 829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, T. , Yamada, K.D. , Tomii, K. and Katoh, K. (2018) Parallelization of MAFFT for large‐scale multiple sequence alignments. Bioinformatics, 34, 2490–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki, E.P. and Eddy, S.R. (2013) Infernal 1.1: 100‐fold faster RNA homology searches. Bioinformatics, 29, 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, D.H. , Barkla, B.J. , Vera‐Estrella, R. , Pantoja, O. , Lee, S.Y. , Bohnert, H.J. and Dassanayake, M. (2015) Cell type‐specific responses to salinity – the epidermal bladder cell transcriptome of Mesembryanthemum crystallinum . New Phytol. 207, 627–644. [DOI] [PubMed] [Google Scholar]
- O'Leary, B. , Park, J. and Plaxton, W.C. (2011) The remarkable diversity of plant PEPC (phosphoenolpyruvate carboxylase): Recent insights into the physiological functions and post‐translational controls of non‐photosynthetic PEPCs. Biochem. J. 436, 15–34. [DOI] [PubMed] [Google Scholar]
- Palansooriya, K.N. , Ok, Y.S. , Awad, Y.M. , Lee, S.S. , Sung, J.K. , Koutsospyros, A. and Moon, D.H. (2019) Impacts of biochar application on upland agriculture: A review. J. Environ. Manage. 234, 52–64. [DOI] [PubMed] [Google Scholar]
- Parra, G. , Bradnam, K. and Korf, I. (2007) CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Pei, Q. , Li, N. , Bai, Y. , Wu, T. , Yang, Q. , Yu, T. , Wang, Z. et al. (2021a) Comparative analysis of the TCP gene family in celery, coriander and carrot (family Apiaceae). Vegetable Res. 1, 5. [Google Scholar]
- Pei, Q. , Yu, T. , Wu, T. , Yang, Q. , Gong, K. , Zhou, R. , Cui, C. et al. (2021b) Comprehensive identification and analyses of the Hsf gene family in the whole‐genome of three Apiaceae species. Hortic. Plant J. 7, 457–468. [Google Scholar]
- Price, A.L. , Jones, N.C. and Pevzner, P.A. (2005) De novo identification of repeat families in large genomes. Bioinformatics, 21(Suppl 1), i351–i358. [DOI] [PubMed] [Google Scholar]
- Price, M.N. , Dehal, P.S. and Arkin, A.P. (2009) FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvera, K. , Santiago, L.S. and Winter, K. (2005) Distribution of crassulacean acid metabolism in orchids of Panama: Evidence of selection for weak and strong modes. Funct. Plant Biol. 32, 397–407. [DOI] [PubMed] [Google Scholar]
- Śliwa‐Cebula, M. , Kaszycki, P. , Kaczmarczyk, A. , Nosek, M. , Lis‐Krzyścin, A. and Miszalski, Z. (2020) The common ice plant (Mesembryanthemum crystallinum L.)‐phytoremediation potential for cadmium and chromate‐contaminated soils. Plants (Basel), 9, 1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, X. , Li, N. , Guo, Y. , Bai, Y. , Wu, T. , Yu, T. , Feng, S. et al. (2021a) Comprehensive identification and characterization of simple sequence repeats based on the whole‐genome sequences of 14 forest and fruit trees. Forestry Res. 1, 7–10. [Google Scholar]
- Song, X. , Sun, P. , Yuan, J. , Gong, K. , Li, N. , Meng, F. , Zhang, Z. et al. (2021b) The celery genome sequence reveals sequential paleo‐polyploidizations, karyotype evolution and resistance gene reduction in apiales . Plant Biotechnol. J. 19, 731–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, X. , Yang, Q. , Bai, Y. , Gong, K. , Wu, T. , Yu, T. , Pei, Q. et al. (2021c) Comprehensive analysis of SSRs and database construction using all complete gene‐coding sequences in major horticultural and representative plants. Hortic Res. 8, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, X. , Liu, H. , Shen, S. , Huang, Z. , Yu, T. , Liu, Z. , Yang, Q. et al. (2022) Chromosome‐level pepino genome provides insights into genome evolution and anthocyanin biosynthesis in Solanaceae. Plant J. 110, 1128–1143. [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. and Morgenstern, B. (2005) AUGUSTUS: A web server for gene prediction in eukaryotes that allows user‐defined constraints. Nucleic Acids Res. 33, W465–W467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, P. , Jiao, B. , Yang, Y. , Shan, L. , Li, T. , Li, X. , Xi, Z. , Wang, X. and Liu, J. (2021) WGDI: A user‐friendly toolkit for evolutionary analyses of whole‐genome duplications and ancestral karyotypes. bioRxiv, 2021.2004.2029.441969. [DOI] [PubMed]
- Suyama, M. , Torrents, D. and Bork, P. (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H. , Bowers, J.E. , Wang, X. , Ming, R. , Alam, M. and Paterson, A.H. (2008) Synteny and collinearity in plant genomes. Science, 320, 486–488. [DOI] [PubMed] [Google Scholar]
- Tarailo‐Graovac, M. and Chen, N. (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. Chapter 4, Unit 4 10. [DOI] [PubMed] [Google Scholar]
- Taybi, T. , Nimmo, H.G. and Borland, A.M. (2004) Expression of phosphoenolpyruvate carboxylase and phosphoenolpyruvate carboxylase kinase genes. Implications for genotypic capacity and phenotypic plasticity in the expression of crassulacean acid metabolism. Plant Physiol. 135, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Williams, B.A. , Pertea, G. , Mortazavi, A. , Kwan, G. , van Baren, M.J. , Salzberg, S.L. et al. (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukagoshi, H. , Suzuki, T. , Nishikawa, K. , Agarie, S. , Ishiguro, S. and Higashiyama, T. (2015) RNA‐seq analysis of the response of the halophyte, Mesembryanthemum crystallinum (ice plant) to high salinity. PLoS One, 10, e0118339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamatsu, A. , Mori, I.C. , Matsuura, T. , Taniwaki, Y. , Ishii, R. and Yoshida, R. (2021) Possible roles for phytohormones in controlling the stomatal behavior of Mesembryanthemum crystallinum during the salt‐induced transition from C3 to crassulacean acid metabolism. J. Plant Physiol. 262, 153448. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Shi, X. , Li, Z. , Zhu, Q. , Kong, L. , Tang, W. , Ge, S. et al. (2006) Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice. BMC Bioinform. 7, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , Debarry, J.D. , Tan, X. , Li, J. , Wang, X. , Lee, T.H. et al. (2012) MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Song, F. , Zhu, J. , Zhang, S. , Yang, Y. , Chen, T. , Tang, B. et al. (2017) GSA: Genome sequence archive. Genom. Proteom. Bioinform. 15, 14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhoff, P. and Gowik, U. (2004) Evolution of c4 phosphoenolpyruvate carboxylase. Genes and proteins: a case study with the genus Flaveria. Ann. Bot. 93, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingett, S. , Ewels, P. , Furlan‐Magaril, M. , Nagano, T. , Schoenfelder, S. , Fraser, P. and Andrews, S. (2015) HiCUP: Pipeline for mapping and processing Hi‐C data. F1000Res, 4, 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter, K. and Holtum, J.A. (2005) The effects of salinity, crassulacean acid metabolism and plant age on the carbon isotope composition of Mesembryanthemum crystallinum L., a halophytic C(3)‐CAM species. Planta, 222, 201–209. [DOI] [PubMed] [Google Scholar]
- Winter, K. and Holtum, J.A. (2007) Environment or development? Lifetime net CO2 exchange and control of the expression of Crassulacean acid metabolism in Mesembryanthemum crystallinum . Plant Physiol. 143, 98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter, K. and Holtum, J.A. (2014) Facultative crassulacean acid metabolism (CAM) plants: powerful tools for unravelling the functional elements of CAM photosynthesis. J. Exp. Bot. 65, 3425–3441. [DOI] [PubMed] [Google Scholar]
- Wu, T. , Feng, S.‐Y. , Yang, Q.‐H. , Bhetariya, P.J. , Gong, K. , Cui, C.‐L. , Song, J. et al. (2021) Integration of the metabolome and transcriptome reveals the metabolites and genes related to nutritional and medicinal value in Coriandrum sativum . J. Integr. Agric. 20, 1807–1818. [Google Scholar]
- Xu, Z. and Wang, H. (2007) LTR_FINDER: An efficient tool for the prediction of full‐length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. (2007) PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yu, T. , Bai, Y. , Liu, Z. , Wang, Z. , Yang, Q. , Wu, T. , Feng, S. et al. (2022) Large‐scale analyses of heat shock transcription factors and database construction based on whole‐genome genes in horticultural and representative plants. Hortic. Res. 9, uhac035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Wu, W. , Xin, X. , Li, X. and Liu, D. (2019a) Extract of ice plant (Mesembryanthemum crystallinum) ameliorates hyperglycemia and modulates the gut microbiota composition in type 2 diabetic Goto‐Kakizaki rats. Food Funct. 10, 3252–3261. [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Zhang, S. , Zhao, Q. , Ming, R. and Tang, H. (2019b) Assembly of allele‐aware, chromosomal‐scale autopolyploid genomes based on Hi‐C data. Nat. Plants, 5, 833–845. [DOI] [PubMed] [Google Scholar]
- Zou, C. , Chen, A. , Xiao, L. , Muller, H.M. , Ache, P. , Haberer, G. , Zhang, M. et al. (2017) A high‐quality genome assembly of quinoa provides insights into the molecular basis of salt bladder‐based salinity tolerance and the exceptional nutritional value. Cell Res. 27, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 K‐mer distribution of the ice plant genome with the K‐mer = 17.
Figure S2 The length and frequency of the main types of repetitive sequences in ice plant genome, including DNA, LINE, LTR, and SINE repeats.
Figure S3 Comparative analysis of CDS length, exon length, exon number, gene length, and intron length in ice plant and other representative species.
Figure S4 The Venn diagram of gene sets evidence in ice plant genome.
Figure S5 The Venn diagram of gene function annotations in ice plant by four databases.
Figure S6 The chromosomal distribution of ice plant ncRNAs, including tRNA, rRNA, miRNA, and snRNA.
Figure S7 Syntenic comparison between ice plant (mcr) and A. hortensis (aho) or C. quinoa (cqu).
Figure S8 Syntenic comparison between ice plant (mcr) and F. tataricum (fta) or V. vinifera (vvi).
Figure S9 Syntenic comparison between ice plant (mcr) and A. cruentus (acr) or B. vulgaris (bvu).
Figure S10 Syntenic comparison between ice plant (mcr) and C. pallidicaule (cpa) or S. oleracea (sol).
Figure S11 Gene retention analysis of ice plant genome comparing with grape.
Figure S12 Gene retention analysis of ice plant genome comparing with A. cruentus.
Figure S13 Gene retention analysis of ice plant genome comparing with A. hortensis.
Figure S14 Gene retention analysis of ice plant genome comparing with B. vulgaris.
Figure S15 Gene retention analysis of ice plant genome comparing with C. pallidicaule.
Figure S16 Gene retention analysis of ice plant genome comparing with C. quinoa.
Figure S17 Gene retention analysis of ice plant genome comparing with F. tataricum.
Figure S18 Gene retention analysis of ice plant genome comparing with S. chinensis.
Figure S19 Gene retention analysis of ice plant genome comparing with S. oleracea.
Figure S20 Maximum‐likelihood trees of CA genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
Figure S21 Maximum‐likelihood trees of MDH genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
Figure S22 Maximum‐likelihood trees of ME genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
Figure S23 Maximum‐likelihood trees of PEPC genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
Figure S24 Maximum‐likelihood trees of PEPCK genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
Figure S25 Maximum‐likelihood trees of PPDK genes that were constructed using the amino acid sequences with 1000 bootstrap repeats in ice plant and other 22 related species.
Table S1 Statistics of sequencing data obtained by illumina Hiseq platform for ice plant genome survey.
Table S2 K‐mer statistics of the genomic characteristics of ice plant obtained by genome survey.
Table S3 Statistics of sequencing data of ice plant by Pacbio HiFi platform.
Table S4 The summary of preliminary assembly of the ice plant genome using illumina and Pacbio HiFi technology.
Table S5 Statistics on the base content of the ice plant genome.
Table S6 The SNP statistics of the ice plant genome.
Table S7 The assembled length and cluster number of each chromosome of ice plant genome.
Table S8 The read coverage statistics of the ice plant genome.
Table S9 The summary of CEGMA assessment of the ice plant genome.
Table S10 The summary of BUSCO assessment of the ice plant genome.
Table S11 The statistics of the repeat sequence classification in ice plant genome.
Table S12 The statistical of gene structure prediction in ice plant genome.
Table S13 Statistics of gene structure of ice plant and other related species.
Table S14 Statistics of gene functional annotations in ice plant genome.
Table S15 The statistics of non‐coding RNA in ice plant genome.
Table S16 The statistics of gene family in the ice plant and other 12 plant genomes.
Table S17 The gene number of each family in the ice plant and other 12 plant genomes.
Table S18 Kernel function analysis of Ks distribution related to duplication events within each genome and between two genomes.
Table S19 The duplication type analysis of each gene in the ice plant and other 12 plant genomes.
Table S20 The table listing the homologous gene sets between grape and other related species. A dot (.) is placed where no homolog is identified in the respective genome.
Table S21 Retained genes in ice plant homologous regions comparing with other related species.
Table S22 The main genes encoding enzymes involved in crassulacean acid metabolism (CAM) pathway according to the KEGG.
Table S23 The expression values (FPKM) of CAM pathway genes in different tissues of the ice plant.
Table S24 The expression values (FPKM) of CAM pathway genes under salt treatment with different days in the ice plant.
Table S25 The expression values (FPKM) of CAM pathway genes under salt treatment with different concentrations in the ice plant.
Table S26 The connections of CAM genes and other genes in the network. The regulated connections (¦PCC¦ >0.95) were used in the network.
Table S27 The edge number of each gene in the network of CAM and other DEG genes. The red color indicated the DEG genes.
Table S28 The KEGG enrichment analysis of DEGs involved in the CAM‐related gene network in ice plant (Q‐value < 0.05).
Table S29 The identified genes involved in crassulacean acid metabolism (CAM) pathway in the 23 species using the Pfam database.
Table S30 The primers sequences used in this study for qRT‐PCR.
Data Availability Statement
The genome sequence and RNA‐seq data sets of ice plant reported in this paper have been deposited in the Genome Sequence Archive (Wang et al., 2017) in BIG Data Center (Members, 2019), Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession numbers CRA005754 and CRA005756 that are publicly accessible at http://bigd.big.ac.cn/gsa. All materials and related data in this study are available upon request.