

Abstract
Twelve patients without therapy-related leukemia were studied after completing TOP2 poison chemotherapy in a high-risk neuroblastoma regimen. One patient harbored an inv(11) that was a KMT2A rearrangement. The KMT2A-MAML2 transcript was expressed at low level. The patient was prospectively followed. The inv(11) was undetectable in ensuing samples. Leukemia never developed after a 12.8-year follow-up period. Enriched etoposide-induced TOP2A cleavage in the relevant MAML2 genomic region supports a TOP2A DNA damage mechanism. After completing TOP2 poison chemotherapies, covert KMT2A-R clones may occur in a small minority of patients; however, not all KMT2A rearrangements herald a therapy-related leukemia diagnosis.
Keywords: KMT2A-MAML2, TOP2 poison chemotherapy, TOP2A cleave-ome, neuroblastoma, therapy-related leukemia
Therapy-related leukemias with balanced translocations attributed to TOP2 poisons, most of which involve KMT2A, were recognized when epipodophyllotoxins came into use in the late 1980s1. Discontinuing the more leukemogenic epipodophyllotoxin teniposide, and dose and schedule modifications lessened risk, but TOP2 poisons are mainstay chemotherapies and TOP2 poison-related leukemias remain a significant problem1.
Therapy-related leukemia occurred following dose-intensive multi-modality MSK metastatic neuroblastoma treatments. MSK-N6, MSK-N7 and MSK-N8 all emphasized dose-intensive TOP2 poison- and alkylating agent-containing induction chemotherapy, but the number of cycles was successively reduced, and post-induction treatments changed2. On MSK-N6 (1990–1993), which used 7 chemotherapy cycles during induction, leukemia incidence was 7%3,4. About 40% of cases exhibited chromosome 5 and/or 7 loss attributed to alkylators; 40% had KMT2A translocations3. On MSK-N7 (1994–1999), which included 7 but later 5 chemotherapy cycles during induction, leukemia incidence was 5.6% after ≥12 years5–7. MSK-N8 (2000–2007) used 5 cycles8–10. After changing to 5 cycles, leukemia incidence was 0 after induction7. Occasional patients requiring additional chemotherapy for relapse developed leukemia/myelodysplasia, but the incidence is <7%5,7. The differences in therapies likely contributed to the decreased incidence of leukemia.
The MSK neuroblastoma experience affords unique opportunities to trace when KMT2A translocations originate relative to treatment and leukemia diagnosis because marrow surveillance is required3,6,7. MSK-N7 specified multi-site marrow samplings at diagnosis, stem cell harvest or second look surgery, end-induction11, every 3–6 months thereafter until 2 years from diagnosis, and periodically for a few more years.
There are five therapy-related KMT2A-R leukemia cases altogether where tracing the translocation in sequential samples was reported12–16. Our studies of two children treated on MSK regimens who developed therapy-related leukemia showed that KMT2A translocations can emerge early or late during treatment, and that latency to leukemia can be short or protracted12,13.
Here we describe a KMT2A rearrangement that emerged and regressed during neuroblastoma treatment without ever manifesting as leukemia. The prolonged prospective follow-up is uniquely longer than for any patients described to date. We investigate how often KMT2A-R occurs during chemotherapy without leukemia by studying 12 patients with metastatic neuroblastoma at an identical treatment timepoint, and study the KMT2A partner gene as a chemotherapy-induced TOP2A cleavage target.
RESULTS AND DISCUSSION
A 17-year-old European Caucasian female was treated on MSK-N76,7 (Fig. 1) for Stage 4N neuroblastoma with disseminated lymph node disease without marrow or extranodal metastases17. Induction chemotherapy comprised four cycles of CAV, and three cycles of PVP. At diagnosis, the marrow karyotype was normal, and lymph node tumor showed a hyper-tetraploid karyotype and 4–5 copies of MYCN by FISH (Fig. 1). At surgical resection three months later, 100% of primary tumor cells showed 4 copies of MYCN by FISH, but all cells after culture showed a 46,XX,t(8;15)(p21;q26) karyotype (Fig. 1). The marrow karyotype after all seven chemotherapy cycles (10 months from diagnosis) demonstrated 46,XX,t(8;15)(p21;q26),inv(11)(q21q23)[cp10];46,XX,t(8;15)(p21;q26)[cp10] (Fig. 1). Except for this sample, there was no karyotypic evidence of inv(11) (Fig. 1). A blood sample before myeloablative radiolabeled 3F8 antibodies and autologous PBSC rescue (11 months from diagnosis) showed 46,XX,t(8;15)(p21;q26)[13];46,XX[3] (Fig. 1); however, marrow was not sampled. The t(8;15) was never detected afterwards (Fig. 1). The conditioning did not include chemotherapy and was not leukemia-directed.
Fig. 1.
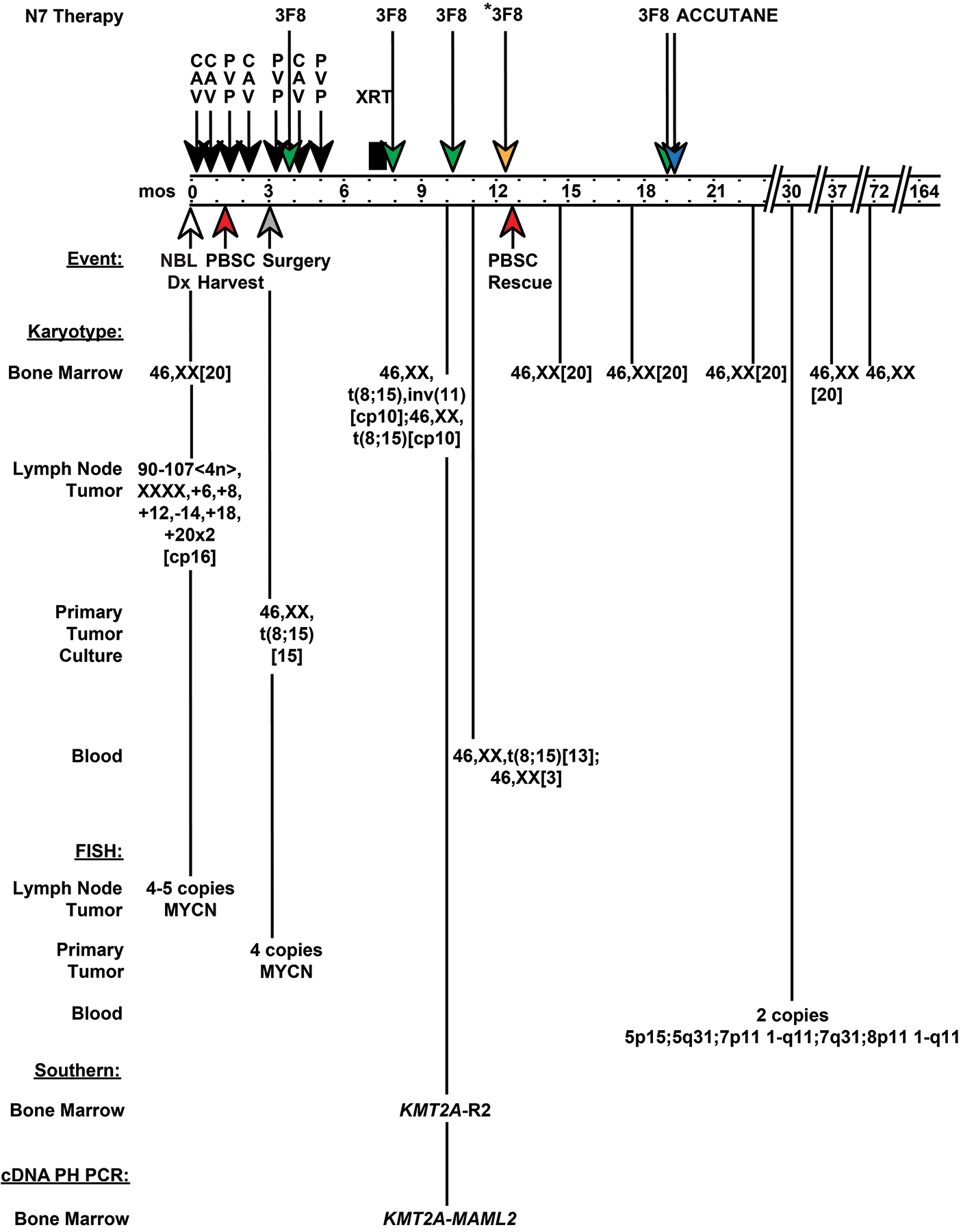
Treatment, clinical course, and timeline of cytogenetic and molecular findings in patient with Stage 4N neuroblastoma enrolled to MSK-N7 trial. Schematic shows when inv(11)(q21q23) resulting in KMT2A-MAML2 rearrangement occurred and disappeared relative to treatment. The treatment included 7 dose-intensive cycles of induction chemotherapy [4 cycles of cyclophosphamide, adriamycin, and vincristine (CAV); 3 cycles of cisplatin and etoposide (PVP)], 3 cycles of cold anti-GD2 monoclonal antibodies (3F8) and local radiation before myeloablation with radiolabeled 3F8 monoclonal antibodies (3F8*) and autologous PBSC rescue. Autologous PBSCs were harvested after cycle 2, which was CAV. Following PBSC rescue, a final cycle of adjuvant 3F8 began, but accutane was substituted for 3F8 due to allergic reaction. At neuroblastoma diagnosis (time 0 months), note hyper-tetraploid karyotype of lymph node tumor tissue, and normal marrow karyotype. At surgical resection (time 3 months) the primary tumor cells contained 4 copies of MYCN by FISH, whereas the karyotype after culture was 46,XX,t(8;15)(p21;q26)[15], suggesting contaminating blood cells in the tumor. After all 7 chemotherapy cycles (time 10 months), all cells in the marrow had a pseudodiploid karyotype with t(8;15)(p21;q26), and half also had inv(11)(q21q23). At time 11 months the peripheral blood karyotype was 46,XX,t(8;15)(p21;q26)[13];46,XX[3]. All subsequent marrow karyotypes were normal. FISH studies and results are shown. Abbreviations: mos, months; CAV, cyclophosphamide 4200 mg/m2, adriamycin 75 mg/m2, vincristine 1.5 mg/m2; PVP, cisplatin 200 mg/m2, etoposide 600 mg/m2; 3F8*, radiolabeled anti-GD2 monoclonal antibody; 3F8, cold anti-GD2 monoclonal antibody; NBL, neuroblastoma; XRT, local radiation therapy to neck and chest and to abdomen and hip; PBSC, peripheral blood stem cell; KMT2A-R2, two rearranged KMT2A bcr fragments; cDNA PH PCR, cDNA panhandle PCR.
These data suggest that the t(8;15) and inv(11) were transient clonal abnormalities, that the clone with t(8;15) and inv(11) was more short-lived than the clone with just t(8;15), and that these abnormalities were acquired during treatment and originated in the marrow. The tumor cells were hyper-tetraploid, but the cells with t(8;15) cultured from the tumor pseudodiploid (Fig. 1), suggesting that the t(8;15) was from contaminating blood cells in the tumor. The two clones with t(8;15) in the marrow 10 months from diagnosis, one of which contained inv(11), suggest that the clone with both abnormalities evolved from the clone with t(8;15) alone. The normal karyotypes of all other marrows, and cells with a normal karyotype besides cells showing t(8;15) in the blood 11 months from diagnosis (Fig. 1), indicate that the t(8;15) was non-constitutional.
During 12.8 years of prospective follow-up from detecting the inv(11) (13.7 years from starting treatment), far beyond the typical latency of therapy-related KMT2A-R leukemias of ~1.5 to 3 years from primary cancer diagnosis18, there were never clinical signs of leukemia, increased blasts or morphologic hematopoietic abnormalities.
Unlike 11 other patients with metastatic neuroblastoma whom we studied after completing all seven chemotherapy cycles who also did not develop leukemia, Southern blot analysis of this patient’s marrow showed KMT2A gene rearrangement (Fig. 2A). Therefore, the inv(11) disrupted KMT2A, and the KMT2A-R clone expanded to Southern blot detection level (using a 32P radiolabeled probe, ~1/100 cells19). cDNA panhandle PCR12 identified the KMT2A partner gene at band 11q21 as MAML2. Two independent cDNA panhandle PCRs were performed using first strand cDNA prepared from bone marrow RNA after chemotherapy cycle 7 (Fig. 1). The transcript joining KMT2A exon 9 to MAML2 exon 2 was found in just one of 94 total subclones (Fig. 2B,C), indicating that expression was low-level.
Fig. 2.
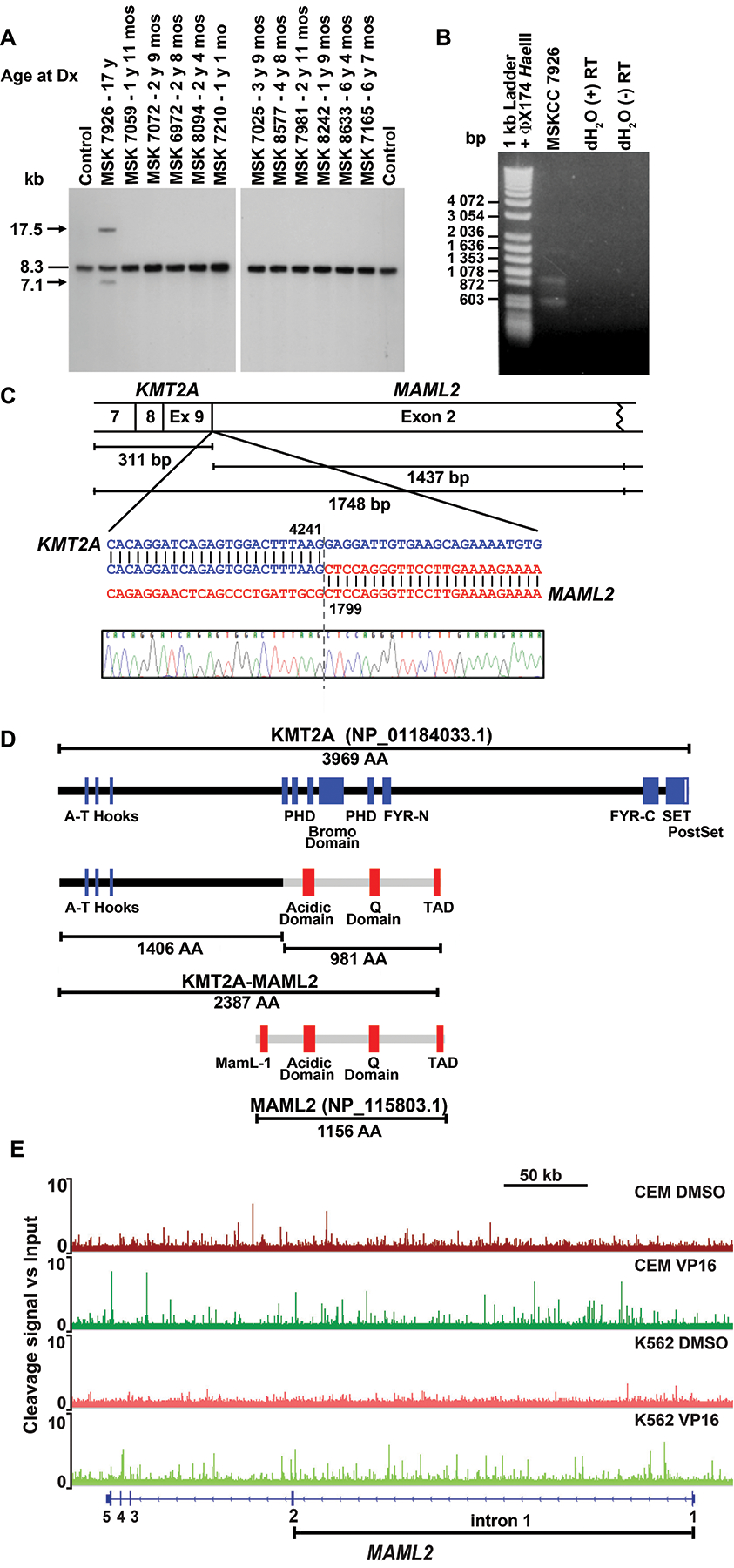
Molecular analysis of KMT2A rearrangement. (A) Southern blot analysis of bone marrow samples of 12 pediatric patients who did not develop leukemia by time of completion of all 7 cycles of MSK-N7 chemotherapy. Arrows show rearrangements; dash, germline bands. Genomic DNA and total RNA were prepared on GITC/CsCl gradients. Southern blot analysis used 5 μg BamHI-digested DNA. B859 fragment of KMT2A cDNA was radiolabeled with 32P by nick translation12,13. Normal adult peripheral WBCs isolated by ACK lysis of red blood cells in whole blood were control. (B) cDNA panhandle PCR12 identification of unknown 5’-KMT2A-partner-3’ transcript. First strand cDNA prepared from bone marrow RNA after chemotherapy cycle 7 using 5’-KMT2A-NNNNNN-3’ primers12, was amplified in two independent cDNA panhandle PCRs. Gel shows products of one reaction, which vary in size as expected12. Products of the two cDNA panhandle PCRs were subcloned by recombination PCR12, 31 and 63 subclones from which, respectively, were sequenced. (C) Fusion transcript sequence compared to native KMT2A (GenBank accession no. NM_001197104.1) and the full-length 5-exon 7115 bp MAML2 (GenBank accession no. NM_032427.3) mRNA reference sequences with coordinates at point of fusion indicated. One subclone contained transcript sequence joining KMT2A exon 9 in-frame to MAML2 exon 2. Exon numbering is according to NCBI. Ninety-two subclones contained KMT2A transcript sequence only, with normally spliced, alternatively spliced and scrambled exons. One subclone was vector only. (D) Schematic of predicted fusion protein (middle) compared to native KMT2A and MAML2 (top and bottom). Amino acids 1 to 1406 from KMT2A are joined to acidic domain, Q domain and carboxyl-terminal TAD of MAML2. The first of three poly-glutamine tracts comprising the Q domain, in which the number of glutamine (Q) repeats is known to vary, contains 30 Q residues in the fusion protein compared to 34 in the reference sequence (GenBank accession no. NM_032427.3). This is accounted for in number of amino acids in the fusion protein. (E) TOP2A cleave-ome data for K56228 and CEM (Yu and Davenport, unpublished) cell lines illustrating cleavage along Chromosome band 11q21 in MAML2 gene body. The Y axis shows fold-change in TOP2A cleavage signal relative to input. Note many peaks in intron 1 (bracketed) in presence of etoposide (VP16) not detected in vehicle treated samples, indicating enriched cleavage with etoposide, of interest because 5’-KMT2A exon 9-MAML2 exon 2–3’ transcript (C) signifies a MAML2 genomic breakpoint upstream to start of exon 2. Fold-change in cleavage signal was plotted and tracts along MAML2 captured using IGV.
Four cases of therapy-related T-cell ALL15,20,21, two cases of therapy-related AML16,22, and one case each of therapy-related MDS23, therapy-related B-cell ALL24, de novo MDS16, and T-cell ALL that underwent lineage switch to AML23 with KMT2A-MAML2 or inv(11)(q21q23) have been reported. KMT2A-MAML2 also occurs in aggressive thymoma subtypes25. The eight patients with therapy-related KMT2A-MAML2 leukemia/MDS differ from this patient. The primary cancer was leukemia15,16,21–23 except in two patients20,24. None had neuroblastoma. Latencies were 25–86 months. The latter is longer than usual for therapy-related KMT2A-R leukemia, but far shorter than the observation time in this patient. Like other KMT2A rearrangements traced in sequential samples manifesting as leukemia12–14, KMT2A-MAML2 remained detectable after first appearing until therapy-related leukemia/MDS diagnosis15,16. In contrast, in this patient, inv(11) never appeared again after first being detected.
MAML2 is one of three Mastermind-Like Notch receptor transcriptional co-activators, each having a conserved amino-terminal basic domain that binds the ankyrin domain within the intracellular domain of Notch receptors, and carboxyl-terminal TAD26,27. The in-frame 5’-KMT2A-MAML2-3’ transcript we identified predicts a fusion protein (Fig. 2D) with potential for Notch signaling perturbation.
We surveyed existing TOP2A cleave-ome datasets for CEM (Yu and Davenport, unpublished) and K56228 cells to gain insight into the MAML2 disruption. Although the genomic breakpoint junction of the inv(11) was not cloned, the MAML2 fusion point in the 5’-KMT2A-MAML2-3’ transcript at the start of exon 2 (Fig. 2C), indicates a genomic breakpoint upstream of exon 2. Accordingly, both cell lines exhibited enriched TOP2A cleavage compared to vehicle along MAML2 intron 1 in the presence of etoposide (Fig. 2E), indicating that MAML2 is a TOP2 poison-induced TOP2A cleavage target. In K562 cells we reported etoposide-enriched TOP2A cleavage along the KMT2A bcr28, further highlighting plausibility of TOP2 poison-induced TOP2A cleavage as the damage mechanism.
Thus, we followed a patient for 12.8 years after KMT2A-MAML2 emerged and regressed during neuroblastoma treatment without onset of leukemia. The case series of 12 patients at the same MSK-N7 treatment timepoint in which just this patient had KMT2A-R, suggests that after receiving TOP2 poisons a small minority of patients harbor clinically silent clonal KMT2A rearrangements. The 11 other patients were not an age-matched cohort (Fig. 2A) because the median age at neuroblastoma diagnosis is 22 months and 90% of cases occur before age 529. The literature suggests that clonal hematopoiesis during pediatric anticancer treatment is uncommon. Targeted next generation sequencing did not detect clonal hematopoiesis in peripheral blood DNA of 84 survivors of heterogeneous pediatric cancers (age at diagnosis 0–25.4 years), including 17 survivors of neuroblastoma (age at diagnosis 0.3–15.4 years), after median follow-up of 6 y30. In pediatric patients with ALL remission (age range 2–16 years), hematopoiesis proved polyclonal31. This is the second reported covert KMT2A-R clone during anticancer treatment without evidence of leukemia. A KMT2A-ARHGEF17 rearrangement occurred after primary pediatric AML treatment in a 5-year old without therapy-related leukemia developing by 30 months from diagnosis32. However, in the patient studied here, the follow-up was much longer. The dynamics of hematopoiesis during chemotherapy in pediatric patients who do and do not develop therapy-related leukemia after TOP2 poisons warrants further study.
Why leukemia did not occur is unknown. The identical transcript occurred in therapy-related leukemias16,21 and thymomas25. Why this transcript was associated with leukemia in other patients, and why expression was low-level are uncertain. Expression is affected by transcriptional and post-transcriptional regulation; decreased production or transcript instability cause low-level expression. MAML2 translocations not involving KMT2A in indolent benign and malignant neoplasms27,33–42 raise questions about oncogenic potency of MAML2 disruption. Additional mutational events may have been required. The marrow milieu or target cell may have been unfavorable. The KMT2A-R clone may have been obliterated immunologically43–49 or by ensuing myeloablative treatment; however, therapy-related KMT2A-R leukemia has occurred on MSK-N75. The latency may prove even longer. Our finding that KMT2A-MAML2 did not herald therapy-related leukemia challenges the paradigm that all KMT2A-R clones connote leukemia. It has important implications about whether careful follow-up vs. aggressive intervention is prudent for clinically silent KMT2A rearrangements. This patient suggests that transient KMT2A-R during chemotherapy may not have the significance that clonal KMT2A rearrangement has historically implied for leukemia prediction.
ACKNOWLEDGEMENTS
This work was supported by NIH grant R01CA85469 (C.A.F.) and Leukemia & Lymphoma Society TRP 6568-19 (C.A.F., B.D.G., J.W.D., X.Y.). C.A.F. is the Joshua Kahan Endowed Chair in Pediatric Leukemia Research. We acknowledge experimental contributions to this research made by Dr. Anastasia Guerriero, a deceased member of the Felix laboratory. We thank Dr. Karen Urtishak for assistance with figure preparation.
ABBREVIATIONS
- 3F8*
Radiolabeled anti-GD2 monoclonal antibody
- 3F8
Cold anti-GD2 monoclonal antibody
- ACK
Ammonium-Chloride-Potassium
- ALL
Acute lymphoblastic leukemia
- AML
Acute myelogenous leukemia
- bcr
breakpoint cluster region
- BM
Bone marrow
- CAV
cyclophosphamide 4200 mg/m2, adriamycin 75 mg/m2, vincristine 1.5 mg/m2
- cDNA PH PCR
cDNA panhandle PCR
- COG
Children’s Oncology Group
- [cp]
Composite karyotype
- Dx
Diagnosis
- FISH
Fluorescence in situ hybridization
- g/m2
grams per square meter
- GITC/CsCl
guanidinium isothiocyanate/Cesium Chloride
- IGV
Integrative Genomics Viewer
- IRB
Institutional Review Board
- KMT2A-R
KMT2A-rearranged
- MDS
myelodysplastic syndrome
- mo
month
- mos
months
- MSK
Memorial Sloan Kettering Cancer Center
- NCBI
National Center for Biotechnology Information
- no.
number
- PBSC
peripheral blood stem cell
- PVP
cisplatin 200 mg/m2, etoposide 600 mg/m2
- TAD
Transcriptional Activation Domain
- TOP2
Topoisomerase II
- VP16
Etoposide
- WBCs
white blood cells
- y
Year/years
- XRT
local radiation therapy to neck and chest and to abdomen and hip
Footnotes
ETHICS STATEMENT
Molecular analyses were performed under IRB approved protocols of the Children’s Hospital of Philadelphia (Protocol 99-001792) and MSK (Clinicaltrials.gov NCT00588068), and written consent was properly documented.
CONFLICT OF INTEREST STATEMENT
C.A.F. and E.F.R. are named inventors on the following issued patent filed by the Children’s Hospital of Philadelphia that has not been licensed: Methods and Kits for Analysis of Chromosomal Rearrangements Associated with Leukemia. United States of America 6,368,791. 2002 April 09. C.A.F. is a named inventor on the following issued patents filed by the Children’s Hospital of Philadelphia that have not been licensed: Compositions and Methods for the Detection of DNA Topoisomerase II Complexes with DNA. United States of America 8,642,265 B2. 2014 February 04. CYP3A4 NFSE Variant and Methods of Use Thereof - United States of America 6,174,684. 2001 January 16. N-K.C. is the inventor and owner of issued patents licensed by MSK to Y-mabs Therapeutics, Biotec Pharmacon, and Abpro-labs. Hu3F8 and 8H9 were licensed by MSK to Y-mabs Therapeutics.
N-K.C. reports receiving commercial research grants for work outside of this work from Y-mabs Therapeutics and Abpro-Labs Inc., holding ownership interest/equity in Y-Mabs Therapeutics Inc., holding ownership interest/equity in Abpro-Labs, and owning stock options in Eureka Therapeutics. Both MSK and N-K.C. have financial interest in Y-mabs. N-K.C. is an advisory board member for Abpro-Labs and Eureka Therapeutics.
DATA SHARING
The KMT2A-MAML2 transcript was deposited in GenBank (Accession no. KY584083).
REFERENCES
- 1.Felix CA. A safer regimen for high-risk neuroblastoma. Pediatr Blood Cancer. 2009;53(1):3–6. [DOI] [PubMed] [Google Scholar]
- 2.Kushner BH, Kramer K, Laquaglia MP, Modak S, Yataghene K, Cheung N-KV. In Reply. Journal of Clinical Oncology. 2005;23(25):6263–6263.16135498 [Google Scholar]
- 3.Kushner BH, Cheung NK, Kramer K, Heller G, Jhanwar SC. Neuroblastoma and treatment-related myelodysplasia/leukemia: the Memorial Sloan-Kettering experience and a literature review. J Clin Oncol. 1998;16(12):3880–3889. [DOI] [PubMed] [Google Scholar]
- 4.Cheung NK, Kushner BH, Cheung IY, et al. Anti-G(D2) antibody treatment of minimal residual stage 4 neuroblastoma diagnosed at more than 1 year of age. J Clin Oncol. 1998;16(9):3053–3060. [DOI] [PubMed] [Google Scholar]
- 5.Kushner BH, Kramer K, LaQuaglia MP, Modak S, Yataghene K, Cheung NK. Reduction from seven to five cycles of intensive induction chemotherapy in children with high-risk neuroblastoma. J Clin Oncol. 2004;22(24):4888–4892. [DOI] [PubMed] [Google Scholar]
- 6.Cheung NK, Kushner BH, LaQuaglia M, et al. N7: a novel multi-modality therapy of high risk neuroblastoma (NB) in children diagnosed over 1 year of age. Med Pediatr Oncol. 2001;36(1):227–230. [DOI] [PubMed] [Google Scholar]
- 7.Kushner BH, Kramer K, Modak S, et al. Reduced risk of secondary leukemia with fewer cycles of dose-intensive induction chemotherapy in patients with neuroblastoma. Pediatr Blood Cancer. 2009;53(1):17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheung IY, Lo Piccolo MS, Kushner BH, Cheung NK. Early molecular response of marrow disease to biologic therapy is highly prognostic in neuroblastoma. J Clin Oncol. 2003;21(20):3853–3858. [DOI] [PubMed] [Google Scholar]
- 9.Kushner BH, Kramer K, LaQuaglia MP, Modak S, Cheung NK. Neuroblastoma in adolescents and adults: the Memorial Sloan-Kettering experience. Med Pediatr Oncol. 2003;41(6):508–515. [DOI] [PubMed] [Google Scholar]
- 10.Cheung NK, Cheung IY, Kushner BH, et al. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol. 2012;30(26):3264–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung NK, Heller G, Kushner BH, Liu C, Cheung IY. Detection of metastatic neuroblastoma in bone marrow: when is routine marrow histology insensitive? J Clin Oncol. 1997;15(8):2807–2817. [DOI] [PubMed] [Google Scholar]
- 12.Megonigal MD, Cheung NK, Rappaport EF, et al. Detection of leukemia-associated MLL-GAS7 translocation early during chemotherapy with DNA topoisomerase II inhibitors. Proc Natl Acad Sci U S A. 2000;97(6):2814–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson BW, Cheung NK, Kolaris CP, et al. Prospective tracing of MLL-FRYL clone with low MEIS1 expression from emergence during neuroblastoma treatment to diagnosis of myelodysplastic syndrome. Blood. 2008;111(7):3802–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanco JG, Dervieux T, Edick MJ, et al. Molecular emergence of acute myeloid leukemia during treatment for acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2001;98(18):10338–10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Metzler M, Staege MS, Harder L, et al. Inv(11)(q21q23) fuses MLL to the Notch co-activator mastermind-like 2 in secondary T-cell acute lymphoblastic leukemia. Leukemia. 2008;22(9):1807–1811. [DOI] [PubMed] [Google Scholar]
- 16.Nemoto N, Suzukawa K, Shimizu S, et al. Identification of a novel fusion gene MLL-MAML2 in secondary acute myelogenous leukemia and myelodysplastic syndrome with inv(11)(q21q23). Genes Chromosomes Cancer. 2007;46(9):813–819. [DOI] [PubMed] [Google Scholar]
- 17.Rosen EM, Cassady JR, Frantz CN, Kretschmar CS, Levey R, Sallen SE. Stage IV-N: a favorable subset of children with metastatic neuroblastoma. Med Pediatr Oncol. 1985;13(4):194–198. [DOI] [PubMed] [Google Scholar]
- 18.Hijiya N, Ness KK, Ribeiro RC, Hudson MM. Acute leukemia as a secondary malignancy in children and adolescents: current findings and issues. Cancer. 2009;115(1):23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zehnbauer BA, Pardoll DM, Burke PJ, Graham ML, Vogelstein B. Immunoglobulin gene rearrangements in remission bone marrow specimens from patients with acute lymphoblastic leukemia. Blood. 1986;67(3):835–838. [PubMed] [Google Scholar]
- 20.Takahashi Y, Terui K, Chinen Y, et al. A pediatric case of secondary T-cell acute lymphoblastic leukemia with KMT2A-MAML2 developing after hepatoblastoma treatment. Pediatr Blood Cancer. 2019:e28033. [DOI] [PubMed] [Google Scholar]
- 21.Mariani RA, Silva M, Caparelli E, et al. Inv(11)(q21q23); KMT2A-MAML2, a Recurrent Genetic Abnormality in T-Cell Therapy-related Acute Lymphoblastic Leukemia. J Pediatr Hematol Oncol. 2019. [DOI] [PubMed] [Google Scholar]
- 22.Obama K, Furukawa Y, Tara M, Niina K. Secondary monocytic leukemia with rearrangement of the MLL gene occurring during the course of adult T-cell leukemia. Int J Hematol. 1998;68(3):323–326. [DOI] [PubMed] [Google Scholar]
- 23.Tang G, Lu X, Wang SA, et al. Homozygous inv(11)(q21q23) and MLL gene rearrangement in two patients with myeloid neoplasms. International journal of clinical and experimental pathology. 2014;7(6):3196–3201. [PMC free article] [PubMed] [Google Scholar]
- 24.Menu E, Beaufils N, Usseglio F, et al. First case of B ALL with KMT2A-MAML2 rearrangement: a case report. BMC Cancer. 2017;17(1):363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Massoth LR, Hung YP, Dias-Santagata D, et al. Pan-Cancer Landscape Analysis Reveals Recurrent KMT2A-MAML2 Gene Fusion in Aggressive Histologic Subtypes of Thymoma. JCO Precision Oncology. 2020(4):109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu L, Sun T, Kobayashi K, Gao P, Griffin JD. Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol Cell Biol. 2002;22(21):7688–7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tonon G, Modi S, Wu L, et al. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet. 2003;33(2):208–213. [DOI] [PubMed] [Google Scholar]
- 28.Yu X, Davenport JW, Urtishak KA, et al. Genome-wide TOP2A DNA cleavage is biased toward translocated and highly transcribed loci. Genome Res. 2017;27(7):1238–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colon NC, Chung DH. Neuroblastoma. Adv Pediatr. 2011;58(1):297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collord G, Park N, Podesta M, et al. Clonal haematopoiesis is not prevalent in survivors of childhood cancer. Br J Haematol. 2018;181(4):537–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Busque L, Ilaria R Jr., Tantravahi R, Weinstein H, Gilliland DG. Clonality analysis of childhood ALL in remission: no evidence of clonal hematopoiesis. Leuk Res. 1994;18(2):71–77. [DOI] [PubMed] [Google Scholar]
- 32.Teuffel O, Betts DR, Thali M, et al. Clonal expansion of a new MLL rearrangement in the absence of leukemia. Blood. 2005;105(10):4151–4152. [DOI] [PubMed] [Google Scholar]
- 33.Acunzo M, Romano G, Wernicke D, et al. Translocation t(2;11) in CLL cells results in CXCR4/MAML2 fusion oncogene. Blood. 2014;124(2):259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Behboudi A, Enlund F, Winnes M, et al. Molecular classification of mucoepidermoid carcinomas-prognostic significance of the MECT1-MAML2 fusion oncogene. Genes Chromosomes Cancer. 2006;45(5):470–481. [DOI] [PubMed] [Google Scholar]
- 35.Winnes M, Molne L, Suurkula M, et al. Frequent fusion of the CRTC1 and MAML2 genes in clear cell variants of cutaneous hidradenomas. Genes Chromosomes Cancer. 2007;46(6):559–563. [DOI] [PubMed] [Google Scholar]
- 36.Okabe M, Miyabe S, Nagatsuka H, et al. MECT1-MAML2 fusion transcript defines a favorable subset of mucoepidermoid carcinoma. Clin Cancer Res. 2006;12(13):3902–3907. [DOI] [PubMed] [Google Scholar]
- 37.Noda H, Okumura Y, Nakayama T, et al. Clinicopathological significance of MAML2 gene split in mucoepidermoid carcinoma. Cancer Sci. 2013;104(1):85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Enlund F, Behboudi A, Andren Y, et al. Altered Notch signaling resulting from expression of a WAMTP1-MAML2 gene fusion in mucoepidermoid carcinomas and benign Warthin’s tumors. Exp Cell Res. 2004;292(1):21–28. [DOI] [PubMed] [Google Scholar]
- 39.Tirado Y, Williams MD, Hanna EY, Kaye FJ, Batsakis JG, El-Naggar AK. CRTC1/MAML2 fusion transcript in high grade mucoepidermoid carcinomas of salivary and thyroid glands and Warthin’s tumors: implications for histogenesis and biologic behavior. Genes Chromosomes Cancer. 2007;46(7):708–715. [DOI] [PubMed] [Google Scholar]
- 40.Fehr A, Roser K, Belge G, Loning T, Bullerdiek J. A closer look at Warthin tumors and the t(11;19). Cancer Genet Cytogenet. 2008;180(2):135–139. [DOI] [PubMed] [Google Scholar]
- 41.Zhu F, Wang W, Hou Y, et al. MAML2 rearrangement in primary pulmonary mucoepidermoid carcinoma and the correlation with FLT1 expression. PLoS One. 2014;9(4):e94399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Behboudi A, Winnes M, Gorunova L, et al. Clear cell hidradenoma of the skin-a third tumor type with a t(11;19)--associated TORC1-MAML2 gene fusion. Genes Chromosomes Cancer. 2005;43(2):202–205. [DOI] [PubMed] [Google Scholar]
- 43.Gyarfas T, Wintgens J, Biskup W, et al. Transient spontaneous remission in congenital MLL-AF10 rearranged acute myeloid leukemia presenting with cardiorespiratory failure and meconium ileus. Molecular and cellular pediatrics. 2016;3(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muller-Schmah C, Solari L, Weis R, et al. Immune response as a possible mechanism of long-lasting disease control in spontaneous remission of MLL/AF9-positive acute myeloid leukemia. Ann Hematol. 2012;91(1):27–32. [DOI] [PubMed] [Google Scholar]
- 45.Chuk MK, McIntyre E, Small D, Brown P. Discordance of MLL-rearranged (MLL-R) infant acute lymphoblastic leukemia in monozygotic twins with spontaneous clearance of preleukemic clone in unaffected twin. Blood. 2009;113(26):6691–6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.D’Orazio JA, Pulliam JF, Moscow JA. Spontaneous resolution of a single lesion of myeloid leukemia cutis in an infant: case report and discussion. Pediatr Hematol Oncol. 2008;25(5):457–468. [DOI] [PubMed] [Google Scholar]
- 47.Hudecek M, Bartsch K, Jakel N, et al. Spontaneous remission of acute myeloid leukemia relapse after hematopoietic cell transplantation in a high-risk patient with 11q23/MLL abnormality. Acta Haematol. 2008;119(2):111–114. [DOI] [PubMed] [Google Scholar]
- 48.Muller CI, Trepel M, Kunzmann R, Lais A, Engelhardt R, Lubbert M. Hematologic and molecular spontaneous remission following sepsis in acute monoblastic leukemia with translocation (9;11): a case report and review of the literature. Eur J Haematol. 2004;73(1):62–66. [DOI] [PubMed] [Google Scholar]
- 49.Granzen B, Bernhard B, Reinisch I, Skopnik H, Mertens R. Transient myeloproliferative disorder with 11q23 aberration in two neonates with Down syndrome. Ann Hematol. 1998;77(1–2):51–54. [DOI] [PubMed] [Google Scholar]