

PURPOSE
Rhabdomyosarcomas (RMS) are rare neoplasms affecting children and young adults. Efforts to improve patient survival have been undermined by a lack of suitable disease markers. Plasma circulating tumor DNA (ctDNA) has shown promise as a potential minimally invasive biomarker and monitoring tool in other cancers; however, it remains underexplored in RMS. We aimed to determine the feasibility of identifying and quantifying ctDNA in plasma as a marker of disease burden and/or treatment response using blood samples from RMS mouse models and patients.
METHODS
We established mouse models of RMS and applied quantitative polymerase chain reaction (PCR) and droplet digital PCR (ddPCR) to detect ctDNA within the mouse plasma. Potential driver mutations, copy-number alterations, and DNA breakpoints associated with PAX3/7-FOXO1 gene fusions were identified in the RMS samples collected at diagnosis. Patient-matched plasma samples collected from 28 patients with RMS before, during, and after treatment were analyzed for the presence of ctDNA via ddPCR, panel sequencing, and/or whole-exome sequencing.
RESULTS
Human tumor-derived DNA was detectable in plasma samples from mouse models of RMS and correlated with tumor burden. In patients, ctDNA was detected in 14/18 pretreatment plasma samples with ddPCR and 7/7 cases assessed by sequencing. Levels of ctDNA at diagnosis were significantly higher in patients with unfavorable tumor sites, positive nodal status, and metastasis. In patients with serial plasma samples (n = 18), fluctuations in ctDNA levels corresponded to treatment response.
CONCLUSION
Comprehensive ctDNA analysis combining high sensitivity and throughput can identify key molecular drivers in RMS models and patients, suggesting potential as a minimally invasive biomarker. Preclinical assessment of treatments using mouse models and further patient testing through prospective clinical trials are now warranted.
INTRODUCTION
Rhabdomyosarcoma (RMS), the most common soft tissue sarcoma in children, is a major cause of pediatric cancer–related death.1 Outcomes for patients with high-risk or relapsed RMS remain particularly poor.2 There is an urgent need to develop accurate prognostic and predictive markers and monitoring tools that can better identify patients at risk of treatment failure. This knowledge can aid in treatment decision making and the identification of patients who may benefit from participation in trials of novel therapeutics.
CONTEXT
Key Objective
Although overall survival for children with rhabdomyosarcoma (RMS) has improved, patients with high-risk and refractory disease continue to experience poor outcomes. This international collaborative pilot study aimed to assess the feasibility of detecting and quantifying circulating tumor DNA (ctDNA) in mouse models of and patients with RMS and investigate its relationship with clinical variables and outcome.
Knowledge Generated
We provide evidence to suggest that ctDNA is a surrogate marker of tumor burden in animal models of RMS and demonstrate feasibility for detecting and quantifying ctDNA in serial plasma samples from patients with RMS via several approaches including whole-exome and targeted sequencing and droplet digital polymerase chain reaction.
Relevance
Our data indicate that ctDNA holds potential as a minimally invasive biomarker in RMS, providing evidence for its assessment in future preclinical animal models and prospective clinical trials.
Molecular profiling of RMS tumors has identified several oncogenic drivers that hold potential as disease biomarkers. Alveolar subtype neoplasms (aRMS) commonly harbor the chromosomal translocations t(2;13) (q35;q14) or t(1;13) (p36;q14), which result in a fusion between the genes FOXO1 and PAX3 or PAX7, respectively.3,4 Crucially, PAX3-FOXO1 fusions are associated with an unfavorable patient prognosis.2,5 By contrast, embryonal RMS (eRMS) is characterized by mutations to key members of the AKT-PI3K and RAS pathways, some of which are predictive for response to certain molecular therapies.6 RMS can also carry copy-number variants such as amplifications of the CDK4 and MYCN genes.6 Hence, there is increasing evidence to support the screening of RMS for clinically relevant molecular alterations.
Recent research has focused on the assessment of blood-based biomarkers, such as cell-free DNA (cfDNA) and its malignant counterpart circulating tumor DNA (ctDNA), as a minimally invasive modality for tumor molecular profiling. This liquid biopsy approach is advantageous over tissue biopsies as it can provide a dynamic measurement of tumor activity in real time, allowing patient response to treatment to be monitored throughout their disease. Many studies have demonstrated the feasibility of using ctDNA for the diagnosis, prognosis, and monitoring of adult cancers.7 However, the assessment of ctDNA in pediatric patients with RMS has thus far been limited. Quantitative polymerase chain reaction (qPCR), targeted sequencing panels, and whole-genome sequencing have previously been used to detect the PAX3-FOXO1 gene fusion in ctDNA from a limited number of patients with aRMS.8-10 However, more evidence is needed (particularly for eRMS or fusion-negative patients) to support the clinical utility of ctDNA in this tumor type.
In this large international collaborative study, we applied several techniques including panel sequencing, whole-exome sequencing (WES), qPCR, and droplet digital PCR (ddPCR) to identify molecular drivers in pediatric RMS and quantify ctDNA in RMS patients and models.
METHODS
Animal Experiments
Three patient-derived xenografts (PDX) were established by implanting RMS patient tumor biopsy samples in immunodeficient non scid gamma (NSG) mice, as previously described (Data Supplement for PDX characteristics).11 For the aRMS PDX experiments, dissociated tumor cells from established xenografts were expanded in culture and labeled with enhanced green fluorescent protein (EGFP) (Data Supplement). One million IC-pPDX-104 EGFP or IC-pPDX-29 EGFP cells were injected orthotopically into the hind limb muscle of seven and five NSG mice, respectively. Tumor size was measured 3 times per week using calipers. Blood (100 μL) was collected via the lateral tail vein every week in IC-pPDX-29–injected mice and from the day tumors started to be visible in IC-pPDX-104–injected mice (day 32) until the end point of the experiment, upon which the mice were anesthetized with a lethal dose of ketamine-xylazine and 250-1,000 μL blood was collected through cardiac puncture. Plasma ctDNA and cfDNA were measured by SYBR Green-based qPCR using hLINE-1 and mPtger2 primer sets, respectively (Data Supplement). For the eRMS PDX experiments in ICR-PDX-RMS008, blood was collected from NSG mice during routine passaging of PDX tumor pieces. These pieces were implanted bilaterally in five NSG mice, with four mice developing tumors and one mouse no tumors. Blood (230-550 μL) was collected through cardiac puncture after lethal anesthetic. Tumor-specific variants in cfDNA were quantified with ddPCR.
Patients and Samples
Blood and tissue samples were obtained from pediatric cancer patients (n = 48) with RMS according to institutional review board–approved protocols. To be included in the study, subjects had to be between age 0 and 18 years with a pathologic diagnosis of RMS. There were no exclusion criteria. Samples were collected after obtaining written informed consent from patients, parents, or legal guardians. Participating institutions included Bambino Gesù Children's Hospital, Rome (protocol number 578); University-Hospital, Padova (4115/AO/17); Institut Curie, Paris (ClinicalTrials.gov identifier: NCT02546453); University Children's Hospital, Zurich (2020-01609); Princess Máxima Centre for Pediatric Oncology, Utrecht (METC2006-148 and PMCLAB2019-053); and The Institute of Cancer Research/Royal Marsden Hospital, London (13/LO/0254, 15/LO/0719 and 18/LO/1860).
Plasma was separated from blood collected in EDTA and DNA extracted from patient's plasma, and fresh, cultured, or formalin-fixed paraffin-embedded tumor tissue according to local standard operating procedures (Data Supplement). Targeted locus amplification, WES, and targeted sequencing with two custom sequencing panels were performed on patient tumor DNA and germline DNA (where available) to identify patient-specific genetic variants of interest (Data Supplement).12
ddPCR
Patient and ICR-PDX-RMS008 cfDNA were assessed for the presence of tumor-specific genetic variants by ddPCR, which was performed on the Bio-Rad QX200 ddPCR system as per manufacturer's instructions (Data Supplement). Plasma ctDNA and cfDNA were measured by assays targeting tumor-specific variants and reference genes, respectively (Data Supplement).
Targeted Sequencing
Baseline cfDNA samples from seven cases with sufficient DNA (10 ng) were analyzed by WES alongside patient-matched germline DNA and tumor DNA from fresh-frozen material, as previously described (Data Supplement).13 Serial cfDNA samples were also sequenced with a targeted sequencing panel that was designed to encompass 196 single-nucleotide variants (SNVs), corresponding to all SNVs observed in WES sequencing and 44 single-nucleotide polymorphisms to identify each sample. Libraries of cfDNA were constructed using a double-capture procedure. Samples were multiplexed for the capture and sequenced with HiSeq reagents (Illumina, Cambridgeshire, UK; expected coverage: 5,000×). Variants were filtered according to an established bioinformatic pipeline.13 For serial plasma samples, variants with < 10 supporting reads were excluded from the final data set.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism v9.0 (GraphPad Software). Pearson's correlation was performed to assess the relationship between mouse plasma ctDNA levels and tumor size or weight. To test whether detection of ctDNA at baseline was associated with clinical features such as tumor size, a two-sided Fisher's exact test was used. A two-tailed Mann-Whitney U test was used to verify the hypothesis that patients with tumors in an unfavorable site (favorable tumor sites include the biliary tract, orbit, head and neck [excluding parameningeal sites] and the genitourinary tract [excluding bladder and prostate]; unfavorable tumor sites are those arising in all other anatomical locations, including [but not limited to] parameningeal sites, the bladder or prostate, and extremities), nodal spread, or metastases had higher pretreatment ctDNA levels than those who did not. The results were considered statistically significant when P < .05.
RESULTS
CtDNA Can Be Detected in Animal Models of RMS and Correlates With Tumor Burden
In PDX models, human tumor DNA can be easily discriminated from host mouse DNA by targeting human-specific or tumor-specific sequences such as the chromosomal translocation PAX3-FOXO1 breakpoint or SNVs. Using serial dilutions of human tumor DNA and mouse plasma cfDNA, we established that hLINE-1 primers were optimal for detecting human DNA and mPtger2 for identifying mouse DNA in aRMS models (Data Supplement). We then tested whether ctDNA could be found in the blood of mice transplanted with aRMS PDXs. Blood samples were collected weekly until mice reached maximal tumor size (Fig 1A). Plasma ctDNA and cfDNA levels were quantified with hLINE-1 and mPtger2 primer sets, respectively. At the earliest time points after tumor injection, ctDNA was detected in only a fraction of the animals, but detection rates increased to 100% at later time points (Figs 1B and 1C). Similar to tumor volumes, ctDNA levels increased during the course of the experiment and ranged from nondetectable up to 25.3 + 2.0 ng/mL blood in IC-pPDX-29 (Fig 1D), and 17.7 ± 2.3 ng/mL blood in IC-pPDX-104 (Fig 1E). A significantly positive Pearson correlation was observed between ctDNA and tumor volume in both aRMS PDXs (Figs 1F and 1G). Importantly, no significant correlation was observed between tumor volume and cfDNA (Figs 1H and 1I), whose levels remained relatively stable during the entire course of the experiment (IC-pPDX-29: 33.9 ± 3.8 ng/mL blood; IC-pPDX-104: 14.4 ± 3.1 ng/mL blood). In the eRMS PDX, tumor-specific variants (Data Supplement) were identified in all four cfDNA samples from tumor-bearing mice, whereas the plasma sample from the mouse that did not grow a tumor had no detectable ctDNA (Data Supplement). These results demonstrate feasibility to detecting human ctDNA in mouse models of RMS and using ctDNA as a marker to monitor tumor growth, providing the rationale for moving forward with patients' samples.
FIG 1.

ctDNA correlates with tumor burden in RMS PDX models. (A) Experimental design. After orthotopic PDX injection, blood was collected weekly until the end point of the experiment. Plasma ctDNA was measured by qPCR using hLINE-1 primer sets; nontumor cfDNA was quantified with mPtger2 primer set. Detection rate of plasma ctDNA at different time points after tumor injection of IC-pPDX-29 (B) or ICpPDX-104 cells (C). The number of mice at the selected time points is indicated. Monitoring of ctDNA concentration and tumor volume over time in mice injected with (D) IC-pPDX-29 or (E) ICpPDX-104 cells. Tumor volume was measured 3 times a week, whereas plasma ctDNA was measured at the selected time points. Data are represented as mean ± SEM of n ≥ 2 animals and connected with an exponential growth curve fit. Correlation between tumor volume and plasma (F and G) ctDNA or (H and I) cfDNA in mice injected with (F and H) IC-pPDX-29 or (G and I) ICpPDX-104 cells. Data points are interpolated with a linear regression. Correlation coefficient (R2), statistical significance (P), and number of data points (n) are indicated. cfDNA, cell-free DNA; ctDNA, circulating tumor DNA; ND, nondetectable; PCR, polymerase chain reaction; PDX, patient-derived xenograft; qPCR, quantitative PCR; RMS, rhabdomyosarcoma.
Patient Cohort
A summary of the samples collected and successfully analyzed is illustrated in Figure 2A. Of the 48 patients, 28 had targetable tumor variants and sufficient cfDNA to analyze (see Tables 1 and 2 for clinical characteristics). Baseline plasma samples (collected at diagnosis) were available for 25/28 (89%) patients (20 frontline and five relapse), whereas serial plasma samples collected during treatment (mean 4, range 2-7) were available for 18/28 (64%) patients (17 frontline and one relapse).
FIG 2.
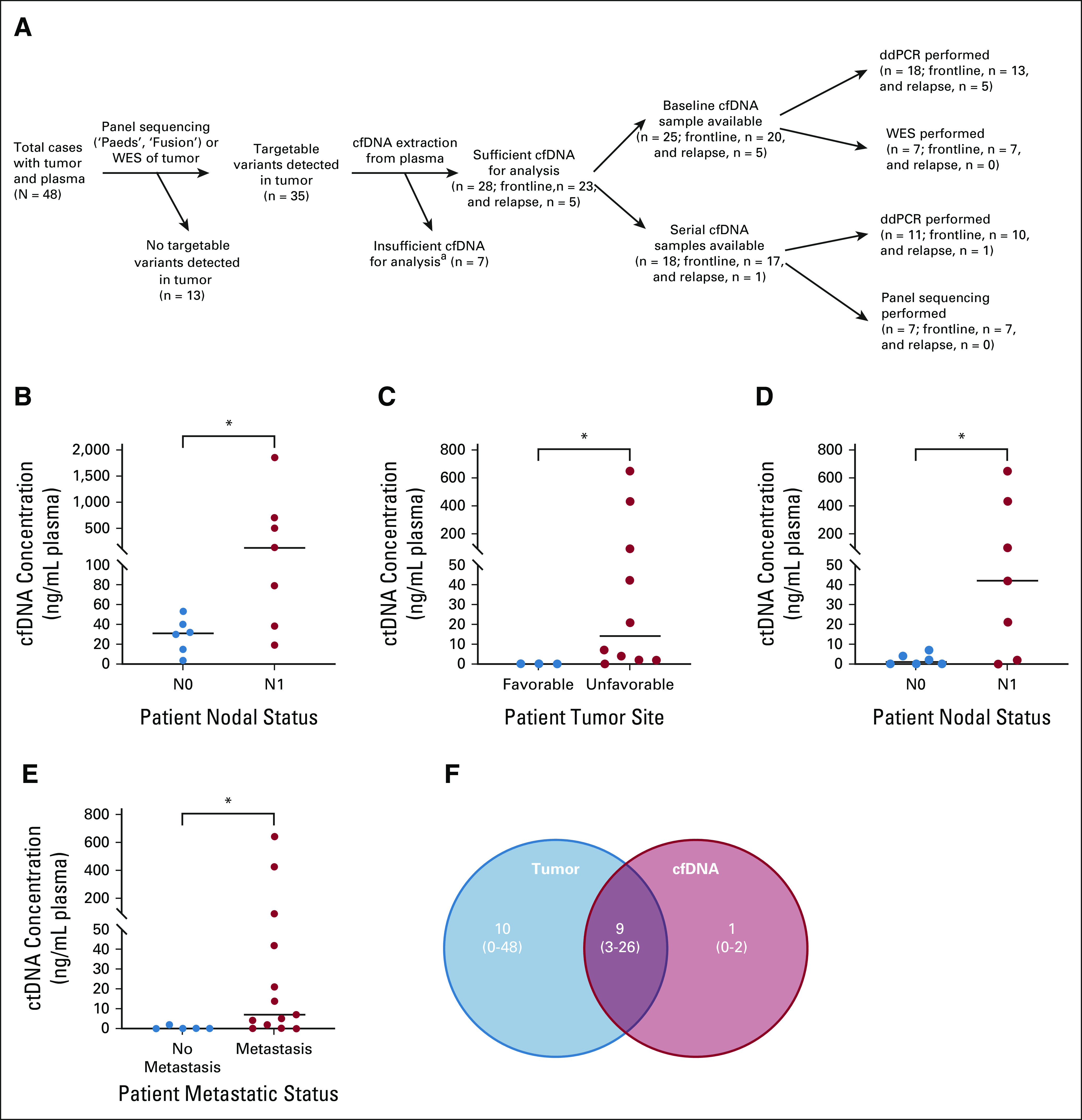
Baseline cfDNA analysis. (A) Overview of samples collected (n = 48 tumor and plasma) and successfully analyzed (n = 28 cfDNA). aInsufficient cfDNA yield is defined as < 1 ng for ddPCR and < 10 ng for WES/panel sequencing. (B) Frontline patients with nodal spread (N1, n = 7) had significantly higher baseline cfDNA yields (ng/mL plasma) compared with those without it (N0, n = 6; P = .035). Baseline ctDNA yields (ng/mL plasma) were significantly higher in frontline patients with (C) tumors in an unfavorable site (n = 10) compared with those with tumors in a favorable site (n = 3; P = .0210) and (D) nodal involvement (N1, n = 7) compared with those without it (N0, n = 6; P = .043). (E) Baseline ctDNA yields (ng/mL plasma) were significantly higher in both frontline and relapsed patients with metastases at diagnosis (n = 13) compared with those without it (n = 5; P = .0201). Median cfDNA or ctDNA yields indicated by horizontal lines on graphs. (F) There was considerable overlap in the molecular profile of matched patient tumor DNA and baseline cfDNA, as illustrated by mean number (and ranges) of variants detected in each via WES. cfDNA, cell-free DNA; ctDNA, circulating tumor DNA; ddPCR, droplet digital polymerase chain reaction.
TABLE 1.
Clinical Characteristics of Patients With Frontline Rhabdomyosarcoma Included in the Study
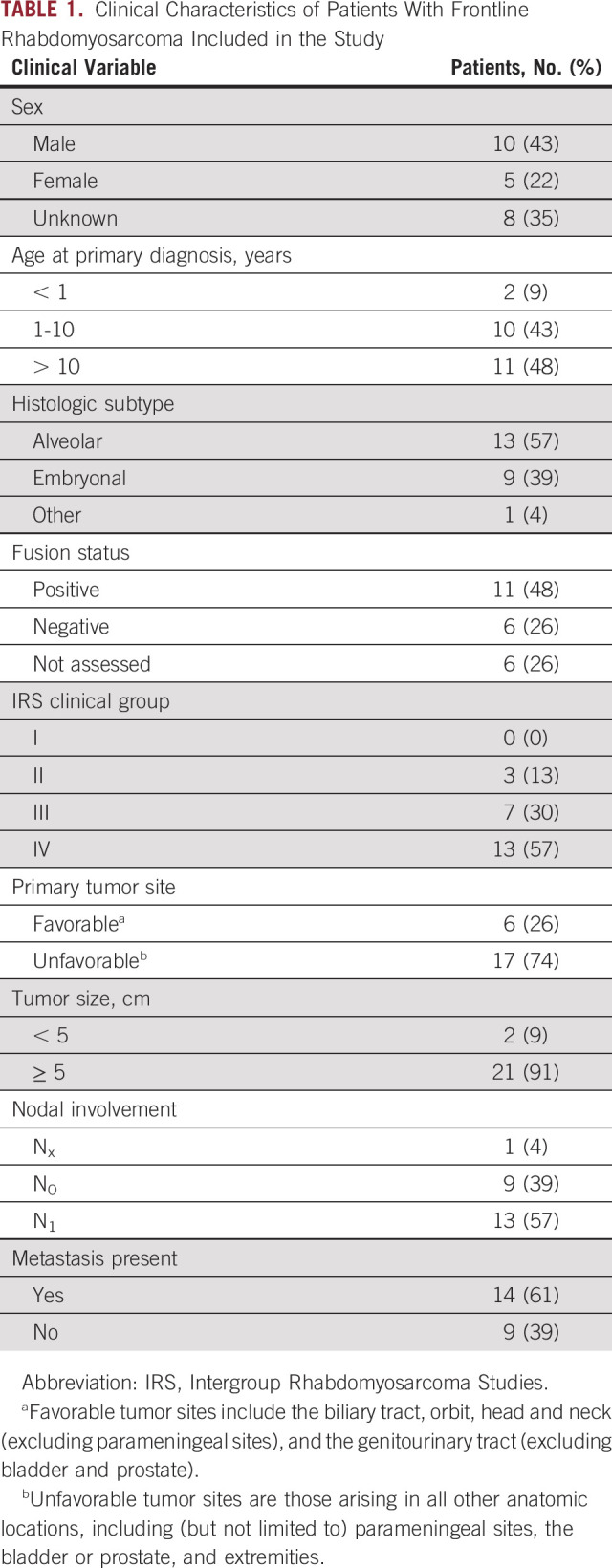
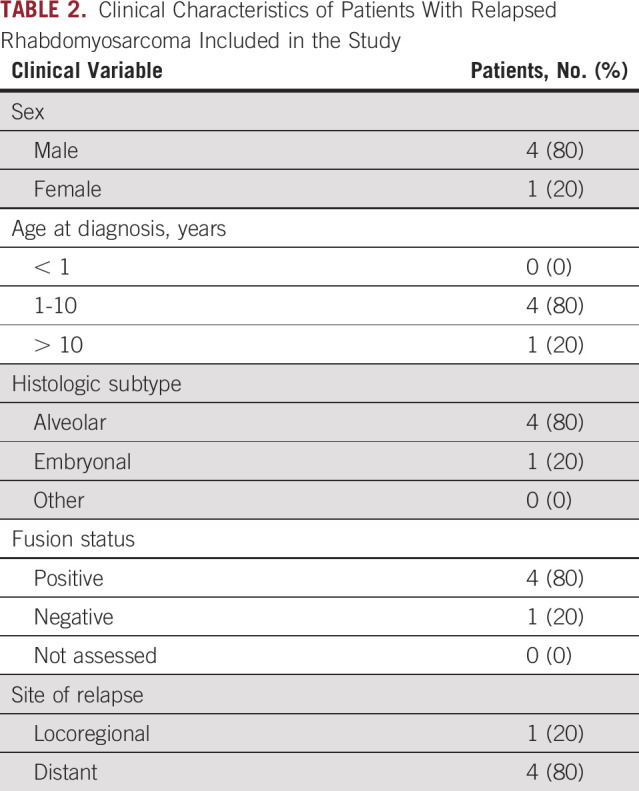
TABLE 2.
Clinical Characteristics of Patients With Relapsed Rhabdomyosarcoma Included in the Study
CtDNA Can Be Detected in Baseline Plasma Samples by ddPCR and Is Associated With Clinical Features in Patients With RMS
Across all baseline plasma samples assessed by ddPCR (n = 18), the median total cfDNA yield was 38.2 ng/mL plasma (range 3.9-1,857.5 ng/mL). Patients with nodal spread (N1) had significantly higher baseline cfDNA compared with those without it (N0; P = .035; Fig 2B), but there was no significant association between plasma cfDNA levels and characteristics such as tumor size, histology, site, or patient clinical risk group.
A tumor-specific variant was detected in 14/18 baseline samples, demonstrating 78% concordance with tumor tissue (Table 3). A patient-specific PAX3/7-FOXO1 fusion was exhibited in 10/11 (91%) baseline cfDNA samples from fusion-positive patients, whereas mutations and copy-number variants were seen in 3 of 5 (60%) and 1 of 2 (50%) patients, respectively.
TABLE 3.
Tumor-Specific Variants Detected in Patient Baseline cfDNA by Droplet Digital Polymerase Chain Reaction

Baseline ctDNA levels were significantly higher in frontline patients with an unfavorable tumor site and positive nodal status (mean = 0, median = 0 v mean = 124.9, median = 13.9 ng/mL plasma for favorable v unfavorable, P = .021, Fig 2C; and mean = 2.2, median = 1.1 v mean = 176.5, median = 41.6 ng/mL plasma for N0 versus N1, P = .043, Fig 2D). Both frontline and relapsed patients with metastasis at diagnosis had significantly higher ctDNA levels at baseline (mean = 97.3, median = 6.6 ng/mL plasma) compared with those without it (mean = 0.5, median = 0 ng/mL plasma, P = .0201, Fig 2E). These results support the utility of ddPCR for the detection of ctDNA in patients with RMS and suggest that diagnostic ctDNA levels are related to disease aggressiveness.
The Molecular Profile of Baseline ctDNA Demonstrates Concordance With That of the Primary Tumor in Frontline RMS Patients
To more comprehensively assess the extent to which the genomic landscape of patient ctDNA reflects that of the primary tumor, we performed WES on seven patients with matched tumor, germline, and baseline cfDNA. ctDNA was detected in all (100%) baseline plasma samples. A mean of nine SNVs per case were common to both the baseline cfDNA and primary tumor (range 3-26 SNVs), with a mean of 1 (range 0-2) SNV detected only in the cfDNA, and a mean of 10 SNVs (range 0-48) seen only in the tumor (Fig 2F). The latter were mainly observed in eRMS. These data demonstrate that patient ctDNA collected at the time of diagnosis largely reflects the molecular profile of the tumor in RMS, and that WES of cfDNA is a useful tool for highlighting variants that may not have been sampled in the tissue biopsy.
CtDNA Levels Reflect the Disease Burden in Patients With RMS Over Time
In cases where serial plasma samples were available (n = 18), we used ddPCR or panel sequencing to track tumor variants over the course of patient treatment. In most of these patients, ctDNA levels decreased after the onset of chemotherapy and remained stable, corresponding with favorable response to therapy (Fig 3A and Data Supplement). However, there were three patients in whom ctDNA was detectable at various time points after treatment commenced, which coincided with disease progression or relapse (Figs 3B-3D). These results provide evidence to support the notion that ctDNA can act as a surrogate marker for disease aggressiveness in patients with RMS and suggest that ctDNA levels reflect patient response to treatment.
FIG 3.
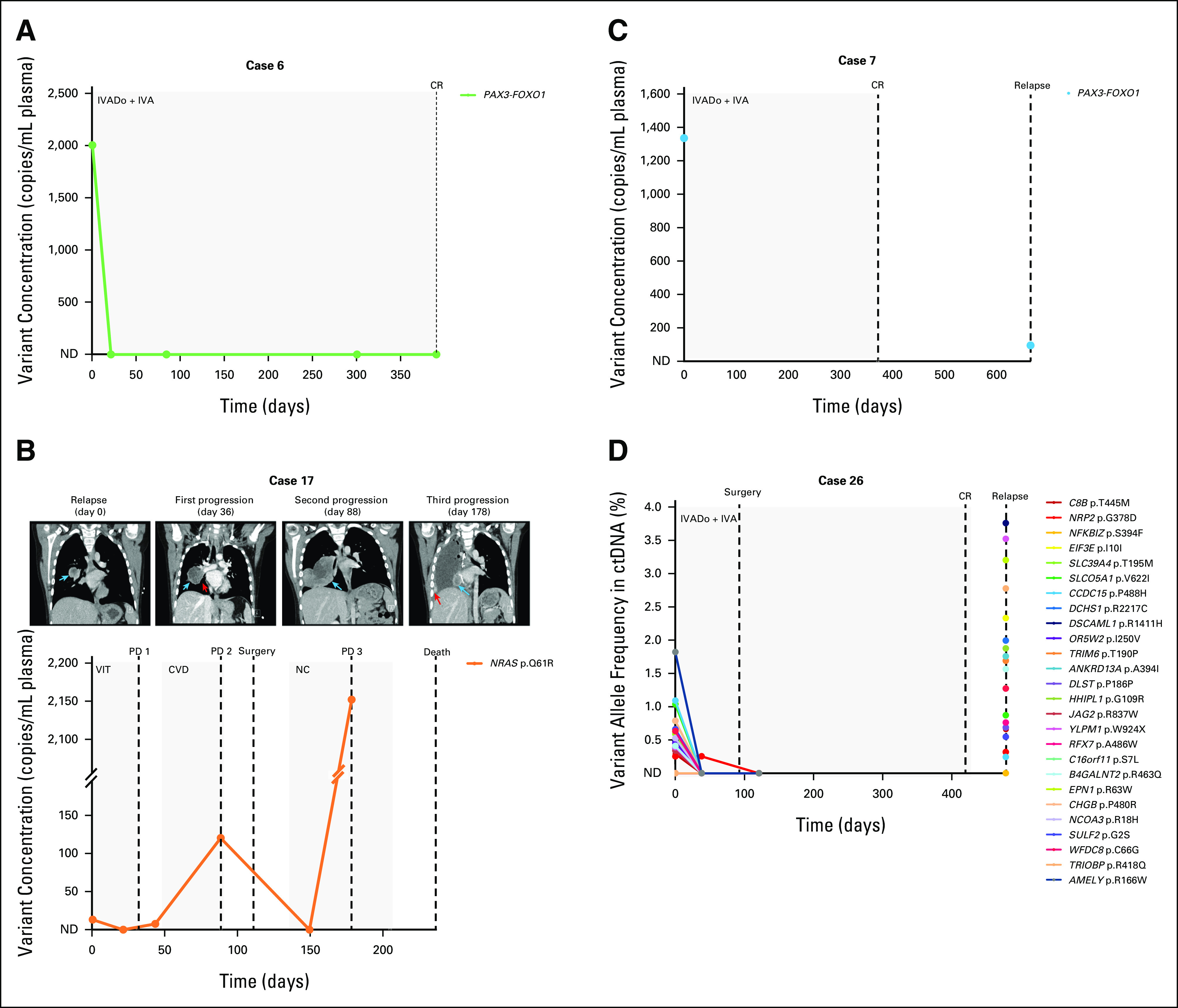
Patient ctDNA levels reflect disease burden over time. (A) A PAX3-FOXO1 rearrangement in the pretreatment ctDNA of a frontline patient with aRMS became undetectable via ddPCR in plasma samples collected during chemotherapy. The patient ended therapy with a complete response. (B) Plasma levels of an NRAS variant in a relapsed eRMS patient with pulmonary metastasis (day 0 CT image, blue arrow) initially decreased after initiation of chemotherapy but increased as the patient's neoplasm enlarged (see enlarged nodule indicated by blue arrow and narrowing of right lower bronchus indicated by red arrow, CT images at days 36 and 88, respectively). The patient was deemed to have disease progression according to the RECIST 1.1.24 Following surgery and adjuvant chemotherapy, ctDNA became undetectable via ddPCR, but a subsequent plasma sample illustrated a re-emergence of the variant, coinciding with further progression in the patient (new pulmonary metastasis in the surgical bed denoted by blue arrow and scattered vascularized ipsilateral pleural deposits indicated by red arrow in CT image day 178; note the broken y-axis of the graph). The patient died 2 months later. (C) A PAX3-FOXO1 fusion initially identified in the pretreatment ctDNA of a patient with frontline aRMS was also detected in a ctDNA sample collected at time of relapse via ddPCR, albeit at a lower concentration. (D) Targeted sequencing of cfDNA from an aRMS patient illustrates an initial response to frontline treatment, as evidenced by decreasing variant allele frequencies (%) in serial plasma samples. However, ctDNA was detected in a blood sample collected 2 months after the completion of treatment, coinciding with clinical relapse. Day 0 for all patients is the day that the pretreatment blood sample was collected. Dots on the line graph correspond to the days in which plasma samples were obtained. Gray boxes indicate the chemotherapy duration. Dashed lines indicate clinical time point (surgery, response as assessed on imaging). cfDNA, cell-free DNA; CR, complete response; CT, computed tomography; ctDNA, circulating tumor DNA; CVD, cyclophosphamide, vincristine, and doxorubicin; ddPCR, droplet digital polymerase chain reaction; eRMS, embryonal RMS; IVA, ifosfamide, vincristine, and actinomycin D; IVADo, ifosfamide, vincristine, actinomycin D, and doxorubicin; NC, navelbine (vinorelbine) and cyclophosphamide; ND, not detected; PD, progressive disease; RMS, rhabdomyosarcoma; VIT, vincristine, irinotecan, and temozolomide.
DISCUSSION
Analysis of ctDNA is rapidly being introduced into the clinic for the diagnosis, prognosis, and monitoring of adult patients with cancer.7 However, its utility for pediatric cancers is yet to be fully realized. In this study, we aimed to assess the feasibility of detecting and quantifying plasma ctDNA in pediatric RMS. Using techniques offering high sensitivity (such as qPCR and ddPCR) and multiplexing of targets (whole-exome and panel sequencing), we have demonstrated that we can detect molecular markers in cfDNA from RMS animal models and patients, including variants of clinical significance, such as PAX3-FOXO1 fusions and MYOD1 mutations.5,6 The detection of mutations is of particular importance, as ctDNA studies of RMS have focused on identifying gene fusions with little evidence for detection of ctDNA in fusion-negative patients.8-10,14 In this study, we have also developed a custom sequencing panel, suitable for formalin-fixed paraffin-embedded tissue, to define the unique PAX3/7-FOXO1 DNA breakpoints. This is more practical for clinical implementation than a requirement for fresh-frozen material.
The sensitivity for detection of ctDNA in diagnostic patient plasma samples (78% and 100% for ddPCR and WES, respectively) was on par with that of previous studies in pediatric sarcomas.9,10,14-17 Interestingly, all three frontline patients in whom baseline ctDNA could not be detected by ddPCR had tumors in a favorable anatomic site (genitourinary tract, excluding the bladder and prostate) and were fusion-negative, which are both positive survival indicators in RMS.2 Two of the three patients had their tumors resected before collecting baseline blood samples, which explains why no ctDNA could be found in them. However, the fourth subject who was ctDNA-negative at baseline had a locoregional recurrence, which is generally associated with longer survival compared with distant relapse.18 This suggests that ctDNA detection at diagnosis may be linked to disease aggressiveness in RMS, although survival data were not available for all patients to test this hypothesis. A recent study by members of our group found that the presence of circulating tumor cells in blood and bone marrow, as detected by an RMS-specific RNA panel at diagnosis, was negatively associated with survival in patients with RMS.19 The identification of novel prognostic markers, such as ctDNA and circulating tumor cells at diagnosis, has the potential to further improve risk stratification for children with RMS, and thus, it will be of great value to assess the prognostic significance of these in future clinical studies.
Plasma ctDNA concentration correlated with tumor size in animal models, suggesting that analysis of ctDNA from models may prove useful for real-time assessment of tumor response to treatment. We believe this approach will better enable the RMS research community to conduct preclinical and coclinical testing of personalized therapies that have the potential to improve patient outcomes. In patients, baseline ctDNA levels were higher in those with advanced disease, supporting the notion that ctDNA acts as a surrogate measure of disease status and, thus, as a minimally invasive biomarker for RMS. This contrasts with nontumor cfDNA levels, which did not correlate with tumor burden in animal models, and was only associated with nodal status in frontline patients (possibly because of increased inflammation, a known trigger of cfDNA release, in cancer-infiltrated lymph nodes).20 Although every effort was made to process blood and extract cfDNA in such a way as to minimize cell lysis and enrich for fragmented DNA, we cannot exclude the possibility of contamination with high-molecular-weight DNA.21 Furthermore, blood collection for this study was only performed ad hoc, resulting in a small sample size, which limits the power of our statistical analysis. As such, these results should be validated in a larger cohort with standardized collection procedures.
We have also provided evidence to support serial monitoring of ctDNA in patients with RMS using both ddPCR and targeted sequencing, alongside current tools such as imaging. Changes in ctDNA levels corresponded to changes in disease burden and are consistent with the frequent initial responsiveness of RMS to current treatments.1 We were also able to detect ctDNA at the time of disease relapse in three patients, indicating that ctDNA analysis has utility in the follow-up of patients after completion of frontline treatment. In this study, ctDNA was collected when relapse was clinically apparent. Future prospective studies will be required to determine whether ctDNA is detectable before imaging modalities and/or onset of disease symptoms in relapse patients, and whether earlier detection and treatment of relapse provides a survival benefit.
We initially used ddPCR for detection of ctDNA as it affords high sensitivity (down to 0.03% frequency in some assays) and absolute quantification of target molecules, enabling direct comparison among serial cfDNA samples. We found it ideal for cases with only one variant (eg, PAX3/7-FOXO1 fusions); however, its capacity for multiplexing targets is limited. In cases with matched fresh-frozen tumor tissue and serial plasma, we instead performed targeted sequencing to assess ctDNA. This allowed for longitudinal monitoring of tumor evolution across multiple genomic targets and the identification of potential treatment-resistant variants that may have been unsampled or below the level of detection in the tumor biopsy, or which arose during therapy. As such, sequencing approaches to monitor ctDNA may be more appropriate for patients who have more than one driver mutation, although for some cases, there were several variants that could not be detected in the ctDNA via WES. Future studies will explore the use of approaches such as ultra-deep panel sequencing for detection of rare variants and/or minimal residual disease.22
Limited starting material can also impact upon the test sensitivity, particularly in pediatric cancers, where blood volumes (and resulting cfDNA yields) may be very small.23 We excluded cases with < 1 ng cfDNA for ddPCR, and < 10 ng for sequencing. However, it is possible that some low-input samples may have generated false-negative results because of limited amplification of target molecules. As such, caution in the interpretation of these results and consideration of other patient variables will be required for clinical application.
In summary, we have demonstrated that we can detect tumor-specific variants in the plasma of children with both aRMS and eRMS, and have provided preliminary evidence for the use of ctDNA to monitor disease burden in these patients. We believe that this approach warrants further investigation in the context of large-scale prospective clinical trials, such as the international Frontline and Relapsed Rhabdomyosarcoma study (ClinicalTrials.gov identifier: NCT04625907).
ACKNOWLEDGMENT
The authors would like to thank all patients and their families for their participation in this study. The authors are also grateful to Harma Feitsma, Ellen Stelloo, Irina Sergeeva, and Max van Min from Cergentis BV (Utrecht, The Netherlands) for performing the TLA procedure, and to Vladimir Kirkin and Gary Box for helping to create the ICR-PDX-RMS008 mouse model and collect blood samples.
Susanne A. Gatz
Consulting or Advisory Role: TESARO, Bayer
Travel, Accommodations, Expenses: AstraZeneca
Julia C. Chisholm
Consulting or Advisory Role: Roche, Bayer, Roche/Genentech, Bayer
Research Funding: Bayer (Inst)
Other Relationship: National Cancer Research Institute—Chair of Children's Research Group, Children's Oncology Group, National Cancer Institute
Isabelle Aerts
Consulting or Advisory Role: AstraZeneca
Travel, Accommodations, Expenses: Jazz Pharmaceuticals
Anna Kelsey
Travel, Accommodations, Expenses: Bayer
Michael Hubank
Honoraria: Boehringer Ingelheim, Incyte (Inst), Qiagen
Consulting or Advisory Role: Guardant Health, Illumina, AstraZeneca, Bristol Myers Squibb Foundation, Bayer, Roche, Lilly, Amgen, Merck Serono, Kuchar, Janssen, Novartis
Gianni Bisogno
Consulting or Advisory Role: Bayer
Gudrun Schleiermacher
Honoraria: BMS
Research Funding: Bristol Myers Squibb (Inst), Pfizer (Inst), MSDavenir (Inst), Roche (Inst)
Travel, Accommodations, Expenses: Roche
No other potential conflicts of interest were reported.
DISCLAIMER
The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
SUPPORT
This project was carried out with the support of the KickCancer Fund, managed by the King Baudouin Foundation and Innovative Therapies for Children with Cancer. O.R. was supported by the children's cancer charity Alice's Arc. N.L. and J.St. were supported by KiKa (Children Cancer Free), grant number 312. S.G.D. received support from the Swiss National Science Foundation grant 31003A_175558 (to B.W.S.). V.D.P. was supported by Fondazione Umberto Veronesi. L.T. was supported by Peter Pan Onlus (Bolzen, Italy). E.I. was supported by Christopher's Smile, the National Institute of Health Research (NIHR) Royal Marsden Biomedical Research Centre (BRC). E.P. is supported by The Royal Marsden Cancer Charity.
This work represents independent research supported by the National Institute for Health Research (NIHR) Biomedical Research Centre at The Royal Marsden NHS Foundation Trust and the Institute of Cancer Research, London (J.C.C., A.W., S.L.G., E.P).
AUTHOR CONTRIBUTIONS
Conception and design: Olivia Ruhen, Nathalie S.M. Lak, Sara G. Danielli, Paul Carter, Elisa Izquierdo, Gianni Bisogno, Angela Di Giannatale, Gudrun Schleiermacher, Beat W. Schäfer, Godelieve A.M. Tytgat, Janet Shipley
Financial support: Louis Chesler, Janet Shipley
Administrative support: Nathalie S.M. Lak, Godelieve A.M. Tytgat
Provision of study materials or patients: Nathalie S.M. Lak, Janine Stutterheim, Lucia Tombolan, Susanne A. Gatz, Julia C. Chisholm, Sally L. George, Louis Chesler, Isabelle Aerts, Gaelle Pierron, Sakina Zaidi, Olivier Delattre, Didier Surdez, Anna Kelsey, Paolo Bonvini, Gianni Bisogno, Angela Di Giannatale, Gudrun Schleiermacher, Godelieve A.M. Tytgat, Janet Shipley
Collection and assembly of data: Olivia Ruhen, Nathalie S.M. Lak, Janine Stutterheim, Sara G. Danielli, Mathieu Chicard, Virginia Di Paolo, Lucia Tombolan, Ewa Aladowicz, Paula Proszek, Sabri Jamal, Reda Stankunaite, Deborah Hughes, Ajla Wasti, Sally L. George, Erika Pace, Louis Chesler, Isabelle Aerts, Gaelle Pierron, Sakina Zaidi, Olivier Delattre, Didier Surdez, Michael Hubank, Paolo Bonvini, Angela Di Giannatale, Gudrun Schleiermacher, Godelieve A.M. Tytgat, Janet Shipley
Data analysis and interpretation: Olivia Ruhen, Nathalie S.M. Lak, Sara G. Danielli, Yasmine Iddir, Alexandra Saint-Charles, Susanne A. Gatz, Paula Proszek, Sabri Jamal, Deborah Hughes, Julia C. Chisholm, Sally L. George, Louis Chesler, Anna Kelsey, Gianni Bisogno, Angela Di Giannatale, Gudrun Schleiermacher, Beat W. Schäfer, Godelieve A.M. Tytgat, Janet Shipley
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Susanne A. Gatz
Consulting or Advisory Role: TESARO, Bayer
Travel, Accommodations, Expenses: AstraZeneca
Julia C. Chisholm
Consulting or Advisory Role: Roche, Bayer, Roche/Genentech, Bayer
Research Funding: Bayer (Inst)
Other Relationship: National Cancer Research Institute—Chair of Children's Research Group, Children's Oncology Group, National Cancer Institute
Isabelle Aerts
Consulting or Advisory Role: AstraZeneca
Travel, Accommodations, Expenses: Jazz Pharmaceuticals
Anna Kelsey
Travel, Accommodations, Expenses: Bayer
Michael Hubank
Honoraria: Boehringer Ingelheim, Incyte (Inst), Qiagen
Consulting or Advisory Role: Guardant Health, Illumina, AstraZeneca, Bristol Myers Squibb Foundation, Bayer, Roche, Lilly, Amgen, Merck Serono, Kuchar, Janssen, Novartis
Gianni Bisogno
Consulting or Advisory Role: Bayer
Gudrun Schleiermacher
Honoraria: BMS
Research Funding: Bristol Myers Squibb (Inst), Pfizer (Inst), MSDavenir (Inst), Roche (Inst)
Travel, Accommodations, Expenses: Roche
No other potential conflicts of interest were reported.
REFERENCES
- 1.Skapek SX, Ferrari A, Gupta AA, et al. : Rhabdomyosarcoma. Nat Rev Dis Primers 5:1, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hibbitts E, Chi YY, Hawkins DS, et al. : Refinement of risk stratification for childhood rhabdomyosarcoma using FOXO1 fusion status in addition to established clinical outcome predictors: A report from the Children's Oncology Group. Cancer Med 8:6437-6448, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barr FG, Galili N, Holick J, et al. : Rearrangement of the PAX3 paired box gene in the pediatric solid tumor alveolar rhabdomyosarcoma. Nat Genet 3:113-117, 1993 [DOI] [PubMed] [Google Scholar]
- 4.Davis RJ, D'Cruz CM, Lovell MA, et al. : Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res 54:2869-2872, 1994 [PubMed] [Google Scholar]
- 5.Missiaglia E, Williamson D, Chisholm J, et al. : PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol 30:1670-1677, 2012 [DOI] [PubMed] [Google Scholar]
- 6.Shern JF, Selfe J, Izquierdo E, et al. : Genomic classification and clinical outcome in rhabdomyosarcoma: A report from an International Consortium. J Clin Oncol 39:2859-2871, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cescon DW, Bratman SV, Chan SM, Siu LL: Circulating tumor DNA and liquid biopsy in oncology. Nat Cancer 1:276-290, 2020 [DOI] [PubMed] [Google Scholar]
- 8.Eguchi-Ishimae M, Tezuka M, Kokeguchi T, et al. : Early detection of the PAX3-FOXO1 fusion gene in circulating tumor-derived DNA in a case of alveolar rhabdomyosarcoma. Genes Chromosomes Cancer 58:521-529, 2019 [DOI] [PubMed] [Google Scholar]
- 9.Klega K, Imamovic-Tuco A, Ha G, et al. : Detection of somatic structural variants enables quantification and characterization of circulating tumor DNA in children with solid tumors. JCO Precis Oncol 2:1-9, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah AT, Azad TD, Breese MR, et al. : A comprehensive circulating tumor DNA assay for detection of translocation and copy-number changes in pediatric sarcomas. Mol Cancer Ther 20:2016-2025, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manzella G, Schreck LD, Breunis WB, et al. : High throughput drug profiling with a living biobank of primary rhabdomyosarcoma cells unravels disease heterogeneity and detects an AKT inhibitor sensitive subgroup. Nat Commun 11:4629, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Izquierdo E, Yuan L, George S, et al. : Development of a targeted sequencing approach to identify prognostic, predictive and diagnostic markers in paediatric solid tumours. Oncotarget 8:112036-112050, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chicard M, Colmet-Daage L, Clement N, et al. : Whole-exome sequencing of cell-free DNA reveals temporo-spatial heterogeneity and identifies treatment-resistant clones in neuroblastoma. Clin Cancer Res 24:939-949, 2018 [DOI] [PubMed] [Google Scholar]
- 14.Tombolan L, Rossi E, Binatti A, et al. : Clinical significance of circulating tumor cells and cell-free DNA in pediatric rhabdomyosarcoma. Mol Oncol 16:2071-2085, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurihara S, Ueda Y, Onitake Y, et al. : Circulating free DNA as non-invasive diagnostic biomarker for childhood solid tumors. J Pediatr Surg 50:2094-2097, 2015 [DOI] [PubMed] [Google Scholar]
- 16.Krumbholz M, Hellberg J, Steif B, et al. : Genomic EWSR1 fusion sequence as highly sensitive and dynamic plasma tumor marker in Ewing sarcoma. Clin Cancer Res 22:4356-4365, 2016 [DOI] [PubMed] [Google Scholar]
- 17.Barris DM, Weiner SB, Dubin RA, et al. : Detection of circulating tumor DNA in patients with osteosarcoma. Oncotarget 9:12695-12704, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heske CM, Mascarenhas L: Relapsed rhabdomyosarcoma. J Clin Med 10:804, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lak NSM, Voormanns TL, Zappeij-Kannegieter L, et al. : Improving risk stratification for pediatric patients with rhabdomyosarcoma by molecular detection of disseminated disease. Clin Cancer Res 27:5576-5585, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin LH, Chang KW, Kao SY, et al. : Increased plasma circulating cell-free DNA could be a potential marker for oral cancer. Int J Mol Sci 19:3303, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Risberg B, Tsui DWY, Biggs H, et al. : Effects of collection and processing procedures on plasma circulating cell-free DNA from cancer patients. J Mol Diagn 20:883-892, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stankunaite R, George SL, Gallagher L, et al. : Circulating tumour DNA sequencing to determine therapeutic response and identify tumour heterogeneity in patients with paediatric solid tumours. Eur J Cancer 162:209-220, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quan PL, Sauzade M, Brouzes E: dPCR: A technology review. Sensors (Basel) 18:1271, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eisenhauer EA, Therasse P, Bogaerts J, et al. : New response evaluation criteria in solid tumors: Revised RECIST guideline (version 1.1). Eur J Cancer 45:228-247, 2009 [DOI] [PubMed] [Google Scholar]