

PURPOSE
Radiation-associated sarcomas (RAS) are rare but aggressive malignancies. We sought to characterize the histology-specific presentation and behavior of soft tissue RAS to improve individualized prognostication.
METHODS
A single-institutional prospectively maintained database was queried for all patients with primary, nonmetastatic RAS treated with surgical resection from 1982 to 2019. Patients presenting with the five most common RAS histologies were propensity-matched to those with sporadic tumors of the same histology. Incidence of disease-specific death (DSD) was modeled using cumulative incidence analyses.
RESULTS
Among 259 patients with RAS, the five most common histologies were malignant peripheral nerve sheath tumor (MPNST; n = 19), myxofibrosarcoma (n = 20), leiomyosarcoma (n = 24), undifferentiated pleomorphic sarcoma (UPS; n = 55), and angiosarcoma (AS; n = 62). DSD varied significantly by histology (P = .002), with RAS MPNST and UPS having the highest DSD. In unadjusted analysis, RAS MPNST was associated with increased DSD compared with sporadic MPNST (75% v 38% 5-year DSD, P = .002), as was RAS UPS compared with sporadic UPS (49% v 28% 5-year DSD, P = .004). Unadjusted DSD was similar among patients with RAS AS, leiomyosarcoma, or myxofibrosarcoma and sporadic sarcoma of the same histology. After matching RAS to sporadic patients within each histology, DSD only differed between RAS and sporadic MPNST (83% v 46% 5-year DSD, P = .013). Patients with RAS AS presented in such a distinct manner to those with sporadic AS that a successful match was not possible.
CONCLUSION
The aggressive presentation of RAS is histology-specific, and DSD is driven by RAS MPNST and UPS histologies. Despite the aggressive presentation, standard prognostic factors can be used to estimate risk of DSD among most RAS. In MPNST, radiation association should be considered to independently associate with markedly higher risk of DSD.
INTRODUCTION
Approximately half of the patients with cancer undergoing therapy will be considered for radiation during the course of their treatment.1 A rare but sinister sequelae of this therapy is radiation-associated sarcoma (RAS). RAS accounts for approximately 3 percent of all sarcomas, and the prevalence in adults receiving prior radiation is estimated to be < 1 percent.2-5
CONTEXT
Key Objective
To describe the histology-specific outcomes of patients presenting with radiation-associated soft tissue sarcoma.
Knowledge Generated
Despite presenting in an aggressive fashion, standard prognostic factors can be used to estimate risk of death from disease among most patients with radiation-associated sarcoma. For patients with malignant peripheral nerve sheath tumors, radiation-association should be considered an independent risk factor.
Relevance
Risk-based treatment decisions may be guided by traditional prognostic factors without independently accounting for radiation association for most histologic types.
The presentation of RAS encompasses a wide range of different histologies, and, similar to sporadic sarcoma, survival is dependent on the histologic type.6-8 Although most series of RAS are limited by small sample sizes and diverse histologic types, RAS are consistently reported to be associated with aggressive presentation and poor overall outcomes.2,5,6,9-13
The prognosis and subsequent management of all sarcoma is guided by traditional clinicopathologic factors: tumor grade, size, depth, location, histologic type, and margin status after resection.14,15 Whether or not the difference in prognosis between RAS and sporadic sarcomas can be accounted for by these clinicopathologic factors remains undefined. There may exist additional, and yet unidentified, intrinsic biological differences in RAS that contribute to the observed poorer outcomes.
This distinction is both biologically interesting and has direct clinical relevance. If classification as a RAS is associated with worse prognosis controlling for all other clinicopathologic factors, radiation association alone may warrant consideration for more aggressive multimodality therapy compared with the same presentation arising sporadically. Alternatively, if the clinical behavior of RAS can be explained by traditional prognostic factors—management may be rationalized to mirror that of sporadic tumors.
Accordingly, we aimed to (1) characterize the histology-specific presentation of RAS to improve individualized risk assessment and (2) compare histology-specific outcomes of patients presenting with RAS to those presenting with sporadic sarcomas, controlling for traditional prognostic clinicopathologic factors.
METHODS
Patient Selection
The prospectively maintained sarcoma database at Memorial Sloan Kettering Cancer Center was queried for patients undergoing surgery for nonmetastatic, primary RAS treated from July 1, 1982, until June 1, 2019. Sarcomas were considered radiation-associated if (1) the patient had a history of radiation exposure at least 6 months before the development of the sarcoma, (2) the sarcoma occurred within the field of prior radiation, and (3) the sarcoma was pathologically distinct from the primary cancer.8 Pathologic re-review of all RAS with equivocal pathology was performed by a sarcoma pathologist (C.R.A. or N.P.A.). The sarcoma database was additionally queried over the same time frame to identify all non–radiation-associated malignant peripheral nerve sheath tumors (MPNST), myxofibrosarcomas (MFS), leiomyosarcomas (LMS), undifferentiated pleomorphic sarcomas (UPS), and angiosarcomas (AS; Data Supplement). This study was undertaken after obtaining approval from the Memorial Sloan Kettering Cancer Center Institutional Review Board.
Clinicopathologic Variables
Time to disease-specific death (DSD), first distant recurrence, and first local recurrence were measured from the date of surgery of RAS until the time of the event or last follow-up. Patients were followed clinically and radiographically as determined by the treating physicians. The typical follow-up protocol for low-grade sarcomas includes examination and scans at 6-month intervals for the first 5 years, yearly for years 5-10, and every 2 years thereafter. High-grade sarcomas are followed more intensively at 4-month intervals for the first 3 years, but subsequently in a similar fashion to low-grade tumors.
Additional clinicopathologic variables included age, sex, tumor site (extremity, retroperitoneal, head and neck, thoracic, and trunk), tumor size (categorized as ≤ 5 cm, > 5 to ≤ 10 cm, > 10 cm, or unknown), depth, resection margin (R0, R1—microscopically positive margins, R2—grossly positive margins), grade (high or low), histologic type of sarcoma, indication for radiation, and time from radiation to sarcoma diagnosis.
Statistical Analyses
Continuous variables were summarized as median and range, and compared between RAS and sporadic groups using Wilcoxon rank sum test. Categorical variables were described with count and percentage and compared using Fisher's exact test. DSD was analyzed in a competing risks framework with death unrelated to sarcoma treated as a competing event. Cumulative incidences were estimated, tested, and modeled using Fine and Gray's test and Fine and Gray regression models.16,17 Given the relative rarity and different patient characteristics of RAS compared with sporadic sarcomas, propensity score (PS) models were built using logistic regression for each of the five most common histologic types. The PS is the predicted probability that given those characteristics the person would be diagnosed with RAS versus sporadic. Age, sex, size, site, grade, and depth were included as covariates in the models, given their known prognostic value across patients with soft tissue sarcoma.18,19 As a supplementary analysis, the match was also performed additionally including margin status (R0 v R1/R2) for each histologic type. Using the PS matching with a caliper of 0.25 standard deviations of the pooled distribution, sporadic patients were matched to patients with RAS in a 2:1 ratio. Histograms were used to visualize the PS overlap between groups before match. Wilcoxon signed rank test and McNemar's test associations were used to assess the balance of covariates used in the PS model between groups after match. In the analyses of matched samples, a Fine and Gray model was used with a robust sandwich estimator to account for matching. SAS version 9.4 software program (SAS institute Inc, Cary, NC) was used for all analyses. All tests were two-sided, and P < .05 was considered significant
RESULTS
Characteristics of All Patients With RAS
From a total of 13,017 patients with soft tissue sarcoma presenting for surgical management, 259 patients with a primary RAS (2.0%) were identified and included in the analysis. The median age at diagnosis was 62 years, and 61% were female. Most were deep to fascia (78%), and low-grade lesions were rare (12%). The median time to development of RAS following radiation therapy was 10 years, although this varied widely (interquartile range, 6-19 years). The most common primary tumors for which radiation was originally administered were breast (40%), lymphoma (17%), and genitourinary tumors (16%; Table 1).
TABLE 1.
RAS Clinicopathologic Features

The median follow-up from date of surgical resection of the RAS was 4.7 years. At 5 years, 38% (95% CI, 32 to 45) of patients had died of disease and 14% (95% CI, 10 to 20) died from other causes (Data Supplement). Multivariable analysis identified size, grade, and histologic type, but not margin status, as significantly associated with disease-specific survival among patients with RAS (Data Supplement). Survival rates by histologic type are shown (Data Supplement). To investigate the behavior of these rare sarcomas in a histology-specific manner, the five most common histologic types were further analyzed by comparison to sporadic sarcomas of the same histologic type.
MPNST
Nineteen patients with a RAS MPNST were identified, 79% of whom received prior radiation for a hematologic malignancy. There were an additional 49 patients with NF1-associated MPNST who were excluded from this analysis as there were no cases of radiation-associated NF1 MPNSTs for comparison. When compared with 113 patients with sporadic MPNST, RAS MPNST were more frequently deep (100% v 80%, P = .044), and more likely to be resected with positive margins (negative margins achieved in 37% v 76%, P = .002; Fig 1A). Death from disease was more common in patients with RAS MPNST (75% at 5 year; 95% CI, 45 to 90) than sporadic MPNST (38% at 5 year; 95% CI, 29 to 48; P = .002; Fig 1B).
FIG 1.
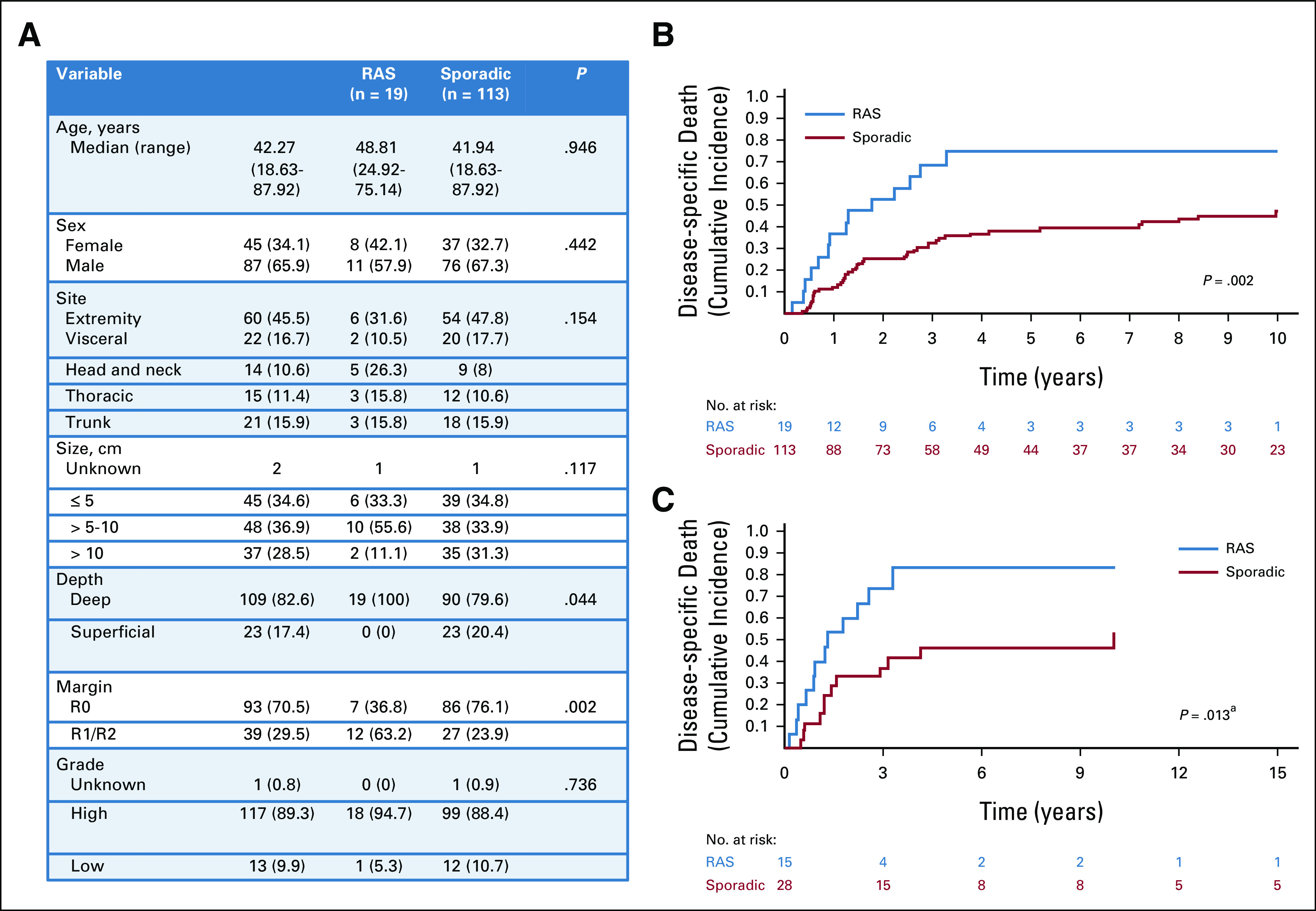
Presentation characteristics and DSD in patients with malignant peripheral nerve sheath tumors. (A) Clinicopathologic factors in all patients presenting with RAS MPNST compared with all patients presenting with sporadic (non–NF1-associated) MPNST. (B) Cumulative incidence of DSD in all patients with RAS MPNST (blue) compared with all patients with sporadic MPNST (red). (C) Cumulative incidence of DSD in propensity-matched patients with RAS (blue) and sporadic (red) MPNST. aPostmatch P value is derived from Cox regression model with sandwich estimator to control for correlation between matched samples rather than directly from the displayed cumulative incidence curve. DSD, disease-specific death; MPNST, malignant peripheral nerve sheath tumor; RAS, radiation-associated sarcoma.
To control for the differences in presentation between RAS and sporadic, a PS match was performed matching 15 RAS MPNST patients to 28 sporadic patients (Data Supplement). In the matched cohort, a significant difference in the cumulative incidence of death from disease between RAS MPNST (83% at 5 year; 95% CI, 38 to 97) and sporadic MPNST (46% at 5 year; 95% CI, 25 to 65) persisted (hazard ratio [HR], 2.58; 95% CI, 1.22 to 5.47; P = .013; Fig 1C). In a supplementary analysis to explore the role of resection margin in explaining this difference, a propensity match including margin status was performed, matching 16 RAS MPNST to 23 sporadic MPNST. DSD was higher in RAS MPNST than sporadic, but not significantly so (70% v 59% 5-year DSD; HR 1.38; 95% CI, 0.70 to 2.70; P = .349) after adjusting for margin status.
Myxofibrosarcoma
Twenty patients with a RAS myxofibrosarcoma were identified and were compared to 550 patients with sporadic myxofibrosarcoma. The presentation of RAS myxofibrosarcomas was not different to those of sporadic with the exception of anatomic site of presentation. RAS myxofibrosarcomas were diagnosed throughout the trunk (25%), chest wall (30%), head and neck (20%), and extremities (25%), whereas extremity location was by far the most common presentation site for sporadic (80%, P < .001; Fig 2A). Death from disease was not different in patients with RAS myxofibrosarcoma (19% at 5 year; 95% CI, 4 to 42) and sporadic myxofibrosarcoma (18% at 5 year; 95% CI, 14 to 21; P = .736; Fig 2B).
FIG 2.
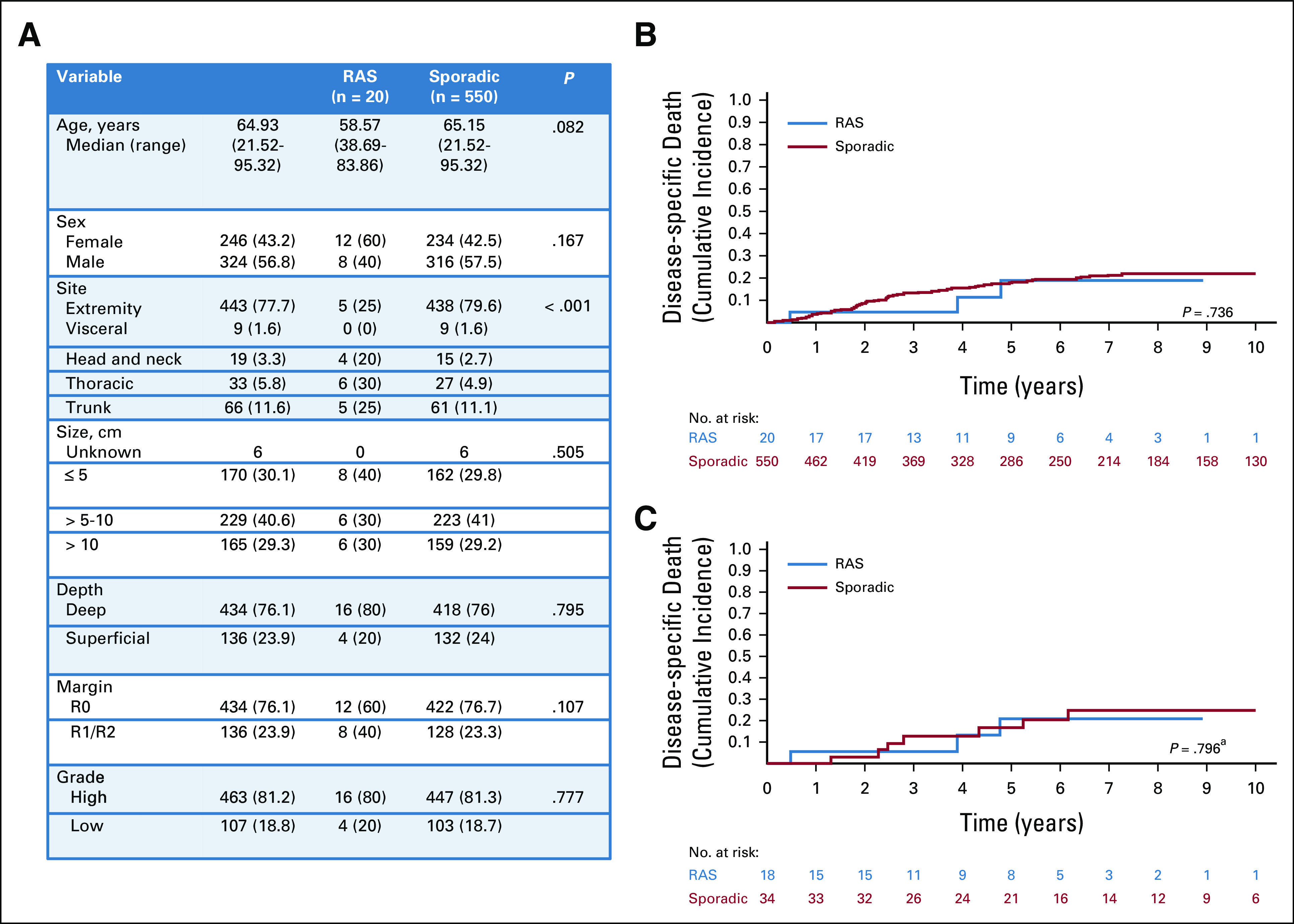
Presentation characteristics and DSD in patients with myxofibrosarcoma. (A) Clinicopathologic factors in all patients presenting with RAS MFS compared with all patients presenting with sporadic MFS. (B) Cumulative incidence of DSD in all patients with RAS MFS (blue) compared with all patients with sporadic MFS (red). (C) Cumulative incidence of DSD in propensity-matched patients with RAS (blue) and sporadic (red) MFS. aPostmatch P value is derived from Cox regression model with sandwich estimator to control for correlation between matched samples rather than directly from the displayed cumulative incidence curve. DSD, disease-specific death; MFS, myxofibrosarcoma; RAS, radiation-associated sarcoma.
Using a PS, 18 patients with RAS myxofibrosarcoma were matched to 34 sporadic patients (Data Supplement). In the matched cohort, no difference in death from disease between RAS myxofibrosarcoma (21% at 5 year; 95% CI, 4 to 46) and sporadic myxofibrosarcoma (17% at 5 year; 95% CI, 6 to 32) was identified (HR, 0.87; 95% CI, 0.30 to 52.51; P = .796; Fig 2C).
LMS
Twenty-four patients with a RAS LMS were identified. The median time to development of RAS LMS was 28 years—more than double that for any other RAS histologic type. When compared with 856 patients with sporadic LMS, the anatomic site of presentation of RAS LMS varied significantly (P < .001). RAS LMS were also more frequently deep (100% v 77%, P = .003; Fig 3A). Death from disease was not different in patients with RAS LMS (30% at 5 year; 95% CI, 11 to 51) and sporadic LMS (25% at 5 year; 95% CI, 22 to 28; P = .129; Fig 3B).
FIG 3.
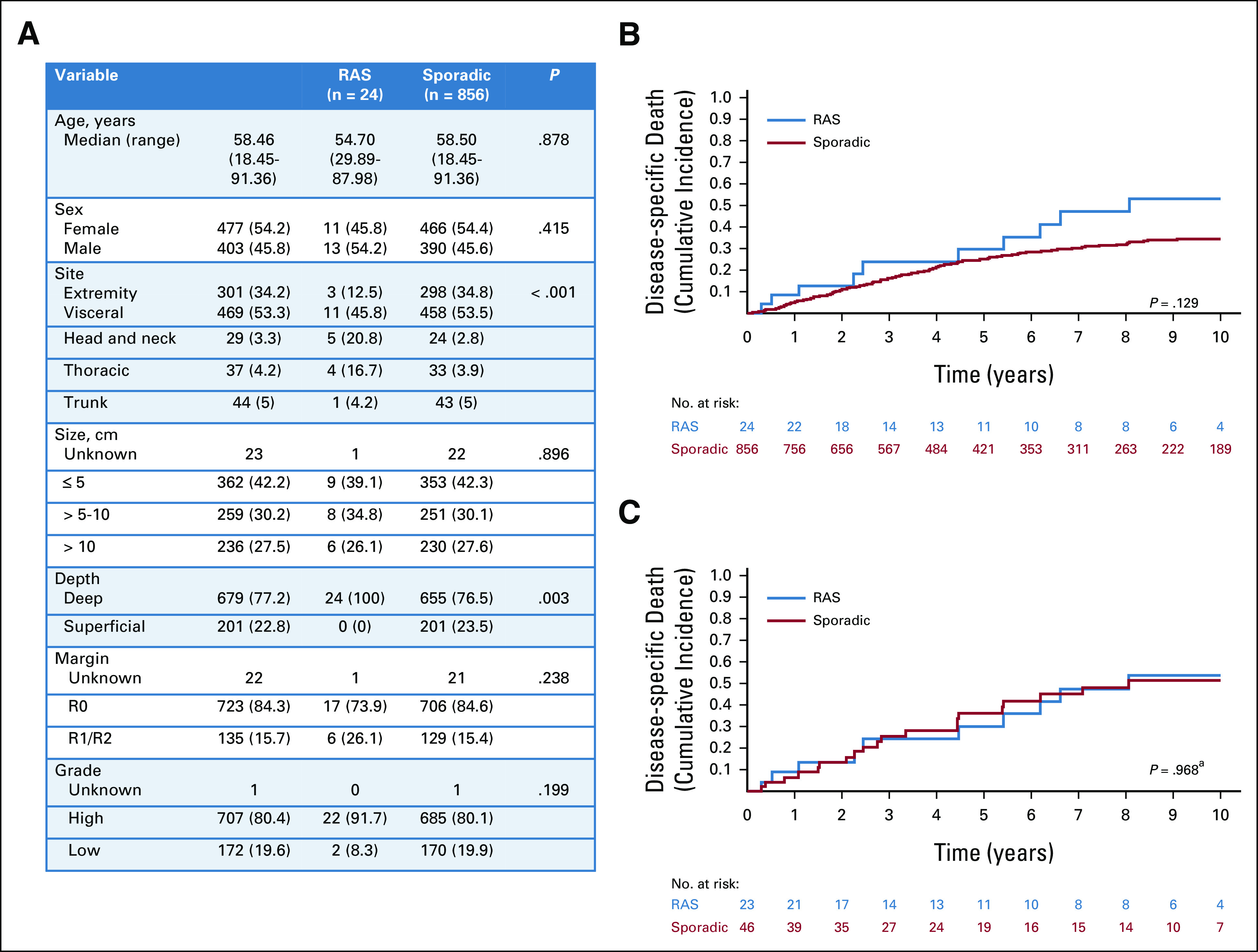
Presentation characteristics and DSD in patients with leiomyosarcoma. (A) Clinicopathologic factors in all patients presenting with RAS LMS compared with all patients presenting with sporadic LMS. (B) Cumulative incidence of DSD in all patients with RAS LMS (blue) compared with all patients with sporadic LMS (red). (C) Cumulative incidence of DSD in propensity-matched patients with RAS (blue) and sporadic (red) LMS. aPostmatch P value is derived from Cox regression model with sandwich estimator to control for correlation between matched samples rather than directly from the displayed cumulative incidence curve. DSD, disease-specific death; LMS, leiomyosarcomas; RAS, radiation-associated sarcoma.
Using a PS, 23 patients with RAS LMS were matched to 46 sporadic patients (Data Supplement). In the matched cohort, no difference in death from disease between RAS LMS (30% at 5 year; 95% CI, 12 to 52) and sporadic LMS (36% at 5 year; 95% CI, 22 to 51) was identified (HR, 1.01; 95% CI, 0.57 to 1.79; P = .968; Fig 3C).
UPS
Fifty-five patients with a RAS UPS were identified and were compared to 572 patients with sporadic UPS. The presentation of patients with RAS UPS differed substantially from sporadic. RAS UPS were less like to present in the extremity (24% RAS v 72% sporadic, P < .001), were smaller in size (50% of RAS < 5 cm v 32% of sporadic, P = .019), were more frequently deep (91% RAS v 73% sporadic, P = .002), and were less likely to be resected with a negative margin (64% of RAS v 84% of sporadic had R0 resection, P < .001; Fig 4A). Death from disease was significantly more common in patients with RAS UPS (49% at 5 year; 95% CI, 33 to 63) than sporadic (28% at 5 year; 95% CI, 24 to 32; P = .004; Fig 4B).
FIG 4.
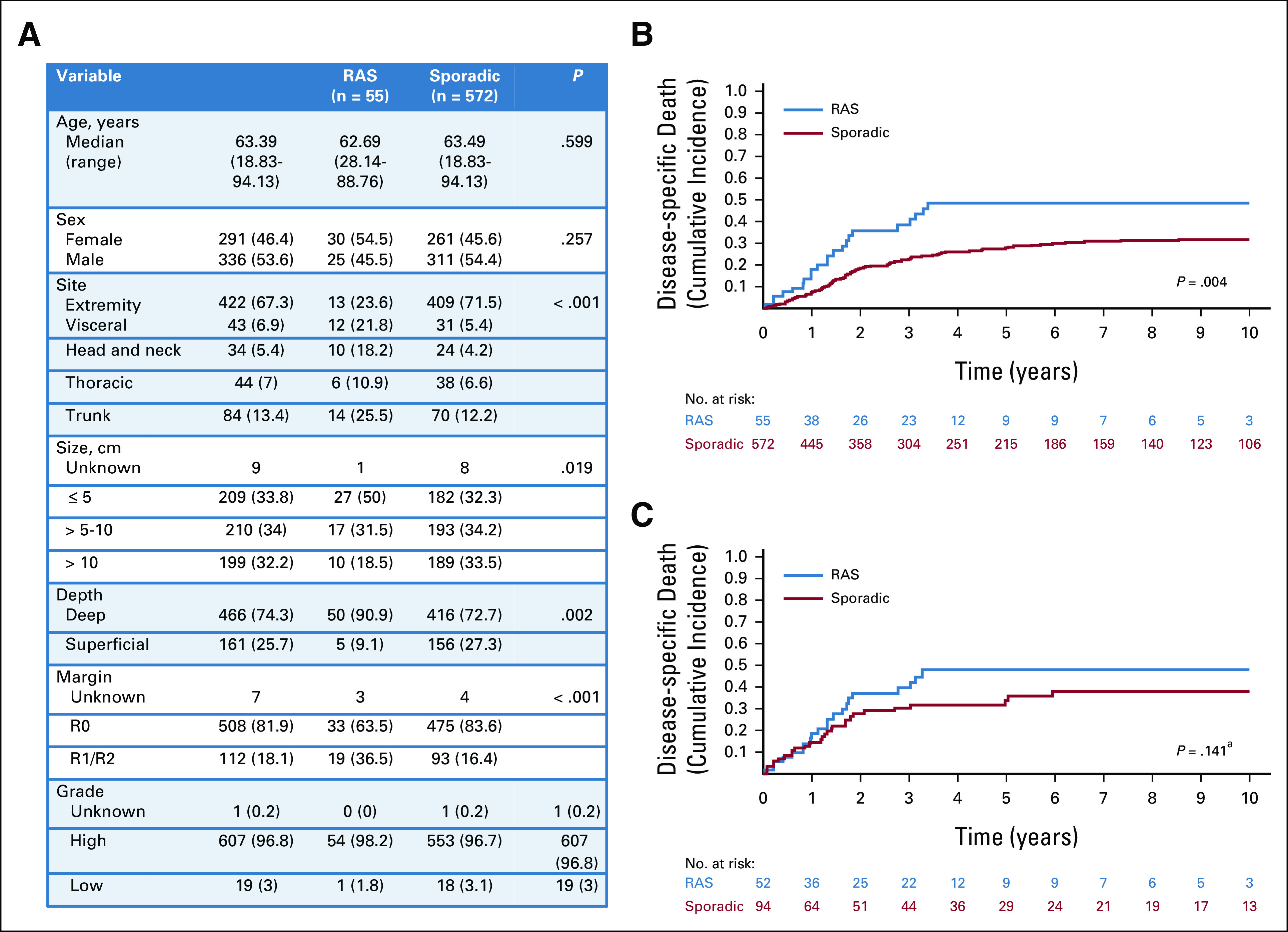
Presentation characteristics and DSD in patients with undifferentiated pleomorphic sarcoma. (A) Clinicopathologic factors in all patients presenting with RAS UPS compared with all patients presenting with sporadic UPS. (B) Cumulative incidence of DSD in all patients with RAS UPS (blue) compared with all patients with sporadic UPS (red). (C) Cumulative incidence of DSD in propensity-matched patients with RAS (blue) and sporadic (red) UPS. aPostmatch P value is derived from Cox regression model with sandwich estimator to control for correlation between matched samples rather than directly from the displayed cumulative incidence curve. DSD, disease-specific death; RAS, radiation-associated sarcoma; UPS, undifferentiated pleomorphic sarcoma.
Using a PS, 52 patients with RAS UPS were matched to 94 sporadic patients (Data Supplement). In the matched cohort, no significant difference in death from disease between RAS UPS (48% at 5 year; 95% CI, 32 to 62) and sporadic UPS (34% at 5 year; 95% CI, 23 to 45) was identified (HR, 1.39; 95% CI, 0.90 to 2.13; P = .141; Fig 4C).
AS
Sixty-two patients were identified with a RAS AS, 89% of whom received radiation for prior breast cancer including ductal carcinoma in situ. When compared with 110 patients with sporadic AS, the presentation of patients with RAS AS differed substantially. Patients with RAS AS were older (median 69 v 59 years, P < .001) and almost exclusively female (97% v 58%, P < .001). The site of presentation of RAS AS varied compared with sporadic AS (P < .001), with the large majority of RAS AS on the trunk (87%). RAS AS were larger (38% v 62% were < 5 cm, P = .01) and more frequently high grade (95% v 83%, P = .03) compared with sporadic ASs (Fig 5A). Despite these many differences, death from disease was not different in patients with RAS AS (40% at 5 year; 95% CI, 26 to 53) than sporadic (38% at 5 year; 95% CI, 28 to 48; P = .837; Fig 5B). A PS match was attempted, but the RAS AS and sporadic populations were so different that a successful match was not possible (Data Supplement).
FIG 5.
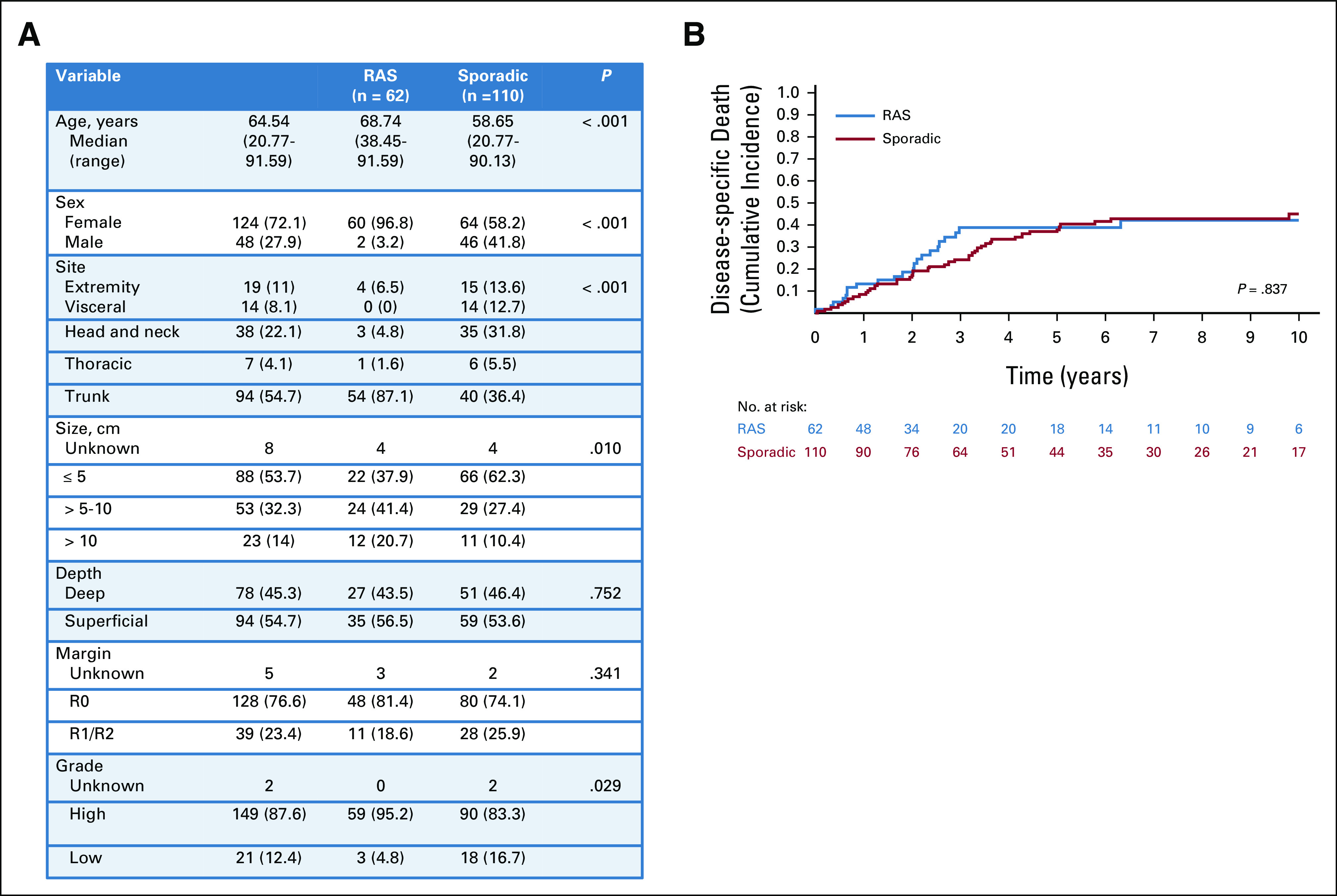
Presentation characteristics and DSD in patients with AS. (A) Clinicopathologic factors in all patients presenting with RAS AS compared with all patients presenting with sporadic AS. (B) Cumulative incidence of DSD in all patients with RAS AS (blue) compared with all patients with sporadic AS (red). Because of the differences in presentation between RAS AS and sporadic AS, a well-matched cohort could not be defined. AS, angiosarcoma; DSD, disease-specific death; RAS, radiation-associated sarcoma.
DISCUSSION
This study examined a large series of patients presenting with primary RAS to better estimate outcomes and improve individualized counseling and treatment recommendations. The findings support the previously described natural history of these tumors as arising typically a decade after treatment with radiation therapy.13,20,21 Although the study supports the generally aggressive nature of presentation for RAS,6,10,11 in this large sample size, clear differences emerge in clinical features at presentation according to the histologic type of RAS. Importantly for the clinician faced with determining appropriate treatment for these rare patients, the similar outcomes after matching among RAS and non-RAS patients suggest that the use of known prognostic factors (size, grade, depth, histologic type, etc) to estimate risk can be similarly applied to patients with RAS. RAS MPNST appears to be an exception to this, with markedly increased risk of death from disease even after accounting for known clinical prognostic factors.
The traditionally aggressive behavior of RAS appears to be a histology-dependent phenomenon. Interestingly, and not previously well described, the survival of patients with RAS MFS, RAS LMS, and RAS AS were comparable with patients presenting with the respective histologies in the sporadic setting, even before matching. However, both RAS MPNST (75% 5-year death from disease) and RAS UPS (49% 5-year death from disease) were associated with roughly double the mortality rate of patients presenting with these malignancies in the sporadic setting (38% and 28% 5-year death from disease, respectively).
Matching of RAS to sporadic patients was performed to determine the extent to which the standard prognostic factors for sarcoma can adjust for, and are appropriate to use, in patients with RAS. The matching revealed that although RAS UPS present in an aggressive manner, when sporadic UPS present in a similar fashion, the prognosis was not significantly different (5-year death from disease 34% for sporadic v 48% for RAS). In a similar sized series of RAS UPS and myxofibrosarcoma, Dineen et al22 report nearly identical survival (48% death from disease at 5-years, n = 55). This study additionally matched to a sporadic cohort, and in that match, a significant survival difference persisted. The methodology of matching, inclusion of myxofibrosarcoma, and the pathologic definition of UPS23 differ somewhat between these studies. It is possible that radiation association independently is associated with modestly increased risk for patients with UPS, but the current study is underpowered or the matching is insufficient to detect this difference.
After matching, MPNST was the one histologic type in which markedly increased risk of DSD was observed in RAS compared with similar sporadic cases (83% v 46% DSD at 5 years). This finding in part can be explained by the lower incidence of negative margin resection in patients with RAS MPNST compared with sporadic (37% v 76%). It is unclear whether RAS MPNST have lower negative resection rates because of anatomic constraints, a locally aggressive biology, or both. Prior series have variably identified margin status as associated with survival among MPNST, but few series have enough RAS MPNST to demonstrate a significant difference in survival compared with sporadic.24-27
Interestingly, patients presenting with RAS AS were so strikingly different in presentation that a match with similar sporadic patients was not possible. The most prominent differences were in age, site, and sex—with differences so clear that RAS AS and sporadic AS may be best considered different disease entities. Because of the inability to match, the similarity in outcomes may be due to confounding variables rather than a reflection of common biology. In this series, AS was the most common histologic type of RAS, which may reflect the increasing incidence of this entity.28 A number of studies have focused on RAS AS in particular.29-31 Molecular changes within RAS AS have also been described, and in particular, MYC amplification has been noted to be associated with RAS AS.32-34 This amplification is not consistently observed in other RAS histologies and, although progress has been made, defining a clear radiation-associated genetic signature has proven challenging.35,36 One potential consideration is the multimodality treatment that these patients receive for their primary malignancy. Whether lymphedema or breast cancer systemic therapies in addition to radiation contribute to the development of RAS AS remains undefined, but it is intriguing to observe AS so clearly associated with breast cancer. Interestingly, RAS MPNST was also strongly associated with prior lymphoma. The anatomy of the field of radiation for each of these primary malignancies is likely to be a primary consideration in this association, but other host factors or prior treatments may also contribute.
For the clinician choosing how to treat the rare patient presenting with RAS, this study provides reassurance that for the majority of histologic types, the known prognostic factors should well assess the risk to the patient. For example, a patient presenting with a 7-cm RAS MFS has the risks similar to those of a sporadic 7-cm MFS and does not appear to warrant more aggressive treatment solely on the basis of the RAS designation. RAS MPNST are particularly high risk and represent an exception whereby standard prognostic factors alone do not appropriately estimate risk.
This study is limited by its retrospective nature and a small sample size for each of the examined histologic types because of the rarity of this disease. The data were collected over a long time period with advances in diagnostics as well as treatment, so may not perfectly reflect current outcomes. It should also clearly be reiterated that this cohort only includes patients treated with surgical resection. Given the aggressive nature of presentation with RAS, many patients may present with unresectable disease. In that setting, the outcomes are reported to be quite poor, although whether they differ from similar presentation of sporadic tumors remains undefined.11 This study provides a best-case outcome, given that the included patients presented with primary, nonmetastatic disease eligible for surgical resection. Further unknown is whether RAS demonstrate a differential response to therapy compared with their sporadic counterparts. One could imagine that the distinct molecular composition of these tumors may respond differently to standard therapies and/or perhaps make them more susceptible to nonconventional therapies such as checkpoint inhibitors.35,37
In conclusion, this study confirms the aggressive presentation of RAS as a group but demonstrates that this phenomenon is histology-dependent. In the primary setting, when prognostic factors are accounted for in a histology-specific manner, RAS sarcomas are not clearly associated with an increased risk of death from disease, with the exception of patients with MPNST. As such, use of traditional prognostic factors to guide treatment and follow-up recommendations in these patients is appropriate.
Edmund K. Bartlett
Honoraria: Excite International
Research Funding: Skyline Diagnostics (Inst)
Ping Chi
Stock and Other Ownership Interests: ORIC Pharmaceuticals
Consulting or Advisory Role: Deciphera, NewBay Pharma, Zai Lab
Research Funding: Deciphera (Inst), Pfizer (Inst), NewBay Pharma (Inst)
Patents, Royalties, Other Intellectual Property: Royalties from ORIC
Li-Xuan Qin
Employment: Viela Bio, Sironax
Leadership: Viela Bio, Sironax
Stock and Other Ownership Interests: Viela Bio, Sironax
No other potential conflicts of interest were reported.
SUPPORT
Supported by the NIH SPORE in Soft Tissue Sarcoma P50 CA140146 and the NIH/NCI Cancer Center Support Grant P30 CA008748.
E.K.B. and A.S. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Edmund K. Bartlett, Avinash Sharma, Bhumika Jadeja, Li-Xuan Qin, Kaled M. Alektiar, Samuel Singer
Financial support: Samuel Singer
Administrative support: Murray F. Brennan, Samuel Singer
Provision of study materials or patients: Cristina R. Antonescu, Ping Chi, Samuel Singer
Collection and assembly of data: Edmund K. Bartlett, Avinash Sharma, Kenneth Seier, Cristina R. Antonescu, Narasimhan P. Agaram, Samuel Singer
Data analysis and interpretation: Edmund K. Bartlett, Avinash Sharma, Kenneth Seier, Cristina R. Antonescu, Bhumika Jadeja, Evan Rosenbaum, Ping Chi, Murray F. Brennan, Li-Xuan Qin, Kaled M. Alektiar, Samuel Singer
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Edmund K. Bartlett
Honoraria: Excite International
Research Funding: Skyline Diagnostics (Inst)
Ping Chi
Stock and Other Ownership Interests: ORIC Pharmaceuticals
Consulting or Advisory Role: Deciphera, NewBay Pharma, Zai Lab
Research Funding: Deciphera (Inst), Pfizer (Inst), NewBay Pharma (Inst)
Patents, Royalties, Other Intellectual Property: Royalties from ORIC
Li-Xuan Qin
Employment: Viela Bio, Sironax
Leadership: Viela Bio, Sironax
Stock and Other Ownership Interests: Viela Bio, Sironax
No other potential conflicts of interest were reported.
REFERENCES
- 1.Delaney G, Jacob S, Featherstone C, Barton M: The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 104:1129-1137, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Bjerkehagen B, Smeland S, Walberg L, et al. : Radiation-induced sarcoma: 25-year experience from the Norwegian Radium Hospital. Acta Oncol 47:1475-1482, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Taghian A, de Vathaire F, Terrier P, et al. : Long-term risk of sarcoma following radiation treatment for breast cancer. Int J Radiat Oncol Biol Phys 21:361-367, 1991 [DOI] [PubMed] [Google Scholar]
- 4.Kirova YM, Vilcoq JR, Asselain B, et al. : Radiation-induced sarcomas after radiotherapy for breast carcinoma: A large-scale single-institution review. Cancer 104:856-863, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Kim KS, Chang JH, Choi N, et al. : Radiation-induced sarcoma: A 15-year experience in a single large tertiary referral center. Cancer Res Treat 48:650-657, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brady MS, Gaynor JJ, Brennan MF: Radiation-associated sarcoma of bone and soft tissue. Arch Surg 127:1379-1385, 1992 [DOI] [PubMed] [Google Scholar]
- 7.Cha C, Antonescu CR, Quan ML, et al. : Long-term results with resection of radiation-induced soft tissue sarcomas. Ann Surg 239:903-909, 2004; discussion 909-910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gladdy RA, Qin LX, Moraco N, et al. : Do radiation-associated soft tissue sarcomas have the same prognosis as sporadic soft tissue sarcomas? J Clin Oncol 28:2064-2069, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mito JK, Mitra D, Doyle LA: Radiation-associated sarcomas: An update on clinical, histologic, and molecular features. Surg Pathol Clin 12:139-148, 2019 [DOI] [PubMed] [Google Scholar]
- 10.Bjerkehagen B, Smastuen MC, Hall KS, et al. : Why do patients with radiation-induced sarcomas have a poor sarcoma-related survival? Br J Cancer 106:297-306, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagrange JL, Ramaioli A, Chateau MC, et al. : Sarcoma after radiation therapy: Retrospective multiinstitutional study of 80 histologically confirmed cases. Radiation therapist and pathologist Groups of the Federation Nationale des Centres de Lutte Contre le Cancer. Radiology 216:197-205, 2000 [DOI] [PubMed] [Google Scholar]
- 12.Riad S, Biau D, Holt GE, et al. : The clinical and functional outcome for patients with radiation-induced soft tissue sarcoma. Cancer 118:2682-2692, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Neuhaus SJ, Pinnock N, Giblin V, et al. : Treatment and outcome of radiation-induced soft-tissue sarcomas at a specialist institution. Eur J Surg Oncol 35:654-659, 2009 [DOI] [PubMed] [Google Scholar]
- 14.Brennan MF, Antonescu CR, Alektiar KM, Maki R: Management of Soft Tissue Sarcoma (ed 2), Cham, Switzerland, Springer, 2016 [Google Scholar]
- 15.Brennan MF, Antonescu CR, Moraco N, Singer S: Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg 260:416-421, 2014. discussion 421-4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fine JP, Gray RJ: A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc 94:496-509, 1999 [Google Scholar]
- 17.Gray RJ: A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat:1141-1154, 1988 [Google Scholar]
- 18.Cahlon O, Brennan MF, Jia X, et al. : A postoperative nomogram for local recurrence risk in extremity soft tissue sarcomas after limb-sparing surgery without adjuvant radiation. Ann Surg 255:343-347, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kattan MW, Leung DH, Brennan MF: Postoperative nomogram for 12-year sarcoma-specific death. J Clin Oncol 20:791-796, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Laskin WB, Silverman TA, Enzinger FM: Postradiation soft tissue sarcomas. An analysis of 53 cases. Cancer 62:2330-2340, 1988 [DOI] [PubMed] [Google Scholar]
- 21.Wiklund TA, Blomqvist CP, Raty J, et al. : Postirradiation sarcoma. Analysis of a nationwide cancer registry material. Cancer 68:524-531, 1991 [DOI] [PubMed] [Google Scholar]
- 22.Dineen SP, Roland CL, Feig R, et al. : Radiation-associated undifferentiated pleomorphic sarcoma is associated with worse clinical outcomes than sporadic lesions. Ann Surg Oncol 22:3913-3920, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee AY, Agaram NP, Qin LX, et al. : Optimal percent myxoid component to predict outcome in high-grade myxofibrosarcoma and undifferentiated pleomorphic sarcoma. Ann Surg Oncol 23:818-825, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anghileri M, Miceli R, Fiore M, et al. : Malignant peripheral nerve sheath tumors: Prognostic factors and survival in a series of patients treated at a single institution. Cancer 107:1065-1074, 2006 [DOI] [PubMed] [Google Scholar]
- 25.Stucky CC, Johnson KN, Gray RJ, et al. : Malignant peripheral nerve sheath tumors (MPNST): The Mayo Clinic experience. Ann Surg Oncol 19:878-885, 2012 [DOI] [PubMed] [Google Scholar]
- 26.Watson KL, Al Sannaa GA, Kivlin CM, et al. : Patterns of recurrence and survival in sporadic, neurofibromatosis type 1-associated, and radiation-associated malignant peripheral nerve sheath tumors. J Neurosurg 126:319-329, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou C, Smith KD, Liu J, et al. : Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg 249:1014-1022, 2009 [DOI] [PubMed] [Google Scholar]
- 28.Bryant AK, Banegas MP, Martinez ME, et al. : Trends in radiation therapy among cancer survivors in the United States, 2000-2030. Cancer Epidemiol Biomarkers Prev 26:963-970, 2017 [DOI] [PubMed] [Google Scholar]
- 29.D'Angelo SP, Antonescu CR, Kuk D, et al. : High-risk features in radiation-associated breast angiosarcomas. Br J Cancer 109:2340-2346, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torres KE, Ravi V, Kin K, et al. : Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol 20:1267-1274, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fodor J, Orosz Z, Szabo E, et al. : Angiosarcoma after conservation treatment for breast carcinoma: Our experience and a review of the literature. J Am Acad Dermatol 54:499-504, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Guo T, Zhang L, Chang NE, et al. : Consistent MYC and FLT4 gene amplification in radiation-induced angiosarcoma but not in other radiation-associated atypical vascular lesions. Genes Chromosomes Cancer 50:25-33, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manner J, Radlwimmer B, Hohenberger P, et al. : MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol 176:34-39, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mentzel T, Schildhaus HU, Palmedo G, et al. : Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: Clinicopathological, immunohistochemical and molecular analysis of 66 cases. Mod Pathol 25:75-85, 2012 [DOI] [PubMed] [Google Scholar]
- 35.Behjati S, Gundem G, Wedge DC, et al. : Mutational signatures of ionizing radiation in second malignancies. Nat Commun 7:12605, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hadj-Hamou NS, Ugolin N, Ory C, et al. : A transcriptome signature distinguished sporadic from postradiotherapy radiation-induced sarcomas. Carcinogenesis 32:929-934, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yarchoan M, Hopkins A, Jaffee EM: Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 377:2500-2501, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]