

Abstract
In this article, we reviewed the current literature studies and our understanding of the parameters that affect the chimeric antigen receptor T cells (CAR-T’s) activation, effector function, in vivo persistence, and antitumour effects. These factors include T cell subsets and their differentiation stages, the components of chimeric antigen receptors (CAR) design, the expression promoters and delivery vectors, and the CAR-T production process. The CAR signalling and CAR-T activation were also studied in comparison to TCR. The last section of the review gave special consideration of CAR design for solid tumours, focusing on strategies to improve CAR-T tumour infiltration and survival in the hostile tumour microenvironment. With several hundred clinical trials undergoing worldwide, the pace of CAR-T immunotherapy moves from bench to bedside is unprecedented. We hope that the article will provide readers a clear and comprehensive view of this rapidly evolving field and will help scientists and physician to design effective CAR-Ts immunotherapy for solid tumours.
Keywords: Adoptive cell transfer, chimeric antigen receptor, T cell engineering, tumour immunotherapy
1. Introduction
Over the last decade, immunotherapies with checkpoint blockades and adoptive cell transfer (ACT) have made remarkable progress in cancer treatment. The success of immunotherapy requires tumour-specific T cells (Ref. 1), which do not exist in most tumours. Engineering patient’s T cells with T cell receptors (TCR) (Refs 2, 3) or chimeric antigen receptors (CAR) (Ref. 4) can provide tumour-specific T cells. ACT of the ‘engineered’ T cells (TCR-T and CAR-T) is a rapidly evolving research field and represents a promising approach of cancer immunotherapy (Refs 5, 6). CARs target surface antigen and do not have MHC restriction. Additionally, the antibody usually has a 1–3 log higher affinity than TCR. While TCR-T is still in the stage of trials,(Ref 7) CAR-Ts are approved for haematological malignancies (Table 1). In this review, we only focused on CAR-Ts. Readers are referred to two excellent recent reviews on TCR-Ts (Refs 7, 8). The complete control of blood cancers by CAR-Ts marks a new era in cancer treatment (Refs 6, 9, 10). There are ~253 ongoing CAR-T trials for different cancers (Ref. 11). CAR molecule is composed of the antigen-binding domain (ABD), a flexible hinge (H), transmembrane (TM), and intracellular signalling domain (SD) that include 1–2 co-stimulatory domain (CD) and CD3ζ (Fig. 1). The ABD and SD are from different sources and form into one artificial receptor (chimaera) that recognise tumour surface antigen independent of MHC. In this article, we will review the factors that affect the function and antitumour efficacy of CAR-Ts. We will cover the T cell subset selection, CAR design, delivery, expression, and the process of CAR-T production. We hope this review will help us to understand the role of each component in the CARs and to design effective CAR-Ts to generate potent and durable antitumour effects, especially for solid tumours.
Table 1.
CAR-Ts approved for treating haematological cancers
| Trade Name | KYMRIAH | YESCARTA | TECARTUS | BREYANZI | ABECMA | ARI-0001 | Relma-cel |
|---|---|---|---|---|---|---|---|
| proper name | tisagenlecleucel | axicabtagene ciloleucel | brexucabtagene autoleucel | lisocabtagene maraleucel | idecabtagene vicleucel | Relmacabtagene Autoleucel | |
| T cell sources | autologous T cells | autologous T cells | autologous T cells | autologous CD4+,CD8+ | autologous T cell | autologous T cell | autologous CD4+ and CD8+ |
| Year approved | 30 August 2017 | 18 October 2017 | 23 July 2020 | 5 February 2021 | 26 March 2021 | 10 February 2021 | 6 September 2021 |
| Approved by | FDA | FDA | FDA | FDA | FDA | AEMPS | NMPA |
| ScFv (Ab) | FMC63 | FMC63 | FMC63 | FMC63 | Murine | A3B1 murine | FMC63 |
| Linker Virus | (GGGGS)3 Lentivirus | (GGGGS)3 Retrovirus | (GGGGS)3 Retrovirus | Lentivirus | Lentivirus | Lentivirus | Lentivirus |
| Promoter | EF1a | MSCV | MSCV | ? | MND | EF1a | |
| Target | CD19 | CD19 | CD19 | CD19 | BCMA | CD19 | CD19 |
| Disease | (R/R) B-ALL, DLBCL, etc. | DLBCL, etc. | (R/R) MCL | (R/R) LBCL, DLBCL, etc. | (R/R) multiple myeloma | (R/R) ALL | (R/R) LBCL |
| H/T domain | CD8a | CD28 | CD28 | IgG4 (H), CD28 (TM) | CD8a | CD8a | |
| Signalling domain | 4–1BB, CD3ζ | CD28, CD3ζ | CD28, CD3ζ | 4–1BB, CD3ζ | 4–1BB, CD3ζ | 4–1BB, CD3ζ | 4–1BB, CD3ζ |
| Other factor Generation | 2nd | 2nd | T cell enrichment 2nd | EGFRt 2nd | 2nd | 2nd | EGFRt 2nd |
| Company | Novartis | Kite Pharma | Kite Pharma | Juno Therapeutics | BMS | Hospital clinic (spain) | JW Therapeutics |
FDA, U.S. Food and Drug Administration; AEMPS, The Spanish Agency of Medicines and Medical Devices; NMPA, the National Medical Products Administration of China.
Figure 1.
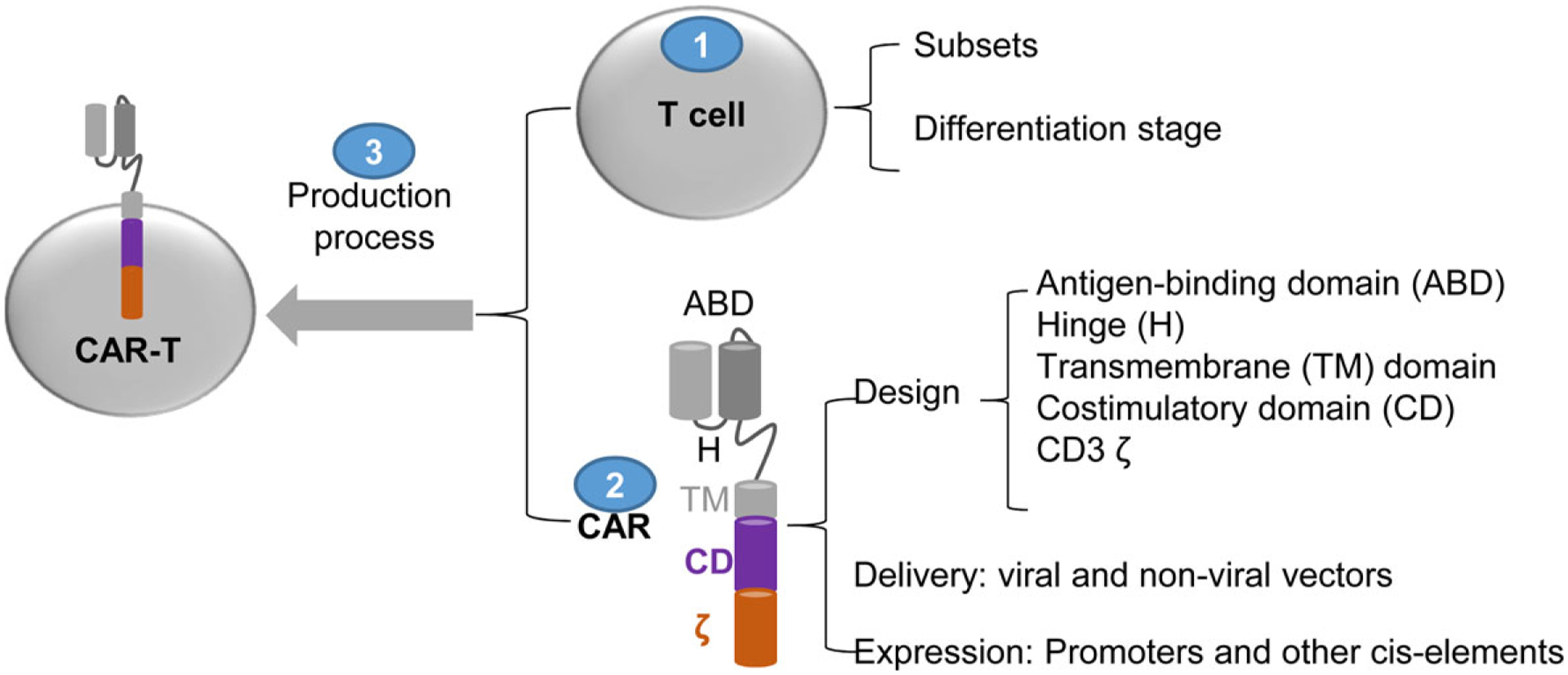
The parameters that affect the function and antitumour effects of CAR-T cells.
1.1. T Cells
Although allogenic CAR-Ts can be pre-made as ’off-the-shelf’ with better standardisation, the graft-vs-host diseases is a big hurdle for their success (Refs 12, 13). Currently, autologous T cells are the main source for CAR-T generation. The CAR-Ts approved by USA, Spain and China are generated from autologous T cells. The subset of T cells and their differentiation can affect CAR-T’s function and antitumour effects.
1.1.1. Subsets
CD4 and CD8 T cells are important in antitumour immunity (Refs 14, 15) and used to generate CAR-Ts, alone or in their unselected composition (Refs 16–19). The cytotoxicity of CD8 CAR-Ts can be higher (Ref. 19), equal (Ref. 16) or lower (Ref. 17) than CD4 counterparts. Although more IFNγ–producing CD8 CAR-Ts were observed upon antigen stimulation (Refs 17, 19), CD4 CAR-Ts produced a higher amount of IFNγ, TNFα and IL-2 (Refs 16, 19). The CD4 CAR-T outperformance was also observed in the glioblastoma-targeted IL13Ra2-specific (IL13–41BBζ) CAR-Ts (Ref. 17), possibly because CD4 cells are less prone to exhaustion than CD8 (Refs 17, 20). The composition of blood CD4 and CD8 cells vary in their activation and differentiation state, especially for cancer patients receiving chemotherapy, which can affect the therapeutic effect and toxicity of CAR-Ts (Refs 18, 21). The antitumour effects of CD4 and CD8 CAR-Ts may vary due to their origin, CAR-T design and tumour types, but CD8 and CD4 CAR-Ts have synergistic antitumour activity (Refs 18, 19). The unselected T cells with both CD4 and CD8 are frequently used to generate CAR-Ts. In this approach, CD8 CAR-Ts can undergo rapid activation and provide a prompt response against the tumour, whereas CD4 CAR-Ts persist longer and generate a durable anti-tumour effect. Together, they work more effectively than their own. Though CAR-Ts are usually generated ex vivo, a recent study reported in vivo selective transduction CD8 and CD4 cells by CAR genes. Using lentivirus with either CD8α-(CD8-LV) or CD4-specific DARPin (designed ankyrin repeat protein) (CD4-LV) as entry receptor, Agarwal et al. selectively delivered CAR gene into CD8 or CD4 T cells in NSG mice (Ref. 20). CD4-LV exhibited superior antitumour effects compared to CD8-LV alone or even in their combination.
1.1.2. Differentiation stages
T cells undergo differentiation from naïve (TN) to stem-like memory (TSCM), central memory (TCM), effector memory (TEM), and effectors (TE) (Ref. 22). In a mouse study, ACT of the activated tumour-specific CD8 cells derived from TCM generated stronger antitumour effects compared to TEM CD8 cells (Ref. 23). Then, they showed that the activated CD8 T cells derived from TN generated stronger antitumour effects than that from TCM (Ref. 24). In a non-human primate study, the CD8 T cells with TCM phenotype persisted longer in vivo (Ref. 25). Similarly, the CAR-T’s proliferation and persistency are affected by their differentiation states as well (Ref. 26). CAR-Ts derived from TN, TSCM and TCM have a greater proliferative potential and longevity (Refs 27, 28). In one study (Ref. 19), investigators found that human CD4/CD8 CD19 CAR-Ts derived from TN had the highest cytokine secretion, followed by TCM and TEM. The in vivo anti-tumour efficacy of the CD4 CAR-Ts derived from different differentiation stages follows the same order: TN > TCM > TEM. In contrast, for CD8 CAR-Ts, those derived from TCM generated longer antitumour effects (TCM > TN = TEM). Importantly, a combination of the TCM-derived CD8 CAR-Ts with the TN-derived CD4 CAR-Ts resulted in complete tumour eradication in treating leukaemia xenografts. Thus, it is generally believed that CAR-Ts generated from defined T cells subsets (TN and/or TCM) may be more effective.
1.1.3. CD26hi T cells
CD26 is a marker for terminal TE. However, in contrast to conventional reasoning, one study showed that the CAR-Ts generated from using CD26hi CD4 T cells persisted and regressed solid tumours (Ref. 29). Further study showed that the CD26high CD4 T cells are a distinct cell population and can be used to develop CAR-Ts with enhanced antitumour effects in treating solid tumours (Ref. 30).
2. CAR design
CAR, the other component of CAR-T, has four major domains: ABD, H, TM, and SD (Fig. 1) (Ref. 21). The selection and design of each domain affect CAR-T activation and antitumour effects.
2.1. ABD
The ABD recognises tumour antigen and confers specificity to CAR-Ts.
2.1.1. Targets
An ideal target should have high coverage to recognise all tumour cells, high specificity to prevent toxicity, and stable expression to maintain durable CAR-T recognition (Ref. 31). However, such an ‘ideal’ target is rare. Instead, shared antigens are frequently targeted. For example, CD19 is on both normal and malignant B cells. Although CD19 CAR-Ts lead to B cell aplasia, this ‘on-target/off-tumour’ toxicity is tolerable and can be managed by immunoglobulin replacement (Ref. 32) or by CAR-T removal (Ref. 33). Loss of antigen is a common mechanism of CAR-T therapy failure (Refs 34, 35). Even initial response was outstanding in multiple trials, long-term follow-up revealed that a notable proportion of patients received KYMRIAH (Ref. 36) or YESCARTA (Ref. 37) relapsed with CD19-negative leukaemia. Other B-cell restricted surface molecules such as CD20, CD22, and CD123 are detected in patients with CD19-negative relapses (Refs 38, 39). CD19 CAR-T failure due to loss of CD19 was overcome by using another single-targeted (Ref. 40), sequential infusion of 2 single-targeted CAR-Ts (Ref. 41), or dual/multi-targeted (Refs 39, 42) CAR-Ts. Antigen density on tumour cells also affects the cytotoxicity and cytokine production of CAR-Ts (Refs 43, 44). Using CD20 CD28ζ CAR-Ts and target cells with various levels of CD20, Watanabe et al. demonstrated that the antigen threshold was ~200 and a few thousand molecules per target cell for lytic activity and cytokine production, respectively (Ref. 44). Similarly, low expression of targets CD19 (Ref. 43), CD22 (Ref. 45), EGFR (Ref. 46), and anaplastic lymphoma kinase (ALK) (Ref. 47) might negatively affect CAR-T function and persistence. The epitope location (membrane-proximal or distal) determines the synapse cleft distance and kinetic segregation of phosphatases like CD45. This will affect the signalling strength and cytotoxic granule delivery (Refs 21, 48). The consensus view is that CAR-Ts targeting membrane-proximal epitopes generate stronger effects. The CAR-Ts targeting the membrane-proximal epitope of mesothelin (Ref. 49) and CD33 (Ref. 50) or membrane-distal epitope of CD22 (Ref. 51) could be modified by regulating the length of the hinge to enhance antitumour function (see section 2.2).
2.1.2. scFv (single-chain variable fragment)
Although other molecules, such as IL13 (Ref. 17), adnectin (Ref. 52), DARPin (Refs 20, 53), and nanobodies (Ref. 54), are used in CAR design, mAb scFv is the most common ABD due to its size, affinity, and specificity (Ref. 55). The scFv consists of variable heavy (VH) and light (VL) chains connected by a flexible linker of 10 to 25AA in the format of VH-linker-VL or VL-linker-VH (Refs 56, 57). The scFv derived from murine mAb (FMC63) is used in 5 approved CD19 CAR-Ts (Table 1). While one report showed that both VH-linker-VL and VL-linker-VH orientations had the similar antigen-binding capacity and CAR-T function (Ref. 58), other found that the antigen recognition could be maintained only in particular VH and VL orientation (Ref. 59). Murine mAbs are used in approved CAR-Ts, they are immunogenic and may induce anti-drug antibodies (ADA) that cause side effects, CAR-T loss, and therapy failure (Ref. 60). Humanised CD19 CAR-Ts (Ref. 61) and fully human anti-CD19 CAR-Ts (Ref. 62) maybe more effective.
2.1.3. Linkers
(G4S)3 and (G4S)4, the 3 and 4 repeats of pentapeptide (Gly–Gly–Gly–Gly–Ser), are the common linkers between VL and VH (Ref. 63). Glycine and serine contribute for flexibility and solubility, which are critical for scFv proper folding and antigen-binding. Recent studies reported that short scFv linker (G4S)1 enhanced ‘tonic signalling’ in CD22-BBζ and CD33-BBζ CAR-Ts (but not CD19-BBζ CAR-Ts) without causing more exhaustion, but leading to better anti-leukaemic function (Ref. 64). In contrast to this beneficial effect, a previous study showed tonic signalling in anti-GD2 CD28ζCAR-Ts resulted in exhaustion and decreased antitumour activity (Ref. 65), which was ameliorated by replacing CD28 with 41BB. These data suggest that the effect of tonic signalling on CAR-T function depends on multi-factors including target, co-stimulatory domain, and even the length of the linker. Thus, tonic signalling is not always bad for CAR-T function, but needs to be taken into consideration in CAR design (Ref. 66).
2.1.4. Affinity and avidity
The affinity (single pair of molecular interaction) of scFv and the avidity (multiple pairs of molecular interaction) can affect CAR-T’s function and therapeutic effect (Refs 67–69). Conventionally, scFvs of high affinity are preferred in CAR-Ts for increased antigen recognition, binding capacity, and tumouricidal effect (Ref. 21). However, CAR-T’s avidity beyond a certain threshold would not increase efficacy (Ref. 70) and even exhibited inferior responses (Ref. 71). A recent report showed that a low-avidity CD19 CAR-T enhanced expansion and persistence in treating refractory acute lymphoblastic leukaemia (Ref. 72). Drent et al. studied 8 anti-CD38 scFvs covering an affinity of 10–1000 folds (Ref. 73). In the study, CD38 CAR-Ts from scFv with ~1000 folds lower affinity could effectively lyse CD38high multiple myeloma cells but not CD38low healthy haematopoietic cells. Recently, how the biophysical properties of CAR (affinity, avidity, and antigen density) contributed to CAR-T function was further studied using CD28ζ CAR-Ts with varying avidity to the same antigen, HLA-A2/Tyrosinase369–377 (Ref. 74). The CAR-T derived from the highest-affinity scFv (4 nM) showed a lower lytic response determined by CD107a, as compared to CAR-Ts from intermediate-affinity scFv (35 and 16 nM). The CAR expression and CAR-T avidity, measured by Tyr/HLA-A2 monomer and multimer binding respectively, correlated with each other in CAR-T function. Increasing avidity resulted in high sensitivity to low antigen concentrations. However, the polyfunctional CAR-Ts (IFNγ, TNFα and IL2 triple-positive) was more in the medium-avidity CAR-Ts. In summary, the intermediate avidity CAR-Ts can reduce the ‘on target/off tumour’ activity by recognising only antigen-high tumour cells (Ref. 73), but also are more polyfunctional, correlating to better clinical efficacy (Refs 62, 63) (Table 2).
Table 2.
Summary of 14 completed clinical trials of solid tumour CAR-T therapy: scFv affinity vs clinical efficacy
| Target | Target binding Domain | ICD | In vitro or Preclinical | Clinical trials |
|---|---|---|---|---|
| FRα (folate binding protein) | scFv based on MOv18 KD: ∼0.2 nM (Ref. 210) | FceRIγ | Dual specific T cells (allo-TCR and FRα CAR) inhibited tumour growth (Ref. 211) | NCT00019136 No reduction in any of 12 patients (Ref. 212) |
| GD2 | scFv from mAb Hu3F8 (Ref. 213) KD: 11 nM | CD28-BB-CD3ζ | GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients (clinical trial show only SD in 4/10 patients. Other 6 died due to PD | NCT02765243 4/10 SD for 1 yr DOI:CSBOLDSTART CSBOLDEND10.21203/rs.3.rs-803629/v1 |
| scFv from 14.G2a (Ref. 213) KD: 77 nM | CD3ζ | EBV-CTL expressing 14.G2a-CD3ζ CAR were expanded and maintained long-term in the presence of EBV-infected B cells (Ref. 214). However, CAR-ATC (activated general T cells) were not expanded by GD2. | NCT00085930: Initial report (Ref. 148) found CAR-CTL (EBV-CTL transduced with CAR) generated better expansion than CAR-ATC, but a later report (Ref. 215) showed that CAR-ATC persists 4 years. Clinical outcome: 3 CR (2 sustained for > 4 years), 1PR, 1SD out of 11 patients. | |
| CD28-OX40- ζ | 14g2a-CD28-OX40-Z Construct Signalling Induces Sustained Clonal Expansion (Ref. 149) | NCT01822652 (Ref. 216). 5/11 SD, among the SD, 2 became CR after salvage treatment. Higher dose plus chemo and PD1 extended survival. | ||
| KM8138 (Ref. 217) KD: 149 nM | CD28- ζ | NCT02761915 (Ref. 218): No response in 12 patients, but some response in bone marrow disease for 1 patient. | ||
| CD171 (L1-CAM) | CE7 scFv KD: 0.01 nM (patent US7354762) | CD3ζ | NCT00006480 (Ref. 219): 1 PR/ 6 patients but only for 56 days. CAR-Ts disappears in a week in high tumour burden and 42 days in limited tumour burden. | |
| CEA | scFv based on hMN14 KD: 3.4 nM (Ref. 220) | CD28-ζ | NCT01373047: Hepatic artery injection of CAR-Ts with (3) or without (3) IL2 support. 1 SD/6 (Ref. 221) | |
| scFv based on MFE23 KD: 2.3 nM (Ref. 222) | CD3ζ | NCT01212887 (Ref. 223). No clinical responses, Low persistence of CAR-Ts | ||
| Claudin 18.2 | hu8E5 scFv (Ref. 224) EC50: 45 nM | CD28-ζ | NCT03159819 (Ref. 225). 1CR, 3PR, 5SD/12 | |
| C-Met | scFvof Onartuzumab KD: 1.2 nM (Ref. 226) | 4–1BB- CD3ζ | NCT01837602 (Ref. 227). Intratumoural injection of mRNA transduced CAR-Ts, no clinical effects | |
| EGFR | scFv based on E10. KD: 20 nM (in nanobody) (Ref. 228) | 4–1BB- CD3ζ | NCT01869166 (Refs 229, 230). 1 CR, 2 PR out of 28 patients | |
| EGFRvIII | scFv based on 3C10–2173 KD of 2173: 101 nM (Ref. 231) | 4–1BB- CD3ζ | Weak antitumour effects in NSG mice (Ref. 231) | NCT02209376 (Ref. 232). No significant clinical effect |
| GPC3 | scFv of GC33 KD (by us): 1.38 nM | CD28- ζ | 2 PR out of 13 patients (Ref. 233). One patient survive more than 2 years. | |
| PMSA | scFv based on 3D8 KD: 22.5 nM (Ref. 234) | CD3ζ | NCT01929239. 2 PR effect but only last 1–2 months (Ref. 235) | |
| ROR1 | scFv based on UC-961 KD: 2 nM (Ref. 236) | 4–1BB- CD3ζ | NCT02706392 (Ref. 237). Limited mix responses in 2 /5 |
The underlying mechanism how intermediate avidity CAR-Ts generated a strong and durable antitumour effect is unclear. Weber et al. reported that in vivo persistence and antitumour effects of CAR-Ts could be improved by temporarily suppressing CAR expression (Ref. 75), suggesting that transient disengagement between CAR-T and tumour may avoid exhaustion or to restore their functionality. Recently, we developed a novel 8F8 mAb that recognised a human glypican 3 epitope next to that of the widely used GC33 mAb, but had 17 times lower affinity (Ref. 76). Despite different avidity, 8F8 and GC33 CAR-Ts possess similar in vitro function. However, in vivo study showed more 8F8-BBζCAR-Ts in the tumour than GC33 CAR-Ts. Importantly, the tumour-infiltrating 8F8 CAR-Ts were less exhausted and better functional than GC33 CAR-Ts, generating durable antitumour effects in treating HCC xenografts. The low-avidity 8F8 CAR-Ts were in a ‘lightly exhausted’ state (PD1lo), whereas high-avidity GC33 CAR-Ts exhibited ‘deeply exhausted’ (PD1hi) (Ref. 77). Compared to high-avidity CAR-Ts, the unstable engagement (Slow on/Fast off) between the low-avidity CAR-Ts and tumour cells may provide a transient break for CAR-Ts and prevent them from exhaustion and apoptosis inside solid tumours.
2.2. Hinge (H)
Hinge is the spacer between scFv to TM. It contributes to the flexibility of scFv and the synaptic distances during CAR-antigen engagement (Refs 21, 78, 79). The optimal hinge length depends on the location of the antigen epitope and steric hindrance on cancer cells. The length of the hinge should provide adequate synaptic cleft distance and enhance epitope binding by scFv (Refs 48, 69). In general, a short hinge is more suitable for CAR targeting membrane-distal epitopes (Refs 69, 78), whereas a long hinge is flexible and more effective for CAR targeting membrane-proximal epitopes that are often sterically hindered (Ref. 80). Two types of the hinge are in current CARs.
2.2.1. IgG-based hinges
Are from human IgG (IgG1 and IgG4) CH2 and CH3 domains (Ref. 57). The CH2 domain of IgG1 and IgG4 can interact with Fcγ receptors, resulting in ‘off-target’ toxicities and activation-induced cell death. Modification or deletion of the CH2 domain could minimise this side effect (Refs 21, 57). The IgG4 hinge is in one FDA-approved CAR-Ts (BREYANZI).
2.2.2. CD8α and CD28 hinges
Are naturally without FcγR binding site and used in most FDA-approved CAR-Ts (Refs 21, 57). Alabanza et al. demonstrated that CD19 CARs with H/TM from human CD28 or CD8α had similar anti-tumours in mice (Ref. 81). However, CARs with CD8α-H/TM may have a clinical advantage due to lower cytokine production. On the other hand, Majzner et al. reported that CD28-H/TM had a more stable and efficient immunological synapse that could enhance CD19 CAR-Ts to recognise low antigen (Ref. 43). In addition, CAR-T with CD28-H showed stronger lytic activity although the CAR level of CD8α-H and CD28-H are similar. CAR-Ts with H/TM from CD8α or CD28 had higher antigen-specific cytotoxicity compared to the basic CD3ζ CAR-T, indicating that TM is another crucial factor to affect CAR-T function. However, it was still difficult to identify which domain (H or TM) contributed to the different CAR-T function as the H and TM were changed simultaneously. In addition, the H length affects CAR-T function. In a recent study, six anti-VEGFR2 CARs were generated in which the H/TM from CD4, CD8α, and CD28 was exchanged individually in the basic CD3ζ CAR (Ref. 82). The data showed that longer H length (CD8α:65AA and CD28:36AA) had a higher expression that CAR with shorter H (CD4:23AA and CD3ζ:9AA). In another study (Ref. 83), a series of anti-CD19 CARs were generated based on CD19 CD8α-H/T BBζ prototype (71aa) (Kymriah) by altering H and intracellular domain, but not TM. The CD19-BBζ CAR (86aa) variant contains 86aa from CD8α with a longer extracellular fragment (55aa versus 45aa) and a longer intracellular sequence (7aa versus 3aa). These CD19-BBζ (86aa) CAR-Ts had a lower level of expansion and cytokine production than Kymriah, but achieved complete or partial remission with no neurotoxicity or CRS in 15/25 patients. This data further indicates that hinge plays a key role in CAR-T function and safety.
2.3. TM
TM was frequently selected together with upstream H and/or downstream SD. It anchors scFv to the cell membrane and contributes to CAR expression, stability, and CAR-T function (Ref. 82). CD19 CAR with CD28-TM domain was more stable on T cell surface than CD19 CAR with a CD3-ζ TM domain (Ref. 84). However, CD28-TM but not CD8 TM domain in the CAR could form a heterodimer with the endogenous CD28, which may affect CAR-T’s function (Ref. 85). The TM can also regulate CAR signalling and CAR-T function by CAR expression level (Ref. 82). H/TM domains from CD8α and CD28 are used in the FDA-approved CAR-Ts KYMRIAH and YESCARTA, respectively. In addition, the inducible co-stimulator (ICOS), including its TM, improved the anti-tumour response and increased persistence of the 3rd generation ICOS-41BBζ CAR-Ts (Ref. 86).
2.4. SD
The chimeric molecule of antibody and TCR was first created in the late 1980s, in which the VL and VH of mAb were linked to TCRα and TCRβ separately (Refs 87, 88). In the following study, scFv was linked to CD3ζ to develop the 1st generation CARs (Ref. 89). The CD3ζ remains an essential component of SD in all following generations of CARs. However, CD3ζ alone did not show significant antitumour effects in vivo (Ref. 90). The addition of 1–2 CD improved CAR-T proliferation, function, persistence, and antitumour effects. The number of CD classify the CARs into 1st (CD3ζ only), 2nd (one CD + CD3ζ), or 3rd (more than one CD + CD3ζ) generation. In general, a CD is placed proximal to the cell membrane, followed by CD3ζ (Ref. 91). Different CD may generate different effects on CAR-T activation, expansion, function, and survival (Refs 92–94).
2.4.1. CD28 and 4–1BB
Are the most frequently used CD in CAR design. All the FDA-approved CAR-Ts use CD28 or 4–1BB CD. In general, CD28 CARs confer greater functionality (Ref. 95) and higher sensitivity (Ref. 43). Using cell lines with different CD19 level, Majzner et al. demonstrated that CD28 CAR outperformed 4–1BB CAR against antigen-low tumours, whereas both CD28 and 4–1BB CARs had a similar effect against tumours with high antigen (Ref. 43). In addition, for low-avidity CAR-Ts, the CD28 domain may enhance their cytotoxicity and cytokine secretion (Ref. 94). However, the higher functionality of CD28 CAR-Ts comes at a cost. They are less persistent in vivo (Ref. 96). In contrast, 4–1BB CAR-Ts have longer persistence with more Tcm (Ref. 97). Metabolically, CD28 enhances glycolysis and 4–1BB increases mitochondrial biogenesis in T cells (Ref. 97). Interestingly, 4–1BBζ CARs can be re-engineered to enhance functional activity against antigen-low tumour and maintain persistence by incorporating 2 copies of CD3ζ or by replacing the CD8α-H/T domain with CD28 H/T domain (Ref. 43). Furthermore, the use of both CD28 and 4–1BB could improve the antitumour effect of CAR-Ts with very-low affinity scFvs (Ref. 94).
2.4.2. Other CDs
From other CD28 and TNFR families were also used in CARs. (1) ICOS belongs to the CD28 family. Incorporation of ICOS resulted in Th17 differentiation and enhanced persistence of both CD4 and CD8 CAR-Ts (Ref. 86), probably due to ICOS-induced PI3 K/Akt signalling (Refs 86, 98, 99). The CD4 CAR-Ts with ICOS have a’ helper effect’ that increase the persistence of CD8 cells expressing either CD28- or 4–1BB–based CARs. Furthermore, the 3rd-generation CAR-Ts using both ICOS and 4–1BB displayed superior antitumour effects and increased persistence in vivo compared to the 2nd-generation with ICOS or 41BB alone (Ref. 86). (2) OX40 is a member of the TNFR family. Different from CD28 required for T cell activation and IL2 production, OX40 is expressed after activation and needed for ongoing T cell proliferation and cytokine production (Ref. 100). Recently, the antigen-independent effect of various co-stimulatory receptors on T cell proliferation was evaluated by transducing T cells with CD20-BBζCAR with an additional P2A-linked full length of 12 different co-stimulatory receptors (Ref. 101). Interestingly, compared to original CD20-BBζCAR-Ts, expression of OX40 showed the greatest increase in proliferation, followed by CD27-expressing, and GITR-expressing CAR-Ts. In addition, the OX40-expressing CAR-Ts possess enhanced cytotoxicity and reduced apoptosis and exhaustion. Moreover, a co-stimulatory combination of CD28 and OX40 benefited the survival and cytotoxicity of the 3rd-generation CAR-Ts (Refs 102, 103). (3) CD27 enhanced effector cytokine production and cytotoxicity, and reduced apoptosis with upregulation of Bcl-XL protein (Ref. 104). In vivo, CD27 signalling enhanced CAR-T’s survival compared to CD28, and generated strong antitumour activity similar to CD28ζ and 4–1BBζ CAR-Ts. In a recent study, the T2–27ζ CAR-Ts targeting trophoblast cell surface antigen 2 (T2) was found to exhibit more durable antitumour activity compared to T2–28ζ CAR-Ts via promoting IL7Rα expression to enhance survival and to reduce exhaustion (Ref. 105). On the other hand, Wang et al. proposed that CD27 co-stimulation might display dual-function: the transient CD27–CD70 interaction between CD27+ CAR-Ts and tumour cells enhanced anti-tumour potency, but constitutively CD27 expression induced CAR-T exhaustion and apoptosis (Ref. 106). (4) CD3ϵ improves CAR-T function by mediating CD3ζ signal (Refs 107, 108). CD3ϵ is the only CD3 chain that downregulates Lck activity and associates with CD3ζ phosphorylation and signal transduction by recruiting inhibitory tyrosine kinase CSK. The incorporation of CD3ϵ cytoplasmic domain into CD19–28ζ CAR at membrane-proximal position results in improved antitumour effect, by negatively regulating excessive cytokine production.
3. CAR delivery vectors and expression promoters
3.1. CAR gene delivery vectors
The majority of the CAR-Ts in development and all seven FDA-approved CAR-Ts were generated by delivering CAR genes with lentivirus or retrovirus (Refs 109, 110). A small fraction of CAR-Ts was developed by transposon such as sleeping beauty (SB) and PiggyBac (PB) (Ref. 111). Compared to viral vectors, gene delivery via transposon is less immunogenic, generating stable genome integration and higher gene expression, which may benefit their clinical application (Ref. 111). However, the transduction efficacy of non-viral vector needs improvement.
3.2. Promoters and CAR expression level
The CAR level can affect CAR avidity and CAR-T function. One study found that targeted insertion of CD19–28z CAR into native TCR loci reduced CAR level by two-fold, generating a better anti-tumour effect by preventing tonic signalling and CAR-T exhaustion (Ref. 112). In contrast, high-level CARs damages CAR-T’s antitumour effects (Ref. 113). The LTR (long terminal repeat) promoter in γ retroviral vector expresses a higher level CAR and causes more CAR-T apoptosis than EF-1α promoter (Ref. 114). On the other hand, in the lentivirus-mediated CAR-T transduction, among the four promoters (CMV, EF-1α, hPGK and RPBSA), EF-1α promoter yielded the strongest cytotoxicity, especially for longer genes (Ref. 115). In another study, Ho, et al. reported that the replacement of EF1α with MND promoter in the CD19-BBζ could reduce CAR density and increase the transduction rate (Ref. 116). The MND-driven CAR-Ts had prolonged activity with low IFNγ and toxic effect in the preclinical study. ABECMA, an MND-driven CAR-T targeting B Cell Maturation Antigen, was recently approved by FDA for refractory/relapsed multiple myeloma.
4. CAR-T production
The CAR-T production process includes T cell isolation from patient peripheral blood, activation by anti-CD3/CD28 coated beads, CAR transduction, and CAR-T expansion. The optimal manufacturing process is an important step to generate high-quality CAR-Ts (Ref. 117). (1) The ratio of anti-CD3/CD28 coated beads versus T cells needs to be optimised so that the T cells are sufficiently activated, but not over-activated to be driven into exhaustion (Ref. 118). (2) In the process of CAR-T production, IL2 is present in media. The use of other common γ chain cytokines (γc-cytokines) such as IL7, IL15 and IL21, alone or in combinations IL7/IL15 (Refs 119–122), IL4/IL7, and IL4/IL7/IL21 (Ref. 123) have been reported to produce superior CAR-Ts with a higher percentage of TCM.
5. CAR signalling and CAR-T function
Proper CAR signalling is critical for CAR-T’s activation, effector function and antitumour effects (Fig. 2). As CAR contains CD3ζ and CD, CAR signalling may follow TCR signalling pathways. Two recent reviews compared the signalling of TCR and CAR (Ref. 124) and among different CARs (Ref. 125). Here, we highlight the main points.
Figure 2.
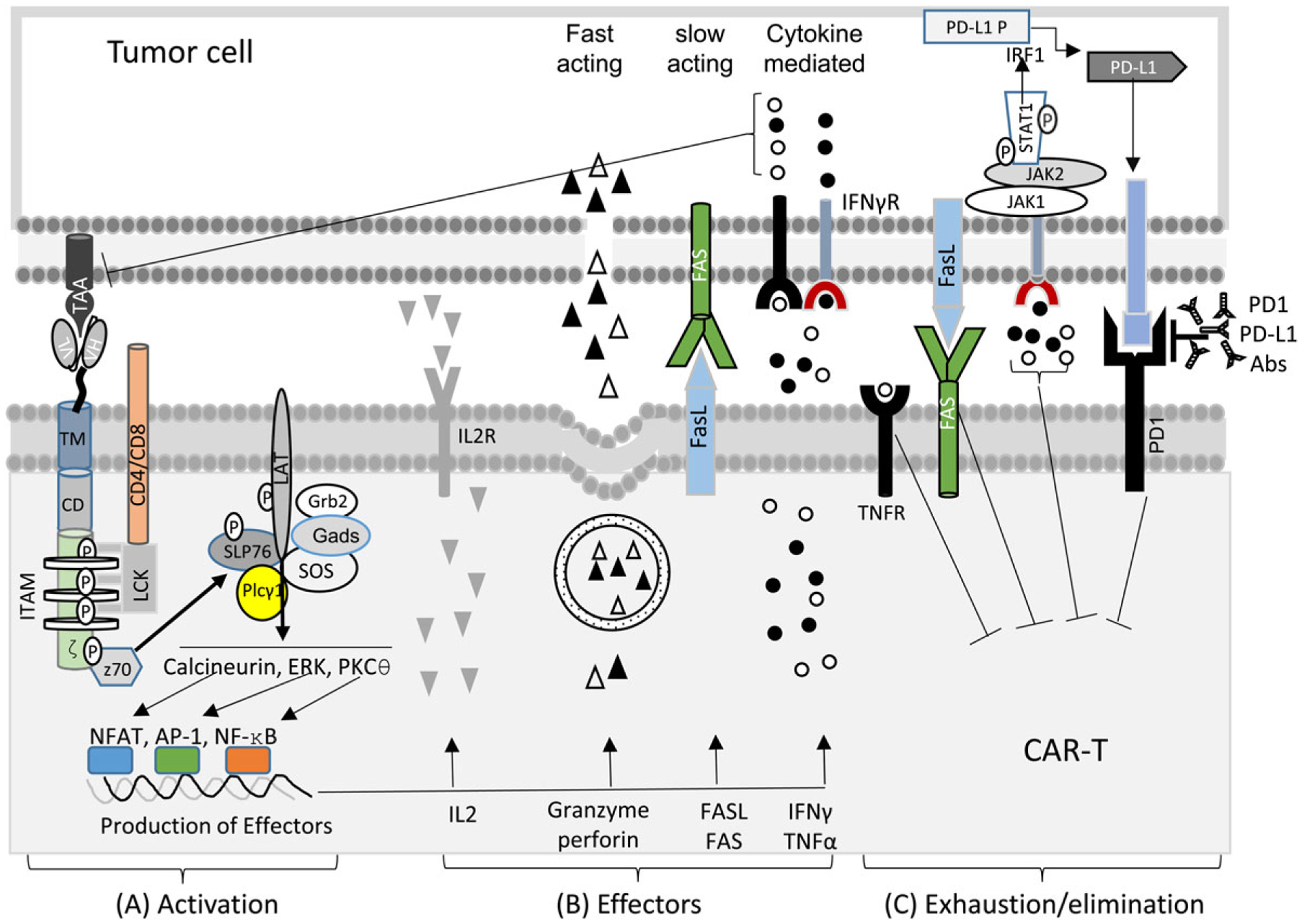
The schematic diagram of CAR signalling and CAR-T activation (A), the effector function of CAR-Ts to kill tumour cells (B), and the exhaustion/death of CAR-Ts by persistent tumour antigen stimulation (C).
5.1. TCR signalling
TCR signalling is well delineated (Refs 126, 127). Briefly, co-receptor CD4 or CD8 helps TCR-MHC/peptide engagement and bring leukocyte-specific tyrosine kinase (Lck) to the TCR complex. The Lck phosphorylates the immuno-tyrosine activation motif (ITAMs) in CD3ζ, CD3δ, CD3ϵ, and CD3γ, which recruit and phosphorylate Zap70. Zap70 phosphorylates a membrane-associated scaffolding protein, linker for activation of T cells (LAT), which become a docking site for other signalling proteins, including Grb2, Sos, Slp-76, and PLC-γ1. The Sos/Grb2 activates RAS guanyl nucleotide-releasing protein (RasGRP)-MAPK-Erk and transcription factor AP-1. The activated PLC-γ1 cleave phosphatidylinositol triphosphate (PIP2) to generate diacylglycerol (DAG) and inositol trisphosphate (IP3). DAG, a membrane-associated lipid, activates protein kinase C θ (PKC-θ), participating in the pathway leading to the nuclear entry of NF-κB. DAG also activate RasGRP-MAPK pathways, leading to activation of AP-1. IP3, on the other hand, stimulates the efflux of Ca2 + from ER to the cytoplasm. Elevated Ca2 + activates calcineurin, which dephosphorylates NFAT. Dephosphorylated NFAT enters the nucleus to join AP-1 and NF-κB to induce specific gene transcription. Productive T cell activation also needs co-stimulation from CD28, CD2, CD5, CD30, 4–1BB, OX40, LFA-1 and ICOS.
5.2. CAR signalling
CAR combines CD3ζsignal 1) and co-stimulatory (signal 2) and may use similar signalling machinery as natural TCR (Refs 124, 125, 128) (Fig. 2A). However, there are several major difference between TCR and CAR signalling. (1) The signal triggering of TCR and CAR may be different (Ref. 124). While TCR does not need clustering (Ref. 129), CAR requires oligomerisation (Ref. 130), to initiate signalling. Thus, soluble antigen does not normally activate CAR-Ts. (2) CAR forms a nonclassical immunological synapse (Ref. 131). (3) The sensitivity of TCR and CAR is different. TCR can detect as few as one MHC/peptide complex, while CAR requires at least hundreds of antigen even though it has a higher affinity (Ref. 44). (4) Though CD28 and 4–1BB CD are associated with PI3 K-Akt and TNFR-associated factors (TRAFs), respectively (Ref. 125), they activated similar signalling proteins based on phosphor-proteomic analysis (Ref. 128). In addition, compared to 4–1BBζ, CD28ζ CARs activated faster and higher-magnitude changes in protein phosphorylation, correlating with effector T cell phenotype and function. (5) The 3rd gen CD28-BBζ CAR-Ts showed enhanced activation by phosphorylation level of the signalling proteins than 2nd gen CAR-Ts (Ref. 132), and may benefit antitumour effect of CAR-Ts with very-low-affinity scFvs (Ref. 93). However, a contradictory report does exist, which showed 2nd gen CAR promoted stronger TCR signalling than 3rd gen CARs (Ref. 133).
5.3. CAR-T function
CAR-Ts may use a similar mechanism as natural T cells to exert their cytotoxicity and to undergo exhaustion after strong and persistent engagement with tumour cells (Ref. 134) (Fig. 2B, 2C). (1) Fast-acting lytic activity. This fast-acting cytotoxicity depends on the perforin/granzyme B (Ref. 135). Granzyme B induces caspase-dependent and independent apoptosis whereas perforin enables granzyme B enters target cells by forming pore (Ref. 136). (2) Slow-acting lytic activity. Fas/FasL mediates caspase-dependent apoptosis (Ref. 136). The Fas/FasL pathway plays a key role in the on-target killing of antigen-positive as well as off-target ‘bystander’ killing of antigen-negative tumour cells (Ref. 137), preventing tumour escape. But, Fas/FasL also cause the death of stimulated T cells (Ref. 138). A recent report showed that CAR-Ts were prone to Fas-mediated cell death (Ref. 139), whereas the use of Fas-DNR (dominant-negative receptors) engineered CAR-Ts resulted in intrinsic disruption of Fas signalling, and leading to longevity and superior antitumour efficacy (Ref. 140). (3) IFNγ and TNFα mediated cell death. Activated CAR-Ts secrete IFNγ and TNFα into the target cell by exocytosis and induce tumour cell death (Ref. 141). Similar to Fas/FasL, TNFα and IFNγ can induce bystander eradication of antigen-lost tumour variants. However, the role of TNFα and IFNγ on tumour progression and immunotherapy is paradoxical. TNFα can promote tumour progression by inducing TNFα-dependent tumour cell dedifferentiation (Ref. 142), triggering TNF/TNFR1 signalling and T cell death (Ref. 143), and promoting Treg (Ref. 144). Similar to TNFα, IFNγ exposure reduced antigen expression (Ref. 145). IFNγ is also a strong inducer of PD-L1 expression on tumour cells, which increase CAR-T exhaustion. Furthermore, the increase of these cytokines may lead to cytokine release syndrome and activation-induced cell death (Refs 146, 147).
6. CAR-Ts for solid tumours
Most human tumours are solid tumours. Scientists and physicians had been studying CAR-T therapy on solid tumours for nearly two decades (Refs 148–150). However, compared to the ~80% complete remission in blood cancers (Ref. 151), CAR-T therapy in solid tumours generated limited effects even to this day. One meta-analysis of 22 clinical trials and 262 patients showed only a 9% of response rate (Ref. 152). In another review of CAR-T solid tumour therapy (Ref. 153), the authors summarised 42 clinical trials of 375 patients. There were 13 complete responses, 35 partial responses, four mixed responses, and 121 stable diseases. The low efficacy of solid tumours may attribute to various reasons, which include antigen heterogeneity, low tumour infiltration, suppressive TME, short CAR-T survival, and life-threatening toxicities. In this section, we are going to review potential strategies to improve the efficacy of CAR-Ts in treating solid tumours.
6.1. Optimise CAR design for better tumour recognition
As discussed in section 2, recognition of tumour cells by CAR-Ts can be optimised by adjusting CAR avidity, epitope location, the hinge, TM, and the signalling domain (Ref. 43). In addition, optimal CAR design includes targeting multiple epitopes on the same antigen (Refs 61, 154), a 2nd different target antigen (Ref. 40), 2 or more antigens sequentially (Tandem CARs) (Ref. 41) or simultaneously (Ref. 155).
6.2. Co-express protein payloads
The 4th generation CAR-Ts, also named as Truck (T cells redirected for antigen-unrestricted cytokine-initiated killing), are CAR-Ts with a constitutive or inducible transgenic ‘payload’ including cytokines (IL7, IL12, IL15, etc.) (Ref. 156) or chemokines (CCL19) (Refs 157–159), which improve TME (Ref. 160), prevent CAR-T exhaustion (Ref. 120), reactivate endogenous innate lymphocytes to exert their anti-tumour responses (Ref. 159), or increase CAR-T trafficking and infiltration (Ref. 161). Furthermore, the 5th generation CAR-Ts were being developed by incorporating the JAK-STAT activation domain between CD28 and CD3ζ, leading to increased proliferation and persistence but decreased differentiation of CAR-Ts (Ref. 162).
6.3. Co-express immunostimulatory RNA
In the latest study (Ref. 163), June and colleagues reported that co-expression of RN7SL1 RNA enhanced CAR-T expansion and persistence, and reduced exhaustion. Importantly, the RN7SL1 RNA could activate myeloid-derived dendritic cells via RIG1/MDA5 pathway and activation of the endogenous T cell responses, generating broader antitumour immune responses to target tumour variants with antigen-loss.
6.4. Develop variants of mAb/TCR chimaera
The native TCR complex contains 10 ITAMs on CD3ζ, γ, ϵ, and δ. In CAR, there are only three ITAMs in the CD3ζ. The use of chimeric VL (VH) with different TCR chains will likely incorporate a native TCR complex to improve CAR-T sensitivity and reduce toxicity. Several studies reported the combination of Fab or scFv with native TCR or co-receptors (Fig. 3).
Figure 3.
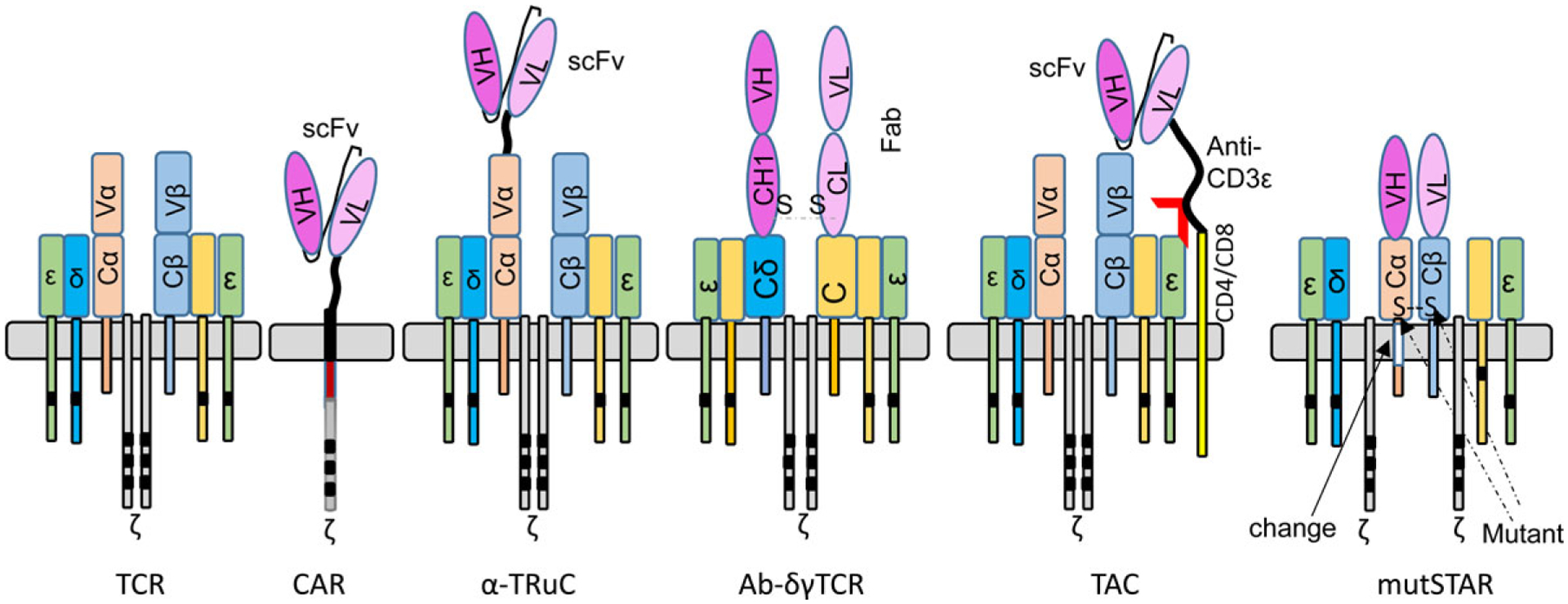
Different variants of chimeric antibody-TCRs.
6.4.1. TRuC (TCR fusion construct)
In TRuC, scFv is fused to one of the intact TCR chains (except ζ chain) (Ref. 164). The data show that some TRuC (ϵ-TRuC, scFv fused to intact CD3ϵ) generated better antitumour effects with less cytokine-mediated toxicity.
6.4.2. Ab-γδTCR
Another format of mAb/TCR chimaera is the Ab-γδTCR, which has the Fab domain of H and L chain links to CD3δ or CD3γ chain (Ref. 165). The CD19 Ab-γδTCR engineered T cells are less exhausted and lower cytokine production and, but generated compared antitumour effects.
6.4.3. TAC (T cell antigen coupler)
TAC has the scFv for antigen binding fused anti-CD3ϵ domain and then linked to CD4 or CD8 molecules (Ref. 166). While the scFv binds tumour cells, its anti-CD3ϵ domain engages with native TCR. In solid tumour models, HER2 TAC-T cells outperformed CD28ζ CAR-Ts with increased tumour infiltration and anti-tumour efficacy and reduced toxicity.
6.4.4. muSTAR
Different from the original CAR design (Ref. 87), in the muSTAR (mutated Synthetic TCR with Antigen Receptor) (Ref. 167), the VL and VH were connected to mutant mouse TCRα or β constant region. The mutant TCR constant region will pair with each other and reduce mispairing with endogenous TCRα and β chains. The muSTAR engineered T cells had overall advantages compared to CAR-Ts. They (1) recognise both antigen high or low tumour cells to avoid ‘tumour escape’; (2) have less auto-activation and tonic signalling; (3) generate stronger tumour killing capacity; (4) have earlier and higher tumour infiltration; (5) are less exhausted. Further improvement in persistence, activity/proliferation of CAR-Ts could be achieved by developing dual targeting muSTAR and by incorporating IL2Rβ/costimulatory domain. Thus, the muSTAR has superior potential for solid tumour treatment by combining the advantage from CAR for antigen-specific recognition and advantage from TCR for proper recruitment of other molecules for signal transduction. The muSTAR leads to increased T cell activity and less toxic effect, probably by becoming an integral part of the TCR complex (Ref. 164).
6.5. Improve CAR-T traffic and infiltration into tumour
For solid tumours, insufficient CAR-T migration, infiltration, and intratumour expansion and accumulation are the likely causes of failure in therapy (Refs 56, 168–171). Solid tumour mass is characterised by abnormal angiogenesis and stromal formation, altered chemokine expression, inhibitory checkpoint receptors and hypoxia environment, which are in favour of recruiting immunosuppressive cells such as Tregs but preventing effector immune cells (Ref. 172). Several approaches can overcome these obstacles to enhance CAR-T tumour infiltration.
6.5.1. Chemo and irradiation
They are an integral part of ACT as they are often used to pre-condition patients. They induce lymphodepletion to free space for incoming CAR-Ts and modify TME to attract T cells and create a favourable milieu for them to exert their function (Ref. 173). A short course of radiotherapy or chemotherapy before CAR-T treatment benefited CAR-T migration/infiltration by depleting the immunosuppressive cells such as Treg in TME (Refs 168, 174). For example, cyclophosphamide treatment generated pro-inflammatory myeloid and T cell signatures in tumours, and enhanced the recruitment of antigen-presenting cells, as well as endogenous and adoptively transferred T cells, resulting in long-term anti-tumour immunity (Ref. 175). In another study, Lin et al. reported that ionising irradiation enhanced IL8 secretion by brain tumour cells and that the CD70-specific CAR-Ts co-expressing IL8 receptors CXCR1 and CXCR2 markedly enhanced CAR-T tumour infiltration and antitumour effects (Ref. 176).
6.5.2. Targeting stroma cells
Facilitates CAR-Ts across the extracellular matrix (ECM) barrier into tumour lesion. Recent studies showed that CAR-Ts co-expressing heparanase (often lost during CAR-T preparation), an enzyme that degrades heparan sulphate proteoglycans, the main components of ECM, significantly increased tumour infiltration (Ref. 177). Another study showed that inhibition of the NADPH oxidase 4 (NOX4), an enzyme upregulated by cancer-associated fibroblast in the stroma, increased CAR-T tumour infiltration (Ref. 178).
6.5.3. Chemokine/chemokine receptors
Play important role in CAR-T migration and infiltration into tumours. Tumour cells can produce certain chemokines in their abnormal growth whereas CAR-Ts often do not express the chemokine receptors. Improved CAR-T infiltration have been reported via the expression of IL7/CCL9 (Ref. 179) that is essential for the recruitment of T cells and DCs from the periphery. In addition, expression of CCR2 (Ref. 180), CXCR2 (Ref. 181) or CCR4 (Ref. 161) on CAR-Ts improved infiltration by binding to the CCL2 or IL8 or CCL17/CCL22 elevated in TME. Other pairs of chemokine/chemokine receptors such as CCL5 and CXCL9 may also improve CD8 CAR-T infiltration (Ref. 182). On the other hand, CXCL12 produced by stromal fibroblasts may prevent T cell infiltration and should be avoided (Ref. 183).
6.6. Improve CAR-T survival
CAR-T survival inside the tumour is a critical factor for generating antitumour efficacy. Several strategies are being studied to enhance their survival. (1) The CAR-Ts derived from TN/TCM have an enhanced cytokine production with longevity (Refs 18–20, 184). (2) The affinity of ABD may be important for CAR-T survival, especially in solid tumour mass. We analysed 14 complete trials of CAR-T therapy in solid tumours (Table 2). We found that CAR-Ts constructed from intermediate affinity scFv (KD from 20–100 nM) generated some clinical responses in all 4 trials. In contrast, 8 other clinical trials with high-affinity CAR-Ts (KD from 0.2–11 nM) showed no clinical responses except one. All the responders have a longer persistence of CAR-Ts. Intermediate avidity CAR-Ts can prevent them from exhaustion and apoptosis due to transient break from continuous stimulation inside solid tumours (Refs 75, 77). (3) 4–1BB enhanced mitochondrial biogenesis that benefit for CAR-T persistence and function in hypoxia environment (Ref. 97). (4) The 3rd generation CAR-Ts with ICOS-4–1BB (Ref. 86) or CD28-OX40 have longer survival than the 2nd generation CAR-Ts (Refs 102, 103). 4) Co-expression of stimulatory cytokines and chemokines: IL4/IL7 (Ref. 185) and IL-4/IL21 (Ref. 186) receptors increase STAT5/STAT3 phosphorylation and improve CAR-T persistence.
6.7. Improve and reprogram TME
Hostile and suppressive TME severely hinder CAR-Ts to exert their function inside tumour lesion. Improving or reprograming TME (Ref. 187) could enhance CAR-Ts efficacy in solid tumours. (1) Counteract immune inhibitory molecules: Co-expression of TGFβ receptor (Ref. 188) (or TGFβ dominant-negative receptor (DNR) (Ref. 189) by CAR-Ts can competitively bind TGFβ to block its immunosuppressive effect. Secondly, co-expression of PD1 DNR (Ref. 190) or Fas DNR (Ref. 140) on CAR-Ts can block the PD1-PD-L1 inhibitory interaction. Similarly, a combination of PD1-PD-L1 checkpoint inhibitor (Ref. 191) and tyrosine kinase inhibitor (dasatinib) (Ref. 75) also enhanced TME and improved CAR-T survival and activity. In addition, CAR-Ts with ‘payload’ cytokines of IL12 (Ref. 192), IL18 (Ref. 193) or IL23 (Ref. 194) inhibited Treg in TME. (2) Deplete or reprogram suppressive cells: Treg could be depleted to increase CD8 T effector function and antitumour immunity (Ref. 195). Interestingly, down-regulation of Helios could convert Treg into T effectors (Ref. 196). Another study showed knockout of Nrp 1 made Treg lost its inhibitory function and promote antitumour immunity (Ref. 197). Secondly, tumour associated macrophages could also be reprogrammed to become tumouricidal (Ref. 198). Thus, reprograming tumour macrophages and other myeloid cells is another strategy to improve TME (Ref. 199). (3) Remodel tumour vasculature: The neovasculature in the tumour lesion can be targeted to change TME. A study showed that angiogenesis inhibitor Bevacizumab could remodel tumour vasculature to increase tumour infiltration of anti-GD2 CARTs and enhance antitumour effects (Ref. 200). (4) Other approaches such as targeting metabolism (Ref. 201) and tumour stroma (Ref. 202) may also improve TME and create a favourable environment for CAR-Ts to exert their antitumour function.
6.8. Reduce CAR-T toxicity
CAR-T toxicities include on-target/off-tumour toxicity, cytokine release syndrome (CRS), and neurotoxicity. Optimising parameters in the ‘CAR design’ (section 2) can reduce CAR-T toxicity. Other strategies are also studied to reduce CAR-T’s toxicity that are well-reviewed (Ref. 203). Here, we focus on two novel strategies in CAR design to reduce toxicity.
6.8.1. The SynNotch switch
Notch is a transmembrane receptor. The transmembrane domain (TM) and its connected part of the extracellular domain form the ‘notch core’. Upon engaging with the ligand, the intracellular domain will be released downstream of the notch core by protease. Lim’s group found that both the extracellular and intracellular domains could be replaced as long as the notch core is retained to create SynNotch (Refs 204, 205). This modular synNotch design provides extraordinary flexibility in engineering T cells with scFv or other ligands targeting specific tumour antigen. Upon engagement, it releases transcriptional factor to drive CAR expression to target another tumour antigen to initiate killing activity. Such design makes sure the CAR-Ts needs to target 2 tumour antigens to initiate killing to avoid toxicity. It will also restrict the cytotoxicity inside the tumour mass (Refs 206, 207).
6.8.2. Co-LOCKR protein switch
Recently, Lajoie et al. designed a co-localisation dependent protein switch, latching orthogonal cage–key protein (LOCKR) (Ref. 208). This Co-LOCKR switch enables AND, OR and NOT Boolean logic operations. (1) They implemented AND gates to redirect T cell target tumour cells expressing two surface antigens while avoiding off-target recognition of single-antigen cells. (2) They also tested the three-input switches that add NOT or OR logic to avoid or include cells expressing the third antigen. Such Co-LOCKR design will increase specificity against tumour cells and thus reduce toxicity.
7. Expert and topical summary
In this review, we gave a comprehensive analysis of different factors that may affect CAR-T’s antitumour efficacy. While a clear strategy for designing effective CAR-Ts for solid tumours is still lacking, we think the following aspects should be considered in CAR design for solid tumours. (1) CAR-Ts capable of strong in situ expansion and persistence inside tumour mass: The CAR-Ts should undergo antigen-driven expansion, persist and exert their tumour killing activity inside tumour lesion, and generate memory immune responses after tumour regression. Based on current analysis, a combination of TN/TCM subsets and intermediate avidity CAR may generate expandable and persistent CAR-Ts. As demonstrated in Table 2, 7 of 8 high avidity CAR-Ts trials ended up with no clinical responses, future clinical trials should test more on the intermediate affinity CAR-Ts. In addition, innovative design of CAR-Ts that allow CAR expression only when they encounter target tumour cells may help avoid CAR-T exhaustion and deletion, achieving potent antitumour effects in solid tumours. (2) Invoking endogenous antitumour immunity: Due to the antigen heterogeneity of solid tumours, it is unlikely that CAR-Ts targeting one antigen will generate persistent antitumour immunity to achieve complete responses. Designing CARs targeting multiple tumour antigens in tandem or simultaneously may not only prevent immune escape and relapse, but also help avoid off-tumour toxicity. In addition, we think one important outcome of CAR-T immunotherapy should include the activation of endogenous immune cells to invoke broad antitumour immunity. In this case, CAR-Ts kill tumour cells and release tumour antigens (shared or neoantigens) that can be taken up by proper dendritic cells to cross-prime a patient’s own immune system. Thus, CAR-Ts will serve as an immune initiator in addition to the direct killing of tumour cells. The incorporation of an innate activator in the CAR-T to activate dendritic cells will facilitate the activation of the patient’s own antitumour immunity. A recent report demonstrated that CAR gene engineered macrophage (CAR-M) could effectively activate endogenous antitumour immune response (Ref. 209), suggesting that CAR-Ts and CAR-M may work together to eradicate tumour mass and invoke endogenous broad spectrum of antitumour immunity. (3) Targeting TME: Co-expression of immune therapeutics may create a favourable TME for CAR-Ts to exert effector function to generate antitumour effects inside tumour lesions. Delivery of immune-stimulatory cytokines and chemokines, immune-stimulatory RNA, or immune checkpoint inhibitors may not only improve TME, but also can achieve direct antitumour effects and invoke endogenous immune system to achieve complete and persistent antitumour immunity.
Acknowledgement.
The authors like to thank current and previous members of Dr He’s laboratory. In addition, over the years, research in He’s lab has been funded by NIH/NCI grants (CA168912 and CA235159) and Augusta University startup package.
References
- 1.Tumeh PC et al. (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Docta RY et al. (2019) Tuning T-cell receptor affinity to optimize clinical risk-benefit when targeting alpha-fetoprotein-positive liver cancer. Hepatology 69, 2061–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu W et al. (2018) Identification of alpha-fetoprotein-specific T-cell receptors for hepatocellular carcinoma immunotherapy. Hepatology 68, 574–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.June CH and Sadelain M (2018) Chimeric antigen receptor therapy. New England Journal of Medicine 379, 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg SA et al. (2008) Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nature Reviews Cancer 8, 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garber K (2018) Driving T-cell immunotherapy to solid tumors. Nature Biotechnology 36, 215–219. [DOI] [PubMed] [Google Scholar]
- 7.Manfredi F et al. (2020) TCR Redirected T cells for cancer treatment: achievements, hurdles, and goals. Frontiers in Immunology 11, 1689. doi: 10.3389/fimmu.2020.01689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Q et al. (2021) Engineered TCR-T cell immunotherapy in anticancer precision medicine: pros and cons. Frontiers in Immunology 12, 658753. doi: 10.3389/fimmu.2021.658753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newick K et al. (2017) CAR T cell therapy for solid tumors. Annual Review of Medicine 68, 139–152. [DOI] [PubMed] [Google Scholar]
- 10.Miliotou AN and Papadopoulou LC (2018) CAR T-cell therapy: a New Era in cancer immunotherapy. Current Pharmaceutical Biotechnology 19, 5–18. [DOI] [PubMed] [Google Scholar]
- 11.June CH et al. (2018) CAR T cell immunotherapy for human cancer. Science (New York, N.Y.) 359, 1361–1365. [DOI] [PubMed] [Google Scholar]
- 12.Depil S et al. (2020) ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nature Reviews. Drug Discovery 19, 185–199. [DOI] [PubMed] [Google Scholar]
- 13.Kim DW and Cho JY (2020) Recent advances in allogeneic CAR-T cells. Biomolecules 10, 263. doi: 10.3390/biom10020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farhood B, Najafi M and Mortezaee K (2019) CD8(+) Cytotoxic T lymphocytes in cancer immunotherapy: a review. Journal of Cellular Physiology 234, 8509–8521. [DOI] [PubMed] [Google Scholar]
- 15.Borst J et al. (2018) CD4( + ) T cell help in cancer immunology and immunotherapy. Nature Reviews Immunology 18, 635–647. [DOI] [PubMed] [Google Scholar]
- 16.Yang Y et al. (2017) TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Science Translational Medicine 9, eaag1209. doi: 10.1126/scitranslmed.aag1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D et al. (2018) Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight 3, e99048. doi: 10.1172/jci.insight.99048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turtle CJ et al. (2016) CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. Journal of Clinical Investigation 126, 2123–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sommermeyer D et al. (2016) Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior anti-tumor reactivity in vivo. Leukemia 30, 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agarwal S et al. (2020) In vivo generation of CAR T cells selectively in human CD4(+) lymphocytes. Molecular Therapy 28, 1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jayaraman J et al. (2020) CAR-T design: elements and their synergistic function. EBioMedicine 58, 102931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Restifo NP and Gattinoni L (2013) Lineage relationship of effector and memory T cells. Current Opinion in Immunology 25, 556–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klebanoff CA et al. (2005) Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proceedings of the National Academy of Sciences of the USA 102, 9571–9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hinrichs CS et al. (2009) Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proceedings of the National Academy of Sciences of the USA 106, 17469–17474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berger C et al. (2008) Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. Journal of Clinical Investigation 118, 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLellan AD and Ali Hosseini Rad SM (2019) Chimeric antigen receptor T cell persistence and memory cell formation. Immunology and Cell Biology 97, 664–674. [DOI] [PubMed] [Google Scholar]
- 27.Busch DH et al. (2016) Role of memory T cell subsets for adoptive immunotherapy. Seminars in Immunology 28, 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadelain M, Riviere I and Riddell S (2017) Therapeutic T cell engineering. Nature 545, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bailey SR et al. (2017) Human CD26(high) T cells elicit tumor immunity against multiple malignancies via enhanced migration and persistence. Nature Communications 8, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson MH et al. (2020) Identification of human CD4(+) T cell populations with distinct antitumor activity. Science Advances 6, eaba7443. doi: 10.1126/sciadv.aba7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei J et al. (2019) Target selection for CAR-T therapy. Journal of Hematology & Oncology 12, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hill JA et al. (2019) CAR-T – and a side order of IgG, to go? – immunoglobulin replacement in patients receiving CAR-T cell therapy. Blood Reviews 38, 100596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paszkiewicz PJ et al. (2016) Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. Journal of Clinical Investigation 126, 4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shalabi H et al. (2018) Sequential loss of tumor surface antigens following chimeric antigen receptor T-cell therapies in diffuse large B-cell lymphoma. Haematologica 103, e215–ee18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majzner RG and Mackall CL (2018) Tumor antigen Escape from CAR T-cell therapy. Cancer Discovery 8, 1219–1226. [DOI] [PubMed] [Google Scholar]
- 36.Grupp SA et al. (2018) Updated analysis of the efficacy and safety of tisagenlecleucel in pediatric and young adult patients with relapsed/refractory (r/r) acute lymphoblastic leukemia. Blood 132(Supplement 1), 895–895. [Google Scholar]
- 37.Oak J et al. (2018) Target antigen downregulation and other mechanisms of failure after axicabtagene ciloleucel (CAR19) therapy. Blood 132(Supplement 1), 4656–4656. [Google Scholar]
- 38.Haso W et al. (2013) Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 121, 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruella M et al. (2016) Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. Journal of Clinical Investigation 126, 3814–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fry TJ et al. (2018) CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nature Medicine 24, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang L et al. (2017) Sequential infusion of anti-CD22 and anti-CD19 chimeric antigen receptor T cells for adult patients with refractory/relapsed B-cell acute lymphoblastic leukemia. Blood 130, 846–846. [Google Scholar]
- 42.Gardner R et al. (2018) Early clinical experience of CD19 × CD22 dual specific CAR T cells for enhanced anti-leukemic targeting of acute lymphoblastic leukemia. Blood 132(Supplement 1), 278–278. [Google Scholar]
- 43.Majzner RG et al. (2020) Tuning the antigen density requirement for CAR T-cell activity. Cancer Discovery 10, 702–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe K et al. (2015) Target antigen density governs the efficacy of anti-CD20-CD28-CD3 zeta chimeric antigen receptor-modified effector CD8+ T cells. Journal of Immunology 194, 911–920. [DOI] [PubMed] [Google Scholar]
- 45.Ramakrishna S et al. (2019) Modulation of target antigen density improves CAR T-cell functionality and persistence. Clinical Cancer Research 25, 5329–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caruso HG et al. (2015) Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Research 75, 3505–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker AJ et al. (2017) Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Molecular Therapy 25, 2189–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Srivastava S and Riddell SR (2015) Engineering CAR-T cells: design concepts. Trends in Immunology 36, 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Z et al. (2019) Modified CAR T cells targeting membrane-proximal epitope of mesothelin enhances the antitumor function against large solid tumor. Cell death & disease 10, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Godwin CD et al. (2021) Targeting the membrane-proximal C2-set domain of CD33 for improved CD33-directed immunotherapy. Leukemia 35, 2496–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Velasco-Hernandez T et al. (2020) Efficient elimination of primary B-ALL cells in vitro and in vivo using a novel 4–1BB-based CAR targeting a membrane-distal CD22 epitope. Journal for Immunotherapy of Cancer 8, e000896. doi: 10.1136/jitc-2020-000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han X et al. (2017) Adnectin-Based design of chimeric antigen receptor for T cell engineering. Molecular Therapy 25, 2466–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hammill JA et al. (2015) Designed ankyrin repeat proteins are effective targeting elements for chimeric antigen receptors. Journal for Immunotherapy of Cancer 3, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie YJ et al. (2019) Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompe-tent mice. Proceedings of the National Academy of Sciences of the USA 116, 7624–7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahmad ZA et al. (2012) scFv antibody: principles and clinical application. Clinical & Developmental Immunology 2012, 980250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rafiq S, Hackett CS and Brentjens RJ (2020) Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nature reviews. Clinical oncology 17, 147–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guedan S et al. (2019) Engineering and design of chimeric antigen receptors. Molecular Therapy. Methods & Clinical Development 12, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Riano-Umbarila L et al. (2020) Comparative assessment of the VH-VL and VL-VH orientations of single-chain variable fragments of scorpion toxin-neutralizing antibodies. Molecular Immunology 122, 141–147. [DOI] [PubMed] [Google Scholar]
- 59.Cheng Y et al. (2016) A VL-linker-VH orientation-dependent single-chain variable antibody fragment against rabies virus G protein with enhanced neutralizing potency in vivo. Protein and Peptide Letters 23, 24–32. [DOI] [PubMed] [Google Scholar]
- 60.Gorovits B and Koren E (2019) Immunogenicity of chimeric antigen receptor T-cell therapeutics. BioDrugs 33, 275–284. [DOI] [PubMed] [Google Scholar]
- 61.Zhao Y et al. (2019) Treatment with humanized selective CD19CAR-T cells shows efficacy in highly treated B-ALL patients who have relapsed after receiving murine-based CD19CAR-T therapies. Clinical Cancer Research 25, 5595–5607. [DOI] [PubMed] [Google Scholar]
- 62.Sommermeyer D et al. (2017) Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia 31, 2191–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stoiber S et al. (2019) Limitations in the design of chimeric antigen receptors for cancer therapy. Cells 8, 472. doi: 10.3390/cells8050472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh N et al. (2021) Antigen-independent activation enhances the efficacy of 4–1BB-costimulated CD22 CAR T cells. Nature Medicine 27, 842–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Long AH et al. (2015) 4–1BB Costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature Medicine 21, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hege K (2021) Context matters in CAR T cell tonic signaling. Nature Medicine 27, 763–764. [DOI] [PubMed] [Google Scholar]
- 67.Lynn RC et al. (2016) High-affinity FRbeta-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia 30, 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Richman SA et al. (2018) High-Affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunology Research 6, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hudecek M et al. (2013) Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clinical Cancer Research 19, 3153–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chmielewski M et al. (2004) T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. Journal of Immunology 173, 7647–7653. [DOI] [PubMed] [Google Scholar]
- 71.Oren R et al. (2014) Functional comparison of engineered T cells carrying a native TCR versus TCR-like antibody-based chimeric antigen receptors indicates affinity/avidity thresholds. Journal of Immunology 193, 5733–5743. [DOI] [PubMed] [Google Scholar]
- 72.Ghorashian S et al. (2019) Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nature Medicine 25, 1408–1414. [DOI] [PubMed] [Google Scholar]
- 73.Drent E et al. (2017) A rational strategy for reducing on-target Off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Molecular Therapy 25, 1946–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Greenman R et al. (2021) Shaping functional avidity of CAR T cells: affinity, avidity, and antigen density that regulate response. Molecular Cancer Therapeutics 20, 872–884. [DOI] [PubMed] [Google Scholar]
- 75.Weber EW et al. (2021) Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science (New York, N.Y.) 372, eaba1786. doi: 10.1126/science.aba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Caraballo Galva LD et al. (2021) Novel low-avidity glypican-3 specific CARTs resist exhaustion and mediate durable antitumor effects against hepatocellular carcinoma. bioRxiv, 2021.07.09.451642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu-Monette ZY et al. (2017) PD-1/PD-L1 blockade: have We found the Key to unleash the antitumor immune response? Frontiers in Immunology 8, 1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hudecek M et al. (2015) The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunology Research 3, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guest RD et al. (2005) The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. Journal of Immunotherapy 28, 203–211. [DOI] [PubMed] [Google Scholar]
- 80.Wilkie S et al. (2008) Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. Journal of Immunology 180, 4901–4909. [DOI] [PubMed] [Google Scholar]
- 81.Alabanza L et al. (2017) Function of novel anti-CD19 chimeric antigen receptors with human variable regions Is affected by hinge and transmembrane domains. Molecular Therapy 25, 2452–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fujiwara K et al. (2020) Hinge and transmembrane domains of chimeric antigen receptor regulate receptor expression and signaling threshold. Cells 9, 1182. doi: 10.3390/cells9051182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ying Z et al. (2019) A safe and potent anti-CD19 CAR T cell therapy. Nature Medicine 25, 947–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Savoldo B et al. (2011) CD28 Costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. Journal of Clinical Investigation 121, 1822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Muller YD et al. (2021) The CD28-transmembrane domain mediates chimeric antigen receptor heterodimerization With CD28. Frontiers in Immunology 12, 639818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guedan S et al. (2018) Enhancing CAR T cell persistence through ICOS and 4–1BB costimulation. JCI Insight 3, e96976. doi: 10.1172/jci.insight.96976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kuwana Y et al. (1987) Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochemical and Biophysical Research Communications 149, 960–968. [DOI] [PubMed] [Google Scholar]
- 88.Gross G, Waks T and Eshhar Z (1989) Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proceedings of the National Academy of Sciences of the USA 86, 10024–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eshhar Z et al. (1993) Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proceedings of the National Academy of Sciences 90, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sadelain M, Riviere I and Brentjens R (2003) Targeting tumours with genetically enhanced T lymphocytes. Nature Reviews Cancer 3, 35–45. [DOI] [PubMed] [Google Scholar]
- 91.Finney HM et al. (1998) Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. Journal of Immunology 161, 2791–2797. [PubMed] [Google Scholar]
- 92.Finney HM, Akbar AN and Lawson AD (2004) Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. Journal of Immunology 172, 104–113. [DOI] [PubMed] [Google Scholar]
- 93.Weinkove R et al. (2019) Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clinical & Translational Immunology 8, e1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Drent E et al. (2019) Combined CD28 and 4–1BB costimulation potentiates affinity-tuned chimeric antigen receptor-engineered T cells. Clinical Cancer Research 25, 4014–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhao Z et al. (2015) Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 28, 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Maude SL et al. (2018) Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New England Journal of Medicine 378, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kawalekar OU et al. (2016) Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 44, 380–390. [DOI] [PubMed] [Google Scholar]
- 98.Parry RV et al. (2003) CD28 And inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. Journal of Immunology 171, 166–174. [DOI] [PubMed] [Google Scholar]
- 99.Fos C et al. (2008) ICOS Ligation recruits the p50alpha PI3 K regulatory subunit to the immunological synapse. Journal of Immunology 181, 1969–1977. [DOI] [PubMed] [Google Scholar]
- 100.Croft M et al. (2009) The significance of OX40 and OX40L to T-cell biology and immune disease. Immunological Reviews 229, 173–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang H et al. (2021) A chimeric antigen receptor with antigen-independent OX40 signaling mediates potent antitumor activity. Science translational medicine 13, eaba7308. doi: 10.1126/scitranslmed.aba7308. [DOI] [PubMed] [Google Scholar]
- 102.Guercio M et al. (2021) CD28.OX40 co-stimulatory combination is associated with long in vivo persistence and high activity of CAR.CD30 T-cells. Haematologica 106, 987–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hombach AA and Abken H (2011) Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. International Journal of Cancer 129, 2935–2944. [DOI] [PubMed] [Google Scholar]
- 104.Song DG et al. (2012) CD27 Costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 119, 696–706. [DOI] [PubMed] [Google Scholar]
- 105.Chen H et al. (2021) CD27 Enhances the killing effect of CAR T cells targeting trophoblast cell surface antigen 2 in the treatment of solid tumors. Cancer Immunology Immunotherapy 70, 2059–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang D et al. (2019) Abstract 2321: dual-function of CD27-CD70 costimulatory signal in CAR T cell therapy. Cancer Research 79(13 Supplement), 2321–2321. [Google Scholar]
- 107.Flemming A (2020) CD3epsilon Tunes CAR T cell anticancer activity. Nature Reviews Immunology 20, 520–521. [DOI] [PubMed] [Google Scholar]
- 108.Wu W et al. (2020) Multiple signaling roles of CD3epsilon and Its application in CAR-T cell therapy. Cell 182, 855–871, e23. [DOI] [PubMed] [Google Scholar]
- 109.Brentjens RJ et al. (2011) Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118, 4817–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Noaks E et al. (2021) Enriching leukapheresis improves T cell activation and transduction efficiency during CAR T processing. Molecular Therapy. Methods & Clinical Development 20, 675–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.D’Aloia MM et al. (2018) CAR-T cells: the long and winding road to solid tumors. Cell death & disease 9, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eyquem J et al. (2017) Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Frigault MJ et al. (2015) Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunology Research 3, 356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gomes-Silva D et al. (2017) Tonic 4–1BB costimulation in chimeric antigen receptors impedes T cell survival and Is vector-dependent. Cell Reports 21, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rad SMA et al. (2020) Promoter choice: who should drive the CAR in T cells? PLoS One 15, e0232915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ho JY et al. (2021) Promoter usage regulating the surface density of CAR molecules may modulate the kinetics of CAR-T cells in vivo. Molecular Therapy. Methods & Clinical Development 21, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stock S, Schmitt M and Sellner L (2019) Optimizing manufacturing protocols of chimeric antigen receptor T cells for improved anticancer immunotherapy. International journal of molecular sciences 20, 6223. doi: 10.3390/ijms20246223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kalamasz D et al. (2004) Optimization of human T-cell expansion ex vivo using magnetic beads conjugated with anti-CD3 and anti-CD28 antibodies. Journal of Immunotherapy 27, 405–418. [DOI] [PubMed] [Google Scholar]
- 119.Zhou J et al. (2019) Chimeric antigen receptor T (CAR-T) cells expanded with IL-7/IL-15 mediate superior antitumor effects. Protein & Cell 10, 764–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu Y et al. (2014) Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19–T cells and are preserved by IL-7 and IL-15. Blood 123, 3750–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Quintarelli C et al. (2018) Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. OncoImmunology 7, e1433518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gargett T and Brown MP (2015) Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy 17, 487–495. [DOI] [PubMed] [Google Scholar]
- 123.Ptáčková P et al. (2018) A new approach to CAR T-cell gene engineering and cultivation using piggyBac transposon in the presence of IL-4, IL-7 and IL-21. Cytotherapy 20, 507–520. [DOI] [PubMed] [Google Scholar]
- 124.Wu L et al. (2020) Signaling from T cell receptors (TCRs) and chimeric antigen receptors (CARs) on T cells. Cellular & Molecular Immunology 17, 600–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lindner SE et al. (2020) Chimeric antigen receptor signaling: functional consequences and design implications. Science Advances 6, eaaz3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Courtney AH, Lo W-L and Weiss A (2018) TCR Signaling: mechanisms of initiation and propagation. Trends in Biochemical Sciences 43, 108–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hwang J-R et al. (2020) Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Experimental & Molecular Medicine 52, 750–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Salter AI et al. (2018) Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Science Signaling 11, eaat6753. doi: 10.1126/scisignal.aat6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Brameshuber M et al. (2018) Monomeric TCRs drive T cell antigen recognition. Nature Immunology 19, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chang ZL et al. (2018) Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nature Chemical Biology 14, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Davenport AJ et al. (2018) Chimeric antigen receptor T cells form non-classical and potent immune synapses driving rapid cytotoxicity. Proceedings of the National Academy of Sciences 115, E2068–E2E76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Karlsson H et al. (2015) Evaluation of intracellular signaling downstream chimeric antigen receptors. PLoS One 10, e0144787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ramello MC et al. (2019) An immunoproteomic approach to characterize the CAR interactome and signalosome. Science Signaling 12, eaap9777. doi: 10.1126/scisignal.aap9777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Benmebarek MR et al. (2019) Killing mechanisms of Chimeric Antigen Receptor (CAR) T cells. International journal of molecular sciences 20, 1283. doi: 10.3390/ijms20061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Voskoboinik I, Smyth MJ and Trapani JA (2006) Perforin-mediated target-cell death and immune homeostasis. Nature Reviews Immunology 6, 940–952. [DOI] [PubMed] [Google Scholar]
- 136.Meiraz A et al. (2009) Switch from perforin-expressing to perforin-deficient CD8(+) T cells accounts for two distinct types of effector cytotoxic T lymphocytes in vivo. Immunology 128, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Upadhyay R et al. (2021) A critical role for Fas-mediated Off-target tumor killing in T-cell immunotherapy. Cancer Discovery 11, 599–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Siegel RM et al. (2000) The multifaceted role of Fas signaling in immune cell homeostasis and autoimmunity. Nature Immunology 1, 469–474. [DOI] [PubMed] [Google Scholar]
- 139.Tschumi BO et al. (2018) CART Cells are prone to Fas- and DR5-mediated cell death. Journal for Immunotherapy of Cancer 6, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yamamoto TN et al. (2019) T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. Journal of Clinical Investigation 129, 1551–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang B et al. (2008) IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. Journal of Clinical Investigation 118, 1398–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Landsberg J et al. (2012) Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 490, 412–416. [DOI] [PubMed] [Google Scholar]
- 143.Bertrand F et al. (2015) Blocking tumor necrosis factor-alpha enhances CD8 T-cell-dependent immunity in experimental melanoma. Cancer Research 75, 2619–2628. [DOI] [PubMed] [Google Scholar]
- 144.Torrey H et al. (2017) Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Science Signaling 10, eaaf8608. doi: 10.1126/scisignal.aaf8608. [DOI] [PubMed] [Google Scholar]
- 145.Le Poole IC et al. (2002) Interferon-gamma reduces melanosomal antigen expression and recognition of melanoma cells by cytotoxic T cells. American Journal of Pathology 160, 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Montfort A et al. (2019) The TNF paradox in cancer progression and immunotherapy. Frontiers in Immunology 10, 1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jorgovanovic D et al. (2020) Roles of IFN-gamma in tumor progression and regression: a review. Biomarker Research 8, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pule MA et al. (2008) Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nature Medicine 14, 1264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pulè MA et al. (2005) A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Molecular Therapy 12, 933–941. [DOI] [PubMed] [Google Scholar]
- 150.Rossig C et al. (2002) Epstein-Barr virus–specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood 99, 2009–2016. [DOI] [PubMed] [Google Scholar]
- 151.Aamir S et al. (2021) Systematic review and meta-analysis of CD19-specific CAR-T cell therapy in relapsed/refractory acute lymphoblastic leukemia in the pediatric and young adult population: safety and efficacy outcomes. Clinical Lymphoma Myeloma and Leukemia 21, e334–ee47. [DOI] [PubMed] [Google Scholar]
- 152.Hou B et al. (2019) Efficiency of CAR-T therapy for treatment of solid tumor in clinical trials: a meta-analysis. Disease Markers 2019, 3425291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Schaft N (2020) The landscape of CAR-T cell clinical trials against solid tumors-A comprehensive overview. Cancers (Basel) 12, 2567. doi: 10.3390/cancers12092567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gu R et al. (2020) Efficacy and safety of CD19 CAR T constructed with a new anti-CD19 chimeric antigen receptor in relapsed or refractory acute lymphoblastic leukemia. Journal of Hematology & Oncology 13, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bielamowicz K et al. (2018) Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology 20, 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hurton LV et al. (2016) Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proceedings of the National Academy of Sciences of the USA 113, E7788–E7E97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chmielewski M and Abken H (2015) TRUCKs: the fourth generation of CARs. Expert Opinion on Biological Therapy 15, 1145–1154. [DOI] [PubMed] [Google Scholar]
- 158.Caraballo Galva LD et al. (2020) Engineering T cells for immunotherapy of primary human hepatocellular carcinoma. Journal of Genetics and Genomics 47, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jin J et al. (2020) Fueling chimeric antigen receptor T cells with cytokines. American Journal of Cancer Research 10, 4038–4055. [PMC free article] [PubMed] [Google Scholar]
- 160.Chmielewski M, Hombach AA and Abken H (2014) Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunological Reviews 257, 83–90. [DOI] [PubMed] [Google Scholar]
- 161.Di Stasi A et al. (2009) T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and anti-tumor activity in a Hodgkin tumor model. Blood 113, 6392–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kagoya Y et al. (2018) A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nature Medicine 24, 352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Johnson LR et al. (2021) The immunostimulatory RNA RN7SL1 enables CAR-T cells to enhance autonomous and endogenous immune function. Cell 184, 4981–4995, e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Baeuerle PA et al. (2019) Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nature Communications 10, 2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Xu Y et al. (2018) A novel antibody-TCR (AbTCR) platform combines Fab-based antigen recognition with gamma/delta-TCR signaling to facilitate T-cell cytotoxicity with low cytokine release. Cell Discovery 4, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Helsen CW et al. (2018) The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nature Communications 9, 3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Liu Y et al. (2021) Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Science Translational Medicine 13, eabb5191. doi: 10.1126/scitranslmed.abb5191. [DOI] [PubMed] [Google Scholar]
- 168.Donnadieu E et al. (2020) Surmounting the obstacles that impede effective CAR T cell trafficking to solid tumors. Journal of Leukocyte Biology 108, 1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hou AJ, Chen LC and Chen YY (2021) Navigating CAR-T cells through the solid-tumour microenvironment. Nature Reviews Drug Discovery 20, 531–550. [DOI] [PubMed] [Google Scholar]
- 170.Martinez M and Moon EK (2019) CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Frontiers in Immunology 10, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tian Y et al. (2020) Gene modification strategies for next-generation CAR T cells against solid cancers. Journal of Hematology & Oncology 13, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Scarfo I and Maus MV (2017) Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment. Journal for Immunotherapy of Cancer 5, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Pocaterra A, Catucci M and Mondino A (2022) Adoptive T cell therapy of solid tumors: time to team up with immunogenic chemo/radiotherapy. Current Opinion in Immunology 74, 53–59. [DOI] [PubMed] [Google Scholar]
- 174.Xu J et al. (2018) Combination therapy: a feasibility strategy for CAR-T cell therapy in the treatment of solid tumors. Oncology Letters 16, 2063–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Murad JP et al. (2021) Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Molecular Therapy 29, 2335–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jin L et al. (2019) CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nature Communications 10, 4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Caruana I et al. (2015) Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nature Medicine 21, 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ford K et al. (2020) NOX4 Inhibition potentiates immunotherapy by overcoming cancer-associated fibroblast-mediated CD8 T-cell exclusion from tumors. Cancer Research 80, 1846–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Adachi K et al. (2018) IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nature Biotechnology 36, 346–351. [DOI] [PubMed] [Google Scholar]
- 180.Craddock JA et al. (2010) Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. Journal of Immunotherapy 33, 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Whilding LM et al. (2019) CAR T-cells targeting the integrin alphav-beta6 and co-expressing the chemokine receptor CXCR2 demonstrate enhanced homing and efficacy against several solid malignancies. Cancers (Basel) 11, 674. doi: 10.3390/cancers11050674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dangaj D et al. (2019) Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell 35, 885–900, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Feig C et al. (2013) Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proceedings of the National Academy of Sciences of the USA 110, 20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tay RE, Richardson EK and Toh HC (2021) Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Therapy 28, 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Leen AM et al. (2014) Reversal of tumor immune inhibition using a chimeric cytokine receptor. Molecular Therapy 22, 1211–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wang Y et al. (2019) An IL-4/21 inverted cytokine receptor improving CAR-T cell potency in immunosuppressive solid-tumor microenvironment. Frontiers in Immunology 10, 1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Datta M et al. (2019) Reprogramming the tumor microenvironment to improve immunotherapy: emerging strategies and combination therapies. American Society of Clinical Oncology Educational Book / Asco. American Society of Clinical Oncology. Meeting 39, 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chang ZL, Hou AJ and Chen YY (2020) Engineering primary T cells with chimeric antigen receptors for rewired responses to soluble ligands. Nature Protocols 15, 1507–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bollard CM et al. (2002) Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood 99, 3179–3187. [DOI] [PubMed] [Google Scholar]
- 190.Cherkassky L et al. (2016) Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. Journal of Clinical Investigation 126, 3130–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rafiq S et al. (2018) Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nature Biotechnology 36, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Pegram HJ et al. (2012) Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 119, 4133–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hu B et al. (2017) Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Reports 20, 3025–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ma X et al. (2020) Interleukin-23 engineering improves CAR T cell function in solid tumors. Nature Biotechnology 38, 448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Buchan SL et al. (2018) Antibodies to costimulatory receptor 4–1BB enhance anti-tumor immunity via T regulatory cell depletion and promotion of CD8 T cell effector function. Immunity 49, 958–970, e7. [DOI] [PubMed] [Google Scholar]
- 196.Nakagawa H et al. (2016) Instability of Helios-deficient Tregs is associated with conversion to a T-effector phenotype and enhanced antitumor immunity. Proceedings of the National Academy of Sciences of the USA 113, 6248–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Overacre-Delgoffe AE et al. (2017) Interferon-γ drives treg fragility to promote anti-tumor immunity. Cell 169, 1130–1141, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Beatty GL et al. (2011) CD40 Agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (New York, N.Y.) 331, 1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Petty AJ et al. (2021) Targeting tumor-associated macrophages in cancer immunotherapy. Cancers 13, 5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bocca P et al. (2017) Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology 7, e1378843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bader JE, Voss K and Rathmell JC (2020) Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Molecular Cell 78, 1019–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Valkenburg KC, de Groot AE and Pienta KJ (2018) Targeting the tumour stroma to improve cancer therapy. Nature Reviews. Clinical Oncology 15, 366–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yu S et al. (2019) Next-generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Molecular Cancer 18, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Roybal KT et al. (2016) Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell 167, 419–432, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Morsut L et al. (2016) Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell 164, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Choe JH et al. (2021) SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Science translational medicine 13, eabe7378. doi: 10.1126/scitranslmed.abe7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hyrenius-Wittsten A et al. (2021) SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Science translational medicine 13, eabd8836. doi: 10.1126/scitranslmed.abd8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lajoie MJ et al. (2020) Designed protein logic to target cells with precise combinations of surface antigens. Science (New York, N.Y.) 369, 1637–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Klichinsky M et al. (2020) Human chimeric antigen receptor macrophages for cancer immunotherapy. Nature Biotechnology 38, 947–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Molthoff CF et al. (1992) Experimental and clinical analysis of the characteristics of a chimeric monoclonal antibody, MOv18, reactive with an ovarian cancer-associated antigen. Journal of Nuclear Medicine 33, 2000–2005. [PubMed] [Google Scholar]
- 211.Kershaw MH, Westwood JA and Hwu P (2002) Dual-specific T cells combine proliferation and antitumor activity. Nature Biotechnology 20, 1221–1227. [DOI] [PubMed] [Google Scholar]
- 212.Kershaw MH et al. (2006) A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clinical Cancer Research 12, 6106–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Cheung NK et al. (2012) Humanizing murine IgG3 anti-GD2 antibody m3F8 substantially improves antibody-dependent cell-mediated cytotoxicity while retaining targeting in vivo. Oncoimmunology 1, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Rossig C et al. (2002) Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood 99, 2009–2016. [DOI] [PubMed] [Google Scholar]
- 215.Louis CU et al. (2011) Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118, 6050–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Heczey A et al. (2017) CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Molecular Therapy 25, 2214–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Nakamura K et al. (2001) Construction of humanized anti-ganglioside monoclonal antibodies with potent immune effector functions. Cancer Immunology Immunotherapy 50, 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Straathof K et al. (2018) Abstract CT145: a cancer research UK phase I trial of anti-GD2 chimeric antigen receptor (CAR) transduced T-cells (1RG-CART) in patients with relapsed or refractory neuroblastoma. Cancer Research 78(13 Supplement), CT145–CCT45. [Google Scholar]
- 219.Park JR et al. (2007) Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Molecular Therapy 15, 825–833. [DOI] [PubMed] [Google Scholar]
- 220.Akamatsu Y et al. (1998) A single-chain immunotoxin against carcinoembryonic antigen that suppresses growth of colorectal carcinoma cells. Clinical Cancer Research 4, 2825–2832. [PubMed] [Google Scholar]
- 221.Katz SC et al. (2015) Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor–modified T-cell therapy for CEA+ liver metastases. Clinical Cancer Research 21, 3149–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Rios X et al. (2019) Immuno-PET imaging and pharmacokinetics of an anti-CEA scFv-based trimerbody and Its monomeric counterpart in human gastric carcinoma-bearing mice. Molecular Pharmaceutics 16, 1025–1035. [DOI] [PubMed] [Google Scholar]
- 223.Thistlethwaite FC et al. (2017) The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunology, Immunotherapy 66, 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Jiang H et al. (2018) Claudin18.2-specific chimeric antigen receptor engineered T cells for the treatment of gastric cancer. JNCI: Journal of the National Cancer Institute 111, 409–418. [DOI] [PubMed] [Google Scholar]
- 225.Zhan X et al. (2019) Phase I trial of claudin 18.2-specific chimeric antigen receptor T cells for advanced gastric and pancreatic adenocarcinoma. Journal of Clinical Oncology 37(15_suppl), 2509–2509. [Google Scholar]
- 226.Merchant M et al. (2013) Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proceedings of the National Academy of Sciences of the USA 110, E2987–E2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Tchou J et al. (2017) Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunology Research 5, 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Nevoltris D et al. (2015) Conformational nanobodies reveal tethered epidermal growth factor receptor involved in EGFR/ErbB2 predimers. ACS Nano 9, 1388–1399. [DOI] [PubMed] [Google Scholar]
- 229.Feng K et al. (2016) Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Science China Life Sciences 59, 468–479. [DOI] [PubMed] [Google Scholar]
- 230.Guo Y et al. (2018) Phase I study of chimeric antigen receptor–modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clinical Cancer Research 24, 1277–1286. [DOI] [PubMed] [Google Scholar]
- 231.Johnson LA et al. (2015) Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Science Translational Medicine 7, 275ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.O’Rourke DM et al. (2017) A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Science Translational Medicine 9, eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Shi D et al. (2020) Chimeric antigen receptor-glypican-3 T-cell therapy for advanced hepatocellular carcinoma: results of phase I trials. Clinical Cancer Research 26, 3979–3989. [DOI] [PubMed] [Google Scholar]
- 234.Choi J et al. (2019) Antigen-binding affinity and thermostability of chimeric mouse-chicken IgY and mouse-human IgG antibodies with identical variable domains. Scientific Reports 9, 19242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Junghans RP et al. (2016) Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. The Prostate 76, 1257–1270. [DOI] [PubMed] [Google Scholar]
- 236.Choi MY et al. (2015) Pre-clinical specificity and safety of UC-961, a first-In-class monoclonal antibody targeting ROR1. Clinical Lymphoma Myeloma and Leukemia 15, S167–SS69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Specht JM et al. (2018) Abstract CT131: a phase I study of adoptive immunotherapy for advanced ROR1+ malignancies with defined subsets of autologous T cells expressing a ROR1-specific chimeric antigen receptor (ROR1-CAR). Cancer Research 78(13 Supplement), CT131–CCT31. [Google Scholar]