

Abstract
Sensory loss leads to widespread cross-modal plasticity across brain areas to allow the remaining senses to guide behavior. While multimodal sensory interactions are often attributed to higher-order sensory areas, cross-modal plasticity has been observed at the level of synaptic changes even across primary sensory cortices. In particular, vision loss leads to widespread circuit adaptation in the primary auditory cortex (A1) even in adults. Here we report using mice of both sexes in which cross-modal plasticity occurs even earlier in the sensory-processing pathway at the level of the thalamus in a modality-selective manner. A week of visual deprivation reduced inhibitory synaptic transmission from the thalamic reticular nucleus (TRN) to the primary auditory thalamus (MGBv) without changes to the primary visual thalamus (dLGN). The plasticity of TRN inhibition to MGBv was observed as a reduction in postsynaptic gain and short-term depression. There was no observable plasticity of the cortical feedback excitatory synaptic transmission from the primary visual cortex to dLGN or TRN and A1 to MGBv, which suggests that the visual deprivation-induced plasticity occurs predominantly at the level of thalamic inhibition. We provide evidence that visual deprivation-induced change in the short-term depression of TRN inhibition to MGBv involves endocannabinoid CB1 receptors. TRN inhibition is considered critical for sensory gating, selective attention, and multimodal performances; hence, its plasticity has implications for sensory processing. Our results suggest that selective disinhibition and altered short-term dynamics of TRN inhibition in the spared thalamic nucleus support cross-modal plasticity in the adult brain.
SIGNIFICANCE STATEMENT Losing vision triggers adaptation of the brain to enhance the processing of the remaining senses, which can be observed as better auditory performance in blind subjects. We previously found that depriving vision of adult rodents produces widespread circuit reorganization in the primary auditory cortex and enhances auditory processing at a neural level. Here we report that visual deprivation-induced plasticity in adults occurs much earlier in the auditory pathway, at the level of thalamic inhibition. Sensory processing is largely gated at the level of the thalamus via strong cortical feedback inhibition mediated through the thalamic reticular nucleus (TRN). We found that TRN inhibition of the auditory thalamus is selectively reduced by visual deprivation, thus playing a role in adult cross-modal plasticity.
Keywords: adult plasticity, cortical plasticity, cross-modal plasticity, inhibitory plasticity, MGB, TRN
Introduction
The adult brain is less plastic than juveniles, yet some forms of plasticity are robustly expressed. In particular, cross-modal sensory deprivation induces a rather rapid functional cortical plasticity across the primary sensory cortices, even in adults (Lee and Whitt, 2015; Ewall et al., 2021). For example, depriving vision of adult mice for a few days potentiates thalamocortical synapses in the primary auditory cortex (A1; Petrus et al., 2014) accompanied by remodeling and refinement of its local circuitry (Meng et al., 2015; Petrus et al., 2015; Meng et al., 2017). These changes correlate with enhanced auditory processing of A1 neurons (Petrus et al., 2014; Solarana et al., 2019) and may be a cellular basis for enhanced auditory performance in blind individuals (Röder et al., 1999; Pang et al., 2020). Cross-modal plasticity observed in the A1 circuit following a week of visual deprivation occurs without significant changes in the external sound environment and vocalization (Petrus et al., 2014), which suggests that the plasticity may be driven by central adaptation of neural circuits involved in propagating auditory information to A1. Auditory information reaching A1 is controlled at the primary auditory thalamus [ventral medial geniculate body (MGBv)] by strong corticothalamic feedback inhibition mediated by the thalamic reticular nucleus (TRN; Zhang et al., 2008). The TRN has been suggested to play a role as a “searchlight” that enables selective sensory gating (Crick, 1984) and has been shown to be critical for attentional control during multisensory tasks (Ahrens et al., 2015; Wimmer et al., 2015). Whether TRN inhibition undergoes plasticity on changes in sensory experience, especially in the adult brain, is unknown. Here we report that a short duration of visual deprivation in adults reduces TRN inhibition selectively to the MGBv without changes to the primary visual thalamus [dorsal lateral geniculate nucleus (dLGN)]. Furthermore, we did not observe any change in the strength of corticothalamic feedback from the primary visual cortex (V1) to dLGN, V1 to TRN, and A1 to MGBv, which suggests that the plasticity is expressed in a modality-specific manner at the level of thalamic inhibition. We also provide evidence that visual deprivation-induced plasticity of TRN inhibition to MGBv may engage endogenous endocannabinoid action via CB1 receptors (CB1-Rs). Our results suggest that the adult thalamic inhibitory circuit is a site of cross-modal plasticity, which can support the processing of the spared sensory modality.
Materials and Methods
Animals.
Both male and female mice of PV-Cre [B6;129P2-Pvalbtm1(cre)Arbr/J; stock# 008069, The Jackson Laboratory; RRID:IMSR_JAX:008069], Ntsr1-Cre [B6.FVB(Cg)-Tg(Ntsr1-cre)GN220Gsat/Mmucd; stock# 030648-UCD, MMRRC; RRID:MMRRC_030648-UCD], Ntsr1-Cre crossed with Ai32 [B6;129S-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze/J; stock# 012569, The Jackson Laboratory; RRID:IMSR_JAX:012569], and SOM-Cre [Ssttm2.1(cre)Zjh/J, stock #013044, The Jackson Laboratory; RRID:IMSR_JAX:013044] crossed with Ai14 [B6.Cog-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J; stock #007914, The Jackson Laboratory; RRID:IMSR_JAX:007914; red] were used in this study. All animals were housed in a normal 12 h light/dark cycle through adulthood with food and water available ad libitum. One week before electrophysiological recordings, animals were randomly split into the following two groups: one group remained in the normal light/dark cycle; and one group underwent dark exposure (DE; see below for details). All animal procedures conform to the guidelines of the US Department of Health and Human Services Office of Laboratory Animal Welfare and are approved by the Institutional Animal Use and Care Committee at Johns Hopkins University.
In vivo stereotaxic injections.
To express channelrhodopsin-2 (ChR2) in TRN, PV-Cre mice were used. Mice were reared normally until postnatal day 25 (P25) to P30 at which point animals underwent surgery to inject double-floxed ChR2 (AAV9.Ef1alpha.dflox.hChR2(H134R)mCherry.WPRE.hGH, Penn Vector Core; stock #20297, Addgene; RRID:Addgene_20297) containing viral vectors selectively into the TRN using stereotaxic pressure injections. Each mouse was anesthetized under 1.5−2% isoflurane/oxygen and head fixed on a stereotactic device (Kopf Instruments) for craniotomy. Surgical procedure was done under aseptic conditions. A small bone drill (Micro Drill, Fine Science Tools) was used to remove the skull just above the injection sites. TRN was targeted bilaterally by two separate injections [coordinates: anteroposterior (AP) = −0.7, mediolateral (ML) = 1.8, dorsoventral (DV) = 3; and AP = −1.22, ML = 1.8, DV = 3]. Mice recovered on a heated pad (30°C) and were returned to the animal colony and housed with approximately two to three same-sex mice until the experimental procedures to allow 8–10 weeks of viral incubation.
To express ChR2 in V1 layer 6 (L6), Ntsr1-cre mice [B6.FVB(Cg)-Tg(Ntsr1-cre)GN220Gsat/Mmucd; stock #030648-UCD, MMRRC; ∼3–5 weeks old] underwent intracranial viral injections using the same procedures as above, except that AAV9-dflox-hChR2(H134R)-EYFP-WPRE-hGH (stock #20298, Addgene; RRID:Addgene_20298) was injected via pressure injection (Pump 11 Elite, Harvard Instruments) into V1 L6. Coordinates for V1 L6 with respect to bregma are as follows: lateral, 2.63; posterior, 3.78; ventral, 1.13. Coordinates were scaled according to the distance between bregma and λ on individual mice. For V1 L6 to dLGN eEPSC short-term plasticity studies (see Fig. 5), we used pups from Ntsr1-Cre cross-bred to Ai32 instead of targeted viral injections.
Figure 5.

DE does not alter V1 L6 corticothalamic excitatory synaptic transmission to dLGN. A, ChR2-evoked-Sr2+-mEPSCs recorded in dLGN. Left, Circuit diagram showing V1 L6 to dLGN input. Second panel from the left, Example traces of ChR2-evoked Sr2+-mEPSCs recorded in dLGN of Ntsr1-Cre;Ai32 mice. A 5 ms LED pulse duration was used. Labels are the same as in Figure 1D. Third panel from the left, Average amplitudes of Sr2+-mEPSCs (NR = 16.4 ± 1.0 pA, n = 10 cells; DE = 15.7 ± 1.3 pA, n = 7 cells; t test: t(15) = 0.4551, p = 0.6626). Bars, Mean ± SEM; gray circles, average value for each cell. Right, Average Sr2+-mEPSC traces from NR and DE. B, Short-term facilitation of V1 L6 excitation to dLGN. Left, Normalized average eEPSC amplitudes across different stimulation frequencies (5, 10, and 15 Hz). A 5 ms LED pulse duration was used. Plot, Mean ± SEM. Right, Average eEPSC traces from NR and DE across different stimulation frequencies. Two-way ANOVA; 5 Hz stimulation train: F(9,144) = 0.7509, p = 0.6617, NR = 7 cells from 2 mice, DE = 11 cells from 3 mice; 10 Hz stimulation train: F(9,135) = 1.732, p = 0.0875, NR = 7 cells, DE = 10 cells; 15 Hz stimulation train: F(9,117) = 1.285, p = 0.2524, NR = 6 cells, DE = 9 cells.
Dark exposure
Between P90 and P115, mice were randomly split into two groups. The experimental group was placed in a completely light-tight dark room for 1 week (7 ± 1 d) for short-term visual deprivation (DE), and the control group was left in a normal 12 h light/dark cycle [normal-reared (NR)] as in previous studies (Petrus et al., 2014, 2015).
Slice preparation.
Each mouse was deeply anesthetized using isoflurane vapors until the absence of corneal reflex and toe pinch response, then transcardially perfused with ice-cold N-methyl-d-glucamine (NMDG) cutting solution as follows (in mm): 92 NMDG, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 12 N-acetyl cysteine, 0.5 CaCl2, and 10 MgSO4, with pH adjusted to 7.4. They were immediately decapitated. The brain was removed and immersed in ice-cold NMDG cutting solution. The brain was then hemisected and mounted with the medial side against a block of 2% agar to stabilize the brain and prevent shearing and jitter. The block containing TRN or MGBv or dLGN was sectioned horizontally into 300-µm-thick slices (Agmon and Connors, 1991) using a vibratome (model VT1200, Leica) set to speed 0.12 and amplitude 1.65. A slow cut speed and large movement amplitude aid in attaining slices with clearly visible cells. Slices were placed in a small chamber filled with dissection buffer and bubbled with carbogen (95% O2/5% CO2). Sections were then transferred to 30°C NMDG buffer and incubated for ∼15 min before being transferred to room temperature ACSF (in mm): 124 NaCl, 5 KCl, 1.25 NaH2PO4 · H2O, 26 NaHCO3, 10 dextrose, 2.5 CaCl2, and 1.5 MgCl2, bubbled with 95% O2/5% CO2 for 1 h.
Electrophysiological recordings.
Brain slices were transferred to a submersion-type recording chamber mounted on the fixed stage of an upright microscope with oblique infrared illumination (model E600FN, Nikon) and were continually supplied with ACSF bubbled with 5% CO2/95% O2 (30°C) with a flow rate of ∼2 ml/min. Whole-cell patch-clamp recordings were performed in MGBv, dLGN, and TRN in cells that were postsynaptic to ChR2-expressing axon terminals.
IPSC recordings were conducted in the presence of 100 µm APV and 5 µm NBQX using CsCl internal solution (in mm): 140 CsCl, 1 EGTA, 6 KCl, four NaCl, 2 MgCl2, 10 HEPES, 3 ATP, 10 Na-phosphocreatine, and 0.5 GTP, at pH 7.4 and 275–285 mOsm, which produces a symmetrical Cl– gradient across the membrane. This allows the recording of IPSCs as inward currents at −80 mV holding potential (Vh). IPSCs from TRN to MGBv or dLGN were evoked by activating ChR2 in the axons by shining blue light (0.05 ms duration; 455 nm LED; Thorlabs) through a 40× objective lens. The use of this short duration of LED pulse is based on our observation from a pilot study (n = 3) that ChR2-expressing PV-Cre TRN neurons reliably generate single action potentials with pulse durations ranging approximately between 0.05 and 0.5 ms across a wide range of LED intensities (LED output, ∼10–500 mA) but start to generate multiple action potentials with a longer pulse duration (5 ms) even at low LED intensities (LED output, ≥5 mA). This differs from principal neurons in V1, which reliably generate single action potentials with a longer LED pulse duration (5 ms; Petrus et al., 2015).
EPSC recordings were conducted in the presence of 100 µm APV and 20 µm bicuculine using Cs-gluconate internal solution (in mm): 130 Cs-gluconate, 10 HEPES, 8 KCl, 1 EGTA, 4 disodium-ATP, 10 disodium-phosphocreatine, 0.5 sodium-GTP, and 5 lidocaine N-ethyl bromide, at pH 7.4 and 275–285 mOsm, at Vh of −80 mV. ChR2-containing axon terminals were activated using a 455 nm wavelength LED (5 ms duration; Thorlabs) through a 40× objective lens.
For recording light-evoked Sr2+-desynchronized miniature IPSCs (Sr2+-mIPSCs) and Sr2+-desynchronized miniature EPSCs (Sr2+-mEPSCs), external Ca2+ was replaced with 4 mm Sr2+, and Mg2+ concentration was raised to 4 mm. In some cases, to ensure that LED-evoked responses were monosynaptic, recordings were performed in the presence of 1 µm TTX and 100 µm 4-AP.
Recordings for assessing short-term plasticity of evoked EPSCs (eEPSCs) and evoked IPSCs (eIPSCs) were performed in normal ACSF (2.5 mm Ca2+, 1.5 mm Mg2+). Trains of stimuli at 5, 10, and 20 Hz were used for eIPSC studies, while 5, 10, and 15 Hz were used for eEPSCs. For ChR2 activation, we used a 5 ms pulse duration of LED for eEPSCs and 0.05–0.5 ms pulse duration of LED for eIPSCs from TRN neurons. For electrical stimulation of inhibitory inputs to MGBv, we used a bipolar glass-stimulating electrode (a theta glass pipette filled with ACSF) with a pulse duration of 0.02 ms and stimulation intensities in the range of 10–100 µA using a stimulation isolation unit (model SIU91A, Cygnus Technology) controlled by a digital stimulator (model PG4000A, Cygnus Technology). Recorded cells were routinely filled with biocytin (1 mg/ml; catalog #B4261, Sigma-Aldrich) to confirm their location post hoc.
All recordings were performed using an amplifier (model 700B, Molecular Devices), digitized at 10 kHz by a data acquisition board (National Instruments), and acquired using a custom-made IGOR program (WaveMetrics).
LED-evoked Sr2+-mIPSC and Sr2+-mEPSC analysis.
Cells exhibiting series resistance ≤25 MΩ and input resistance ≥200 MΩ were used for final analysis. For LED-evoked Sr2+-mEPSC recordings, rms noise >2 were excluded from analysis, and the threshold for detecting events was set at three times the rms noise. In the case of LED-evoked Sr2+-mIPSC recordings, we did not set an rms noise cutoff, and the detection threshold was set at three times the rms noise. LED-evoked desynchronized Sr2+-mIPSCs and Sr2+-mEPSCs were analyzed following previous studies (Petrus et al., 2014, 2015; Chokshi et al., 2019). In brief, a 400 ms window before the LED stimulation was used for collecting spontaneous mIPSCs or mEPSCs from each trace. Another 400 ms window was set 50 ms following the LED stimulation to collect LED-evoked desynchronized mEPSCs or mIPSCs. From each cell, ∼50–200 events were collected from pre-LED and post-LED analysis windows. Cells with a <2 Hz difference in the frequency of events before and after LED stimulation were excluded from the final analysis since those reflect cells with insufficient LED-evoked desynchronized events. Spontaneous events (pre-LED events) were mathematically subtracted from the post-LED events as detailed in prior studies (Petrus et al., 2014, 2015; Chokshi et al., 2019) to obtain the average amplitude of LED-evoked events using the following equation:
where, Apost is the average amplitude of events post-LED, Fpost is the average frequency of events post-LED, Apre is the average amplitude of events pre-LED, and Fpre is the average frequency of events pre-LED.
Brain section processing and immunostaining for confocal imaging.
To verify the expression of ChR2, some brain slices were fixed in 10% formalin (catalog #HT5014, Sigma-Aldrich) immediately after recording and left at 4°C overnight. Tissue was rinsed and permeabilized in 2% Triton X-100 in PBS at pH 7.4 and incubated in DAPI solution (0.1 µg/ml) for 10 min. Sections were rinsed with PBS and mounted with ProLong Gold Antifade Mounting Medium (catalog #P36930, Thermo Fisher Scientific).
For verification of Cre-lines, brains were fixed via transcardial perfusion, postfixed overnight at 4°C, and sectioned at 40 µm thickness on a vibratome (Ted Pella). Sections were collected in PBS and processed for immunohistochemical staining. To stain for parvalbumin (PV)-positive neurons, sections were incubated first in blocking solution (10% normal donkey serum, 0.1% Triton X-100 in PBS) for 30 min at room temperature then incubated in primary antibody solution (dilution, 1:1000 in blocking solution; anti-parvalbumin antibody; #ab11427, Abcam; RRID:AB_298032) at 4°C overnight. After 4× 5 min washes in PBS, sections were incubated in the secondary antibody solution (anti-rabbit Alexa Fluor 488; 1:200 dilution in 1.5% normal donkey serum in PBS) for 2 h at room temperature. After 4× 5 min washes in PBS, sections were counterstained with DAPI (0.1 µg/ml, 10 min) and rinsed again with PBS before mounted with ProLong Gold Antifade Mounting Medium (catalog #P36930, Thermo Fisher Scientific).
Fluorescence images of brain sections were acquired using a confocal microscope (model LSM700, Zeiss).
Osmotic pump surgery.
Alzet osmotic pumps (model 1007D; 7 d delivery) were filled the day before the surgery with either AM251 (100 mm AM251 dissolved in DMSO diluted in saline to deliver 3 mg/kg/d) or a vehicle solution (DMSO in saline). Filled pumps were primed in sterile saline solution overnight at 30°C. On the day of the surgery, mice were anesthetized under 1.5−2% isoflurane/oxygen and head fixed on a stereotactic device (Kopf Instruments). Fur around the back of the neck was removed using Nair. The area was cleaned using iodine before a small incision was made. A small space for the pump was created in the incision site using a hemostat after injection of warm sterile saline. The pump was inserted subcutaneously with the modulator facing away from the incision site. The incision site was sutured with dissolvable sutures (5–1 PDS II violet 18 inch P-3 cutting sutures, eSutures). Antibiotic ointment was applied over the closed incision site. Mice are given a dose of meloxicam (5 mg/kg, s.c.) and recovered on a 30°C heat pad.
Experimental design and statistical analyses.
The basic experimental design used in this study is a two-group comparison of measured variables between NR (control group) and DE (experimental group). Details of the experimental design are provided in the main text and figure legends. All statistical analyses were performed using Prism 7.0 (GraphPad Software), and data are presented as the mean ± SEM. N represents the number of recorded neurons. Unpaired two-tailed Student's t test was used to compare recordings between NR and DE groups. For statistical comparison of short-term dynamics, two-way ANOVA was used to determine the interaction between sensory manipulation (NR and DE) and synaptic responses to a train of stimuli. In all cases, p < 0.05 was considered statistically significant.
Data availability.
All data reported have been deposited at Mendeley Data (doi: 10.17 632/5z34kbp93j.1) and are publicly available as of the date of publication.
Results
To determine whether the plasticity of thalamic inhibition is involved in cross-modal plasticity, we examine whether visual deprivation could induce plasticity of TRN inhibition to MGBv (Fig. 1). We used the PV-Cre transgenic mouse line to drive ChR2 expression in TRN neurons. We verified that PV-Cre line targets TRN neurons effectively by crossing it to Ai14 line to express tdTomato in TRN neurons and observed axonal projections with tdTomato expression in MGBv (Fig. 1A). We also determined that PV-positive TRN neurons partially overlap with somatostatin (SOM)-expressing neurons, and there is no somatic expression of PV in MGBv or dLGN (Fig. 1B). The latter observation is consistent with prior studies reporting intrinsic GABAergic neurons in dLGN as being ontologically distinct from PV neurons (Golding et al., 2014; Jager et al., 2016) and suggests that using PV-Cre to drive gene expression in TRN will not cause expression of the construct in the primary thalamic nuclei in case of viral spillover. To limit the expression of ChR2 to TRN, we performed targeted injections of Cre-inducible ChR2 (AAV-DIO-ChR2-mCherry) in adult PV-Cre mice using stereotactic coordinates and verified the expression of ChR2 within the TRN (Fig. 1C). At the approximate age of P90 to P100, a group of mice was visually deprived for 1 week via DE, and controls remained in a normal 12 h light/dark cycle (NR). Whole-cell recordings were performed in MGBv of acute slices. To quantify the strength of TRN inhibition onto MGBv neurons, we substituted extracellular Ca2+ with Sr2+ and measured light-evoked Sr2+-mIPSCs, which are desynchronized single-vesicle events triggered from ChR2 activation of TRN axons; hence, the average Sr2+-mIPSC amplitude reflects the average strength of individual synapses (Petrus et al., 2015). DE significantly decreased the average amplitude of Sr2+-mIPSCs evoked from TRN axons compared with that in NR controls (Fig. 1D). This result demonstrates that TRN inhibition is plastic in adults and DE induces depression of TRN inhibition to MGBv.
Figure 1.
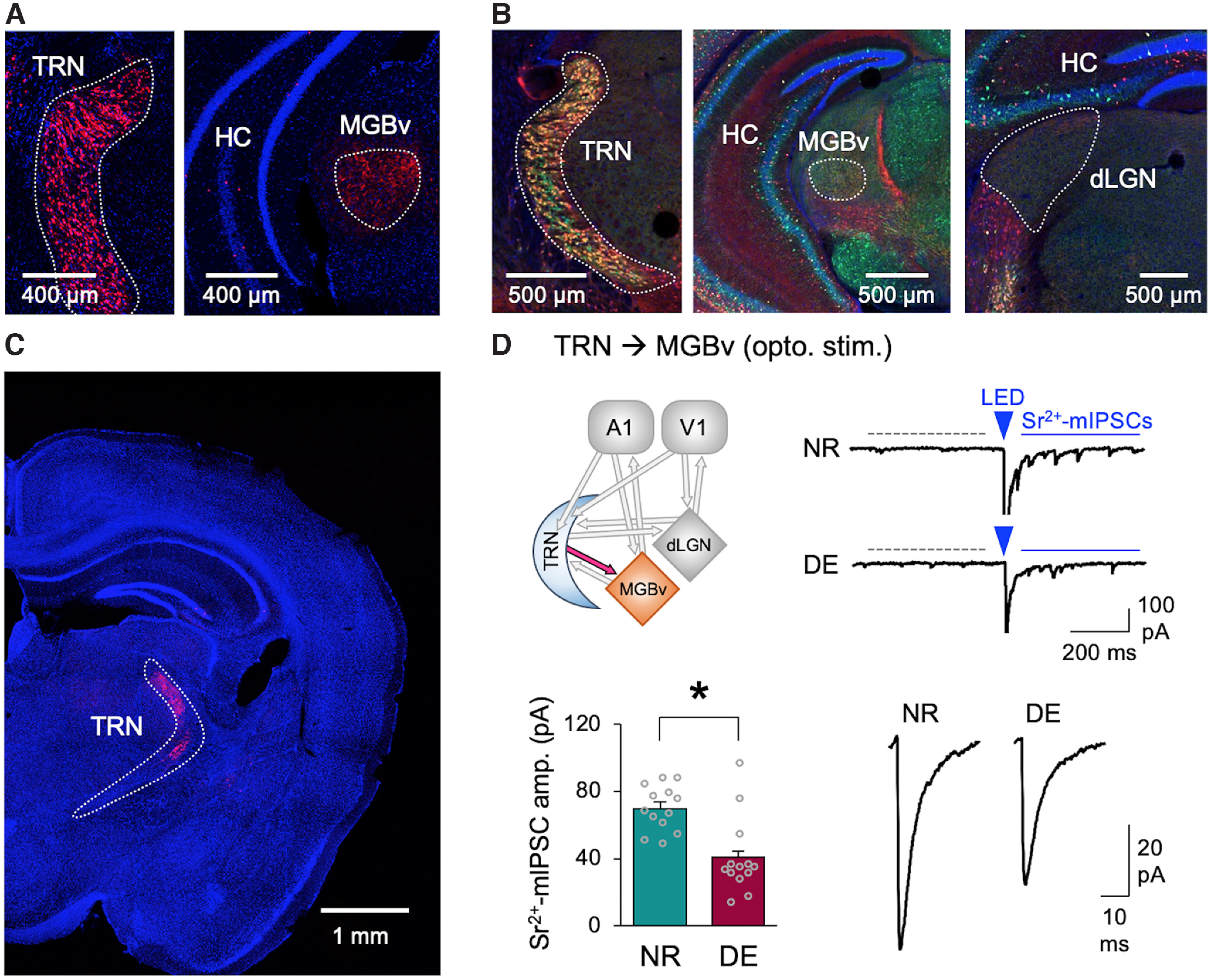
Depriving the vision of adults by DE reduces the strength of TRN inhibition in MGBv. A, PV-Ai14 mouse expresses tdTomato (red) in TRN neurons (left) and axons in MGBv (right). Sections were counterstained with DAPI (blue). TRN and MGBv are outlined in each panel (white dotted line). B, Brain sections from SOM-Cre mice crossed with Ai14 (tdTomato expression shown in red) stained with antibody against PV (green) and DAPI (blue). Left, SOM-Cre drives the expression of tdTomato (red) in a subset of PV-positive neurons in TRN. MGBv (middle) and dLGN (right) show PV-stained (green) and SOM-Cre-driven tdTomato labeled (red) axons. No cell bodies are labeled within these structures. This is consistent with the fact that rodent MGBv has sparse intrinsic inhibitory neurons (Ito et al., 2011), and GABAergic neurons intrinsic to dLGN are ontologically distinct from PV or SOM neurons (Golding et al., 2014; Jager et al., 2016). TRN, MGBv, and dLGN are outlined in each panel. C, Using PV-Cre to drive the expression of ChR2 in TRN neurons. Confocal images of a brain section of a PV-Cre mouse with targeted injections of AAV-DIO-ChR2-mCherry to TRN. Note ChR2-mCherry expression (red) in TRN. The section was counterstained with DAPI (blue). TRN is outlined. D, DE decreases the amplitude of ChR2-evoked Sr2+-mIPSCs recorded in MGBv. Top left, Circuit diagram highlighting TRN to MGBv input. Top right, Example of LED-evoked Sr2+-mEPSC traces from NR and DE mice. Dotted gray line, A 400 ms time window used for collecting spontaneous mEPSCs. Blue solid line shows a 400 ms time window used for collecting post-LED events, which include LED-evoked desynchronized Sr2+-mEPSCs. The arrowhead is the time point when LED (5 ms duration) was activated to evoke release. Bottom left, Average amplitudes of ChR2-evoked-Sr2+-mIPSCs (NR = 69.8 ± 3.8 pA, n = 13 cells from 6 mice; DE = 40.6 ± 6.4 pA, n = 13 cells from 7 mice; t test, t(24) = 3.938, p = 0.0006). Bars, Mean ± SEM. Gray circles, Average value for each cell. Bottom right, Average ChR2-evoked-Sr2+-mIPSC traces.
Another aspect of synaptic transmission is its short-term dynamics and in particular that of TRN inhibition has been shown to control the temporal profile of activity propagation through the primary sensory thalamus (Crandall et al., 2015). We tested whether DE alters the short-term dynamics of TRN inhibition in MGBv. To quantify the short-term dynamics using optogenetics, we first determined the LED duration and intensity range that evokes reliable single action potentials in ChR2-expressing PV-positive TRN neurons. Unlike cortical pyramidal neurons, which generate reliable single action potentials with a 5 ms LED pulse duration (Petrus et al., 2015), a much shorter duration of LED light pulse (∼0.05–0.5 ms) was needed to generate reliable single action potentials across a wide range of light intensities. Using this short-duration light pulses to activate ChR2-expressing TRN axons, we recorded ChR2-eIPSCs from MGBv neurons in normal external Ca2+ in response to a short train of stimulation at different frequencies (5, 10, and 20 Hz; Fig. 2A). We observed short-term depression of TRN inhibition to MGBv in NR controls, which was significantly reduced following a week of DE (Fig. 2A). A reduction in short-term depression reflects reduced presynaptic release (Regehr, 2012), but also indicates a shift in temporal properties of inhibition by causing a relative increase in inhibition during the later phase of a stimulus train while reducing the initial inhibition (Jiang et al., 2010). There is a possibility that altered Ca2+ dynamics at ChR2-expressing axon boutons might affect the outcome of short-term synaptic dynamics. Based on a report that rodent MGBv has a low proportion of intrinsic inhibitory neurons (Ito et al., 2011), we recorded electrical stimulation-induced eIPSCs from MGBv neurons by placing a stimulation electrode within MGBv. A similar change in short-term dynamics was observed for eIPSCs by electrical stimulation (Fig. 2B), suggesting that the DE-induced changes are not likely because of the experimental method that can potentially alter Ca2+ dynamics with presynaptic ChR2 expression (Jackman et al., 2014). These results suggest that DE reduces the inhibitory synaptic transmission of TRN to MGBv and alters its short-term dynamics.
Figure 2.

DE reduces the short-term depression of TRN inhibition onto MGBv. A, DE reduces short-term depression of TRN inhibition to MGBv across stimulation frequencies (5, 10, and 20 Hz). ChR2-expressing TRN axons were activated by LED pulses of 0.05 ms duration. Left, Average eIPSC amplitudes normalized to the first response for 5, 10, and 20 Hz stimulation. Plot, Mean ± SEM. Right, Average eIPSC traces from example neurons of NR and DE. Two-way ANOVA statistics: 5 Hz stimulation train: F(9,171) = 2.892, p = 0.0033; 10 Hz stimulation train: F(9,171) = 3.763, p = 0.0002; 20 Hz stimulation train: F(9,171) = 2.325, p = 0.0171. NR: n = 11 cells from 4 mice; DE: n = 10 cells from 4 mice. B, DE reduces short-term depression of electrical stimulation-evoked IPSCs to MGBv across stimulation frequencies (5, 10, and 20 Hz). Left, Average eIPSC amplitudes normalized to the first response. Plot, Mean ± SEM. Right, Average eIPSC traces. Two-way ANOVA statistics: 5 Hz stimulation train: F(9,126) = 2.480, p = 0.0122; 10 Hz stimulation train: F(9,126) = 2.915, p = 0.0036; 20 Hz stimulation train: F(9,126) = 1.636, p = 0.1117; NR, n = 7 cells from 5 mice; DE, n = 9 cells from 5 mice.
Next, we examined whether the DE-induced decrease in TRN inhibition is specific to MGBv or it is also observed in the visual thalamus (dLGN; Fig. 3). We used the same experimental setup using PV-Cre to target ChR2 expression in TRN, except recordings were performed in dLGN (Fig. 3A). We confirmed that PV-Cre does not drive expression in dLGN neurons (Fig. 3B). When recording from dLGN neurons, we found that the amplitude of ChR2-evoked Sr2+-mIPSCs did not change with DE (Fig. 3C). We also examined whether DE would alter the short-term dynamics of TRN inhibition in dLGN but did not observe significant changes (Fig. 4). Collectively, these results suggest that DE-induced plasticity of TRN inhibition is selective to MGBv and absent in dLGN.
Figure 3.

DE does not alter the strength of TRN inhibition to dLGN. A, Circuit diagram highlighting TRN to dLGN input. B, PV-Ai14 mouse expresses tdTomato (red) in TRN neurons and axons in dLGN. The section is counterstained with DAPI (blue). Left, A tiled confocal image of a brain section containing TRN and dLGN. White boxes outlined in TRN and dLGN are zoomed in to show tdTomato-expressing cell bodies in TRN (middle), and axons and boutons in dLGN (right). Note the absence of PV-positive neurons in dLGN. C, ChR2-evoked Sr2+-mIPSCs recorded in dLGN. Left, Example ChR2-evoked Sr2+-mIPSC traces. The labels are the same as in Figure 1D. Middle, Average amplitudes of ChR2-evoked Sr2+-mIPSCs (NR = 57.8 ± 9.4 pA, n = 14 cells from 7 mice; DE = 52.2 ± 8.0 pA, n = 14 cells from 6 mice; t test: t(26) = 0.4509, p = 0.6558). Bars, Mean ± SEM; gray circles, average value for each cell. Right, Average ChR2-evoked Sr2+-mIPSC traces.
Figure 4.

DE does not alter the short-term depression of TRN inhibition to dLGN. Short-term depression measured by optogenetic activation of ChR2-expressing TRN axons in dLGN. Left, Normalized average eIPSC amplitudes across different stimulation frequencies (5, 10, and 20 Hz). Plot, Mean ± SEM. Right, Example average eIPSC traces across different stimulation frequencies from NR and DE. Two-way ANOVA; 5 Hz stimulation train: F(9,234) = 0.6041, p = 0.7930; 10 Hz stimulation train: F(9,234) = 0.5854, p = 0.8086; 20 Hz stimulation train: F(9,234) = 1.128, p = 0.3438. NR, n = 14 cells from 3 mice; DE, n = 14 cells from 4 mice.
Previous studies demonstrate that DE produces widespread synaptic plasticity within V1 circuitry (Whitt et al., 2014), including plasticity of excitatory synapses onto L6 neurons (Petrus et al., 2011). Furthermore, the visual experience-dependent plasticity seen in dLGN during the cortical critical period has been shown to depend on the corticothalamic feedback excitation (Thompson et al., 2016). Therefore, we investigated whether corticothalamic feedback from V1 L6 to dLGN undergo plasticity with DE in adults. To examine this, we used adult Ntsr1-Cre (L6-Cre);Ai32 mice, which express ChR2 in corticothalamic L6 neurons. Whole-cell recordings were performed in dLGN neurons in acute brain slices. DE did not alter the average amplitude of ChR2-evoked Sr2+-mEPSCs or short-term facilitation of EPSCs evoked with a short train of optical stimulation to activate ChR2 in V1 L6 axons (Fig. 5). These results suggest that corticothalamic excitation in adult dLGN is not plastic to visual deprivation. V1 L6 also provides corticothalamic excitation to TRN, but we did not observe significant changes in the average amplitude of ChR2-evoked Sr2+-mEPSCs recorded in TRN neurons (Fig. 6). Since DE-induced plasticity of TRN inhibition was specific to the auditory thalamus, we examined whether corticothalamic input to MGBv undergo plasticity. To do this, we injected AAV-DIO-ChR2-YFP into A1 of adult Ntsr1-Cre (L6-Cre) mice and recorded ChR2-evoked-Sr2+-mEPSCs from MGBv neurons. DE did not significantly alter the strength of A1 L6 synapses on MGBv neurons (Fig. 7). Collectively, these results suggest that DE does not trigger the plasticity of corticothalamic feedback excitatory synaptic transmission in adults.
Figure 6.

DE does not alter the strength of corticothalamic feedback excitatory synaptic transmission from V1 L6 to TRN. A, Circuit diagram highlighting V1 input to TRN. B, Confocal image of a horizontal brain slice of an Ntsr1-Cre (L6-Cre) mouse injected with AAV-dflox-ChR2-EYFP (green) in V1 L6. The section is counterstained with DAPI (blue). Note ChR2-EYFP-expressing axons in TRN. Left, Lower magnification showing ChR2-EYFP-expressing axons in TRN. LGv, Ventral lateral geniculate nucleus; HC, hippocampus. Right, Higher magnification of TRN. Bright small puncta correspond to boutons. Cell bodies visibly lack EYFP signals. C, ChR2-evoked Sr2+-mEPSCs recorded from TRN neurons. Left, Example traces from NR and DE mice. A 5 ms LED pulse duration was used. Labels are the same as in Figure 1D. Middle, Comparison of the average amplitude of ChR2-evoked Sr2+-mEPSCs (NR: 25.0 ± 2.9 pA, 16 cells from 8 mice; DE: 25.5 ± 2.9 pA, 12 cells from 5 mice; t test: t(26) = 0.1039, p = 0.918). Note that spontaneous mEPSCs recorded from TRN neurons (16 NR cells) display fast kinetics (decay tau = 1.92 ± 0.31 ms, 10–90% rise time = 0.57 ± 0.08 ms) and high frequency (19.1 ± 0.3 Hz) typical of PV-positive inhibitory neurons. Bars, Mean ± SEM; gray circles, average value for each cell. Right, Average ChR2-evoked Sr2+-mEPSC traces from NR and DE.
Figure 7.

DE does not alter the strength of A1 L6 feedback excitatory synaptic transmission to MGBv. A, Circuit diagram highlighting A1 L6 inputs to MGBv. B, Confocal tiled image of a coronal section showing ChR2-YFP-expressing A1 L6 neurons and axonal labeling in MGBv (green). The section was counterstained with DAPI (blue). C, ChR2-evoked Sr2+-mEPSCs recorded from MGBv neurons. Left, Example traces from NR and DE mice. A 5 ms LED pulse duration was used. Labels are the same as in Figure 1D. Middle, Comparison of the average amplitude of ChR2-evoked Sr2+-mEPSCs (NR: 12.3 ± 0.4 pA, 16 cells from 8 mice; DE: 11.5 ± 0.5 pA, 12 cells from 5 mice; t test: t(26) = 1.267, p = 0.2165). Bars, Mean ± SEM; gray circles, average value for each cell. Right, Average ChR2-evoked Sr2+-mEPSC traces from NR and DE.
Last, we examined the potential mechanism for the DE-induced depression of TRN inhibition to MGBv. One mechanism that is known to mediate depression of inhibitory synaptic transmission across multiple brain regions is endocannabinoid via its action on presynaptic CB1-Rs (Chevaleyre et al., 2006). CB1-R mRNA is expressed in TRN neurons while mostly absent in thalamic neurons (see the Allen Mouse Brain in situ hybridization database, http://mouse.brain-map.org). Hence, we tested whether CB1-Rs may play a role in DE-induced changes in the short-term dynamics of TRN inhibition in MGBv neurons. To do this, we bilaterally expressed AAV-DIO-ChR2-YFP in the TRN of PV-Cre mice and surgically implanted an osmotic pump in each mouse to subcutaneously deliver a CB1-R antagonist (AM215 3 mg/kg/d in DMSO in saline) or a vehicle solution (DMSO in saline) during the 1 week of DE. We then prepared brain slices from these mice to compare the short-term dynamics of ChR2-evoked IPSCs in MGBv neurons using whole-cell recordings (Fig. 8A). We found that the AM251-treated group shows larger short-term depression of eIPSCs compared with saline controls (Fig. 8B), which is indicative of higher release probability. We confirmed that the ChR2-eIPSCs are mediated by GABAA receptors by bath-applying bicuculline (Fig. 8C). Our results suggest that the reduced short-term dynamics of GABAergic synaptic inhibition from TRN to MGBv following DE may be mediated by endogenous CB1-Rs.
Figure 8.

CB1-R antagonist increases short-term depression of TRN inhibition onto MGBv of DE mice. A, Confocal images verifying ChR2 expression in TRN of PV-Cre mice. Left, TRN neurons expressing ChR2-YFP (green). Middle, A low-magnification tiled image showing ChR2-YFP expressing TRN axons (green) and an MGBv neuron filled with biocytin during whole-cell recording (red). The section is counterstained with DAPI (blue). DG, Dentate gyrus. Right, A zoomed-in image of the yellow boxed area shown in the middle. B, Short-term depression of ChR2-evoked IPSCs recorded from TRN to MGBv neurons in mice treated with osmotic pump containing AM251 (dark purple) or saline (light purple) during a week of DE (two-way ANOVA statistics: F(9,306) = 6.48, p < 0.0001; saline, n = 15 cells from 5 mice; AM251, n = 21 cells from 5 mice). Left, Comparison of the normalized eIPSC amplitude recorded during a 10 Hz train (10 pulses) optical stimulation. Plot, Mean ± SEM. Right, Example eIPSC traces from saline control and AM251-treated groups. C, ChR2-eIPSCs are blocked by bath application of bicuculline (10 μm). Overlay of 5 consecutive traces before (black) and after (red) bicuculline application. Bicuculline blocked both evoked and spontaneous IPSCs.
Discussion
Thalamic plasticity has been thought to be restricted to a narrow window before the critical period for cortical plasticity (Hooks and Chen, 2020), but recent studies highlight some degree of plasticity during the cortical critical period (from approximately P21 to P35) that is mostly mediated by corticothalamic feedback (Thompson et al., 2016) and inhibition within the sensory thalamus (Sommeijer et al., 2017). Our study extends the age for thalamic plasticity (in the range of P90 to P120) and is novel in that the plasticity is not at the level of corticothalamic feedback within the deprived modality (Fig. 5) but is observed in TRN inhibition specifically for the spared sensory modality (Figs. 1–4). We did not examine corticothalamic feedback in the spared modality, hence whether there is plasticity in that circuit will require further investigation. We observe that the DE-induced reduction in the short-term depression of TRN inhibition to MGBv may be mediated by CB1-R activity (Fig. 8). Selective disinhibition of the MGBv could allow compensatory adaptation of the auditory circuit in the absence of vision.
How the loss of vision leads to selective disinhibition of MGBv is unclear. TRN is divided into different sensory modality sectors based on its connectivity (Guillery et al., 1998; Crabtree, 1999), but some TRN neurons are multimodal (Kimura, 2014) and bridge functional connections across multiple thalamic nuclei (Crabtree and Isaac, 2002; Crabtree, 2018), allowing cross-modal interactions. Also, it has been demonstrated that a large fraction of unimodal TRN neurons is modulated by subthreshold influences from other sensory inputs (Kimura, 2014). Therefore, TRN neurons are in a position to integrate and be modulated by multisensory information. How loss of vision leads to the plasticity of TRN inhibition to MGBv will require further studies. While we did not observe plasticity at V1 inputs to TRN (Fig. 6), visual deprivation-induced plasticity of V1 circuit (Whitt et al., 2014) might alter the pattern of corticothalamic activity to TRN and drive plasticity of TRN inhibition onto MGBv. In addition, TRN is a target of multiple neuromodulatory systems (McCormick, 1992; Varela, 2014), many of which are implemented in promoting synaptic plasticity (Kirkwood, 2007). In either case, the plasticity we observe is restricted to the TRN inhibition in MGBv, and not to dLGN, which suggests that the postsynaptic target may play a role in enabling the plasticity.
We made an observation that implicates CB1-R activity in regulating GABAergic synaptic transmission from TRN to MGBv with visual deprivation (Fig. 8). Endocannabinoids acting through CB1-Rs have been shown to mediate presynaptic forms of long-term depression (LTD) of GABAergic synaptic transmission [inhibitory LTD (iLTD)] across many brain regions (Chevaleyre et al., 2006; Huang et al., 2010; Jiang et al., 2010; Castillo, 2012; Zhao et al., 2015). Endocannabinoids are retrograde messengers produced by the postsynaptic neuron upon increased activity and act via presynaptic CB1-Rs to reduce presynaptic release (Chevaleyre et al., 2006). In V1, CB1-R-mediated iLTD has been shown to be critical for the maturation of inhibition by visual experience (Jiang et al., 2010) and a period of visual deprivation in adults restores iLTD (Huang et al., 2010). We surmise that the reduction in the short-term depression of TRN inhibition in MGBv accompanying visual deprivation may reflect an iLTD of GABAergic synaptic transmission. Endocannabinoid-dependent iLTD has been shown to require activation of postsynaptic metabotropic glutamate receptors (Chevaleyre and Castillo, 2003) or tyrosine receptor kinase-B (Zhao et al., 2015), which then leads to endocannabinoid synthesis and release. Such a mechanism would allow postsynaptic target-specific depression of TRN inhibition as observed in our study (Figs. 1–4). The detailed mechanisms of our proposed iLTD of TRN inhibition would require follow-up studies.
In the somatosensory system, corticothalamic feedback control through TRN inhibition has been shown to depend on cortical activity such that low-frequency stimulation of L6 neurons predominantly inhibits the primary somatosensory thalamus [ventral posteromedial (VPM) nuclei] while higher frequency leads to activation of VPM nucleus (Crandall et al., 2015). The frequency-dependent control is because of differences in the short-term dynamics of corticothalamic synapses in TRN compared with the primary sensory thalamic nucleus, where the former exhibits short-term depression while the latter undergo short-term facilitation. The visual system also displays a similar difference in short-term dynamics for the corticothalamic circuit, where synapses from V1 to TRN and dLGN facilitate (Jurgens et al., 2012), while those from TRN to dLGN depress (Campbell et al., 2020). Consistent with these prior studies, we observe that TRN inhibition to dLGN displays short-term depression (Fig. 4), while V1 input to dLGN exhibits short-term facilitation (Fig. 5). The loss of visually driven activity in V1 would then likely cause a net inhibition of dLGN because of reduced corticothalamic feedback activity. On the other hand, visual deprivation increased the gain of A1 neurons (Petrus et al., 2014), which could result in higher corticothalamic activity in the auditory pathway. This, coupled with the selective reduction in TRN inhibition in MGBv (Figs. 1, 2) without plasticity of A1 L6 inputs to MGBv (Fig. 7), would aid in activity propagation through the auditory thalamus. We surmise that such enhancement of activity transmission through MGBv with visual deprivation could mediate potentiation of the feedforward circuit in A1 (Petrus et al., 2014, 2015) by promoting correlated activity between MGBv and A1. This is consistent with the proposed role of TRN in mediating the attentional searchlight that can enable correlation-based synaptic plasticity (Crick, 1984). In addition, DE-induced change in the short-term dynamics of TRN inhibition is also expected to alter the temporal pattern of activity propagation through the circuit (Abbott and Regehr, 2004; Jiang et al., 2010; Bridi et al., 2020), such as to place emphasis on the initial part of a train of action potentials arriving at MGBv neurons. How such adaptation leads to changes in auditory processing to benefit function will require further studies. In conclusion, our results suggest that the adult thalamic circuit undergoes cross-modal plasticity to selectively reduce the inhibitory gating of the spared sensory modality.
Footnotes
This work was supported by National Institutes of Health Grants R01-EY-022720 and R01-DC-018790 to H.-K.L. and P.O.K.
The authors declare no competing financial interests.
References
- Abbott LF, Regehr WG (2004) Synaptic computation. Nature 431:796–803. 10.1038/nature03010 [DOI] [PubMed] [Google Scholar]
- Agmon A, Connors BW (1991) Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41:365–379. 10.1016/0306-4522(91)90333-j [DOI] [PubMed] [Google Scholar]
- Ahrens S, Jaramillo S, Yu K, Ghosh S, Hwang GR, Paik R, Lai C, He M, Huang ZJ, Li B (2015) ErbB4 regulation of a thalamic reticular nucleus circuit for sensory selection. Nat Neurosci 18:104–111. 10.1038/nn.3897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridi MS, Shin S, Huang S, Kirkwood A (2020) Dynamic recovery from depression enables rate encoding in inhibitory synapses. iScience 23:100940. 10.1016/j.isci.2020.100940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PW, Govindaiah G, Masterson SP, Bickford ME, Guido W (2020) Synaptic properties of the feedback connections from the thalamic reticular nucleus to the dorsal lateral geniculate nucleus. J Neurophysiol 124:404–417. 10.1152/jn.00757.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE (2012) Presynaptic LTP and LTD of excitatory and inhibitory synapses. Cold Spring Harb Perspect Biol 4:a005728. 10.1101/cshperspect.a005728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2003) Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38:461–472. 10.1016/s0896-6273(03)00235-6 [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE (2006) Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci 29:37–76. 10.1146/annurev.neuro.29.051605.112834 [DOI] [PubMed] [Google Scholar]
- Chokshi V, Gao M, Grier BD, Owens A, Wang H, Worley PF, Lee HK (2019) Input-Specific Metaplasticity in the Visual Cortex Requires Homer1a-Mediated mGluR5 Signaling. Neuron 104:736–748.e6. 10.1016/j.neuron.2019.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JW (1999) Intrathalamic sensory connections mediated by the thalamic reticular nucleus. Cell Mol Life Sci 56:683–700. 10.1007/s000180050462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JW (2018) Functional diversity of thalamic reticular subnetworks. Front Syst Neurosci 12:41. 10.3389/fnsys.2018.00041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JW, Isaac JT (2002) New intrathalamic pathways allowing modality-related and cross-modality switching in the dorsal thalamus. J Neurosci 22:8754–8761. 10.1523/JNEUROSCI.22-19-08754.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall SR, Cruikshank SJ, Connors BW (2015) A corticothalamic switch: controlling the thalamus with dynamic synapses. Neuron 86:768–782. 10.1016/j.neuron.2015.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F (1984) Function of the thalamic reticular complex: the searchlight hypothesis. Proc Natl Acad Sci U|S|A 81:4586–4590. 10.1073/pnas.81.14.4586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewall G, Parkins S, Lin A, Jaoui Y, Lee H-K (2021) Cortical and subcortical circuits for cross-modal plasticity induced by loss of vision. Front Neural Circuits 15:665009. 10.3389/fncir.2021.665009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding B, Pouchelon G, Bellone C, Murthy S, Di Nardo AA, Govindan S, Ogawa M, Shimogori T, Lüscher C, Dayer A, Jabaudon D (2014) Retinal input directs the recruitment of inhibitory interneurons into thalamic visual circuits. Neuron 81:1443. 10.1016/j.neuron.2014.03.005 [DOI] [PubMed] [Google Scholar]
- Guillery RW, Feig SL, Lozsádi DA (1998) Paying attention to the thalamic reticular nucleus. Trends Neurosci 21:28–32. 10.1016/S0166-2236(97)01157-0 [DOI] [PubMed] [Google Scholar]
- Hooks BM, Chen C (2020) Circuitry underlying experience-dependent plasticity in the mouse visual system. Neuron 106:21–36. 10.1016/j.neuron.2020.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Gu Y, Quinlan EM, Kirkwood A (2010) A refractory period for rejuvenating GABAergic synaptic transmission and ocular dominance plasticity with dark exposure. J Neurosci 30:16636–16642. 10.1523/JNEUROSCI.4384-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Bishop DC, Oliver DL (2011) Expression of glutamate and inhibitory amino acid vesicular transporters in the rodent auditory brainstem. J Comp Neurol 519:316–340. 10.1002/cne.22521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman SL, Beneduce BM, Drew IR, Regehr WG (2014) Achieving high-frequency optical control of synaptic transmission. J Neurosci 34:7704–7714. 10.1523/JNEUROSCI.4694-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager P, Ye Z, Yu X, Zagoraiou L, Prekop HT, Partanen J, Jessell TM, Wisden W, Brickley SG, Delogu A (2016) Tectal-derived interneurons contribute to phasic and tonic inhibition in the visual thalamus. Nat Commun 7:13579. 10.1038/ncomms13579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Huang S, de Pasquale R, Millman D, Song L, Lee HK, Tsumoto T, Kirkwood A (2010) The maturation of GABAergic transmission in visual cortex requires endocannabinoid-mediated LTD of inhibitory inputs during a critical period. Neuron 66:248–259. 10.1016/j.neuron.2010.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens CW, Bell KA, McQuiston AR, Guido W (2012) Optogenetic stimulation of the corticothalamic pathway affects relay cells and GABAergic neurons differently in the mouse visual thalamus. PLoS One 7:e45717. 10.1371/journal.pone.0045717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A (2014) Diverse subthreshold cross-modal sensory interactions in the thalamic reticular nucleus: implications for new pathways of cross-modal attentional gating function. Eur J Neurosci 39:1405–1418. 10.1111/ejn.12545 [DOI] [PubMed] [Google Scholar]
- Kirkwood A (2007) Neuromodulation of cortical synaptic plasticity. In: Monoaminergic modulation of cortical excitability (Tseng KY, Atzori M, eds). Boston: Springer. [Google Scholar]
- Lee HK, Whitt JL (2015) Cross-modal synaptic plasticity in adult primary sensory cortices. Curr Opin Neurobiol 35:119–126. 10.1016/j.conb.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA (1992) Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Prog Neurobiol 39:337–388. 10.1016/0301-0082(92)90012-4 [DOI] [PubMed] [Google Scholar]
- Meng X, Kao JP, Lee HK, Kanold PO (2015) Visual deprivation causes refinement of intracortical circuits in the auditory cortex. Cell Rep 12:955–964. 10.1016/j.celrep.2015.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Kao JP, Lee HK, Kanold PO (2017) Intracortical circuits in thalamorecipient layers of auditory cortex refine after visual deprivation. eNeuro 4:ENEURO.0092-17.2017. 10.1523/ENEURO.0092-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang W, Xing H, Zhang L, Shu H, Zhang Y (2020) Superiority of blind over sighted listeners in voice recognition. J Acoust Soc Am 148:EL208. 10.1121/10.0001804 [DOI] [PubMed] [Google Scholar]
- Petrus E, Anguh TT, Pho H, Lee A, Gammon N, Lee HK (2011) Developmental switch in the polarity of experience-dependent synaptic changes in layer 6 of mouse visual cortex. J Neurophysiol 106:2499–2505. 10.1152/jn.00111.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrus E, Isaiah A, Jones AP, Li D, Wang H, Lee HK, Kanold PO (2014) Crossmodal induction of thalamocortical potentiation leads to enhanced information processing in the auditory cortex. Neuron 81:664–673. 10.1016/j.neuron.2013.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrus E, Rodriguez G, Patterson R, Connor B, Kanold PO, Lee HK (2015) Vision loss shifts the balance of feedforward and intracortical circuits in opposite directions in mouse primary auditory and visual cortices. J Neurosci 35:8790–8801. 10.1523/JNEUROSCI.4975-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG (2012) Short-term presynaptic plasticity. Cold Spring Harb Perspect Biol 4:a005702. 10.1101/cshperspect.a005702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röder B, Teder-Sälejärvi W, Sterr A, Rösler F, Hillyard SA, Neville HJ (1999) Improved auditory spatial tuning in blind humans. Nature 400:162–166. 10.1038/22106 [DOI] [PubMed] [Google Scholar]
- Solarana K, Liu J, Bowen Z, Lee HK, Kanold PO (2019) Temporary visual deprivation causes decorrelation of spatiotemporal population responses in adult mouse auditory cortex. eNeuro 6:ENEURO.0269-19.2019. 10.1523/ENEURO.0269-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommeijer JP, Ahmadlou M, Saiepour MH, Seignette K, Min R, Heimel JA, Levelt CN (2017) Thalamic inhibition regulates critical-period plasticity in visual cortex and thalamus. Nat Neurosci 20:1715–1721. 10.1038/s41593-017-0002-3 [DOI] [PubMed] [Google Scholar]
- Thompson AD, Picard N, Min L, Fagiolini M, Chen C (2016) Cortical feedback regulates feedforward retinogeniculate refinement. Neuron 91:1021–1033. 10.1016/j.neuron.2016.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela C (2014) Thalamic neuromodulation and its implications for executive networks. Front Neural Circuits 8:69. 10.3389/fncir.2014.00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitt JL, Petrus E, Lee HK (2014) Experience-dependent homeostatic synaptic plasticity in neocortex. Neuropharmacology 78:45–54. 10.1016/j.neuropharm.2013.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer RD, Schmitt LI, Davidson TJ, Nakajima M, Deisseroth K, Halassa MM (2015) Thalamic control of sensory selection in divided attention. Nature 526:705–709. 10.1038/nature15398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Liu CH, Yu YQ, Fujimoto K, Chan YS, He J (2008) Corticofugal projection inhibits the auditory thalamus through the thalamic reticular nucleus. J Neurophysiol 99:2938–2945. 10.1152/jn.00002.2008 [DOI] [PubMed] [Google Scholar]
- Zhao L, Yeh ML, Levine ES (2015) Role for endogenous BDNF in endocannabinoid-mediated long-term depression at neocortical inhibitory synapses. eNeuro 2:ENEURO.0029-14.2015. 10.1523/ENEURO.0029-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data reported have been deposited at Mendeley Data (doi: 10.17 632/5z34kbp93j.1) and are publicly available as of the date of publication.