

Abstract
The crystal structure of the dopamine D3 receptor (D3R) in complex with eticlopride inspired the design of bitopic ligands that explored 1) N-alkylation of eticlopride’s pyrrolidine ring, 2) shifting the position of the pyrrolidine nitrogen, 3) expanding the pyrrolidine ring system, and 4) incorporating O-alkylations at the 4-position. Structure Activity Relationships (SAR) revealed that moving the N- or expanding the pyrrolidine ring was detrimental to D2R/D3R binding affinities. Small pyrrolidine N-alkyl groups were poorly tolerated, but addition of a linker and secondary pharmacophore (SP) improved affinities. Moreover, O-alkylated analogues showed higher binding affinities compared to analogously N-alkylated compounds, e.g., O-alkylated 33 (D3R, 0.436 nM and D2R, 1.77 nM) vs. the N-alkylated 11 (D3R, 6.97 nM and D2R, 25.3 nM). All lead molecules were functional D2R/D3R antagonists. Molecular models confirmed that 4-position modifications would be well-tolerated for future D2R/D3R bioconjugate tools that require long linkers and or sterically bulky groups.
Keywords: Bitopic, Dopamine D2/D3 receptor antagonist, eticlopride, functional efficacy, molecular modeling
INTRODUCTION
Dopamine D2-like receptors, comprised of D2, D3 and D4 receptor subtypes (D2R, D3R, and D4R, respectively) are notable for their clinical relevance and have been targeted for the development of medications to treat neurological and neuropsychiatric disorders.1 For example, D2-like receptor agonists serve as anti-Parkinsonian agents whereas all current antipsychotic medications have the common mechanism of D2-like receptor antagonism or weak partial agonism.2–5 Nevertheless, these drugs can produce multiple undesirable side effects that can limit their therapeutic tolerability.6 Indeed, inhibiting and activating D2R, which is highly expressed in the brain and periphery, may trigger serious side effects including cardiovascular hypertension and undesirable motor effects, some of which may be mediated by “on target” actions.7,8
D2R and D3R subtypes have high sequence identity (78%) in their transmembrane domains, which creates a challenge to develop highly selective D2R or D3R ligands. The high-resolution structural information of D2R, D3R, and D4R, in both the inactive and more recently active states have provided solid bases to facilitate structure-based lead optimization.9–11 Based on these structures, we have identified a highly conserved orthosteric binding site (OBS) in which the primary pharmacophores (PP) of D2-like drugs and also the endogenous neurotransmitter dopamine (DA) bind that does not offer wide options to achieve highly selective ligands.12,13 Hence, one strategy to improve subtype selectivity and modify functional efficacy is to design bitopic ligands. Such molecules would bind concurrently to both the OBS as well as a secondary binding pocket (SBP), which may also serve as an allosteric binding site (ABS) and confer allosteric pharmacology, subtype selectivity or improved affinity to the molecule.14–17 Bitopic ligands have the potential to further modulate receptor function with altered efficacy, binding kinetics and/or functional selectivity, as well as potentially reduce “on target” side effects that plague current medications.8,18 This strategy has successfully identified compounds with D2R or D3R subtype selectivity, including the identification of highly selective D3R antagonists and partial agonists,19–22 and more recently, potent Go-protein biased full D2R agonists23 and D3R-selective agonists.24
Eticlopride is a selective and high affinity D2R/D3R antagonist/inverse agonist and has served as a critical tool used to understand D2-like receptor function, associated behavior, and the influence of receptor antagonism in many preclinical models.25 In 2010, a high-resolution crystal structure of D3R in complex with eticlopride revealed that it binds in the OBS of D3R.10 Further, computational studies with (R)-PG648 (1, Figure 1) revealed that the 2,3-diCl-phenylpiperazine overlaid onto eticlopride in the OBS establishing this moiety as the PP, where its indole amide terminus interacts with a SBP, the shape and size of which was later discovered to differ from D2R.12,13,26,27 These differences are largely determined by a single extra glycine residue on the extracellular loop 1 of D3R, rendering 1 highly D3R selective and confirming the indole amide as the SP.10,13,28 Subsequent SAR studies have further defined the SBP and established the indole amide as a privileged structure for D2-like receptors.29–31 Additional studies have revealed interesting allostery that has been attributed to both the PP, SP and importantly, the length, composition and chirality of the linking chain between them.14,29,32,33
Figure 1.
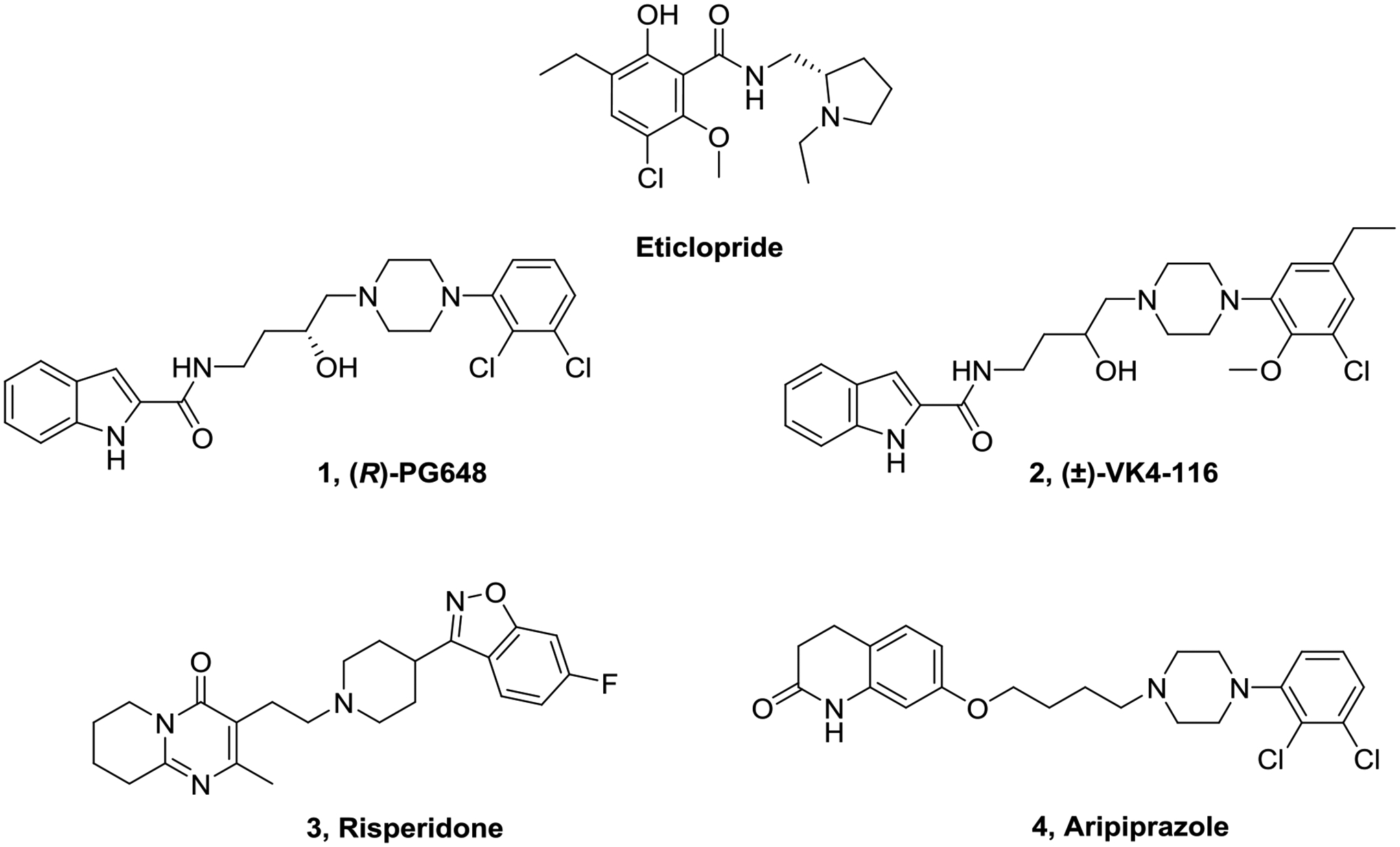
Chemical structures of representative dopamine D2R/D3R antagonists/partial agonists.
Highly D3R selective antagonists and partial agonists have been discovered (e.g., (±)-VK4–116 (2, Figure 1)) that were inspired by eticlopride and the D3R crystal structure.10,20,28 These bitopic ligands have low to subnanomolar D3R affinity, >1000-fold selectivity over D2R, and are effective in a number of animal models of substance use disorders (SUD).20,34–37 Nevertheless, while highly D3R-selective antagonists have been discovered, it has been far more challenging to discover highly D2R-selective antagonists/partial agonists.38–40 In 2018, the crystal structure of D2R in complex with the antipsychotic drug risperidone (3, Figure 1) revealed an unexpected mode of binding to D2R, further illuminating important structural features for the actions of risperidone and related drugs at D2R.9 This challenge prompted us to design new bitopic ligands using the eticlopride scaffold as the PP on which we identified the positions to link an SP to achieve high affinity binding and to potentially identify an SBP of unique pharmacological interest. Further, our aim was to discover a position on the eticlopride scaffold that was amenable to functionalization with structurally bulky groups as new molecular tools with which to study structure and function of D2R and D3R.
Our design includes four modifications to the eticlopride PP as shown in Figure 2. We first synthesized a few N-alkyl substituted eticlopride analogues, ultimately appending SPs (e.g., indole, benzofuran, and dihydroquinolone from the antipsychotic drug, aripiprazole (4, Figure 1)), a well-characterized partial agonist at D2-like receptors.41–43 We then shifted the N- in the pyrrolidine ring to enable linking a SP from the 3-position. As this change was not well tolerated, we expanded the ring to a piperidine. Finally, we appended an -OH group to the 4-position of the pyrrolidine ring and then modified this position with various linked SPs (Figure 2).
Figure 2.
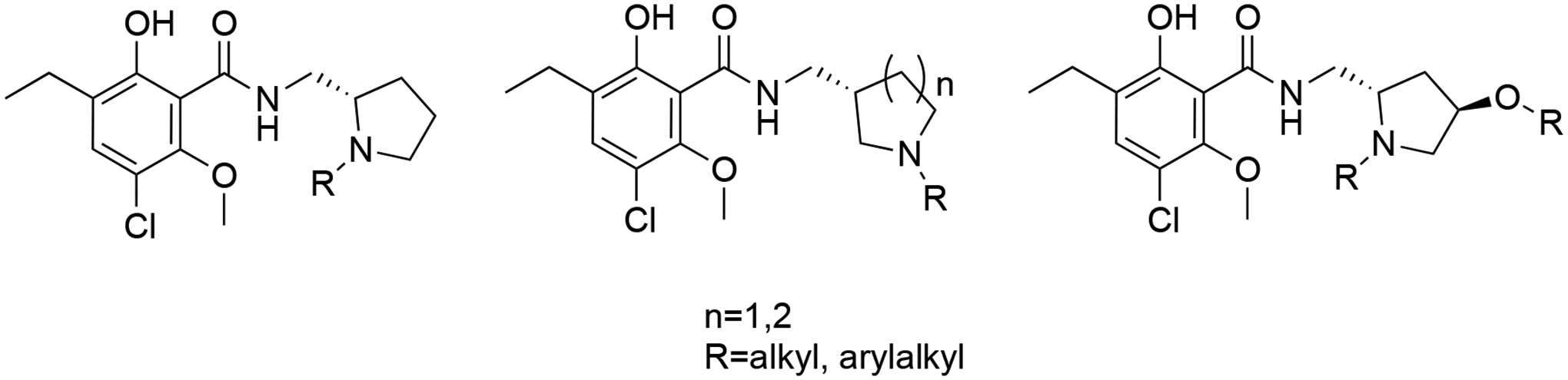
Eticlopride analogues design strategy
RESULTS AND DISCUSSION
Chemistry
The synthetic strategy of N-alkylated eticlopride analogues is shown in Scheme 1. To investigate SAR at the pyrrolidine ring, alkyl, aromatic and heteroaromatic SPs were introduced with different alkyl chain lengths to give N-alkylated bitopic analogues as depicted in Figure 2. All the compounds in Scheme 1 were synthesized by using hydrochloride salts of (S)-nor-eticlopride (5; provided by the NIDA Drug Supply Program). In the presence of inorganic bases such as potassium carbonate (K2CO3) or sodium bicarbonate (NaHCO3), compound 5 underwent N-alkylation with 1-iodopropane yielding the simple alkyl chain compound 6. Similarly, 5 reacted with benzyl bromide to give 7 and 8, respectively. Alkylations to obtain compounds 9-11 were likewise performed. Intermediate 12 (N-(4-bromobutyl)benzofuran-2-carboxamide) was prepared by employing the Appel reaction on N-(2-hydroxyethyl)benzofuran-2-carboxamide.30 Subsequently, 5 underwent N-alkylation with 12 to give 13. However, the synthesis of 15 did not proceed under base-catalyzed N-alkylation with N-(4-bromobutyl)-1H-indole-2-carboxamide due to undesired intramolecular cyclization. Hence, N-(4-oxobutyl)-1H-indole-2-carboxamide (14) was prepared from the corresponding alcohol23 and then subjected to reductive amination with 5 in the presence of sodium triacetoxyborohydride (NaBH(OAc)3)24 to give 15.
Scheme 1.
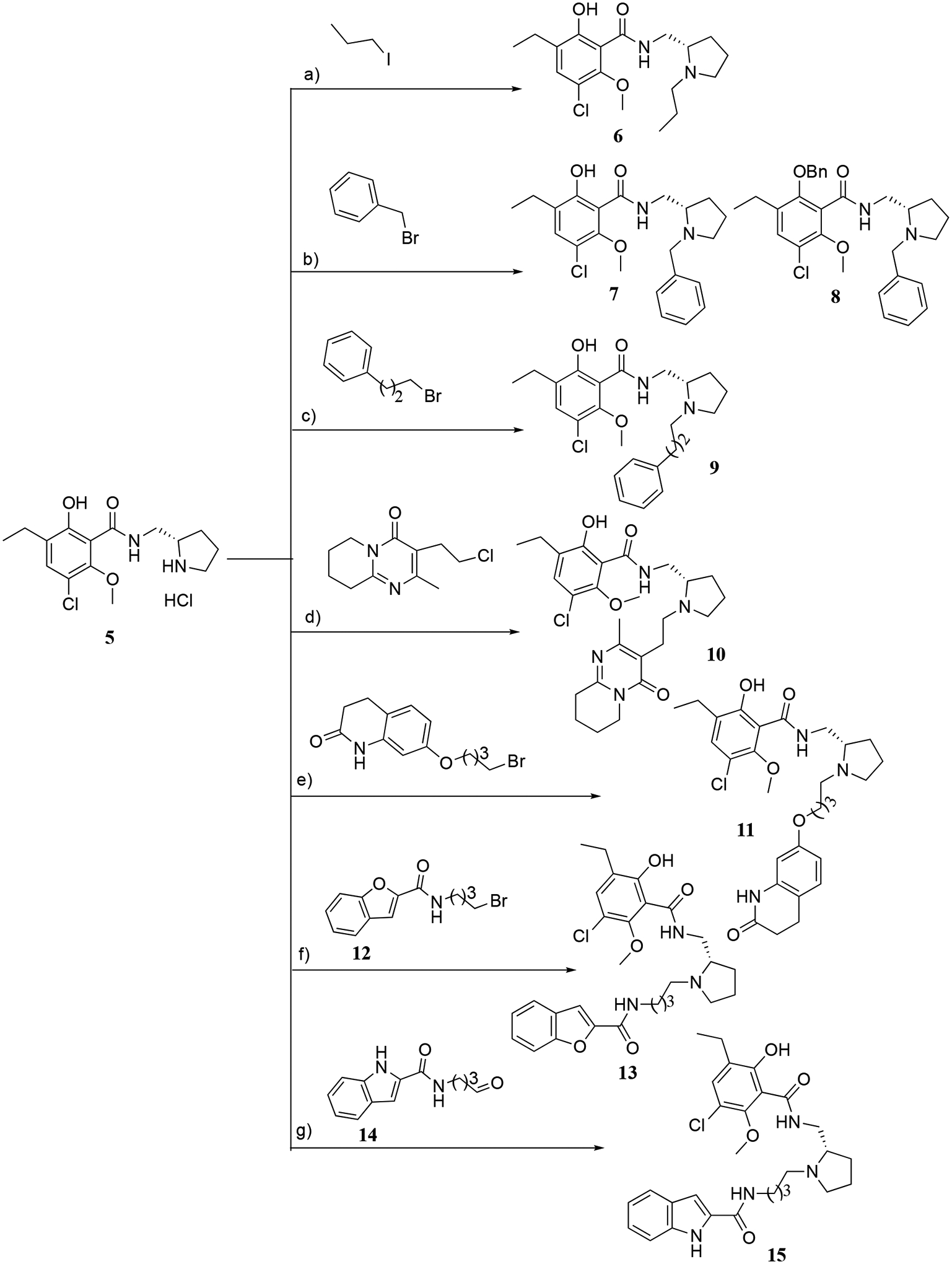
Reagents and conditions: a) K2CO3/acetonitrile, reflux, 12 h; b) NaHCO3/DMF, RT, 3–4 h; c) K2CO3/DMF, RT, 3–4 h; d) NaI, K2CO3/acetone, reflux, 8 h; e) K2CO3/DMF, RT, 3–4 h; f) 1230, NaHCO3/acetonitrile, reflux, 12 h; g) 14,23 NaBH(OAc)3/DCE/AcOH, RT, 5 h.
The synthesis of three N-alkylated eticlopride analogues where the position of the N- in the pyrollidine ring is moved (17a-c) is depicted in Scheme 2. 3-Chloro-5-ethyl-6-hydroxy-2-methoxybenzoic acid (16), was prepared as previously described,44 and converted to the acid chloride intermediate, with thionyl chloride (SOCl2), followed by treatment with the respective N-alkylated pyrrolidine amines: (1-methylpyrrolidin-3-yl)methanamine for 17a, (1-ethylpyrrolidin-3-yl)methanamine for 17b, and (1-propylpyrrolidin-3-yl)methanamine for 17c, in the presence of triethylamine (TEA). A second strategy employed 1,1’-carbonyldiimidazole (CDI) in THF to react 16 with the respective amines to give the desired products 17a-c.
Scheme 2.
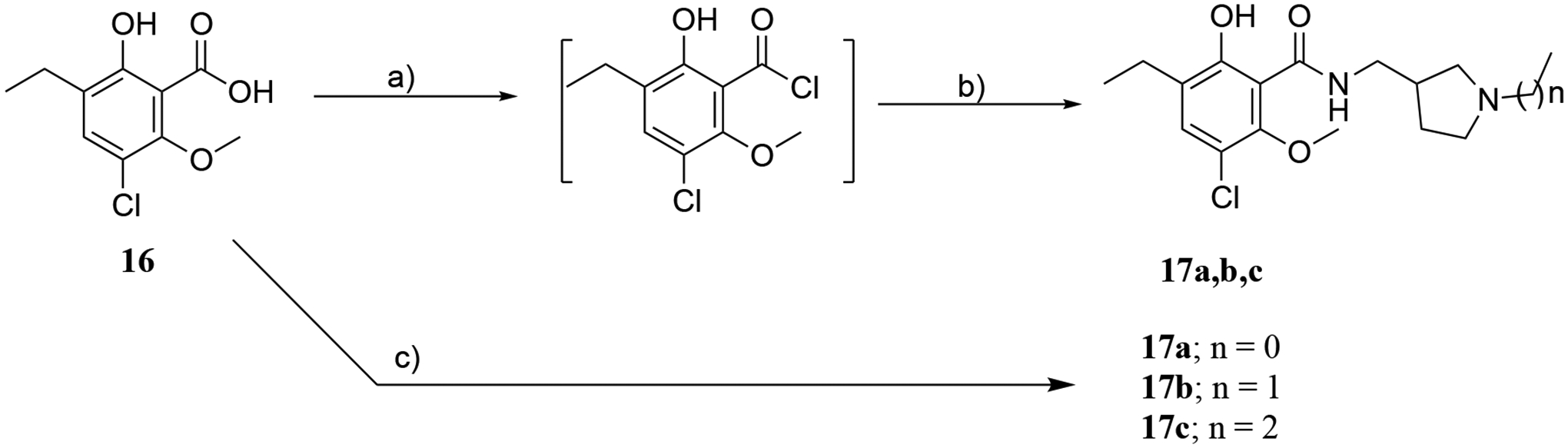
Reagents and conditions: a) SOCl2, toluene, DMF, RT, 12 h. (b) (1-methylpyrrolidin-3-yl)methanamine for 17a, (1-ethylpyrrolidin-3-yl)methanamine for 17b, (1-propylpyrrolidin-3-yl)methanamine for 17c, CHCl3, TEA, 0 °C to RT, 48 h. (c) CDI/THF, RT, overnight.
The synthetic strategy for 24a,b is outlined in Scheme 3. Initially, 20 was prepared from 2-(4-bromobutyl)isoindoline-1,3-dione (18) and commercially available tert-butyl (piperidin-3-ylmethyl)carbamate (19) by N-alkylation under base catalyzed conditions. Removal of the phthalimide protecting group in compound 20 afforded the primary amine 21, which was coupled with indole- or benzofuran-2-carboxylic acids in the presence of CDI to afford 22a,b, respectively. Subsequently, deprotection with 2M HCl in diethyl ether gave primary amine intermediates 23a,b, which underwent amidation with 16 in the presence of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) to yield the bitopic analogues 24a,b.
Scheme 3.
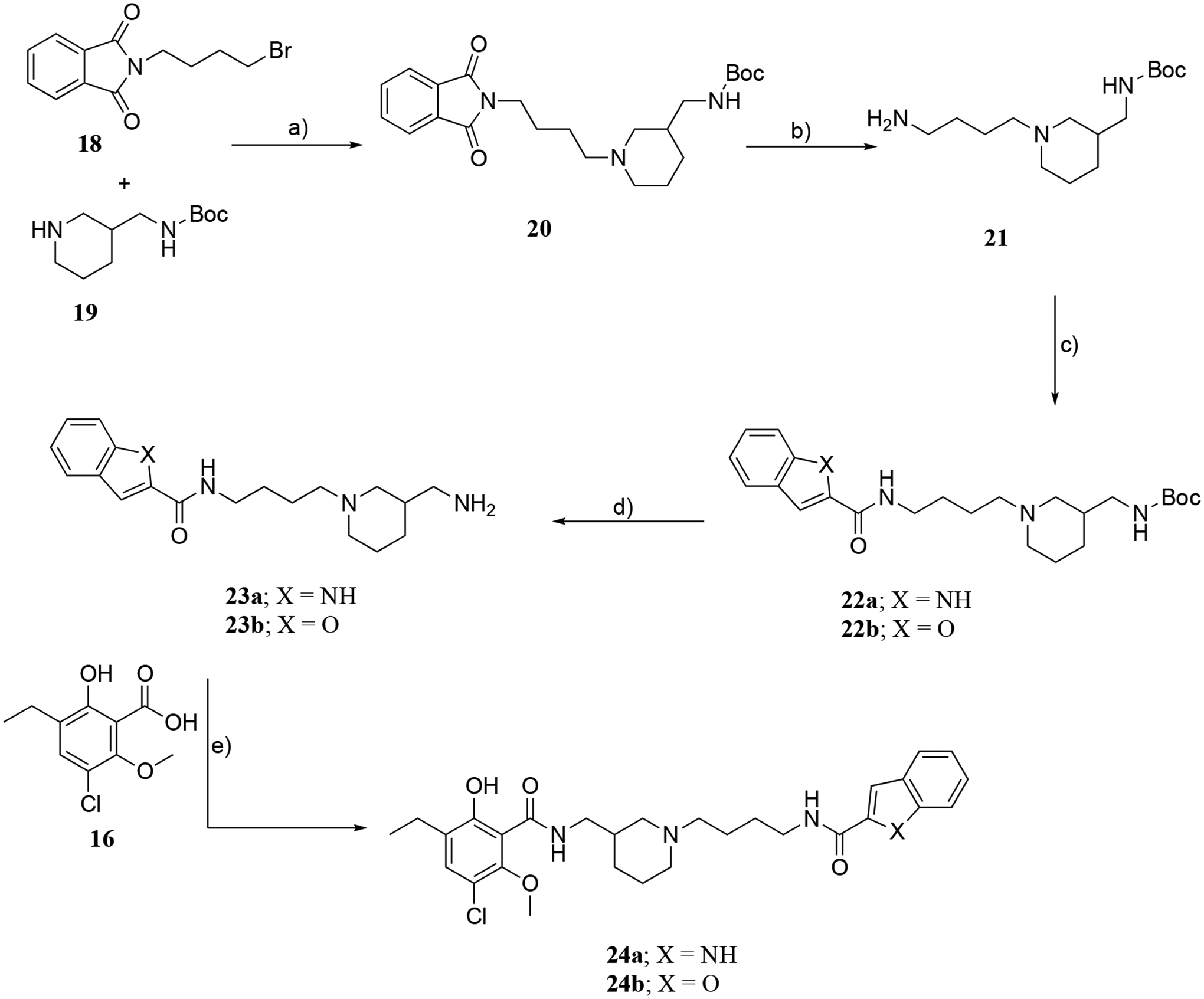
Reagents and conditions: a) K2CO3, NaI/acetone, reflux, 22 h; b) N2H4/ethanol, reflux, 4 h; c) indole-2-carboxylic acid for 22a, benzofuran-2-carboxylic acid for 22b, CDI/THF, 0 °C to RT, overnight; d) 2M HCl in diethyl ether, MeOH, reflux, 3 h; e) 16, EDC/DCM, Na2CO3, RT, 24 h
The synthetic strategies for compounds incorporating O-alkylations at the 4-position of the pyrrolidine ring are depicted in Schemes 4–6. Initially, as shown in Scheme 4, the carboxylic acid intermediate 27 was obtained when (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid (25) and 7-(2-bromoethoxy)-3,4-dihydroquinolin-2(1H)-one (26) underwent O-alkylation in the presence of NaH at 0 °C.45,46 Next, 27 underwent Steglich esterification with benzyl alcohol in the presence of EDC, affording the corresponding benzyl ester intermediate 28 that was selectively reduced to the primary alcohol intermediate 29 by LiBH4 at −15 °C. Subsequently, under Mitsunobu conditions, 29 was reacted with phthalimide to give 30, followed by removal of the phthalimide group resulting in the primary amine intermediate 31. This intermediate was coupled with 16 in the presence of O-(6-Chloro-1-hydrocibenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU) to give carboxamide 32, which was treated with trifluoroacetic acid (TFA) to give the target bitopic eticlopride analogue 33 having the 3,4-dihydroquinolin-2(1H)-one moiety as the SP. Finally, reductive amination at the pyrrolidine nitrogen of 33 with propionaldehyde yielded the desired N-propyl bitopic compound 34.
Scheme 4.
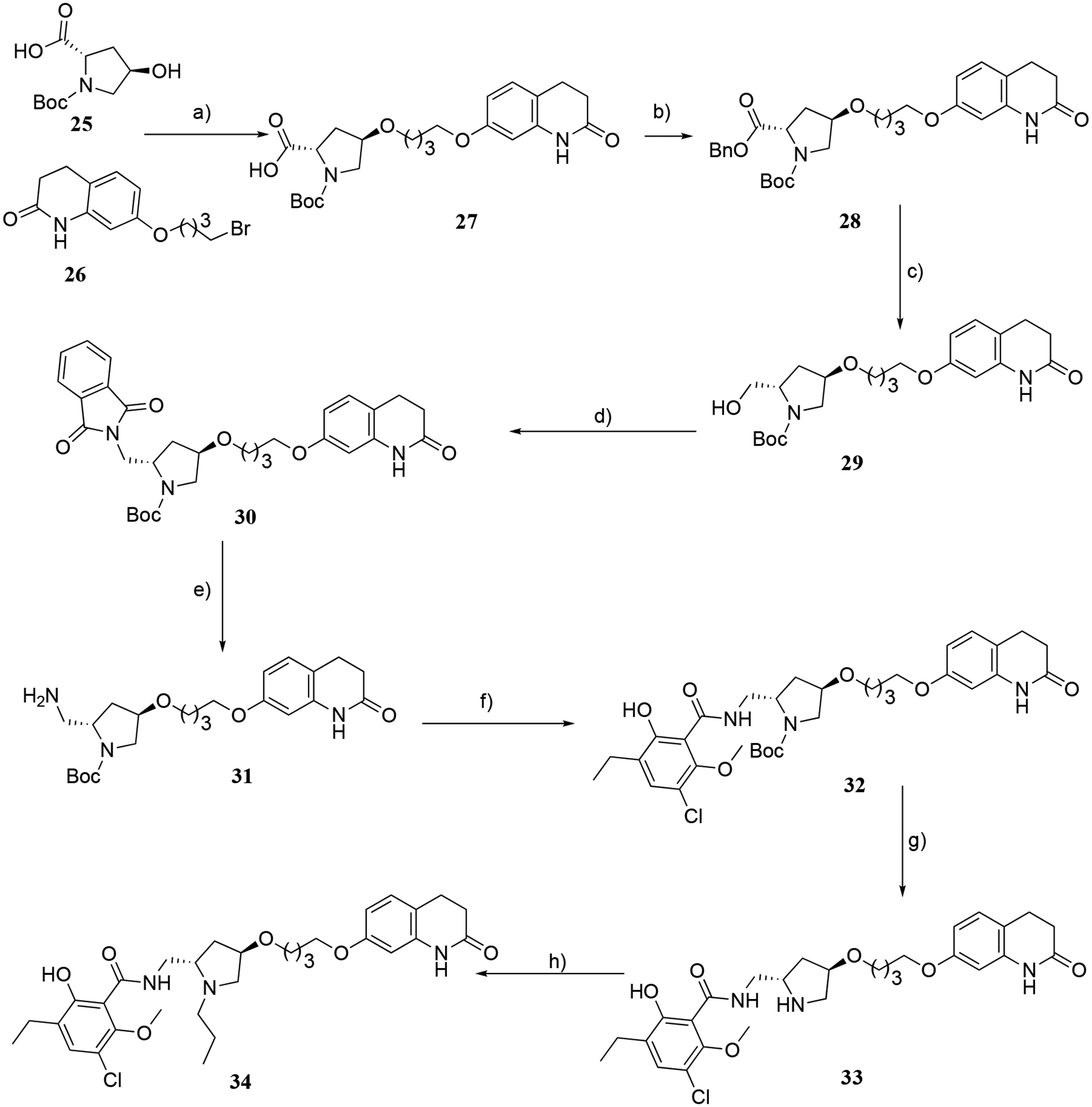
Reagents and conditions a) NaH/DMF, 0 °C, 2–3 h; b) benzyl alcohol, EDC/DMAP/DCM, RT, 5 h; c) LiBH4/THF, −15 °C to RT, 1 h; d) phthalimide, TPP/DEAD/THF, 0 °C to RT, 8 h; e) N2H4/ethanol, reflux, 3–4 h; f) 16, HCTU/CHCl3, RT, 2–3 h; g) TFA/DCM, RT, 8 h; h) CH3CH2CHO/NaBH(OAc)3, DCE/AcOH, RT, 5 h.
Scheme 6.
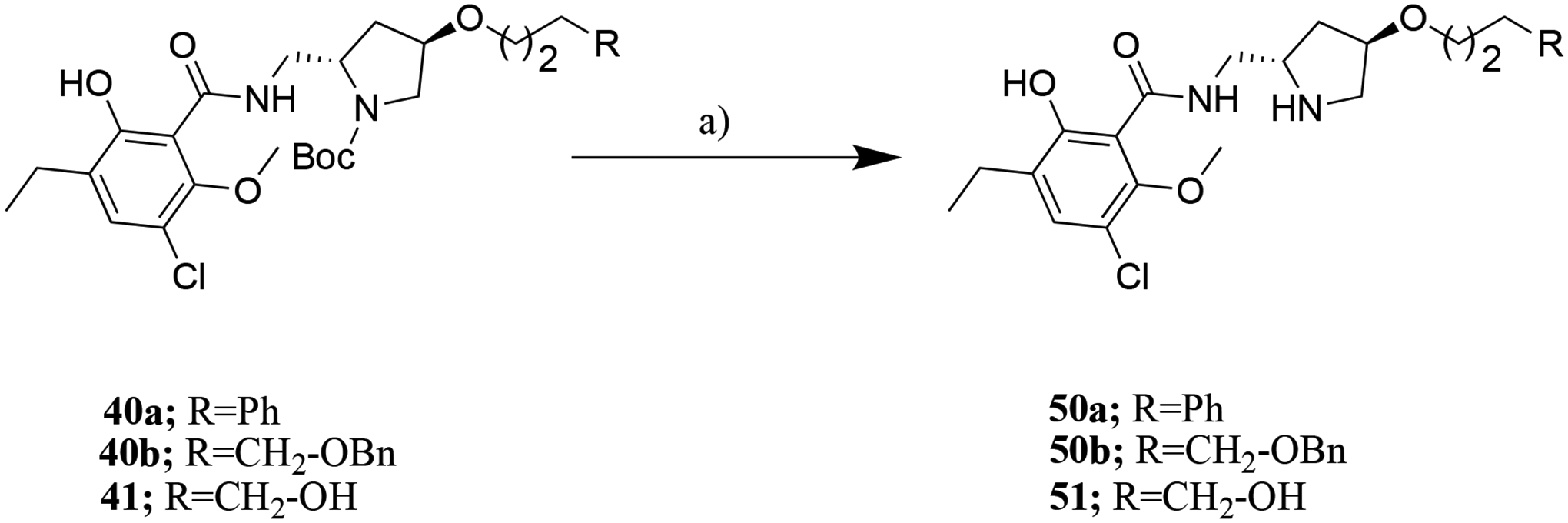
Reagents and conditions: a) TFA/DCM, RT, 8 h.
In Scheme 5, we further introduced benzofuran- or indole-2-carboxamide as the SP through O-alkylation at the 4-position of the pyrrolidine ring. The carboxylic acid intermediates 36a,b were synthesized starting from different alkyl halides 35a,b and 25 through the O-alkylation method as shown in Scheme 4. Subsequently, the carboxylic acid intermediates were directly reduced to the primary alcohols (37a,b) in the presence of borane dimethylsulfide (BMS). Under Mitsunobu reaction conditions, 37a,b was reacted with phthalimide to yield 38a,b followed by removal of the phthalimide protecting group, affording the primary amine intermediates (39a,b). These primary amines were coupled with 16, in the presence of HCTU, as described earlier, to yield carboxamides (40a,b). Further, 40b underwent debenzylation by Pd/C catalyzed hydrogenation to produce the primary alcohol intermediate 41, which was reacted with phthalimide under Mitsunobu conditions to afford 42. Subsequently, phthalimide deprotection gave primary amine 43 that was coupled with indole 2- or benzofuran-2-carboxylic acids to give carboxamides 44, 45. After treatment with TFA compounds 46 and 47 were isolated and subjected to reductive amination with acetaldehyde producing the N-ethyl analogues 48 and 49 comprising an indole or benzofuran as the SPs, respectively.
Scheme 5.
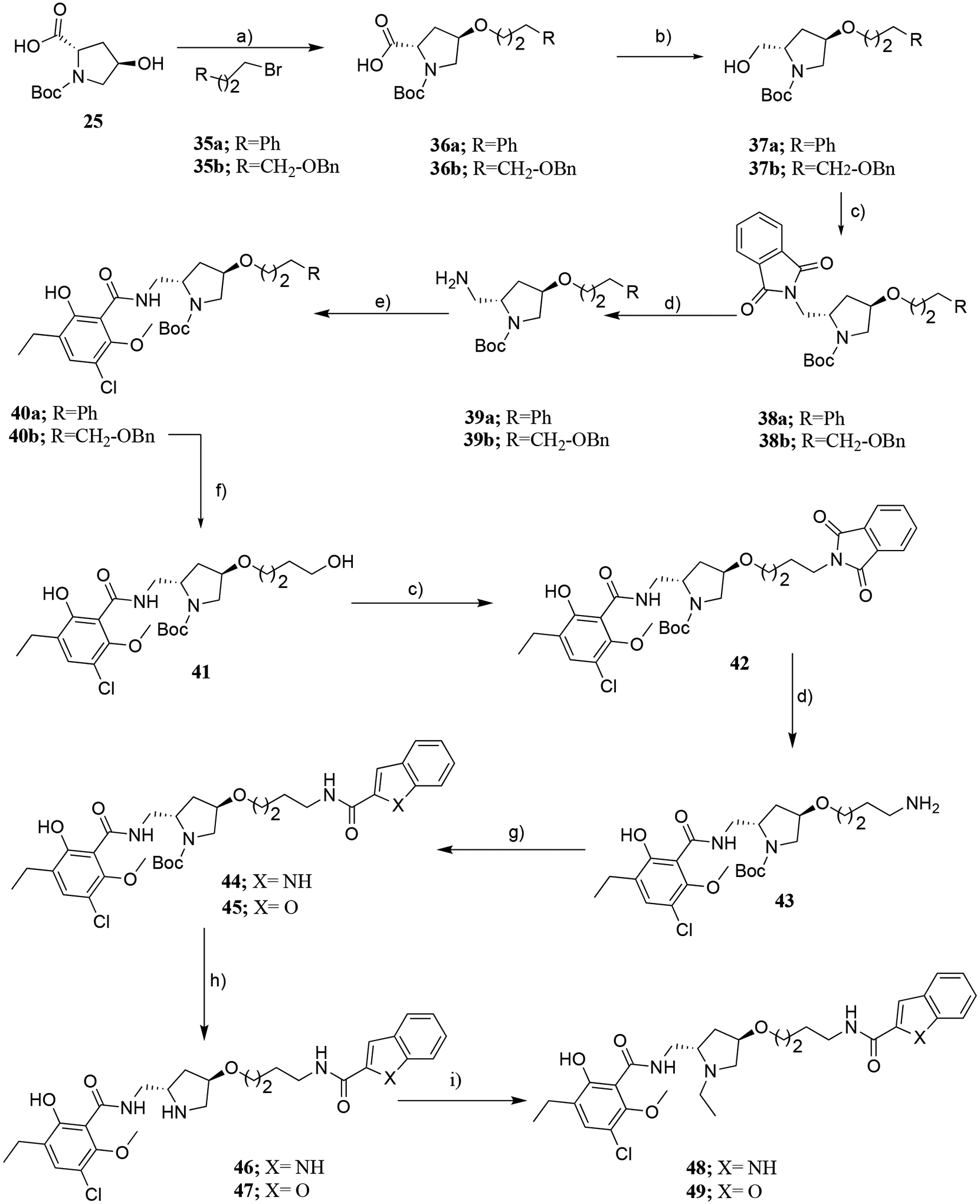
Reagents and conditions: a) NaH/DMF, 0 °C, 2–3 h; b) Borane dimethylsulfide, THF, RT, 5 h; c) Phthalimide, TPP/DEAD/THF, 0 °C to RT, 8 h; d) N2H4/ethanol, reflux, 3–4 h; e) 16, HCTU/DCM, RT, 2–3 h, f) 10% Pd/C, H2 (45 psi), MeOH:EtOAc (1:1), 1 h; g) 1H-indole-2-carboxylic acid for 44, benzofuran-2-carboxylic acid for 45, HCTU/CHCl3, RT, 2–3 h; h) TFA/DCM RT, 8 h; i) CH3CHO, NaBH(OAc)3, DCE/AcOH, RT, 5 h.
In Scheme 6, 40a,b and 41 underwent deprotection in the presence of TFA to give the additional target compounds 50a,b and 51 respectively.
SAR of the eticlopride analogues at D2R and D3R
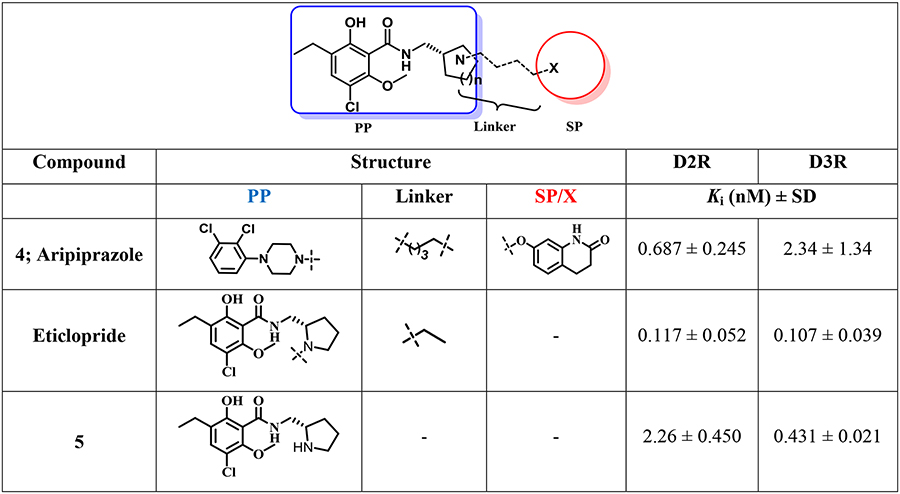
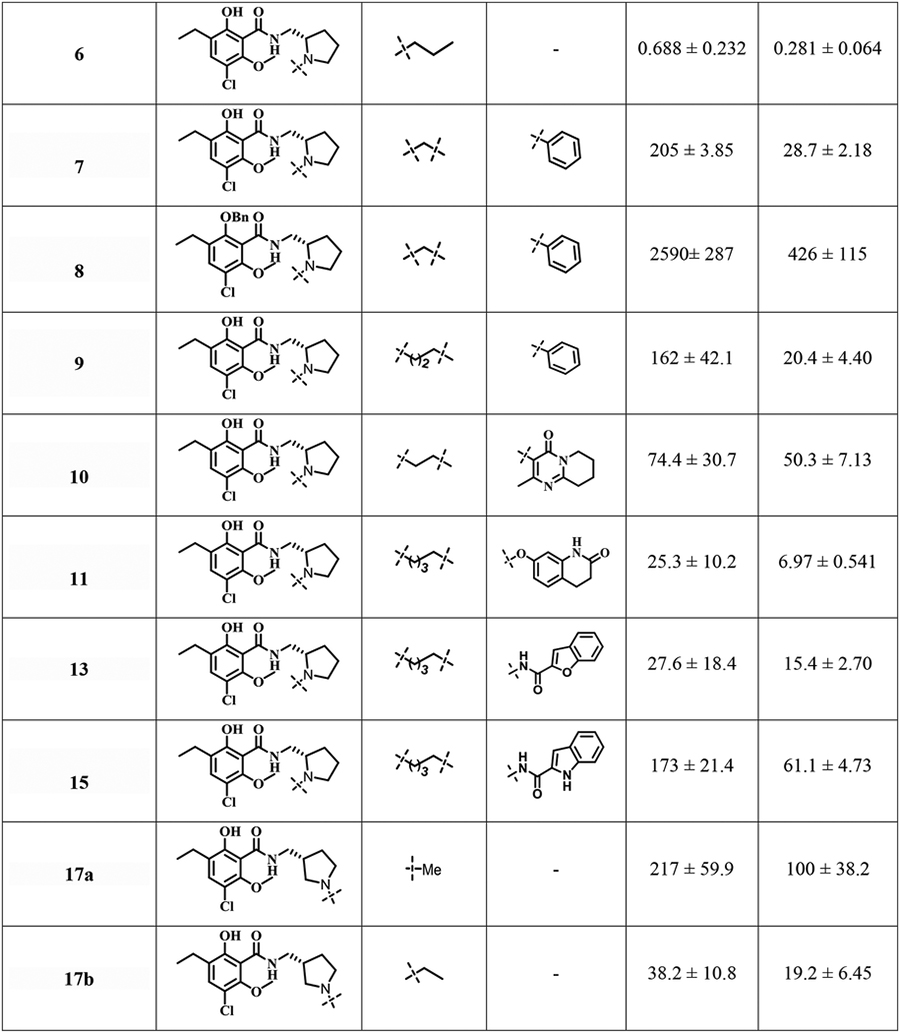
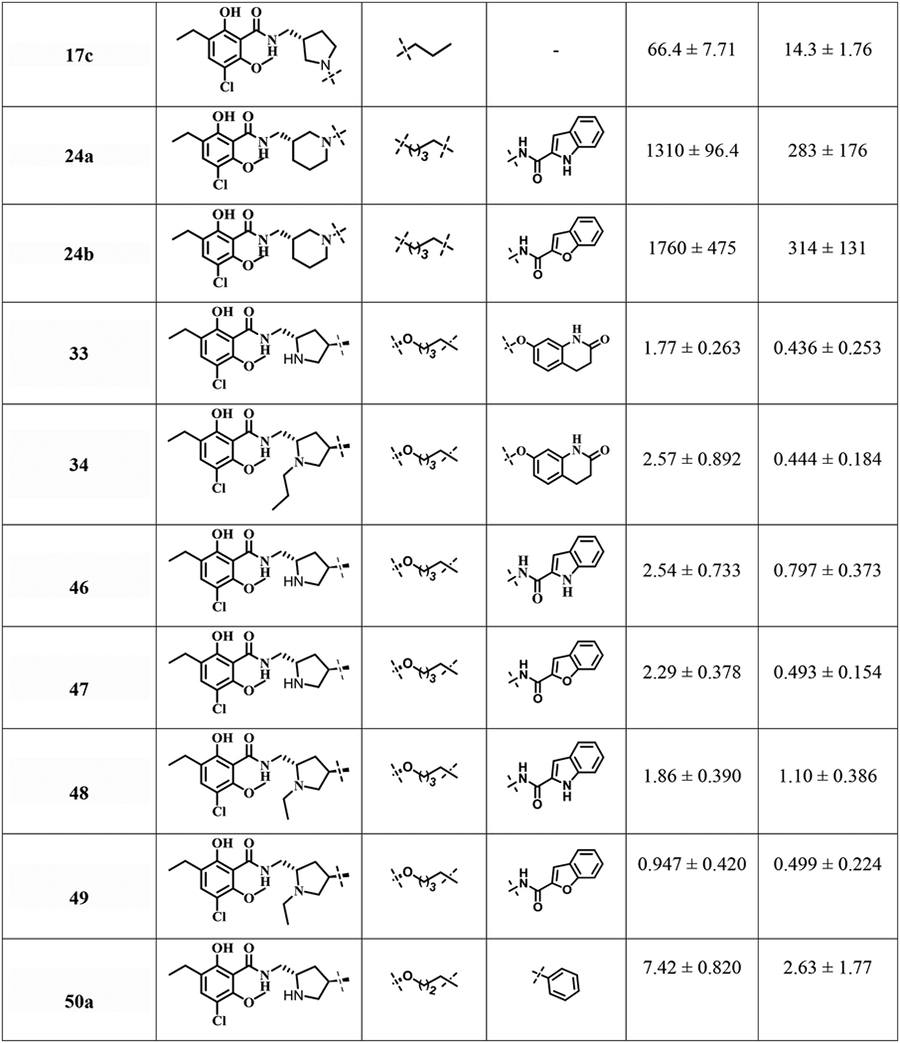
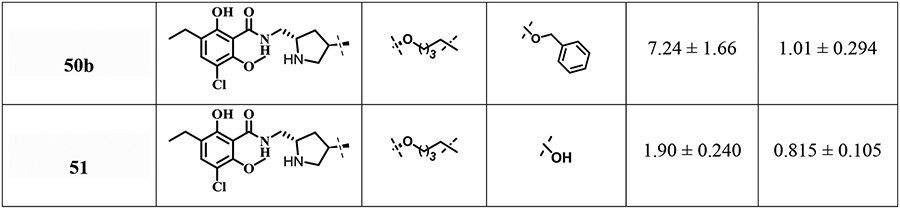
To investigate SAR at D2R and D3R in this series of eticlopride analogues where the SP was linked to various positions on the eticlopride PP, we determined their binding affinities at D2R and D3R. The radioligand [3H]N-methylspiperone was employed in all the competition binding experiments, using membrane preparations from stably transfected HEK293 cells expressing human D2L and D3 receptors. Ki values are listed in Table 1.
Table 1.
Human dopamine D2-like receptors (D2R and D3R) binding data in HEK cells for bitopic eticlopride analoguesa
|
|
|
|
The values represent the arithmetic mean ± SEM obtained from at least three independent experiments, each performed in triplicate. IC50 values for each compound were determined from dose-response curves and Ki values were calculated by the Cheng-Prusoff equation47 using GraphPad Prism version 6.00 for Macintosh.
Removing the N-ethyl substituent from eticlopride to give nor-eticlopride (5) resulted in a 4-fold decrease in binding affinity at D3R but a remarkable 19-fold decrease at D2R. In contrast, when the N-alkyl chain was extended to N-propyl in compound 6, D2R and D3R binding affinities were modestly decreased 6- and 2.6-fold, respectively, whereas the N-benzyl analogue (7) showed dramatic decreases at both D3R (268-fold) and D2R (1800-fold) compared to eticlopride. As expected, the di-benzylated side product, 8, showed low affinity, especially at D2R confirming the necessity of the phenolic-OH function. The N-phenylpropyl analog 9 showed similar affinities as compared to the N-benzyl analogue, 7. Compound 10, inspired by the atypical antipsychotic drug risperidone bearing the 2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one as the SP linked with a 2C linker was equiactive at D2R and D3R, but compound 11, whose SP was the same as the D2-like receptor partial agonist aripiprazole showed a 3.6-fold preference for D3R (Ki=6.97 nM) v. D2R (Ki=25.3 nM). Nevertheless, this modest D3R preference was not seen for compound, 13, but was recapitulated in 15, whose SP is considered a privileged structures for D3R. However, 15 showed relatively low affinities (Ki=173 nM and 61.1 nM) for D2R and D3R, respectively.
To further investigate SAR, we moved the N in the pyrrolidine ring to the meta-position relative to the attached PP, to explore the possibility of accessing an SBP in that direction at D2R and D3R. The three simple alkylated analogues 17a-c showed dramatically reduced binding affinities at both D2R and D3R compared to eticlopride. Thus, we abandoned that series and expanded the pyrrolidine ring to a piperidine ring. Nevertheless, incorporation of a 2-indole or 2-benzofuran carboxamide with a 4C linker (24a,b) both showed even lower binding affinities (Ki>1000 nM and ~300 nM) at D2R and D3R, respectively.
We then turned to O-alkylation at the 4-position of the pyrrolidine ring. Interestingly, compounds 33 and 34 bearing a 4C linked dihydroquinolinone as the SP with either N-H (D2R, Ki = 1.77 nM; D3R, Ki = 0.436 nM) or N-propyl (D2R, Ki = 2.57 nM; D3R, Ki = 0.444 nM) groups in the pyrrolidine ring demonstrated high binding affinities. Similarly, N-nor-analogues 46 and 47 anchoring the indole- or benzofuran-2-carboxamide as the SPs showed high affinity for both D2R, (Ki = 2.54 and 2.29 nM) and D3R, (Ki = 0.797 and 0.493 nM) respectively. Moreover, incorporation of the N-ethyl group in compounds 48 and 49 (as in the parent compound eticlopride) were also well tolerated at D2R and D3R (Ki<2 nM). Interestingly, by simply appending either a terminal phenyl group in 50a or a hydroxyl group in 51 or a 4C linked O-benzyl 50b as SPs the affinities at both D2R and D3R remained in the low nanomolar range.
Functional efficacy study
We then selected a set of five compounds (13, 15, 17b, 48, 49) and used bioluminescence resonance energy transfer (BRET) assays, which measure conformational changes of the Go protein heterotrimer following activation by D2R or D3R, to evaluate functional efficacy.21,23 Our results indicated that like the parent compound eticlopride, and in contrast to the D2R/D3R agonist, quinpirole, all analogues tested were antagonists or very weak partial agonists (Figure 3).
Figure 3.
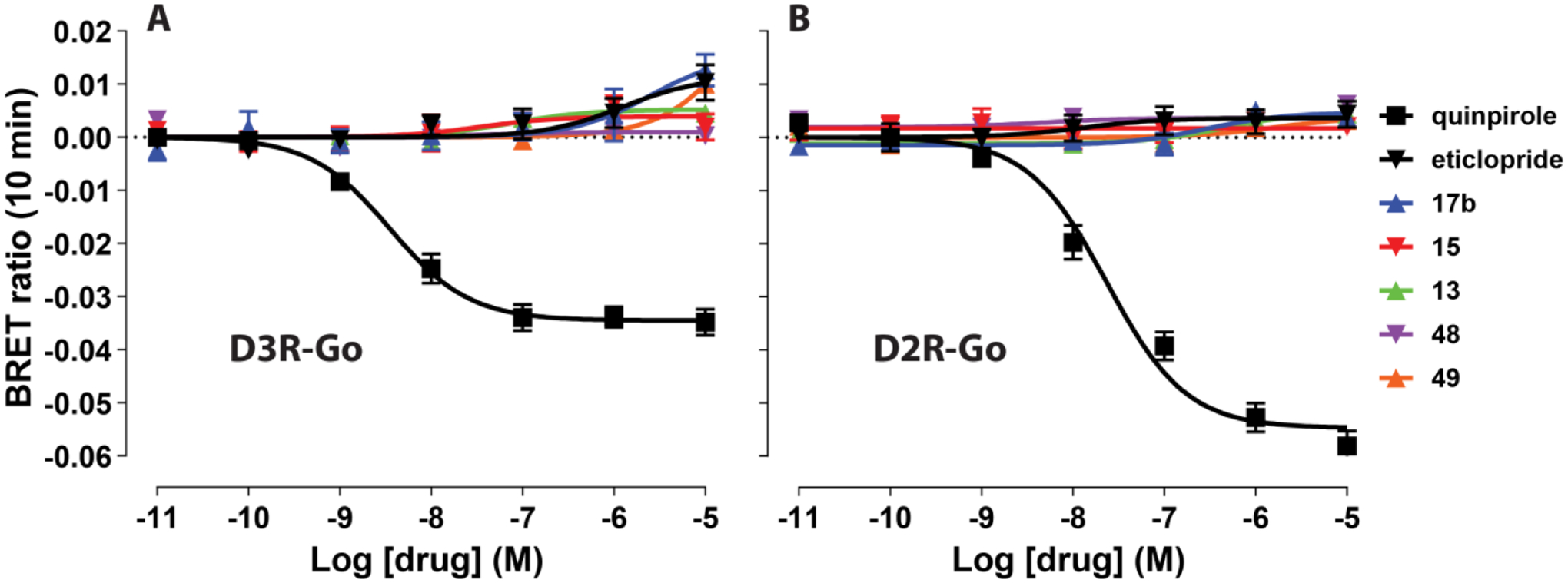
The eticlopride analogues 13, 15, 17b, 48, 49 were all antagonists or very weak partial agonists at both D3R and D2R. The Go BRET activation assays show that these compounds have very low efficacies at both D3R (A) and D2R (B) similar to levels of the reference antagonist eticlopride, compared to the full agonist quinpirole.
Molecular modeling study
To understand the structural basis of the impact of different SPs and linkers on the affinities at D3R, we carried out a molecular docking study for the parent molecule, eticlopride, and five selected ligands (13, 15, 17b, 48, 49) with different SPs or linkers. As the results of our functional assays indicated that these analogues were antagonists or very weak partial agonists (Figure 3), but their binding affinities did not show much selectivity for D3R over D2R, we chose to use only the D3R structure in the inactive state for these docking studies.
The only difference between 17b and eticlopride is the position of the charged nitrogen. From the results of molecular docking, we found that the charged nitrogen of 17b points to Tyr7.35. Thus, it would not be expected to effectively form any ionic interaction with Asp3.32, compared to that of eticlopride (Figure 4), which would contribute to its significantly lower affinity at D3R, as compared to the parent ligand.
Figure 4.
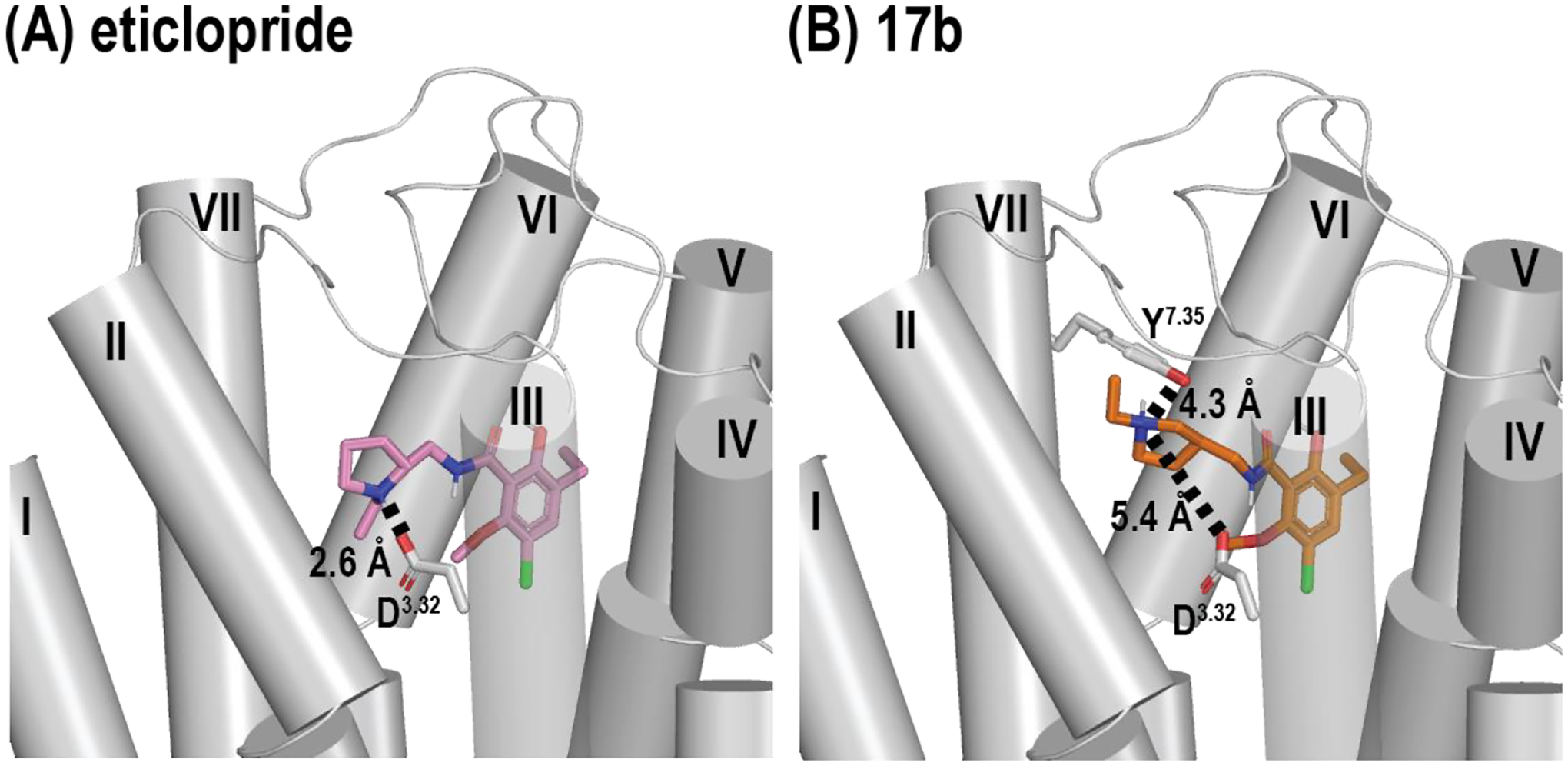
Docking poses of (A) eticlopride and (B) 17b shows Asp3.32 has a different interaction strength depending on the position of the charged nitrogen.
We then examined the docking poses of the ligands with different SPs and linkers: 13, 15, 48, and 49. Note that 13 and 15 commonly have a N-alkylated linker, while 48 and 49 have a O-alkylated linker at the C4-position. The common SP for 15 and 48 is indoleamide, while that for 13 and 49 is benzofuran. The molecular docking results suggest that the SPs of these compounds interact with several residues in the extracellular vestibule, a location different from the previously identified binding pockets responsible for D3R ligand selectivity.27 In particular, the aromatic ring of the SPs stack with the aromatic rings of His6.55 and Tyr7.35, two conserved residues between D3R and D2R (Figure 5). We further evaluated the effect of different SPs with the same linker (ΔΔG = ΔG(compound_with_benzofuran) - ΔG(compound_with_indoleamide)) with Molecular Mechanics with Generalized Born and Surface Area solvation (MM-GB/SA). The ΔΔG value is −0.49 kcal/mol for the pair with the N-alkylated linker (ΔG13-ΔG15) and −0.95 kcal/mol for the pair with the O-alkylated linker (ΔG49-ΔG48). These relatively small ΔΔG values are consistent with their correspondingly small Ki differences, suggesting that with the same linker, the different indole and benzofuran SP have only a small impact on binding affinity. From the MM-GB/SA calculations of the compounds with different linkers but the same SP (ΔΔG = ΔG(compound_with_O-alkylated_linker) - ΔG(compound_with_N-alkylated_linker)), the ΔΔG is −11.47 kcal/mol for the pair 48 and 15 (ΔG48-ΔG15) and −11.93 kcal/mol for the pair 49 and 13 (ΔG49-ΔG13). These large ΔΔG values agree with their correspondingly significant Ki differences, i.e., ~56- and ~31-fold, respectively. This is likely due to the ligands with an O-alkylated linker forming stronger interactions with Asp3.32 compared to those with the N-alkylated linker (Figure 5).
Figure 5.
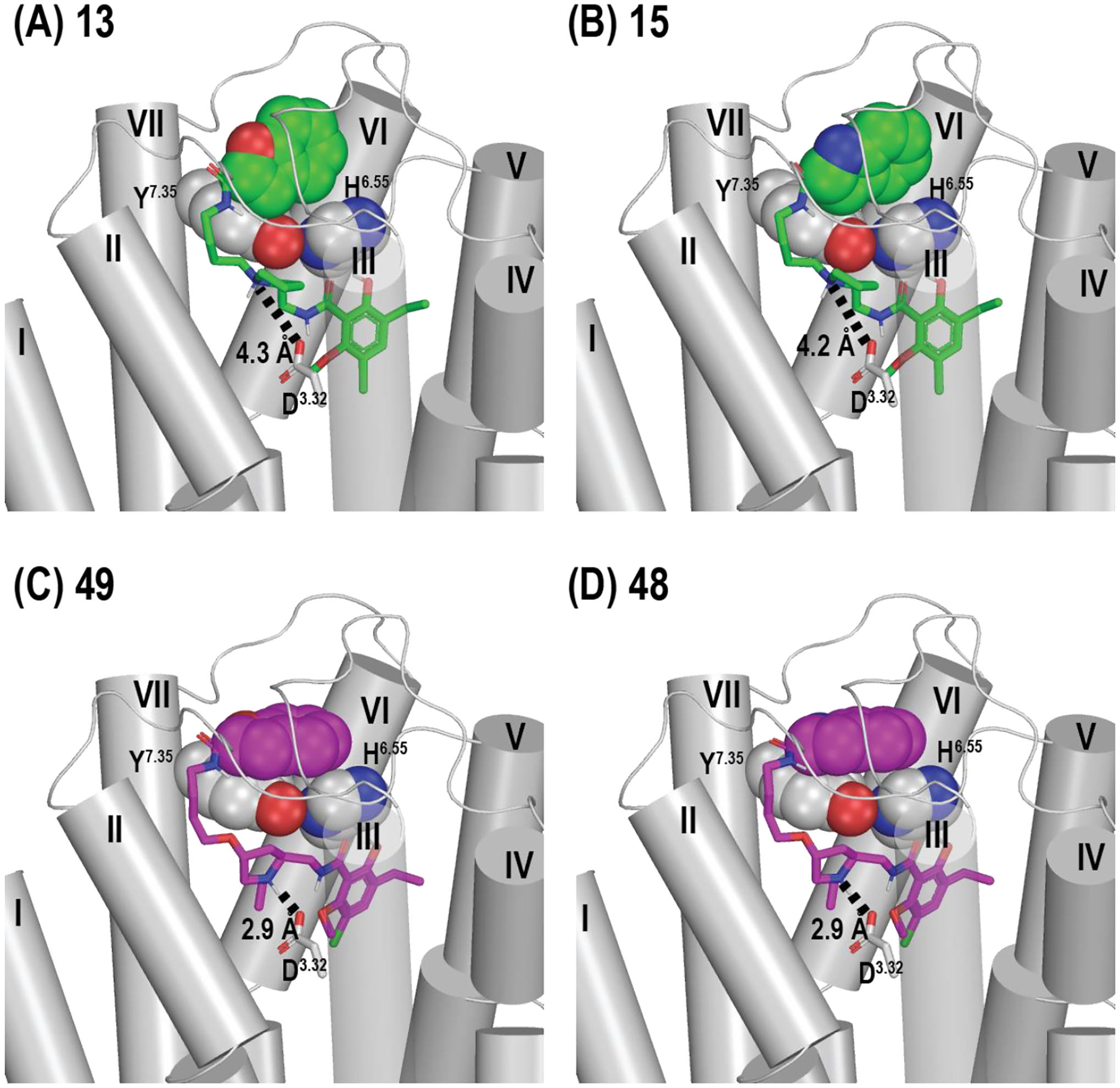
Docking poses of (A) 13, (B) 15, (C) 49, and (D) 48 at D3R. The carbon atoms of 13 and 15, which commonly have an N-alkylated linker, are colored in green; those of 48 and 49, which commonly have an O-alkylated linker, are colored in purple. In these poses, 48 and 49 have stronger interactions with Asp3.32 than 13 and 15, as indicated by the distances between their charged nitrogen atoms and the sidechain of Asp3.32. The SPs of these ligands, i.e., indoleamide and benzofuran, as well as the aromatic sidechains of His6.55 and Tyr7.35 are shown as spheres.
CONCLUSION
In summary, a series of eticlopride analogues were synthesized by introducing the SP with linker at the 2- (N) or 4- (C) positions of the pyrrolidine ring of eticlopride. Various SPs were investigated for their effect on D2R and D3R binding affinities. We discovered that O-alkylated analogues were preferable in terms of high affinity binding at both D2R and D3R as compared to their equally N-alkylated analogues e.g., the O-alkylated 33, 34, 46, and 47 showed higher binding affinities than the comparable N-alkylated 11, 13, and 15. Moreover, in comparing eticlopride with its N-nor analogue, 5, the N-ethyl group contributes to binding affinities at D3R and particularly D2R. However, N-alkylation no longer plays an important role in the 4-substituted bitopic ligands e.g., 33 v. 34, 46 v. 48, 47 v. 49.
A subset of analogues was evaluated in BRET assays for functional activity. All of these analogues were antagonists or very weak partial agonists. Molecular models confirmed SAR showing that the SPs on the higher affinity O-alkylated analogs were tolerated at both D2R and D3R and they did not access an SBP that was different between the subtypes. This is in contrast to previously reported and highly D3R selective antagonists and partial agonists.20,22 Importantly, this study provides the necessary SAR to position long linker chains and/or sterically bulky groups on the eticlopride molecule to create bioconjugate D2R/D3R tools of interest such as fluorescent ligands and Drugs Acutely Restricted by Tethering (DARTs).48 Further, the design of novel dual target mu opioid receptor and D3R partial agonists as potentially nonaddictive analgesics has already been inspired by these novel eticlopride ligands.49
EXPERIMENTAL METHODS
The reaction conditions and yields were not optimized. Reagents and anhydrous solvents were purchased from Sigma-Aldrich, AK Scientific, TCI America, Chem Impex and Alfa Aesar, and were used without further purification. Spectroscopic data and yields are reported for the compounds as free bases. Flash chromatographic purifications were performed on silica gel, either manually (EMD Chemicals, Inc.; 230–400 mesh, 60 Å), or using a Teledyne ISCO CombiFlash Rf instrument. 1H, 13C spectra were acquired using a Varian Mercury Plus 400 spectrometer. 1H chemical shifts are reported as parts per million (δ ppm) relative to tetramethylsilane (0.00 ppm). All the Coupling constants are measured in Hz. Chemical shifts for 13C NMR spectra are reported as parts per million (δ ppm) relative to deuterated solvents. Chemical shifts, multiplicities and coupling constants (J) have been reported and calculated using Vnmrj Agilent-NMR 400MR or MNova 9.0. Gas chromatography-mass spectrometry (GC/MS) data were acquired (where obtainable) using an Agilent Technologies (Santa Clara, CA) 6890N GC equipped with an HP-5MS column (cross-linked 5% PH ME siloxane, 30 m × 0.25 mm i.d. × 0.25 μm film thickness) and a 5973 mass-selective ion detector in electron-impact mode. Ultrapure grade helium was used as the carrier gas at a flow rate of 1.2 mL/min. The injection port and transfer line temperatures were 250 and 280 °C, respectively, and the oven temperature gradient used was as follows: the initial temperature (100 °C) was held for 3 min and then increased to 295 °C at 15 °C/min over 13 min, and finally maintained at 295 °C for 10 min. Combustion analysis was performed by Atlantic Microlab, Inc. (Norcross, GA) and the results agree within ±0.4% of calculated values (Table S1). HRMS (mass error within 5 ppm) and MS/MS fragmentation analysis were performed on LTQ-Orbitrap Velos (Thermo-Scientific, San Jose, CA) coupled with an ESI source in positive ion mode. HPLC analysis was performed using an Agilent system coupled with DAD (Diode Array Detector). Separation of the analyte, purity and enantiomeric or diastereomeric excess determinations was achieved employing the methods described in the experimental section and supplementary information for each compound analyzed. HPLC analytical columns used were CHIRALCEL OD-H (Daicel Corporation CPI Company) column (4.6 × 250 mm, 5 μm) and CHIRALPAK AD-H (Daicel Corporation CPI Company) column (4.6 × 250 mm, 5 μm). The column temperature was maintained at 40 °C, and 20 μl of 0.5 mg/mL sample solution were injected in the HPLC for the analyses. Analyte detection was performed with DAD wavelength set at 254 nm. Infrared (IR) spectra were obtained (neat) on a Perkin Elmer Spectra Two FTIR spectrometer version 10.4.4. Melting point determination was conducted using an OptiMelt automated melting point system and are uncorrected. Based on NMR, HPLC and combustion analysis data, all final compounds are ≥95% pure, unless otherwise stated.
General N-alkylation method.
To the (S)-3-chloro-5-ethyl-6-hydroxy-2-methoxy-N-(pyrrolidin-2-ylmethyl)benzamide hydrochloride, (5) (1.0 equiv) dissolved in DMF. K2CO3 (8.0 equiv) or NaHCO3 (8.0 equiv) was added at room temperature (RT), and stirred for 5 min. To this mixture appropriate alkyl halides (1.2 equiv) were added and stirred for 3–4 h. The progress of the reaction was monitored by TLC. After reaction completion, cold water (50–100 mL) was added to the reaction mixture and the product was extracted with EtOAC (x 3). The combined organic fractions were washed (x 2) with cold water, dried over anhydrous Na2SO4 or anhydrous MgSO4, filtered and then evaporated to afford the crude N-alkylated analogues.
(S)-3-Chloro-5-ethyl-6-hydroxy-2-methoxy-N-((1-propylpyrrolidin-2-yl)methyl)benzamide (6).
A mixture of 5, (0.200 g, 0.57 mmol), 1-iodopropane (0.07 mL, 0.74 mmol) and K2CO3 (0.634 g, 4.58 mmol) in acetonitrile (8 mL) was stirred at reflux under argon at 80 °C for 12 h. The mixture was diluted with H2O (50 mL) and extracted with CHCl3 (3×100 mL). The combined organic layers were collected and dried over anhydrous MgSO4, filtered and concentrated to afford the crude product. The crude product was purified by flash column chromatography (5% CMA; 95% CHCl3, 5% MeOH, 0.1% ammonium hydroxide) to give the product (160 mg, 79% yield) as a light yellow oil. 1H NMR (400 MHz, CDCl3) δ 13.86 (s, 1H), 8.83 (d, J = 7.1 Hz, 1H), 7.20 – 7.18 (m, 1H), 3.86 (s, 3H), 3.79–3.74 (m, 1H), 3.31–3.27 (m, 1H), 3.18 (dd, J = 9.5, 5.3 Hz, 1H), 2.75 – 2.53 (m, 4H), 2.22 – 2.09 (m, 2H), 1.96 – 1.84 (m, 1H), 1.77 – 1.66 (m, 2H), 1.65 – 1.43 (m, 3H), 1.17 (t, J = 7.5 Hz, 3H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 169.5, 160.1, 152.5, 132.8, 130.7, 115.99, 108.2, 62.3, 61.37, 56.2, 53.9, 40.4, 28.1, 22.6, 22.1, 13.4, 11.9; IR: 3366.3 (s, br) cm−1; GC-MS (EI): m/z 354 (M+). Anal(C18H27ClN2O3) C, H, N.
(S)-N-((1-Benzylpyrrolidin-2-yl)methyl)-3-chloro-5-ethyl-6-hydroxy-2-methoxybenzamide (7).
The compound was prepared from 5 (0.300 g, 0.86 mmol), benzylbromide (0.176 g, 1.03 mmol) and NaHCO3 (0. 58g, 6.88 mmol) according to the general N-alkylation procedure. The crude product was purified by flash chromatography using 20% acetone/CHCl3 as eluent to give the desired product as a yellow solid (0.246 g, 71% yield). 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 7.38 (s, 1H), 7.32 (d, J = 7.9 Hz, 5H), 7.23 (s, 1H), 4.02 (d, J = 12.5 Hz, 2H), 3.83 (s, 3H), 3.33 (d, J = 13.0 Hz, 1H), 3.00 (s, 1H), 2.80 (s, 1H), 2.62 (q, J = 7.5 Hz, 2H), 2.25 (d, J = 6.5 Hz, 1H), 1.98 (s, 1H), 1.70 (s, 2H), 1.41 – 1.06 (m, 5H). 13C NMR (101 MHz, CDCl3) δ 169.72, 160.32, 152.58, 139.16, 132.92, 130.97, 130.71, 128.74, 128.28, 127.24, 116.06, 108.39, 61.97, 61.42, 58.41, 54.24, 46.23, 40.19, 28.20, 25.43, 22.53, 13.43. The HCl salt was precipitated from acetone; Mp 72–74 °C. Anal (C22H27ClN2O3 •HCl•0.25H2O) C, H, N. Compound 7 was analyzed by HPLC: tR=4.912 min, ee was >99% (detailed method reported in SI; Figure S1).
(S)-2-(Benzyloxy)-N-((1-benzylpyrrolidin-2-yl)methyl)-5-chloro-3-ethyl-6-methoxybenzamide (8).
The compound was prepared from 5 (0.300 g, 0.86 mmol), benzylbromide (0.293 g, 1.72 mmol) and NaHCO3 (0. 58g, 6.88 mmol) according to the general N-alkylation procedure. The crude product was purified by flash chromatography using 20% acetone/CHCl3 as eluent to give the desired product as a colorless solid (0.275 g, 65% yield). 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.38 (d, J = 6.5 Hz, 5H), 7.30 (d, J = 7.5 Hz, 5H), 7.22 (s, 1H), 6.34 (s, 2H), 4.95 (s, 2H), 3.90 (s, 3H), 3.84 (d, J = 12.8 Hz, 1H), 3.75 (s, 1H), 3.26 (s, 1H), 3.20 (d, J = 12.8 Hz, 1H), 2.86 – 2.80 (m, 1H), 2.71 (s, 1H), 2.63 (d, J = 7.4 Hz, 1H), 2.15 (d, J = 8.0 Hz, 1H), 1.84 (s, 1H), 1.56 (s, 2H), 1.21 (t, J = 7.6 Hz, 3H). The HCl salt was precipitated from acetone; Mp 71–73 °C. Anal (C29H33ClN2O3•HCl•1.5H2O) C, H, N.
(S)-3-Chloro-5-ethyl-6-hydroxy-2-methoxy-N-((1-(3-phenylpropyl)pyrrolidin-2-yl)methyl)benzamide (9).
The compound was prepared from 5 (0.250 g, 0.72 mmol), (3-bromopropyl)benzene (0.171 g, 0.86 mmol) and K2CO3 (0.796 g, 5.76 mmol) according to the general N-alkylation procedure. The crude product was purified by flash chromatography using 10% acetone/CHCl3 as eluent to give the desired product as a colorless solid (0.215 g, 70% yield). 1H NMR (400 MHz, CDCl3) δ 13.85 (br s, 1H), 8.83 (s, 1H), 7.24 (s, 2H), 7.22 (s, 2H), 7.18 (s, 1H), 7.16 (s, 1H), 3.86 (s, 3H), 3.80 – 3.67 (m, 1H), 3.34 – 3.17 (m, 2H), 2.86 – 2.67 (m, 2H), 2.67 – 2.56 (m, 2H), 2.32 – 2.14 (m, 2H), 1.89 (m, 2H), 1.78 – 1.70 (m, 2H), 1.69 – 1.52 (m, 2H), 1.35 – 1.24 (m, 2H), 1.20 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 169.74, 160.25, 152.59, 142.11, 132.91, 130.89, 128.30, 125.79, 116.04, 108.39, 62.40, 61.59, 53.95, 53.95, 40.45, 34.02, 30.45, 28.17, 22.51, 13.59. HRMS (ESI): m/z calcd for (C24H31ClN2O3+H+); 431.2023, found 431.2087 (M+H+). Compound 9 was analyzed by HPLC: tR=4.591 min, ee was >98% (detailed method reported in SI; Figure S2).
(S)-3-Chloro-5-ethyl-6-hydroxy-2-methoxy-N-((1-(2-(2-methyl-4-oxo-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-3-yl)ethyl)pyrrolidin-2-yl)methyl)benzamide (10).
The compound was prepared from 5 (0.300 g, 0.86 mmol), commercially available 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (0.233 g, 1.03 mmol), and K2CO3 (1.14 g, 8.24 mmol). The reaction mixture was stirred at reflux in acetone (15 mL) for 8 h in the presence of sodium iodide (NaI) (0.125 g, 0.50 mmol). The progress of the reaction was monitored by TLC. After reaction completion, the solvent was removed then water added to the reaction mixture and the product was extracted with EtOAC (3×100 mL). The combined organic fractions were dried over anhydrous Na2SO4, filtered and evaporated to afford crude compound. The crude product was purified by flash chromatography using 30% MeOH/CHCl3 as eluent to give the desired product as a colorless solid (0.168 g, 39 % yield). 1H NMR (400 MHz, CDCl3) δ 13.86 (s, 1H), 8.79 (s, 1H), 7.20 (s, 1H), 3.87 (s, 3H), 3.72 (dd, J = 13.5, 6.6 Hz, 2H), 3.33 (s, 3H), 3.03 – 2.90 (m, 1H), 2.79 (s, 3H), 2.72 (s, 2H), 2.67 – 2.55 (m, 2H), 2.49 – 2.32 (m, 2H), 2.27 (d, J = 7.9 Hz, 3H), 1.91 (s, 3H), 1.86 – 1.69 (m, 4H), 1.69 – 1.52 (m, 1H), 1.19 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 169.46, 162.51, 160.12, 158.35, 155.89, 152.56, 132.83, 130.67, 119.07, 116.02, 108.16, 61.92, 61.46, 53.91, 51.72, 42.62, 40.42, 31.34, 28.13, 25.71, 22.77, 22.50, 21.93, 21.21, 19.19, 13.41. The HCl salt was precipitated from acetone; Mp 131–132 °C. Anal (C26H35ClN4O4•2HCl•2H2O) C, H, N. Compound 10 was analyzed by HPLC: tR=8.002 min, ee was >97% (detailed method reported in SI; Figure S3).
(S)-3-Chloro-5-ethyl-6-hydroxy-2-methoxy-N-((1-(4-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)oxy)butyl)pyrrolidin-2-yl)methyl)benzamide (11).
The compound was prepared from 5 (0.3 g, 0.86 mmol), 7-(4-bromobutoxy)-3,4-dihydroquinolin-2(1H)-one 5e (0.307 g 1.031 mmol), and K2CO3 (0.950 g, 6.88 mmol) according to the general N-alkylation procedure. The crude product was purified by flash chromatography using 30% acetone/CHCl3 as eluent to give the desired product as a brown oil (0.180 g, 40% yield). 1H NMR (400 MHz, CDCl3) δ 13.86 (s, 1H), 8.82 (s, 1H), 7.75 (s, 1H), 7.21 (s, 1H), 7.01 (d, J = 8.4 Hz, 1H), 6.47 (dd, J = 8.3, 2.4 Hz, 1H), 6.26 (d, J = 2.4 Hz, 1H), 3.91 (t, J = 6.1 Hz, 1H), 3.86 (s, 3H), 3.73 (dt, J = 22.9, 11.2 Hz, 1H), 3.38 – 3.15 (m, 2H), 2.93 – 2.83 (m, 2H), 2.80 (dd, J = 13.8, 5.7 Hz, 2H), 2.70 – 2.49 (m, 5H), 2.32 – 2.13 (m, 2H), 1.93 – 1.52 (m, 8H), 1.19 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.37, 169.57, 160.09, 158.59, 152.47, 138.04, 132.90, 130.82, 128.64, 116.05, 115.69, 108.57, 108.17, 102.01, 67.78, 62.31, 61.39, 53.79, 53.59, 40.40, 31.08, 28.19, 26.98, 25.35, 24.58, 22.58, 22.50, 13.40. The HCl salt was precipitated from acetone; Mp 79–81 °C Anal (C28H36ClN3O5•HCl•H2O) C, H, N. HRMS (ESI): m/z calcd for (C28H36ClN3O5+H+); 530.2343, found, 529.2435 (M+H+). Compound 11 was analyzed by HPLC: tR=12.065 min, ee was >99% (detailed method reported in SI; Figure S4).
(S)-N-(4-(2-((3-Chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)pyrrolidin-1-yl)butyl) benzofuran-2-carboxamide (13).
A mixture of 5 (0.2 g, 0.57 mmol), N-(4-bromobutyl)benzofuran-2-carboxamide 1230 (0.17 g, 0.57 mmol), and NaHCO3 (0.048 mg, 0.57 mmol) in acetonitrile (10 mL) was stirred at reflux under argon at 80 °C for 12 h. The mixture was purified by flash column chromatography (30% EtOAc:Hexanes) followed by 5% CMA to yield a light pink oil as pure product (210 mg, 70% yield). 1H NMR (400 MHz, CDCl3) δ 13.80 (s, 1H), 8.81 (d, J = 6.9 Hz, 1H), 7.64 (dd, J = 7.8, 1.3, Hz, 1H), 7.49 – 7.34 (m, 3H), 7.31 – 7.25 (m, 1H), 7.14 (d, J = 0.8 Hz, 1H), 6.73 (s, 1H), 3.84 (s, 3H), 3.76–72 (m, 1H), 3.55 – 3.42 (m, 2H), 3.33–3.28 (m, 1H), 3.20 (dt, J = 9.6, 4.5 Hz, 1H), 2.79 (dt, J = 12.0, 7.7 Hz, 1H), 2.62–2.54 (m, 3H), 2.31 – 2.13 (m, 2H), 1.91 (dt, J = 12.2, 7.9 Hz, 1H), 1.81 – 1.53 (m, 7H), 1.16 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 169.5, 160.1, 158.9, 154.6, 152.4, 148.8, 132.8, 130.8, 127.6, 126.7, 123.6, 122.7, 116.0, 111.7, 110.2, 108.1, 62.4, 61.4, 53.8, 53.7, 40.3, 39.2, 28.2, 27.6, 26.2, 22.5, 22.49, 13.4; IR: 3351.2 (s, br) cm−1; Anal. (C28H34ClN3O5) C, H, N.
General reductive amination method
To the carbaldehydes, (1.0 equiv) dissolved in dichloroethane (DCE), corresponding free amines (1.0 equiv) were added at RT. Subsequently, 2–4 drops of glacial acetic acid (AcOH) was added and allowed to stir for 30 min. Subsequently, NaBH(OAc)3 (1.25 equiv) was added portionwise, and the stirring continued at RT for 5 h. The progress of the reaction was monitored by TLC. The excess acetic acid was removed under vacuum and water (50–200 mL) was added to the reaction mixture. The pH of the solution was adjusted to 9 by using saturated aq NaHCO3 solution and the crude compounds were extracted (x3) with CHCl3. The combined organic fractions were dried over anhydrous Na2SO4, filtered and then evaporated to afford the crude products.
(S)-N-(4-(2-((3-Chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)pyrrolidin-1-yl)butyl)-1H-indole-2-carboxamide (15).
The compound was prepared from 5 (0.3 g, 0.86 mmol), N-(4-oxobutyl)-1H-indole-2-carboxamide 1423 (0.2 g, 0.87 mmol), and NaBH(OAc)3 (0.227 g 1.08 mmol) according to the general reductive amination method. The crude product was purified by flash chromatography using 40% acetone/CHCl3 as eluent to give the desired product as a colorless solid (0.285 g, 63% yield). 1H NMR (400 MHz, CDCl3) δ 13.67 (br s, 1H), 9.65 (brs, 1H), 8.81 (d, J = 4.3 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.43 (dd, J = 8.3, 0.8 Hz, 1H), 7.30 – 7.22 (m, 1H), 7.17 (s, 1H), 7.12 (ddd, J = 8.0, 7.1, 0.9 Hz, 1H), 6.82 (dd, J = 2.1, 0.7 Hz, 1H), 6.39 (s, 1H), 3.84 (s, 3H), 3.74 (ddd, J = 13.9, 7.1, 2.4 Hz, 1H), 3.50 (dd, J = 12.9, 6.7 Hz, 2H), 3.32 (ddd, J = 13.9, 4.7, 3.1 Hz, 1H), 3.26 – 3.12 (m, 1H), 2.88 – 2.72 (m, 1H), 2.69 – 2.50 (m, 3H), 2.31 – 2.21 (m, 2H), 1.92 (ddd, J = 16.5, 11.9, 8.1 Hz, 1H), 1.73 (dt, J = 18.1, 9.6 Hz, 3H), 1.62 (ddd, J = 16.1, 12.9, 5.9 Hz, 4H), 1.17 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 169.54, 161.74, 160.10, 152.37, 136.31, 132.89, 130.84, 130.78, 127.61, 124.29, 121.82, 120.50, 116.04, 111.96, 108.14, 101.88, 62.49, 61.44, 53.88, 40.39, 39.45, 28.16, 27.59, 26.04, 22.55, 22.50, 13.36. The HCl salt was precipitated from acetone; Mp 110–112 °C. Anal (C28H35ClN4O4•HCl•0.5H2O) C, H, N. HRMS (ESI): m/z calcd for (C28H35ClN4O4+H+), 527.2347; found 527.2449 (M+H+). Compound 15 was analyzed by HPLC: tR=18.155 min, ee was >99% (detailed method reported in SI; Figure S5).
General amidation method (A).
Thionyl chloride (3 equiv) was added to a solution of 3-chloro-5-ethyl-6-hydroxy-2-methoxybenzoic acid, 16 (1 equiv) in toluene (15 mL/1 mmol) and DMF (4 μL/1 mmol). The mixture was stirred at RT for 12 h under argon. Excess SOCl2 was removed under vacuum to give a brown solid product (3-chloro-5-ethyl-6-hydroxy-2-methoxybenzoyl chloride) (VI), which was dissolved in dry CHCl3 (6 mL), followed by addition of triethylamine (2.4 equiv), in an ice bath. Then the corresponding primary amine (1 equiv) was added to the mixture and stirred under argon for 48 h at RT. The solvent was removed under reduced pressure, and the product was purified by column chromatography (5% CMA).
General amidation method (B)
CDI (1.2 equiv), was added to a solution of 3-chloro-5-ethyl-6-hydroxy-2-methoxybenzoic acid, 16 (1 equiv) in THF. The reaction mixture was stirred at RT for 2 h under argon. The corresponding primary amine (1 equiv) was added to the reaction mixture and stirred at RT under argon overnight. The solvent was removed under reduced pressure, and the product was purified by column chromatography (5% CMA).
3-Chloro-5-ethyl-6-hydroxy-2-methoxy-N-((1-methylpyrrolidin-3-yl)methyl)benzamide (17a).
This compound was synthesized according to the general amidation method (A) by using 16 (250 mg, 1.1 mmol) and (1-methylpyrrolidin-3-yl)methanamine (113.9 mg, 1.0 mmol). The product 17a (120 mg, 46% yield) was obtained as a light yellow oil. For method (B), 16 (461 mg, 2.0 mmol) and (1-methylpyrrolidin-3-yl)methanamine (228 mg, 2.0 mmol) were used. The product 17a (340 mg, 52% yield) was obtained as a light yellow oil. The free base was converted to the HCl salt as a foam. 1H NMR (400 MHz, CDCl3) δ 13.63 (s, 1H), 8.66 (s, 1H), 7.20 (t, J = 0.7 Hz, 1H), 3.84 (s, 3H), 3.44 (dt, J = 6.6, 5.4 Hz, 2H), 2.65 – 2.38 (m, 7H), 2.35 (s, 3H), 2.06 – 2.04 (m, 1H), 1.58 – 1.54 (m, 1H), 1.17 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 169.4, 159.98, 152.2, 132.9, 130.9, 116.1, 108.1, 61.5, 60.3, 55.98, 44.2, 42.02, 37.3, 29.0, 22.5, 13.4; IR: 3361.3 (s, br) cm−1; GC-MS (EI): m/z 326 (M+). Anal. (C16H23ClN2O3•HCl•1.25H2O) C, H, N
3-Chloro-5-ethyl-N-((1-ethylpyrrolidin-3-yl)methyl)-6-hydroxy-2-methoxybenzamide (17b).
This compound was synthesized according to the general amidation method (A) by using 16 (200 mg, 0.80 mmol) and (1-ethylpyrrolidin-3-yl)methanamine (103 mg, 0.80 mmol). The product 17b (110 mg, 40% yield) was obtained as a light brown oil. For method (B), 16 (490 mg, 2.1 mmol) and (1-ethylpyrrolidin-3-yl)methanamine (272 mg, 2.1 mmol) were used. The product 17b (360 mg, 50% yield) was obtained as a yellow oil. The free base was converted to the HCl salt as a foam. 1H NMR (400 MHz, CDCl3) δ 13.60 (s, 1H), 8.66 (s, 1H), 7.21 (t, J = 0.7 Hz, 1H), 3.85 (s, 3H), 3.48–3.44 (m, 2H), 2.81 – 2.46 (m, 9H), 2.45 – 2.03 (m, 1H), 1.58 – 1.53 (m, 1H), 1.18 (t, J = 7.5 Hz, 3H), 1.11 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 169.4, 159.97, 152.2, 132.9, 130.9, 116.2, 108.2, 61.5, 58.0, 53.5, 50.1, 44.2, 36.6, 28.4, 22.5, 13.8, 13.4; IR: 3368.9 (s, br) cm−1; GC-MS (EI): m/z 340 (M+). Anal. (C17H25ClN2O3•HCl•1.75H2O) C, H, N
3-Chloro-5-ethyl-6-hydroxy-2-methoxy-N-((1-propylpyrrolidin-3-yl)methyl)benzamide (17c).
This compound was synthesized according to the general amidation method (A) by using 16 (200 mg, 0.80 mmol) and (1-propylpyrrolidin-3-yl)methanamine (114 mg, 0.80 mmol). The product 17c (135 mg, 48% yield) was obtained as a light yellow oil. For method (B), 16 (461 mg, 2.0 mmol) and (1-propylpyrrolidin-3-yl)methanamine (284 mg, 2.0 mmol) were used. The product 17c (360 mg, 51% yield) was obtained as a yellow oil. The free base was converted to the HCl salt as a foam. 1H NMR (400 MHz, CDCl3) δ 13.60 (s, 1H), 8.66 (s, 1H), 7.21 (t, J = 0.7 Hz, 1H), 3.85 (s, 3H), 3.49–3.45 (m, 2H), 2.81 – 2.30 (m, 10H), 2.12 – 2.00 (m, 2H), 1.18 (t, J = 7.5 Hz, 4H), 1.11 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 169.4, 159.95, 152.2, 132.9, 130.9, 116.2, 108.2, 61.5, 58.0, 53.5, 50.1, 44.2, 36.6, 28.4, 22.6, 13.8, 13.4; IR: 3366.3 (s, br) cm−1; GC-MS (EI): m/z 354 (M+). Anal. (C18H27ClN2O3•HCl•1.25H2O) C, H, N
tert-Butyl ((1-(4-(1,3-dioxoisoindolin-2-yl)butyl)piperidin-3-yl)methyl)carbamate (20).
K2CO3 (8.51 g, 61.59 mmol) and NaI (4.20 g, 27.99 mmol) were added to a solution of 2-(4-bromobutyl)isoindoline-1,3-dione 18 (6.0 g, 27.99 mmol) and 3-(Boc-aminomethyl)piperidine 19 (9.87 g, 34.99 mmol) in acetone (60 mL). The resulting suspension was stirred at reflux at 50–60 °C for 22 h. After cooling the reaction mixture to RT, K2CO3 was removed via filtration and the filtrate was concentrated. The crude product was purified by flash column chromatography (5% CMA) to give pure 20 (10.21 g, 70% yield) as a thick yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.87 – 7.76 (m, 2H), 7.74 – 7.65 (m, 2H), 4.83 (s, 1H), 3.24 (t, J = 6.6 Hz, 2H), 3.06 (br s, 2H), 2.83 (d, J = 8.4 Hz, 2H), 2.44 (br, 1H), 2.27 (br, 2H), 1.98 (d, J = 7.6 Hz, 3H), 1.90 – 1.63 (m, 5H), 1.40 (s, 9H), 1.15 (d, J = 11.6 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 168.4, 156.1, 134.0, 131.95, 123.4, 79.6, 57.3, 56.4, 53.1, 43.5, 36.8, 35.3, 28.4, 28.3, 26.8, 26.1, 22.5, 21.6, 20.1.
tert-Butyl ((1-(4-aminobutyl)piperidin-3-yl)methyl)carbamate (21).
Anhydrous hydrazine (2.13 mL, 67.88 mmol) was added to a solution of 20 (10.22 g, 24.59 mmol) in ethanol (120 mL). The solution was stirred under reflux at 90 °C for 4 h. The reaction mixture was cooled and concentrated. The crude reaction mixture was partitioned between CHCl3 (300 mL) and 20% K2CO3 (100 mL) aq solution, and the organic layer was collected and dried over MgSO4. The organic layer was filtered and concentrated to give 21 (4.21 g, 60% yield) as a light yellow oil. 1H NMR (400 MHz, CDCl3) δ 4.88 (br s, 1H), 3.24 (br s, 3H), 2.94 (d, J = 6.2 Hz, 2H), 2.85 – 2.72 (m, 2H), 2.68 (t, J = 6.4 Hz, 1H), 2.27 (t, J = 7.0 Hz, 2H), 1.84 (dd, J = 11.3, 2.7 Hz, 1H), 1.70–1.60 (m, 4H), 1.56 – 1.42 (m, 4H), 1.38 (s, 9H), 0.90 (d, J = 11.3 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 156.1, 78.99, 58.8, 57.8, 54.1, 44.5, 41.5, 36.8, 30.8, 28.5, 28.4, 24.7, 24.3; GC-MS (EI): m/z 285 (M+).
General amidation method (C).
CDI (1.02 equiv) was added to the solution of carboxylic acid (1.0 equiv) in THF (10 mL/mmol). The solution was cooled to 0 °C and the amine substrate (1.0 equiv) in THF (3 mL/mmol) was added dropwise. The reaction mixture was allowed to warm to RT and stirred overnight. The reaction mixture was concentrated, and the crude product was diluted with CHCl3 (20 mL/mmol) and washed with saturated aq NaHCO3 solution (2 × 10 mL). The organic layer was dried over MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography 5% CMA), or as indicated.
tert-Butyl ((1-(4-(1H-indole-2-carboxamido)butyl)piperidin-3-yl)methyl)carbamate. (22a).
This compound was synthesized according to general amidation Method (C), using 21 (1.08 g, 3.78 mmol) and commercially available indole-2-carboxylic acid (600 mg, 3.72 mmol). The pure product (1.60 g, 99% yield) was obtained as a brown solid; 1H NMR (400 MHz, CDCl3) δ 9.90 (s, 1H), 7.70 – 7.56 (m, 1H), 7.46 – 7.36 (m, 1H), 7.28 – 7.19 (m, 1H), 7.14 – 7.04 (m, 1H), 7.03 – 6.94 (m, 1H), 6.90 (q, J = 0.9 Hz, 1H), 4.72 (s, 1H), 3.47 (q, J = 6.3 Hz, 2H), 2.99 (d, J = 5.7 Hz, 2H), 2.82–2.81 (m, 2H), 2.36 (d, J = 9.0 Hz, 2H), 1.93 (t, J = 11.0 Hz, 1H), 1.80 – 1.46 (m, 9H), 1.41 (s, 9H), 0.95 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 161.9, 156.2, 136.4, 135.1, 131.1, 127.6, 124.2, 121.8, 120.4, 112.0, 105.2, 102.4, 79.2, 58.1, 57.6, 54.06, 44.3, 39.3, 36.7, 28.4, 28.3, 27.4, 24.43, 23.97.
tert-Butyl ((1-(4-(benzofuran-2-carboxamido)butyl)piperidin-3-yl)methyl)carbamate (22b).
This compound was synthesized according to general amidation method (C), using 21 (1.07 g, 3.76 mmol) and commercially available benzofuran-2-carboxylic acid (600 mg, 3.70 mmol). The pure product 22b (1.52 g, 94% yield) was obtained as a yellow solid; 1H NMR (400 MHz, CDCl3) δ 7.65–7.64 (m, 1H), 7.50 – 7.42 (m, 2H), 7.40 –7.36 (m, 1H), 7.30 – 7.23 (m, 1H), 7.03 (br, 1H), 4.64 (s, 2H), 3.48 (q, J = 6.6 Hz, 2H), 3.02 (br, 2H), 2.88 (br, 2H), 2.42 (br, 2H), 2.01 (d, J = 9.8 Hz, 2H), 1.89 – 1.55 (m, 7H), 1.41 (s, 9H), 0.98 (d, J = 8.8 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 158.9, 156.1, 154.7, 148.9, 127.6, 126.7, 123.6, 122.7, 111.7, 110.2, 79.2, 58.2, 57.6, 54.1, 44.3, 39.0, 36.7, 28.4, 28.2, 27.4, 24.4, 23.9.
N-(4-(3-(Aminomethyl)piperidin-1-yl)butyl)-1H-indole-2-carboxamide (23a).
Compound 22a (1.43 g, 3.34 mmol)) was dissolved in MeOH (30 mL) and 2M HCl in diethyl ether (19.11 mL) was added slowly dropwise. The reaction mixture was stirred at reflux for 3 h. The solvent was removed in vacuo to yield the crude product as the HCl salt. The salt was dissolved in water (100 mL) and saturated aq K2CO3 (100 mL) solution and extracted with CHCl3 (3×75 mL). The combined organic layers were collected and dried over MgSO4, filtered and concentrated to give 23a as the free base (0.910 g, 93% yield) as a yellow solid which was used for the next step without further purification; 1H NMR (400 MHz, CDCl3) δ 10.07 (s, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 8.2 Hz, 1H), 7.31 – 7.17 (m, 1H), 7.10 (t, J = 7.5 Hz, 1H), 6.80 (d, J = 11.9 Hz, 2H), 3.49 (q, J = 6.2 Hz, 2H), 3.01 – 2.75 (m, 2H), 2.62 – 2.45 (m, 2H), 2.34 (t, J = 6.9 Hz, 2H), 1.87–1.72 (m, 2H), 1.68 – 1.49 (m, 8H), 1.45 – 1.23 (m, 2H), 0.88 (qd, J = 12.1, 4.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 161.8, 136.4, 131.1, 127.6, 124.2, 121.7, 120.4, 112.0, 101.9, 58.5, 58.2, 54.4, 46.4, 39.7, 39.6, 28.7, 27.6, 25.1, 24.5; GC-MS (EI): m/z 328 (M+).
N-(4-(3-(Aminomethyl)piperidin-1-yl)butyl)benzofuran-2-carboxamide (23b).
The compound was synthesized by employing the same procedure described for 23a, by using 22b (1.32 g, 3.07 mmol). The organic layer was filtered and concentrated to give the free base 23b (0.88 g, 90% yield) as a yellow oil which was used in the next step without further purification; 1H NMR (400 MHz, CDCl3) δ 7.59 – 7.56 (m, 1H), 7.42 – 7.39 (m, 2H), 7.34 – 7.30 (m, 1H), 7.26 – 7.18 (m, 2H), 3.56 – 3.11 (m, 4H), 2.83 (dd, J = 38.8, 10.3 Hz, 2H), 2.52 – 2.50 (m, 2H), 2.28 (br, 2H), 1.80 (t, J = 11.5 Hz, 1H), 1.71 – 1.68 (m, 1H), 1.57 (br, 8H), 0.83 (q, J = 11.7 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 158.9, 154.6, 148.9, 127.6, 126.7, 123.6, 122.6, 111.6, 110.1, 58.4, 57.97, 54.3, 45.9, 39.1, 38.9, 28.6, 27.5, 24.9, 24.2; GC-MS (EI): m/z 329 (M+).
General amidation method (D).
To the solution of 3-chloro-5-ethyl-6-hydroxy-2-methoxybenzoic acid 16 (1.0 equiv) in CH2Cl2 (10 mL), Na2CO3 (1.0 equiv) dissolved in water (3 mL) was added, followed by EDC (1.25 equiv) and corresponding amines 23a or 23b (1.5 equiv). The reaction mixture was stirred at RT for 24 h. The aq layer was extracted twice with 1:1 THF/diethyl ether and the combined organic extracts were washed with saturated NaHCO3. The organic layer was dried over MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography (5% CMA).
N-(4-(3-((3-Chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)piperidin-1-yl)butyl)-1H-indole-2-carboxamide (24a).
This compound was prepared according to the general amidation method (D) by using 16 (0.39 g, 1.69 mmol) and 23a (0.64 g, 1.95 mmol). The crude product was purified by flash column chromatography to obtain the pure 24a as a viscous oil (0.32 g, 31% yield). 1H NMR (400 MHz, CD3OD) δ 7.57 (d, J = 8.1, Hz, 1H), 7.42 (d, J = 8.2, Hz, 1H), 7.26 (s, 1H), 7.26–7.17 (m, 1H), 7.08 – 7.00 (m, 2H), 3.84 (s, 3H), 3.43 (q, J = 6.1, 5.6 Hz, 4H), 3.16 – 3.08 (m, 2H), 2.86–2.76 (m, 2H), 2.63 – 2.48 (m, 2H), 2.21 (br, 1H), 2.01–1.65 (m, 9H), 1.43 – 1.27 (m, 1H), 1.14 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 169.6, 162.97, 158.9, 152.5, 136.9, 132.9, 130.6, 130.4, 127.5, 123.7, 121.3, 119.8, 116.3, 111.6, 108.8, 102.9, 101.2, 60.9, 52.6, 37.9, 29.2, 26.5, 22.1, 21.1, 12.6. IR: 3363.8 (s, br) cm−1. The HCl salt was precipitated from 2-propanol/acetone. Mp. 182–184 °C. Anal (C29H37ClN4O4·HCl) C, H, N.
N-(4-(3-((3-Chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)piperidin-1-yl)butyl)benzofuran-2-carboxamide (24b).
This compound was prepared according to the general amidation method (D) by using 16 (0.250 g, 1.08 mmol) and 23b (0.41 g, 1.25 mmol). The crude product was purified by flash column chromatography to obtain the pure product 24b (0.160 g, 24% yield) as a viscous oil. 1H NMR (400 MHz, CDCl3) δ 13.59 (s, 1H), 8.47 (t, J = 5.7 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.46 – 7.42 (m, 2H), 7.38–7.34 (m, 1H), 7.27 – 7.22 (m, 1H), 7.19 (t, J = 0.7 Hz, 1H), 7.00 – 6.97 (m, 1H), 3.83 (s, 3H), 3.47 (q, J = 6.4 Hz, 2H), 3.43 – 3.27 (m, 2H), 2.89–2.82 (m, 2H), 2.59 (qd, J = 7.5, 0.7 Hz, 2H), 2.37 (t, J = 6.9 Hz, 2H), 2.04 – 1.89 (m, 2H), 1.82 – 1.56 (m, 8H), 1.17 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, cdcl3) δ 169.3, 159.96, 158.9, 154.6, 152.1, 148.9, 132.95, 130.98, 127.6, 126.7, 123.6, 122.7, 116.1, 111.6, 110.2, 108.1, 61.7, 58.4, 57.98, 54.1, 43.1, 39.1, 36.4, 28.7, 27.5, 24.7, 24.3, 22.5, 13.4; IR: 3351.2 (s, br) cm−1.The HCl salt was precipitated from 2-propanol/acetone. Mp. 172–173 °C; Anal. (C29H36ClN3O5 ·HCl·1.5H2O) C, H, N.
General O-alkylation Procedure.
Sodium hydride (NaH) (3.0 equiv) (60% dispersion in mineral oil) was washed three times with hexane to remove mineral oil, then dried in vacuo. To the dried NaH in a round bottom flask, dry DMF (30 mL) was added and cooled to 0 °C. To this mixture, (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid (25, 1.0 equiv) was added, then the reaction was allowed to stir for 30 min. The corresponding alkyl halide (1.2 equiv), dissolved in DMF (5.0 mL), was added dropwise while maintaining the temperature at 0 °C. The reaction mixture was then allowed to stir for additional 2–3 h at 0 °C. Reaction progress was monitored by TLC. After confirming the reaction completion, ice cold water (100–200 mL) was added, and the aq layer washed with EtOAC to remove unreacted alkyl halide. The separated aq layer was acidified using aq 10% citric acid solution to pH 2, followed by extraction with 100–200 mL of EtOAC. The combined organic fractions were washed twice with cold water, dried over anhydrous Na2SO4, filtered and then evaporated to afford the corresponding crude O-alkylated carboxylic acid intermediates, then purified by flash column chromatography.
General amidation method (E).
To the corresponding carboxylic acids (1.2 equiv) dissolved in CHCl3 (10–30 mL), HCTU (1.3 equiv) was added at RT. After 10 min, the corresponding alkyl amine (1.0 equiv) was added, and the mixture continued to stir at RT for 2–3 h. The reaction progress was monitored by TLC. After completion of the reaction, the mixture was basified to pH 9 with saturated aq NaHCO3 and the compounds were extracted with CHCl3 (4×50 mL). The combined organic fractions were dried over anhydrous Na2SO4, filtered and then concentrated to afford the corresponding crude amide products. All final products were purified by flash column chromatography eluting with CHCl3/acetone or EtOAC/hexane solvent systems, as described.
(2S,4R)-1-(tert-Butoxycarbonyl)-4-(4-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)oxy)butoxy)pyrrolidine-2-carboxylic acid (27).
The compound was prepared from (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid 25 (5.0 g, 22 mmol), 7-(4-bromobutoxy)-3,4-dihydroquinolin-2(1H)-one (7.74 g, 26.0 mmol) 26 and NaH (1.56 g, 64.92 mmol) in DMF (40 mL) according to the general O-alkylation procedure. The crude product was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as a clear wax (7.18 g, 74% yield). Caution: this compound was corrosive and should be handled with care. Rotamers observed in NMR, 1H NMR (400 MHz, CDCl3) δ 9.26 (s, 1H; rotamer A), 8.86 (s, 1H; rotamer B), 7.03 (d, J = 8.3 Hz, 1H), 6.52 (d, J = 8.2 Hz, 1H), 6.36 (d, J = 9.5 Hz, 1H), 4.39 (dt, J = 38.4, 7.4 Hz, 1H), 4.12 (dd, J = 14.1, 6.9 Hz, 1H), 3.94 (t, J = 6.2 Hz, 2H), 3.58 – 3.39 (m, 2H), 2.89 (t, J = 7.5 Hz, 2H), 2.62 (t, J = 7.5 Hz, 2H), 2.46 – 2.08 (m, 2H), 1.91 – 1.78 (m, 2H), 1.79 – 1.65 (m, 2H), 1.47 (d, J = 6.2 Hz, 5H), 1.42 (s, 6H).
2-Benzyl 1-(tert-butyl) (2S,4R)-4-(4-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)oxy)butoxy)pyrrolidine-1,2-dicarboxylate (28).
Compound 27 (4.10 g, 91.5 mmol) was dissolved in DCM (40 mL) and cooled to 0 °C. To this solution, EDC (2.19 g, 11.4 mmol) and DMAP (1.40 g, 11.4 mmol) were added and allowed to stir for 10 min. Benzyl alcohol (1.24 g, 11.4 mmol) dissolved in DCM (10 mL) was added dropwise, and the mixture stirred at RT for 5 h. The reaction progress was monitored by TLC and after reaction completion, saturated aq, NaHCO3 (200 mL) and water (200 mL) were added to the reaction mixture, followed by extraction with EtOAC (3×300 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated. The crude product was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as a yellow oil (3.80 g, 77% yield). 1H NMR (400 MHz, CDCl3) δ 7.88 (s, 1H), 7.40 – 7.30 (m, 5H), 7.03 (d, J = 8.3 Hz, 1H), 6.50 (dd, J = 8.3, 2.4 Hz, 1H), 6.30 (dd, J = 7.9, 2.3 Hz, 1H), 5.28–5.08 (m, 2H), 4.41 (dt, J = 35.9, 7.8 Hz, 1H), 4.04 (s, 2H), 3.92 (td, J = 6.2, 2.3 Hz, 2H), 3.66 – 3.53 (m, 1H), 3.45 (t, J = 6.3 Hz, 2H), 2.88 (t, J = 7.5 Hz, 2H), 2.60 (t, J = 7.3 Hz, 2H), 2.32 (dt, J = 12.0, 10.0 Hz, 1H), 2.11 – 1.96 (m, 1H), 1.80 (dd, J = 13.8, 6.3 Hz, 2H), 1.70 (dt, J = 10.3, 6.7 Hz, 2H), 1.43 (d, J = 17.0 Hz, 3H), 1.34 (s, 6H).
tert-Butyl (2S,4R)-2-(hydroxymethyl)-4-(4-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)oxy)butoxy)pyrrolidine-1-carboxylate (29).
Compound 28 (3.80 g, 7.060 mmol) was dissolved in anhydrous THF (40 mL) and cooled to −15 °C. To this solution, LiBH4 (0.230 g, 10.59 mmol) was added slowly and continued to stir at RT for 1 h. The reaction progress was monitored by TLC and after reaction completion, water (300 mL) was added, followed by extraction with EtOAC (3×300 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The crude product was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as a yellow oil (2.30 g, 75% yield). 1H NMR (400 MHz, CDCl3) δ 7.86 (s, 1H), 7.03 (d, J = 8.4 Hz, 1H), 6.51 (d, J = 8.4 Hz, 1H), 6.30 (s, 1H), 4.74 (d, J = 7.5 Hz, 1H), 4.07 (s, 1H), 3.94 (s, 3H), 3.70 (s, 1H), 3.57 (d, J = 10.6 Hz, 2H), 3.42 (dd, J = 30.9, 8.5 Hz, 3H), 2.89 (t, J = 7.5 Hz, 2H), 2.61 (t, J = 7.3 Hz, 2H), 2.06 (d, J = 35.0 Hz, 1H), 1.88 – 1.78 (m, 2H), 1.73 (d, J = 6.3 Hz, 2H), 1.47 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.48, 158.59, 138.14, 128.65, 115.73, 108.73, 102.00, 80.51, 68.43, 67.83, 67.06, 59.09, 52.91, 34.48, 31.08, 28.43, 26.42, 26.28, 24.58.
tert-Butyl (2S,4R)-2-((1,3-dioxoisoindolin-2-yl)methyl)-4-(4-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)oxy)butoxy)pyrrolidine-1-carboxylate (30).
Anhydrous THF (20 mL) and triphenylphosphine (TPP; 1.82 g, 6.94 mmol) were added to a round bottom flask and cooled to 0 °C. After 5 min, diethyl azodicarboxylate (DEAD; 1.40 g, 6.9 mmol) was added dropwise to the reaction mixture. The stirring continued for 30 min at the same temperature, followed by dropwise addition of 29 (2.0 g 4.6 mmol) dissolved in THF (10 mL). After 10 min phthalimide (0.82 g, 5.6 mmol) was added portion wise to the reaction mixture, the reaction was then warmed to RT and continued to stir for 8 h. Reaction progress was monitored by TLC, after reaction completion, 10% NaOH aq solution (100 mL) and water (200 mL) were added followed by extraction with EtOAC (3 × 250 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The crude product was purified by flash chromatography using 40% EtOAC/hexane as eluent to give the desired product as a yellow oil (1.43 g, 55% yield). 1H NMR (400 MHz, CDCl3) δ 7.84 (m, 2H), 7.69 (m, 2H), 7.03 (d, J = 8.5 Hz, 2H), 6.49 (d, J = 8.3 Hz, 1H), 6.33 (s, 1H), 4.36 (m, 1H), 4.15 – 4.09 (m, 1H), 3.96 – 3.83 (m, 3H), 3.73 (s, 2H), 3.44 (s, 3H), 2.89 (t, J = 7.4 Hz, 2H), 2.61 (t, J = 7.5 Hz, 2H), 2.02 (m, 2H), 1.81 (s, 2H), 1.70 (s, 2H), 1.38 (s, 3H), 1.31 – 1.15 (m, 6H).
tert-Butyl (2S,4R)-2-(aminomethyl)-4-(4-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)oxy)butoxy)pyrrolidine-1-carboxylate (31).
Compound 30 (1.43 g, 2.54 mmol) was dissolved in ethanol (10 mL). To the reaction mixture, anhydrous hydrazine (N2H4) (0.326 g, 10.2 mmol) was added and allowed to stir at reflux for 3–4 h. After reaction completion, 20% aq K2CO3 (20 mL) was added then extracted with CHCl3 (3 × 250 mL). The product obtained was a pure yellow oil and taken as such for the next step without further purification (0.850 g, 85% yield). 1H NMR (400 MHz, CDCl3) δ 8.83 (s, 1H, rotamer A), 8.77 (s, 1H, rotamer B), 7.01 (d, J = 8.2 Hz, 1H), 6.54 – 6.45 (m, 1H), 6.39 (s, 1H), 3.99 (s, 2H), 3.93 (s, 2H), 3.73 (s, 1H), 3.48 (m, 2H), 3.34 (s, 1H), 2.86 (t, J = 7.2, 13.1 Hz, 4H), 2.59 (t, J = 7.4 Hz, 2H), 2.05 (dd, J = 8.6, 7.0 Hz, 1H), 1.95 (t, J = 9.6 Hz, 1H), 1.80 (d, J = 6.7 Hz, 2H), 1.71 (d, J = 5.7 Hz, 2H), 1.61 (s, 2H), 1.45 (s, 9H).
tert-Butyl (2S,4R)-2-((3-chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)-4-(4-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)oxy)butoxy)pyrrolidine-1-carboxylate (32).
The compound was prepared by using 31 (0.85 g, 1.96 mmol), 16 (0.542 g, 2.35 mmol) and HCTU (1.05 g, 2.55 mmol) according to the general amidation method (E). The crude product was purified by flash chromatography using 40% EtOAC/hexane as eluent to give the desired product as brown oil (0.800 g, 63% yield). Rotamers observed in 1H NMR. 1H NMR (400 MHz, CDCl3) δ 13.70 (s, 1H, rotamer A), 13.58 (s, 1H, rotamer B), 8.90 (s, 1H, rotamer A), 8.57 (s, 1H, rotamer B), 8.04 (s, 1H), 7.22 (s, 1H), 7.03 (d, J = 8.0 Hz, 1H), 6.50 (d, J = 8.0 Hz, 1H), 6.29 (s, 1H), 4.12 (d, J = 6.9 Hz, 1H), 3.94 (d, J = 16.7 Hz, 3H), 3.86 (s, 3H), 3.64 (s, 1H), 3.45 (s, 2H), 3.41 – 3.24 (m, 1H), 2.88 (t, J = 7.1 Hz, 2H), 2.68 – 2.53 (m, 4H), 2.12 (s, 1H), 1.89 (brs, 1H), 1.85 – 1.80 (m, 3H), 1.74 – 1.68 (m, 3H), 1.47 (s, 9H), 1.19 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.40, 169.72, 160.05, 158.60, 155.44, 152.56, 138.17, 133.10, 130.91, 128.64, 116.13, 115.70, 108.70, 108.07, 101.99, 68.50, 67.81, 61.47, 56.13, 52.64, 42.18, 35.57, 31.09, 30.88, 28.42, 26.39, 26.23, 24.59, 22.51, 13.39. HRMS (ESI): m/z calcd for (C33H44ClN3O8+H+), 646.2817; found 646.2893 (M+H+). Compound 32 was analyzed by HPLC: tR=19.660 min, diastereomeric excess was >96% (detail method reported in SI; Figure S6).
3-Chloro-5-ethyl-6-hydroxy-2-methoxy-N-(((2S,4R)-4-(4-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)oxy)butoxy)pyrrolidin-2-yl)methyl)benzamide (33).
The compound was prepared from 32 (0.80 g, 1.23 mmol) in the presence of 20% TFA/DCM (20 mL). The reaction mixture stirred at RT for 8 h and monitored by TLC. The mixture was basified to pH 9 with saturated aq NaHCO3 solution. The product was extracted with CHCl3 (4 X 200 ml), the combined organic fractions were dried over anhydrous Na2SO4, filtered and evaporated to afford the crude material. The crude product was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as a brown oil (0.560 g, 83% yield). 1H NMR (400 MHz, CDCl3) δ 13.66 (brs, 1H), 8.93 (brs, 1H), 8.60 (br s, 1H), 7.20 (s, 1H), 7.01 (d, J = 8.3 Hz, 1H), 6.49 (d, J = 8.3 Hz, 1H), 6.36 (s, 1H), 4.02 (s, 1H), 3.93 (t, J = 6.2 Hz, 2H), 3.87 (s, 3H), 3.62 (dd, J = 9.7, 5.4 Hz, 2H), 3.43 (t, J = 10.9 Hz, 2H), 3.27 (dd, J = 12.4, 7.0 Hz, 1H), 3.07 (d, J = 12.1 Hz, 1H), 2.97 (dd, J = 12.2, 3.6 Hz, 1H), 2.86 (t, J = 7.4 Hz, 2H), 2.59 (q, J = 7.4 Hz, 4H), 2.12 – 1.99 (m, 1H), 1.82 (dt, J = 13.4, 6.7 Hz, 3H), 1.76 – 1.65 (m, 3H), 1.62 – 1.49 (m, 1H), 1.18 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 171.82, 169.28, 160.04, 158.60, 152.41, 138.16, 132.93, 130.78, 128.57, 116.14, 115.64, 108.77, 108.12, 102.07, 80.28, 68.36, 67.82, 61.54, 56.34, 51.79, 43.17, 36.01, 31.03, 26.43, 26.14, 24.54, 22.49, 13.38; Mp 128–130 °C. The HCl salt was precipitated from acetone; Mp 123–125 °C. Anal (C28H36ClN3O6•HCl•0.5H2O) C, H, N. HRMS (ESI): calcd for (C28H36ClN3O6+H+), 546.2293; found, 546.2354 (M +H+). Compound 33 was analyzed by HPLC: tR=25.268 min, diastereomeric excess was >99% (detailed method reported in SI; Figure S7).
3-Chloro-5-ethyl-6-hydroxy-2-methoxy-N-(((2S,4R)-4-(4-((2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)oxy)butoxy)-1-propylpyrrolidin-2-yl)methyl)benzamide (34).
The compound was prepared from 33 (0.200 g, 0.36 mmol) and propionaldehyde (0.032 g, 0.54 mmol) using the general reductive amination method. Both the reactants were dissolved in DCE (10 mL) and allowed to stir for 10 min at RT in the presence of 2–3 drops of glacial acetic acid. To the reaction mixture, NaBH(OAc)3 (0.116 g, 0.55 mmol) was added portionwise and the reaction mixture stirred at RT for 5 h. After completion of the reaction, the solution was basified to pH 9 with saturated aq NaHCO3. The product was extracted with CHCl3 (3×100 mL) and the combined organic fractions were dried over anhydrous Na2SO4, filtered and then evaporated to afford crude material. This crude compound was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as a brown oil (0.125 g, 58% yield). 1H NMR (400 MHz, CDCl3) δ 13.81 (s, 1H), 8.80 (s, 1H), 8.16 (s, 1H), 7.21 (s, 1H), 7.03 (d, J = 8.3 Hz, 1H), 6.50 (d, J = 8.1 Hz, 1H), 6.32 (s, 1H), 3.93 (t, J = 6.1 Hz, 2H), 3.86 (s, 3H), 3.79 (s, 1H), 3.42 (td, J = 15.6, 8.6 Hz, 2H), 3.32 (s, 1H), 2.88 (t, J = 7.5 Hz, 2H), 2.70 (s, 1H), 2.61 (dd, J = 13.0, 6.3 Hz, 3H), 2.27 (s, 2H), 2.17 (d, J = 0.6 Hz, 2H), 2.02 (d, J = 13.0 Hz, 1H), 1.96 – 1.76 (m, 2H), 1.76 – 1.65 (m, 1H), 1.49 (d, J = 49.9 Hz, 2H), 1.32 – 1.22 (m, 2H), 1.19 (t, J = 7.5 Hz, 3H), 0.91 (t, J = 7.3 Hz, 3H), 0.83 (d, J = 4.3 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 206.83, 171.60, 169.60, 160.13, 158.62, 152.49, 138.10, 132.95, 130.81, 128.62, 116.04, 115.71, 108.57, 108.10, 102.14, 68.76, 67.82, 61.44, 59.69, 55.91, 39.52, 35.27, 31.07, 30.88, 26.50, 26.14, 24.57, 22.51, 13.40, 11.90. HRMS (ESI): m/z calcd for (C31H42ClN3O6+H+); 588.2762, found, 588.2822 (M+H+). Compound 34 was analyzed by HPLC: tR=20.839 min, diastereomeric excess was >99% (Detailed method reported in SI; Figure S8).
(2S,4R)-1-(tert-Butoxycarbonyl)-4-(3-phenylpropoxy)pyrrolidine-2-carboxylic acid (36a).
The compound was prepared by using (3-bromopropyl)benzene (35a; 1.03 g, 5.19 mmol), and (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid 25 (1.00 g, 4.33 mmol) and NaH (0.311 g, 12.9 mmol) according to the general O-alkylation procedure. The crude product was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as a yellow oil (1.30 g, 86% yield). Rotamers observed in NMR, 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 7.4 Hz, 2H), 7.18 (t, J = 8.5 Hz, 3H), 4.43 (dd, J = 27.1, 20.2 Hz, 1H), 4.09 – 3.91 (m, 1H), 3.49 (t, J = 5.1 Hz, 1H), 3.42 (dd, J = 15.7, 6.4 Hz, 3H), 2.67 (t, J = 7.6 Hz, 2H), 2.49 – 2.32 (m, 1H), 2.15 (dt, J = 50.7, 15.4 Hz, 1H), 1.88 (dt, J = 13.4, 6.5 Hz, 2H), 1.49 (s, 9H).
(2S,4R)-4-(4-(Benzyloxy)butoxy)-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid (36b).
The compound was prepared with ((4-bromobutoxy)methyl)benzene (5.00 g, 20.66 mmol) 35b, and (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid 25 (3.80 g, 16.528 mmol) and NaH (1.19 g 49.58 mmol) according to the general O-alkylation procedure. The crude product was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as a yellow oil (4.20 g, 52% yield). Rotamers observed in NMR, 1H NMR (400 MHz, CDCl3) δ 7.38 – 7.27 (m, 5H), 4.50 (s, 2H), 4.39 (dt, J = 42.0, 7.6 Hz, 1H), 4.06 – 3.93 (m, 1H), 3.54 – 3.35 (m, 5H), 2.51 – 2.30 (m, 1H), 2.23 – 2.05 (m, 1H), 1.65 (t, J = 3.1 Hz, 4H), 1.48 (s, 6H), 1.42 (s, 3H).
tert-Butyl (2S,4R)-2-(hydroxymethyl)-4-(3-phenylpropoxy)pyrrolidine-1-carboxylate (37a).
The compound was prepared by using 36a (1.30 g, 3.73 mmol), and borane dimethyl sulfide complex (0.424 g, 5.593 mmol) according to the procedure described for 37b. The resulting crude product was then taken for the next step without further purification (1.0 g, 80% yield). 1H NMR (400 MHz, CDCl3) δ 7.31 – 7.24 (m, 2H), 7.18 (t, J = 6.8 Hz, 3H), 4.83 (s, 1H), 4.11 (dt, J = 12.5, 6.3 Hz, 1H), 3.90 (s, 1H), 3.69 (d, J = 6.2 Hz, 1H), 3.55 (dd, J = 11.1, 6.9 Hz, 2H), 3.39 (d, J = 5.3 Hz, 3H), 2.68 (dd, J = 15.5, 7.9 Hz, 2H), 2.10 (dd, J = 12.1, 7.3 Hz, 1H), 1.94 – 1.79 (m, 2H), 1.63 (s, 1H), 1.47 (s, 9H).
tert-Butyl (2S,4R)-4-(4-(benzyloxy)butoxy)-2-(hydroxymethyl)pyrrolidine-1-carboxylate (37b).
To the carboxylic acid intermediate 36b (4.10 g, 10.43 mmol) anhydrous THF (40 mL) was added and cooled to 0°C. To this solution borane dimethyl sulfide complex (1.18 g, 15.64 mmol) was added drop wise and allowed to stir for 5 h at RT. After completion of the reaction 200 mL of saturated aq NaHCO3 was added drop wise followed by 200 mL cold water added and extracted with EtOAC (3×400 mL). The combined organic fractions were dried over anhydrous Na2SO4, filtered and evaporated to afford the desired product as a colorless solid (3.30 g, 83% yield). The resulting crude product was then taken for the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 7.35 – 7.31 (m, 5H), 4.50 (s, 2H), 4.08 – 3.86 (m, 2H), 3.74 – 3.63 (m, 1H), 3.59 – 3.51 (m, 2H), 3.48 (t, J = 5.9 Hz, 2H), 3.39 (t, J = 9.4 Hz, 3H), 1.74 – 1.56 (m, 6H), 1.46 (s, 9H).
tert-Butyl (2S,4R)-4-(4-(benzyloxy)butoxy)-2-((1,3-dioxoisoindolin-2-yl)methyl)pyrrolidine-1-carboxylate (38b).
This compound was prepared from 37b (3.29 g, 8.69 mmol), TPP (3.42 g, 13.04 mmol), DEAD (2.63 g, 13.04 mmol) and phthalimide (1.60 g, 10.86 mmol) according to the procedure described for 30. The crude product was partially purified by flash chromatography using 25% EtOAC/hexane as eluent to give the desired product as a yellow oil, the compound was used for the next step, without further purification.
tert-Butyl (2S,4R)-2-((1,3-dioxoisoindolin-2-yl)methyl)-4-(3-phenylpropoxy) pyrrolidine-1-carboxylate (38a).
This compound was prepared from 37a (1.00 g, 2.99 mmol), TPP (1.17 g, 4.49 mmol), DEAD (0.91 g, 4.48 mmol) and phthalimide (0.528 g, 3.59 mmol) according to the procedure described for 38b. The crude product was partially purified by flash chromatography using 25% EtOAC/hexane as eluent to give the desired product as a yellow oil and used in the next step without further purification. (1.10 g, 80% yield). 1H NMR (400 MHz, CDCl3) δ 7.84 (s, 2H), 7.72 (s, 2H), 7.30 – 7.22 (m, 2H), 7.15 (d, J = 7.4 Hz, 3H), 4.98 (dt, J = 12.7, 6.4 Hz, 2H), 4.36 (m, 1H), 4.15 – 4.01 (m, 1H), 3.90 (m, 1H), 3.67 (s, 2H), 3.39 (s, 3H), 2.64 (s, 2H), 2.10 – 1.92 (m, 1H), 1.84 (s, 1H), 1.58 (s, 9H).
tert-Butyl (2S,4R)-2-(aminomethyl)-4-(3-phenylpropoxy)pyrrolidine-1-carboxylate (39a).
This compound was prepared from 38a (1.10 g, 2.37 mmol) using the same procedure described for 31. The crude product obtained was a yellow oil and taken as such for the next step without further purification.
tert-Butyl (2S,4R)-2-(aminomethyl)-4-(4-(benzyloxy)butoxy)pyrrolidine-1-carboxylate (39b).
This compound was prepared from 38b (3.00 g, 5.91 mmol) using the same procedure described for 31. The crude product obtained was a yellow oil which was used for the next step without further purification. (1.904 g, 85% yield). 1H NMR (400 MHz, CDCl3) δ 7.35 – 7.31 (m, 5H), 4.50 (br s, 2H), 3.98 (br s, 1H), 3.89 (br s, 1H), 3.68 (br s, 1H), 3.48 (d, J = 5.6 Hz, 2H), 3.41 (br s, 3H), 2.88 – 2.72 (m, 2H), 2.04 (br s, 1H), 1.94 (br s, 1H), 1.65 (br s, 4H), 1.46 (s, 9H).
tert-Butyl (2S,4R)-2-((3-chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)-4-(3-phenylpropoxy)pyrrolidine-1-carboxylate (40a).
The compound was prepared by using 39a (0.56 g, 1.67 mmol), 16 (0.46 g, 2.01 mmol) and HCTU (1.04 g, 2.51 mmol) according to the general amidation method (E). The crude product was purified by flash chromatography using 30% EtOAC/hexane as eluent to give the desired product as brown oil (0.57 g, 62% yield). 1H NMR (400 MHz, CDCl3) δ 13.71 (s, 1H), 8.84 (s, 1H, rotamer A), 8.50 (s, 1H, rotamer B), 7.26 (s, 2H), 7.22 (s, 1H), 7.16 (d, J = 7.0 Hz, 3H), 4.11 (m, 1H), 3.95 (s, 1H), 3.87 (s, 3H), 3.63 (s, 2H), 3.38 (s, 3H), 2.63 (dt, J = 14.6, 7.6 Hz, 3H), 2.17 – 2.02 (m, 2H), 1.97 – 1.78 (m, 3H), 1.57 (s, 9H), 1.26 (s, 1H), 1.19 (t, J = 7.5 Hz, 3H).
tert-Butyl (2S,4R)-4-(4-(benzyloxy)butoxy)-2-((3-chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)pyrrolidine-1-carboxylate (40b).
The compound was prepared by using 39b, (1.50 g, 3.96 mmol), 16, (1.09 g, 4.75 mmol) and HCTU (2.45 g, 5.94 mmol) according to the general amidation method (E). The crude product was purified by flash chromatography using 30% EtOAC/hexane as eluent to give the desired product as brown oil (1.47 g, 63% yield). 1H NMR (400 MHz, CDCl3) δ 13.67 (s, 1H, rotamer A), 13.54 (s, 1H, rotamer B), 8.85 (s, 1H, rotamer A), 8.50 (s, 1H, rotamer B), 7.32 (br s, 5H), 7.21 (s, 1H), 4.49 (s, 2H), 4.10 (d, J = 16.1 Hz, 1H), 3.93 (d, J = 16.4 Hz, 1H), 3.86 (s, 3H), 3.81 – 3.70 (m, 2H), 3.62 (s, 1H), 3.47 (s, 2H), 3.39 (s, 3H), 2.61 (q, J = 14.9, 7.5 Hz, 2H), 2.11 (s, 1H), 1.88 (s, 1H), 1.61 (d, J = 27.5 Hz, 4H), 1.47 (s, 9H), 1.19 (t, J = 7.5 Hz, 3H).
tert-Butyl (2S,4R)-2-((3-chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)-4-(4-hydroxybutoxy)pyrrolidine-1-carboxylate (41).
A mixture of 40b (1.00 g, 1.69 mmol) and 10% Pd/C (0.100 g) in MeOH:EtOAC (1:1), (30 mL) was shaken on a Parr hydrogenator apparatus, under an atmosphere of hydrogen gas (H2, 45 psi) at RT, for 1 h. The reaction mixture was filtered through a celite pad and evaporated under vacuum. The crude product was purified by flash chromatography using 5% MeOH/CHCl3 as eluent to give the desired product as a yellow oil (0.676 g, 80% yield). 1H NMR (400 MHz, CDCl3) δ 13.76 – 13.46 (m, 1H), 8.88 (s, 1H, rotamer A), 8.55 (s, 1H, rotamer B), 7.21 (s, 1H), 4.08 (t, J = 16.2, 5.9 Hz, 1H), 3.97 (s, 2H), 3.85 (s, 3H), 3.78 (dd, J = 13.3, 8.7 Hz, 1H), 3.62 (t, J = 5.6 Hz, 2H), 3.44 (d, J = 14.4 Hz, 3H), 2.59 (q, J = 7.5 Hz, 2H), 2.10 (d, J = 23.7 Hz, 2H), 1.96 – 1.84 (m, 2H), 1.68 – 1.57 (m, 4H), 1.46 (s, 9H), 1.18 (t, J = 7.5 Hz, 3H).
tert-Butyl (2S,4R)-2-((3-chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)-4-(4-(1,3-dioxoisoindolin-2-yl)butoxy)pyrrolidine-1-carboxylate (42).
This compound was prepared from 41 (0.900 g, 1.79 mmol), TPP (0.705 g, 2.69 mmol), DEAD (0.544 g, 2.69 mmol) and phthalimide (0.330 g, 2.245 mmol) according to the procedure described for 30. The crude product was purified by flash chromatography using 30% EtOAC/hexane as eluent to give the desired product as a pale yellow solid (0.644 g, 57% yield). 1H NMR (400 MHz, CDCl3) δ 13.65 (m, 1H), 8.90 (s, 1H, rotamer A), 8.55 (s, 1H, rotamer B), 7.85 – 7.79 (m, 2H), 7.73 – 7.66 (m, 2H), 7.21 (s, 1H), 4.08 – 4.04 (m, 1H), 3.95 (s, 1H), 3.86 (s, 3H), 3.69 (t, J = 7.1 Hz, 2H), 3.56 (m, 1H), 3.40 (s, 3H), 2.60 (q, J = 7.5 Hz, 2H), 2.13 (s, 1H), 1.87 (s, 1H), 1.73 (dt, J = 14.3, 6.4 Hz, 2H), 1.63 (s, 2H), 1.62 – 1.53 (m, 2H), 1.45 (s, 9H), 1.25 (t, J = 7.1 Hz, 3H).
tert-Butyl (2S,4R)-4-(4-aminobutoxy)-2-((3-chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)pyrrolidine-1-carboxylate (43).
This compound was prepared from 42 (0.64 g, 1.02 mmol) according to the procedure described for 31. The compound obtained as a pure yellow oil (0.36 g, 70% yield). 1H NMR (400 MHz, CDCl3) δ 13.90 – 13.14 (m, 1H), 8.85 (s, 1H, rotamer A), 8.51 (s, 1H, rotamer B), 7.17 (s, 1H), 4.03 (d, J = 5.1 Hz, 1H), 3.91 (s, 1H), 3.81 (s, 3H), 3.74 (s, 1H), 3.53 (m, 3H), 3.33 (s, 2H), 2.67 (s, 1H), 2.56 (q, J = 7.5 Hz, 2H), 2.09 (d, J = 7.7 Hz, 1H), 1.84 (s, 1H), 1.56 – 1.49 (m, 4H), 1.42 (s, 9H), 1.39 (s, 2H), 1.14 (t, J = 7.5 Hz, 3H).
tert-Butyl (2S,4R)-4-(4-(1H-indole-2-carboxamido)butoxy)-2-((3-chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)pyrrolidine-1-carboxylate (44).
The compound was prepared by using 43 (0.140 g, 0.281 mmol), indole-2-carboxylic acid (54.3 mg, 0.337 mmol) and HCTU (0.174 g, 0.421 mmol) according to the general amidation method (E). The crude product was purified by flash chromatography using 40% EtOAC/hexane as eluent to give the desired product as brown oil (0.120 g, 65% yield).
1H NMR (400 MHz, CDCl3) δ 13.58 (s, 1H, rotamer A), 13.70 (s, 1H, rotamer B), 9.41 (s, 1H), 8.90 (s, 1H, rotamer A), 8.56 (s, 1H, rotamer B), 7.64 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.28 (d, J = 7.2 Hz, 1H), 7.22 (s, 1H), 7.13 (t, J = 7.5 Hz, 1H), 6.87 (d, J = 31.1 Hz, 1H), 6.47 (m, 1H), 4.12 (s, 1H), 3.97 (s, 1H), 3.86 (s, 3H), 3.63 (s, 1H), 3.55 – 3.42 (m, 3H), 3.28 (s, 1H), 2.80 (s, 2H), 2.61 (q, J = 7.5 Hz, 2H), 2.14 (s, 1H), 1.90 (d, J = 6.7 Hz, 2H), 1.67 (s, 3H), 1.47 (s, 9H), 1.29 (d, J = 32.5 Hz, 1H), 1.19 (t, J = 7.5 Hz, 3H).
tert-Butyl (2S,4R)-4-(4-(benzofuran-2-carboxamido)butoxy)-2-((3-chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)pyrrolidine-1-carboxylate (45).
The compound was prepared by using 43 (0.200 g, 0.40 mmol), benzofuran-2-carboxylic acid (0.081 g, 0.50 mmol) and HCTU (0.248 g, 0.601 mmol) according to the general amidation method (E). The crude product was purified by flash chromatography using 40% EtOAC/hexane as eluent to give the desired product as brown oil (0.160 g, 62% yield).
1H NMR (400 MHz, CDCl3) δ 13.70 (s, 1H, rotamer A), 13.58 (s, 1H, rotamer B), 8.90 (s, 1H, rotamer A), 8.56 (s, 1H, rotamer B), 7.65 (d, J = 7.4 Hz, 1H), 7.48 (dd, J = 8.3, 0.8 Hz, 1H), 7.45 (d, J = 0.9 Hz, 1H), 7.39 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 7.32 – 7.27 (m, 1H), 7.21 (s, 1H), 6.78 (s, 1H), 3.98 (s, 1H), 3.85 (s, 3H), 3.84 (m, 1H), 3.62 (s, 1H), 3.48 (dd, J = 15.5, 6.0 Hz, 4H), 3.32 (s, 1H), 2.60 (q, J = 7.6 Hz, 2H), 2.16 (s, 1H), 1.90 (s, 1H), 1.78 – 1.60 (m, 5H), 1.46 (s, 9H), 1.27 (m, 1H), 1.19 (t, J = 10.2 Hz, 3H).
N-(4-(((3R,5S)-5-((3-Chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)pyrrolidin-3-yl)oxy)butyl)-1H-indole-2-carboxamide (46).
The compound was prepared from 44 (0.120 g, 0.186 mmol) according to the procedure described for 33. The crude product was purified by flash chromatography using 10% MeOH/ CHCl3 as eluent to give the desired product as brown oil (0.076 g, 74% yield). 1H NMR (400 MHz, CDCl3) δ 13.95 – 13.36 (br s, 1H), 9.51 (br s, 1H), 8.91 (br s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.27 (d, J = 10.9 Hz, 1H), 7.20 (s, 1H), 7.12 (t, J = 7.4 Hz, 1H), 6.81 (s, 1H), 6.41 (s, 1H), 4.01 (m, 1H), 3.87 (s, 3H), 3.68 – 3.56 (m, 2H), 3.50 (d, J = 6.4 Hz, 2H), 3.46 (s, 2H), 3.33 – 3.19 (m, 1H), 3.07 (d, J = 12.2 Hz, 1H), 3.01 – 2.89 (m, 1H), 2.59 (q, J = 7.4 Hz, 2H), 2.04 (dd, J = 13.5, 6.8 Hz, 1H), 1.80 – 1.61 (m, 5H), 1.54 (td, J = 13.0, 6.1 Hz, 1H), 1.18 (t, J = 7.7 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 169.16, 161.46, 159.89, 152.26, 136.08, 132.79, 130.66, 130.64, 127.44, 124.25, 121.65, 120.46, 116.00, 111.80, 107.96, 101.60, 80.26, 68.05, 61.41, 56.13, 51.60, 43.15, 39.18, 35.86, 26.88, 26.62, 22.34, 13.23. HRMS (ESI): m/z calcd for (C28H35ClN4O5+H+); 543.2296, found, 543.2377 (M +H+). Compound 46 was analyzed by HPLC: tR=29.407 min, diastereomeric excess was >99% (detailed method reported in SI; Figure S9).
N-(4-(((3R,5S)-5-((3-Chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)pyrrolidin-3-yl)oxy)butyl)benzofuran-2-carboxamide (47).
The compound was prepared from 45 (0.160 g, 0.248 mmol) according to the procedure described for 33. The crude product was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as brown oil (0.092 g, 68% yield). 1H NMR (400 MHz, CDCl3) δ 13.88 – 13.38 (br s, 1H), 8.97 (s, 1H), 7.64 (d, J = 7.7 Hz, 1H), 7.47 (dd, J = 8.3, 0.7 Hz, 1H), 7.43 – 7.37 (m, 1H), 7.36 (d, J = 0.7 Hz, 1H), 7.31 – 7.27 (m, 1H), 7.17 (s, 1H), 6.80 (s, 1H), 4.04 (m, 1H), 3.90 (s, 3H), 3.79 – 3.59 (m, 2H), 3.56 – 3.37 (m, 4H), 3.36 – 3.23 (m, 1H), 3.13 (d, J = 12.3 Hz, 1H), 3.03 (dd, J = 12.3, 3.9 Hz, 1H), 2.64 – 2.46 (m, 2H), 2.09 (dd, J = 13.6, 7.0 Hz, 1H), 1.80 – 1.52 (m, 5H), 1.25 (s, 1H), 1.15 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 169.40, 160.06, 158.90, 154.65, 152.50, 148.66, 132.95, 130.70, 127.59, 126.82, 123.66, 122.71, 116.13, 111.59, 110.30, 108.06, 80.17, 68.14, 61.56, 56.44, 51.49, 43.05, 39.00, 35.92, 26.89, 26.76, 22.43, 13.31. HRMS (ESI): m/z calcd for (C28H34ClN3O6+H+); 544.2136, found 544.2208 (M+H+). Compound 47 was analyzed by HPLC: tR=15.702 min, diastereomeric excess was >99% (detailed method reported in SI; Figure S10).
N-(4-(((3R,5S)-5-((3-Chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)-1-ethylpyrrolidin-3-yl)oxy)butyl)-1H-indole-2-carboxamide (48).
This compound was prepared from 46 (0.080 g, 0.15 mmol), acetaldehyde (0.013 g, 0.29 mmol), NaBH(OAc)3 (0.040 g, 0.188 mmol) and 2 drops of acetic acid according to the procedure described for 34. The crude product was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as brown oil (0.042 g, 50% yield). 1H NMR (400 MHz, CDCl3) δ 13.80 (br s, 1H), 9.31 (s, 1H), 8.81 (d, J = 5.9 Hz, 1H), 7.63 (d, J = 8.1 Hz, 1H), 7.43 (d, J = 8.2 Hz, 1H), 7.29 (d, J = 7.2 Hz, 1H), 7.22 (s, 1H), 7.13 (t, J = 7.2 Hz, 1H), 6.80 (d, J = 1.3 Hz, 1H), 6.34 (s, 1H), 3.96 (m, 1H), 3.85 (s, 3H), 3.81 – 3.72 (m, 1H), 3.51 (dd, J = 13.0, 6.7 Hz, 3H), 3.44 (td, J = 9.3, 3.4 Hz, 2H), 3.36 – 3.24 (m, 1H), 2.86 (dt, J = 19.2, 7.4 Hz, 2H), 2.62 (q, J = 7.6 Hz, 2H), 2.30 (dt, J = 10.0, 6.2 Hz, 2H), 2.09 – 1.97 (m, 1H), 1.98 – 1.79 (m, 2H), 1.79 – 1.56 (m, 3H), 1.20 (t, J = 5.7 Hz, 3H), 1.10 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 169.56, 161.52, 160.14, 152.45, 139.25, 136.13, 132.93, 130.84, 127.63, 124.43, 121.82, 120.65, 116.04, 114.03, 111.88, 108.12, 101.57, 68.71, 61.42, 60.41, 59.12, 47.32, 39.49, 39.31, 35.45, 27.17, 26.68, 22.67, 22.52, 14.09, 13.48, 13.41. HRMS (ESI) calcd for (C30H39ClN4O5+H+); m/z 571.2609, found 571.2716 (M +H+). Compound 48 was analyzed by HPLC: tR=20.495 min, diastereomeric excess was >96% (detailed method reported in SI; Figure S11).
N-(4-(((3R,5S)-5-((3-chloro-5-ethyl-6-hydroxy-2-methoxybenzamido)methyl)-1-ethylpyrrolidin-3-yl)oxy)butyl)benzofuran-2-carboxamide (49).
The compound was prepared from 47 (0.100 g, 0.18 mmol), acetaldehyde (0.016 g, 0.36 mmol), NaBH(OAc)3 (0.048 g, 0.225 mmol)) and 2 drops of acetic acid according to the procedure described for 34. The crude product was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as brown oil (0.054 g, 52% yield). 1H NMR (400 MHz, CDCl3) δ 13.81 (s, 1H), 8.81 (d, J = 5.9 Hz, 1H), 7.67 – 7.63 (m, 1H), 7.47 (dd, J = 8.3, 0.8 Hz, 1H), 7.45 (d, J = 0.9 Hz, 1H), 7.39 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 7.30 – 7.27 (m, 1H), 7.21 (s, 1H), 6.78 (s, 1H), 3.96 (dt, J = 9.5, 5.9 Hz, 1H), 3.85 (s, 3H), 3.76 (ddd, J = 14.1, 7.4, 2.0 Hz, 1H), 3.54 – 3.47 (m, 3H), 3.42 (tt, J = 11.7, 4.5 Hz, 2H), 3.31 (ddd, J = 14.1, 4.6, 2.6 Hz, 1H), 2.96 – 2.79 (m, 2H), 2.61 (q, J = 7.5 Hz, 2H), 2.36 – 2.24 (m, 2H), 1.88 (qdd, J = 15.4, 7.8, 5.1 Hz, 2H), 1.78 – 1.60 (m, 4H), 1.19 (t, J = 8.9 Hz, 3H), 1.10 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 169.56, 160.14, 158.83, 154.65, 152.46, 148.85, 132.92, 130.82, 127.63, 126.76, 123.65, 122.69, 116.03, 111.60, 110.21, 108.11, 68.64, 61.42, 60.41, 59.10, 47.34, 39.48, 38.99, 35.45, 27.21, 26.58, 22.51, 14.09, 13.41. HRMS (ESI): m/z calcd for (C30H38ClN3O6+H+); 572.2449, found 572.2551 (M +H+). 49 was analyzed by HPLC: tR=14.506 min, diastereomeric excess was >99% (detailed method reported in SI; Figure S12).
3-Chloro-5-ethyl-6-hydroxy-2-methoxy-N-(((2S,4R)-4-(3-phenylpropoxy)pyrrolidin-2-yl)methyl)benzamide (50a).
The compound was prepared from 40a (0.540 g, 0.987 mmol) according to the procedure described for 33. The crude product was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as brown oil (0.326 g, 74% yield). 1H NMR (400 MHz, CDCl3) δ 13.67 (s, 1H), 8.90 (s, 1H), 7.32 – 7.24 (m, 2H), 7.21 (s, 1H), 7.17 (d, J = 6.9 Hz, 3H), 3.98 (s, 1H), 3.90 (s, 3H), 3.59 (d, J = 6.7 Hz, 2H), 3.37 (t, J = 6.3 Hz, 3H), 3.23 (s, 1H), 3.03 (d, J = 12.2 Hz, 1H), 2.91 (d, J = 12.4 Hz, 1H), 2.76 – 2.55 (m, 3H), 2.08 – 1.97 (m, 1H), 1.95 – 1.80 (m, 2H), 1.54 (dd, J = 13.5, 6.5 Hz, 1H), 1.26 (s, 1H), 1.19 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 169.18, 160.07, 152.42, 141.77, 132.90, 130.80, 128.39, 128.30, 125.80, 116.14, 108.17, 80.54, 67.84, 61.54, 56.16, 51.93, 43.40, 36.10, 32.36, 31.28, 22.51, 13.40. HRMS (ESI): m/z calcd for (C24H31ClN2O4+H+) 447.1972, found 447.2028 (M +H+). Compound 50a was analyzed by HPLC: tR=7.574 min, diastereomeric excess was >99% (detailed method reported in SI; Figure S13).
N-(((2S,4R)-4-(4-(Benzyloxy)butoxy)pyrrolidin-2-yl)methyl)-3-chloro-5-ethyl-6-hydroxy-2-methoxybenzamide (50b).
The compound was prepared from 40b (0.300 g, 0.50 mmol) according to the procedure described for 33. The crude product was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as brown oil (0.192 g, 77% yield).
1H NMR (400 MHz, CDCl3) δ 13.92 – 13.25 (br s, 1H), 7.34 (d, J = 2.2 Hz, 1H), 7.33 (s, 5H), 7.21 (s, 1H), 4.50 (s, 2H), 3.99 (s, 1H), 3.89 (s, 3H), 3.68 – 3.54 (m, 2H), 3.48 (t, J = 6.2 Hz, 2H), 3.38 (t, J = 5.9 Hz, 2H), 3.32 – 3.18 (m, 1H), 3.05 (d, J = 12.4 Hz, 1H), 2.93 (dd, J = 12.3, 4.2 Hz, 1H), 2.61 (q, J = 7.5 Hz, 2H), 2.03 (dd, J = 13.7, 7.0 Hz, 1H), 1.74 – 1.59 (m, 3H), 1.60 – 1.46 (m, 1H), 1.19 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 169.28, 160.07, 152.43, 138.54, 132.94, 130.81, 128.33, 127.59, 127.50, 116.14, 108.12, 80.21, 72.89, 70.07, 68.54, 61.57, 56.32, 51.76, 43.18, 36.00, 26.66, 26.55, 22.50, 13.39. HRMS (ESI): m/z calcd for (C26H35ClN2O5+H+); 491.2234, found 491.2310 (M +H+). Compound 50b was analyzed by HPLC: tR=6.885 min, diastereomeric excess was >99% (detailed method in SI; Figure S14).
3-Chloro-5-ethyl-6-hydroxy-N-(((2S,4R)-4-(4-hydroxybutoxy)pyrrolidin-2-yl)methyl)-2-methoxybenzamide (51).
The compound was prepared from 41 (0.300 g, 0.60 mmol) according to the procedure described for 33. The crude product was purified by flash chromatography using 10% MeOH/CHCl3 as eluent to give the desired product as brown oil (0.175 g, 73% yield). 1H NMR (400 MHz, CDCl3) δ 13.31 (br s, 1H), 9.02 (m, 1H), 7.21 (s, 1H), 4.74 (s, 1H), 4.09 (s, 1H), 3.95 (dd, J = 11.2, 6.8 Hz, 1H), 3.86 (s, 3H), 3.81 (dd, J = 10.0, 4.3 Hz, 1H), 3.66 – 3.52 (m, 3H), 3.40 (d, J = 4.7 Hz, 2H), 3.31 (d, J = 12.6 Hz, 1H), 3.18 (dd, J = 12.4, 3.8 Hz, 1H), 2.59 (q, J = 7.5 Hz, 2H), 2.23 (dd, J = 13.7, 6.8 Hz, 1H), 1.80 – 1.68 (m, 1H), 1.59 (m, 4H), 1.18 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.10, 159.98, 152.42, 133.30, 130.75, 116.07, 107.61, 78.19, 68.64, 62.43, 61.50, 57.58, 50.30, 41.39, 35.08, 29.33, 26.35, 22.38, 13.25. HRMS (ESI): m/z calculated for (C19H29ClN2O5+H+); 401.1765, found, 401.1837 (M +H+). Compound 51 was analyzed by HPLC: tR=9.091 min, diastereomeric excess was >99% (detailed method in SI; Figure S15).
Radioligand binding assays.
Binding at dopamine D2-like receptors was determined similarly to previously described methods.22 Membranes were prepared from HEK293 cells expressing human D2LR or D3R grown in a 50:50 mix of DMEM and Ham’s F12 culture media, supplemented with 20 mM HEPES, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1X antibiotic/antimycotic, 10% heat-inactivated fetal bovine serum, and 200 μg/mL hygromycin (Life Technologies, Grand Island, NY) and kept in an incubator at 37 °C and 5% CO2. Upon reaching 80–90% confluence, cells were harvested using pre-mixed Earle’s Balanced Salt Solution (EBSS) with 5 mM EDTA (Life Technologies) and centrifuged at 3,000 rpm for 10 min at 21 °C. The supernatant was removed, and the pellet was resuspended in 10 mL hypotonic lysis buffer (5 mM MgCl2 · 6 H2O, 5 mM Tris, pH 7.4 at 4 °C) and centrifuged at 14,500 rpm (~25,000 g) for 30 min at 4 °C. The pellet was then resuspended in fresh EBSS binding buffer made from 8.7 g/L Earle’s Balanced Salts without phenol red (US Biological, Salem, MA), 2.2 g/L sodium bicarbonate, pH to 7.4. A Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration and membranes were diluted to 500 μg/mL and stored in a −80 °C freezer for later use.
Radioligand competition binding experiments were conducted using thawed membranes on test day, each test compound was diluted into 10 half-log serial dilutions using 30% DMSO vehicle, starting from 1 mM or 100 μM concentration. When it was necessary to assist solubilization of the drugs at the highest tested concentration, 0.1% or 0.01% acetic acid (final concentration v/v), respectively, was added alongside the vehicle. Previously frozen membranes were diluted in fresh EBSS to a 200 μg/mL (for hD2LR or hD3R) stock for binding. Radioligand competition experiments were conducted in 96-well plates containing 300 μl fresh binding buffer, 50 μl of diluted test compound, 100 μl of membranes (20 μg/well total protein for hD2LR and hD3R, and 50 μl of [3H]N-methylspiperone radioligand diluted in binding buffer (0.4 nM final concentration; Perkin Elmer). Nonspecific binding was determined using 10 μM (+)-butaclamol (Sigma-Aldrich, St. Louis, MO) and total binding was determined with 30% DMSO vehicle. All compound dilutions were tested in triplicate and the reaction incubated for 1 h at RT. The reaction was terminated by filtration through Perkin Elmer Uni-Filter-96 GF/B, presoaked for 1 h in 0.5% polyethylenimine, using a Brandel 96-Well Plates Harvester Manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed (3 × 1 mL/well) of ice-cold binding buffer. Perkin Elmer MicroScint 20 Scintillation Cocktail (65 μL) was added to each well and filters were counted using a Perkin Elmer MicroBeta Microplate Counter. IC50 values for each compound were determined from dose-response curves and Ki values were calculated using the Cheng-Prusoff equation.47 When a complete inhibition couldn’t be achieved at the highest tested concentrations, Ki values have been extrapolated by constraining the bottom of the dose-response curves (= 0% residual specific binding) in the non-linear regression analysis. These analyses were performed using GraphPad Prism versions 6.00–8.00 for Macintosh (GraphPad Software, San Diego, CA).
Ki values were determined from at least 3 independent experiments and are reported as mean ± SD. Kd values for the radioligand were obtained from homologous competition binding experiments (D2R Kd = 0.378 nM and D3R Kd = 0.431 nM), using the same assay conditions and vehicle reported above.
Bioluminescence resonance energy transfer (BRET) assays.
The Go-protein activation assay uses a set of BRET-based constructs previously described.21 Briefly, HEK293T cells were transiently co-transfected with pcDNA3.1 vectors encoding i) D3R or D2R, ii) GαoA fused to Renilla luciferase 8 (Rluc8; provided by Dr. S. Gambhir, Stanford University, Stanford, CA) at residue 91, iii) untagged Gβ1, and iv. Gγ2 fused to mVenus. Transfections were performed using polyethyleneimine (PEI) at a ratio of 2:1 (PEI:total DNA; wt:wt), and cell culture was maintained as described previously.23 After ~48 h of transfection, cells were washed with PBS and resuspended in PBS + 0.1% glucose + 200 μM Na Bisulfite buffer. Approximately 200,000 cells were then distributed in each well of the 96-well plates (White Lumitrac 200, Greiner bio-one). Coelenterazine H (5 μM), a luciferase substrate for BRET, was then added followed by addition of vehicle and test compounds using an automated stamp transfer protocol (Nimbus, Hamilton Robotics) from an aliquoted 96-well compound plate. The reference ligands – quinpirole and eticlopride were from Tocris Bioscience. mVenus emission (530 nm) over RLuc 8 emission (485 nm) were then measured after 10 min of ligand incubation at 37 °C using a PHERAstar FSX plate reader (BMG Labtech). BRET ratio was then determined by calculating the ratio of mVenus emission over RLuc 8 emission.
Data were collected from at least 3 independent experiments. Concentration response curves (CRCs) were generated using a non-linear sigmoidal dose-response analyses using Prism 8 (GraphPad Software). CRCs are presented as mean drug-induced BRET ± SEM.
Molecular modeling
The crystal structure of D3R in complex with eticlopride (PDB ID: 3PBL)10 was first processed by the protein preparation wizard of the Schrodinger Suite (release 2020–4) after the thermostabilizing mutation Leu119 was mutated back to wildtype Trp. We then carried out pKa predictions of the compounds to be docked using both Jaguar50 and Epik51,52 programs in Schrodinger suite. The results of these predictions show that those compounds all have a charged nitrogen, which is supposed to interact with Asp3.32.
The induced-fit docking (IFD) protocol53 was used for molecular docking with core restraint of m-xylene on eticlopride (i.e., Cc1cccc(C)c1). For compounds with SP, the docking was carried out in two steps: we first dock the primary pharmacophore in the orthosteric binding site (OBS) and then use the resulting docking pose with the best IFD score as the core restraint to dock the full-length compound, which allowed adequate sampling of the linkers and SPs beyond the OBS. The final docking poses of the ligands were selected based on the IFD score.
To estimate binding affinity, the MM-GB/SA protocol54 was used. To compare the relative binding affinity for ligands with same linker but different SP, the ΔΔG was calculated as the difference between the ΔG of two compared compounds.
Supplementary Material
ACKNOWLEDGEMENT
Support for this research was provided to ABS, CAB, AB, FB, and AHN (ZIA DA000424), and to LC, RC, SR, KHL, and LS (Z1A DA000609) by the National Institute on Drug Abuse Intramural Research Program. The authors thank Dr. Ludovic Muller from the Structural Biology Core at NIDA-IRP for high-resolution MS analyses.
ABBREVIATIONS USED
- CDCl3
deuterated chloroform
- CDI
N,N’-carbonyldiimidazole
- CD3OD
deuterated methanol
- CMA
chloroform/methanol/ammonium hydroxide
- 95% CMA
(95% chloroform, 4% methanol, 1% ammonium hydroxide)
- DA
dopamine
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- HPLC
high-performance liquid chromatography
- MM-GB/SA
molecular mechanics with generalized born and surface area solvation
- NMR
nuclear magnetic resonance
- OBS
orthosteric binding site
- RT
room temperature
- SAR
structure activity relationship
- SBP
secondary binding pocket
- TFA
Trifluoroacetic acid
- THF
tetrahydrofuran
- TM
transmembrane
- tR
retention time
Footnotes
Supporting information. Elemental analysis results and HPLC chromatograms of the final compounds are reported. This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES:
- (1).Ye N; Neumeyer JL; Baldessarini RJ; Zhen X; Zhang A Update 1 of: Recent progress in development of dopamine receptor subtype-selective agents: potential therapeutics for neurological and psychiatric disorders. Chem. Rev 2013, 113, PR123–178. [DOI] [PubMed] [Google Scholar]
- (2).Creese I; Burt DR; Snyder SH Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 1976, 192, 481–483. [DOI] [PubMed] [Google Scholar]
- (3).Beaulieu JM; Gainetdinov RR The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev 2011, 63, 182–217. [DOI] [PubMed] [Google Scholar]
- (4).Martel JC; Gatti McArthur S Dopamine receptor subtypes, physiology and pharmacology: new ligands and concepts in schizophrenia. Front. Pharmacol 2020, 11, 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Aringhieri S; Carli M; Kolachalam S; Verdesca V; Cini E; Rossi M; McCormick PJ; Corsini GU; Maggio R; Scarselli M Molecular targets of atypical antipsychotics: from mechanism of action to clinical differences. Pharmacol. Ther 2018, 192, 20–41. [DOI] [PubMed] [Google Scholar]
- (6).Muench J; Hamer AM Adverse effects of antipsychotic medications. Am Fam Physician 2010, 81, 617–622. [PubMed] [Google Scholar]
- (7).Klein Herenbrink C; Sykes DA; Donthamsetti P; Canals M; Coudrat T; Shonberg J; Scammells PJ; Capuano B; Sexton PM; Charlton SJ; Javitch JA; Christopoulos A; Lane JR The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun 2016, 7, 10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sykes DA; Moore H; Stott L; Holliday N; Javitch JA; Lane JR; Charlton SJ Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D2 receptors. Nat. Commun 2017, 8, 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wang S; Che T; Levit A; Shoichet BK; Wacker D; Roth BL Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Chien EY; Liu W; Zhao Q; Katritch V; Han GW; Hanson MA; Shi L; Newman AH; Javitch JA; Cherezov V; Stevens RC Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wang S; Wacker D; Levit A; Che T; Betz RM; McCorvy JD; Venkatakrishnan AJ; Huang XP; Dror RO; Shoichet BK; Roth BL D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 2017, 358, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Michino M; Beuming T; Donthamsetti P; Newman AH; Javitch JA; Shi L What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol Rev 2015, 67, 198–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Newman AH; Beuming T; Banala AK; Donthamsetti P; Pongetti K; LaBounty A; Levy B; Cao J; Michino M; Luedtke RR; Javitch JA; Shi L Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem 2012, 55, 6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Newman AH; Battiti FO; Bonifazi A 2016 Philip S. Portoghese medicinal chemistry lectureship: designing bivalent or bitopic molecules for G-protein coupled receptors. The whole is greater than the sum of its parts. J. Med. Chem 2020, 63, 1779–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Valant C; Sexton PM; Christopoulos A Orthosteric/allosteric bitopic ligands: going hybrid at GPCRs. Mol. Interv 2009, 9, 125–135. [DOI] [PubMed] [Google Scholar]
- (16).Valant C, Robert Lane J, Sexton PM, Christopoulos A. The best of both worlds? bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol 2012, 52, 153–178. [DOI] [PubMed] [Google Scholar]
- (17).Lane JR; Sexton PM; Christopoulos A Bridging the gap: bitopic ligands of G-protein-coupled receptors. Trends Pharmacol Sci 2013, 34, 59–66. [DOI] [PubMed] [Google Scholar]
- (18).Fyfe TJ; Kellam B; Sykes DA; Capuano B; Scammells PJ; Lane JR; Charlton SJ; Mistry SN Structure-kinetic profiling of haloperidol analogues at the human dopamine D2 receptor. J. Med. Chem 2019, 62, 9488–9520. [DOI] [PubMed] [Google Scholar]
- (19).Tan L; Zhou Q; Yan W; Sun J; Kozikowski AP; Zhao S; Huang XP; Cheng J Design and synthesis of bitopic 2-phenylcyclopropylmethylamine (PCPMA) derivatives as selective dopamine D3 receptor ligands. J. Med. Chem 2020, 63, 4579–4602. [DOI] [PubMed] [Google Scholar]
- (20).Kumar V; Bonifazi A; Ellenberger MP; Keck TM; Pommier E; Rais R; Slusher BS; Gardner E; You ZB; Xi ZX; Newman AH Highly selective dopamine D3 receptor (D3R) antagonists and partial agonists based on eticlopride and the D3R crystal structure: new leads for opioid dependence treatment. J. Med Chem 2016, 59, 7634–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Michino M; Boateng CA; Donthamsetti P; Yano H; Bakare OM; Bonifazi A; Ellenberger MP; Keck TM; Kumar V; Zhu C; Verma R; Deschamps JR; Javitch JA; Newman AH; Shi L Toward understanding the structural basis of partial agonism at the dopamine D3 receptor. J. Med Chem 2017, 60, 580–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shaik AB; Kumar V; Bonifazi A; Guerrero AM; Cemaj SL; Gadiano A; Lam J; Xi ZX; Rais R; Slusher BS; Newman AH Investigation of novel primary and secondary pharmacophores and 3-substitution in the linking chain of a series of highly selective and bitopic dopamine D3 receptor antagonists and partial agonists. J. Med. Chem 2019, 62, 9061–9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bonifazi A; Yano H; Guerrero AM; Kumar V; Hoffman AF; Lupica CR; Shi L; Newman AH, Novel and potent dopamine D2 receptor Go-protein biased agonists. ACS. Pharmacol. Transl. Sci, 2019, 2, 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Battiti FO; Cemaj SL; Guerrero AM; Shaik AB; Lam J; Rais R; Slusher BS; Deschamps JR; Imler GH; Newman AH; Bonifazi A The significance of chirality in drug design and synthesis of bitopic ligands as D3 receptor (D3R) selective agonists. J. Med. Chem 2019, 62, 6287–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Martelle JL; Nader MA A review of the discovery, pharmacological characterization, and behavioral effects of the dopamine D2-like receptor antagonist eticlopride. CNS Neurosci. Ther 2008, 14, 248–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Verma RK; Abramyan AM; Michino M; Free RB; Sibley DR; Javitch JA; Lane JR; Shi L The E2.65A mutation disrupts dynamic binding poses of SB269652 at the dopamine D2 and D3 receptors. PLoS. Comput. Biol 2018, 14, e1005948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Michino M; Donthamsetti P; Beuming T; Banala A; Duan L; Roux T; Han Y; Trinquet E; Newman AH; Javitch JA; Shi L A single glycine in extracellular loop 1 is the critical determinant for pharmacological specificity of dopamine D2 and D3 receptors. Mol. Pharmacol 2013, 84, 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Keck TM; Burzynski C; Shi L; Newman AH Beyond small-molecule SAR: using the dopamine D3 receptor crystal structure to guide drug design. Adv. Pharmacol 2014, 69, 267–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kumar V; Moritz AE; Keck TM; Bonifazi A; Ellenberger MP; Sibley CD; Free RB; Shi L; Lane JR; Sibley DR; Newman AH Synthesis and pharmacological characterization of novel trans-cyclopropylmethyl-binked bivalent ligands that exhibit selectivity and allosteric pharmacology at the dopamine D3 receptor (D3R). J. Med. Chem 2017, 60, 1478–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zou MF; Keck TM; Kumar V; Donthamsetti P; Michino M; Burzynski C; Schweppe C; Bonifazi A; Free RB; Sibley DR; Janowsky A; Shi L; Javitch JA; Newman AH Novel analogues of (R)-5-(Methylamino)-5,6-dihydro-4H-imidazo-[4,5,1-ij]quinolin-2(1H)-one (Sumanirole) provide clues to dopamine D2/D3 receptor agonist selectivity. J. Med. Chem 2016, 59, 2973–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kopinathan A; Draper-Joyce C; Szabo M; Christopoulos A; Scammells PJ; Lane JR; Capuano B Subtle modifications to the indole-2-carboxamide motif of the negative allosteric modulator N-((trans)-4-(2-(7-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)cyclohexyl)-1H-indole-2-carboxamide (SB269652) yield dramatic changes in pharmacological activity at the dopamine D2 receptor. J. Med. Chem 2019, 62, 371–377. [DOI] [PubMed] [Google Scholar]
- (32).Moritz AE; Bonifazi A; Guerrero AM; Kumar V; Free RB; Lane JR; Verma RK, Shi L; Newman AH; Sibley DR Evidence for a stereoselective mechanism for bitopic activity by extended-length antagonists of the D3 dopamine receptor. ACS. Chem. Neurosci 2020, 11, 3309–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Mistry SN; Shonberg J; Draper-Joyce CJ; Klein Herenbrink C; Michino M; Shi L; Christopoulos A; Capuano B; Scammells PJ; Lane JR Discovery of a novel class of negative allosteric modulator of the dopamine D2 receptor through fragmentation of a bitopic ligand. J. Med Chem 2015, 58, 6819–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).You ZB; Bi GH; Galaj E; Kumar V; Cao J; Gadiano A; Rais R; Slusher BS; Gardner EL; Xi ZX; Newman AH Dopamine D3R antagonist VK4–116 attenuates oxycodone self-administration and reinstatement without compromising its antinociceptive effects. Neuropsychopharmacology 2019, 44, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jordan CJ; Humburg BA; Thorndike EB; Shaik AB; Xi ZX; Baumann MH; Newman AH; Schindler CW Newly developed dopamine D3 receptor antagonists, R-VK4–40 and R-VK4–116, do not potentiate cardiovascular effects of cocaine or oxycodone in rats. J. Pharmacol. Exp Ther 2019, 71, 602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).de Guglielmo G; Kallupi M; Sedighim S; Newman AH; George O Dopamine D3 receptor antagonism reverses the escalation of oxycodone self-administration and decreases withdrawal-induced hyperalgesia and irritability-like behavior in oxycodone-dependent heterogeneous stock rats. Front. Behav. Neurosci 2020, 13, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Jordan CJ; He Y; Bi GH; You ZB; Cao J; Xi ZX; Newman AH (±)VK4–40, a novel dopamine D3 receptor partial agonist, attenuates cocaine reward and relapse in rodents. Br J Pharmacol 2020, 177, 4796–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ågren R; Zeberg H; Stępniewski TM; Free RB; Reilly S; Luedtke RR; Århem P; Ciruela F; Sibley DR; Mach RH; Selent J; Nilsson J; Sahlholm K Ligand with two modes of interaction with the dopamine D2 receptor-an induced-fit mechanism of insurmountable antagonism. ACS. Chem. Neurosci 2020, 11, 3130–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Vangveravong S; Zhang Z; Taylor M; Bearden M; Xu J; Cui J; Wang W; Luedtke RR; Mach RH Synthesis and characterization of selective dopamine D₂ receptor ligands using aripiprazole as the lead compound. Bioorg Med Chem 2011, 19, 3502–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Grundt P; Prevatt KM; Cao J; Taylor M; Floresca CZ; Choi JK; Jenkins BG; Luedtke RR; Newman AH Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: potential substance abuse therapeutic agents. J. Med. Chem 2007, 50, 4135–4146. [DOI] [PubMed] [Google Scholar]
- (41).Burris KD; Molski TF; Xu C; Ryan E; Tottori K; Kikuchi T; Yocca FD; Molinoff PB. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J. Pharmacol. Exp Ther 2002, 2, 381–389. [DOI] [PubMed] [Google Scholar]
- (42).Klein Herenbrink C; Verma R; Lim HD; Kopinathan A; Keen A; Shonberg J; Draper-Joyce CJ; Scammells PJ; Christopoulos A; Javitch JA; Capuano B; Shi L; Lane JR Molecular determinants of the intrinsic efficacy of the antipsychotic aripiprazole. ACS. Chem. Biol 2019, 14, 1780–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lane JR; Abramyan AM; Adhikari P; Keen AC; Lee KH; Sanchez J; Verma RK; Lim HD; Yano H; Javitch JA; Shi L Distinct inactive conformations of the dopamine D2 and D3 receptors correspond to different extents of inverse agonism. Elife 2020, 9, e52189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).de Paulis T; Kumar Y; Johansson L; Rämsby S; Florvall L; Hall H; Angeby-Möller K; Ogren SO. Potential neuroleptic agents. 3. chemistry and antidopaminergic properties of substituted 6-methoxysalicylamides. J. Med. Chem 1985, 28, 1263–1269. [DOI] [PubMed] [Google Scholar]
- (45).Northfield SE; Mountford SJ; Wielens J; Liu M; Zhang L; Herzog H; Holliday ND; Scanlon MJ; Parker MW; Chalmers DK; and Thompson PE Propargyloxyprolines regio- and stereoisomers for click-conjugation of peptide: synthesis and application in linear and cyclic peptides. Aust. J. Chem 2015, 68, 1365–1372 [Google Scholar]
- (46).Mihali V; Foschi F; Penso M; Pozzi G Chemoselective Synthesis of N‐Protected Alkoxyprolines under Specific Solvation Conditions. Eur. J. Org. Chem 2014, 24, 5351–5355. [Google Scholar]
- (47).Cheng Y; Prusoff WH Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (48).Shields BC; Kahuno E; Kim C; Apostolides PF; Brown J; Lindo S; Mensh BD; Dudman JT; Lavis LD; Tadross MR Deconstructing behavioral neuropharmacology with cellular specificity. Science 2017, 356, 6333. [DOI] [PubMed] [Google Scholar]
- (49).Bonifazi A; Battiti FO; Sanchez J; Zaidi SA; Bow E; Makarova M; Cao J; Shaik AB; Sulima A; Rice KC; Katritch V; Canals M; Lane JR; Newman AH Novel dual target Mu opioid (MOR) and dopamine D3 receptors (D3R) ligands as potential non-addictive pharmacotherapeutics for pain management. J. Med. Chem 2021, 64, 7778–7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bochevarov AD; Harder E; Hughes TF; Greenwood JR; Braden DA; Philipp DM; Rinaldo D; Halls MD; Zhang J; Friesner RA Jaguar: A high‐performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum. Chem 2013, 113, 2110–2142. [Google Scholar]
- (51).Shelley JC; Cholleti A; Frye LL; Greenwood JR; Timlin MR; Uchimaya M Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des 2007, 21, 681–691. [DOI] [PubMed] [Google Scholar]
- (52).Greenwood JR; Calkins D; Sullivan AP; Shelley JC Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aq solution. J. Comput. Aided. Mol Des 2010, 24, 591–604. [DOI] [PubMed] [Google Scholar]
- (53).Sherman W; Beard HS; Farid R Use of an induced fit receptor structure in virtual screening. Chem Biol. Drug Des 2006, 67, 83–84. [DOI] [PubMed] [Google Scholar]
- (54).Li J; Abel R; Zhu K; Cao Y; Zhao S; Friesner RA The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.