

Abstract
SARS-CoV-2 is currently causing an unprecedented pandemic. While vaccines are massively deployed, we still lack effective large-scale antiviral therapies. In the quest for antivirals targeting conserved structures, we focused on molecules able to bind viral RNA secondary structures. Aminoglycosides are a class of antibiotics known to interact with the ribosomal RNA of both prokaryotes and eukaryotes and have previously been shown to exert antiviral activities by interacting with viral RNA. Here we show that the aminoglycoside geneticin is endowed with antiviral activity against all tested variants of SARS-CoV-2, in different cell lines and in a respiratory tissue model at non-toxic concentrations. The mechanism of action is an early inhibition of RNA replication and protein expression related to a decrease in the efficiency of the −1 programmed ribosomal frameshift (PRF) signal of SARS-CoV-2. Using in silico modeling, we have identified a potential binding site of geneticin in the pseudoknot of frameshift RNA motif. Moreover, we have selected, through virtual screening, additional RNA binding compounds, interacting with the same site with increased potency.
Abbreviations: SARS-CoV-2, Severe actute respiratory syndrome - Coronavirus -2; PRF, Programmed ribosomal frameshifting; FSE, Frameshifting element; CC50, 50% cytotoxic concentration; EC50, 50% effective concentration
1. Introduction
Since the beginning of the SARS-CoV-2 pandemic, a huge effort has been made for the identification of effective vaccines and antivirals. The vaccines program has been an immense success with the approval of three vaccines in less than one year, and the vaccination, at the time of writing, of 68% of the world population. (Statistic and Research Coronavirus Vaccinations, https://ourworldindata.org/covid-vaccinations, accessed October 2022). The drug discovery effort has also led to the identification of three antiviral drugs, Remdesivir, Molnupiravir and Paxlovid, which have been fully approved by the FDA in the first case, or approved for emergency use (Parums, 2022). However, the emergence of new SARS-COV 2 variants, which can potentially escape the vaccine-mediated immunity and the effectiveness of therapies, highlights the importance to identify new potential pan antiviral agents against SARS-CoV-2.
RNA structure elements represent an attractive target for antiviral drug discovery. Viral genomes contain highly conserved RNA elements that play a critical role in gene regulation and viral replication. These RNA elements are directly involved in the viral infection process, interacting with proteins, DNA or other RNAs, modulating their activity (Embarc-Buh et al., 2021). The function and activity of these RNA molecules are based on the complex three-dimensional structure they can adopt (Ganser et al., 2019). Due to its conserved nature and its well-defined structure, the RNA provides potentially unique interaction sites for selective small-molecule ligands that affect viral replication. The high conservation of untranslated regions reduces the possibility of a drug-resistant mechanism, increasing the effectiveness of potential antiviral drugs (Warner et al., 2018). Any change in nucleotide sequence can result in inactive elements through misfolding the RNA structure, as recently demonstrated with the programmed −1 ribosomal frameshifting element (−1 PRF) of SARS-CoV-2 (Bhatt et al., 2021). Programmed ribosomal frameshifting is one of the strategies commonly used by RNA viruses, such as flaviviruses, coronaviruses, influenza A viruses and HIV, to regulate the relative expression level of two proteins encoded on the same messenger RNA (mRNA) (Brierley and dos Ramos, 2006; Firth et al., 2012; Penn et al., 2020). This strategy is rarely used by human cells, making it an attractive therapeutic target for antiviral drug development. Several studies have proposed the frameshifting element (FSE) as a target for disruption of virus replication (Ahn et al., 2011; de Wit et al., 2016; Haniff et al., 2020; Park et al., 2011). The SARS-CoV-2 FSE is a small region between the open reading frame (ORF) 1a and the ORF 1b. The ORF1b encodes all the enzymes necessary for viral RNA replication, including the RNA-dependent RNA polymerase. The frameshifting events depend on the flexibility of the RNA structure and its ability to interact with the ribosome. A small molecule that can alter the structural organization of the FSE can block the frameshifting event and consequently the viral replication. An example of -1PRF inhibitor is merafloxacin, a fluoroquinolone antibiotic, identified by screening compounds with a dual luciferase system with the -1PRF sequence of SARS-CoV-2 intercalated. The compound retained activity against the full virus and other coronaviruses (Sun et al., 2020). Furthermore, independent research groups confirmed its antiviral activity (Bhatt et al., 2021).
In the quest of additional inhibitors of -1PRF of SARS-CoV-2 we focused on aminoglycosides since they are among the molecules known to interact with secondary or tertiary structures on RNA. This class of antibiotics is known to interact with the ribosomal RNA of prokaryotes and eukaryotes (Garreau De Loubresse et al., 2014; Vicens and Westhof, 2003) in particular with the tRNA recognition site, blocking a conformational switch of the ribosomal A site. The affinity for RNA makes this class of molecules potentially interact with additional RNA structures as shown for RNA HIV dimerization sites, or for a riboswitch sequence in the 5’ leader RNA of a resistance gene in bacteria (Jia et al., 2013). Additionally an aminoglycoside showed activity against the frameshifting element of HIV (Staple et al., 2008).
Among the different aminoglycosides, geneticin is one of the few for which the cells are permeable, and it is commonly used in cell lines as a selective agent due to its alteration in eukaryotic protein synthesis when administered at high doses for a prolonged time (Davies and Jimenez, 1980). However, the drug proved to be effective as well against multiple viruses (Bovine Viral Diarrhea Virus, Dengue Virus and Hepatitis C virus (HCV)) (AlexanderBirk1 et al., 2008; Ariza-Mateos et al., 2016; Zhang et al., 2009) at non-toxic concentrations. In particular, in the evaluation of the antiviral activity of geneticin against HCV, a specific interaction with a double-stranded RNA switch structure in the 5′UTR of the virus was shown (Ariza-Mateos et al., 2016), its binding resulted in a stabilization of the open conformation leading to inhibition of the production of non-structural protein 3 (NS3) and viral replication in cell lines.
Here we show that geneticin is active against SARS-CoV-2 through an early inhibition in its life cycle and an alteration of the −1 PRF efficiency. The activity in the micromolar range is maintained against multiple variants, in different cell lines, and in respiratory tissues and has a high barrier to resistance. Importantly, we identified a putative binding site for geneticin on the −1 PRF sequence of SARS-CoV-2 through in silico modeling and we validated it with dual luciferase assays. After a screening of RNA binding molecules interacting with the same site, we identified compounds displaying antiviral activity at lower half-maximal effective concentrations (EC50) than geneticin, paving the road for the future development of SARS-CoV-2 antivirals.
2. Results
2.1. Geneticin is active against different variants of SARS-CoV-2 at non-toxic concentrations
Antiviral activity of geneticin against several variants of SARS-CoV-2 was assessed in Vero-E6 cells with the addition of the molecule post-infection. Merafloxacin, a molecule previously shown to inhibit SARS-CoV-2 (Hoffmann et al., 2020), and sotrovimab were tested against B.1.1.7 as control. Importantly the seven different variants tested, including the alpha (B.1.1.7), the beta (B.1.135), the delta (B.1.617.2) and the omicron (BA.1) were directly isolated from clinical specimens at the University Hospital of Lausanne with minimal passaging in cell lines to avoid any cell adaptation (Mathez and Cagno, 2021). We observed dose-response activity in the micromolar range for all the variants tested (Table 1 ). Analysis of the sequences did not reveal any particular cell adaptation, nor common changes in the variants showing higher EC50s if compared to the others (Supplementary Fig. 1). Moreover, the activity of geneticin was conserved against HCoV-229e in Huh7 cells (Table 1), while we observed a lack of activity against an unrelated virus, namely Influenza A virus (H1N1) (Table 1). Importantly, we excluded that the antiviral activity is linked to a toxic effect of geneticin on the cell with viability, cytotoxicity, and apoptosis assays (Supplementary Fig. 2A-B-C). Moreover, we verified that at the highest concentration used in the antiviral assays (600 μM) the protein synthesis in the cell is not impaired, in opposition with cycloheximide, a known elongation blocker (Supplementary Fig. 2D).
Table 1.
Antiviral activity of geneticin against SARS-CoV-2. EC50: half-maximal effective concentration. EC90: 90% effective concentration. 95% CI: confidence interval 95%.
| Virus | Variant | EC50 (95% CI) [μM] | EC90 (95% CI) [μM] | CC50 [μM] | |
|---|---|---|---|---|---|
| Geneticin | SARS-CoV-2 | B.1.1.7 (Alpha) | 47.2 (35.1–62.4) | 245 (149–460) | 3951 |
| B.1.351 (Beta) | 129 (84.7–188) | 542 (273–1537) | 3951 | ||
| B.1.617.2 (Delta) | 32.8 (21.3–48.2) | 201 (100–505) | 3951 | ||
| BA.1 (Omicron) | 25.7 (16.9–38.9) | 155 (60–528) | 934 | ||
| B.1.258 | 42.4 (29.7–60.4) | 163 (86–377) | 3951 | ||
| B.1.160 | 31.6 (21.2–44.8) | 150 (74–368) | 3951 | ||
| B.1.177 | 101.7 (74.8–138) | 407 (222–852) | 3951 | ||
| HCoV-229e | 7.21 (5.25–9.58) | 23.9 (12.1–62.3) | 684 | ||
| Influenza virus A | >600 | 5841 | |||
| Merafloxacin | SARS-CoV-2 | B.1.1.7 (Alpha) | 23.7 (16.8–33.8) | 116 (52.9–458) | >100 |
| Sotrovimab | SARS-CoV-2 | B.1.1.7 (Alpha) | 0.00013 (8.76 * 10−5 – 0.0002) | 0.0042 (0.0018–0.011) | >0.68 |
2.2. The antiviral activity is maintained in human respiratory cell lines and in tissues
To assess the antiviral activity in more relevant cell models, we evaluated the antiviral activity in dose response of geneticin, in parallel with merafloxacin and sotrovimab, in Calu3 cells, a lung adenocarcinoma cell line, which was previously shown to mimic faithfully SARS-CoV-2 infection in a respiratory cell line (Thi Nhu Thao et al., 2020). The results evidenced a sustained antiviral activity (EC50 164.0 μM) also in this cellular model in absence of toxicity (Table 2 ).
Table 2.
Antiviral activity of geneticin in Calu3 cells against SARS-CoV-2. EC50: half-maximal effective concentration. EC90: 90% effective concentration. 95% CI: confidence interval 95%.
| Virus | Variant | EC50 (95% CI) [μM] | CC50 [μM] | |
|---|---|---|---|---|
| Geneticin | SARS-CoV-2 | B.1.1.7 (Alpha) | 164.0 (107.3–232.9) | >600 |
| Merafloxacin | SARS-CoV-2 | B.1.1.7 (Alpha) | 21.76 (14.62–31.53) | >100 |
| Sotrovimab | SARS-CoV-2 | B.1.1.7 (Alpha) | 0.0041 (0.0028–0.0058) | >0.68 |
We then tested the activity in a pseudostratified model of the human respiratory tract (Mucilair, Epithelix). This tissue model is composed of the typical cells of the human upper respiratory tract, namely ciliated, goblet and basal cells. In this infection model, we aimed to mimic a possible treatment with the molecule by starting the treatment 24 h post-infection (hpi) when the infection of the tissue was already well established and we used viral stocks produced in the same tissue and never passaged in cell lines to exclude any adaptation. The treatment was performed apically by adding 30 μl of geneticin at 1 mg/ml (2 mM) concentration (resulting in 30 μg/tissue), and the infection was monitored up to 4 days post-infection by collecting an apical wash and performing either a qPCR or a titration in Vero E6 (Fig. 1 ). The results evidenced significant protection from viral infections with both B.1.1 (Fig. 1B) and omicron BA.1 (Fig. 1C) variants, without a decrease in viability for the tissues (Supplementary Fig. 3).
Fig. 1.

The activity of geneticin in maintained in human-derived tissues. A) and B) Mucilair tissues were infected with A) SARS-CoV-2 B.1.1 106 RNA copies or B) SARS-CoV-2 Omicron BA.1 105 RNA copies, the following day the apical treatment with 30 μg/tissue started. Every 24 h an apical wash was performed and collected after 20 min at 37 °C. The supernatant was then used for viral RNA quantification (solid lines) or for plaque assay (dashed lines). The results are the mean and SEM of two to three independent experiments performed in duplicate. P values < 0.0332 (*), <0.0021 (**), <0.0002 (***), <0.0001 (****).
2.3. Geneticin has a high barrier to resistance
To evaluate the barrier for resistance, we passaged the virus in the presence or absence of increasing doses of geneticin for 11 passages (Supplementary Fig. 4A). At the end of the experiment, we evaluated the EC50s of the viruses grown in presence of geneticin in comparison to the untreated viruses. We did not observe any significant change in the EC50s (Fig. 2 A). We verified through Next Generation Sequencing (NGS) the presence of mutations in the untreated and geneticin treated viruses. We observed the typical features of viruses passaged in cell lines such as the inactivation or deletion of the furin cleavage site (R685H in the untreated, del 679-85 in the geneticin treated), and we could identify a mutation in the ORF1a in the geneticin treated not present in the untreated at passage 11 (Fig. 2B). However, the same mutation was not present in the duplicate condition (Supplementary Fig. 4B).
Fig. 2.

High barrier of resistance of geneticin A) The EC50s of geneticin were evaluated against the B.1.1.7 stock, viruses are grown in Vero E6 without treatment for 11 passages, or in presence of increasing doses of geneticin. B) The mutations observed at passage 11 as compared to the original B.1.1.7 stock (created with biorender.com).
2.4. Geneticin inhibits the -1PRF of SARS-CoV-2
In order to assess the mechanism of action and the stage of viral replication of SARS-CoV-2 inhibited by geneticin, we first assessed viral protein expression. We exploited a GFP expressing SARS-CoV-2 previously generated (Thi Nhu Thao et al., 2020) evaluating the GFP expression in the presence or absence of the drug at 24h and 48hpi (Supplementary Fig. 5). The results evidenced, as expected, a marked reduction in the number of infected cells and in addition, the GFP intensity was significantly reduced in the infected cells treated with geneticin, when compared to the untreated control (Supplementary Fig. 5). Moreover, we analyzed the amount of viral nucleoprotein and cellular tubulin in cells infected and treated for 24 or 48h by Western blot, confirming the marked selective reduction in viral protein expression (Fig. 3 A and Supplementary Fig. 6). We included as control merafloxacin, a compound previously shown to interfere with the −1 PRF signal. The results show a decrease in viral protein production for both compounds, suggesting a block of infection at an initial stage of the viral life cycle. To evaluate if the inhibition of protein expression was related to a block of translation or viral replication, we then monitored viral replication through an RT-qPCR measuring the viral RNA replication at different time points. As shown in Fig. 3B, the addition of geneticin or merafloxacin, results in inhibition of viral RNA replication at 4h, 8h and 24h post-infection, demonstrating a rapid inhibition of viral replication by the two drugs. Finally, we assessed the ability of geneticin, in comparison with merafloxacin, to interfere with the programmed ribosomal frameshifting element of SARS-CoV-2 with a dual luciferase assay. The −1 PRF signal was cloned between Renilla and Firefly luciferase and the relative expression of the luciferases was evaluated in the presence or absence of the drugs as described in (Bhatt et al., 2021) and depicted in Supplementary Fig. 7. The results of Fig. 3C show a reduction in the −1 PRF efficiency in presence of both compounds suggesting a direct interaction of the drug with the -1PRF sequence resulting in impaired replication (Fig. 3B) and protein production (Fig. 3A). Additionally, we tested mutated PRF as described in Bhatt et al.; (Fig. 3C and D). The results showed a similar inhibitory profile for geneticin and merafloxacin, suggesting a common binding pocket on the −1 PRF.
Fig. 3.

Mechanism of action of geneticin. A) Vero-E6 cells were infected with B.1.1.7 SARS-CoV-2 at MOI 0.1 and at MOI 0.01 (24hpi and 48hpi conditions respectively). Cells were treated post-infection with geneticin (600 μM). Cells were lysed 24hpi or 48hpi and protein quantification was done by Western Blot (Supplementary Fig. 6). Values are expressed by the ratio of the intensity of SARS-CoV-2 nucleocapsid over alpha tubulin quantified by ImageJ. B) Vero-E6 were infected with SARS-CoV-2 at MOI 0.1 for 1 h at 37 °C. After the removal of the inoculum, geneticin (600 μM) or merafloxacin (100 μM) were added to the well. At 0, 4, 8 and 24 h post-infection cells were lysed and viral RNA was quantified. C) Dual luciferase evaluation was performed at 24 h post-transfection, as depicted in Supplementary Fig. 7, in Vero-E6 cells treated with geneticin (600 μM) or merafloxacin (50 μM)). The results are mean and SEM of three independent experiments performed in duplicate. P values < 0.0332 (*), <0.0021 (**), <0.0002 (***), <0.0001 (****). D) SARS-CoV-2 RNA frameshift-stimulatory element sequence and the mutant sequences.
2.5. In silico modeling and prediction of geneticin binding site
To further rationalize the results obtained by dual-luciferase and antiviral assays, the cryo-EM structure of the RNA frameshift-stimulatory element (FSE) was used to investigate the Geneticin-FSE binding complex (Zhang et al., 2021). The cryo-EM RNA structure shows a λ-like tertiary arrangement composed of a three-stemmed H-type pseudoknot structure with three loops. Starting from the 5′-end and proceeding to the 3′-end, the cryo-EM structure begins with a slippery site, followed by Stem 1 (S1), which leads to the Loop (L1), and it continues to Stem 2 (S2) (Fig. 4 A). From the second stem (S2), the RNA strands continue to form a hairpin region (S3), followed by an unpaired segment J3/2, which leads back to Stem 2 and closes the Stem 1-Stem 2 pseudoknot (Fig. 4A). The cryo-EM data also suggested alternative conformations due to the structural flexibility at the 5′-ends, which appeared poorly resolved (Zhang et al., 2021). Moreover, the cryo-EM structure was resolved at low-mid resolution 6.9 Å, which can affect the assignment of the atom position with high certainty. Molecular dynamic (MD) simulations have proven useful in refining macromolecular structures, particularly unveiling the atomic details for low-resolution regions of the cryo-EM map (Bissaro et al., 2020; McGreevy et al., 2016; Nierzwicki and Palermo, 2021). In this study, we initially refined the cryo-EM FSE structure by 100 ns molecular dynamics simulation using the GROMACS software package (Abraham et al., 2015). Overall, after an initial 40 ns of equilibration, the structural fluctuation of the RNA reduced, with the simulation system converging around a fixed RMSD value of 1.5 Å. This RMSD value was chosen as a cut-off for selecting a series of different conformers, which were successively clustered to select a representative structure (Fig. 4B). The comparison between the cryo-EM and our model showed a similar structure rearrangement with minimum RMSD variations in nucleotide position, except for the slippery site and S3 region, which displayed a higher level of flexibility (Fig. 4B). These results are in line with previous studies conducted by Omar et al. and Rangan et al., showing that stem 3 could adopt multiple conformations (Omar et al., 2021a; Rangan et al., 2021).
Fig. 4.

Comparison of the cryo-EM RNA structure (A) and the refined RNA structure by molecular dynamic simulation (B). (C) Different sequential mutations in the FSE structure: point mutations in Loop1 (in red); deletion of loop1 (in green); deletion of A and ACA in J3/2 (turquoise and yellow respectively). (D) The 3 binding sites identified by RNAsite. The binding site 1 (ring site); 2 (J3/2) and 3 (stem 2) are highlighted in red, blue and green, respectively. The 2D figures show geneticin interactions with the surrounding nucleotides in the binding sites. Turquoise dashed lines indicate weak H-bond; green dashed lines indicate strong H-bond, and purple dashed lines indicate electrostatic interactions.
Mutational studies showed that the virus replication is highly sensitive to any conformational change in the pseudoknots region, as evident by the point mutation of guanidine to adenine in loop 1 (Fig. 3, Fig. 4C), which reduced the frameshifting efficiency to 60%. (Fig. 3C). According to the mutation results and the uncertainty of the S3 region, we hypothesized that geneticin could significantly alter and disrupt the FSE conformational plasticity and consequently the viral replication, directly binding the S1/S2 -J3/2 pseudoknots region. A previous study showed that geneticin can interact with tertiary RNA structures through hydrogen bonds and electrostatic interactions (Prokhorova et al., 2017; Vicens and Westhof, 2003). The binding affinity of geneticin for the RNA structures is mainly due to the presence of four amino groups which are positively charged at physiological pH and can form strong electrostatic interaction with the negatively charged phosphates in the nucleic acid backbone (Fig. 4D). Furthermore, the presence of seven hydroxyl groups can stabilize the RNA-binding complex through a series of hydrogen bonds with the base atoms and phosphate oxygen atoms of the nucleic acid. Several studies demonstrated the preference of aminoglycoside compounds to bind RNA helix and junction sites (Aradi et al., 2020). We first investigated if there were potential geneticin-binding sites in FSE regions using the refined cryo-EM structure. The binding site analyses, performed by the RNAsite module (Su et al., 2021a), identified 3 different potential active sites situated between stem 1, stem 2 and junction site (Fig. 4D), which partially confirmed the results obtained by Zhang and collaborators, who reported the presence of a ‘ring site’, a ‘J3/2 site’ and the ‘slippery hairpin binding site (Zhang et al., 2021). Our results showed that two potential binding sites, 1 and 2; which were located in close proximity, sharing 3 nucleotide residues (G18, G19 and G20), similar to the ring site and J3/2 site reported by Zhang and collaborators (Zhang et al., 2021). However, contrary to Zhang and collaborators, we could not detect any suitable binding site on the slippery site, instead, a new potential pocket (binding site 3) was located at the beginning of stem 2 (Fig. 4D).
The geneticin-binding affinity was evaluated against all the three potential binding sites using an in silico protocol, which comprises three steps: firstly, the compound was docked using XP GLIDE module (Maestro, Schrodinger), then the docked poses were refined using MM-GBSA module, and lastly the refined poses were rescored using two scoring functions optimized specifically for RNA-ligand complex, Annapurna and Amber score function (DOCK6). The purpose of multiple scoring functions was to ascertain the most potentially accurate ligand poses and avoid any possible bias associated with using a single docking program/scoring function. The docking results showed that although geneticin can be well accommodated inside all three binding sites in different rational configurations, it has a slighter higher affinity for site 1 compared to sites 2 and 3 (ΔGmm-gbsa −102.98, −90.34, −80.77 kcal/mol, respectively). Site 2 and 3 showed the largest surface area, but are solvent-exposed, which affect the ligand-RNA interaction: geneticin was only partially in contact with the RNA surface while the rest of the molecule was exposed to solvent (Fig. 4D). On the other hand, site 1 showed a smaller surface area, but it was surrounded by nucleotides (G18, G19, G20, G43, G44, G46, U75 and A76), which form a tunnel-like binding site. Geneticin can well occupy the active site with the streptamine core inside the tunnel cavity, interacting with G19, U20, U45 and A74 through hydrogen bonds (H-bonds) and with U75 by electrostatic interactions between the amino group chain and phosphate groups of U75 (Fig. 4D). To confirm the results obtained by the MM-GBSA analysis, the refined docked poses were rescored using Annapurna and DOCK6 score function. In both software, the top-ranked binding poses were predicted to site 1 (Supplementary Table 1), suggesting that this site might be more accessible and druggable than the other two binding sites. Interestingly, merafloxacin can also bind site 1, showing similar binding interactions of Geneticin (H-bonds with U45 and A74 and electrostatic interaction with U75) (Supplementary Fig. 8).
In order to confirm this binding pocket, we designed specific mutations on these three nucleotides (U45, A74, U75) to modify the size of the drug-binding pocket, and occlude the binding of geneticin (Fig. 5 A and B). In the dual luciferase assay, this combination of mutations (T- > A in S3 and AT- > TA in J3/2) resulted in a significant loss of -1PRF inhibition for both compounds without affecting the general frameshift (Fig. 5C) confirming the binding pocket for geneticin and merafloxacin.
Fig. 5.

Effect of point mutations in the binding pocket (A natural nucleosides B mutated nucleoside) C) Dual luciferase evaluation was performed at 24 h post-transfection in Vero-E6 cells treated with geneticin (600 μM) or merafloxacin (50 μM) (Supplementary Fig. 7). The results are mean and SEM of three independent experiments performed in duplicate. P values < 0.0332 (*), <0.0021 (**), <0.0002 (***), <0.0001 (****). D) SARS-CoV-2 RNA frameshift-stimulatory element sequence and the mutant sequences.
2.6. Identification of -1PRF binding compounds
To test the druggability of the binding site, we screened an RNA-targeted library (Enamine, ChemDIV), which contains 44,520 commercially available RNA-binding compounds, against site 1. The virtual screening was performed using the previously described protocol. Firstly, the XP glide docking mode was employed to virtually screen the RNA-target library. The best 10% of docked poses to this initial screening were refined and rescored through MM-GBSA. To validate the top-scored docking results, the compounds were rescored using Annapurna and DOCK6 scoring functions. After applying a consensus score procedure, 132 molecules were chosen, which were further evaluated by visual inspection considering the ability of compounds to occupy the binding site and the number of interactions formed between the compounds and the target. At the end of this workflow, twenty compounds were selected, purchased and evaluated in antiviral assays (Supplementary Fig. 9 and Supplementary Table 2). Among them, three compounds could inhibit the virus replication with an EC50 in the micromolar range, with higher potency than geneticin (Table 3 ).
Table 3.
Antiviral activity of geneticin analogs. EC50: half-maximal effective concentration, CC50: half-maximal cytotoxic concentration.
| Analog | Structure | EC50 [μM] | CC50 [μM] |
|---|---|---|---|
| 1 (AB-3234) | 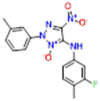 |
13.0 | >100 |
| 2 (AB-3241) | 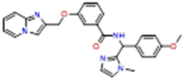 |
25.2 | >100 |
| 3 (AB-3285) | 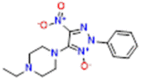 |
12.0 | >100 |
The most potent compound was further analyzed for kinetics of RNA expression (Fig. 6 A) and dual luciferase (Fig. 6B) confirming a similar activity to geneticin. The in-silico results showed that could completely occupy the tunnel-binding site, forming a cation-pi with G19 and H-bonds with G19, G18, C43 and G44 (Fig. 6C).
Fig. 6.

Antiviral activity of site 1 -1 PRF binders against SARS-CoV-2. A,B,C) Mechanism of action of AB-3285. A) Vero-E6 were infected with SARS-CoV-2 at MOI 0.1 for 1 h at 37 °C. After the removal of the inoculum, AB-3285 (250 μM) was added to the well. At 0, 4, 8 and 24 h post.-infection cells were lysed and viral RNA was quantified. B) Dual luciferase evaluation was performed at 24 h post-transfection in Vero-E6 cells treated with AB-3285 (500 μM). The frameshift efficiency was normalized compared to untreated. C) Binding pose of AB-3285 in the PRF binding site 1. The results are mean and SEM of at least two independent experiments performed in duplicate. P values < 0.0332 (*), <0.0021 (**), <0.0002 (***), <0.0001 (****).
More recently, the SARS-COV-2 FSE structure solved by x-ray confirmed the cryo-EM three-stemmed H-type pseudoknot structure, but it showed different tertiary arrangements: the cryo-EM structure has a λ-like tertiary arrangement, meanwhile, the x-ray adopts a vertical conformation (Roman et al., 2021) Although the x-ray shows a higher resolution of 2.09 Å, it lacks the 5’- slippery site sequence, which might affect the tertiary arrangement. These different arrangements of the FSE have also been supported by previous chemical probing, mutational, and NMR studies demonstrating that the arrangement of stem 1 and stem 2 relative to stem 3 can be flexible (Schlick et al., 2021a). The superposition of the x-ray structure and our model showed a similar binding site in the experimental structure, as also revealed by RNAsite, which overlaps our identified binding site 1 (Supplementary Fig. 10).
3. Discussion
The alteration of the flexibility of the FSE of SARS-CoV-2 is detrimental to the replication of the virus (Bhatt et al., 2021; Huston et al., 2021; Manfredonia et al., 2020; Omar et al., 2021b; Schlick et al., 2021b). If the viral RNA cannot interact correctly with the ribosome, the −1 PRF is altered and ORF 1 ab cannot be expressed at the correct ratio, resulting in a lack of production of the viral polymerase and a consequent reduction of the replication (Bhatt et al., 2021). The FSE of SARS-CoV-2 was previously shown to be a possible target for antiviral development with basic modeling (Park et al., 2011) or with empiric screening with dual luciferase assays (Su et al., 2021). In the last few years, several inhibitors of different viral PRF have been identified. These compounds are characterized by high structural and physicochemical properties diversity, and a modest antiviral activity. Furthermore, most of them show poor drug-like properties, which limits their translation into clinical applications. Interestingly, during the course of this project, a recent study conducted by Munshi and collaborators (Munshi et al., 2022) identified a series of small molecules, which were able to inhibit coronaviruses −1 PRF, confirming the frameshift element is a promising target developing pan-coronavirus antivirals. The precise druggable pockets of the FSE however were not previously identified. With the aim of identifying new molecules interacting with viral RNA, we tested geneticin, an aminoglycoside known to interact with RNA secondary structures.
The compound proved to be effective against multiple variants of SARS-CoV-2 (Table 1). The range of EC50s determined (Table 1) might be linked to the fitness of the variants in the Vero E6 and their plaque-forming ability. To exclude any bias, we verified as well the activity of geneticin in Calu3 cells (Table 2), and we tested both B.1.1 and omicron BA.1 variants in a human respiratory airway model. In all conditions, we confirmed the antiviral activity of geneticin (Fig. 1). Furthermore, the compound showed activity as well against HCoV-229e, demonstrating a broad-spectrum activity against coronaviruses, while it was not active against an unrelated RNA virus, Influenza A virus (Table 1), proving that the mechanism of action is not related to a general effect on the ribosome that will impair the replication of all viruses. The absence of toxicity at the antiviral tested doses is demonstrated as well by the toxicity analysis (Supplementary Fig. 1) and by the Western blot analysis in which we observe a significant decrease of viral nucleoprotein and an absence of effect on cellular tubulin (Supplementary Fig. 6). However, we cannot exclude completely a role of the molecule directly on the ribosome, considering the load of viral protein synthesis ongoing in a cell during a viral infection.
We then verified an early inhibition in the life cycle with reduced viral protein expression and RNA replication (Fig. 3A and B), and we tested the activity on the PRF through dual luciferase assays (Fig. 3C) on wild-type and mutated sequences, in which geneticin and merafloxacin behaved similarly supporting the same mechanism of action and possibly the same binding site. These results are further supported by the substitution of 3 nucleotides in the binding pocket leading to a significant loss of activity of both compounds (Fig. 5). However, both compounds retain a partial activity also in presence of these mutations. This can be related to additional mechanisms of action, as discussed above, or to the dual luciferase assay, in which the −1 PRF sequence is cloned in between non-viral sequences and the readout is the luciferase activity that might be in a non-linear relation with the luciferase expression.
Targeting a highly conserved sequence in the RNA, the development of resistance is intrinsically limited, however, we verified it by growing the virus in presence of increasing concentrations of geneticin; after 11 passages we failed to observe any difference in the EC50s nor the appearance of specific mutations (Fig. 2). These results are in line with the conserved activity of geneticin when we mutated residues in the –1PRF sequence (Fig. 3C). For antivirals in use, in contrast to geneticin, resistance was selected. SARS-CoV-2 developed resistance to remdesivir after 13 passages (Szemiel et al., 2021) and in a patient in only one week (Gandhi et al., 2022), similarly to what reported for Nirmatrelvir (Paxlovid) (Iketani et al., ; Jochmans et al.,). However, considering the different experimental settings, we cannot exclude that with an increased number of passages, or alternative models for selection, resistance to geneticin could be selected.
The high flexibility and plasticity of the FSE is an essential requirement for its biological activity (Bhatt et al., 2021.; Huston et al., 2021; Manfredonia et al., 2020; Omar et al., 2021b; Schlick et al., 2021b). This unique characteristic is also supported by cryo-EM and x-ray structures recently published (Bhatt et al., 2021.; Roman et al., 2021; Zhang et al., 2021). In particular, the pseudoknot structure seems to be highly dynamic before encountering the ribosome. However, the unique 3-stem architecture of the FSE (Fig. 4) and its mechanism made the FSE a viable target for small molecules. Our computational studies confirmed the presence of a suitable binding site in the pseudoknot structure, originally identified by Zhang and collaborators (Zhang et al., 2021). This binding site is located between J3/2 and stem 3 regions, and it is large enough to accommodate geneticin, merafloxacin, and small ligands. Interestingly, this pocket is close to the S3 region of the FSE. Our molecular dynamic simulation studies revealed that the S3 region is particularly flexible, showing higher fluctuations than the other regions. According to these results, we hypothesized that the S3 region might play a critical role in the conformational change of the FSE, necessary for the frameshifting event. Hence, geneticin could exert its antiviral activity by altering the flexibility of this region, and consequently interfering with the conformational changes between the two main FSE structures.
The resulting antiviral activity is however linked to several limitations: the activity is in the micromolar range, and further studies should focus on the identification of more potent compounds. The antiviral activity is at non-toxic concentrations, also in human-derived respiratory tissues (Supplementary Fig. 3), however, the selectivity index of geneticin is narrow since it is known to bind eukaryotic ribosomes and it is associated with toxicity in cell culture. Although the administration in a viral infection is most likely to be for a short duration, future work should be directed to the identification of compounds devoid of interaction with ribosomal RNA. Moreover, aminoglycosides are associated with nephrotoxicity and ototoxicity when administered systemically, therefore a topical administration should be envisaged for compounds similar to geneticin.
To overcome these limitations and to validate the druggability of the binding pocket identified, we used a virtual screening simulation to identify additional molecules, from a library of RNA binders. Our in silico screening against the “J3/2- stem 3” site revealed that the architecture of the pocket might be sufficiently complex to be targeted by more specific ligands. Through our simulations, we have identified molecules that might engage the FSE targeting the J3/2- stem 3 pocket, enhancing or reducing the pseudoknot stability. The identification of compound 3 with increased potency, reduction of RNA replication, and alteration of the -1PRF (Table 3, Fig. 6) demonstrates the feasibility of our approach. However the activity is still in the micromolar range and the compound has an N-nitroso group, which is often categorized as a structural alert due to the potential carcinogenic effect. Future work will be directed toward the identification of analogs with increased potency, retaining the same mechanism of action, but suitable pharmacological properties.
4. Conclusion
Programmed −1 ribosomal frameshifting plays major functional and regulatory roles in the SARS-CoV-2 replication. Thus, the frameshifting element is an attractive target for the development of new potential antiviral drugs. In this study, we have shown that geneticin, a well-known aminoglycoside antibiotic, could inhibit the viral replication engaging the frameshifting element, similarly to merafloxacin, a structurally different molecule, previously reported in the literature. The mode of action was confirmed by three different biological assays: the inhibition of RNA synthesis, the reduction of viral protein production, and the luciferase expression assays under the control of the -1PRF in presence of WT and mutated sequences. Moreover, we have identified a potentially targetable pocket in the FSE structure, which can well accommodate the geneticin as evident by the high in silico binding affinity and reduced activity in presence of targeted mutations. The druggability of the binding pocket identified was confirmed through an in silico screening of a small library of RNA-binding small molecules. Among them, one compound showed higher antiviral activity than geneticin.
5. Material and methods
5.1. Cells
Vero C1008 (clone E6) (ATCC CRL-1586), Huh7, MDCK and A549 cells were a kind gift from Prof Gary Kobinger, Dr Sylvia Rothenberger, Prof. Mirco Schmolke and Prof. Caroline Tapparel respectively. Calu-3 were purchased from ATCC. Cells were propagated in DMEM High Glucose (or MEM for Calu-3) + Glutamax supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptavidin (pen/strep).
5.2. Viruses
hCoV-19/Switzerland/GE-SNRCI-29949586/2020 (B.1.1) was isolated from a clinical specimen in the University Hospital in Geneva in Vero-E6 and passaged twice before the experiments. SARS-CoV-2 GFP was a kind gift from Prof Volker Thiel (Thi Nhu Thao et al., 2020). The other clinical strains (hCoV-19/Switzerland/VD-CHUV-GEN3159/2021 (B.1.1.7), hCoV-19/Switzerland/VD-GEN3343/2021 (B.1.351), hCoV-19/Switzerland/VD-CHUV-GEN5521/2021 (B.1.617.2), hCoV-19/Switzerland/VD-CHUV-GEN8840/2021 (BA.1), hCoV-19/Switzerland/VD-GEN3642/2021 (B.1.160), hCoV-19/Switzerland/VD-GEN3807/2021 (B.1.177), hCoV-19/Switzerland/VD-GEN3770/2021 (B.1.258)) were isolated from clinical specimens from the University Hospital of Lausanne (CHUV) as described in (Mathez and Cagno, 2021). The supernatant of infected cells was collected, clarified, aliquoted, and frozen at −80 °C and subsequently titrated by plaque assay in Vero-E6. Human coronavirus 229E was bought from ATCC (VR-740) and cultured on Huh7 cells. Influenza virus A/Netherlands/602/2009 was a kind gift from Prof. Mirco Schmolke and cultured on MDCK.
5.3. Cell toxicity assay
Cell viability was measured by the MTT assay or MTS assay (Promega) for tissues. Confluent cell cultures seeded in 96-well plates were incubated with different concentrations of geneticin in duplicate under the same experimental conditions described for the antiviral assays. Absorbance was measured using a Microplate Reader at 570 nm. The effect on cell viability at different concentrations of geneticin and additional compounds was expressed as a percentage, by comparing the absorbance of treated cells with the one of cells incubated with equal concentrations of solvent in medium. The 50% cytotoxic concentrations (CC50) and 95% confidence intervals (CIs) were determined using Prism software (Graph-Pad Software, San Diego, CA). For LDH assays the supernatants of the cells treated with geneticin as described before were analyzed with cytotoxicity detection kit (CyQUANT™ LDH Cytotoxicity Assay, Thermofisher) and the 100% was calculated with a well in which the supernatant contained 0.05% Triton. For apoptosis assay, the cells were analyzed with the Caspase-3 assay kit (Biovision) according to the manufacturer instructions.
5.4. Bioorthogonal noncanonical amino acid tagging (BONCAT)
A549 cells (350′000 cells/well) were seeded in 6 well plate incubated with 600 μM of geneticin or 50 μg/ml of cycloheximide in medium without amino acids supplemented with 1% NEAA, 1% glutamine, 50 μM L-cysteine and 50 μM L-Azidohomoalanine. Following 24h, 48h or 72h incubation wells were detached, pelleted fixed and subjected to click reaction with TAMRA-alkyne according to (Dieterich et al., 2007). The TAMRA signal was quantified with Cytoflex instrument and quantified with FlowJo.
5.5. Antiviral assay in Vero-E6 cells
Vero-E6 cells (105 cells per well) were seeded in 24-well plate. Cells were infected with SARS-CoV-2 (MOI, 0.001 PFU/cell) for 1 h at 37 °C. The monolayers were then washed and overlaid with medium supplemented with 5% FBS containing serial dilutions of compounds for the experiments with SARS-CoV-2 expressing GFP. For experiments with the different SARS-CoV-2 variants and analogs of geneticin, Vero-E6 cells were overlaid instead with 0.4% avicel gp3515 in medium containing 2.5% FBS. Two days after infection, cells were fixed with 4% formaldehyde and stained with crystal violet solution containing ethanol. Plaques were counted, and the percent inhibition of virus infectivity was determined by comparing the number of plaques in treated wells with the number in untreated control wells. 50% effective concentration (EC50) was calculated with Prism 9.1 (GraphPad).
5.6. Antiviral assay in Calu3 cells
Calu-3 cells (4 × 104 cells per well) were seeded in 96-well plate. Cells were infected with B.1.1.7 SARS-CoV-2 (MOI 0.1 PFU/cell) for 1 h at 37 °C. The monolayers were then washed and overlaid with medium containing serial dilutions of geneticin. At 24 hpi, supernatant was collected and viral RNA was extracted with EZNA total RNA kit (Omega Bio-tek). SARS-CoV-2 RNA was quantified by RT-qPCR with the QuantiTect Kit (Qiagen, 204443) with Sarbeco E gene primers and probe in a QuantStudio 3 thermocycler (Applied Biosystems). Percent inhibition of virus infectivity was determined by comparing viral load in treated wells with the viral load in untreated control wells. EC50 was calculated with Prism 9.1 (GraphPad).
5.7. Resistance selection and next generation sequencing
Vero-E6 cells (3.5 × 105 per well) were seeded in 6-well plate. At the first passage, cells were infected with B.1.1.7 SARS-CoV-2 (MOI 0.01) for 1 h at 37 °C. The inoculum was removed and an overlay with 2.5% FBS in DMEM was added. Half of EC50 concentration of geneticin (40 μM) was added in 2 wells. Two other wells were left untreated. Supernatant was collected 3 days post-infection and clarified at 2 × 103 rpm for 5 min. Each sample was quantified by plaque assay in Vero-E6 cells (105 cells per well) with an overlay of 0.6% avicel gp3515 in 2.5% FBS DMEM. For the following passages, cells were infected with the previous corresponding passage (MOI 0.01). The concentration of geneticin was doubled up to a final concentration of 600 μM.
RT-PCR targeting the PRF sequence was done for each condition at passage 10 (see below). At passage 11, untreated and treated conditions were used for dose-response with geneticin as described above. A sample per condition was lysed with TRK Lysis Buffer (Omega Bio-tek) for next generation sequencing as previously described (Jacot et al., 2021). Briefly, SARS-CoV-2 genome was amplified with the CleanPlex® SARS-CoV-2 FLEX panel. The tiled amplicons were then sequenced with 2 × 150 bp on a MiSeq instrument (Illumina, San Diego, USA). Reads were analyzed with GENCOV https://github.com/metagenlab/GENCOV), a pipeline modified from CoVpipe (https://gitlab.com/RKIBioinformaticsPipelines/ncov_minipipe). Variant calling was performed with Freebayes (Garrison and Marth, 2012) (parameters: min-alternate-fraction 0.1 –min-coverage 10 –min-alternate-count 9) and consensus sequences were obtained using bcftools (Danecek et al., 2021) based on variants supported by at least 70% of reads. Lineages were assigned to the consensus sequence using Pangolin (O'Toole et al., 2021).
5.8. Kinetics of RNA expression
Vero-E6 cells were seeded in 24-well plates at a density of 105 cells per well and infected in duplicate with B.1.1.7 SARS-CoV-2 at MOI 0.1 PFU/cell for 1 h at 37 °C. After the removal of the inoculum the treatment was started and cells were lysed with TRK buffer (Omega Biotech) at 0, 4, 8 and 24 h post infection. RNA was extracted with the Total RNA kit (Omega Biotech) and amplified with the E-sarbeco primers for SARS-CoV-2.
5.9. Western blot
Vero-E6 were seeded in 6-well plates at a density of 3.5*105 cells per well. Cells were infected with B.1.1.7 SARS-CoV-2 with a different MOI for each time point (MOI 0.1 for 24hpi and MOI 0.01 for 48hpi). After 1 h of infection at 37 °C, inoculum was removed and fresh DMEM 2.5% FBS was added. Geneticin 600 μM and merafloxacin 50 μM were used as post-treatment. One or two days after infection, cells were lysed at 4 °C for 30min with RIPA buffer (0.001% SDS, 0.01% Triton, 0.1% sodium deoxycholate, 5 μM NaCl, 0.0025% Tris HCl, 2 nM EDTA, protease inhibitors) and clarified at 13′000 rpm for 30min. Supernantants were collected and quantified with Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Twenty μg of proteins were loaded in an 8% acrylamide gel (8% acrylamide, 0.05% SDS, 422 mM Tris HCl, 0.1%APS, 0.001% TEMED) and separated at 150V for 2 h in running buffer (0.1% SDS, 25 mM Tris, 190 mM glycine). Proteins were transferred on a nitrocellulose membrane after 1 h at 100V in transfer buffer (20% methanol, 50 mM Tris, 40 mM glycine, 0.037% SDS). Nitrocellulose membrane was blocked with 5% milk diluted in TTBS (0.05% Tween, 20 mM Tris HCl, 500 mM NaCl) for 30min at room temperature. The membrane was incubated overnight at 4 °C with 1:5000 alpha-tubulin and 1:5000 SARS-CoV-2 nucleocapsid antibodies in TTBS with 5% milk. After three washes in TTBS, 1:2000 anti-mouse IgG and 1:2000 anti-rabbit IgG HRP-linked antibodies was added on the membrane. The membrane were developed with WesternBright ECL (Advansta). Intensity of alpha tubulin and SARS-CoV-2 nucleocapsid were quantified by ImageJ.
5.10. Flow cytometry analysis
Vero-E6 cells were seeded in 24-well plates at a density of 105 cells per well and infected in duplicate with SARS-CoV-2 GFP at an MOI of 0.01 PFU/cell for 1 h at 37 °C. The cells were then treated with geneticin and incubated at 37 °C for additional 24 or 48 h. Supernatant was collected, cells washed once and detached with trypsin. Once in suspension cells were pelleted and then fixed with paraformaldehyde 4% in PBS. Percentages of GFP positive cells and mean GFP value for each positive cell was evaluated with an Accuri C6 cytometer (BD biosciences).
5.11. Dual luciferase
pSGDLuc v3.0 was modified to include the −1 PRF signal of SARS-CoV-2 as described in (Bhatt et al., 2021). The wild type, deletion of loop 1, mutation of G- > A in loop 1, mutation of G- > C in loop 1, deletion of ACA in J3/2, deletion of A in J3/2 and their corresponding in frame control were kindly given by Prof Loughran Gary. The three substitutions mutations (T- > A in S3/AT- > TA in J3/2) were made with Q5 Site-Directed Mutagenesis Kit (BioLabs). Vero-E6 cells were seeded 24 h in advance in 96-well plates (104 cells per well), treated with geneticin, merafloxacin or geneticin analog and transfected with Lipofectamine 3000 (Thermofisher) and the plasmid containing the -1PRF sequence or the in frame control. Luciferase was evaluated 24 h post transfection with the Dual Luciferase Reporter Assay System (Promega). The percentage of ribosomal frameshift was calculated as described in (Bhatt et al., 2021).
5.12. PRF sequencing
RNA was extracted from isolated clinical SARS-CoV-2 with E.Z.N.A total RNA (Omega Bio-Tek). Maxima H Minus cDNA Synthesis (Thermofisher) and Platinium II Taq (Thermofisher) were used as RT-PCR kits with designed primers (Fwd 5′-GCC ACA GTA CGT CTA CAA GC-3′, Rev 5′-GGC GTG GTT TGT ATG AAA TC-3′). PCR products were Sanger sequenced by Microsynth.
5.13. MucilAir antiviral assays
Tissues were obtained from Epithelix (Geneva, Switzerland). For all experiments, epithelia were prepared with different single donor's biopsies. Before inoculation with the viruses, MucilAir tissues were incubated in 250 μL of PBS Ca2+Mg2+ (PBS++) for 45 min at 37 °C. Infection was done with 106 RNA copies/tissue with B.1.1 SARS-CoV-2 or 105 RNA copies with SARS-CoV-2 BA.1 (omicron). At 4 h after incubation at 33 °C, tissues were rinsed three times with MucilAir medium to remove non-adsorbed virus and cultures were continued in the air-liquid interface. Every 24 h, 200 μL of MucilAir medium was applied to the apical face of the tissue for 20 min at 33 °C for sample collection, followed by apical treatment with geneticin (30 μg/tissue) starting at 24 hpi. Viral load was determined by qPCR as described previously. At the same time point, the basal medium was replaced with 500 μL of fresh MucilAir medium. At the end of the experiments, tissues were fixed and subjected to immunofluorescence.
5.14. Statistics and data analysis
Experiments were performed in duplicate and from two to four independent experiments as stated in the figure legends. Results are shown as mean and SEM. The EC50 and CC50 values for inhibition curves were calculated by regression analysis using the program GraphPad Prism version 9.1 to fit a variable slope sigmoidal dose-response curve as described in (Mathez and Cagno, 2021).One-way or Two-ways Anova followed by multiple comparison analysis was used as statistical tests to compare grouped analysis. Unpaired t-test was used to compare two different conditions. Area under the curve analysis followed by unpaired t-test or one-way ANOVA was done to compare curves.
5.15. Molecular modelling
All molecular modelling experiments were performed on Asus WS X299 PRO Intel® i9-10980XECPU @ 3.00 GHz × 36 running Ubuntu 18.04 (graphic card: GeForce RTX 2080 Ti). Molecular Operating Environment (MOE, 2019.10, Montreal, QC, Canada); Maestro (Schrödinger Release, 2021-1, New York, NY, USA); GROMACS (2020.4) (Abraham et al., 2015); Dock6 (Lang et al., 2009); Annapurna (Stefaniak and Bujnicki, 2021); RNAsite (Su et al., 2021a), were used as molecular modelling software. A library of commercially available RNA-targeting compounds was downloaded from Enamine and ChemDiv website.
5.16. Molecular dynamic simulations
MD simulations were performed with Gromacs software package. The ff99+bsc0+χOL3 force field was used for MD simulation since this is the most validated and recommended FFs for RNA system (Aytenfisu et al., 2017). The cryo-EM of the SARS-COV-2 FSE was download from PDB (http://www.rcsb.org/; PDB entry 6xrz). The structure was solvated with 14,0812 TIP4P-Ew waters and 87 Na + counterions to neutralise the charge on the RNA. All the molecular dynamics simulations were performed for 100ns on the isothermal-isobaric ensemble, using the stochastic velocity rescaling thermostat at 300 K and the Berendsen barostat with an isotropic pressure coupling of 1 bar. The RMSD, was used to verify the stability of the simulated systems during the MD simulation. The conformations obtained after 40 ns were extracted, and RMSD between all structures was used to perform a cluster analysis to group the different RNA conformations and to select a representative structure.
5.17. Binding site identification and molecular docking
The refined cryo-EM structure was prepared for further refinement with the Schrödinger Protein Preparation Wizard. Protonation states of RNA nucleotides were calculated considering a temperature of 300 K and a pH of 7.4, and restrained energy minimisation of the added hydrogens using the OPLS4 force field was performed. The Geneticin and the RNA-targeting compounds were prepared using the Maestro LigPrep tool by energy minimising the structures (OPLS4 force field), generating possible ionisation states at pH 7 ± 2 (Epik), tautomers and stereoisomers per each ligand. RNAsite was employed to identify a potential binding site using the refined structure(Su et al., 2021b). An 11 Å docking grid was prepared using as the centroid the predicted binding pocket previously identified by RNAsite. A Glide XP precision was employed to screen the compounds keeping the default parameters and setting 3 as the number of output poses per input ligand. The best-docked poses were then refined using MM-GBSA module. The docking poses obtained were then rescored using Annapurna and amber DOCK6 scoring functions. The values of the three different scoring functions for each docking pose were then analyzed together (consensus score) and only the Docking poses falling in the top 25% of the score value range in all the three scoring functions were selected for the final visual inspection. The visual inspection process, conducted as the last step of the structure-based virtual screening, was performed using MOE 2019.10. The 2D interaction plot was generated using Flare.
Funding
This work was supported by the Swiss National Science Foundation [PZ00P3_193,289 to V.C.] and the University Hospital of Geneva PRD financing to V.C and L.K., and by the Welsh Government Office for Science Sêr Cymru Tackling COVID-19 grant to C.V.. Funding for open access charge: Swiss National Science Foundation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
We thank the diagnostics of the Institute of Microbiology from the University Hospital of Lausanne for providing the clinical specimens of SARS-CoV-2. We thank also Dr. Kate Marie O'Connor for the plasmids of the dual luciferase assay.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.antiviral.2022.105452.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Raw data are available at 10.6084/m9.figshare.20338695.
References
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindah E. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. Software. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Ahn D.G., Lee W., Choi J.K., Kim S.J., Plant E.P., Almazán F., Taylor D.R., Enjuanes L., Oh J.W. Interference of ribosomal frameshifting by antisense peptide nucleic acids suppresses SARS coronavirus replication. Antivir. Res. 2011;91:1–10. doi: 10.1016/j.antiviral.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander V Birk1, Dubovi Edward J., Zhang Xianchao, Szeto Hazel H. Antiviral activity of geneticin against bovine viral diarrhoea virus. Antivir. Chem. Chemother. 2008;19:33–40. doi: 10.1177/095632020801900105. [DOI] [PubMed] [Google Scholar]
- Aradi K., di Giorgio A., Duca M. Aminoglycoside conjugation for RNA targeting: antimicrobials and beyond. Chem. Eur J. 2020 doi: 10.1002/chem.202002258. [DOI] [PubMed] [Google Scholar]
- Ariza-Mateos A., Díaz-Toledano R., Block T.M., Prieto-Vega S., Birk A., Gómez J. Geneticin stabilizes the open conformation of the 5′ region of hepatitis C virus RNA and inhibits viral replication. Antimicrob. Agents Chemother. 2016;60:925–935. doi: 10.1128/AAC.02511-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aytenfisu A.H., Spasic A., Grossfield A., Stern H.A., Mathews D.H. Revised RNA dihedral parameters for the amber force field improve RNA molecular dynamics. J. Chem. Theor. Comput. 2017;13:900–915. doi: 10.1021/acs.jctc.6b00870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt P.R., Scaiola A., Loughran G., Leibundgut M., Kratzel A., Meurs R., Dreos R., O'Connor K.M., McMillan A., Bode J.W., Thiel V., Gatfield D., Atkins J.F., Ban N. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science. 2021;372:1306–1313. doi: 10.1126/science.abf3546. 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissaro M., Sturlese M., Moro S. Exploring the RNA-recognition mechanism using supervised molecular dynamics (SuMD) simulations: toward a rational design for ribonucleic-targeting molecules? Front. Chem. 2020;8 doi: 10.3389/fchem.2020.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I., dos Ramos F.J. Programmed ribosomal frameshifting in HIV-1 and the SARS-CoV. Virus Res. 2006;119:29–42. doi: 10.1016/j.virusres.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10 doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J., Jimenez A. A new selective agent for eukaryotic cloning vectors. Am. J. Trop. Med. Hyg. 1980;29:1089–1092. doi: 10.4269/ajtmh.1980.29.1089. [DOI] [PubMed] [Google Scholar]
- de Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016 doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich D.C., Lee J.J., Link A.J., Graumann J., Tirrell D.A., Schuman E.M. Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat. Protoc. 2007;2:532–540. doi: 10.1038/nprot.2007.52. [DOI] [PubMed] [Google Scholar]
- Embarc-Buh A., Francisco-Velilla R., Martinez-Salas E. Rna-binding proteins at the host-pathogen interface targeting viral regulatory elements. Viruses 13. 2021 doi: 10.3390/v13060952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth A.E., Jagger B.W., Wise H.M., Nelson C.C., Parsawar K., Wills N.M., Napthine S., Taubenberger J.K., Digard P., Atkins J.F. Ribosomal frameshifting used in influenza A virus expression occurs within the sequence UCC-UUU-CGU and is in the +1 direction. Open Biol. 2012;2 doi: 10.1098/rsob.120109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S., Klein J., Robertson A.J., Peña-Hernández M.A., Lin M.J., Roychoudhury P., Lu P., Fournier J., Ferguson D., Mohamed Bakhash S.A.K., Catherine Muenker M., Srivathsan A., Wunder E.A., Kerantzas N., Wang W., Lindenbach B., Pyle A., Wilen C.B., Ogbuagu O., Greninger A.L., Iwasaki A., Schulz W.L., Ko A.I. De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: a case report. Nat. Commun. 2022;13 doi: 10.1038/s41467-022-29104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser L.R., Kelly M.L., Herschlag D., Al-Hashimi H.M. The roles of structural dynamics in the cellular functions of RNAs. Nat. Rev. Mol. Cell Biol. 2019 doi: 10.1038/s41580-019-0136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garreau De Loubresse N., Prokhorova I., Holtkamp W., Rodnina M.v., Yusupova G., Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014;513:517–522. doi: 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- Garrison E., Marth G. Haplotype-based variant detection from short-read sequencing. 2012. [DOI]
- Haniff H.S., Tong Y., Liu X., Chen J.L., Suresh B.M., Andrews R.J., Peterson J.M., O'Leary C.A., Benhamou R.I., Moss W.N., Disney M.D. Targeting the SARS-COV-2 RNA genome with small molecule binders and ribonuclease targeting chimera (RiboTAC) degraders. ACS Cent. Sci. 2020;6:1713–1721. doi: 10.1021/acscentsci.0c00984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Mösbauer K., Hofmann-Winkler H., Kaul A., Kleine-Weber H., Krüger N., Gassen N.C., Müller M.A., Drosten C., Pöhlmann S. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature. 2020;585:588–590. doi: 10.1038/s41586-020-2575-3. [DOI] [PubMed] [Google Scholar]
- Huston N.C., Wan H., Strine M.S., de Cesaris Araujo Tavares R., Wilen C.B., Pyle A.M. Comprehensive in vivo secondary structure of the SARS-CoV-2 genome reveals novel regulatory motifs and mechanisms. Mol. Cell. 2021;81:584–598. doi: 10.1016/j.molcel.2020.12.041. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iketani, S., Mohri, H., Culbertson, B., Jung Hong, S., Duan, Y., Luck, I., Annavajhala, M.K., Guo, Y., Sheng, Z., Uhlemann, A.-C., Goff, S.P., Sabo, Y., Yang, H., Chavez, A., Ho, D.D., n.d. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. 10.1101/2022.08.07.499047. [DOI] [PMC free article] [PubMed]
- Jacot D., Pillonel T., Greub G., Bertelli C. Assessment of SARS-CoV-2 genome sequencing: quality criteria and low-frequency variants. J. Clin. Microbiol. 2021;59 doi: 10.1128/JCM.00944-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X., Zhang J., Sun W., He W., Jiang H., Chen D., Murchie A.I.H. Riboswitch control of aminoglycoside antibiotic resistance. Cell. 2013;152:68–81. doi: 10.1016/j.cell.2012.12.019. [DOI] [PubMed] [Google Scholar]
- Jochmans , D., Liu, C., Donckers, K., Stoycheva, A., Boland, S., Stevens, S.K., de Vita, C., Vanmechelen, B., Maes, P., Trüeb, B., Ebert, N., Thiel, V., de Jonghe, S., Vangeel, L., Bardiot, D., Jekle, A., Blatt, L.M., Beigelman, L., Symons, J.A., Raboisson, P., Chaltin, P., Marchand, A., Neyts, J., Deval, J., Vandyck, K., n.d. The substitutions L50F, E166A and L167F in SARS-CoV-2 3CLpro are selected by a protease inhibitor in vitro and confer resistance to nirmatrelvir. 10.1101/2022.06.07.495116. [DOI] [PMC free article] [PubMed]
- Lang P.T., Brozell S.R., Mukherjee S., Pettersen E.F., Meng E.C., Thomas V., Rizzo R.C., Case D.A., James T.L., Kuntz I.D. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA. 2009;15:1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredonia I., Nithin C., Ponce-Salvatierra A., Ghosh P., Wirecki T.K., Marinus T., Ogando N.S., Snijder E.J., van Hemert M.J., Bujnicki J.M., Incarnato D. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Res. 2020;48:12436–12452. doi: 10.1093/nar/gkaa1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathez G., Cagno V. Clinical severe acute respiratory syndrome coronavirus 2 isolation and antiviral testing. Antivir. Chem. Chemother. 2021;29 doi: 10.1177/20402066211061063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGreevy R., Teo I., Singharoy A., Schulten K. Advances in the molecular dynamics flexible fitting method for cryo-EM modeling. Methods. 2016;100:50–60. doi: 10.1016/j.ymeth.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi S., Neupane K., Ileperuma S.M., Halma M.T.J., Kelly J.A., Halpern C.F., Dinman J.D., Loerch S., Woodside M.T. Identifying inhibitors of −1 programmed ribosomal frameshifting in a broad spectrum of coronaviruses. Viruses 14. 2022 doi: 10.3390/v14020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nierzwicki Ł., Palermo G. Molecular dynamics to predict cryo-EM: capturing transitions and short-lived conformational states of biomolecules. Front. Mol. Biosci. 2021;8 doi: 10.3389/fmolb.2021.641208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omar S.I., Zhao M., Sekar R.V., Moghadam S.A., Tuszynski J.A., Woodside M.T. Modeling the structure of the frameshift-stimulatory pseudoknot in SARS-CoV-2 reveals multiple possible conformers. PLoS Comput. Biol. 2021;17 doi: 10.1371/journal.pcbi.1008603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole Á., Scher E., Underwood A., Jackson B., Hill V., McCrone J.T., Colquhoun R., Ruis C., Abu-Dahab K., Taylor B., Yeats C., du Plessis L., Maloney D., Medd N., Attwood S.W., Aanensen D.M., Holmes E.C., Pybus O.G., Rambaut A. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021;7 doi: 10.1093/ve/veab064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.J., Kim Y.G., Park H.J. Identification of rna pseudoknot-binding ligand that inhibits the - 1 ribosomal frameshifting of SARS-coronavirus by structure-based virtual screening. J. Am. Chem. Soc. 2011;133:10094–10100. doi: 10.1021/ja1098325. [DOI] [PubMed] [Google Scholar]
- Parums D.v. Medical Science Monitor; 2022. Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non- Hospitalized Patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn W.D., Harrington H.R., Schlebach J.P., Mukhopadhyay S. 2020. Annual Review of Virology Regulators of Viral Frameshifting: More than RNA Influences Translation Events. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokhorova I., Altman R.B., Djumagulov M., Shrestha J.P., Urzhumtsev A., Ferguson A., Chang C.W.T., Yusupov M., Blanchard S.C., Yusupova G., Puglisi J.D. Aminoglycoside interactions and impacts on the eukaryotic ribosome. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E10899–E10908. doi: 10.1073/pnas.1715501114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangan R., Watkins A.M., Chacon J., Kretsch R., Kladwang W., Zheludev I.N., Townley J., Rynge M., Thain G., Das R. De novo 3D models of SARS-CoV-2 RNA elements from consensus experimental secondary structures. Nucleic Acids Res. 2021;49:3092–3108. doi: 10.1093/nar/gkab119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman C., Lewicka A., Koirala D., Li N.-S., Piccirilli J.A. The SARS-CoV-2 programmed −1 ribosomal frameshifting element crystal structure solved to 2.09 Å using chaperone-assisted RNA crystallography. ACS Chem. Biol. 2021 doi: 10.1021/acschembio.1c00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlick T., Zhu Q., Dey A., Jain S., Yan S., Laederach A. To knot or not to knot: multiple conformations of the SARS-CoV-2 frameshifting RNA element. J. Am. Chem. Soc. 2021;143:11404–11422. doi: 10.1021/jacs.1c03003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staple D.W., Venditti V., Niccolai N., Elson-Schwab L., Tor Y., Butcher S.E. Guanidinoneomycin B recognition of an HIV-1 RNA helix. Chembiochem. 2008;9:93–102. doi: 10.1002/cbic.200700251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefaniak F.4, Bujnicki J.M. AnnapuRNA: A scoring function for predicting RNA-small molecule binding poses. Plos computational biology. 2021 doi: 10.1371/journal.pcbi.1008309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H., Peng Z., Yang J. Recognition of small molecule–RNA binding sites using RNA sequence and structure. Bioinformatics. 2021;37:36–42. doi: 10.1093/bioinformatics/btaa1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Y., Abriola, L., Niederer, R.O., Pedersen, S.F., Alfajaro, M.M., Monteiro, S., Wilen, C.B., Ho, Y.-C., Gilbert, W. v, Surovtseva, Y. v, Lindenbach, B.D., Guo, J.U.. Restriction of SARS-CoV-2 replication by targeting programmed −1 ribosomal frameshifting. 10.1073/pnas.2023051118/-/DCSupplemental. [DOI] [PMC free article] [PubMed]
- Szemiel A.M., Merits A., Orton R.J., MacLean O.A., Pinto R.M., Wickenhagen A., Lieber G., Turnbull M.L., Wang S., Furnon W., Suarez N.M., Mair D., da Silva Filipe A., Willett B.J., Wilson S.J., Patel A.H., Thomson E.C., Palmarini M., Kohl A., Stewart M.E. In vitro selection of Remdesivir resistance suggests evolutionary predictability of SARS-CoV-2. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thi Nhu Thao T., Labroussaa F., Ebert N., V’kovski P., Stalder H., Portmann J., Kelly J., Steiner S., Holwerda M., Kratzel A., Gultom M., Schmied K., Laloli L., Hüsser L., Wider M., Pfaender S., Hirt D., Cippà V., Crespo-Pomar S., Schröder S., Muth D., Niemeyer D., Corman V.M., Müller M.A., Drosten C., Dijkman R., Jores J., Thiel V. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature. 2020;582:561–565. doi: 10.1038/s41586-020-2294-9. [DOI] [PubMed] [Google Scholar]
- Vicens Q., Westhof E. Crystal structure of geneticin bound to a bacterial 16 S ribosomal RNA A site oligonucleotide. J. Mol. Biol. 2003;326:1175–1188. doi: 10.1016/S0022-2836(02)01435-3. [DOI] [PubMed] [Google Scholar]
- Warner K.D., Hajdin C.E., Weeks K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018;17:547–558. doi: 10.1038/nrd.2018.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Zheludev I.N., Hagey R.J., Haslecker R., Hou Y.J., Kretsch R., Pintilie G.D., Rangan R., Kladwang W., Li S., Wu M.T.-P., Pham E.A., Bernardin-Souibgui C., Baric R.S., Sheahan T.P., D'Souza V., Glenn J.S., Chiu W., Das R. Cryo-EM and antisense targeting of the 28-kDa frameshift stimulation element from the SARS-CoV-2 RNA genome. Nat. Struct. Mol. Biol. 2021;28:747–754. doi: 10.1038/s41594-021-00653-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.G., Mason P.W., Dubovi E.J., Xu X., Bourne N., Renshaw R.W., Block T.M., Birk A.v. Antiviral activity of geneticin against dengue virus. Antivir. Res. 2009;83:21–27. doi: 10.1016/j.antiviral.2009.02.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are available at 10.6084/m9.figshare.20338695.