

SUMMARY
Genotype-phenotype associations for common diseases are often compounded by pleiotropy and metabolic state. Here we devised a pooled human organoid-panel of steatohepatitis to investigate the impact of metabolic status on genotype-phenotype association. En masse population-based phenotypic analysis under insulin insensitive conditions predicted key non-alcoholic steatohepatitis (NASH)-genetic factors including the glucokinase regulatory protein (GCKR)-rs1260326:C>T. Analysis of NASH clinical cohorts revealed that GCKR-rs1260326-T allele elevates disease severity only under diabetic state but protects from fibrosis under non-diabetic states. Transcriptomic, metabolomic and pharmacological analyses indicate significant mitochondrial dysfunction incurred by GCKR-rs1260326, which was not reversed with metformin. Uncoupling oxidative mechanisms mitigated mitochondrial dysfunction and permitted adaptation to increased fatty acid supply while protecting against oxidant stress, forming a basis for future therapeutic approaches for diabetic NASH. Thus, in-a-dish genotype-phenotype association strategies disentangled the opposing roles of metabolic-associated gene variant functions, and offer a rich mechanistic, diagnostic, and therapeutic inference toolbox toward precision hepatology.
Graphical Abstract
eTOC/In Brief:
Human organoid modeling combined with genotype-phenotype association studies disentangles the unique opposing roles of a steatohepatitis susceptible gene variant
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease affecting billions of people worldwide. The prevalence of NAFLD globally is 25.24%, with wide geographical variation, ranging from 6.3% to 33% depending on the population, ethnicity, lifestyle, and method of diagnosis (Bellentani et al., 2010; Everhart and Bambha, 2010; Rinella and Charlton, 2016; Vernon et al., 2011). Even in children, hepatic steatosis has become increasingly recognized, from 13–17% depending on age group, and, interestingly, without the increase of visceral adipose tissue for those less than the age of 9.8, indicating potential genetic contributors. Given that steatosis in childhood can lead to a higher incidence of mortality from non-alcoholic steatohepatitis (NASH), liver diseases, and malignancy (Paik et al., 2022), earlier prediction and detection of pathogenic NAFLD is a priority public health challenge. In adults, susceptibility to NAFLD/NASH is highly variable as not all individuals who are obese develop steatosis, and most cases of steatosis do not progress to chronic liver disease. Thus, there remains a critical need to better understand the susceptibility of vulnerable populations toward future preventive and therapeutic strategies (Tomita et al., 2017).
Although our understanding of the genetic underpinnings in many diseases has advanced (Hirschhorn and Daly, 2005), known risk variants explain only a modest fraction of heritability in common metabolic disorders such as NAFLD (Loomba et al., 2015). Genetic pleiotropy, when intersected with metabolic traits and disorders, further complicates the genetic interpretation of pathogenicity. The major comorbidity of NAFLD is Type 2 diabetes (T2D), a prevalent and rapidly growing metabolic disease. In the US, the co-prevalence of NAFLD and T2D ranges from 32% to 90%, depending on the age group (Younossi et al., 2020). Since NAFLD and T2D are often present in the same patients, the contributions of the many different genetic variants identified to date for each clinical phenotype remain to be established.
The glucokinase regulatory protein (GCKR) rs1260326:C>T SNP (single nucleotide polymorphism) is one such coding variant. GCKR is expressed almost exclusively in liver hepatocytes (Chambers et al., 2011; Speliotes et al., 2011) and is recognized to play a critical role in glucose utilization and lipogenesis (Nozaki et al., 2020). SNP rs1260326 association with NAFLD is widely debated (Anstee et al., 2020; Gao et al., 2019) partly due to its genetic pleiotropy (Yeh et al., 2022): e.g. protection from T2D, chronic kidney disease, with an increased risk of NAFLD, hypertriglyceridemia, hyperuricemia, gout, and metabolic syndrome (Zain et al., 2015) (Xia et al., 2019). In support, a recent unbiased deep-phenotype genome-wide association study (GWAS) identified the same GCKR variant as one of only a handful of genomic variants (MHC, ALDH2, ATXN2/SH2B3, ABO loci) that was associated with broader traits (28 and 20 traits in European and Japanese populations, respectively) including T2D (Sakaue et al., 2021). Hence, a complete understanding of the pleiotropic roles of candidate variants like GCKR-rs1260326 is essential for more insightful diagnosis and prognosis, particularly for highly heterogeneous metabolic diseases like NAFLD.
The utility of In-a-dish organ systems is being envisioned as models that allow genotype and phenotype association studies under a defined metabolic context in the absence of other major confounding factors. Recent GWAS-in-a-dish approaches, using metabolic cell types differentiated from induced pluripotent stem cells (iPSCs), have validated human gene expression variation such as gene expression quantitative trait loci (eQTL) (Kilpinen et al., 2017). Emerging organoid-based approaches can further increase the potential of this in-a-dish strategy as organoids emulate anatomical and physiological characteristics of in vivo organs in health and disease (Takebe and Wells, 2019). For example, human liver organoid cultures consist of apico-basolaterally polarized hepatocytes with bile canaliculi-like architecture and can maintain the directional bile acid excretion pathway for several weeks (Ramli et al., 2020; Shinozawa et al., 2021). This microarchitecture enabled the modeling of cholestatic damage induced by drugs and was purposed for a population-based study to delineate novel polygenic architectures of drug-Induced-Liver-Injury (DILI) (Koido et al., 2020). These studies highlight the potential of in-a-dish based genomics approach in predicting metabolic dysfunction associated with human phenotype at a targeted organ level.
We previously established organoid-based simple steatosis (Kimura et al., 2018) and steatohepatitis (Ouchi et al., 2019) models that contain essential stromal lineages involving stellate- and Kupffer-like cells. Herein, we devised a pooled human population organoid panel (PoP) for steatohepatitis genotype-phenotype association studies. Steatohepatitis-like organoids from multiple genotyped individuals could be phenotyped under insulin-insensitive conditions, enabling efficient evaluation of genetic association with diabetic NAFLD. Organoid-informed genetic and molecular mechanisms were integrated with extensive clinical data obtained during three randomized controlled trials of therapies for patients with advanced fibrosis due to NASH (Harrison et al., 2020; Loomba et al., 2021). This revealed that the pleiotropic effects of the GCKR-rs1260326:C>T variant (Shen et al., 2009),(Pollin et al., 2011) are clinically significant for concomitant NAFLD and T2D, with revelation from organoid models that the functional significance of the variant is dependent on the metabolic status and influences the inflammatory milieu. We propose that metabolically-resolved genetic and phenotypic assessments are critical to identifying appropriate biomarkers and tailoring interventional strategies.
RESULTS
A steatohepatitis human liver organoid panel informs known genotype-phenotype associations for non-alcoholic fatty liver disease (NAFLD).
Numerous common genomic SNP variants associated with NAFLD/NASH have been identified by GWAS, with the significance most evident for the PNPLA3-rs738409 SNP. However, for the vast majority of SNPs, the significance remains unclear. We recently reported that human liver organoids (HLOs) from induced pluripotent stem cells (iPSC) are effective for modeling inflammatory diseases such as DILI (Shinozawa et al., 2021) and NAFLD (Kimura et al., 2018; Ouchi et al., 2019). Here we posit that the HLOs could be used to screen for clinically relevant NAFLD/NASH phenotype-genotype associations in different metabolic states. To assess the genotype-phenotype correlation between known significant GWAS SNPs for steatosis, and lipid accumulation phenotypes in our steatohepatitis-like HLO (referred to as sHLO for “steatohepatitis-like HLO”), we evaluated an HLO panel of 24 donor iPSC lines of varying genotypes (Table S1). This number of lines was based on a recent Monte Carlo simulation that suggested for an event predicted to occur 1 in 10 patients, a cohort of 24 human iPSC lines would yield a 92% probability that the event will be identified (Fermini et al., 2018).
To expedite and facilitate comparative analyses of 24 donor HLOs, we improved our iPSC differentiation protocol to develop an en masse strategy (schematically depicted in Figure 1A). We discovered that dissociating iPSC-derived foregut progenitor clusters to single cells before embedding in Matrigel can generate single donor-derived HLO. The single foregut progenitor cells, furthermore, could be cryopreserved without affecting viability or differentiation capabilities. Together with improved culturing conditions, we took advantage of our clonal differentiation protocol to generate a population organoid panel or PoP by a mixed population of cryopreserved foregut progenitors from 24 donors. The multicellular composition of the pooled HLO was evaluated by scRNAseq (Figure 1A and S1A). Consistent with our previous publications (Ouchi et al., 2019; Shinozawa et al., 2021), our protocols enabled paralleled production of hepatocyte-like, macrophage-like, and stellate-like cells (Figure S1A, B).
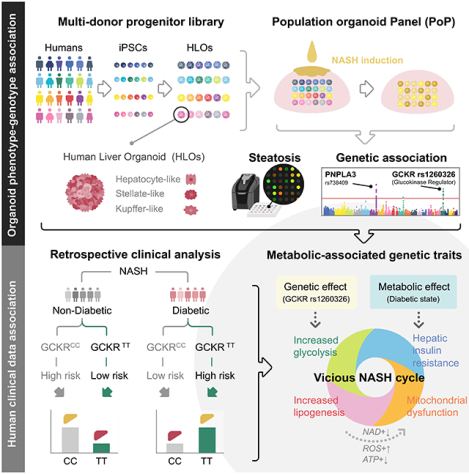
Figure 1. A steatohepatitis organoid panel informs known genotype-phenotype associations for hepatic steatosis.
(A) Schematic diagram of the developing genetically diverse population organoid panel (PoP). A mixture of cryopreserved foregut progenitors from multiple donors enabled parallel and clonal production of multicellular human liver organoids. Oleic acid (OA) treatment induces steatohepatitis phenotype. PoP based genotype-phenotype association analysis was performed en masse. (B) Transcriptome analysis of HLOs from 24 donors. (C) Graph shows the mean ± sem of hepatosteatosis index in each donor determined by fluorescent-guided PoP steatosis screening. SNP rs1260326 zygosity is as indicated. (D) SNP genotype profiles associated with NAFLD in the 24 donors of PoP. Dark green indicates 2 variant alleles, light green indicates 1 variant allele. (E) The odds ratios (ORs) for the 24-donor sHLO model, based on the fat accumulation phenotype. The OR was calculated for major NAFLD-related SNPs. Error bars represent 95% confidence intervals. (F) Comparison of diagnostic odds ratios in clinical trials to odds ratios of HLO models for PNPLA3-rs738409. The sample size (n) and minor allele frequency (MAF), as indicated. (G) Comparison of diagnostic odds ratios in clinical trials to odds ratios of HLO models for GCKR-rs1260326.
Derivation of pooled HLOs from single donor cells was confirmed in each organoid by (a) discernable identity based on unique SNP PCR genotyping of the donors (Figure S1C, D) and consistent morphology (Figure S1E); and (b) lack of organoid chimerism where >90% of HLOs carrying single donor-derived SNPs (Figure S1F). Further gene expression analysis of the differentiated pooled 24 FG progenitors showed relatively minimal donor-dependent variations of hepatic gene markers, which were distinct from iPSC and FG, but similar to primary hepatocytes (Figure 1B). Expression of the markers in the clonally-differentiated HLOs, moreover, was consistent with the individual donor-derived HLOs (Figure 1B). Although there were some variations amongst individual donor-derived HLO in the expression of genes encoding ATP-binding cassette transporters for drug clearance (BSEP = ABCB11; MRP3 = ABCC3), expression of the key hepatic markers, albumin (ALB), a member of cytochrome P450 superfamily CYP2C9, and tryptophan 2.3-dioxygenase TDO2, were comparable (Figure 1B). Thus, we developed the PoP as a viable strategy for enabling en masse generation of the genetically diverse human liver organoid panel.
To generate a NAFLD phenotype in the PoP HLOs, we cultured them with the common fatty acid, oleic acid (OA), which reproducibly induced steatohepatitis-like pathologies of lipid and metabolite accumulation (Figure S2A–G), and inflammation (Figure S2H, I) as previously reported (Ouchi et al., 2019). There was a marked difference in the amount of cholesterol and TG secreted into the culture supernatant (Figure S2F). Moreover, mass-spec analysis quantified an increase in lipogenesis and lipid oxidation markers in the sHLO including both acetyl-CoA (lipogenic precursor) and palmitate (saturated lipid), as well as mid- and long-chain acyl-carnitines, e.g., octanoyl-carnitine (C8 – medium chain) and palmitoyl-carnitine (C16–- long chain), indicating impaired lipid oxidation (Figure S2G). Enhanced inflammatory cytokine production consistent with a steatohepatitis-like phenotype was also detected in OA-treated HLOs (Figure S2H, S2I). Moreover, the OA-treated sHLO exhibited perturbed gluconeogenesis (Figure S2J, K) and impaired production of glucose response to insulin (Figure S2L, M), reminiscent of NASH patients who often show hepatic insulin resistance (Samuel and Shulman, 2019). Collectively, our OA-induced PoP model replicates insulin-insensitive NASH-like traits accompanied by increased lipogenesis, impaired fatty acid oxidation, and elevated inflammation.
With evidence supporting steatohepatitis-like phenotype in the sHLO, we next sought to leverage our en masse steatosis quantification strategy to determine whether the organoid-based genotype-phenotype association could predict common NAFLD-associated SNP genotypes. Live BODIPY imaging of lipid droplets staining in OA-treated PoP HLOs allowed us to quantitate a hepatosteatosis index, and distinguish lipid-low accumulating HLOs from lipid-high accumulators (Figure 1C). Each donor carried multiple SNPs (Figure 1D) with odds ratio (OR) analyses (combination of 2 and 1 alleles) showing strong correlations between HLO lipid accumulation phenotype and the most reported risk SNPs, specifically, the PNPLA3-rs738409 (Donati et al., 2016), and GCKR-rs780094 and -rs1260326 (Keebler et al., 2010) risk alleles (Figure 1E). Only GCKR-rs1260326 allelic status is shown in Figure 1C. The oRs were statistically insignificant for other reported SNPs in our HLO model system (Figure 1E) including the TM6SF2-rs58542926 risk alleles. Since our HLO sample size is small, it is possible that the very low allelic frequency of some of these known SNPs may not be readily captured (e.g., for TM6SF2-rs58542926, frequency T=0.065365, GnomAD exome).
It was notable that none of the 24 donors carried two alleles of the well-established PNPLA3-rs738409 risk SNP. Yet, the OR determined by heterozygosity was still statistically significant and comparable to those calculated from hundreds to thousands of patients in multiple clinical studies (Figure 1F). Interestingly, in our HLO model, the OR of GCKR-rs1260326 was as significant as PNPLA3-rs738409 (Figure 1E). Clinical significance of GCKR-rs1260326, in contrast, ranged from relatively modest to not significant and had calculated OR decidedly less significant than our HLO model (Figure 1G). These results indicate that our PoP model offers a human-based system to evaluate the pathophysiological significance of potential risk SNPs and, importantly, can inform steatotic genotype-phenotype correlations for NAFLD/NASH.
GCKR-rs1260326 TT genotype (p.Pro446Leu) increases susceptibility to de novo lipid accumulation in HLO
GCKR-rs1260326:C>T is a functional coding, SNP, c.1337C>T, in which the C to T substitution alters the proline at position 446 to leucine (p.Pro446Leu), while GCKR-rs780094, which is in strong linkage disequilibrium with rs1260326 (Santoro et al., 2012; Speliotes et al., 2011), is intronic and non-functional. In the liver, GCKR competes with glucose for binding to GCK and, upon binding, inactivates GCK, in part by retaining GCK in the nucleus (schematically depicted in Figure 2A). The GCKR-rs1260326 TT variant, which has a reduced ability to bind GCK (Beer et al., 2009), has been proposed to constitutively activate hepatic glucose uptake and glycolysis with the subsequent generation of excess acetyl-CoA, a rate-limiting substrate for lipogenesis (Beer et al., 2009).
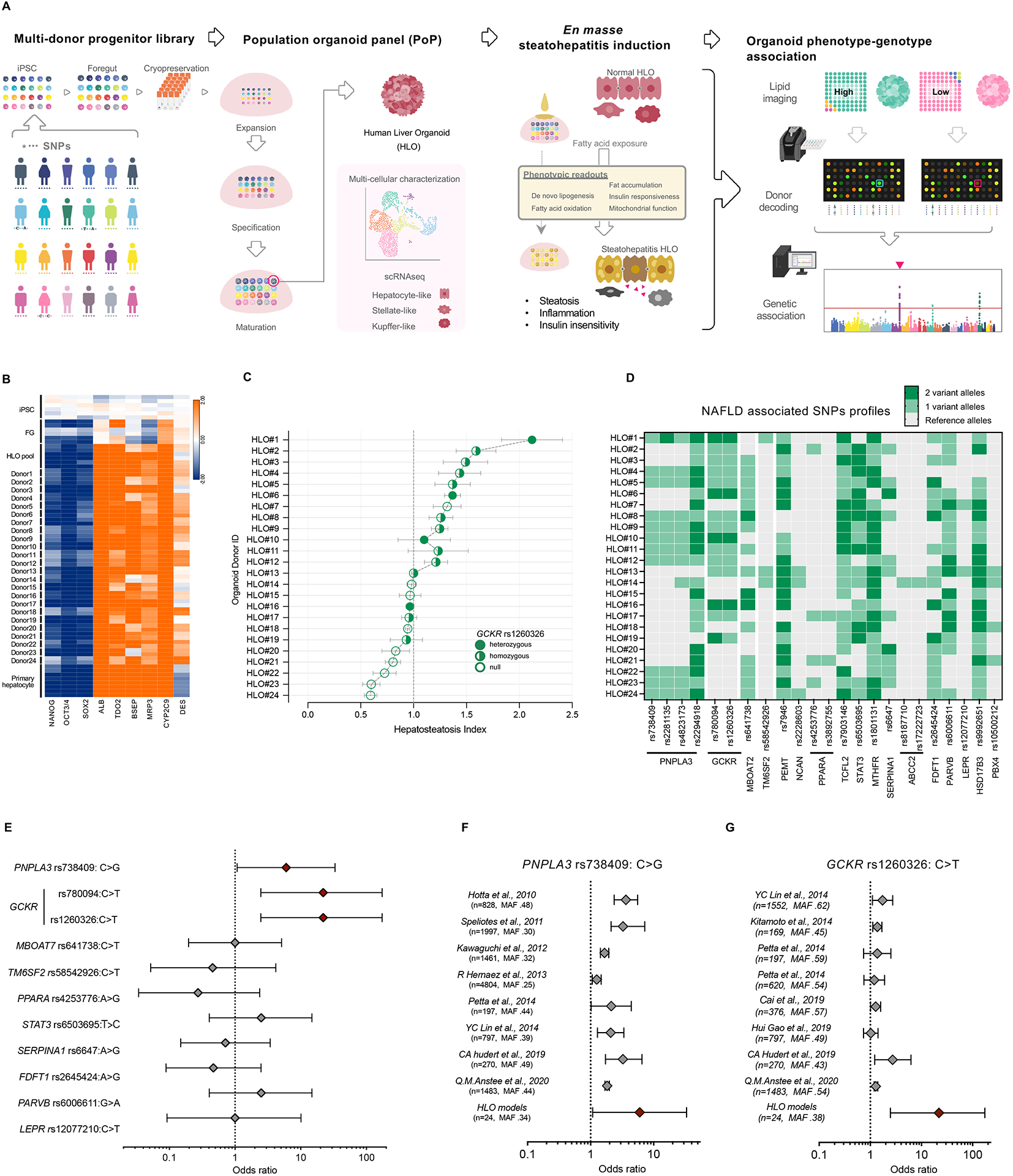
Figure 2. GCKR-rs1260326 TT genotype confers susceptibility to de novo lipid accumulation in HLO.
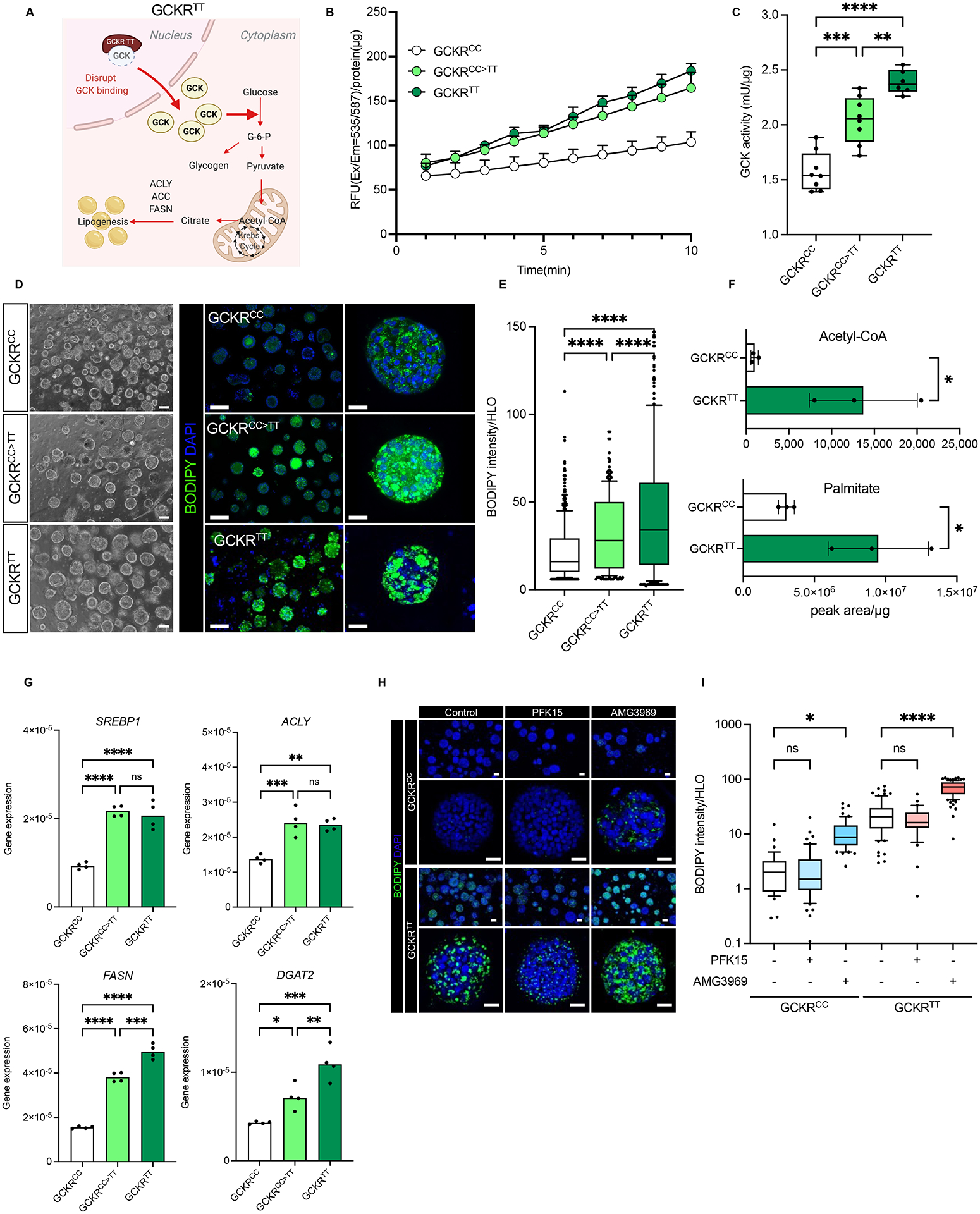
(A) Schematic diagram of GCKR variant association with glucokinase (GCK). GCKR functions as an inhibitor of GCK in the liver. The TT variant of GCKR-rs1260326 has a reduced ability to bind GCK and is less effective in suppressing GCK activities. (B) The time-course dynamics of GCK activity in HLOs carrying GCKRCC, GCKRCC>TT (gene-edited), GCKRTT. Data are shown as means ± SD (error bars), n=4. (C) Measurement of GCK activity in GCKRCC, GCKRCC>TT, and GCKRTT-HLOs. Data are shown as mean ± SD (Error bars), n=4, in triplicate. Unpaired t-test; **p < 0.01, ***p < 0.001, ****p < 0.0001. (D) Representative images of de novo lipid accumulation in GCKRCC, GCKRCC>TT, and GCKRTT-HLOs. Images were stained with BODIPY for fat accumulation (Green) and Hoechst 33342 for the nucleus (Blue). Scale bars, low magnification: 300μm, high magnification: 50μm. (E) Quantification of de novo lipid accumulation in GCKRCC, GCKRCC>TT, and GCKRTT-HLOs. The intensity of lipid was normalized to nuclear signals (mean ± SD, n = 8 independent experiments). Unpaired t-test; ****p < 0.0001. (F) Mass-spec analysis of protein content-normalized levels of acetyl-CoA and palmitate in HLOs. Data are shown as mean ± SD (Error bars), n=3, Unpaired t-test; *p < 0.05. (G) Comparison of lipogenesis-associated gene expression in GCKRCC, GCKRCC>TT, and GCKRTT-HLOs. Data are shown as means ± SD. (error bars), n=4–8. Unpaired t-test; *p < 0.05, **p < 0.01, ***p < 0.001, ***p < 0.0001. (H) Imaging of de novo lipid accumulation in GCKRCC and GCKRTT-HLOs, treated with PFKFB3 inhibitor (PFK15), and GCK-GCKR disruptor (AMG3969). Images were stained with BODIPY for fat accumulation and Hoechst 33342 for the nucleus. Scale bars, low magnification: 100μm, high magnification: 50μm. (I) Quantification of de novo lipid accumulation in GCKRCC and GCKRTT HLOs treated with PFK15 or AMG3969. The intensity of lipid was normalized to nuclear signals (mean ± SD, n = 8 independent experiments). Unpaired t-test; *p < 0.05, ****p < 0.0001.
To assess the pathogenesis of the GCKR-rs1260326 TT variant (hereafter referred to as GCKRTT) in our HLO model, we selected three GCKRTT donor iPSC lines for further analyses, comparing to four GCKR-rs1260326 CC variant (GCKRCC) iPSC lines. We also gene-edited several GCKRCC iPSC lines to GCKRTT (GCKRCC>TT) to specifically test the causative impact of this SNP in an isogenic background (Figure S3). Three GCKRCC>TT donor iPSC lines (#038 and #039 and #040) were selected for further analyses, and compared to their cognate GCKRCC donor iPSC lines (Table S1). GCK activity in the GCKRTT and GCKRCC>TT HLOs was consistently higher than in the isogenic GCKRCC HLOs (Figure 2B, C). We next evaluated whether the higher GCK activity enhanced de novo lipogenesis (DNL). Steady-state hepatic lipid metabolism under standard high glucose and high insulin culture conditions without exogenous fatty acid loading required prolonged culturing time, and thus, HLOs were analyzed on day 30. BODIPY imaging analysis showed that GCKRTT and GCKRCC>TT HLOs generated and accumulated significantly more lipid droplets compared to GCKRCC HLOs (Figure 2D, E). The fact that GCKRTT HLO has more lipid than GCKRCC>TT indicates the contribution of other disease modifiers. Mass-spec analysis further showed that acetyl-CoA and palmitate were significantly increased in GCKRTT HLOs (Figure 2F), which correlated with the enhanced expression of lipogenesis genes including Sterol Regulatory Element-Binding Protein 1 (SREBP1), ATP citrate lyase (ACLY), Fatty Acid Synthase (FASN), and Diacylglycerol O-Acyltransferase 2 (DGAT2) (Figure 2G). Thus, the GCKR-rs1260326 TT variant causatively facilitated GCK enzyme activation, lipid droplet formation, and unsaturated long-chain fatty acids accumulation, concordant with increased expression of genes associated with de novo lipogenesis due to excess acetyl-CoA.
To further evaluate the pathogenesis of dysfunctional GCK-GCKR binding, we treated HLOs with PFK15, a 6-phosphofructo-2-kinase (PFKB3) inhibitor, and AMG3969, which disrupts GCK-GCKR binding, and evaluated the effects on de novo fat accumulation (Figure 2H and I). PFK15 should increase Fructose 6-phosphate (F6P) and consequently suppresses GCK activity and inhibits hepatic glycolysis (Beer et al., 2009), while AMG3969 treatment should result in the release and migration of GCK into the cytoplasm from the nucleus, independent of glucose availability, thus enhancing glycolysis and lipogenesis (Lloyd et al., 2013). As predicted, inhibition of GCK activity prevented inhibited lipid accumulation, although this was more obvious with GCKRTT HLOs, which had higher basal lipid profiles (Figure 2I). Inhibition of GCK-GCKR complex formation by AMG3969, on the contrary, significantly enhanced lipid droplet accumulation in both GCKRCC and GCKRTT HLOs (Figure 2H and I). Notably, the maximum lipid droplets detected in GCKRCC HLO were consistently below untreated GCKRTT HLO (Figure 2I). Our results suggest that, in HLO models, the GCKRTT variant, independent of PNPLA3-rs738409, is functionally associated with de novo fat accumulation phenotype in the absence of exogenous fat-induced lipogenesis.
GCKR-rs1260326 TT allele enhanced Lobular Inflammation in NAFLD patients with type 2 diabetes
The discrepant metabolic impact of the GCKRTT variant between our sHLO model and highly variable clinical reports raised the question of whether other co-morbid metabolic traits, such as the onset of diabetes-like symptoms including insulin resistance, should be taken into consideration when evaluating the clinical impacts of GCKR-rs1260326 alleles. Indeed, the cultured conditions (high glucose and high insulin) for our HLO models are most consistent with NAFLD/NASH and T2D phenotypes and explain the blunted responses to insulin under steatotic conditions, independent of risk variant status (Figure S2).
For in vivo assessments, we performed a retrospective analysis on a cohort of 1091 adults diagnosed with biopsy-proven NASH (STELLAR-3 trial, NCT03053050, and ATLAS-trial, NCT03449446) for which demographic, biomarkers, and liver histology of clinical samples were available (Table S2). Demographics were predominantly Caucasian, middle-aged, females with high BMI in obese range. Correlation analyses between NAFLD clinical parameters and genetic risk variants in the cohort focused on the GCKR-rs1260326 variant compared to the most prevalent risk variants in PNPLA3, MBOAT7, and TM6SF2 (Table 1, Figure 3A). Measurements include accepted markers of liver injury (ALT, alanine aminotransferase), histological grading of NAFLD/NASH pathology (NAFLD Activity Score, NAS; Lobular Inflammation, LI; steatosis, activity, fibrosis, SAF score, for ballooning and LI). The well-established pathogenic PNPLA3-rs738409 GG risk variant (c.444C>G, p.I148M), as expected, is associated with higher ALT levels (although still within normal ranges) when compared to the reference CC variant (Table 1) but were comparable between reference and risk variants in GCKR, MBOAT7 and TM6SF2. All variants’ histological analyses were indistinguishable and within pathological ranges (Table 1).
Table 1. Genotype-phenotype association in a cohort of 1089 NASH patients.
The dataset included 1089 adults from the STELLAR-3 (NCT03053050) and ATLAS (NCT03449446) trials who were diagnosed with NAFLD and characterized demographics, biomarkers, and liver histology of clinical samples. Demographics were predominantly Caucasian, middle-aged, females with high BMI in obese range. Hemoglobin A1C values were not available for two samples out of a total of 1091 sample information. P values were analyzed for reference allele and alternate allele.
| Genotype | ALT (U/L) | P value | NAS (0–10) | P value | Lobular Inflam. (0–3) | P value | SAF score (1–4) | P value | ||
|---|---|---|---|---|---|---|---|---|---|---|
| PNPLA3 | CC(284) | 26.10% | 51.59 | 5.31 | 2.42 | 3.72 | ||||
| rs738409 | CG(473) | 43.40% | 54.82 | 5.3 | 2.44 | 3.7 | ||||
| (n=1089) | GG(332) | 30.50% | 60.85 | 8.0E–04** | 5.39 | 2.40E–01 | 2.5 | 1.60E–01 | 3.79 | 9.70E–02 |
| HbA1c (%) | ||||||||||
| Normal | CC(51) | 20.30% | 51.74 | 5.25 | 2.33 | 3.73 | ||||
| <5.7(n=251) | CG(103) | 41.00% | 51.7 | 5.17 | 2.36 | 3.57 | ||||
| GG(97) | 38.60% | 57.04 | 3.90E–01 | 5.3 | 7.70E–01 | 2.43 | 3.50E–01 | 3.74 | 8.40E–01 | |
| Pre-diabetic | CC(69) | 24.90% | 49.04 | 5.39 | 2.57 | 3.71 | ||||
| 5.7–6.4(n=277) | CG(127) | 45.80% | 56.19 | 5.2 | 2.36 | 3.69 | ||||
| GG(81) | 29.20% | 61.32 | 2.3E–02* | 5.36 | 8.20E–01 | 2.53 | 7.30E–01 | 3.72 | 9.40E–01 | |
| Diabetic | CC(164) | 29.20% | 52.68 | 5.3 | 2.4 | 3.73 | ||||
| >6.4(n=561) | CG(243) | 43.30% | 55.51 | 5.42 | 2.53 | 3.77 | ||||
| GG(154) | 27.50% | 63 | 6.7E–03** | 5.47 | 7.10E–02 | 2.52 | 9.50E–02 | 3.86 | 1.7E–02* | |
| GCKR | CC(324) | 29.80% | 55.97 | 5.28 | 2.42 | 3.72 | ||||
| rs1260326 | CT(500) | 45.90% | 56.55 | 5.37 | 2.48 | 3.73 | ||||
| (n=1089) | TT(265) | 24.30% | 54.21 | 5.10E–01 | 5.33 | 4.40E–01 | 2.45 | 6.50E–01 | 3.76 | 4.40E–01 |
| HbA1c (%) | ||||||||||
| Normal | CC(64) | 25.50% | 59.7 | 5.5 | 2.55 | 3.81 | ||||
| <5.7(n=251) | CT(126) | 50.20% | 53.8 | 5.21 | 2.37 | 3.63 | ||||
| TT(61) | 24.30% | 47.49 | 2.9E–02* | 5.03 | 4.5E–03** | 2.23 | 6.6E–03** | 3.61 | 3.6E–02* | |
| Pre-diabetic | CC(90) | 32.50% | 53.94 | 5.29 | 2.49 | 3.69 | ||||
| 5.7–6.4(n=277) | CT(118) | 42.60% | 57.46 | 5.3 | 2.47 | 3.7 | ||||
| TT(69) | 24.90% | 55.83 | 7.20E–01 | 5.3 | 9.10E–01 | 2.42 | 5.10E–01 | 3.72 | 6.70E–01 | |
| Diabetic | CC(170) | 30.30% | 55.64 | 5.19 | 2.34 | 3.71 | ||||
| >6.4(n=561) | CT(256) | 45.60% | 57.57 | 5.49 | 2.54 | 3.8 | ||||
| TT(135) | 24.10% | 56.53 | 8.10E–01 | 5.49 | 2.5E–03** | 2.56 | 3.3E–03** | 3.84 | 2.7E–02* | |
| MBOAT7 | CC(381) | 35.00% | 57.37 | 5.36 | 2.48 | 3.73 | ||||
| rs641738 | CT(509) | 46.70% | 54.69 | 5.34 | 2.46 | 3.74 | ||||
| (n=1089) | TT(199) | 18.30% | 55.7 | 5.80E–01 | 5.26 | 1.80E–01 | 2.4 | 1.40E–01 | 3.74 | 8.90E–01 |
| HbA1c (%) | ||||||||||
| Normal | CC(82) | 32.70% | 52.73 | 5.34 | 2.41 | 3.76 | ||||
| <5.7(n=251) | CT(121) | 48.20% | 52.47 | 5.21 | 2.4 | 3.61 | ||||
| TT(48) | 19.10% | 58.82 | 3.20E–01 | 5.15 | 2.30E–01 | 2.29 | 3.00E–01 | 3.67 | 3.40E–01 | |
| Pre-diabetic | CC(98) | 35.40% | 63.14 | 5.38 | 2.48 | 3.69 | ||||
| 5.7–6.4(n=277) | CT(125) | 45.10% | 53.16 | 5.32 | 2.5 | 3.73 | ||||
| TT(54) | 19.50% | 49.16 | 1.5E–02* | 5.09 | 6.60E–02 | 2.35 | 2.50E–01 | 3.67 | 7.70E–01 | |
| Diabetic | CC(201) | 35.80% | 56.45 | 5.36 | 2.51 | 3.74 | ||||
| >6.4(n=561) | CT(263) | 46.90% | 56.57 | 5.43 | 2.48 | 3.8 | ||||
| TT(97) | 17.30% | 57.79 | 7.50E–01 | 5.4 | 6.70E–01 | 2.47 | 6.70E–01 | 3.81 | 2.20E–01 | |
| TM6SF2 | CC(842) | 77.30% | 56.15 | 5.37 | 2.48 | 3.76 | ||||
| rs58542926 | CT(229) | 21.00% | 55.72 | 5.23 | 2.37 | 3.68 | ||||
| (n=1089) | TT(18) | 1.70% | 41.19 | 6.50E–02 | 5.06 | 1.40E–01 | 2.44 | 8.30E–01 | 3.39 | 2.2E–03** |
| HbA1c (%) | ||||||||||
| Normal | CC(191) | 76.10% | 41.9 | 5.2 | 2.37 | 3.64 | ||||
| <5.7(n=251) | CT(53) | 21.10% | 57.15 | 5.4 | 2.42 | 3.79 | ||||
| TT(7) | 2.80% | 53.27 | 3.50E–01 | 5.14 | 8.80E–01 | 2.43 | 8.30E–01 | 3.43 | 3.40E–01 | |
| Pre-diabetic | CC(214) | 77.30% | 37.5 | 5.33 | 2.47 | 3.73 | ||||
| 5.7–6.4(n=277) | CT(61) | 22.00% | 55.94 | 5.2 | 2.43 | 3.62 | ||||
| TT(2) | 0.70% | 56.07 | 4.60E–01 | 4.5 | 1.80E–01 | 2.5 | 9.50E–01 | 3 | 4.1E–02* | |
| Diabetic | CC437) | 77.90% | 41.44 | 5.46 | 2.53 | 3.82 | ||||
| >6.4(n=561) | CT(115) | 20.50% | 55.14 | 5.18 | 2.33 | 3.68 | ||||
| TT(9) | 1.60% | 57.47 | 1.70E–01 | 5.11 | 2.10E–01 | 2.44 | 6.90E–01 | 3.44 | 1.5E–02* |
Hemoglobin A1C values were not available for only two samples out of a total of 1091 sample information.
P values were analyzed for reference allele and alternate allele.
P<.05 or <.001 are noted as * or **.
Figure 3. GCKR-rs1260326 TT genotype confers inverse risk for inflammation dependent on HbA1c levels.
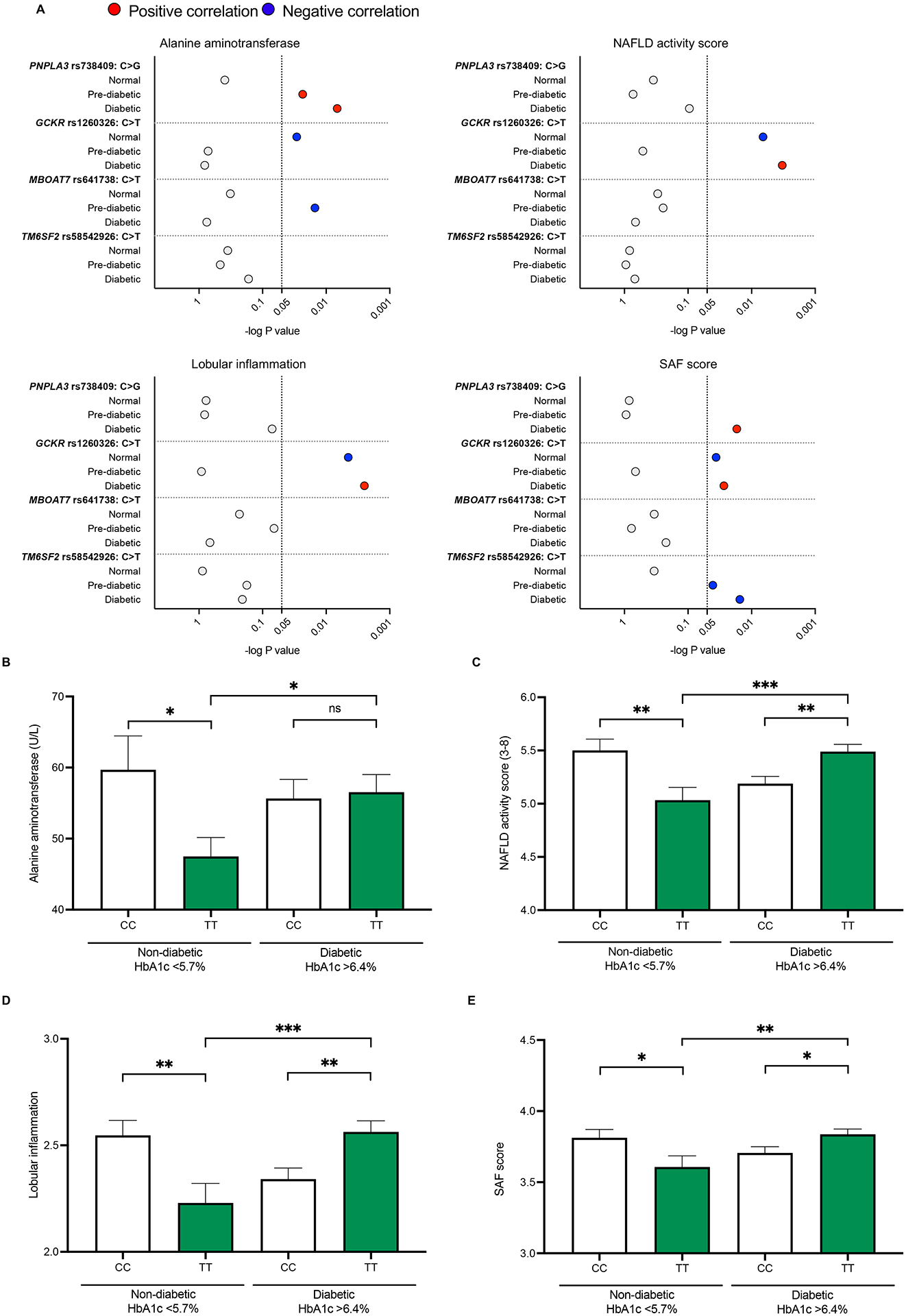
Impact of HbA1c values (normal, <5.7%, versus diabetic, >6.4%) on clinical measurements (see Table 1), graphically depicted. (A) Red and blue dots indicate significant differences. Coloring shows NAFLD exacerbating (red), and protective (blue) associations. The scale of the x-axis corresponds to the P value in the log10 scale for each SNP genotype. The dashed vertical line indicates p= 0.05. (B-E) NAFLD phenotype associated with GCKR-rs1260326 genotype: (B) ALT measurements; (C) NAFLD activity score (NAS); (D) Lobular inflammation scores; and (E) SAF score.
It is of note that fasting glucose (normal: <99 mg/dL; prediabetes: 100 – 125 mg/dL; diabetes: >126 mg/dL) and fasting insulin levels (normal: <25 uIU/ml) were generally above normal ranges, irrespective of variants, consistent with indications of insulin resistance (Table S2). We, therefore, performed additional association analysis with hemoglobin A1c (HbA1c) data available for 1089 of the 1091 subjects (Table 1). HbA1c is a common measurement in T2D diagnosis. HbA1c measures glycated hemoglobin and, unlike fasting glucose, reflects a weighted average of blood glucose levels in the preceding 2–4 months. Specifically, HbA1c cutoff values recommended for adults by the CDC (Center for Disease Control), USA, were used, where below 5.7% is within normal ranges; 5.7%–6.4%, pre-diabetic; and >6.4%, diabetic. Analyses showed that when HbA1c values were in the diabetic >6.4% ranges for PNPLA3-rs728409, the GG risk variant (compared to reference CC) was significantly associated with increased ALT, increased SAF score, but histological NAS and Lobular inflammation (LI) were not significantly different (Table 1, Figure 3A).
In patients carrying the GCKRTT variant, ALT, NAS, LI and SAF scores were significantly better (i.e., lower scoring) than reference GCKRCC when HbA1c values were within normal <5.7% ranges (Table 1, Figure 3A–E). In stark contrast, when HbA1c values were in the diabetic >6.4% range, scoring was higher (i.e., indicative of worsened pathology) in GCKRTT versus GCKRCC cohorts (Figure 3A–E). This worsening of NAFLD/NASH inflammatory pathology was most pronounced within the GCKRTT cohort, when comparing the non-diabetic HbA1c <5.7% to the diabetic HbA1c >6.4% sub-cohorts (Figure 3B–E). For the GCKRCC variant, diabetic HbA1c >6.4% conditions did not worsen inflammatory pathology but trended towards improved (lower) scores (Figure 3B–E). In sum, based on differential HbA1c values, the GCKRTT variant appears to confer unexpected inverse risks for inflammatory pathology, trends not observed with the other genetic risk variants evaluated (Table 1, Figure 3A), such as, for example, the PNPLA3-rs728409 GG risk variant (Figure S4A–D). Thus, for patients carrying the GCKRTT variant, HbA1c measurements may have prognostic value for delineating the severity of NAFLD-associated inflammatory pathology.
Mitochondrial dysregulation is associated with GCKRTT related metabolic assaults
To assess GCKRTT cellular impacts, we performed an unbiased transcriptomic analysis of available RNA-seq datasets from genotyped patient hepatocyte samples (Table S3, S4). In GCKRTT NASH hepatocytes compared to hepatocytes carrying GCKRCC or CT, differential expression gene (DEG) analyses by edgeR (FC >1.50, FDR <0.05) revealed upregulated genes including lipogenic genes (Figure 4A). This is remarkably consistent with OA-induced sHLO models comparing GCKRCC and TT (Figure S5A–E; Table S4). Intriguingly, down-regulated genes included multiple subunits of the mitochondrial ATP synthase (Figure 4A), a membrane multimeric complex which utilize the electrochemical proton gradient during oxidative phosphorylation to catalyze ATP synthesis from ADP. The dysregulation of mitochondrial gene sets was also identified by GSEA (gene set enrichment analysis)-REACTOME analysis (Figure 4B, C). While upregulated pathways included inflammatory gene sets in our sHLO models (Figure S5F– H; Table S4), several mitochondrial-related pathways were downregulated with NES values amongst the top downregulated REACTOME pathways (Figure 4B). When comparing these GCKRTT NASH hepatocyte samples to GCKRTT HLO models, a similar subset of GSEA-REACTOME pathways was significantly downregulated (Figure 4C), most prominent of which were the mitochondrial-related REACTOME pathways (Figure 4D). The downregulation of respiratory electron transport ATP synthesis pathways was also supported by Enrichment Plot analyses demonstrating that these metabolic gene sets were significantly different between GCKRTT and GCKRCC with p<0.001, in both NASH hepatocytes from patients and the HLO models (Figure 4E).
Figure 4. Comparative clinical and organoid transcriptomic signatures associated with GCKR-rs1260326.
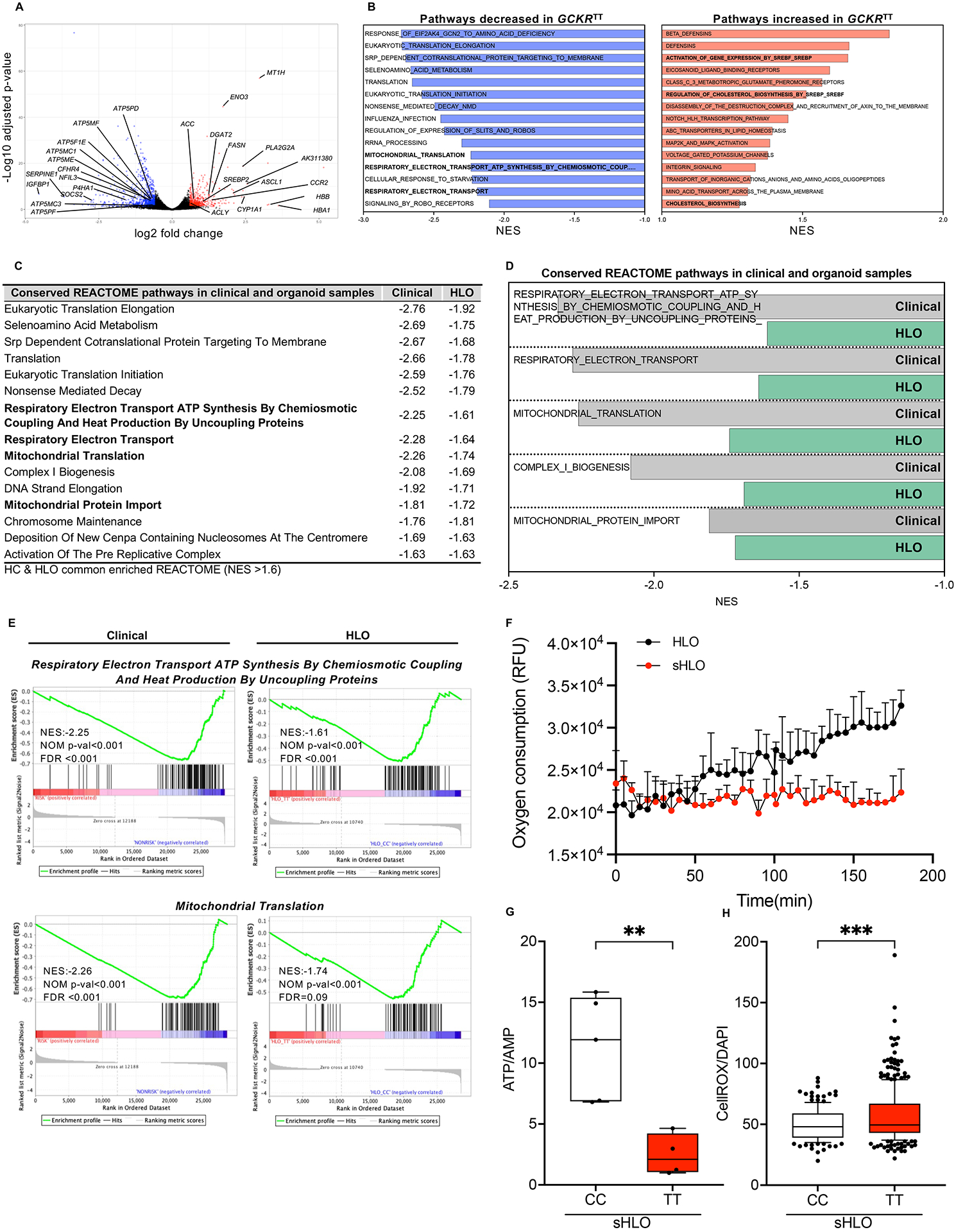
(A) Volcano plot of differentially expressed gene (DEGs) analysis (edge R) in primary NASH hepatocytes comparing GCKRTT to CC variants. Fold change >1.5, P-value <0.05. (B) Unbiased gene set enrichment analysis (GSEA). REACTOME pathways up-regulated and down-regulated in primary NASH hepatocytes, GCKRTT relative to GCKRCC. Normalized enrichment scores (NES) are presented in descending order. (C) Conserved GSEA-REACTOME pathways in primary NASH hepatocytes (clinical samples) and HLOs (GCKRTT relative to GCKRCC). NES less than −1.6 are shown. (D) Conserved GSEA-REACTOME mitochondrial-related pathways in clinical (primary NASH hepatocytes) and HLO models. (E) Enrichment plots of selected gene-expression profiles based on GSEA-REACTOME evaluations. (F) Oxygen consumption rate (OCR) analysis (Extracellular Oxygen Consumption Assay, a fluorescence-based assay) of GCKRTT-HLO and -sHLO. Data are shown as means ± SD. (error bars), n=3. (G) The ratio of ATP/AMP of GCKRTT and GCKRCC-sHLO was analyzed by NMR (nuclear magnetic resonance) spectroscopy. Data are shown as means ± SD (error bars), n for GCKRCC-HLO = 5, n for GCKRTT-HLO =4 donors. Unpaired t-test; **p < 0.01. (H) Quantifications of reactive oxidant species (ROS) production in GCKRTT-sHLOs versus GCKRCC-sHLOs. ROS production was detected with CellROX live staining and Hoechst 33342 for the nucleus. The intensity of ROS was normalized to nuclear signals. Analysis was performed in >50 organoids per line, three independent experiments. Unpaired t-test; ***p < 0.001.
Given the transcriptomic indications of mitochondrial dysregulation associated with GCKRTT variant compared to GCKRCC, and the correlation between clinical GCKRTT NASH hepatocytes and our HLO models, we sought to verify if the mitochondrial function was perturbed in our HLO models. Typically, mitochondrial aerobic respiration relies on electron transfer and a proton gradient to drive ATP production, with ROS (reactive oxygen species) as natural, tightly controlled by-products. We hypothesize that in our HLO systems, mitochondrial dysregulation was a consequence of enhanced oxidative stress created by chronic ROS production, in GCKRTT variant cells, which was exacerbated by fatty acid accumulation. We showed that oxygen consumption rates (OCR), determined by a fluorescence-based assay, were significantly compromised in GCKRTT sHLO compared to GCKRTT HLO (Figure 4F). Further, ATP/AMP ratios determined by intracellular metabolite profiling were dramatically reduced in GCKRTT sHLO compared to GCKRCC sHLO (Figure 4G). These results were consistent with enhanced ROS, quantified by live cell staining, in GCKRTT sHLO compared to GCKRCC sHLO (Figure 4H).
To evaluate the effect on mitophagy in GCKRTT sHLO by quantifying intracellular Mitophagy Dye, sHLO exhibited similar fluorescence intensity to controls (Figure S6A). Flow cytometry indicated that the relative protein levels of mitophagy-related proteins parkin (PRKN) and mitofusin 2 (MFN2) were increased in sHLO although only the increase in MFN2 was statistically significant (Figure S6B). We also evaluated mtDNA copy number to assess the possibility of mitochondrial disposal by mitophagy in steatosis condition. Transmission electron microscopy and mitochondrial DNA copy number analyses confirmed the increased presence of mitochondria in OA-treated sHLOs (Figure S6C, D). Altogether, our HLO and sHLO models revealed mitochondrial dysregulation rather than reduction, driven by genetic GCKRTT risk factors and exogenous fatty acid perturbations.
NR and NTZ, but not metformin, mitigates mitochondrial dysfunctions of GCKRTT sHLO and reduce inflammatory gene expression
Metformin, the first line of medication used to treat T2D associated with obesity, has been shown to improve mitochondrial respiratory activities via the AMPK pathway in mouse models (Wang et al., 2019). However, patients carrying GCKRTT did not show improvement in multiple phenotypic measurements after 48 weeks of metformin treatment, in contrast to patients carrying GCKRCC or CT (Table S5, Figure S4E). Hence, we infer that in vivo, metabolic dysfunction associated with GCKRTT, including potential mitochondrial dysregulation, is unlikely to improve with metformin treatment. To determine if our HLO model reflected these in vivo observations, we evaluated the effects of metformin treatment in sHLO and assessed whether controlling elevated ROS in GCKRTT sHLO can better modulate the undesirable effects of excessive fatty acid accumulation.
Among the mechanisms to control the unwanted generation of ROS, we focused on uncoupling the oxidative phosphorylation process. Specifically, GCKRTT sHLO were supplemented with an NAD+ precursor, nicotinamide riboside (NR), in combination with nitazoxanide (NTZ), an FDA-approved anti-parasitic and anti-viral drug recently shown to possess mitochondrial uncoupling, and respiration-enhancing, activities (Amireddy et al., 2017; Sahdeo et al., 2014). We first demonstrated the effects of NR and NTZ in our GCKRTT HLO systems. GCKRTT sHLO exposed to fatty acids dramatically reduced OCR, which NR and NTZ co-treatment reversed (Figure 5A). This restoration of energy consumption in GCKRTT sHLO to comparable levels detected in GCKRTT HLO was due to the synergistic effects of NR and NTZ in increasing NAD+ availability (Figure 5B). NAD+, the metabolic co-factor involved in redox reactions, is known to protect hepatocytes from the harmful effects of ROS (Guarino and Dufour, 2019).
Figure 5. Mitochondrial dysregulation is associated with GCKR-rs1260326 related metabolic assaults.
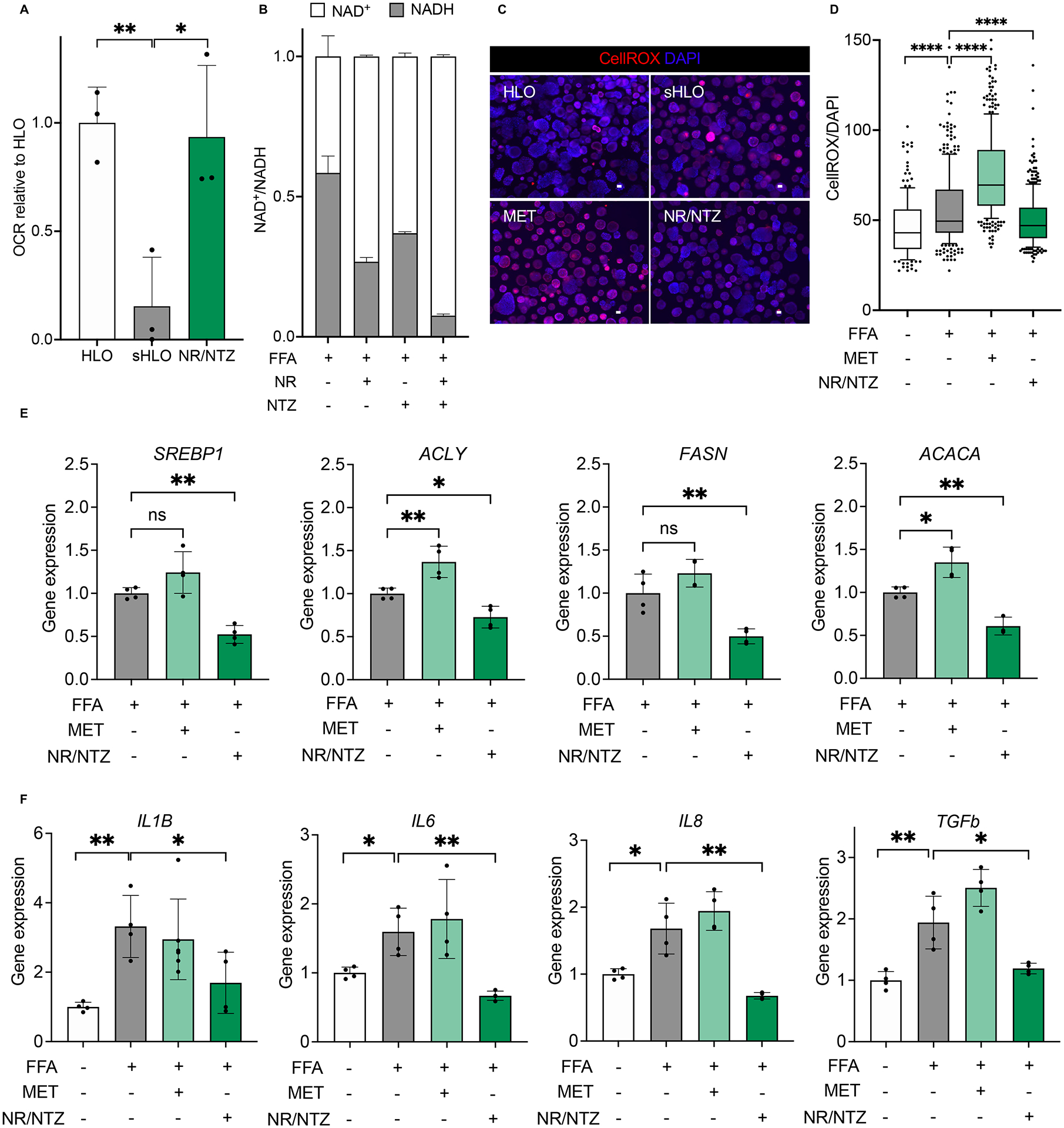
(A) Oxygen consumption rate (OCR) analysis of GCKRTT-HLO (white), -sHLO (gray), and -sHLO treated with nicotinamide riboside (NR), nitazoxanide (NTZ) combination (green). Data are shown as means ± SD (error bars), n=3, Unpaired t-test; *p < 0.05, **p < 0.01. (B) NAD+/NADH ratios in GCKRTT-sHLO treated with NR, NTZ, or combination. (C) Representative images of ROS production in GCKRTT-HLO, -sHLO (FFA treated), and -sHLO treated with metformin (MET) or a combination of NR/NTZ. Images were stained with CellROX for ROS and Hoechst 33342 for the nucleus. Scale bars, 300μm. (D) Quantifications of ROS production in GCKRTT-HLO, -sHLO (FFA treated), and -sHLO treated with metformin (MET) or a combination of NR/NTZ. ROS production was detected with CellROX live staining and Hoechst 33342 for the nucleus. The intensity of ROS was normalized to nuclear signals. Analysis was performed in >50 organoids per line, three independent experiments. Unpaired t-test; ****p < 0.0001. (E) Lipogenic gene expression in GCKRTT-sHLO (FFA treated) and -sHLO treated with metformin (MET) or NR/NTZ combination were compared to GCKRTT-HLO. Data are shown as means ± SD normalized by internal standard 18S (error bars), n=4. Unpaired t-test; *p<0.05, **p<0.01. (F) Relative gene expressions of proinflammatory cytokine in GCKRTT-sHLO (FFA treated) and -sHLO treated with metformin (MET) or NR/NTZ combination, compared to GCKRTT-HLO, which was arbitrarily assigned a value of 1. Data are shown as means ± SD. (error bars), n=4. Unpaired t-test; *p < 0.05, **p < 0.01.
We confirmed that ROS production was significantly higher in GCKRTT sHLO than in GCKRTT HLO (Figure 5C, D). In contrast, metformin treatment did not reduce ROS but actually increased ROS, which may exacerbate the disease phenotype (Figure 5D). In contrast to metformin, co-treatment with NR and NTZ significantly decreased cellular ROS (Figure 5D) without affecting mtDNA copy number (Figure S6D). To investigate the effect of NR/NTZ on HLOs in the absence of FFAs, cotreatment of NR and NTZ was performed. The results showed that HLOs without FFAs reduced mitochondrial ROS production (Figure S6E). We also observed a concomitant decreased DNL gene expression (Figure 5E) and suppression of fatty acid-induced inflammatory gene expression (Figure 5F). Collectively, the susceptibility to mitochondrial dysfunction conferred by the GCKRTT variant and fatty acid exposure could be mitigated by oxidative uncoupling that permits adaptation to increased fatty acid supply while granting protection against oxidant stress.
DISCUSSION
‘GWAS in-a-dish’ is a potential strategy to determine the personalized phenotypes in a collection of cells from multiple individuals (Kimura et al., 2018). This report integrated the GWAS in-a-dish concept with a PoP-based functional approach (Takebe and Wells, 2019) to capture pathological genetic variations associated with NAFLD/NASH. Our in vitro manipulatable approach for evaluating heritable variants circumvented the numerous in vivo non-heritable confounders, including lifestyle and nutrition, which perturb NAFLD/NASH and other metabolism-dependent diseases, leading to controversial interpretations of discovered variants. The PoP strategy provides an alternative to conventional laboratory-scale protocols, which are generally ill-equipped to perform large-scale phenotypic analyses as the cost is prohibitive and procedures are labor intensive. Our improved differentiation methodologies led to successful parallel and clonal differentiation of the pooled foregut progenitors into HLOs. The clonally-derived PoP was conducive to en masse screening for quantifying donor-specific intra-hepatocytic lipid levels, an early pathophysiological manifestation of NAFLD. Hence our pooled iPSC-derived foregut progenitors enabled: 1. application of identical pathologic insults; 2. live tracking and sorting of organoids utilizing phenotypic readouts; and 3. SNP profiling is associated with the organoid-of-origin encompassing phenotypic information. As a proof-of-principle, our PoP genotype-phenotype association studies informed the impact of key, GWAS identified, NAFLD risk alleles on liver steatosis phenotype (Anstee et al., 2020; Hernaez et al., 2013; Hotta et al., 2010; Hudert et al., 2019; Kawaguchi et al., 2012; Lin et al., 2014; Petta et al., 2014; Speliotes et al., 2011). Collectively, we propose that our pooling strategy represents an organoid level ‘forward cellomics’ platform (Kimura et al., 2018) to interrogate genotype-driven phenotypic association in human organoid models.
One key advantage of our organoid models is viable human-based systems for evaluating in-depth phenotypic impacts of an identified variant, independent of patient metabolic status. We demonstrated that one of the most pleiotropic variants, GCKR-rs1260326 TT variant (Sakaue et al., 2021), in pooled and individually assessed HLOs, was biologically significant under culturing conditions that mimic T2D insulin resistance. In addition to enhanced fatty-acid-induced TG accumulation with correlating inflammatory signatures, we demonstrated that DNL, insulin resistance, and mitochondrial dysfunction were distinguishable from HLOs carrying non-risk GCKR variants. This differential functional evidence of metabolic perturbations in HLOs provided insights into the contribution of the GCKRTT variant in NALFD and is indicative of the vast potential of human organoids for mechanistic studies.
In vivo, the prognostic value of a T2D phenotype for patients carrying GCKRTT variant was highlighted by our discovery that HbA1c measurements, a diagnostic indicator for T2D, uniquely delineated the severity of NAFLD/NASH-associated inflammatory pathologies. Patients with T2D diabetic HbA1c values (>6.4%) were associated with more severe pathologies than those with normal HbA1c values (<5.7%). This differentiation factor was not observed for other genetic risk variants, particularly the well-established PNPLA3-rs728409 GG risk variant. Our study firmly supports the inclusion of HbA1c measurements, which are often missing in clinical studies of GCKR-rs1260326 cohorts (Anstee et al., 2020; Cai et al., 2019; Gao et al., 2019; Hudert et al., 2019; Kitamoto et al., 2014; Lin et al., 2014; Petta et al., 2014). Interestingly, a search of the publicly available database of Phenome-wide association studies (Japanese patients; https://pheweb.jp) indicated a strong correlation between GCKR-rs1260326 and HbA1c, amongst liver-related phenotype and other metabolism-related markers, The influence of ethnicity, gender, age, and BMI status on differential HbA1c values in GCKR-rs1260326 cohorts, remains to be further determined.
Since T2D complication in NAFLD patients present with hepatic insulin resistance, the GCKR-rs1260326 dependent subgrouping by HbA1c values may be informative for precision patient management strategies. We noted that the subgroup of patients who had non-diabetic HbA1c values (<5.7%), exhibited improved pathologies upon metformin therapy, while patients with T2D indications (HbA1c >6.4%), and our insulin insensitive HLO models, were poorly responsive to metformin. One implication for risk carriers, is the possibility that reduction of dietary fat supplementation may alleviate hepatic substrate-dependent lipogenesis, improving insulin resistance and suppressing lipid deposition. Such non-medical treatments, i.e., lifestyle modification and weight loss, which continue to be recommended as alternatives to medication (The Lancet Gastroenterology, 2020) despite high variability in outcomes (Pollin et al., 2011) (Belalcazar et al., 2016), could be beneficial when integrated with understanding the genotype-driven physiological condition of the patient. For the GCKR-rs1260326 TT variant, we conclude that its contribution to NAFLD/NASH is highly dependent on the diabetic status informed by HbA1c measurements and, thus, may be of prognostic value for GCKR-rs1260326 TT carriers. Our results also pave the way for designing focused approaches to better address the T2D complication in populations of GCKR-TT carriers, by optimally controlling substrate intake and de novo lipogenesis via lifestyle modification and/or drug exposure. Collectively, the integration of in vitro HLO models with in vivo clinical data provided insights to improve capturing the highly variable in vivo pathogenesis of NAFLD/NASH risk variants.
The GCKR-rs1260326 polymorphism (Chambers et al., 2011; Speliotes et al., 2011), commonly found in non-African population (Santoro et al., 2012; Wessel and Marrero, 2016), is correlated with fatty liver-associated hepatic insulin resistance (Nozaki et al., 2020). However, the resultant expressed GCKR p.Pro446Leu protein, with loss of ability to interact and modulate GCK activities, is also recognized to protect against T2D as hepatic glucose utilization is enhanced independent of insulin (Zain et al., 2015) (Xia et al., 2019). This “double-edged sword” feature of GCKR actions hampered the initial therapeutic promise of biologics disrupting GCK-GCKR complex interactions, as undesirable side-effects of hypoglycemia, increased hepatic steatosis (as we also observed in our HLO model) were observed, and loss of efficacy was presumably related to the development of hepatic insulin resistance (Hale et al., 2015). Our transcriptomic and functional analyses revealed that, in addition to modulating GCK activities, the GCKR-rs1260326 TT variant was strongly associated with mitochondrial dysregulation. Evidence from our GCKRTT HLO studies indicated potently enhanced persistent mitochondrial ROS and reduced OCR, which was exacerbated by exposure to fatty acid. Most intriguingly, treating our GCKRTT HLO steatohepatitis-like models with oxidative uncouplers (NR/NTZ) but not metformin normalized mitochondrial functions and suppressed fatty-acid induced inflammatory responses, suggesting potential therapeutic pathways.
Emerging therapeutic approaches to treat NAFLD and T2D have focused on disrupting metabolic and inflammatory pathways that interconnect the two conditions. Efficacy, however, has remained poor, limited or controversial, due, in part, to still imperfect understanding of these complex interconnections (Ferguson and Finck, 2021) and the contributions of genomic SNPs. For carriers of the GCKR-rs1260326 (patients and HLO models), metformin, and developing NASH drugs obeticholic acid and CCR2/5 inhibitor (not shown), demonstrated poor pharmacological benefits in suppressing NASH-associated inflammatory phenotypes. Our findings that an FDA-approved drug NTZ in concert with NR was capable of suppressing lipid-induced inflammation in GCKRTT-HLO, suggest the possibility of effective pharmacological intervention for at least a subset of patients with NAFLD. Since the GCKR-rs1260326 TT variant represents almost 40% of the US population (Orho-Melander et al., 2008), with African ethnicity having the lowest (app. 10%) allelic frequency (Rich et al., 2018), our study provides a better understanding of the mechanism in which risk variants contribute to NAFLD and indicate the need to tailor therapeutic intervention depending on both genetic and metabolic status of the patients.
In conclusion, our present study integrating organoid modeling and clinical analyses highlights methodological advances and emphasizes insights for a better understanding of the personalized basis of complex, common diseases such as NAFLD/NASH. Improving patient stratification will enable earlier identification and implementation of preventive and therapeutic strategies. With NAFLD affecting nearly one billion people globally, early identification of susceptible individuals with a rigorous interventional design, including lifestyle management and refining treatment options, is critical for precision hepatology.
Limitations of the Study
First, in this study, metabolic dysfunction incurred by a genetic variant was predominantly studied in a static state. Dynamic effects such as those inferred from lipogenic flux studies were not addressed and may shed more light on lipogenic defects in genetically-defined organoids. Second, although this is the en masse investigative approach for performing GWAS in a dish with organoids to dissect complex genetic contributions, the scale achieved (24 patients) is modest and still requires a significant amount of “hand-to-hand combat” which remains a substantial hurdle for the field of human genetics. Future extensive improvements in throughput for organoid generation and phenotypic callouts will be key to the overall scale of the approach. This can be facilitated by combinatorial use of bioprinter, organoid sorter, and functional genomics technologies. Third, we limited in-depth characterization of hepatocytes in our system, as hepatocytes are responsible for the most upstream cause of NAFLD. Given that our en masse protocols allow for multicellular lineage induction from the same donor, unlike primary-cell derived epithelial organoid system, parsing out multi-cellular pathogenesis and the gene-environment interactions will be of further interest to predicate the utility of the system for subsequent downstream events such as inflammation and fibrosis.
STAR★Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Takanori Takebe (takanori.takebe@cchmc.org)
Material Availability
Plasmids generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
The processed gene expression data in this paper have been deposited into the NCBI GEO database: GSE213932 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE213932).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
iPSC cell lines and cell culture.
Human iPSC lines used in this study are summarized in Table S1. Patient cells, where applicable, were obtained with consent in compliance with ethics guidelines (Institutional Review Board, Cincinnati Children’s Hospital Medical Center) and reprogrammed into iPSC by the CCHMC Pluripotent Stem Cell Facility. All human iPSC lines were maintained as described previously (Takebe et al., 2017). Briefly, undifferentiated hiPSCs were cultured on Laminin 511E8-fragment (Nippi, Japan) coated dishes in Stem Fit medium (Ajinomoto Co, Japan) with 100ng/ml bFGF (R&D Systems, MN, USA) at 37°C in 5% CO2 with 95% air.
Clinical analysis.
A retrospective analysis of a cohort of 1091 adults diagnosed with NAFLD (STELLAR-3 trial, NCT03053050 (Younossi et al., 2019); ATLAS-trial, NCT03449446 (Loomba et al., 2021)) was performed with demographics, biomarkers, and liver histology available for clinical samples. The STTELLAR-3 trial was phase 3 study of selonsertib enroll 808 patients with bridging fibrosis attributable to NASH. The ATLAS trial was phase 2b study enrolled 395 patients with bridging fibrosis or compensated cirrhosis attributable to NASH. Both trials were an international, multi-center, randomized, placebo-controlled, allocation-concealed. The protocols conformed to ethical guidelines and were approved by the appropriate national and institutional review committees. All patients provided written informed consent.
Sample-size estimation.
For en masse organoid phenotyping, the number of donors was determined on a recent Monte Carlo simulation that a cohort of 24 human iPSC lines would yield a 92% probability that can predict an event occurring 1 in 10 patients (Fermini et al., 2018). Retrospective clinical analysis was carried out based on predetermined samples, therefore, no sample size estimation was applied.
METHOD DETAILS
Induction and cryopreservation of the foregut.
Human iPSCs were differentiated into foregut using previously described methods . In brief, hiPSCs were detached by Accutase (Thermo Fisher Scientific Inc., MA, USA) and were seeded on Laminin coated tissue culture plate with 50,000 cells/cm2. The medium was changed to RPMI 1640 medium (Life Technologies) containing 100 ng/mL Activin A (R&D Systems) and 50 ng/mL bone morphogenetic protein 4 (BMP4; R&D Systems) at day 1, 100 ng/mL Activin A and 0.2% fetal calf serum (FCS; Thermo Fisher Scientific Inc.) at day 2, and 100 ng/mL Activin A and 2% FCS at day 3. For 4–6 days, cells were cultured in Advanced DMEM/F12 (Thermo Fisher Scientific Inc.) with B27 (Life Technologies) and N2 (Gibco, CA, USA) containing 500 ng/ml fibroblast growth factor 4 (FGF4; R&D Systems) and 3 μM CHIR99021 (Stemgent, MA, USA). Cells were maintained at 37 °C in 5% CO2 with 95% air and the medium was replaced daily. The foregut cells were detached by Accutase and cryopreserved in Cell Banker 1 media (Nippon Zenyaku Kogyo Co., Ltd., Japan), at −150 °C.
Generation of HLO and pooled organoid panel.
The cryopreserved foregut cells were thawed quickly and gently centrifuged at 800 rpm for 3 minutes. Cells were suspended with Matrigel™ matrix (Corning Inc., NY, USA) on ice, for a final concentration of 750,000 cells/mL. Details of the analysis of the pooled organoid panel are described in Figure 1A. In brief, the frozen foregut cells derived from each iPSC cell line were mixed and resuspended in Matrigel on ice. The mixture of cells and Matrigel was embedded in 50μl drops on dishes in advanced DMEM/F12 with 2% B27, 1% N2, 10 mM HEPES, 1% Glutamax, 1% Pen/Strep, 5 ng/mL fibroblast growth factor 2 (FGF2), 10 ng/mL vascular endothelial growth factor (VEGF), 20 ng/mL epidermal growth factor (EGF), 3 μM CHIR99021, 0.5 μM A83–01, and 50 μg/mL ascorbic acid, and incubated in the CO2 incubator for 4 days with medium changed every 2 days. The medium was then switched to advanced DMEM/F12 with 2% B27, 1% N2, 10 mM HEPES, 1% Glutamax, 1% Pen/Strep, and 2 μM retinoic acid (RA), and incubated in the CO2 incubator for further 4 days with medium changed every 2 days. The final media switch was to the hepatocyte culture medium (HCM; Lonza, MD, USA) and the cells were incubated in a CO2 incubator for 6 days, changing the medium every 2 days.
Induction of steatohepatitis HLO (sHLO) and measurement of lipid accumulation.
HLO was isolated from Matrigel and washed with 1xPBS, then cultured with HCM media containing 5 μg/ml insulin and 300 μM sodium oleate (Sigma) on ultra-low attachment 6 multi-well plates (Corning) to induce sHLO. sHLO were collected at day 3 for lipids accumulation and day 3 or 7 for evaluating markers of inflammation. Accumulation of lipid in HLOs was measured using BODIPY® 493/503 (ThermoFisher Scientific). Briefly, sHLOs were rinsed three times with warm PBS to remove residual oleic acid from the cell surface. Lipids accumulated in sHLOs and nuclei were stained with 2 μM BODIPY® 493/503 and NucBlue™ Live ReadyProbes™ Reagent (ThermoFisher Scientific). After staining, sHLOs were scanned using a Nikon A1 inverted confocal microscope (Japan) and Keyence BZ-X710 automated fluorescence microscope (Japan). The lipid droplet volume was calculated by using Analysis Application Hybrid cell count (Keyence) and normalized with each nucleus signal. Hepatosteatosis index was calculated by donor distribution in high- and low- fat accumulator groups, determined by donor-specific SNP-PCR. The hepatosteatosis index of 1 means that the indicated donor is equally distributed between high- and low-groups.
Donor identification and phenotypic screening of pooled organoid panels.
Details of the donor identification of pooled organoid panels are described in Figure S1F. In brief, donor-specific SNP genotypes are used to detect the ratio of each donor in a multi-donor HLO panel. The gDNA of each donor-derived iPSC was extracted and the SNP profile was obtained by SNP array. Based on the SNP profile, each donor-specific SNP was selected from TaqMan SNP Genotyping Assays (Thermo Fisher Scientific Inc). Standard curves for each donor were generated using donor gDNA mixed in arbitrary ratios. The gDNA of the multi-donor HLO panel was extracted in batches and the ratio of each donor was determined using the generated standard curve. For screening of lipid accumulation phenotypes in the multi-donor HLO panel, the high and low lipid accumulation phenotype groups were separated under fluorescence microscopy from 24 donor population organoid panel of fatty acid-induced sHLO. The cutoff value of fluorescence intensity was set to 50 using BZ-X710 automated fluorescence microscope and Analysis Application Hybrid cell count (Keyence). The gDNA from the two isolated groups was extracted with DNeasy Blood & Tissue Kit (Qiagen), and the distribution of each donor-specific SNP was measured using SNP donor identification method as described.
Single Cell RNA Sequencing of a pooled organoid panel.
Single-cell RNA sequencing of the pooled organoid panel was performed as previously described (Ouchi et al., 2019). In brief, a pooled organoid panel consisting of 24 donors at day 27 was dissociated to single cells by Trypsin-EDTA (0.05%) in phenol red (GIBCO) for 10 min and washed with 1xPBS. 17,500 cells underwent single cell RNA-seq library preps and were sequenced with the 10X Genomics Chromium platform. Sequenced reads were processed using the Cell Ranger gene expression pipelines mkfastq and count, starting with demultiplexing and conversion of barcode and read data to fastq files. Raw reads were aligned to the Hg19 genome and filtered, creating gene-barcode matrices. Analyses were performed in AltAnalyze, where gene and cell clusters were identified through unsupervised analysis to identify predominant sample groups via expression clustering. Following the removal of outliers, clusters were restricted to having > 5 cells, a minimum Pearson correlation of 0.5, and a between-cluster fold change > 4. Cell cycle effects were removed. Cluster-based heatmaps and plots were generated using Seurat R package.
GCKR-rs1260326 SNP editing of hiPSC.
To derive GCKR-rs1260326 SNP edited hiPSC, we chose the YD8–1 hiPSC as a parental cell line since this hiPSC line has no NAFLD-associated risk variants in PNPLA3-rs738409, GCKR-rs1260326, and TM6SF2-rs58542926. Introduction of GCKR-rs1260326 TT risk variant in YD8–1 hiPSC was performed using the microhomology-assisted excision (MhAX) method (Kim et al., 2018) with minor modifications, summarized in Supplementary Figure 3. In brief, donor plasmids for the introduction of each allele were created by PCR amplification of the homology arms from YD8–1 hiPSC genomic DNA using the primers listed in Table S6. In-Fusion cloning (Clontech) was used to assemble the arms with tdTomato-P2A-PUR selection cassette and pAAVS1-P-CAG-mCh plasmid backbone (Addgene). PCR-amplified regions and In-Fusion junctions were verified by Sanger sequencing. Oligonucleotides encoding sgRNA protospacer sequences targeting GCKR and ps1 (for cassette excision) (Table S6) were annealed and cloned into pSpCas9(BB)-2A-GFP (pX458) and pX330-U6-Chimeric_BB-CBh-hSpCas9 (pX330), respectively (Addgene). sgRNAs were verified by Sanger sequencing. For gene targeting, the donor plasmid (8 μg) and the GCKR-targeting Cas9/sgRNA expression plasmid (pX458-based, 2 μg) were co-transfected by electroporation into 1 × 106 cells of YD8–1 hiPSC, followed by plating under feeder-free conditions for 48 h in AK02N medium containing 10 μM Y-27632. Antibiotic selection using 0.5 μg/ml of puromycin was initiated 3 days post electroporation. Ten days after plating, puromycin-resistant cells were pooled and passaged. Single-cell-derived colonies were isolated, cultured, and processed for genomic DNA isolation under feeder-free conditions in 96-well format. The clones positive for PCR genotyping and sequencing were further expanded as populations for subsequent cassette excision. For cassette excision, 5 μg of pX330-based expression plasmid targeting ps1 was transfected into 1 × 106 gene-targeted cells followed by growth without selection for 7 days. tdTomato-negative cells were isolated by FACS on a BD FACSAria III cell sorter, and plated into a 96-well plate followed by the growth of single-cell-derived clones. Isolated clones were cultured and then genotyped for cassette excision by PCR and Sanger sequencing. The selected, SNP-edited clones were subjected to karyotyping with G-band analysis.
Measurement of GCK activity.
GCK activity of HLOs was measured using the PicoProbe™ Glucokinase Activity Assay Kit (BioVision inc., CA, USA) according to the manufacturer’s protocol. HLOs were homogenized with 100 μl ice-cold GCK Assay Buffer containing 2.5 mM DTT and kept on ice for 10 min. The samples were centrifuged at 12,000 × g at 4 C° for 10 min and the supernatant was collected. Fluorescence was measured using a BioTek™ Synergy™ H1 hybrid multi-mode monochromator fluorescence microplate reader (BioTek, VT, USA). The total protein content was also measured, and fluorescence intensity was normalized to total protein.
Mass-spec analyses.
Metabolomic analyses were conducted at the Northwestern University Metabolomics Core. For acetyl-CoA and palmitate analyses, metabolites were extracted using 10% trichloroacetic and solid phase extraction using Oasis HLB 1cc (30mg) cartridges (Waters Corporation, Milford, MA). For acyl-carnitine extraction, 80:20 methanol/water extraction was used. Protein quantity for normalization of mass-spec values was analyzed in pellets produced during metabolite solubilization procedures. The following standards were employed: 16:0(d4) Coenzyme A: palmitoyl(11,11,12,12-d4) Coenzyme A (AvantiLipids), Acetyl-Coenzyme A (sodium salt; Cayman Chemicals), Palmitic Acid-d9 MaxSpec® Standard (Cayman Chemicals), NSK-B labeled carnitine standards set B (Cambridge Isotope Labs). Samples were analyzed by High-Performance Liquid Chromatography and High-Resolution Mass Spectrometry and Tandem Mass Spectrometry (HPLC-MS/MS). Specifically, the system consisted of a Thermo Q-Exactive in line with an electrospray source and an Ultimate3000 (Thermo) series HPLC consisting of a binary pump, degasser, and auto-sampler outfitted with an Xbridge Amide column (Waters; dimensions of 4.6 mm × 100 mm and a 3.5 μm particle size). The mobile phase A contained 95% (vol/vol) water, 5% (vol/vol) acetonitrile, 20 mM ammonium hydroxide, 20 mM ammonium acetate, pH = 9.0; B was 100% Acetonitrile. The gradient was as following: 0 min, 15% A; 2.5 min, 30% A; 7 min, 43% A; 16 min, 62% A; 16.1–18 min, 75% A; 18–25 min, 15% A with a flow rate of 400 μL/min. The capillary of the ESI source was set to 275 °C, with sheath gas at 45 arbitrary units, auxiliary gas at 5 arbitrary units, and the spray voltage at 4.0 kV. In positive/negative polarity switching mode, an m/z scan range from 70 to 850 was chosen, and MS1 data was collected at a resolution of 70,000. The automatic gain control (AGC) target was set at 1 × 106 and the maximum injection time was 200 ms. The top 5 precursor ions were subsequently fragmented, in a data-dependent manner, using the higher energy collisional dissociation (HCD) cell set to 30% normalized collision energy in MS2 at a resolution power of 17,500. The sample volumes of 25 μl were injected. Data acquisition and analysis were carried out by Xcalibur 4.0 software and Tracefinder 2.1 software, respectively (both from Thermo Fisher Scientific). Metabolite levels were analyzed as peak area normalized to total ion content and to protein quantity. Metabolite analysis was performed blinded to experimental groups.
PFKB3 inhibitor (PFK15) and GCK-GCKR disruptor (AMG3969) treatment of HLO.
GCKRCC and GCKRTT HLOs were cultured in HCM media in the presence or absence of 200μM PFK15 and 10μM AMG3969 for 5 days. To assess the impact on lipid accumulation in HLO, BODIPY staining was performed and scanned using a Nikon A1 inverted confocal microscope. The lipid droplet volume was calculated by using Analysis Application Hybrid cell count and normalized with each nucleus signal.
Genotyping of NASH clinical trial participants.
DNA of consenting subjects enrolled in NASH clinical trials (Harrison et al., 2020) was submitted for whole genome sequencing (performed by WuXi NextCODE). Genomes were sequenced to an average coverage of 20X using paired 150nt reads. DNA sequencing reads were aligned to the human genome (GRCh38) using BWA-MEM software (Li and Durbin, 2009). Genotypes were called for each sample using a GATK4.0-compatible computational pipeline (Kendig et al., 2019). Tests for associations between genotypes and clinical measures of interest were carried out using R package “finalfit” using default parameters.
RNA-seq data and informatics.
The DonorMatched™ RNASeq Characterization Data Set (Samsara Sciences, #RSDP) was used for the four human hepatocyte transcriptome datasets (n = 3 per donor). Whole-transcriptome RNA sequencing of HLOs generated from two donors of GCKRTT variant and three donors of GCKRCC variant (n = 2 each per donor) was performed by Novogene (China) on an Illumina Novaseq S4 platform. RNA sequencing parameters were 150bp pair-end sequencing at a depth of 20M reads per sample. Clean data were generated from the raw data that was filtered by data-processing steps, including removal of adapter sequences, reads with more than 10% N, and low-quality sequences (the percentage of low-quality bases of quality value ≤ 5 is greater than 50% in a read). All the Fastq read files for each sample, for both human hepatocyte and HLOs were then aligned to hg19 version of the human genome using the Computational Suite for Bioinformaticians and Biologists version 3.0 (CSBB-v3.0, https://github.com/praneet1988/Computational-Suite-For-Bioinformaticians-and-Biologists) to obtain raw transcript counts. The trimmed mean of M-values (TMM) normalized Log2 Counts-per-Million (CPM) values were obtained and analyzed for differential expression with the interactive Gene Expression Analysis Kit (Choi and Ratner, 2019) and Gene Set Enrichment Analysis (Subramanian et al., 2005) (GSEA). For differential expression, statistical and biological significance was set at p < 0.05, fold-change > 1.5, Benjamini-Hochberg procedure was used for multiple testing, with a minimum of 0.5 CPM in one of the samples. The GCKR variant was determined from the RNA-seq data using the Genome Analysis Toolkit v4.0 haplotype caller (GATK HC) after merging the triplicate data. The variants were filtered using GATK variantfilteration step and were annotated using ANNOVAR (Wang et al., 2010). Curated gene sets of Reactome (https://reactome.org/), a general-purpose public database of human pathways, were used for GSEA. The pathways with a significant difference of p < 0.05 were selected for further analysis and ordered by normalized enrichment score (NES).
Measurement of Oxygen Consumption rate (OCR).
OCR of HLOs and sHLOs was measured using the Extracellular Oxygen Consumption Assay (Abcam, Cambridge, UK) according to the manufacturer’s protocol. About 300 organoids were distributed into each well of black flat- and clear-bottomed 96-well microplate (Corning). Fluorescence intensity (380 nm excitation and 650 nm emission) was measured kinetically for 180 minutes using BioTek™ Synergy™ H1 hybrid multi-mode monochromator fluorescence microplate reader with a time-resolved fluorescence mode. The delay time was set as 30 μs and the total time of windows was 100 μs. OCR was calculated from the linear portion of the fluorescence intensity versus time plot, and then normalized to the total HLO number counted by Keyence BZ-X710 automated fluorescence microscope with cell count Analysis Application (Keyence).
NMR-based metabolomics analysis.
To obtain both polar and non-polar fractions of the HLOs for NMR analysis, all sample preparation was completed as previously described (Watanabe et al., 2016). Briefly, the appropriate volumes of solvents (final constant ratio of 2:2:1.8 of chloroform: methanol: water) were added to HLO samples. The hydrophilic extract and non- polar fraction were dried in a vacuum centrifuge at room temperature and stored at −80°C until further preparation for NMR data collection. On the day of the data collection, dried polar extracts were re-hydrated with 220 μL of NMR buffer containing 100mM phosphate buffer, pH7.3, 1mM TMSP (3-Trimethylsilyl 2,2,3,3-d4 propionate), 1mg/mL sodium azide) prepared in D2O. The non-polar extracts were suspended with 220uL D-chloroform with 0.3 v/v TSP. The final volume of 200 uL of each sample was transferred into a 103.5 mm × 3 mm NMR tube (Bruker Biospin, Germany) for data collection. NMR spectra were recorded and processed as previously described (Matrka et al., 2017) on a Bruker Avance II 600 MHz spectrometer with BBO Prodigy probe. Metabolite assignments and quantification were performed using Chenomx® NMR Suite profiling software (Chenomx Inc. version 8.4) based on the internal standard, TMSP. The metabolite abundances were normalized to total protein prior to statistical analysis.
Live imaging of reactive oxygen species (ROS).
ROS production in HLO and sHLOs and nuclei were stained with 5 μM CellROX™ Orange Reagent, for oxidative stress detection (ThermoFisher Scientific) and NucBlue™ Live ReadyProbes™ Reagent, respectively. Mitochondrial superoxide was detected using MitoSOX Red mitochondrial superoxide indicator (ThermoFisher Scientific). After staining, HLOs and sHLOs were scanned using a Nikon A1 inverted confocal microscope (Japan) and Keyence BZ-X710 automated fluorescence microscope (Japan). The ROS production was calculated by using Analysis Application Hybrid cell count and normalized with each nucleus signal.
Measurement of NAD/NADH.
NAD/NADH in HLOs and sHLOs were measured using the NAD/NADH Quantitation Kit (Sigma, MO, USA) according to the manufacturer’s protocol. HLOs were rinsed with cold PBS and extracted with 500 μL of NADH/NAD Extraction Buffer by homogenization. The sample was mixed vigorously by vortexing for 30 sec, and then centrifuged at 13,000 × g at 4 °C for 10 minutes to remove the insoluble fraction. Fluorescence was measured using a BioTek™ Synergy™ H1 hybrid multi-mode monochromator fluorescence microplate reader (BioTek, VT, USA). The fluorescence intensity was normalized to the total protein concentration of the respective sample.
Metformin (MET), nicotinamide riboside (NR), and nitazoxanide (NTZ) treatments.
sHLOs were cultured in HCM media in the presence or absence of 250μg/ml MET or 1mM NR and 3μM NTZ. NR and NTZ were dissolved in dimethyl sulfoxide (DMSO) for stock solutions of 1000 mM and 30 mM, respectively. The final working concentration of DMSO in culture medium was 0.1% DMSO, and controls received 0.1% DMSO as the vehicle. To assess the impact on lipid accumulation in sHLO, BODIPY staining was performed. These HLOs were further assayed for reactive oxygen species (ROS) by imaging and RT-qPCR.
IL1b and TNFα ELISA.
Inflammation cytokines secreted from HLOs and sHLOs were measured using the MSD V-PLEX Proinflammatory Panel assay kit (Meso Scale Diagnostics, MD, USA) according to the manufacturer’s protocol. To measure secreted IL1b and TNFα, culture supernatants were collected after 72 h of culture. HLO in the analyzed wells was quantified using the CellTiter-Glo® 3D Cell Viability Assay (Promega, WI, USA) to normalize secreted cytokine.
Measurement of glucose production.
Glucose production in HLOs and sHLOs were measured using the Glucose Colorimetric/Fluorometric Assay Kit (BioVision inc., CA, USA) according to the manufacturer’s protocol. HLOs and sHLOs were rinsed three times with cold PBS, suspended in DMEM without glucose, supplemented with 100 μM sodium pyruvate (Gibco), and seeded onto ultra-low attachment 6 multi-well plates (Corning). After 12 hours, the culture supernatant was collected and used for glucose production measurement. Fluorescence intensity was measured with a BioTek™ Synergy™ H1 hybrid multi-mode monochromator fluorescence microplate reader (BioTek, VT, USA). HLOs in the analyzed wells were quantified using the CellTiter-Glo® 3D Cell Viability Assay (Promega) to normalize secreted glucose. To assess insulin responsiveness in HLOs, HLOs and sHLOs were incubated in an AdDMEM medium for 12 hours in the presence or absence of 1 μM insulin. The amount of glucose in the collected supernatant was measured.
Measurement of triglyceride and cholesterol secretion.
Triglyceride and cholesterol secretion in HLOs and sHLOs were measured using the Triglyceride Quantification Colorimetric/Fluorometric kit (BioVision) and Cholesterol Quantification Assay kit (Sigma) according to the manufacturer’s protocol. HLOs and sHLOs were rinsed with cold PBS and seeded onto ultra-low attachment 6 multi-well plates (Corning) in HCM medium. After 24 hours, the culture supernatant was collected for triglyceride and cholesterol measurements. Fluorescence intensity was measured with a BioTek™ Synergy™ H1 hybrid multi-mode monochromator fluorescence microplate reader (BioTek, VT, USA). HLOs in the analyzed wells were quantified using the CellTiter-Glo® 3D Cell Viability Assay (Promega) to normalize secreted triglyceride and cholesterol.
Measurement of insulin responsiveness in sHLO by Western Blot (WB) analysis.
HLOs and sHLOs were starved with 0.2% FCS/DMEM/F12 for 18 h and then stimulated with insulin (170 ng/ml) for 20 min. Treated HLOs were washed twice with PBS and lysed with M-PER™ Mammalian Protein Extraction Reagent (Thermo Fisher Scientific Inc.). Protein quantification was measured using the Pierce™ Rapid Gold BCA Protein Assay Kit (Thermo Fisher Scientific Inc.). For WB analysis, 10 μg protein were separated on 8% SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and probed with anti-AKT (Clone C67E7, Cell signaling technologies, MA, USA), anti-phospho-AKT (Clone C31E5E, Cell signaling technologies), anti-α-tubulin (Clone DM1A, Cell signaling technologies) as indicated.
Detection of mitophagy in sHLO.
Mitophagy was evaluated using Mitophagy dye (#MD01, Dojindo Molecular Technology) according to the manufacturer’s protocol. Briefly, HLOs and sHLOs were rinsed with warm HCM, then treated with 100 nM of Mitophagy dye for 30 minutes at 37 °C. The cells were rinsed twice with warm HCM and incubated at 37 °C and 5% CO2. After 24 hours, cells were washed with warm HCM and stained with NucBlue™ Live ReadyProbes™ Reagent (ThermoFisher Scientific). After staining, the cells were observed on a Keyence BZ-X800 automated fluorescence microscope (Japan). The mitophagy levels were calculated using Analysis Application Hybrid cell count (Keyence) and normalized to each nucleus signal.
Flow Cytometry analysis for mitophagy-related proteins.
Flow cytometry analysis of HLO was performed as previously described (Ouchi et al., 2019). In brief, HLOs were dissociated to single cells with Trypsin-EDTA (0.05%) in phenol red (GIBCO) for 15 min. After PBS wash, cells were stained with primary antibodies (mouse anti-Parkin antibody, rabbit anti-Mitofusin 2 antibody) and corresponding secondary antibodies, and were subjected to flow cytometry. Dead cells were excluded by propidium iodide staining. All analysis was performed by BD FACSCanto3 (BD Biosciences) and FlowJo (FlowJo, LLC).
Quantifying mitochondrial DNA copy number in HLOs.
The copy number of mitochondrial DNA was determined by amplifying mitochondrial tRNA-Leu(UUR) and nuclear-encoded β2m (beta-2-microglobulin) genes as previously described (Rooney et al., 2015). Total DNAs were extracted from HLOs and sHLOs using DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer’s protocol, then quantified with the Qubit dsDNA HS Assay Kit by a Qubit 2.0 Fluorometer (ThermoFisher Scientific). Amplification was carried out using SYBR Green PCR Master Mix (Thermo Fisher Scientific). Cycling was performed at 50°C for 2 minutes, 95°C for 10 minutes, followed by 45 cycles of 95°C for 15 seconds and 62°C for 1 minute. PCR amplification used the following primers: tRNA-Leu (F: 5’-CACCCAAGAACAGGGTTTGT-3’, R: 5’-TGGCCATGGGTATGTTGTTA-3’); β2m (F: 5’-TGCTGTCTCCATGTTTGATGTATCT-3’, R: 5’-TCTCTGCTCCCCACCTCTAAGT-3’). The relative level of mtDNA copy number was calculated using the 2−ΔΔCt method.
Quantification and statistical analysis.
Statistical analyses were mainly carried out using GraphPad Prism 9.0 (GraphPad Software, Inc., CA, USA) using unpaired two-tailed Student’s t-test, Dunn-Holland-Wolfe test, or Welch’s t-test. P values < 0.05 were considered statistically significant. N-value specified in figure legend refers to biologically independent replicates. For comparisons between unpaired 2 groups, when groups were independent and the variances were unequal, a non-parametric Brunner-Munzel test was performed, unless noted otherwise. No intentional randomization nor blinding was applied however, experimenters were not aware of the nature of the samples due to en masse nature of the assays. No exclusion was made when analyzing the data.
Supplementary Material
Figure S1 Development of a multi-cellular human liver organoid panel from mixed foregut progenitors, Related to Figure 1. (A) UMAP plot and heatmap analysis by scRNAseq of population organoid panel (PoP). (B) Cell type-specific marker analysis of PoP. (C) Schematic diagram of the Donor decoding method of PoP using donor-specific SNP genotyping. The gDNA of each donor-derived iPSC was extracted and the SNP profile was obtained by SNP array. Based on the SNP profile, donor-specific SNP profiles were constructed. (D) The presence of 20 donors in the PoP was confirmed by donor-specific SNPs. (E) Individual HLO morphology in PoP. (F) Clonality analysis of PoP.
Figure S2. Human organoid modeling of NAFLD-like phenotype induced by fatty acid loading, Related to Figure 1 and 3. (A, B) Live imaging of accumulating lipid droplets in fatty acid-loaded HLO. Shown, HLO without fatty acid loading and sHLO treated with fatty acid for 72 hours. Lipid droplets stained green with BODIPY and nuclei stained with Hoechst 33342. Scale bars, 50μm. (C, D) Imaging of lipid droplets by transmission electron microscopy (TEM), with (C) representing HLO and (D) representing sHLO. Yellow arrows indicate accumulated lipid droplets. Scale bars, 10μm. (E) Analysis of non-polar metabolites of sHLO by nuclear magnetic resonance (NMR) spectroscopy. Metabolites of sHLO induced by fatty acid treatment for 72 h were measured. Quantifications were performed in five independent donor iPSC lines. Unpaired t-test: *p < 0.05, ****p < 0.0001. (F) Supernatant analysis of secreted triglyceride and cholesterol from sHLO after 72 hours of fatty acid treatment. Data are shown as means ± SD (error bars), n=6. Unpaired t-test; ****p < 0.0001. (G) Mass-spec analysis of protein content-normalized levels of acetyl-CoA, palmitate (C16 – long chain saturated fatty acid), octanoyl-carnitine (C8 – medium chain acyl-carnitine) and palmitoyl-carnitine (C16 - long-chain acyl-carnitine). (H) ELISA analysis of secreted pro-inflammatory cytokines from sHLO after 72 hours of fatty acid treatment. Data are shown as means ± SD (error bars), n=9. Unpaired t-test; **p < 0.01, ****p < 0.0001. (I) Analysis of pro-inflammatory cytokine gene expressions in sHLO. Proinflammatory cytokine gene expression was measured by qPCR in sHLOs after 72 hours of fatty acid treatment. Data are shown as means ± SD. (error bars), n=4, Unpaired t-test; **p < 0.01, ***p < 0.001. (J) Measurement of glucose production in HLO and sHLO. The amount of glucose was normalized to the CellTiter-Glo value (mean ± SD, n = 9 independent experiments per line). Unpaired t-test; ****p < 0.0001. (K) Gene expression analysis (qPCR) of PCK1 in HLO and sHLO (mean ± SD, n = 8 independent experiments). Unpaired t-test; ****p < 0.0001. (L) Western blot analysis to determine insulin responsiveness of sHLO. HLOs and sHLOs were starved for 24 hours and treated with 1 μg/ml of insulin for 20 minutes. (M) Glucose production assay to determine insulin responsiveness of sHLO. HLOs and sHLOs were starved for 24 hours and treated with 1μM of insulin for 12 hours. The amount of glucose in the collected supernatant was measured. The amount of glucose was normalized to the CellTiter-Glo value (mean ± SD, n = 9 independent experiments per line). Unpaired t-test; ***p < 0.001.
Figure S3. Gene-targeting for GCKR-rs1260326 locus in hiPSC, Related to Figure 2. (A) Schematic of the gene-targeting procedure for GCKR-rs1260326 mutations using the MhAX method. Details of the targeting and genotyping procedures are provided in B, C, and E. (B) Design of guide RNA targeting GCKR locus. The sgRNA sequence is colored in purple. The red letter indicates the target base for editing no-risk C allele into risk G allele. The blue letter indicates the non-sense mutation found in the genome of the parental human iPSC line. (C) Detailed schematic of GCKR targeting vector. Homology arm (HA) overlap generates a 51 bp tandem microhomology (μH, blue), with the mutation (C>T, red). The two target sequences by ps1 sgRNA (green), which is used for cassette excision, are inserted next to the μH-T and μH-C sequences. (D) Representative FACS plot for the targeting and excision steps. (E) Sanger sequencing revealed the genotype of GCKR edited iPSC clones obtained from a single excision step. The arrows indicate rs1260326 C/T single nucleotide variation. (F) Representative bright field views of the GCKR edited iPSC clones. Scale bar, 500 μm. (G) Representative immunofluorescent images of pluripotency marker Oct4 in the GCKR edited iPSC clones. Scale bar, 100 μm. (H) Normal karyotype (46, XX) in the GCKR edited iPSC clones by chromosomal G-banding analysis.
Figure S4. Impact of PNPLA3 rs738409 variant in NASH patients stratified by HbA1c levels, Related to Figure 3 and Figure5. Graphic depiction of selected clinical parameters from Table 1, delineated by PNPLA3 variant and HbA1c values. (A) ALT measurements. (B) NAFLD activity score (NAS). (C) Lobular inflammation scores. (D) SAF activity score. (E) GCKR-rs1260326 confers differential metformin sensitivity in NAFLD patients. Graphic depiction of the effects of metformin treatment on NAS. Patients carrying GCKRTT did not show improvement in NAS after 48 weeks of metformin treatment, in contrast to patients carrying GCKRCC or CT.
Figure S5. GCKR-rs1260326 TT risk variant impacts lipid accumulation and enhances inflammatory responses in sHLO, Related to Figure 4. (A) Live imaging of accumulating lipid droplets in sHLO with GCKRCC or GCKRTT alleles, denoted as “CC” or “TT”. Images were stained with BODIPY for fat accumulation (Green) and Hoechst 33342 for the nucleus (Blue). Scale bars, 100μm, 50μm. (B) Quantification of lipid droplets in sHLOs. The intensity of lipid was normalized to nuclear signals. Unpaired t-test; ****p < 0.0001. (C - E) RNA sequence analysis of lipid-associated genes, glucose/fatty acid uptake genes, and fatty acid oxidation genes in sHLO. Data are shown as means ± SD (error bars), n=6. Unpaired t-test; *p < 0.05, **p < 0.001, ***p < 0.001, ****p < 0.0001. (F) Pathways enriched in GCKRTT HLOs, compared to GCKRCC HLOs, with NES > 1.2. (G) GSEA evaluation of gene-expression profile enriched in GCKRTT sHLO. REACTOME FATTY ACIDS (NES 1.68, p-value=0.002, FDR <0.3039), REACTOME INTERFERON GAMMA SIGNALING (NES 1.77, p-value<0.001 FDR <0.068). (H) qPCR analysis of pro-inflammatory cytokine gene expressions in sHLO. Data are shown as means ± SD (error bars), n=4, three independent experiments. Unpaired t-test; **p < 0.01, ***p < 0.001, ****p < 0.0001.
Figure S6. Mitochondrial dysregulation is associated with GCKR-rs1260326 related metabolic assaults, Related to Figure 5. (A) Quantifications of mitophagy events in GCKRTT HLO and GCKRTT sHLO (FFA treated). Relative levels of mitophagy were expressed as the ratio between Mitophagy Dye and Hoechst 33342 fluorescence intensity. Analysis was performed in >50 organoids per line, two independent experiments. (B) Relative protein levels of mitophagy-related proteins parkin (PRKN) and mitofusin 2 (MFN2) were analyzed by flow cytometry. Analysis was performed in >50 organoids per line, five independent experiments. Data in HLO was arbitrarily assigned a value of 1 and shown as means ± SD (error bars). Unpaired t-test; **p < 0.01. (C) Transmission electron microscopy (TEM) analysis. Representative electron micrographs identified the mitochondrial presence in GCKRTT-HLO (left) and GCKRTT-sHLO (right) samples. Mitochondrial areas are indicated by yellow circles. (D) Mitochondrial DNA copy number of GCKRTT HLO (white), -sHLO (gray), and -sHLO treated with nicotinamide riboside (NR), nitazoxanide (NTZ) combination (green). The mtDNA copy number was divided by the total amount of nuclear DNA, then each data was normalized to fold change over the HLO samples. Data are shown as means ± SD. (error bars), n=4, Tukey test; *p < 0.05, **p < 0.01. (E) Quantifications of ROS production in GCKRTT HLO and GCKRTT HLO treated with a combination of NR/NTZ. Mitochondrial ROS production was detected with MitoSOX live staining and Hoechst 33342 for the nucleus. The intensity of mitochondrial ROS was normalized to nuclear signals. Analysis was performed in >50 organoids per line. Unpaired t-test; ***p < 0.001.
Table S1. iPSC line information in this study. Related to Figure 1 and Star Methods.
Table S2. Demographic and baseline characteristics in a cohort of 1091 NAFLD patients. Related to Table 1 and Figure 3.
Table S3. Clinical sample information provided in the RNA sequence. Related to Figure 5.
Table S4. A list of Differentially Expressed Genes (DEG) by GCKR variants of the clinical and HLO/sHLO samples analyzed in this study. Related to Figure 5 and S5.
Table S5. Liver tests and histology after 48 weeks of metformin treatment in NAFLD patients. Related to Table 1 and Figure S4.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Recombinant Anti-Parkin antibody [PRK8] | Abcam | Cat# ab77924 |
| Recombinant Anti-Mitofusin 2 antibody [EPR19796] | Abcam | Cat# ab205236 |
| Phospho-Akt (Thr308) (C31E5E) Rabbit mAb | Cell signaling | Cat# 2965 |
| Akt (pan) (C67E7) Rabbit mAb | Cell signaling | Cat# 4691 |
| α-Tubulin (DM1A) Mouse mAb | Cell signaling | Cat# 3873 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RPMI 1640 | Gibco | Cat# 11875093 |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | Cat# 15140148 |
| HEPES (1 M) | Gibco | Cat# 15630080 |
| Advanced DMEM/F-12 | Gibco | Cat# 12634028 |
| B-27™ Supplement (50X), serum-free | Gibco | Cat# 17504044 |
| N-2 Supplement (100X) | Gibco | Cat# 17502048 |
| GlutaMAX™ Supplement | Gibco | Cat# 35050061 |
| HCMTM Hepatocyte Culture Medium BulletKitTM | Lonza | Cat# CC-3198 |
| dFBS | Hyclone | Cat# SH30070.02 |
| Laminin 511E8-fragment | Nippi | Cat# 892011 |
| Matrigel® Basement Membrane Matrix | Corning | Cat# 356237 |
| Human/Mouse/Rat Activin A | Shenandoah | Cat# 800–01; GenPept:P08476 |
| Human BMP4 | Shenandoah | Cat# 314-BP-050; GenPept:Q53XC5 |
| Human FGF4 | Shenandoah | Cat# 100–31; GenPept:P08620 |
| CHIR99021 | Peprotech | Cat# 2520691; CAS:252917-06-9 |
| Retinoic acid | Sigma-Aldrich | Cat# R2625–50MG; CAS:302-79-4 |
| Human FGF basic | R&D Systems | Cat# 233-FB-01M; GenPept:P09038 |
| Human VEGF165 | Peprotech | Cat# 100–20; GenPept:P15692.2 |
| Human EGF | Shenandoah | Cat# 100–26; GenPept:P01133.2 |
| A83-01 | Tocris | Cat# 2939; CAS:909910-43-6 |
| Ascorbic acid | Sigma-Aldrich | Cat# A4544; GCAS:50-81-7 |
| Human HGF | PeproTech | Cat# 100-39; GenPept:P14210.2 |
| Dexamethasone | Sigma-Aldrich | Cat# D4902; CAS:50-02-2 |
| Human Oncostatin M | Peprotech | Cat# 300–10; GenPept:P13725 |
| Sodium oleate | Sigma-Aldrich | Cat# O7501; CAS:143-19-1 |
| Insulin Solution Human | Sigma-Aldrich | Cat# I9278; CAS:11061-68-0 |
| Critical Commercial Assays | ||
| PicoProbe™ Glucokinase Activity Assay Kit | Biovision | Cat# K969 |
| BODIPY® 493/503 | Thermo-Fisher | Cat# D3922 |
| CellROX™ Orange Reagent | Thermo-Fisher | Cat# C10443 |
| MitoSOX™ Red Mitochondrial Superoxide Indicator | Thermo-Fisher | Cat# M36008 |
| Extracellular Oxygen Consumption Assay | Abcam | Cat# ab197243 |
| Triglyceride Quantification Colorimetric/Fluorometric kit | Biovision | Cat# K622–100 |
| Cholesterol Quantification Assay kit | Sigma-Aldrich | Cat# CS0005 |
| Mitochondria Isolation Kit for Tissue | Abcam | Cat# ab110168 |
| V-PLEX Human Proinflammatory Panel II (4-Plex) | MSD | Cat# K15053D-1 |
| CellTiter-Glo® 3D Cell Viability Assay | Promega | Cat# #G9681 |
| Cholesterol Quantification Assay kit | Sigma-Aldrich | Cat# CS0009 |
| Glucose Colorimetric/Fluorometric Assay Kit | Biovision | Cat# K606 |
| Mitophagy Detection Kit | Fisher Scientific | Cat# NC1133251 |
| Mitochondria Isolation Kit for Tissue | Abcam | Cat# ab110168 |
| 10x Genomics Chromium Single Cell 3’ | 10X Genomics | PN-1000075 |
| Deposited Data | ||
| RAW and analyzed RNA sequence data | This study | GSE213932 |
| Experimental models: Cell lines | ||
| Human induced pluripotent stem cells (iPSCs) | Table S1 | Table S1 |
| Oligonucleotides | ||
| Gene expression analysis | This study | Table S6 |
| Unique SNPs for donor identification | This study | Table S6 |
| GCKR rs1260326 SNP editing | This study | Table S6 |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP (pX458) | (Ran et al., 2013) | Addgene_48138 |
| pAAVS1-P-CAG-mCh | (Oceguera-Yanez et al., 2016) | Addgene_80492 |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | (Cong et al., 2013) | Addgene_42230 |
| Software and Algorithms | ||
| R 4.2.0 | The R Foundation | http://www.r-project.org/ |
| Seurat R package | (Stuart et al., 2019) | http://satijalab.org/seurat/ |
| Cell Ranger | 10x Genomics | https://support.10xgenomics.com |
| Image J | ImageJ software | https://imagej.nih.gov/ij/L |
| Prism 9.3.1 | Graphpad Prism | https://www.graphpad.com |
| FlowJo software | FlowJo, LLC | https://www.flowjo.com/ |
| Analysis Applications Hybrid Cell Count | KEYENCE CORPORATION | https://www.keyence.com |
Highlights.
En masse liver organoid analysis informs NASH genotype-phenotype associations
Gene-editing in organoids delineate GCKR-rs1260326 impact on glycolysis and lipogenesis
GCKR variant has opposing impacts on NASH severity with or without diabetes.
Mitochondrial dysfunction is associated with GCKR variant-defined diabetic NASH
ACKNOWLEDGMENTS
We would like to express sincere gratitude to members of the Takebe groups, James Wells and Aaron Zorn groups for their support and excellent technical assistance. We also thank for strategic guidance by Spyros Goulas and the Stellar Science Foundation team and Dr. Yukinari Okada for his critical reading of the manuscript. Dr. Peng Gao conducted mass-spec analyses at the Northwestern University Metabolomics Core. This work was supported by Cincinnati Children’s Research Foundation grant, CURE award, NIH Director’s New Innovator Award (DP2 DK128799-01), and CREST (JP22gm1210012) grant from Japan Agency for Medical Research and Development (AMED). This work was also supported by an NIH grant UG3/UH3 DK119982, Cincinnati Center for Autoimmune Liver Disease Fellowship Award, PHS Grant P30 DK078392 (Integrative Morphology Core and Pluripotent Stem Cell and Organoid Core) of the Digestive Disease Research Core Center in Cincinnati, the Falk Transformational Awards Program, JST Moonshot R&D Grant Number JPMJMS2033 and JPMJMS2022, Takeda Science Foundation award, Mitsubishi Foundation award and AMED JP20fk0210037, JP20bm0704025, JP21fk0210060, JP21bm0404045, JP22fk0210091, JP22fk0210106, JST JPMJPS2033, JPJSBP220203101, and JSPS JP18H02800. TT is a New York Stem Cell Foundation – Robertson Investigator. This work was also supported by NIH grants DK121875, HL158531 and DK130908 and AG078174 and CCHMC grants Trustee Award, Heart Translational Funds and CuSTOM pilot grant to MQ.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
R.M., M.S. and T.T. are equity holders for The Liver Company, inc. M.K. and T.T. are co-inventors for the technology disclosed in the manuscript.
REFERENCES
- Amireddy N, Puttapaka SN, Vinnakota RL, Ravuri HG, Thonda S, and Kalivendi SV (2017). The unintended mitochondrial uncoupling effects of the FDA-approved anti-helminth drug nitazoxanide mitigates experimental parkinsonism in mice. J Biol Chem 292, 15731–15743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstee QM, Darlay R, Cockell S, Meroni M, Govaere O, Tiniakos D, Burt AD, Bedossa P, Palmer J, Liu YL, et al. (2020). Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort(). J Hepatol 73, 505–515. [DOI] [PubMed] [Google Scholar]
- Beer NL, Tribble ND, McCulloch LJ, Roos C, Johnson PR, Orho-Melander M, and Gloyn AL (2009). The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet 18, 4081–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belalcazar LM, Papandonatos GD, Erar B, Peter I, Alkofide H, Balasubramanyam A, Brautbar A, Kahn SE, Knowler WC, Ballantyne CM, et al. (2016). Lifestyle Intervention for Weight Loss and Cardiometabolic Changes in the Setting of Glucokinase Regulatory Protein Inhibition: Glucokinase Regulatory Protein-Leu446Pro Variant in Look AHEAD. Circ Cardiovasc Genet 9, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellentani S, Scaglioni F, Marino M, and Bedogni G (2010). Epidemiology of non-alcoholic fatty liver disease. Dig Dis 28, 155–161. [DOI] [PubMed] [Google Scholar]
- Cai W, Weng DH, Yan P, Lin YT, Dong ZH, Mailamuguli, and Yao H (2019). Genetic polymorphisms associated with nonalcoholic fatty liver disease in Uyghur population: a case-control study and meta-analysis. Lipids Health Dis 18, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P, Holm H, Sanna S, Kavousi M, Baumeister SE, et al. (2011). Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet 43, 1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K, and Ratner N (2019). iGEAK: an interactive gene expression analysis kit for seamless workflow using the R/shiny platform. BMC Genomics 20, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati B, Motta BM, Pingitore P, Meroni M, Pietrelli A, Alisi A, Petta S, Xing C, Dongiovanni P, del Menico B, et al. (2016). The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology 63, 787–798. [DOI] [PubMed] [Google Scholar]
- Everhart JE, and Bambha KM (2010). Fatty liver: think globally. Hepatology 51, 1491–1493. [DOI] [PubMed] [Google Scholar]
- Ferguson D, and Finck BN (2021). Emerging therapeutic approaches for the treatment of NAFLD and type 2 diabetes mellitus. Nature Reviews Endocrinology 17, 484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermini B, Coyne ST, and Coyne KP (2018). Clinical Trials in a Dish: A Perspective on the Coming Revolution in Drug Development. SLAS Discov 23, 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Liu S, Zhao Z, Yu X, Liu Q, Xin Y, and Xuan S (2019). Association of GCKR Gene Polymorphisms with the Risk of Nonalcoholic Fatty Liver Disease and Coronary Artery Disease in a Chinese Northern Han Population. J Clin Transl Hepatol 7, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarino M, and Dufour JF (2019). Nicotinamide and NAFLD: Is There Nothing New Under the Sun? Metabolites 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale C, Lloyd DJ, Pellacani A, and Veniant MM (2015). Molecular targeting of the GK-GKRP pathway in diabetes. Expert Opin Ther Targets 19, 129–139. [DOI] [PubMed] [Google Scholar]
- Harrison SA, Wong VW, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, Kohli A, Sarin S, Caldwell SH, Alkhouri N, et al. (2020). Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J Hepatol 73, 26–39. [DOI] [PubMed] [Google Scholar]
- Hernaez R, McLean J, Lazo M, Brancati FL, Hirschhorn JN, Borecki IB, Harris TB, Nguyen T, Kamel IR, Bonekamp S, et al. (2013). Association between variants in or near PNPLA3, GCKR, and PPP1R3B with ultrasound-defined steatosis based on data from the third National Health and Nutrition Examination Survey. Clin Gastroenterol Hepatol 11, 1183–1190.e1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn JN, and Daly MJ (2005). Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6, 95–108. [DOI] [PubMed] [Google Scholar]
- Hotta K, Yoneda M, Hyogo H, Ochi H, Mizusawa S, Ueno T, Chayama K, Nakajima A, Nakao K, and Sekine A (2010). Association of the rs738409 polymorphism in PNPLA3 with liver damage and the development of nonalcoholic fatty liver disease. BMC Medical Genetics 11, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudert CA, Selinski S, Rudolph B, Bläker H, Loddenkemper C, Thielhorn R, Berndt N, Golka K, Cadenas C, Reinders J, et al. (2019). Genetic determinants of steatosis and fibrosis progression in paediatric non-alcoholic fatty liver disease. Liver Int 39, 540–556. [DOI] [PubMed] [Google Scholar]
- Kawaguchi T, Sumida Y, Umemura A, Matsuo K, Takahashi M, Takamura T, Yasui K, Saibara T, Hashimoto E, Kawanaka M, et al. (2012). Genetic polymorphisms of the human PNPLA3 gene are strongly associated with severity of non-alcoholic fatty liver disease in Japanese. PLoS One 7, e38322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keebler ME, Deo RC, Surti A, Konieczkowski D, Guiducci C, Burtt N, Buxbaum SG, Sarpong DF, Steffes MW, Wilson JG, et al. (2010). Fine-mapping in African Americans of 8 recently discovered genetic loci for plasma lipids: the Jackson Heart Study. Circ Cardiovasc Genet 3, 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendig KI, Baheti S, Bockol MA, Drucker TM, Hart SN, Heldenbrand JR, Hernaez M, Hudson ME, Kalmbach MT, Klee EW, et al. (2019). Sentieon DNASeq Variant Calling Workflow Demonstrates Strong Computational Performance and Accuracy. Front Genet 10, 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen H, Goncalves A, Leha A, Afzal V, Alasoo K, Ashford S, Bala S, Bensaddek D, Casale FP, Culley OJ, et al. (2017). Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 546, 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Azuma M, Zhang RR, Thompson W, Mayhew CN, and Takebe T (2018). Digitalized Human Organoid for Wireless Phenotyping. iScience 4, 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto A, Kitamoto T, Nakamura T, Ogawa Y, Yoneda M, Hyogo H, Ochi H, Mizusawa S, Ueno T, Nakao K, et al. (2014). Association of polymorphisms in GCKR and TRIB1 with nonalcoholic fatty liver disease and metabolic syndrome traits. Endocr J 61, 683–689. [DOI] [PubMed] [Google Scholar]
- Koido M, Kawakami E, Fukumura J, Noguchi Y, Ohori M, Nio Y, Nicoletti P, Aithal GP, Daly AK, Watkins PB, et al. (2020). Polygenic architecture informs potential vulnerability to drug-induced liver injury. Nat Med 26, 1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Chang PF, Chang MH, and Ni YH (2014). Genetic variants in GCKR and PNPLA3 confer susceptibility to nonalcoholic fatty liver disease in obese individuals. Am J Clin Nutr 99, 869–874. [DOI] [PubMed] [Google Scholar]
- Lloyd DJ, St Jean DJ Jr., Kurzeja RJ, Wahl RC, Michelsen K, Cupples R, Chen M, Wu J, Sivits G, Helmering J, et al. (2013). Antidiabetic effects of glucokinase regulatory protein small-molecule disruptors. Nature 504, 437–440. [DOI] [PubMed] [Google Scholar]
- Loomba R, Noureddin M, Kowdley KV, Kohli A, Sheikh A, Neff G, Bhandari BR, Gunn N, Caldwell SH, Goodman Z, et al. (2021). Combination Therapies Including Cilofexor and Firsocostat for Bridging Fibrosis and Cirrhosis Attributable to NASH. Hepatology 73, 625–643. [DOI] [PubMed] [Google Scholar]
- Loomba R, Schork N, Chen CH, Bettencourt R, Bhatt A, Ang B, Nguyen P, Hernandez C, Richards L, Salotti J, et al. (2015). Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 149, 1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrka MC, Watanabe M, Muraleedharan R, Lambert PF, Lane AN, Romick-Rosendale LE, and Wells SI (2017). Overexpression of the human DEK oncogene reprograms cellular metabolism and promotes glycolysis. PLoS One 12, e0177952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki Y, Petersen MC, Zhang D, Vatner DF, Perry RJ, Abulizi A, Haedersdal S, Zhang XM, Butrico GM, Samuel VT, et al. (2020). Metabolic control analysis of hepatic glycogen synthesis in vivo. Proc Natl Acad Sci U S A 117, 8166–8176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orho-Melander M, Melander O, Guiducci C, Perez-Martinez P, Corella D, Roos C, Tewhey R, Rieder MJ, Hall J, Abecasis G, et al. (2008). Common missense variant in the glucokinase regulatory protein gene is associated with increased plasma triglyceride and C-reactive protein but lower fasting glucose concentrations. Diabetes 57, 3112–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi R, Togo S, Kimura M, Shinozawa T, Koido M, Koike H, Thompson W, Karns RA, Mayhew CN, McGrath PS, et al. (2019). Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab 30, 374–384.e376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik JM, Kabbara K, Eberly KE, Younossi Y, Henry L, and Younossi ZM (2022). Global burden of NAFLD and chronic liver disease among adolescents and young adults. Hepatology 75, 1204–1217. [DOI] [PubMed] [Google Scholar]
- Petta S, Miele L, Bugianesi E, Cammà C, Rosso C, Boccia S, Cabibi D, Di Marco V, Grimaudo S, Grieco A, et al. (2014). Glucokinase regulatory protein gene polymorphism affects liver fibrosis in non-alcoholic fatty liver disease. PLoS One 9, e87523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollin TI, Jablonski KA, McAteer JB, Saxena R, Kathiresan S, Kahn SE, Goldberg RB, Altshuler D, Florez JC, and Diabetes Prevention Program Research, G. (2011). Triglyceride response to an intensive lifestyle intervention is enhanced in carriers of the GCKR Pro446Leu polymorphism. J Clin Endocrinol Metab 96, E1142–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramli MNB, Lim YS, Koe CT, Demircioglu D, Tng W, Gonzales KAU, Tan CP, Szczerbinska I, Liang H, Soe EL, et al. (2020). Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 159, 1471–1486 e1412. [DOI] [PubMed] [Google Scholar]
- Rich NE, Oji S, Mufti AR, Browning JD, Parikh ND, Odewole M, Mayo H, and Singal AG (2018). Racial and Ethnic Disparities in Nonalcoholic Fatty Liver Disease Prevalence, Severity, and Outcomes in the United States: A Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol 16, 198–210 e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinella M, and Charlton M (2016). The globalization of nonalcoholic fatty liver disease: Prevalence and impact on world health. Hepatology 64, 19–22. [DOI] [PubMed] [Google Scholar]
- Rooney JP, Ryde IT, Sanders LH, Howlett EH, Colton MD, Germ KE, Mayer GD, Greenamyre JT, and Meyer JN (2015). PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol Biol 1241, 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahdeo S, Tomilov A, Komachi K, Iwahashi C, Datta S, Hughes O, Hagerman P, and Cortopassi G (2014). High-throughput screening of FDA-approved drugs using oxygen biosensor plates reveals secondary mitofunctional effects. Mitochondrion 17, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, Narita A, Konuma T, Yamamoto K, Akiyama M, et al. (2021). A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet 53, 1415–1424. [DOI] [PubMed] [Google Scholar]
- Samuel VT, and Shulman GI (2019). Nonalcoholic Fatty Liver Disease, Insulin Resistance, and Ceramides. N Engl J Med 381, 1866–1869. [DOI] [PubMed] [Google Scholar]
- Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, Dykas DJ, Bale AE, Giannini C, Pierpont B, et al. (2012). Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 55, 781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Pollin TI, Damcott CM, McLenithan JC, Mitchell BD, and Shuldiner AR (2009). Glucokinase regulatory protein gene polymorphism affects postprandial lipemic response in a dietary intervention study. Hum Genet 126, 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozawa T, Kimura M, Cai Y, Saiki N, Yoneyama Y, Ouchi R, Koike H, Maezawa M, Zhang RR, Dunn A, et al. (2021). High-Fidelity Drug-Induced Liver Injury Screen Using Human Pluripotent Stem Cell-Derived Organoids. Gastroenterology 160, 831–846 e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, Eiriksdottir G, Garcia ME, Launer LJ, et al. (2011). Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 7, e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebe T, Sekine K, Kimura M, Yoshizawa E, Ayano S, Koido M, Funayama S, Nakanishi N, Hisai T, Kobayashi T, et al. (2017). Massive and Reproducible Production of Liver Buds Entirely from Human Pluripotent Stem Cells. Cell Rep 21, 2661–2670. [DOI] [PubMed] [Google Scholar]
- Takebe T, and Wells JM (2019). Organoids by design. Science 364, 956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Lancet Gastroenterology H. (2020). Headway and hurdles in non-alcoholic fatty liver disease. Lancet Gastroenterol Hepatol 5, 93. [DOI] [PubMed] [Google Scholar]
- Tomita K, Kohli R, MacLaurin BL, Hirsova P, Guo Q, Sanchez LHG, Gelbard HA, Blaxall BC, and Ibrahim SH (2017). Mixed-lineage kinase 3 pharmacological inhibition attenuates murine nonalcoholic steatohepatitis. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon G, Baranova A, and Younossi ZM (2011). Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 34, 274–285. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research 38, e164–e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, An H, Liu T, Qin C, Sesaki H, Guo S, Radovick S, Hussain M, Maheshwari A, Wondisford FE, et al. (2019). Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep 29, 1511–1523 e1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Roth TL, Bauer SJ, Lane A, and Romick-Rosendale LE (2016). Feasibility Study of NMR Based Serum Metabolomic Profiling to Animal Health Monitoring: A Case Study on Iron Storage Disease in Captive Sumatran Rhinoceros (Dicerorhinus sumatrensis). PLoS One 11, e0156318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel J, and Marrero D (2016). Do Genes Determine Our Health? Implications for Designing Lifestyle Interventions and Drug Trials. Circ Cardiovasc Genet 9, 2–3. [DOI] [PubMed] [Google Scholar]
- Xia MF, Bian H, and Gao X (2019). NAFLD and Diabetes: Two Sides of the Same Coin? Rationale for Gene-Based Personalized NAFLD Treatment. Front Pharmacol 10, 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh KH, Hsu LA, Teng MS, Wu S, Chou HH, and Ko YL (2022). Pleiotropic Effects of Common and Rare GCKR Exonic Mutations on Cardiometabolic Traits. Genes (Basel) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Stepanova M, Younossi I, and Racila A (2019). Validation of Chronic Liver Disease Questionnaire for Nonalcoholic Steatohepatitis in Patients With Biopsy-Proven Nonalcoholic Steatohepatitis. Clin Gastroenterol Hepatol 17, 2093–2100.e2093. [DOI] [PubMed] [Google Scholar]
- Younossi ZM, Tampi RP, Racila A, Qiu Y, Burns L, Younossi I, and Nader F (2020). Economic and Clinical Burden of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes in the U.S. Diabetes Care 43, 283–289. [DOI] [PubMed] [Google Scholar]
- Zain SM, Mohamed Z, and Mohamed R (2015). Common variant in the glucokinase regulatory gene rs780094 and risk of nonalcoholic fatty liver disease: a meta-analysis. J Gastroenterol Hepatol 30, 21–27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Development of a multi-cellular human liver organoid panel from mixed foregut progenitors, Related to Figure 1. (A) UMAP plot and heatmap analysis by scRNAseq of population organoid panel (PoP). (B) Cell type-specific marker analysis of PoP. (C) Schematic diagram of the Donor decoding method of PoP using donor-specific SNP genotyping. The gDNA of each donor-derived iPSC was extracted and the SNP profile was obtained by SNP array. Based on the SNP profile, donor-specific SNP profiles were constructed. (D) The presence of 20 donors in the PoP was confirmed by donor-specific SNPs. (E) Individual HLO morphology in PoP. (F) Clonality analysis of PoP.
Figure S2. Human organoid modeling of NAFLD-like phenotype induced by fatty acid loading, Related to Figure 1 and 3. (A, B) Live imaging of accumulating lipid droplets in fatty acid-loaded HLO. Shown, HLO without fatty acid loading and sHLO treated with fatty acid for 72 hours. Lipid droplets stained green with BODIPY and nuclei stained with Hoechst 33342. Scale bars, 50μm. (C, D) Imaging of lipid droplets by transmission electron microscopy (TEM), with (C) representing HLO and (D) representing sHLO. Yellow arrows indicate accumulated lipid droplets. Scale bars, 10μm. (E) Analysis of non-polar metabolites of sHLO by nuclear magnetic resonance (NMR) spectroscopy. Metabolites of sHLO induced by fatty acid treatment for 72 h were measured. Quantifications were performed in five independent donor iPSC lines. Unpaired t-test: *p < 0.05, ****p < 0.0001. (F) Supernatant analysis of secreted triglyceride and cholesterol from sHLO after 72 hours of fatty acid treatment. Data are shown as means ± SD (error bars), n=6. Unpaired t-test; ****p < 0.0001. (G) Mass-spec analysis of protein content-normalized levels of acetyl-CoA, palmitate (C16 – long chain saturated fatty acid), octanoyl-carnitine (C8 – medium chain acyl-carnitine) and palmitoyl-carnitine (C16 - long-chain acyl-carnitine). (H) ELISA analysis of secreted pro-inflammatory cytokines from sHLO after 72 hours of fatty acid treatment. Data are shown as means ± SD (error bars), n=9. Unpaired t-test; **p < 0.01, ****p < 0.0001. (I) Analysis of pro-inflammatory cytokine gene expressions in sHLO. Proinflammatory cytokine gene expression was measured by qPCR in sHLOs after 72 hours of fatty acid treatment. Data are shown as means ± SD. (error bars), n=4, Unpaired t-test; **p < 0.01, ***p < 0.001. (J) Measurement of glucose production in HLO and sHLO. The amount of glucose was normalized to the CellTiter-Glo value (mean ± SD, n = 9 independent experiments per line). Unpaired t-test; ****p < 0.0001. (K) Gene expression analysis (qPCR) of PCK1 in HLO and sHLO (mean ± SD, n = 8 independent experiments). Unpaired t-test; ****p < 0.0001. (L) Western blot analysis to determine insulin responsiveness of sHLO. HLOs and sHLOs were starved for 24 hours and treated with 1 μg/ml of insulin for 20 minutes. (M) Glucose production assay to determine insulin responsiveness of sHLO. HLOs and sHLOs were starved for 24 hours and treated with 1μM of insulin for 12 hours. The amount of glucose in the collected supernatant was measured. The amount of glucose was normalized to the CellTiter-Glo value (mean ± SD, n = 9 independent experiments per line). Unpaired t-test; ***p < 0.001.
Figure S3. Gene-targeting for GCKR-rs1260326 locus in hiPSC, Related to Figure 2. (A) Schematic of the gene-targeting procedure for GCKR-rs1260326 mutations using the MhAX method. Details of the targeting and genotyping procedures are provided in B, C, and E. (B) Design of guide RNA targeting GCKR locus. The sgRNA sequence is colored in purple. The red letter indicates the target base for editing no-risk C allele into risk G allele. The blue letter indicates the non-sense mutation found in the genome of the parental human iPSC line. (C) Detailed schematic of GCKR targeting vector. Homology arm (HA) overlap generates a 51 bp tandem microhomology (μH, blue), with the mutation (C>T, red). The two target sequences by ps1 sgRNA (green), which is used for cassette excision, are inserted next to the μH-T and μH-C sequences. (D) Representative FACS plot for the targeting and excision steps. (E) Sanger sequencing revealed the genotype of GCKR edited iPSC clones obtained from a single excision step. The arrows indicate rs1260326 C/T single nucleotide variation. (F) Representative bright field views of the GCKR edited iPSC clones. Scale bar, 500 μm. (G) Representative immunofluorescent images of pluripotency marker Oct4 in the GCKR edited iPSC clones. Scale bar, 100 μm. (H) Normal karyotype (46, XX) in the GCKR edited iPSC clones by chromosomal G-banding analysis.
Figure S4. Impact of PNPLA3 rs738409 variant in NASH patients stratified by HbA1c levels, Related to Figure 3 and Figure5. Graphic depiction of selected clinical parameters from Table 1, delineated by PNPLA3 variant and HbA1c values. (A) ALT measurements. (B) NAFLD activity score (NAS). (C) Lobular inflammation scores. (D) SAF activity score. (E) GCKR-rs1260326 confers differential metformin sensitivity in NAFLD patients. Graphic depiction of the effects of metformin treatment on NAS. Patients carrying GCKRTT did not show improvement in NAS after 48 weeks of metformin treatment, in contrast to patients carrying GCKRCC or CT.
Figure S5. GCKR-rs1260326 TT risk variant impacts lipid accumulation and enhances inflammatory responses in sHLO, Related to Figure 4. (A) Live imaging of accumulating lipid droplets in sHLO with GCKRCC or GCKRTT alleles, denoted as “CC” or “TT”. Images were stained with BODIPY for fat accumulation (Green) and Hoechst 33342 for the nucleus (Blue). Scale bars, 100μm, 50μm. (B) Quantification of lipid droplets in sHLOs. The intensity of lipid was normalized to nuclear signals. Unpaired t-test; ****p < 0.0001. (C - E) RNA sequence analysis of lipid-associated genes, glucose/fatty acid uptake genes, and fatty acid oxidation genes in sHLO. Data are shown as means ± SD (error bars), n=6. Unpaired t-test; *p < 0.05, **p < 0.001, ***p < 0.001, ****p < 0.0001. (F) Pathways enriched in GCKRTT HLOs, compared to GCKRCC HLOs, with NES > 1.2. (G) GSEA evaluation of gene-expression profile enriched in GCKRTT sHLO. REACTOME FATTY ACIDS (NES 1.68, p-value=0.002, FDR <0.3039), REACTOME INTERFERON GAMMA SIGNALING (NES 1.77, p-value<0.001 FDR <0.068). (H) qPCR analysis of pro-inflammatory cytokine gene expressions in sHLO. Data are shown as means ± SD (error bars), n=4, three independent experiments. Unpaired t-test; **p < 0.01, ***p < 0.001, ****p < 0.0001.
Figure S6. Mitochondrial dysregulation is associated with GCKR-rs1260326 related metabolic assaults, Related to Figure 5. (A) Quantifications of mitophagy events in GCKRTT HLO and GCKRTT sHLO (FFA treated). Relative levels of mitophagy were expressed as the ratio between Mitophagy Dye and Hoechst 33342 fluorescence intensity. Analysis was performed in >50 organoids per line, two independent experiments. (B) Relative protein levels of mitophagy-related proteins parkin (PRKN) and mitofusin 2 (MFN2) were analyzed by flow cytometry. Analysis was performed in >50 organoids per line, five independent experiments. Data in HLO was arbitrarily assigned a value of 1 and shown as means ± SD (error bars). Unpaired t-test; **p < 0.01. (C) Transmission electron microscopy (TEM) analysis. Representative electron micrographs identified the mitochondrial presence in GCKRTT-HLO (left) and GCKRTT-sHLO (right) samples. Mitochondrial areas are indicated by yellow circles. (D) Mitochondrial DNA copy number of GCKRTT HLO (white), -sHLO (gray), and -sHLO treated with nicotinamide riboside (NR), nitazoxanide (NTZ) combination (green). The mtDNA copy number was divided by the total amount of nuclear DNA, then each data was normalized to fold change over the HLO samples. Data are shown as means ± SD. (error bars), n=4, Tukey test; *p < 0.05, **p < 0.01. (E) Quantifications of ROS production in GCKRTT HLO and GCKRTT HLO treated with a combination of NR/NTZ. Mitochondrial ROS production was detected with MitoSOX live staining and Hoechst 33342 for the nucleus. The intensity of mitochondrial ROS was normalized to nuclear signals. Analysis was performed in >50 organoids per line. Unpaired t-test; ***p < 0.001.
Table S1. iPSC line information in this study. Related to Figure 1 and Star Methods.
Table S2. Demographic and baseline characteristics in a cohort of 1091 NAFLD patients. Related to Table 1 and Figure 3.
Table S3. Clinical sample information provided in the RNA sequence. Related to Figure 5.
Table S4. A list of Differentially Expressed Genes (DEG) by GCKR variants of the clinical and HLO/sHLO samples analyzed in this study. Related to Figure 5 and S5.
Table S5. Liver tests and histology after 48 weeks of metformin treatment in NAFLD patients. Related to Table 1 and Figure S4.
Data Availability Statement
The processed gene expression data in this paper have been deposited into the NCBI GEO database: GSE213932 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE213932).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.