

Abstract
Background
Liver fibrosis is a pathological wound-healing response caused by chronic liver damage. Mitochondria regulate hepatic energy metabolism and oxidative stress. Accumulating evidence has revealed that increased mitochondrial oxidative stress contributes to the activation of fibrogenesis. However, the roles and underlying mechanisms of mitochondrial oxidative stress in liver fibrosis remain unknown.
Methods and results
In this study, C57BL/6 mice were used to establish a model of liver fibrosis via oral gavage with CCl4 treatment for 8 weeks. Furthermore, intervention experiments were achieved by CCl4 combined with the intraperitoneal injection of mitoquinone mesylate (mitoQ). We demonstrated that the chronic CCl4 exposure resulted in severe hepatic fibrogenesis and significantly promoted the production of reactive oxygen species (ROS) and mitochondrial abnormalities. Besides, JNK/YAP pathway was also activated. By contrast, the administration of mitoQ markedly inhibited the expression of pro-fibrogenic transforming growth factor-β as well as type I collagen. The antifibrotic effects of mitoQ were also confirmed by hematoxylin and eosin staining and Sirius red staining. Moreover, mitoQ substantially reduced CCl4-induced mitochondrial damage and the release of ROS. Further studies suggested that this protection against liver fibrosis was mechanistically related to the inhibition of phosphorylation of JNK and the nuclear translocation of YAP.
Conclusion
In conclusion, these findings revealed that mitoQ attenuated liver fibrosis by inhibiting ROS production and the JNK/YAP signaling pathway. Selective targeting JNK/YAP may serve as a therapeutic strategy for retarding progression of chronic liver disease.
Keywords: mitochondrial oxidative stress, liver fibrosis, JNK/YAP, MitoQ
1. Introduction
Liver fibrosis characterized by the deposition of extracellular matrix (ECM) is a reversible wound-healing response triggered by acute or chronic liver injury.1,2 Hepatic stellate cell (HSC) is the primary source of ECM in liver upon injury, and their activation represents a critical event in fibrosis.3 Cirrhosis is the terminal stage of progressive liver fibrosis, which is estimated to affect 1–2% of global population and results in over 1 million deaths annually worldwide.4,5 However, there is still a huge unmet medical need for anti-fibrotic therapies to prevent liver disease progression and hepatocellular carcinoma (HCC) development. In recent years, researches have shown that oxidative stress plays an important role in the occurrence and development of liver fibrosis, and many of the factors that cause liver injury are associated with the increase of oxidative level or the decrease of antioxidant capacity in varying degrees.6 Therefore, the relationship between oxidative stress and liver fibrosis’ pathological mechanism is attracting more and more attention.
Mitochondrial oxidative damage contributes to a range of pathologies including steatosis and insulin resistance (IR), diabetes, nonalcoholic steatohepatitis (NASH), or various stages of alcoholic steatohepatitis.7–10 Previous study has demonstrated that liver tissues from patients with obesity and NASH have ultrastructural damage to the mitochondria,11,12 involving in the mechanism of high mitochondrial levels of reactive oxygen species (ROS) and ROS-mediated mitochondrial DNA (mtDNA) damage.10,13 Low and intermediate levels of ROS are physiologically important as cell signaling molecules, whereas high ROS concentrations that preclude cell clearance cause oxidative stress, mitochondrial dysfunction, and cellular and DNA damage that can result in inflammation and cell death.14,15 More importantly, ROS are believed to play an important role in inducing the proliferation of HSCs, the migration of HSCs, collagen accumulation, and the formation of liver fibrosis.16,17 It implies that ROS generation may be considered as one of the mechanisms of CCl4-induced liver fibrosis.18
The roles for mitochondrial ROS-induced MAPK signaling pathways in the development of NAFLD, NASH, fibrosis, and IR have been investigated in animal models—sustained activation and mitochondrial translocation of c-Jun N-terminal kinase (JNK) mediate liver injury.19 Phosphorylated JNK translocate to mitochondria where it binds to MOM and MIM scaffold proteins to inhibit MRC, increasing ROS formation.19,20 Furthermore, reducing production of mitochondrial ROS and inhibiting interactions between phosphorylated JNK and mitochondria reduce liver injury in mouse models.19 Besides, recent studies have shown that JNK is an upstream molecule of YAP and regulate the activation of YAP.21,22 For example, one study demonstrated that the JNK inhibitor (SP600125) blocked mechanical-tension-induced YAP activation.22 Importantly, activation of YAP promotes fibrosis in multiple organs, including the liver, kidney, skin, and lung.23–25 Mechanistic studies have revealed that YAP drives HSC activation and fibrotic programs regulated by Hedgehog signaling,26,27 and it is an important regulator of biology in liver diseases.28 Targeting YAP in fibroblasts has been shown to attenuate fibrosis.29,30 However, the mechanism link between mitochondrial oxidative stress and JNK/YAP pathway in liver fibrosis remains largely unclear.
Mitoquinone mesylate (mitoQ), mitochondria-targeted antioxidant, comprises coenzyme Q10 and TPP cations, which makes it several hundred-fold more potent than an untargeted antioxidant with respect to the prevention of mitochondrial oxidative damage.31 MitoQ plays a protective role in various diseases, such as neurodegenerative disease, diabetic kidney disease, and liver fibrosis.32–35 Rehman et al.33 demonstrated that mitoQ ameliorates liver fibrosis in vivo and directly inhibits HSC activation in vitro. However, its exact role and mechanism remain unclear, especially whether mitoQ is related to JNK/YAP signaling pathway in the process of suppressing CCl4-induced liver fibrosis.
This study was designed to investigate the roles of mitochondrial oxidative stress on HSC activation driven by chronic CCl4 exposure and to determine the potential mechanisms by investigating the roles of mitochondrial oxidative stress in JNK/YAP pathway. We found that CCl4 administration induced the mass production of ROS and mitochondrial damage. Moreover, JNK/YAP pathway was significantly activated by chronic CCl4 exposure, thereby increasing HSC activation. More importantly, our results revealed that the inhibition of mitochondrial oxidative stress effectively prevented mitochondrial damage and the occurrence of CCl4-induced liver fibrosis. Mechanistically, mitoQ significantly inhibited the activation of HSCs through regulating the phosphorylation levels of JNK and nuclear transcription of YAP. Therefore, our study demonstrates that mitochondrial oxidative damage involves in liver fibrosis and may be attributed to the modulation of JNK/YAP pathway, which underline the hepatoprotective effects of mitoQ against chronic liver disease.
2. Materials and methods
2.1 Materials
CCl4 (purity ≥99%) was purchased from Shanghai Macklin Biochemical Co., Ltd. MitoQ (purity ≥98.0%) was purchased from Med Chem Express (Monmouth Junction, NJ). Horseradish peroxidase (HRP)-conjugated goat anti-mouse/rabbit IgG SABC kit (SA1020) and diaminobenzidine (DAB) chromogenic reagent kit (AR1027-3) were purchased from Boster Biological Technology, Wuhan, China. Primary antibodies used in this study include anti-COL1A1 (CST, #91144), α-SMA (CST, #19245s), JNK (Santa Cruz, sc-7345), p-JNK (CST, #4668), LATS1 (CST, #3477), YAP (CST, #4912) and YAP (Santa Cruz, sc-101199), p-YAP (CST, #4911), transforming growth factor-β (TGF-β, CST, 3711s), and TGF-β (LBI1E12). Monoclonal anti-β-actin, peroxidase-conjugated goat anti-rabbit and goat anti-mouse IgG secondary antibodies, and protease inhibitor cocktail were purchased from Sigma-Aldrich Chemical Co. Alexa Fluor 568 goat anti-mouse IgG (H + L), Alexa Fluor 488 goat anti-mouse IgG (H + L), Alexa Fluor® 568 goat anti-rabbit IgG (H + L), Alexa Fluor® 488 Goat Anti-Rabbit IgG (H + L), Hoechst 33343 are from Life Technologies (Carlsbad, CA). NE-PER nuclear and cytoplasmic extraction reagents kits, BCA protein assay kit, and SuperSignal® West Pico chemiluminescent substrate kit were purchased from Pierce Biotechnology, Inc. (Rockford, IL). All other chemicals were of analytical grade from commercial supplier.
2.2 Animals and treatments
Male C57BL/6 mice were purchased from the Experimental Animal Center of Shandong University. The animals were kept in an air-conditioned room at 23–25 °C with a 12 h dark/light cycle for 1 week prior to the initiation of the experiments. All the animals received appropriate care during the study with free access to food and water. Fifty male C57BL/6 mice were randomly divided into five groups to obtain the time-response model (n = 10 mice/group): (i) control group, (ii) 2-week CCl4 group, (iii) 4-week CCl4 group, (iv) 6-week CCl4 group, (v) 8-week CCl4 group. Mice receive escalating doses of CCl4 (50/50 vol. mixed with corn oil) three times per week via oral gavage for 8 weeks, starting with 0.437 ml/kg (first to third dose, week 1), 0.656 ml/kg (fourth dose, week 2), 0.875 ml/kg (fifth to ninth dose, week 2–4), and 1.25 ml/kg (10th–24th dose, week 4–8). Control mice received the same volume of corn oil via oral gavage. Then, animals were sacrificed every 2 weeks. Furthermore, another 40 C57BL/6 mice were divided into four groups for mitoQ intervention experiment (n = 10 mice/group): (i) control group, the mice were oral gavage with corn oil for 8 weeks; (ii) 2 mg/kg mitoQ group, (iii) CCl4 group, the mice were oral gavage with CCl4 for 8 weeks, (iv) CCl4 + 2 mg/kg mitoQ group, the mice were administered both mitoQ and CCl4 for 8 weeks. MitoQ was dissolved in 5% polyethylene glycol 400, 5% Tween 80 and was given intraperitoneally to animals of intervention groups. The mice were euthanized, and liver samples were removed and snap-frozen in liquid nitrogen for storage at −80 °C. An additional section was fixed in 4% paraformaldehyde for 24 h and embedded in paraffin. Serum and liver tissue of all groups were collected for further analysis. All experiments were carried out in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and followed the principles in the “Use of Animals in Toxicology”. The use of animals was approved by the Ethics Committee of Shandong University Institute of Preventive Medicine, and Ethics Committee number is 20190201.
2.3 Serum analyses and histological examination
The blood samples were kept at room temperature for 1 h. After centrifugation at 3,000 g for 15 min, serum was collected to measure the levels of alanine transaminase (ALT) and aspartate transferase (AST) with kits according to the manufacturer’s instructions. Furthermore, a segment of liver was fixed in 4% neutral buffered formalin solution at least 24 h. Then, the liver tissues were dehydrated with a sequence of ethanol solutions, embedded in paraffin wax, and sectioned at 4-μm thickness. At last, the liver tissue sections were stained with hematoxylin and eosin (H&E) for morphological evaluation; Sirius Red staining were used to assess the degree of liver fibrosis and then observed to evaluate histopathological changes under light microscopy. Quantification of the liver damage area and percentage of collagen per field with Sirius Red staining used Image J.
2.4 Immunohistochemical staining
The liver tissues embedded in paraffin were sliced into 4–6-μm pieces, deparaffinized, and serially dehydrated in ethanol. The sections were boiled for 3 min while immersed in EDTA-Tris (pH 9.0) for antigen retrieval to nuclear staining and then treated with hydrogen peroxide for 30 min to inactivate the endogenous peroxidase. The sections were incubated overnight at 4 °C with primary antibody α-SMA. The sections were subsequently incubated with the appropriate biotinylated secondary (goat anti-rabbit /mouse IgG) for 30 min at room temperature, stained with DAB, and counterstained with hematoxylin. The positive areas were colored a brownish yellow and relative α-SMA expression was quantified by Image J.
2.5 Immunofluorescence staining of livers
Paraffin sections of mice livers (5 μm) were firstly deparaffinized and hydrated. Then, antigen retrieval was performed by submerging the sections in citrate buffer and microwaving for 10 min. For immunostaining, the sections were treated with 10% serum, followed by overnight incubation with primary antibodies. The next day, sections were incubated with fluorescent secondary antibodies for 4 h at room temperature. Nuclei were stained with Hoechst 33343 and mounted with SlowFade Antifade Mountant. Finally, stained sections were observed and photographed with Zeiss fluorescence microscope. The relative fluorescence intensity was quantified by Image J.
2.6 Tissue preparation and immunoblot analysis
Total proteins were extracted from mouse liver using RIPA lysis buffer. Tissue homogenates were centrifuged at 12,000 g for 15 min, and then the supernatants were kept for western analysis of target protein. Mitochondrial proteins were extracted from mice livers using the tissue mitochondria isolation kit. The cytosolic and nuclear fractions were prepared as previously described with mild modification.36 Briefly, mice liver was homogenized in the hypotonic buffer (1.5 mM MgCl2, 10 mM KCl, 1 mM dithiothreitol, 10 mM HEPES, 1 mM PMSF, 1 mM Na3VO4, 5 mM NaF, 1% cocktail protein protease inhibitor, pH 7.9). The homogenates were centrifuged at 1,000 × g for 10 min, and the supernatants were collected as the cytosolic fraction. The pellet was lysed in another buffer (1.5 mM MgCl2, 5 mM HEPES, 0.2 mM EDTA, 0.5 mM DTT, 400 mM NaCl, 26% glycerol, 1% cocktail protein protease inhibitor, pH 7.9) and then centrifuged at 24,000 g for 20 min. The supernatant was collected as the nuclear protein fraction. Equivalent amounts of protein samples were separated by SDS-PAGE electrophoresis and transferred to polyvinylidene fluoride(PVDF) membranes. Then the membranes were blocked in nonfat milk for 1 h at room temperature, incubated with primary antibody overnight at 4 °C, and incubated with HRP-conjugated secondary antibody at the room temperature. Subsequently, the membranes were detected by chemiluminescence kit. Finally, the immunoreactive bands of proteins were scanned with Agfa Duoscan T1200 scanner, and the digitize data were quantified as integrated optical density using Image-Pro Plus software.
2.7 mtDNA copy number
mtDNA in the cytosolic fraction was analyzed by quantitative polymerase chain reaction (qPCR). Briefly, liver tissue was lysed with RIPA lysis buffer, homogenized, and incubated on ice for 15 min. The lysates were centrifuged at 13,000 rpm at 4 °C for 15 min. The cytosolic mtDNA from supernatant was detected by DNeasy blood and tissue kit according to manufacturer’s protocol. The relative expression of the genes was detected using the SYBR Premix Ex Taq II kit via real-time fluorescent qPCR. qPCR was carried out in a 20 μl reaction volume containing 10 μl LightCycler 480 SYBR Green I Master (Roche, Shanghai, China), 1 μl forward primer, 1 μl reverse primer, 2 μl DNA, and 6 μl RNase Free dH2O. PCR was carried out in LightCycler 480II qPCR system (Roche). Threshold cycles were determined and used for quantification of initial inputs. The hepatic mtDNA copy number was assessed by determining the copy number of the mtDNA relative to the copy number of the nuclear DNA (18 S rRNA) using quantitative PCR as previously described. qPCR was performed in MicroAmp optical 96-well plates using StepOne PCR system (Applied Biosystems). Primer sequences were:
mtDNA forward—5′-CACCCAAGAACAGGGTTTGT-3′, mtDNA reverse—5′-TGGCCATGGGTATGTTGTTAA-3′,
18 s rRNA forward—5′-TAGAGGGACAAGTGGCGTTC-3′, 18 s rRNA reverse—5′-CGCTGAGCCAGTCAGTGT-3′.
2.8 Statistical analysis
All data were expressed as the mean ± standard deviation. Multiple statistical comparisons between CCl4 groups and control groups were performed by one-way analysis of variance using SPSS software, version 20.0. P value <0.05 was considered statistically significant.
3. Results
3.1 CCl4 induces a severe fibrosis in mice liver
Chronic CCl4 exposure caused severe liver fibrosis, as indicated by the markers of hepatocellular damage and liver fibrogenesis. As shown in Figure 1a, histological analysis with H&E staining showed that liver sections from the CCl4 groups exhibited various degrees of several alterations, including hepatocellular vacuolation, extensive necrosis, and mild inflammatory cell filtration. Sirius staining suggested that CCl4 exposure mice had an obvious increase in collagen deposition and fibrosis compared with control group (Fig. 1a). Consistent with the severity of liver fibrosis detected by Sirius staining, immunofluorescence staining and immunoblot analysis demonstrated that expression of TGFβ and collagen1 were also significantly increased (Fig. 1a and e). The results demonstrated that 8-week CCl4 exposure caused a progressive liver fibrosis.
Fig. 1.

CCl4 induces a severe fibrosis in mice liver. a) Histopathological analysis of representative HE-stained liver samples of CCl4-treated mice with b) the liver damage area quantified (n > 5). Scale bars = 50 μm. Collagen deposition was evaluated by Sirius Red staining with c) percentage of collagen per field quantified (n > 5). Scale bars = 200 μm. The expression of TGF-β was detected by immunofluorescence staining (magnification ×200) with d) relative fluorescence intensity quantified (n > 5). Scale bars = 100 μm. e) Western blots showing the fibrosis protein expression in livers from control or CCl4 treatment mice. The data of target proteins in the experiment groups were normalized to β-actin and then expressed as the mean percentage of the control (100%), n = 6. The data are representative of three independent experiments. *P < 0.05 compared with control group; #P < 0.05 compared with 4-week group.
3.2 CCl4 induces production of ROS and mitochondrial damage in mice
Excessive ROS interferes with the normal function of liver-specific cells and presumably plays a role in the pathogenesis of liver fibrosis.37,38 To assess the effect of CCl4 exposure on liver oxidative stress, we determined the ROS levels using the fluorogenic dye Dihydroethidium(DHE). As shown in Figure 2a, CCl4 challenge led to a significant increase in ROS production, as shown by the strong red fluorescence observed under a fluorescence microscope. Additionally, excessive activation of ROS leads to mitochondrial damage. We next tested the cytoplasmic mtDNA content, which is potential sensitive biomarker to mitochondrial damage. The results showed that the cytoplasmic mtDNA content gradually increased with the aggravation of liver fibrosis (Fig. 2c).
Fig. 2.

The changes of production of ROS and mitochondrial damage in livers of mice treated with CCl4. a) Representative microphotographs of DHE staining in liver sections in each group with b) relative fluorescence intensity quantified (n > 5). Scale bars = 100 μm. c) The cytoplasmic mtDNA contents (n = 6). The data are representative of three independent experiments. *P < 0.05 compared with control group; #P < 0.05 compared with 4-week group.
3.3 CCl4 induces upregulation of JNK/YAP pathway and HSCs activation in mice
Oxidative stress-induced mitochondrial damage has been demonstrated to drive the activity of JNK signaling.19 Therefore, we investigated whether chronic CCl4 exposure induced JNK signaling. As shown in Figure 3a, CCl4 challenge resulted in increased JNK and p-JNK protein levels. In addition, we test downstream signaling of JNK, and the result showed that CCl4 led to a significant increase in YAP nuclear transcription, but the protein levels of LATS1 and p-YAP were decreased (Fig. 3a).
Fig. 3.

The changes of JNK/YAP pathway and HSCs activation in livers of mice treated with CCl4. a) Immunoblot analysis for JNK, p-JNK, LATS1, YAP, and p-YAP in mice liver lysates. The data of target proteins in the experiment groups were normalized to β-actin or Lamin B, then expressed as the mean percentage of the control (100%), n = 6. b) The HSCs activation was detected by α-SMA immunohistochemical staining with relative expression quantified (n > 5). Scale bars = 100 μm. The data are representative of three independent experiments. *P < 0.05 compared with control group; #P < 0.05 compared with 4-week group.
The activation of HSCs is a key event for liver fibrosis. In normal liver, quiescent HSCs secrete a minute amount of α-SMA, which is upregulated shortly after liver fibrosis. Therefore, α-SMA usually acts as an HSC activation marker.39,40 In this study, immunohistochemistry staining revealed no obvious positive staining in the normal control, but the number of α-SMA positive cells was significantly increased in the CCl4 group in a time-dependent manner (Fig. 3b). In summary, the results suggested that CCl4 chronic exposure induced the upregulation of JNK/YAP pathway and the activation of HSCs.
3.4 MitoQ ameliorates liver fibrosis damage in mice challenged with CCl4
Based on above findings, we explored whether mitochondrial oxidative damage was an early event during the development of liver fibrosis. To this end, mice were treated with mitoQ, an inhibitor of mitochondrial oxidative stress. As shown in Figure 4a, the fresh mice livers in the model group were obviously swollen, hard in texture, dark red in color, rough and uneven on the surface, and some had granular protrusions with blunt edges. By contrast, inhibition of mitoQ apparently reversed these changes. In the meantime, treatment with mitoQ significantly attenuated the increase in ALT and AST activities and decreased the areas of liver extensive necrosis and mild inflammatory cell filtration in CCl4-treated mice (Fig. 4b and c). In particular, the activity of serum ALT and AST in mice treated with mitoQ was markedly reduced to the margin level of normal mice. Sirius staining showed that mitoQ-treated mice had a marked reduction in collagen deposition and fibrosis compared with CCl4 exposure group (Fig. 4a). Consistent with histological observation, immunofluorescence and immunoblot analysis further revealed mitoQ significantly attenuated CCl4-induced liver fibrosis by the expression of TGFβ and collagen I (Fig. 4c and g). Taken together, these results support that suppression of mitochondrial oxidative stress attenuates CCl4-induced liver fibrosis.
Fig. 4.
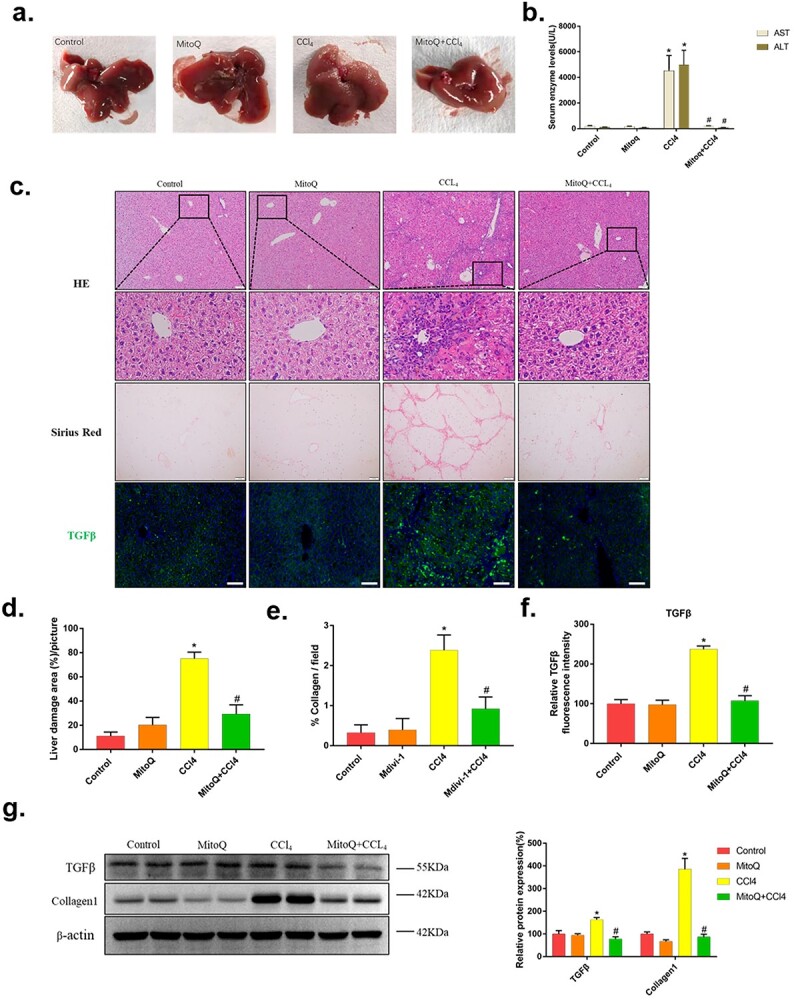
Biochemical assessment, histopathological and liver fibrosis analysis of mice in intervention experiments with mitoQ. a) Observation of fresh liver tissue. b) Serum ALT and AST activities in mitoQ intervention experiment. c) Histopathological analysis of representative HE-stained liver samples of mitoQ-treated mice with d) the liver damage area quantified (n > 5). Scale bars = 50 μm. Collagen deposition was evaluated by Sirius Red staining with e) percentage of collagen per field quantified (n > 5). Scale bars = 200 μm. The expression of TGF-β was detected by immunofluorescence staining (magnification ×200) with f) relative fluorescence intensity quantified (n > 5). Scale bars = 100 μm. g) Western blots showing the fibrosis protein expression in livers from mitoQ intervention mice. The data of target proteins in the experiment groups were normalized to β-actin and then expressed as the mean percentage of the control (100%), n = 6. The data are representative of three independent experiments. *P < 0.05 compared with control group; #P < 0.05 compared with CCl4 group.
3.5 MitoQ suppresses upregulation of JNK/YAP pathway and prevents HSCs activation in CCl4-induced mice
Given the protective role of mitoQ in liver fibrosis, we next sought to further explore possible mechanism. As shown in Figure 5a, CCl4 challenge resulted in increased phosphorylation JNK protein levels, which was significantly suppressed by mitoQ. YAP activity is regulated by phosphorylation, which causes YAP inactivation and nuclear to cytoplasmic translocation. Activation of YAP is thus associated with its translocation from the cytoplasm into the nucleus.41 We found that mitoQ led to a significant increase in the protein levels of LATS1 and p-YAP compared with CCl4-treated liver. In addition, western blotting results showed that fibrous livers had elevated levels of nuclear YAP and decreased levels of cytoplasm YAP. Importantly, mitoQ significantly restored the levels of nuclear YAP and cytoplasm YAP (Fig. 5a). Immunofluorescence result further confirmed that mitoQ effectively blocked CCl4-induced YAP nuclear localization (Fig. 5b). α-SMA, the marker of HSCs activation, was also reduced by mitoQ treatment by the results of immunohistochemistry staining and western blotting (Fig. 5c). These results provided possible evidence that mitoQ inhibit the activation of HSCs by regulating JNK/YAP pathway.
Fig. 5.

The changes in JNK/YAP pathway and HSCs activation in mice liver with mitoQ treatment. a) Immunoblot analysis for JNK, p-JNK, LATS1, YAP, and p-YAP in mice liver lysates. The data of target proteins in the experiment groups were normalized to β-actin or Lamin B, then expressed as the mean percentage of the control (100%), n = 6. b) YAP into the nucleus in mice liver sections by immunofluorescence analysis with YAP-nucleated cells per picture quantified (n > 5). YAP, red; Hoechst 33343, blue. Scale bars = 20 μm. c) The HSCs activation was detected by α-SMA immunohistochemical staining with relative expression quantified (n > 5) and immunoblot analysis in mice liver lysates. Scale bars = 100 μm. The data of target proteins in the experiment groups were normalized to β-actin and then expressed as the mean percentage of the control (100%), n = 6. The data are representative of three independent experiments. *P < .05 compared with control group; #P < .05 compared with CCl4 group.
3.6 MitoQ restores ROS generation and ameliorates mitochondrial damage in CCl4-induced mice
As shown in Figure 6a, we found that mitoQ administration markedly decreased CCl4-induced ROS generation, as shown by no apparent fluorescence under a fluorescence microscope, and the quantitative data showed similar fluorescence intensity between control and mitoQ-treated mice liver (Fig. 6b). Furthermore, mitoQ significantly suppressed the CCl4-induced increase in the cytoplasmic mtDNA production (Fig. 6c), suggesting mitochondrial damage was relieved.
Fig. 6.

The changes in production of ROS and mitochondrial damage in mice liver with mitoQ treatment. a) Representative microphotographs of DHE staining in liver sections in each group with b) relative fluorescence intensity quantified (n > 5). Scale bars = 100 μm. c) The cytoplasmic mtDNA contents (n = 6). The data are representative of three independent experiments. *P < 0.05 compared with control group; #P < 0.05 compared with CCl4 group.
4. Discussion
Previous studies have demonstrated that mitochondrial oxidative stress plays critical roles in the development of liver fibrosis.33,42 Accumulating evidence has also demonstrated that the activation of HSCs is the key cause of liver fibrosis.3,43 ROS, which are produced by mitochondria, induce the transdifferentiation of HSCs and increase the production of collagen; again, antioxidants prevent these effects,44 but the underlying mechanism remains to be elucidated. In this study, we used chronic CCl4 exposure-driven in vivo fibrosis models to mimic liver fibrosis in chronic liver diseases. We found that CCl4 challenge significantly increased the excretion of ROS and mitochondrial damage in mice liver and promoted the activation of JNK/YAP signaling pathway and HSCs. However, mitoQ attenuated CCl4-driven mitochondrial oxidative damage in mouse livers. More importantly, mitoQ effectively suppressed CCl4-induced the increase in the phosphorylation level of JNK and the significant nuclear transcription of YAP and eventually inhibited the activation of HSCs. These results clearly demonstrated that mitoQ ameliorated the CCl4-driven liver fibrosis in mice liver, suggesting that the protective effect of mitoQ against chronic liver diseases might be related to the regulation of JNK/YAP pathway.
Mitochondria play a pivotal role in energy metabolism and are the major site of ROS generation through the electron transport chain.45,46 Oxidative stress is a redox state caused by an imbalance between the production and detoxification of ROS.47 As previously shown by others’ studies, chronic liver diseases are associated with increased oxidative stress.48,49 In this study, we found that chronic CCl4 challenge led to a significant increase in ROS production, as shown by the strong DHE red fluorescence. Additionally, excessive activation of ROS leads to mitochondrial damage revealed by the result of increased cytoplasmic mtDNA content. Considering that elevated oxidative stress in liver fibrosis is one of the major mechanisms leading to HSC activation and that mitochondria are central sites where ROS are produced,50 the present study evaluated the effects of the mitochondria-targeted antioxidant mitoQ on oxidative stress. Our study confirmed that mitoQ effectively inhibited ROS generation and mitochondrial damage in chronic CCl4 exposure-induced liver fibrosis, consistent with the study by Vilaseca et al.51
A previous study reported that excessive ROS may directly damage mtDNA and activate the MAPK signaling pathway.52 Specifically, ROS oxidizes Trx to dissociate from ASK-1 for its activation, resulting in the activation of JNK and p38 pathways.53 In addition, MAPK signaling pathway, especially JNK, is activated in response to different stresses, and this signaling pathway has important roles in the development of liver diseases and injuries, such as NAFLD, ALD, viral hepatitis, fibrosis, inflammation, carcinogenesis, and drug-induced hepatotoxicity.19,54 In the current study, we found that CCl4-treated mice liver exhibited increased protein levels of JNK and phosphorylated JNK. Furthermore, mitoQ significantly suppressed the phosphorylation of JNK, suggesting that mitochondrial oxidative damage resulted in JNK activation in liver fibrosis injury. JNK signaling is considered as a major modulator of liver fibrosis.55 And one study confirmed that JNK1 promotes cholestasis- and CCl4-induced liver fibrosis,56 which is similar to our research. Therefore, it is demonstrated that mitochondrial oxidative stress might play a vital role in CCl4-induced mice liver fibrosis by regulating the activation of JNK.
JNK contributes to YAP nuclear expression.22 YAP is a transcription coactivator known to be a key nuclear effector of HSCs activation.57 Under active conditions, the phosphorylated form of YAP localizes in the cytoplasm and the dephosphorylated form of YAP localizes in the nucleus, where it interacts with other transcription factors.58 In this study, we detected hardly any nuclear YAP expression in the livers of control mice. By contrast, CCl4 administration significantly promoted YAP nuclear location and prevented the expression of phosphorylated form of YAP. Our further research found that the nuclear accumulation of YAP was completely blocked by mitoQ treatment in fibrosis liver. And the phosphorylation of YAP returned to control levels. Our findings are in agreement with previous studies, which reported that JNK and p38 MAPK signaling cascades regulated YAP nuclear expression. And their results demonstrated that JNK inhibitor SP600125 shifted YAP from a predominantly nuclear to predominantly cytoplasmic localization.21,22,59 Therefore, studies from our lab and others have revealed that mitochondrial damage-induced JNK activation did promote nuclear expression of YAP in CCl4-induced liver fibrosis. More importantly, as shown by recent researches that YAP played a vital role in regulating HSCs activation.25,27 Continued activation of stellate cells during chronic liver damage causes excessive matrix deposition and the formation of pathological scar tissue leading to fibrosis.25 Furthermore, transforming growth factor β1 is closely related to HSCs activation and is considered as one of the most important markers of HSCs activation.60,61 We found that CCl4 exposure significantly increased protein levels of TGFβ, which suggesting HSCs activation. Besides, increased TGFβ expression in CCl4-challenged mice was abolished by mitoQ treatment. Liver-specific YAP gene knockout improved IR-induced liver fibrosis and liver dysfunction.62 These results suggest that inactivation of JNK/YAP pathway contributes to the suppression of HSCs activation by mitoQ treatment.
In conclusion, this study demonstrated the novel beneficial effects of mitoQ on liver fibrosis using chronic CCl4 exposure model. The mechanism underlying its effects involve restoring mitochondrial oxidative damage, thereby inhibiting phosphorylation of JNK and nuclear localization of YAP, which eventually results in decreased collagen fiber production in HSCs and liver fibrosis regression (Fig. 7). These results suggest that the mitochondrial-targeted antioxidant mitoQ is an attractive agent for preventing liver fibrosis injury.
Fig. 7.

MitoQ improves CCl4-induced liver fibrosis in mice by inhibiting JNK/YAP pathway. TAZ is a YAP paralog in mammals similarly regulated by the Hippo pathway.63 CCl4 challenge significantly increased the excretion of ROS and mitochondrial damage in mice liver and promoted the activation of JNK/YAP signaling pathway and HSCs. By contrast, mitoQ attenuated CCl4-driven mitochondrial oxidative damage, thereby effectively suppressed CCl4-induced the increase in the phosphorylation level of JNK and the significant nuclear transcription of YAP and eventually inhibited the activation of HSCs and liver fibrosis injury.
Funding
This work was supported by Key Research and Development Plan of Shandong Province (2018GSF118013) and National Natural Science Foundation of China (No. 82173552).
Conflict of interest. The authors declare no conflict of interest.
Authors’ contributions
Fuyong Song conceived of the study. Shulin Shan performed data collection, data analysis. Zhaoxiong Liu produced the figures and finally wrote the initial draft, with overall guidance from Fuyong Song. All authors reviewed and edited the manuscript.
Contributor Information
Shulin Shan, Department of Toxicology and Nutrition, School of Public Health, Cheeloo College of Medicine, Shandong University, 44 West Wenhua Road, Jinan, Shandong 250012, P. R. China.
Zhaoxiong Liu, Department of Toxicology and Nutrition, School of Public Health, Cheeloo College of Medicine, Shandong University, 44 West Wenhua Road, Jinan, Shandong 250012, P. R. China.
Zhidan Liu, Department of Toxicology and Nutrition, School of Public Health, Cheeloo College of Medicine, Shandong University, 44 West Wenhua Road, Jinan, Shandong 250012, P. R. China.
Cuiqin Zhang, Department of Toxicology and Nutrition, School of Public Health, Cheeloo College of Medicine, Shandong University, 44 West Wenhua Road, Jinan, Shandong 250012, P. R. China.
Fuyong Song, Department of Toxicology and Nutrition, School of Public Health, Cheeloo College of Medicine, Shandong University, 44 West Wenhua Road, Jinan, Shandong 250012, P. R. China.
References
- 1. Roeb E. Matrix metalloproteinases and liver fibrosis (translational aspects). Matrix Biol. 2018:68-69:463–473. [DOI] [PubMed] [Google Scholar]
- 2. Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011:6(1):425–456. [DOI] [PubMed] [Google Scholar]
- 3. Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017:121:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mohammad H Forouzanfar, Ashkan Afshin, Lily T Alexander et al. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet (London, England). 2016:388(10053):1659–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ginès P, Krag A, Abraldes JG, Solà E, Fabrellas N, Kamath PS. Liver cirrhosis. Lancet (London, England). 2021:398(10308):1359–1376. [DOI] [PubMed] [Google Scholar]
- 6. Cichoż-Lach H, Michalak A. Oxidative stress as a crucial factor in liver diseases. World J Gastroenterol. 2014:20(25):8082–8091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pessayre D, Berson A, Fromenty B, Mansouri A. Mitochondria in steatohepatitis. Semin Liver Dis. 2001:21(01):57–69. [DOI] [PubMed] [Google Scholar]
- 8. Tell G, Vascotto C, Tiribelli C. Alterations in the redox state and liver damage: hints from the EASL Basic School of Hepatology. J Hepatol. 2013:58(2):365–374. [DOI] [PubMed] [Google Scholar]
- 9. Larosche I, Lettéron P, Fromenty B, Vadrot N, Abbey-Toby A, Feldmann G, Pessayre D, Mansouri A. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J Pharmacol Exp Ther. 2007:321(2):526–535. [DOI] [PubMed] [Google Scholar]
- 10. Pessayre D. Role of mitochondria in non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2007:22(Suppl 1):S20–S27. [DOI] [PubMed] [Google Scholar]
- 11. Pessayre D, Mansouri A, Fromenty B. Nonalcoholic steatosis and steatohepatitis. V. Mitochondrial dysfunction in steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2002:282(2):G193–G199. [DOI] [PubMed] [Google Scholar]
- 12. Sanyal AJ, Campbell–Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001:120(5):1183–1192. [DOI] [PubMed] [Google Scholar]
- 13. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015:21(5):739–746. [DOI] [PubMed] [Google Scholar]
- 14. Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011:194(1):7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lobo V, Patil A, Phatak A, Chandra N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn Rev. 2010:4(8):118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nieto N, Friedman SL, Cederbaum AI. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome P450 2E1-derived reactive oxygen species. Hepatology (Baltimore, MD). 2002:35(1):62–73. [DOI] [PubMed] [Google Scholar]
- 17. Sánchez-Valle V, Chávez-Tapia NC, Uribe M, Méndez-Sánchez N. Role of oxidative stress and molecular changes in liver fibrosis: a review. Curr Med Chem. 2012:19(28):4850–4860. [DOI] [PubMed] [Google Scholar]
- 18. Cheng Q, Li C, Yang CF, Zhong YJ, Wu D, Shi L, Chen L, Li YW, Li L. Methyl ferulic acid attenuates liver fibrosis and hepatic stellate cell activation through the TGF-β1/Smad and NOX4/ROS pathways. Chem Biol Interact. 2019:299:131–139. [DOI] [PubMed] [Google Scholar]
- 19. Win S, Than TA, Zhang J, Oo C, Min RWM, Kaplowitz N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology (Baltimore, MD). 2018:67(5):2013–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Win S, Than TA, Le BH, García-Ruiz C, Fernandez-Checa JC, Kaplowitz N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J Hepatol. 2015:62(6):1367–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Codelia VA, Sun G, Irvine KD. Regulation of YAP by mechanical strain through Jnk and Hippo signaling. Curr Biol. 2014:24(17):2012–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Z, Wu H, Jiang K, Wang Y, Zhang W, Chu Q, Li J, Huang H, Cai T, Ji H, et al. MAPK-mediated YAP activation controls mechanical-tension-induced pulmonary alveolar regeneration. Cell Rep. 2016:16(7):1810–1819. [DOI] [PubMed] [Google Scholar]
- 23. Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V, Probst CK, Hiemer SE, Sisson TH, Horowitz JC, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cellular Mol Physiol. 2015:308(4):L344–L357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Szeto SG, Narimatsu M, Lu M, He X, Sidiqi AM, Tolosa MF, Chan L, de Freitas K, Bialik JF, Majumder S, et al. YAP/TAZ Are Mechanoregulators of TGF-β-Smad signaling and renal fibrogenesis. J Am Soc Nephrol. 2016:27(10):3117–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LFR, Hoorens A, Reynaert H, Halder G, van Grunsven LA. The hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol. 2015:63(3):679–688. [DOI] [PubMed] [Google Scholar]
- 26. Swiderska-Syn M, Xie G, Michelotti GA, Jewell ML, Premont RT, Syn WK, Diehl AM. Hedgehog regulates yes-associated protein 1 in regenerating mouse liver. Hepatology (Baltimore, MD). 2016:64(1):232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, Dalton GD, Thelen E, Rizi BS, Jung Y, et al. Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology. 2018:154(5):1465–1479.e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Manmadhan S, Ehmer U. Hippo signaling in the liver - a long and ever-expanding story. Front Cell Dev Biol. 2019:7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liang M, Yu M, Xia R, Song K, Wang J, Luo J, Chen G, Cheng J. Yap/Taz deletion in Gli(+) cell-derived myofibroblasts attenuates fibrosis. J Am Soc Nephrol. 2017:28(11):3278–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haak AJ, Kostallari E, Sicard D, Ligresti G, Choi KM, Caporarello N, Jones DL, Tan Q, Meridew J, Diaz Espinosa AM, et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci Transl Med. 2019:11(516):eaau6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wani WY, Gudup S, Sunkaria A, Bal A, Singh PP, Kandimalla RJL, Sharma DR, Gill KD. Protective efficacy of mitochondrial targeted antioxidant MitoQ against dichlorvos induced oxidative stress and cell death in rat brain. Neuropharmacology. 2011:61(8):1193–1201. [DOI] [PubMed] [Google Scholar]
- 32. Oyewole AO, Birch-Machin MA. Mitochondria-targeted antioxidants. FASEB J. 2015:29(12):4766–4771. [DOI] [PubMed] [Google Scholar]
- 33. Rehman H, Liu Q, Krishnasamy Y, Shi Z, Ramshesh VK, Haque K, Schnellmann RG, Murphy MP, Lemasters JJ, Rockey DC, et al. The mitochondria-targeted antioxidant MitoQ attenuates liver fibrosis in mice. Int J Physiol Pathophysiol Pharmacol. 2016:8(1):14–27. [PMC free article] [PubMed] [Google Scholar]
- 34. Yin X, Manczak M, Reddy PH. Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington's disease. Hum Mol Genet. 2016:25(9):1739–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang D, Wang J, Qin Y, Liu Y, Tang C, et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017:11:297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yao P, Nussler A, Liu L, Hao L, Song F, Schirmeier A, Nussler N. Quercetin protects human hepatocytes from ethanol-derived oxidative stress by inducing heme oxygenase-1 via the MAPK/Nrf2 pathways. J Hepatol. 2007:47(2):253–261. [DOI] [PubMed] [Google Scholar]
- 37. Luangmonkong T, Suriguga S, Mutsaers HAM, Groothuis GMM, Olinga P, Boersema M. Targeting oxidative stress for the treatment of liver fibrosis. Rev Physiol Biochem Pharmacol. 2018:175:71–102. [DOI] [PubMed] [Google Scholar]
- 38. Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol. 2014:20(39):14205–14218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. An P, Wei LL, Zhao S, Sverdlov DY, Vaid KA, Miyamoto M, Kuramitsu K, Lai M, Popov YV. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat Commun. 2020:11(1):2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alsamman S, Christenson SA, Yu A, Ayad NME, Mooring MS, Segal JM, Hu JKH, Schaub JR, Ho SS, Rao V, et al. Targeting acid ceramidase inhibits YAP/TAZ signaling to reduce fibrosis in mice. Sci Transl Med. 2020:12(557):eaay8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ota M, Sasaki H. Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development (Cambridge, England). 2008:135(24):4059–4069. [DOI] [PubMed] [Google Scholar]
- 42. Dou SD, Zhang JN, Xie XL, Liu T, Hu JL, Jiang XY, Wang MM, Jiang HQ. MitoQ inhibits hepatic stellate cell activation and liver fibrosis by enhancing PINK1/parkin-mediated mitophagy. Open Med (Warsaw, Poland). 2021:16(1):1718–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol. 2013:3(4):1473–1492. [DOI] [PubMed] [Google Scholar]
- 44. Nieto N, Friedman SL, Cederbaum AI. Cytochrome P450 2E1-derived reactive oxygen species mediate paracrine stimulation of collagen I protein synthesis by hepatic stellate cells. J Biol Chem. 2002:277(12):9853–9864. [DOI] [PubMed] [Google Scholar]
- 45. Batandier C, Fontaine E, Kériel C, Leverve XM. Determination of mitochondrial reactive oxygen species: methodological aspects. J Cell Mol Med. 2002:6(2):175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007:12(5):913–922. [DOI] [PubMed] [Google Scholar]
- 47. Wang X, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci. 2010:2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muriel P. Role of free radicals in liver diseases. Hepatol Int. 2009:3(4):526–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Halliwell B. Antioxidant defence mechanisms: from the beginning to the end (of the beginning). Free Radic Res. 1999:31(4):261–272. [DOI] [PubMed] [Google Scholar]
- 50. Murphy MP, Smith RA. Drug delivery to mitochondria: the key to mitochondrial medicine. Adv Drug Deliv Rev. 2000:41(2):235–250. [DOI] [PubMed] [Google Scholar]
- 51. Vilaseca M, García-Calderó H, Lafoz E, Ruart M, López-Sanjurjo CI, Murphy MP, Deulofeu R, Bosch J, Hernández-Gea V, Gracia-Sancho J, et al. Mitochondria-targeted antioxidant mitoquinone deactivates human and rat hepatic stellate cells and reduces portal hypertension in cirrhotic rats. Liver Int. 2017:37(7):1002–1012. [DOI] [PubMed] [Google Scholar]
- 52. Yu L, Liu Z, Qiu L, Hao L, Guo J. Ipatasertib sensitizes colon cancer cells to TRAIL-induced apoptosis through ROS-mediated caspase activation. Biochem Biophys Res Commun. 2019:519(4):812–818. [DOI] [PubMed] [Google Scholar]
- 53. Katagiri K, Matsuzawa A, Ichijo H. Regulation of apoptosis signal-regulating kinase 1 in redox signaling. Methods Enzymol. 2010:474:277–288. [DOI] [PubMed] [Google Scholar]
- 54. Meakin PJ, Chowdhry S, Sharma RS, Ashford FB, Walsh SV, McCrimmon R, Dinkova-Kostova AT, Dillon JF, Hayes JD, Ashford ML. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol Cell Biol. 2014:34(17):3305–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology (Baltimore, MD). 2015:61(3):1066–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kluwe J, Pradere JP, Gwak GY, Mencin A, de Minicis S, Österreicher CH, Colmenero J, Bataller R, Schwabe RF. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 2010:138(1):347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu Y, Lu T, Zhang C, Xu J, Xue Z, Busuttil RW, Xu N, Xia Q, Kupiec-Weglinski JW, Ji H. Activation of YAP attenuates hepatic damage and fibrosis in liver ischemia-reperfusion injury. J Hepatol. 2019:71(4):719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007:21(21):2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Reginensi A, Scott RP, Gregorieff A, Bagherie-Lachidan M, Chung C, Lim DS, Pawson T, Wrana J, McNeill H. Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet. 2013:9(3):e1003380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Aydın MM, Akçalı KC. Liver fibrosis. Turkish J Gastroenterol. 2018:29(1):14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bissell DM, Roulot D, George J. Transforming growth factor beta and the liver. Hepatology (Baltimore, MD). 2001:34(5):859–867. [DOI] [PubMed] [Google Scholar]
- 62. Dai Y, Hao P, Sun Z, Guo Z, Xu H, Xue L, Song H, Li Y, Li S, Gao M, et al. Liver knockout YAP gene improved insulin resistance-induced hepatic fibrosis. J Endocrinol. 2021:249(2):149–161. [DOI] [PubMed] [Google Scholar]
- 63. Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, Zhao S, Xiong Y, Guan KL. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008:28(7):2426–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]