

Abstract
Retinal diseases exhibit extensive genetic heterogeneity and complex etiology with varying onset and severity. Mutations in over 200 genes can lead to photoreceptor dysfunction and/or cell death in retinal neurodegeneration. To deduce molecular pathways that initiate and/or drive cell death, we adopted a temporal multiomics approach and examined molecular and cellular events in newborn and developing photoreceptors before the onset of degeneration in a widely-used Pde6brd1/rd1 (rd1) mouse, a model of autosomal recessive retinitis pigmentosa caused by PDE6B mutations. Transcriptome profiling of neonatal and developing rods from the rd1 retina revealed early downregulation of genes associated with anabolic pathways and energy metabolism. Quantitative proteomics of rd1 retina showed early changes in calcium signaling and oxidative phosphorylation, with specific partial bypass of complex I electron transfer, which precede the onset of cell death. Concurrently, we detected alterations in central carbon metabolism, including dysregulation of components associated with glycolysis, pentose phosphate and purine biosynthesis. Ex vivo assays of oxygen consumption and transmission electron microscopy validated early and progressive mitochondrial stress and abnormalities in mitochondrial structure and function of rd1 rods. These data uncover mitochondrial overactivation and related metabolic alterations as determinants of early pathology and implicate aberrant calcium signaling as an initiator of higher mitochondrial stress. Our studies thus provide a mechanistic framework with mitochondrial damage and metabolic disruptions as early drivers of photoreceptor cell death in retinal degeneration.
Introduction
Neurodegenerative diseases are characterized by progressive dysfunction and death of post-mitotic neuronal cells that lead to severe cognitive and/or sensory defects, greatly impacting the quality of life. These largely untreatable pathologies typically afflict specific neurons, with divergent clinical manifestations caused by heterogenous genetic factors (1,2). Genes and pathways implicated in neurodegeneration participate in energy metabolism, signaling, stress response, and autophagy, among other homeostatic processes (3–5). Mitochondrial dysfunction is arguably a key factor in neurodegenerative disease (6–8). However, the concept of increased stress of functional mitochondria (9) has been largely underappreciated. One-carbon and serine metabolism pathways are also implicated in health and pathology and may provide novel therapeutic targets (10,11). To date, we still lack knowledge of the precise cellular events, especially integrative molecular cues convergent at the onset of pathology that determine the natural history of neuronal cell death in degenerative disease.
The retina, the most accessible part of the central nervous system, provides an excellent model for examining neurodegeneration. Retinal degenerative diseases constitute a major cause of incurable vision impairment worldwide, with pathogenic mutations identified in almost 300 genes (https://sph. uth.edu/retnet/). Extensive genetic and phenotypic heterogeneity associated with Mendelian retinal diseases (12,13) make the design of gene-based therapies particularly challenging (14,15). The death of photoreceptor cells, predominantly by apoptosis, is the primary end-point that leads to vision loss in most retinal diseases (16,17), and thus antiapoptotic interventions have been attempted to restore vision (18). The ‘one-hit model’ of cell death in different models of photoreceptor degeneration and other neurodegenerative diseases argues against the cumulative damage hypothesis and suggests that mutations create an adaptive state of equilibrium and cell death is initiated by a single random event (19). We and others previously hypothesized convergence in pathways leading to pathology in divergent inherited retinal diseases (20,21). Regardless, the pathological events and critical mutant steady state preceding photoreceptor cell death are poorly understood.
Spontaneous and transgenic mouse mutants can elegantly phenocopy human retinal disease (22,23); albeit, the onset and severity may vary. We chose a widely-studied retinal neurodegeneration model, the retinal degeneration 1 mouse (rd1; also referred as Pde6brd1/rd1), to investigate early molecular and cellular events that may trigger photoreceptor cell death. The rd1 mouse carries a homozygous loss of function mutation in the Pde6b gene, which encodes the rod photoreceptor-specific cGMP phosphodiesterase 6 b subunit (PDE6b) (24,25). PDE6b is an evolutionarily conserved key component which is localized in rod outer segments (26) and mediates phototransduction by controlling cGMP levels and facilitating the opening and closing of cGMP-gated ion channels (27). The rod cell death in rd1 mouse retina is histologically evident starting at postnatal day (P)10, with a complete loss of the photoreceptor outer nuclear layer (ONL: composed of rod and cone nuclei) by P30. Mutations in the human PDE6B gene lead to a spectrum of autosomal recessive retinal degeneration phenotypes (28). Since its discovery nearly a century ago, the rd1 mouse has been a popular model of choice for investigating the mechanism of retinal degeneration. Over the years, numerous studies have reported global transcriptome analysis of the rd1 retina and indicated specific pathway alterations (29–33). Proteomic analyses of the rd1 retina have also identified changes in biochemical pathways (34,35). However, these studies focused on stages on or after the onset of degeneration or on late stages after a significant loss of photoreceptor population, and hence missed the very early pathological alterations in the rd1 retina.
Here, we report widespread predegeneration pathophysiological changes in the neonatal rd1 retina using an integrative multiomics approach that included temporal transcriptomics of purified rod photoreceptors along with proteomic and metabolomic analysis of whole retinas in combination with structural and functional assays. These data demonstrate a higher mitochondrial stress as early as P6; much before photoreceptor cell death is evident in the mutant retina. We show, for the first time, that calcium signaling defects which drive mitochondrial and metabolic alterations constitute the predeath mutant state in rod photoreceptors. These results are consistent with the known high metabolic demand and activity of photoreceptors (36–38). Our findings of predegeneration molecular and functional dysfunction in mitochondria and their associated cell signaling regulation are the earliest detected etiology of rod cell death and support the proposed convergence of cellular events during retinal neurodegeneration (2,20,21).
Results
Early transcriptomic divergence in the rd1 rods reveals altered metabolic and signaling pathways
Histological changes and cell death markers are detected in rod photoreceptors of rd1 retina starting at or around P10, and early pathology is characterized by shorter outer segments and progressive thinning of the ONL (Fig. 1A). Since the genetic and biochemical defect is limited to these retinal cells, we first performed transcriptome profiling of newborn and developing rod photoreceptors. We generated rd1-GFP mice by mating the Pde6brd1/rd1 mice to TgNrlp-EGFP (WT) (39) mice, used flow cytometry to purify GFP+ rods at P2, P4, P6, P8 and P10 (before rod cell death is evident), and performed RNA-seq analysis (Fig. 1A). After rigorous filtering and normalization, we captured expression of 14 534 genes from temporal transcriptome profiles of rd1 and WT photoreceptors. Principal component analysis (PCA) of rd1 and WT samples showed progressively divergent RNA profiles as early as P2 (the peak of rod birth is P0–P2), the earliest time we sampled the purified rods (Fig. 1B and C). Heat maps of a total of 1474 differentially expressed genes (DEGs) displayed distinct patterns (shown as clusters C1–C10: see below) in rd1 rods at predegeneration states (Fig. 1D).
Figure 1.
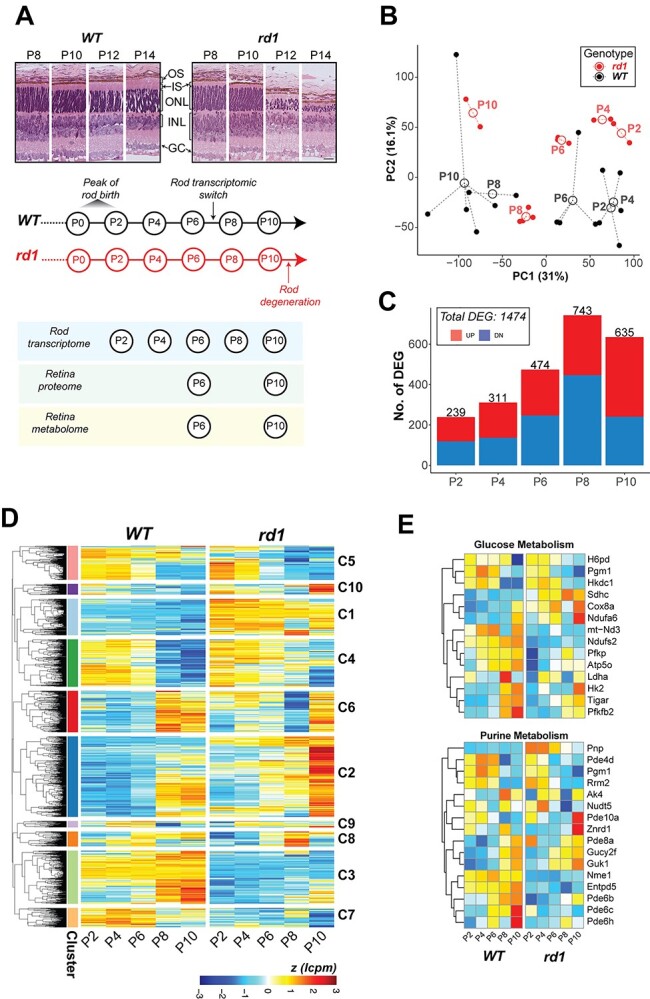
Transcriptome dynamics of rod photoreceptors prior to cell death in the rd1 retina. (A) Pathology in the rd1 retina and study design. Representative histology pictures of retinal cross sections from WT and rd1 mice at ages ranging from P8 to P14 (top panel). A schematic summary of timeline showing development in WT retina and onset of degeneration in rd1 (middle panel). Experimental plan and ages of sampling for each assay (bottom panel). Scale bar = 20 μm. OS: outer segment, IS: inner segment, ONL: outer nuclear layer, INL: inner nuclear layer, GC: ganglion cell. (B) Principal component analysis (PCA) plot summarizing the transcriptomic landscape of rod photoreceptors of WT and rd1 retina. (C) Trend of differential gene expression between WT and rd1 rods. The number atop each bar represents the total number of significantly differentially expressed genes (DEGs) in a pairwise comparison between age-matched WT and rd1 samples. (D) Heatmap of gene expression of all DEGs that are significant across age-matched comparisons. Log CPM (lcpm) values are row scaled to z-scores for plotting. See also Supplementary Material, Figure S1D. (E) Heatmaps of gene expression of significant DEGs that participate in glucose metabolism (glycolysis, oxidative phosphorylation, pentose phosphate) and purine metabolism pathways. Log CPM (lcpm) values are row scaled to z-scores for plotting. Color scale bar is same as in Figure 1D. See also Supplementary Material, Figure S2.
KEGG pathway enrichment analysis of DEGs revealed that central carbon metabolism and signaling pathways were prominently impacted in the early stages of developing rd1 rods (Figs 1E, Supplementary Material, S1 and S2). Metabolism related DEGs at P2, P4 and P6 included those associated with carbohydrate metabolism, cellular anabolic processes and fatty acid biosynthesis (Fig. 1E, Supplementary Material, S1A–C and S2) and corresponded primarily to glycolysis (e.g. Pfkp, Tigar and Pfkfb2), oxidative phosphorylation (OXPHOS; e.g. Ndufs2, Atp5o and mt-Nd3), pentose phosphate pathway (PPP) (e.g. H6pd), purine metabolism (e.g. Pnp) and glycogenolysis (e.g. Agl and Pgm1) (Fig. 1E, Supplementary Material, S1B and S2). A number of lipid metabolism genes exhibited higher expression; e.g. fatty acid biosynthesis gene Acot1 and several phospholipid metabolism genes (Abhd4, Lpcat2 and Samd8) (Supplementary Material, Figs S1B and S2). Additionally, homocysteine and one-carbon metabolism genes—Ahcy, Mthfr and Mthfd2—were among those showing the highest fold change differences (Supplementary Material, Fig. S1B, Supplementary Material, Table S1). The gene coding for myoferlin (Myof) was markedly under-expressed in rd1 rods (Supplementary Material, Fig. S1B), even though its consequence in neuronal cells remains unspecified. Transcriptional modulators, such as Duxbl1, Sox30 and Hmga1b, and mitochondrial stress regulator Atf4 were also significantly differentially expressed in the early developing rods (Supplementary Material, Fig. S2). Importantly, we observed differential expression of calcium-related signaling genes (e.g. Hcn, S100a10 and Atf4) in rd1 rods at early stages, much before the onset of degeneration at P10 (Supplementary Material, Fig. S1C and S2). Finally, genes involved in the phototransduction pathway demonstrated significantly altered expression at these early ages (Supplementary Material, Fig. S1A).
Rd1 rod DEGs could be grouped into 10 clusters (C1–C10). Three of the larger clusters showed concordance with changes in distinct pathways (Fig. 1D, Supplementary Material, S1D and E). Cluster 1 (C1) genes consistently demonstrated higher expression in rd1 rods starting at P2 and included metabolic genes for one-carbon pool, purine metabolism and fatty acid metabolism. Conversely, genes in C3 cluster with low expression in rd1 rods belonged to phototransduction and Hedgehog signaling. Cluster C2 showed sharp induction of gene expression at the onset of degeneration (P10) and contained fatty acid biosynthesis, ferroptosis as well as phototransduction genes.
Reduced expression of mitochondrial NADH dehydrogenase complex and signaling proteins in rd1 retina
In developing mouse retina, the photoreceptor differentiation is concordant with major changes in the transcriptome between P6 and P10 (40). We therefore wanted to ascertain whether disease-specific trends in rd1 gene expression further translate to proteome changes that precede developmental milestones and onset of degeneration. Mass spectrometry-based proteome analysis of P6 and P10 rd1 and WT retina identified a total of 4955 proteins. Of these, 621 were differentially expressed in P6 rd1 retina and an additional 630 showed altered expression at P10 (Fig. 2A and Supplementary Material, S3A). Gene set enrichment analysis (GSEA) using Fast Gene Set Enrichment Analysis (fgsea) uncovered mitochondrial electron transport chain (ETC) as the most downregulated pathway in both P6 and P10 rd1 retina (Fig. 2B and Supplementary Material, S3B) with specific reductions in the subunits of NADH dehydrogenase (complex I) (Fig. 2C). Further targeted analysis, using a curated GSEA, confirmed a significant negative enrichment of the OXPHOS pathway at P6 and P10 (Supplementary Material, Fig. S3C). This trend begins a negative course at P6 with further reductions by P10, clearly showing a correlation of OXPHOS impairment with age in rd1 retina. Leading-edge analysis identified complex I subunits to be the driver of OXPHOS impairment, with other ETC complexes showing insignificant changes (Fig. 2D). Complex I enzymatic activity assay in mitochondria-enriched retinal extracts also showed lower activity in rd1 retina between P2 and P8 (Supplementary Material, Fig. S3D), confirming an impairment of complex I function.
Figure 2.

Proteomics identifies dysregulation of Electron Transport Chain complex I in the rd1 retina before degeneration onset. (A) Venn diagram comparison of differentially expressed proteins in P6 and P10 rd1 retina. (B) Summary of pathway enrichment analysis that identifies differential regulation at early stages (P6) in the rd1 retina. Ridge plots show fold change (rd1 versus WT) distribution of leading-edge proteins from significant genesets. The top and bottom panels show over- and under-enriched pathways in red and blue colors respectively. See also Supplementary Material, Figure S3B for late stage (P10) data. (C) Proteomic fold change (FC) of mitochondrial electron transport chain complexes. Black dots inside violin plots represent significant differential expression of an individual protein (rd1 versus WT) of a specific complex. Gray dots inside violin plots represent non-significant differential expression. (D) Dumb bell plot showing change in protein expression from P6 to P10 for leading-edge proteins of oxidative phosphorylation. Dark red and purple dots represent significantly different proteins at P6 and P10, respectively. Grayed dots represent proteins with expression less than statistical significance threshold. See also Supplementary Material, Figure S3C.
Manual curation of significantly altered mitochondrial proteins uncovered overexpression of several ribosomal proteins, tRNA ligases, import/processing subunits and ubiquinone biosynthesis proteins at P6 and P10, suggesting a more active mitochondrial biogenesis in rd1 retina (Fig. 3A). For example, the most overexpressed mitochondrial protein (SUPV3L1, FCP6 = 4.98) is an RNA helicase associated with the RNA surveillance system in mitochondria (41), suggesting enhanced transcription of mitochondrial DNA. In addition, several proteins involved in assembly of respiratory complexes were increased in rd1 retina (ATPAF1, TTC19, AIFM1 and NLN). Furthermore, increased expression of amino acid degradation enzymes (GLDC, IVD, MCCC2) suggested an augmented degradation of mitochondria in rd1 retina, resulting in a faster mitochondrial turnover due to a parallel increase in biogenesis. In addition, mitochondria in the mutant retina seem to exhibit a higher capacity to donate electrons directly into the ubiquinone pool bypassing complex I, as suggested by higher expression of enzymes involved in fatty acid oxidation (MCEE, ECI2, HADHA, ECH1, ECI1), leucine degradation (IVD, MCCC2) and pyrimidine metabolism (DHODH).
Figure 3.
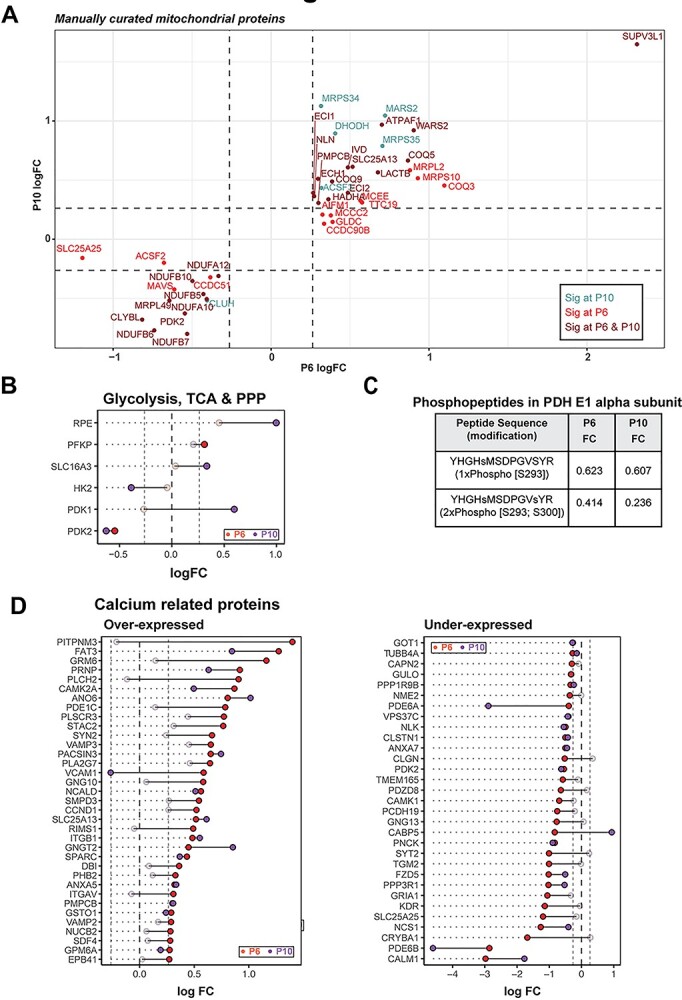
rd1 retinas harbor abnormal abundance of mitochondrial and calcium-related proteins before degeneration. (A) Scatter plot of manually curated mitochondrial proteins show differential regulation of mitochondrial metabolic and signaling proteins at P6 and P10. Proteins with abundances significantly different at P6, P10 and both P6 and P10 are labeled in red, blue and brown, respectively. (B) Dumb bell plot showing change in protein expression from P6 to P10 for significantly differential glycolysis, TCA and PPP proteins. Dark red and purple dots denote proteins above significance threshold at P6 and P10, respectively. Gray dots represent proteins with non-significant differential expression. (C) Phosphopeptides detected in pyruvate dehydrogenase (PDH) E1 alpha subunit. (D) Dumb bell plot showing change in protein expression from P6 to P10 for significantly differential calcium-related proteins. Plot on the left summarizes proteins overexpressed in the rd1 retina, whereas plot on the right depicts under-expressed proteins. Color codes are same as in Figure 3B.
Curiously, a similar analysis of the strategically upstream glycolytic, PPP and TCA cycle proteins did not show a pathway-wide consistent difference, although a few specific proteins including PFKP, SLC16A3, HK2, PDK1 and PDK2 were significantly different at one or both ages (Fig. 3B). Consistent with the significant under-expression of the pyruvate dehydrogenase kinase PDK2 at P6 and P10, we observed reduction in ratios of single- and double-phosphorylated peptides from subunit E1α of the pyruvate dehydrogenase complex (PDC) in rd1 at both P6 and P10 (Fig. 3C). When analyzing the differential protein expression of all calcium-associated proteins, robust and consistent calcium dysregulation was evident in the rd1 retina (Fig. 3D). One of the most downregulated proteins in the rd1 retina was calmodulin (CALM1) (Supplementary Material, Fig. S3A), which serves as the primary intracellular calcium sensor and initiator of downstream signaling events that correspond to changing calcium levels. In addition, calcium-binding mitochondrial carrier proteins, such as SLC25A13 (fold change at P6, FCP6 = 1.48) and SLC25A25 (FCP6 = 0.43), were significantly altered. Interestingly, the highest overexpressed protein in rd1 retina was PITPNM3 (FCP6 = 2.63), a membrane-associated phosphatidylinositol transfer protein implicated in autosomal dominant cone rod dystrophy (42). These findings strongly indicate abnormal calcium signaling in the rd1 retina at early stages before the onset of degeneration.
Metabolite imbalance, especially of central carbon pathways, in rd1 retina
Altered expression of enzymatic proteins may not directly or proportionally translate to changes in corresponding metabolic flux. Hence, we examined 116 metabolites in P6 and P10 rd1 and WT retina (Fig. 4 and Supplementary Material, S4) to investigate disruption in metabolic homeostasis. We detected alterations in glycolytic and PPP intermediates as well as nucleotides at both stages (Fig. 4A and Supplementary Material, S4A). Carnosine, a dipeptide involved in resistance to oxidative stress and divalent ion chelation (43) was the most elevated metabolite in the P6 rd1 retina. We generated a metabolite-pathway network and identified biosynthesis of amino acids, carbon metabolism, ATP-binding-cassette transporters, purine metabolism and PPP as those with the most difference in metabolites (Fig. 4B).
Figure 4.
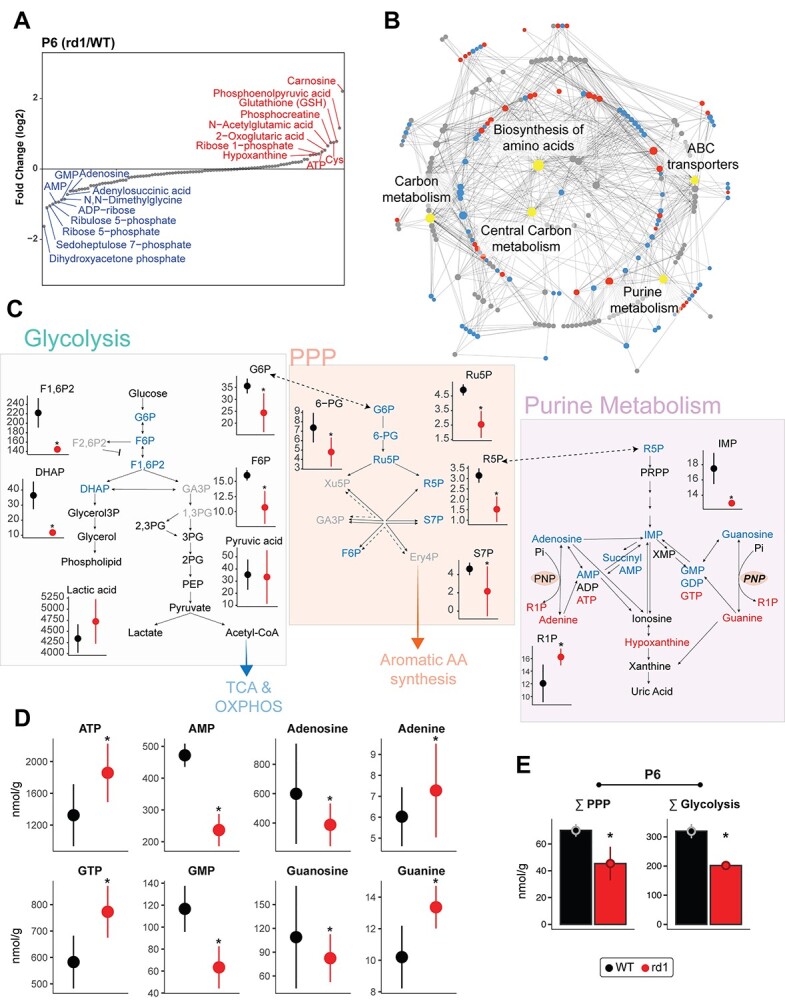
Global metabolite profiling identifies aberrant central carbon metabolism. (A) Relative abundance plot for the metabolome of the rd1 retina relative to WT, at P6, showing the highest and least abundant metabolites. See also Supplementary Material, Figure S4A for P10 data. (B) Mapping of identified metabolites to KEGG pathways. Significantly altered metabolites at P6 in rd1 relative to WT retinas are colored in either red or blue corresponding to increased or decreased abundance, respectively, whereas pathways are shown in gray. The top five pathways with the most significantly different metabolites are labeled and highlighted in yellow. Gray dots are metabolites with differential abundance level less than statistical significance threshold. (C) Annotated pathway diagram and connections between glycolysis, PPP, and purine metabolism, with differential metabolites at P6 highlighted. Metabolites with significant increase or decrease in abundance are highlighted in red or blue, respectively. Black colored metabolites are detected but do not show differential abundance, whereas gray colored metabolites were not detected in the metabolomic analysis. Red and black colors in metabolite abundance plots represent rd1 and WT, respectively. Significantly altered metabolites are marked with an asterisk (see Methods for details). (D) Abundance levels of adenine and guanine and their corresponding nucleotides at P6. Color and significance features are same as Figure 4C. (E) Sum of metabolites involved in glycolysis and PPP in the P6 WT and rd1 retina. Color and significance features are same as Figure 4C. See also Supplementary Material, Figure S4B for P10 data.
We then focused on central carbon metabolism (i.e. glycolysis, PPP and purine metabolism) because of significant aberrations in the P6 rd1 retina. We observed a significant decrease in the upstream intermediates (glucose 6-phosphate, fructose 6-phosphate, fructose 1,6-biphosphate and dihydroxyacetone phosphate) (Fig. 4C), yet downstream glycolytic intermediates and products such as glycerol 3-P and glycerol (required for phospholipid synthesis) as well as pyruvate and lactate were relatively unchanged. Together, these data suggest that the net glycolytic flux did not decrease at this early stage (i.e. by P6). Our results also demonstrated significant depletion of most PPP intermediates in the rd1 retina. In contrast, we observed a marked increase of purine triphosphates and their respective nucleobases, apparently at the expense of early purine biosynthesis intermediates including adenosine and guanosine and their monophosphorylated forms (Fig. 4D). Thus, significant reductions of total metabolite concentrations in glycolysis and PPP were observed in the rd1 retina as early as P6 and even at P10 (Fig. 4E and Supplementary Material, S4B). These metabolic changes suggest a shift in carbon utilization rather than a decrease in metabolic flux through these pathways.
Predegeneration functional and morphological defects in rd1 mitochondria
Higher ATP concentrations in P6 rd1 retina (see Fig. 4D) suggested higher OXPHOS activity. To directly assess mitochondrial respiration coupled to ATP production in relation to maximal (uncoupled) respiratory activity, we measured oxygen consumption rate (OCR) in acutely isolated, ex vivo retinal punches from rd1 and WT mice (Fig. 5A) using a previously optimized protocol (9). We noted that basal mitochondrial OCR (coupled to ATP production) decreased in WT retina during development, reaching a low point at P10, followed by an increase at eye opening on P14 (Fig. 5B). In contrast, the rd1 retinas showed a trend of higher basal OCR compared to WT as early as P3 (Fig. 5B). With the onset of degeneration after P10 (see Fig. 1A), basal mitochondrial OCR in rd1 retina was reduced significantly at P14 (Fig. 5B) when as many as 50% of the rod photoreceptors are lost. After adding a mitochondrial uncoupler (Bam15) to obtain the maximal OCR, the mitochondrial reserve capacity (MRC) was obtained by calculating the additional percentage of maximal OCR that is not used for ATP production under basal conditions. In developing WT retina, MRC progressively increased and peaked to ~50% around P8 and P10 before decreasing to ~30% at P14 (Fig. 5B), consistent with our previous observations of low MRC in healthy adult retina (9). Interestingly, rd1 retinas were characterized by lower MRC values from P3 through P10 before overt rod degeneration was observed at P14. MRC was markedly increased at P14 by 50–60%, as previously reported (9) (Fig. 5B).
Figure 5.

Early functional aberrations in mitochondria before the onset of photoreceptor degeneration in rd1 retina. (A) Traces of mitochondrial OCR in retinal punches isolated from WT and rd1 mice at various ages from P3 to P14 (n = 12–29 retinal punches, for each group). BAM15 and rotenone were added at indicated time during experiments. Data are represented as mean ± SEM. (B) Basal mitochondrial OCR in WT and rd1 retinal punches at each age tested. Values were taken at 36 min, right before addition of BAM15. Plot of mitochondrial reserve capacity in WT and rd1 retinal punches at each age tested. Data are represented as mean ± SEM. Student t test, *P < 0.05, **P < 0.01, ***P < 0.001.
To assess whether enhanced oxygen consumption might result from increased number of mitochondria in the rd1 retina, we examined the Mit/Nu DNA ratio (44). Quantitative qPCR analysis demonstrated a trend of lower Mit/Nu DNA ratios in rd1 retina, suggesting relatively fewer mitochondria in P2 to P8 rd1 retina compared to age-matched WT retina (Supplementary Material, Fig. S5).
Ultrastructural analysis by transmission electron microscopy (TEM) revealed delayed organization of the rod inner segments and morphological defects in mitochondria cristae structure as early as P6 in the rd1 retina (Fig. 6). Delayed elongation of inner and outer segments in rd1 photoreceptors persisted into P8 and P10. At P8, abnormal mitochondria were more prevalent in inner segments of rd1 rod photoreceptors and showed loss of inner membrane cristae and a swollen vesicular appearance. However, a majority of mitochondria at this stage still exhibited normal gross morphology. Swollen mitochondria were larger and more numerous in rd1 rods at P10, indicating a disrupted mitochondrial network even before the histological rod cell death was observed. With extensive degeneration evident at P14, few mitochondria in rd1 rods had a normal gross or ultrastructural morphology, and the retina exhibited a disrupted overall structure and lamination (Supplementary Material, Fig. S6).
Figure 6.
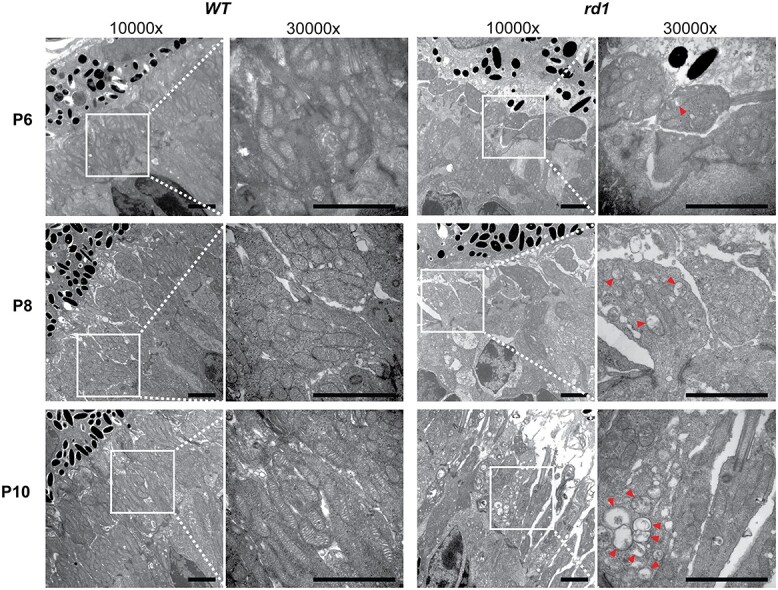
Mitochondrial ultrastructural alterations at early stages before photoreceptor degeneration in rd1 retinas. (A) TEM of photoreceptor inner segment area from P6 to P10 WT and rd1 retina are shown at magnifications of 10 000× and 30 000× (zoomed-in area, indicated by white square box). Red arrowheads indicate abnormal mitochondria with swollen cristae. Scale bar = 2 mm. See also Supplementary Material, Figure S6.
Proposed model of molecular and cellular events leading to photoreceptor cell death
Temporal dynamics of transcriptome changes, together with proteomic and energy metabolism defects detected at early stages in the rd1 retina, enabled us to construct a model to explain the etiology of rod photoreceptor cell death in the rd1 mouse (Fig. 7). Important features identified in our study include progressively increasing metabolic alterations and mitochondrial deficiencies that were initiated prior to any observed morphological and/or functional defects in degenerating rod photoreceptors of the rd1 mouse. The null mutation in Pde6b gene leads to increased stress and global alterations in gene expression as early as P2. Homocysteine and one-carbon metabolism genes, Ahcy and Mthfr, presented the highest fold change among the DEGs, suggesting aberrant nucleic acid synthesis and cellular methylation reactions. We hypothesize that a functional imbalance in pathways associated with one-carbon metabolism and energy homeostasis, as indicated by RNA profiles of newborn rod photoreceptors, results in reduction of complex I and abnormalities in mitochondrial structure and function, related to chronically high intracellular calcium levels. Initially, increased cytosolic calcium would likely stimulate mitochondrial oxidation of pyruvate by activating pyruvate dehydrogenase phosphatase (PDP), which dephosphorylates PDC to its catalytically active state. A faster consumption of pyruvate would result in the observed depletion of the glycolytic and pentose pathway metabolite pools. However, a sustained increase in cytosolic calcium would also cause mitochondrial swelling and rupture over time. A more rapid replacement of damaged mitochondria, in an attempt to maintain the number of functional mitochondria, would concurrently accelerate degradation and biogenesis. The resulting enhanced biosynthesis of mitochondrial membrane phospholipids would further deplete glycolytic intermediates to obtain the glycerol 3-phosphate required for the head groups of various mitochondrial glycerophospholipids. The observed lower complex I expression and activity appear to be compensated by increased electron transfer from fatty acid, aspartate and leucine metabolism into the ubiquinone pool. These changes, together with a direct stimulation of oxidative phosphorylation by calcium, are consistent with the enhanced OCR, reduced MRC, and increased level of high-energy phosphates (ATP and GTP) we detected in our studies. In concordance, expression levels of a key intracellular calcium regulator, calmodulin, are greatly reduced leading to dysregulation of mitochondrial calcium uptake and calcium-dependent signaling pathways (e.g. the activation of PDC through calcium-activated dephosphorylation by PDP). Notably, disease-related activity of the one-carbon cycle and homocysteine metabolism can aggravate cellular distress by potentially altering NADPH levels, consistent with observed differences in purine metabolism. Later accumulation of cGMP in rd1 mutants may also lead to differential abundance of purine molecules, reflecting the rod cell’s attempts to control purine production by metabolic reorganization, which can further contribute to initiation of molecular pathology. Thus, an increasing and accumulating imbalance of energy metabolism pathways coupled with a progressive increase in intracellular and mitochondrial calcium levels result in elevated mitochondria stress and calcium overload, eventually leading to mitochondria swelling and rod photoreceptor cell death in the rd1 retina.
Figure 7.
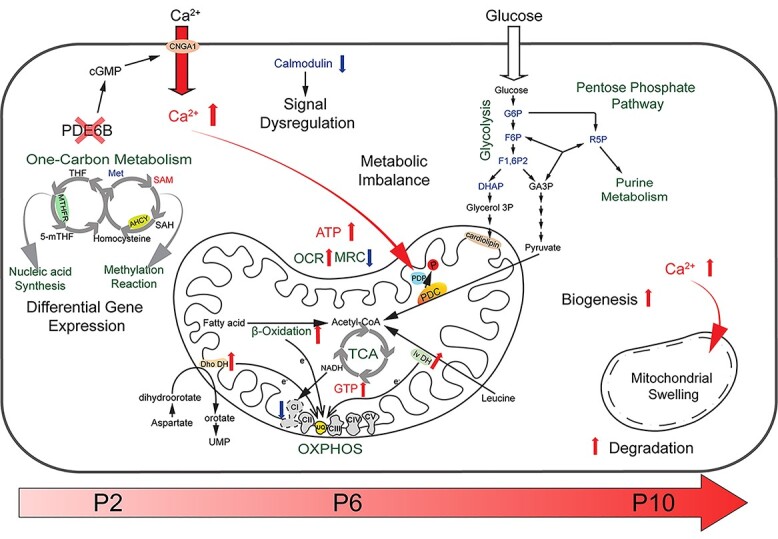
An integrated mitochondria-centered model of molecular events leading to photoreceptor cell death in retinal degeneration. Altered gene expression starts in newborn rd1 rod photoreceptors carrying the Pde6b mutation. Differentially expressed key enzymes (MTHFR and AHCY) affect the balance of one-carbon metabolism, which in turn influences nucleic acid synthesis and pan-methylation status. Concurrently, reduced calmodulin expression leads to dysregulation of calcium-mediated signaling. Increased intracellular calcium enters mitochondria, activates PDP, which in turn stimulates PDC by enhancing its dephosphorylation. Faster consumption of pyruvate triggers an imbalance of the metabolite pool in glycolysis and pentose pathways. Higher calcium also increases damage to mitochondria, which are replaced more rapidly by accelerated degradation and biogenesis (turnover). Enhanced biosynthesis of mitochondrial membrane phospholipids would further deplete glycolytic intermediates. Lower complex I activity is compensated by increased electron transfer from fatty acid, aspartate and leucine metabolism into the UQ pool. These changes lead to enhanced OCR, reduction in MRC and increase in cytosolic ATP. With progressive imbalance in metabolism and augmented stress, mitochondria and their cristae swell and rupture, eventually committing the cell to a death fate. UQ: ubiquinone, Dho DH: dihydroorotate dehydrogenase, Iv DH: isovaleryl-CoA dehydrogenase, PDC: pyruvate dehydrogenase complex, PDP: pyruvate dehydrogenase phosphatase, PDK: pyruvate dehydrogenase kinase.
Discussion
The lack of regenerative potential in the mammalian retina makes photoreceptor cell dysfunction and/or death a leading cause of incurable blindness. Rod photoreceptors are specifically vulnerable to degeneration, and their deficiency is generally followed by the loss of cones in inherited retinal diseases (21). Even in a multifactorial disorder affecting the macula (namely age-related macular degeneration, AMD), rod cell death precedes cone loss (45). Convergence to rod degeneration despite extensive clinical and genetic heterogeneity suggests commonalities in molecular and cellular pathways and offers opportunities for gene-agnostic intervention in retinal and macular diseases (20,21). As an example, a thioredoxin-like protein RdCVF has been shown to protect cone function in multiple distinct models of retinal degeneration by inducing glucose uptake and aerobic glycolysis (46). We hypothesized that the presence of a genetic mutation would impact molecular/biochemical pathways in target cells early in development and that progressive and varying deterioration of cellular responses would eventually lead to cell death (20). By applying three ‘omics’ technologies, here we demonstrate that the loss of PDE6b by an inherited mutation results in extensive adaptations in metabolic and mitochondrial components of newborn and developing rod photoreceptors, much before the established critical requirement of PDE6b in signaling during phototransduction. Our studies identify early convergence of multiple cellular responses to mitochondria-related metabolic and signaling pathways and point to disruptions or reallocations of energy resources and calcium signaling in early stages preceding the onset of retinal neurodegeneration.
The rd1 mouse represents arguably the most studied model of retinal degeneration. PDE6b is associated with controlling cGMP levels, and cGMP accumulation has been implicated in driving the death of rod photoreceptors in the rd1 retina as well as in phototransduction-associated inherited retinal diseases (17,47,48). cGMP is a key second messenger in multiple cellular signaling pathways. In photoreceptors, cGMP-gated channels control the flux of ions in light and dark conditions contributing to Ca2+ homeostasis (49). Remarkably however, photoreceptor cell death begins in rd1 retina around P10 and before eye opening in mouse (at P14). Although most studies focused on the disease progression at or after onset of rod death (after P10), our transcriptome profiling of neonatal (P2–P6) rd1 rods shows major and highly specific abnormalities in expression of genes associated with central carbon metabolism. These early changes likely impact energy homeostasis, redistribute metabolites within one-carbon pathways, and initiate a cascade of signaling events. Two of the earliest DEGs—Ahcy (lower expression) and Mthfr (high expression)—regulating the one-carbon metabolism are significantly altered with high magnitude and influence nucleic acid synthesis and the balance of S-adenosylmethionine to S-adenosylhomocysteine (SAM/SAH) ratio. Continued differential expression of Ahcy and Mthfr is reflected by an increase in SAM in the metabolome data from P10 rd1 retina, suggesting a shift in pan-methylation status. The differential gene expression trends suggest that mutant photoreceptors never undergo normal development despite their normal morphological appearance in early stages. Proteomic and metabolomic data from P6 rd1 retina further supports these findings and implicates mitochondria and related metabolic and calcium signaling pathways as the early mediators of the cellular response in retinal neurodegeneration.
Photoreceptors have high-energy requirements for maintaining their physiological state in dark and light (50). Like other neurons, glucose provides the primary source of energy in photoreceptors (9,51), and the stimulation of glucose uptake and/or metabolic intermediates can be neuroprotective in photoreceptor degeneration (46,52–54). Specialized cristae architecture, low reserve capacity and high OXPHOS of mitochondria (9,51,55) validate their primary role in providing the energy requirements of photoreceptors. Distinctive reduction in complex I subunits and alterations in several mitochondrial proteins (including others associated with ETC complexes) in P6 rd1 retina strongly argue in favor of altered mitochondrial structure and function being early indicators of photoreceptor stress. Predictably, we detected lower complex I activity in rd1 retina, consistent with a previous report (56). However, a partial deficiency in complex I activity can be compensated by alternate pathways that donate electrons at the level of the ubiquinone pool. Several of these pathways are augmented in rd1 retina as indicated by increase in proteins associated with fatty acid oxidation (MCEE, ECI2, HADHA, ECH1, ECI1), leucine degradation (IVD, MCCC2) and pyrimidine metabolism (DHODH). These results of partial bypassing of complex I electron transfer are in accordance with the higher basal mitochondrial OCR used for ATP production and enhanced ATP content.
As an indicative parameter of mitochondrial health/stress, the MRC can be affected by several factors, including the ability of the cell to deliver substrate(s) to mitochondria and functional capacity of enzymes involved in electron transport (57). Reduced MRC in rd1 retina as early as P3 suggests that a fraction of mitochondria in mutant photoreceptor cells have undergone uncoupling, as reported in cells during injury and aging as an adaptive response to decrease reactive oxygen species (ROS) (58). Changes in MRC are also correlated with oxidative stress, calcium overload and cone cell death (59). A lower MRC and higher OCR in the rd1 retina are consistent with chronically higher calcium concentrations in photoreceptors, as reported as early as P5 (60), which would at first stimulate OXPHOS (61) and ATP production but then cause swelling and mitochondrial damage by increasing ROS formation and inner membrane depolarization. Moreover, a faster flow of electrons to the ubiquinone pool to compensate for partial complex I deficiency may also result in enhanced ROS. Our ultrastructural observations show progressive mitochondrial damage in developing rd1 rods. The higher content of proteins involved in biogenesis and degradation suggests a faster turnover to regenerate functional mitochondria. The observed lower Mit/Nu DNA ratios in rd1 retina supported this hypothesis. With progressive calcium overload, mitochondrial respiration is inhibited and accompanied by cytochrome c release, decreased ATP production and disruption of the mitochondrial matrix and cristae (62).
High aerobic glycolysis is a characteristic feature of photoreceptor cells and crucial for anabolic demands and functional homeostasis (37,63). In our study, glycolytic abnormalities in predegeneration stage rd1 photoreceptors are noted by significant overexpression of two important regulators: Tigar and Pfkfb2. TIGAR is a negative regulator of glycolysis, whereas PFKFB2 modulates the pathway by controlling the pool of fructose-2,6-bisphosphate (64). Though we did not detect significant change in pyruvate, lactate or acetyl-CoA, the evidence from proteomic profiling indicates reprogramming of the tricarboxylic acid (TCA) cycle in the rd1 photoreceptors. Mitochondrial PDC links glycolysis and the TCA cycle, modulating the overall rate of oxidation of carbohydrate fuels under aerobic conditions (65). Significantly lower content of one isoform of PDC kinase (PDK2) and reduced phosphorylation of the PDC E1 alpha subunit in P6 and P10 rd1 retina suggest a faster-working PDC, which can potentially accelerate flux through the TCA cycle. Our data are concordant with previously reported prolonged photoreceptor survival in mouse models of retinal degeneration by increasing glycolysis flux through inhibition of SIRT6 (53) or by promoting lactate catabolism as fuel by Txnip administration (54). We propose that the faster PDC activity, besides being the consequence of higher mitochondrial calcium, could be an adaptive response of mutant photoreceptors by fine tuning TCA cycle in dealing with the stress. A recent report demonstrating slowdown of disease progression in Pde6α and RhoP23H mouse models by dietary supplementation of TCA cycle intermediates, a-ketoglutarate and citrate, also supports our hypothesis (66).
Elevated intracellular calcium cannot be explained by PDE6b’s role in phototransduction at early stages before eye opening. Notably, our metabolomic data do not reveal a significant difference in cGMP concentration between P6 or P10 WT and rd1 retina, even though mitochondrial structural defects were observed as early as P6 and P8. Interestingly, calmodulin, a key regulator of numerous calcium-sensitive proteins, is nearly undetectable at P6 in the rd1 retina, suggesting a dysregulation of calcium-dependent signaling at an early stage. Given that calmodulin directly interacts with PDE (67), we propose that the loss of PDE6b compromises calmodulin stability or activity, and in turn, affects other calcium-dependent processes. Indeed, our proteomic data reveal altered expression of a number of calcium-regulated proteins in the rd1 retina. Furthermore, calmodulin controls the sensitivity of rod cGMP channels to cGMP in a calcium-dependent manner, and with a decrease in calmodulin, cGMP channels exhibit a higher affinity for cGMP and more readily open to allow the influx of calcium into the inner segments (68). This is a more feasible explanation for the initiation of calcium-mediated mitochondria injury that we detect as early as P6. The absence of PDE6b activity during phototransduction likely amplifies the damage at eye opening (P14), resulting in rapid photoreceptor loss in the rd1 retina.
Our studies argue against the cumulative damage and support an adaptive mutant state in concordance with the ‘one-hit’ model (19). Identification of early disease-associated trends reported herein establishes mitochondria as an integrative node in the cellular response to genetic mutations with a potentially decisive role before the onset of neurodegeneration. We surmise that a mitochondrial link to rd1 specific PDE6b mutation, prior to the physiological regulation of cGMP channels, likely results from altered energy homeostasis and abnormal metabolic and calcium signaling. Our findings provide a plausible framework for the etiology of retinal degeneration by incorporating unique photoreceptor ‘omics’ and physiology in concert with our observations from the rd1 mutant retina. In addition, these studies are consistent with the mechanistic underpinning of neurodegenerative diseases (69,70) and should have broad implications for deciphering molecular and cellular drivers of disease pathology. Discovery of metabolic and signaling pathways that are altered in anticipation of disease sets the foundation for drug discovery and interventions at an early stage, thereby improving outcomes in clinical management.
Materials and Methods
Animal models
Mice were housed in 12-hour light–dark housing conditions and were cared for following recommendations of the Guide for the Care and Use of Laboratory Animals, Institute of Laboratory Animal Resources, the Public Health Service Policy on Humane Care and Use of Laboratory Animals. All mouse protocols have been approved by the Animal Care and Use Committee of the National Eye Institute (ASP#650). Pde6brd1/rd1 mice were originally obtained from Jackson Labs (Cat. #000635). The Pde6brd1/rd1; TgNrlp-EGFP mice (referred to as rd1) were generated by crossing Pde6brd1/rd1 mice to TgNrlp-EGFP mice (39). Both TgNrlp-EGFP and Pde6brd1/rd1; TgNrlp-EGFP mice were backcrossed with C57BL/6 J mice for >10 generations and then until maintained by sibling crosses. The rd1 and TgNrlp-EGFP alleles were maintained as homozygous. TgNrlp-EGFP mice served as wild type (WT) control in all experiments. Both male and female mice were used in this study.
Retinal histology
After enucleation, eyes were fixed with 4% glutaraldehyde for 30 min at room temperature and then with 4% paraformaldehyde overnight at 4°C. Eyes were then embedded in methacrylate and sectioned at 1-micron thickness. Representative sections from the superior central retina were stained with standard hematoxylin and eosin (H&E) staining protocol. Pictures of retinal section were taken at 40×.
Transcriptomic profiling
Flow sorting of rod photoreceptors
Rod photoreceptors from Pde6brd1/rd1; TgNrlp-GFP (rd1) and TgNrlp-GFP (WT) mice were isolated as previously described (40). In brief, dissected retinas were treated with papain (Worthington Biochemical, NJ, USA) and dissociated singe cells were collected by centrifugation at 800 × g for 5 min at 4°C. Cell pellets were resuspended in ice-cold PBS. GFP-positive photoreceptor cells were isolated using FACS Aria II (Becton Dickinson, CA, USA) with a stringent precision setting which maximized the purity of sorted cells.
RNA sequencing
Rod photoreceptor cells were lysed with TRIzol LS (Invitrogen), and the RNA was isolated following the manufacturer’s instructions. RNAseq data were generated using TruSeq Stranded mRNA Sample Prep Kit (Illumina), as previously described (71), and 125 base pair-end reads were generated on HiSeq 2500 platform (Illumina).
Differential RNA expression and downstream analysis
Unprocessed fastq files from the sequencer were quality checked, trimmed and aligned to the Mus musculus Ensembl annotation (v98) using a robust pipeline as described before (71). Briefly, the count matrix was normalized, and batch corrected to generate abundance summary in counts per million using the Bioconductor packages edgeR and limma. Additionally, to remove artefacts we set an expression filter that required genes to have >5 CPM in minimum two replicates of any sample before qualifying for differential expression analysis. Statistical tests for differential expression between age-matched samples of rd1 and WT were performed with voom transformed counts fit to a linear model using functions in limma. Finally, a cutoff of 10% false discovery rate (FDR) and 1.5-fold change difference was decided to group genes as DEG. Clusters of DEG were determined from a k-means algorithm after setting a value of k = 10. Functional enrichment analysis of DEG groups was performed with gProfileR and custom scripts where KEGG pathways were ordered by pathway impact, as defined by the ratio between overlap and pathway size. Gene sets for volcano plots were obtained from gProfileR (Reactome), Mouse Genomics Institute (Gene Ontology) and Mitocarta.
Proteomic profiling
Retinal tissue lysate processing
Retinal tissues from WT and rd1 mice at P6 and P10 were subject for whole proteomic analysis, performed by core facility at National Heart Lung and Blood Institute. Retinas from nine non-littermate mice of each sample group were frozen and thawed after dissection, divided in replicates of six retinas for each strain and age (n = 3 for each group,12 samples total), and resuspended in 100 μl of lysis buffer containing 6 M urea, 2 M thiourea and 4% CHAPS (Thermo Fisher Scientific). Each combined replicate of samples was loaded onto a QIAshredder spin column (QIAGEN) placed in a 2 ml collection tube and spun for 2 min at maximum speed in a microcentrifuge to aid in tissue disruption. The flow-through for each sample was collected and protein was quantified using the Pierce detergent compatible Bradford assay kit (Thermo Fisher Scientific).
A volume corresponding to 100 μg of protein from each replicate, as well as two normalization loading controls generated by pooling 5.55 μg of each replicate were taken to 100 μl by adding the necessary volume of lysis buffer. Each resulting sample was reduced by mixing with 5 μl of the 200 mM DTT and incubating at room temperature for 1 h, and then alkylated by adding 5 μl of 375 mM iodoacetamide and incubating for 30 min protected from light at room temperature. Protein in each sample was precipitated by adding 600 μl of prechilled acetone and storing overnight at −20°C. Samples were then centrifuged at 8000 × g for 10 min at 4°C, and acetone was carefully decanted without disturbing the white pellet, which was allowed to dry for 2–3 min. Each protein pellet was resuspended in 100 μl of a buffer containing 100 mM triethylammonium bicarbonate (TEAB) and 0.1% of Progenta anionic acid-labile surfactant I (AALSI, Protea Biosciences). Each sample was proteolyzed by adding 10 μl of 1.25 μg/μl sequencing grade modified trypsin (Promega) dissolved in 100 mM TEAB and incubated overnight at 37°C. Each sample was labeled with a different TMT label reagent (Thermo Fisher Scientific) by adding the contents of each label tube after dissolving with 41 μl of acetonitrile. The reaction was allowed to proceed for 1 h at room temperature and was quenched by adding 8 μl of 5% hydroxylamine to each sample and incubating for 15 min.
All seven samples from each strain (six replicates plus a normalization loading control) were combined in a new microcentrifuge tube, which was dried under vacuum until all acetonitrile was removed. Contaminating CHAPS was removed using 2 ml Pierce detergent removal columns (thermo Fisher Scientific) and residual AALSI was cleaved by adding 30 μl of 10% trifluoroacetic acid. The combined samples were desalted using an Oasis HLB column (Waters) and dried under vacuum.
Offline HPLC peptide fractionation
High-pH reversed-phase liquid chromatography was performed on an offline Agilent 1200 series HPLC. Approximately 1 mg of desalted peptides were resuspended in 0.1 ml of 10 mM triethyl ammonium bicarbonate with 2% (v/v) acetonitrile. Peptides were loaded onto an Xbridge C18 HPLC column (Waters; 2.1 mm inner diameter × 100 mm, 5 μm particle size), and profiled with a linear gradient of 5–35% buffer B (90% acetonitrile, 10 mM triethyl ammonium bicarbonate) over 60 min, at a flowrate of 0.25 ml/min. The chromatographic performance was monitored by sampling the eluate with a diode array detector (1200 series HPLC, Agilent) scanning between wavelengths of 200 and 400 nm. Fractions were collected at 1 min intervals followed by fraction concatenation (72). Fifteen concatenated fractions were dried and resuspended in 0.01% formic acid, 2% acetonitrile. Approximately 500 ng of peptide mixture was loaded per liquid chromatography-mass spectrometry run.
Mass spectrometry
All fractions were analyzed on an Ultimate 3000-nLC coupled to an Orbitrap Fusion Lumos Tribrid instrument (Thermo Fisher Scientific) equipped with a nanoelectrospray source. Peptides were separated on an EASY-Spray C18 column (75 μm × 50 cm inner diameter, 2 μm particle size and 100 Å pore size, Thermo Fisher Scientific). Peptide fractions were placed in an autosampler and separation was achieved by 90 min gradient from 4% to 35% buffer B (100% ACN and 0.1% formic acid) at a flow rate of 300 nl/min. An electrospray voltage of 1.9 kV was applied to the eluent via the EASY-Spray column electrode. The Lumos was operated in positive ion data-dependent mode, using Synchronous Precursor Selection (SPS-MS3) (73).
Full scan MS1 was performed in the Orbitrap with a precursor selection range of 375–1275 m/z at 1.2 × 105 normal resolution. The AGC target and maximum accumulation time settings were set to 4 × 105 and 50 ms, respectively. MS2 was triggered by selecting the most intense precursor ions above an intensity threshold of 5 × 103 for collision induced dissociation (CID)-MS2 fragmentation with an AGC target and maximum accumulation time settings of 1 × 104 and 60 ms, respectively. Mass filtering was performed by the quadrupole with 0.7 m/z transmission window, followed by CID fragmentation in the linear ion trap with 35% normalized collision energy in turbo scan mode and parallelizable time option was selected. SPS was applied to coselect 10 fragment ions for HCD-MS3 analysis. SPS ions were all selected within the 400–1200 m/z range and were set to preclude selection of the precursor ion and TMTC ion series. The AGC target and maximum accumulation time were set to 1 × 105 and 125 ms (respectively) and parallelizable time option was selected. Coselected precursors for SPS-MS3 underwent HCD fragmentation with 65% normalized collision energy and were analyzed in the Orbitrap with nominal resolution of 5 × 104. The number of SPS-MS3 spectra acquired between full scans was restricted to a duty cycle of 3 s.
Mass spectrometry data processing
Raw data files were processed using Proteome Discoverer (v2.4, Thermo Fisher Scientific), using both Mascot (v2.6.2, Matrix Science) and Sequest HT (Thermo Fisher Scientific) search algorithms. All the peak lists were searched against the UniProtKB/Swiss-Prot protein database released 2020_02 with M. musculus taxonomy (17 010 sequences) and concatenated with reversed copies of all sequences. The following search parameters were set as static modifications; carbamidomethylation of cysteine, TMT 10-plex modification of lysine and peptide N-terminus. The variable modifications were set as; oxidation of methionine, deamidation of aspartamine and glutamine. For SPS-MS3 the precursor and fragment ion tolerances of 12 ppm and 0.5 Da were applied, respectively. Up to two-missed tryptic cleavages were permitted. Percolator (v3.02.1, University of Washington) algorithm was used to calculate the FDR of peptide spectrum matches, set to q-value 0.05 (74,75). TMT 10-plex quantification was also performed by Proteome Discoverer v.2.4 by calculating the sum of centroided ions within 20 ppm window around the expected m/z for each of the 10 TMT reporter ions. Spectra with at least 50% of SPS masses matching to the identified peptide are considered as quantifiable PSMs. Quantification was performed at the MS3 level where the median of all quantifiable PSMs for each protein group was used for protein ratios.
Differential expression analysis of the proteome
Proteins identified from the MS experiment were filtered to retain only those with at least one unique peptide and that have minimum two peptide spectral matches from either of the two algorithms, MASCOT or Sequest. Further, to classify proteins as differentially expressed, a threshold of 1.2-fold change in both directions of over- and under-expression was decided for age-matched comparisons between rd1 and WT.
Pathway enrichment analysis of the proteome
GSEA for the proteomic dataset was performed using fgsea with ranked fold change values and Reactome and Gene Ontology annotations downloaded from the gProfileR webserver. Fgsea significant pathways (P < 0.05) were further collapsed to address redundancy. Ridge plots were generated using custom scripts and functions in the tidyverse and ggridges packages. Additionally, a list of OXPHOS proteins and ETC complexes were curated from literature for use in various analyses.
Metabolomic profiling
Retinal metabolome profiling
Targeted quantitative analysis was performed on retinal samples from WT and rd1 mice of P6 and P10 to detect cationic and anionic metabolites, using capillary electrophoresis mass spectrometry (CE-TOFMS and CE-QqQMS) in cation and anion analysis modes, respectively, with internal standard (Human Metabolome Technologies). Retinal tissue samples (30–45 mg) were mixed with 50% (v/v) acetonitrile and homogenized and the supernatant obtained after brief centrifugation was filtrated through 5 kDa cut-off filter to remove macromolecules. Filtrate was centrifugally concentrated and resuspended in water for mass spectrometry measurement. Metabolite concentrations were calculated by normalizing to internal standard and quantity of sample used. Primary analysis of metabolite detection and abundance estimates were performed by Human Metabolome Technologies, Boston MA, USA.
Retinal metabolome analysis
For analysis, metabolites were linked to their corresponding KEGG pathways and visualized in a network diagram created with igraph (R package) and Cytoscape. To classify differential abundance of metabolites we used two sets of thresholds: (a) significance differential abundance with P < 0.05 from pairwise tests of age-matched samples at P6 and P10; and (b) metabolites with fold change of minimum 1.2 times in either over or under abundance in rd1 versus WT comparisons.
In vitro and ex vivo analysis of mitochondrial activity
Complex I activity assay
The protocol for complex I activity assay was adopted from previous publications (76). In brief, dissected retinas were homogenized in SETH buffer containing 0.25 M sucrose, 2 mM EDTA and 10 mM Tris pH 7.4. The lysate was first spun at 600 × g for 10 min at 4°C followed by a second spin of supernatant at 14 000 × g for 10 min at 4°C. Mitochondria pellet was resuspended in 10 mM Tris pH 7.4. Complex I oxidizes NADH and produces an electron. The produced electron is then used to reduce an artificial substrate decylubiquinone, which subsequently passes the electron to the terminal electron receptor DCIP that is blue in color. Reduction of DCIP concentration can be followed spectrophotometrically at 600 nm. Rotenone is added to terminate the reaction and reveal any residue non-complex I related reduction of DCIP.
Mitochondrial respiration and oxygen consumption
OCR of retinal punches were measured using Seahorse XF24 Bioanalyzer with XF24 Islet Fluxpaks (Agilent) following our published protocol (9,77). In brief, eyes were enucleated and placed in ice-cold PBS for dissection. Cornea and lens were removed, and the retinal cup was gently separated away from the scleral layer. Three to four of 1 mm diameter punches equidistant from the optic nerve head were taken from each retina for the measurement. Ames’ buffer (Sigma) containing 120 mM NaCl, 10 mM HEPES, pH 7.4 was used in the assay. After steady basal OCR was established, mitochondrial uncoupler (2-fluorophenyl)(6-[(2-fluorophenyl)amino](1,2,5-oxadiazolo[3,4-e]pyrazin-5-yl))amine (BAM15; Timtec, Newark, DE, USA) was added at a final concentration of 5 mM to uncouple the electrochemical gradient of protons, thereby uncoupling ATP production from oxygen consumption and causing mitochondrial OXPHOS to run at full capacity. The complex I inhibitor rotenone was used at a final concentration of 1 mM to inhibit the entire ETC and thereby revealing the residual non-mitochondrial oxygen consumption.
Raw OCR values were computed by the Seahorse KSV algorithm, per manufacturer’s recommendations and non-mitochondrial oxygen consumption, the residual readings obtained after addition of rotenone, was subtracted from all points. Basal mitochondrial OCR (OCRbasal) was taken at 36 min, the last measuring point before BAM15 addition, whereas the highest value from uncoupled points was used for maximal OCR (OCRmax). Mitochondrial reserve capacity (MRC) was calculated as the percentage difference between maximal uncontrolled OCRmax and the initial OCRbasal: 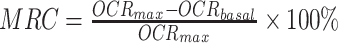 . A range of 4–8 non-littermate animals were included in each age group of WT and rd1 mice, and data from 12 to 29 retinal punches were included for analysis in each age/genotype group. Group means ± SEMs are plotted for each age group and strain.
. A range of 4–8 non-littermate animals were included in each age group of WT and rd1 mice, and data from 12 to 29 retinal punches were included for analysis in each age/genotype group. Group means ± SEMs are plotted for each age group and strain.
Transmission electron microscopy
The mitochondrial ultrastructure was evaluated by TEM following standard preparation procedures (78). Eyes were enucleated and immersion fixed in 1% paraformaldehyde, 2.5% glutaraldehyde in PBS (pH 7.4), followed by treatment with 1% osmium tetroxide. Fixed samples were dehydrated through a series of ethanol gradient and in propylene oxide, and then embedded in epoxy resin. The retina samples were sectioned at 80–90 nm, mounted on copper grids, and doubly stained with uranyl acetate and lead citrate. Sections were imaged at the NEI Histology Core with JEOL JEM-1010 TEM. Photographs were taken for the inner segment region of the photoreceptors at 10 000× and 30 000× magnifications.
qPCR and Mit/Nu DNA ratio
The Mit/Nu DNA ratio were determined on P2 to P10 retinal samples from WT and rd1 mice using a published protocol (44). Whole genomic DNA was isolated from retinal tissues and quantitative PCR on the genomic DNA abundance of mitochondria-encoded Nd1 and nucleus-encoded Hk2 was performed to evaluate copy number of mitochondrial and nuclear genome respectively. The mtDNA to nuDNA ratio was calculated by a ΔΔCt method.
Quantification and statistical analysis
Unless specified otherwise, all statistical analyses and data visualizations were done using the R statistical platform. Differences from WT were regarded as significant if P < 0.05.
Supplementary Material
Acknowledgements
We are grateful to Yide Mi, Ashley Yedlicka and Megan Kopera for mouse colony management; Julie Laux, Jessica Albrecht and Rafael Villasmil of the flow cytometry core for their assistance; and Thad Whitaker, Maria Campos, Mones Abu-Asab and the histology core for histology and transmission electron microscopy. Drs Raul Covian and Anand Swaroop equally contributed to conceptual development of this project.
Conflict of Interest Statement: The authors declare no conflict of interest.
Contributor Information
Ke Jiang, Neurobiology, Neurodegeneration & Repair Laboratory, National Eye Institute, National Institutes of Health, 6 Center Drive, Bethesda, MD 20892, USA.
Anupam Kumar Mondal, Neurobiology, Neurodegeneration & Repair Laboratory, National Eye Institute, National Institutes of Health, 6 Center Drive, Bethesda, MD 20892, USA.
Yogita K Adlakha, Neurobiology, Neurodegeneration & Repair Laboratory, National Eye Institute, National Institutes of Health, 6 Center Drive, Bethesda, MD 20892, USA; Translational Health Science and Technology Institute, National Capital Region Biotech Cluster, Faridabad, India.
Jessica Gumerson, Neurobiology, Neurodegeneration & Repair Laboratory, National Eye Institute, National Institutes of Health, 6 Center Drive, Bethesda, MD 20892, USA.
Angel Aponte, Proteomics Core Facility, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Linn Gieser, Neurobiology, Neurodegeneration & Repair Laboratory, National Eye Institute, National Institutes of Health, 6 Center Drive, Bethesda, MD 20892, USA.
Jung-Woong Kim, Neurobiology, Neurodegeneration & Repair Laboratory, National Eye Institute, National Institutes of Health, 6 Center Drive, Bethesda, MD 20892, USA; Department of Life Science, College of Natural Sciences, Chung-Ang University, Seoul 06974, Republic of Korea.
Alexis Boleda, Neurobiology, Neurodegeneration & Repair Laboratory, National Eye Institute, National Institutes of Health, 6 Center Drive, Bethesda, MD 20892, USA; Center for Bioinformatics and Computational Biology, University of Maryland, College Park, MD 20740, USA.
Matthew J Brooks, Neurobiology, Neurodegeneration & Repair Laboratory, National Eye Institute, National Institutes of Health, 6 Center Drive, Bethesda, MD 20892, USA.
Jacob Nellissery, Neurobiology, Neurodegeneration & Repair Laboratory, National Eye Institute, National Institutes of Health, 6 Center Drive, Bethesda, MD 20892, USA.
Donald A Fox, Neurobiology, Neurodegeneration & Repair Laboratory, National Eye Institute, National Institutes of Health, 6 Center Drive, Bethesda, MD 20892, USA.
Robert Balaban, Laboratory of Cardiac Energetics, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA.
Raul Covian, Laboratory of Cardiac Energetics, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA.
Anand Swaroop, Neurobiology, Neurodegeneration & Repair Laboratory, National Eye Institute, National Institutes of Health, 6 Center Drive, Bethesda, MD 20892, USA.
Funding
The Intramural Research Program of the National Eye Institute (ZIAEY000450 and ZIAEY000546) and utilized the high-performance computational capabilities of the Biowulf Linux cluster at National Institutes of Health (http://biowulf.nih.gov).
References
- 1. Parikshak, N.N., Gandal, M.J. and Geschwind, D.H. (2015) Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet, 16, 441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gan, L., Cookson, M.R., Petrucelli, L. and La Spada, A.R. (2018) Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci., 21, 1300–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Menzies, F.M., Fleming, A. and Rubinsztein, D.C. (2015) Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci., 16, 345–357. [DOI] [PubMed] [Google Scholar]
- 4. Verdin, E. (2015) NAD(+) in aging, metabolism, and neurodegeneration. Science, 350, 1208–1213. [DOI] [PubMed] [Google Scholar]
- 5. Wolozin, B. and Ivanov, P. (2019) Stress granules and neurodegeneration. Nat. Rev. Neurosci., 20, 649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wright, A.F., Jacobson, S.G., Cideciyan, A.V., Roman, A.J., Shu, X., Vlachantoni, D., McInnes, R.R. and Riemersma, R.A. (2004) Lifespan and mitochondrial control of neurodegeneration. Nat. Genet., 36, 1153–1158. [DOI] [PubMed] [Google Scholar]
- 7. Lin, M.T. and Beal, M.F. (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443, 787–795. [DOI] [PubMed] [Google Scholar]
- 8. Hasson, S.A., Kane, L.A., Yamano, K., Huang, C.H., Sliter, D.A., Buehler, E., Wang, C., Heman-Ackah, S.M., Hessa, T., Guha, R. et al. (2013) High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature, 504, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kooragayala, K., Gotoh, N., Cogliati, T., Nellissery, J., Kaden, T.R., French, S., Balaban, R., Li, W., Covian, R. and Swaroop, A. (2015) Quantification of oxygen consumption in retina ex vivo demonstrates limited reserve capacity of photoreceptor mitochondria. Invest Ophthal Vis Sci, 56, 8428–8436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ducker, G.S. and Rabinowitz, J.D. (2017) One-carbon metabolism in health and disease. Cell Metab., 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang, L., Garcia Canaveras, J.C., Chen, Z., Wang, L., Liang, L., Jang, C., Mayr, J.A., Zhang, Z., Ghergurovich, J.M., Zhan, L. et al. (2020) Serine catabolism feeds NADH when respiration is impaired. Cell Metab., 31, 809, e806–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hanany, M., Rivolta, C. and Sharon, D. (2020) Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. U.S.A., 117, 2710–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Perea-Romero, I., Gordo, G., Iancu, I.F., Del Pozo-Valero, M., Almoguera, B., Blanco-Kelly, F., Carreno, E., Jimenez-Rolando, B., Lopez-Rodriguez, R., Lorda-Sanchez, I. et al. (2021) Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci. Rep., 11, 1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roska, B. and Sahel, J.A. (2018) Restoring vision. Nature, 557, 359–367. [DOI] [PubMed] [Google Scholar]
- 15. Garafalo, A.V., Cideciyan, A.V., Heon, E., Sheplock, R., Pearson, A., WeiYang Yu, C., Sumaroka, A., Aguirre, G.D. and Jacobson, S.G. (2020) Progress in treating inherited retinal diseases: early subretinal gene therapy clinical trials and candidates for future initiatives. Prog. Retin. Eye Res., 77, 100827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Travis, G.H. (1998) Mechanisms of cell death in the inherited retinal degenerations. Am. J. Hum. Genet., 62, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Power, M., Das, S., Schutze, K., Marigo, V., Ekstrom, P. and Paquet-Durand, F. (2020) Cellular mechanisms of hereditary photoreceptor degeneration - focus on cGMP. Prog. Retin. Eye Res., 74, 100772. [DOI] [PubMed] [Google Scholar]
- 18. Sanges, D., Comitato, A., Tammaro, R. and Marigo, V. (2006) Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc. Natl. Acad. Sci. U.S.A., 103, 17366–17371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clarke, G., Collins, R.A., Leavitt, B.R., Andrews, D.F., Hayden, M.R., Lumsden, C.J. and McInnes, R.R. (2000) A one-hit model of cell death in inherited neuronal degenerations. Nature, 406, 195–199. [DOI] [PubMed] [Google Scholar]
- 20. Swaroop, A., Kim, D. and Forrest, D. (2010) Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat. Rev. Neurosci., 11, 563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wright, A.F., Chakarova, C.F., Abd El-Aziz, M.M. and Bhattacharya, S.S. (2010) Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet, 11, 273–284. [DOI] [PubMed] [Google Scholar]
- 22. Veleri, S., Lazar, C.H., Chang, B., Sieving, P.A., Banin, E. and Swaroop, A. (2015) Biology and therapy of inherited retinal degenerative disease: insights from mouse models. Dis. Model. Mech., 8, 109–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Collin, G.B., Gogna, N., Chang, B., Damkham, N., Pinkney, J., Hyde, L.F., Stone, L., Naggert, J.K., Nishina, P.M. and Krebs, M.P. (2020) Mouse models of inherited retinal degeneration with photoreceptor cell loss. Cell, 9, 931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bowes, C., Li, T., Danciger, M., Baxter, L.C., Applebury, M.L. and Farber, D.B. (1990) Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature, 347, 677–680. [DOI] [PubMed] [Google Scholar]
- 25. Pittler, S.J. and Baehr, W. (1991) Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc. Natl. Acad. Sci. U. S. A., 88, 8322–8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baehr, W., Devlin, M.J. and Applebury, M.L. (1979) Isolation and characterization of cGMP phosphodiesterase from bovine rod outer segments. J. Biol. Chem., 254, 11669–11677. [PubMed] [Google Scholar]
- 27. Palczewski, K. (2014) Chemistry and biology of the initial steps in vision: the Friedenwald lecture. Invest Ophthal Vis Sci, 55, 6651–6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McLaughlin, M.E., Sandberg, M.A., Berson, E.L. and Dryja, T.P. (1993) Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat. Genet., 4, 130–134. [DOI] [PubMed] [Google Scholar]
- 29. Hackam, A.S., Strom, R., Liu, D., Qian, J., Wang, C., Otteson, D., Gunatilaka, T., Farkas, R.H., Chowers, I., Kageyama, M. et al. (2004) Identification of gene expression changes associated with the progression of retinal degeneration in the rd1 mouse. Invest Ophthal Vis Sci, 45, 2929–2942. [DOI] [PubMed] [Google Scholar]
- 30. Azadi, S., Paquet-Durand, F., Medstrand, P., van Veen, T. and Ekstrom, P.A. (2006) Up-regulation and increased phosphorylation of protein kinase C (PKC) delta, mu and theta in the degenerating rd1 mouse retina. Mol. Cell. Neurosci., 31, 759–773. [DOI] [PubMed] [Google Scholar]
- 31. Punzo, C. and Cepko, C. (2007) Cellular responses to photoreceptor death in the rd1 mouse model of retinal degeneration. Invest Ophthal Vis Sci, 48, 849–857. [DOI] [PubMed] [Google Scholar]
- 32. Dickison, V.M., Richmond, A.M., Abu Irqeba, A., Martak, J.G., Hoge, S.C., Brooks, M.J., Othman, M.I., Khanna, R., Mears, A.J., Chowdhury, A.Y. et al. (2012) A role for prenylated rab acceptor 1 in vertebrate photoreceptor development. BMC Neurosci., 13, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou, J., Rasmussen, M. and Ekstrom, P. (2021) cGMP-PKG dependent transcriptome in normal and degenerating retinas: novel insights into the retinitis pigmentosa pathology. Exp. Eye Res., 212, 108752. [DOI] [PubMed] [Google Scholar]
- 34. Hauck, S.M., Ekstrom, P.A., Ahuja-Jensen, P., Suppmann, S., Paquet-Durand, F., van Veen, T. and Ueffing, M. (2006) Differential modification of phosducin protein in degenerating rd1 retina is associated with constitutively active Ca2+/calmodulin kinase II in rod outer segments. Mol. Cell. Proteomics, 5, 324–336. [DOI] [PubMed] [Google Scholar]
- 35. Cavusoglu, N., Thierse, D., Mohand-Said, S., Chalmel, F., Poch, O., Van-Dorsselaer, A., Sahel, J.A. and Leveillard, T. (2003) Differential proteomic analysis of the mouse retina: the induction of crystallin proteins by retinal degeneration in the rd1 mouse. Mol. Cell. Proteomics, 2, 494–505. [DOI] [PubMed] [Google Scholar]
- 36. Punzo, C., Xiong, W. and Cepko, C.L. (2012) Loss of daylight vision in retinal degeneration: are oxidative stress and metabolic dysregulation to blame? J. Biol. Chem., 287, 1642–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kanow, M.A., Giarmarco, M.M., Jankowski, C.S., Tsantilas, K., Engel, A.L., Du, J., Linton, J.D., Farnsworth, C.C., Sloat, S.R., Rountree, A. et al. (2017) Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. elife, 6, e28899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park, K.S., Xu, C.L., Cui, X. and Tsang, S.H. (2018) Reprogramming the metabolome rescues retinal degeneration. Cell. Mol. Life Sci., 75, 1559–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Akimoto, M., Cheng, H., Zhu, D., Brzezinski, J.A., Khanna, R., Filippova, E., Oh, E.C., Jing, Y., Linares, J.L., Brooks, M. et al. (2006) Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors. Proc. Natl. Acad. Sci. U. S. A., 103, 3890–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim, J.W., Yang, H.J., Brooks, M.J., Zelinger, L., Karakulah, G., Gotoh, N., Boleda, A., Gieser, L., Giuste, F., Whitaker, D.T. et al. (2016) NRL-regulated transcriptome dynamics of developing rod photoreceptors. Cell Rep., 17, 2460–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Borowski, L.S., Szczesny, R.J., Brzezniak, L.K. and Stepien, P.P. (2010) RNA turnover in human mitochondria: more questions than answers? Biochim. Biophys. Acta, 1797, 1066–1070. [DOI] [PubMed] [Google Scholar]
- 42. Kohn, L., Kadzhaev, K., Burstedt, M.S., Haraldsson, S., Hallberg, B., Sandgren, O. and Golovleva, I. (2007) Mutation in the PYK2-binding domain of PITPNM3 causes autosomal dominant cone dystrophy (CORD5) in two Swedish families. Eur. J. Hum. Genet., 15, 664–671. [DOI] [PubMed] [Google Scholar]
- 43. Boldyrev, A.A., Aldini, G. and Derave, W. (2013) Physiology and pathophysiology of carnosine. Physiol. Rev., 93, 1803–1845. [DOI] [PubMed] [Google Scholar]
- 44. Quiros, P.M., Goyal, A., Jha, P. and Auwerx, J. (2017) Analysis of mtDNA/nDNA ratio in mice. Curr Protoc Mouse Biol, 7, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jackson, G.R., Owsley, C. and Curcio, C.A. (2002) Photoreceptor degeneration and dysfunction in aging and age-related maculopathy. Ageing Res. Rev., 1, 381–396. [DOI] [PubMed] [Google Scholar]
- 46. Ait-Ali, N., Fridlich, R., Millet-Puel, G., Clerin, E., Delalande, F., Jaillard, C., Blond, F., Perrocheau, L., Reichman, S., Byrne, L.C. et al. (2015) Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell, 161, 817–832. [DOI] [PubMed] [Google Scholar]
- 47. Xu, J., Morris, L., Thapa, A., Ma, H., Michalakis, S., Biel, M., Baehr, W., Peshenko, I.V., Dizhoor, A.M. and Ding, X.Q. (2013) cGMP accumulation causes photoreceptor degeneration in CNG channel deficiency: evidence of cGMP cytotoxicity independently of enhanced CNG channel function. J. Neurosci., 33, 14939–14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang, T., Tsang, S.H. and Chen, J. (2017) Two pathways of rod photoreceptor cell death induced by elevated cGMP. Hum. Mol. Genet., 26, 2299–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vinberg, F., Chen, J. and Kefalov, V.J. (2018) Regulation of calcium homeostasis in the outer segments of rod and cone photoreceptors. Prog. Retin. Eye Res., 67, 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Okawa, H., Sampath, A.P., Laughlin, S.B. and Fain, G.L. (2008) ATP consumption by mammalian rod photoreceptors in darkness and in light. Curr. Biol., 18, 1917–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rueda, E.M., Johnson, J.E., Jr., Giddabasappa, A., Swaroop, A., Brooks, M.J., Sigel, I., Chaney, S.Y. and Fox, D.A. (2016) The cellular and compartmental profile of mouse retinal glycolysis, tricarboxylic acid cycle, oxidative phosphorylation, and ~P transferring kinases. Mol. Vis., 22, 847–885. [PMC free article] [PubMed] [Google Scholar]
- 52. Venkatesh, A., Ma, S., Le, Y.Z., Hall, M.N., Ruegg, M.A. and Punzo, C. (2015) Activated mTORC1 promotes long-term cone survival in retinitis pigmentosa mice. J. Clin. Invest., 125, 1446–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang, L., Du, J., Justus, S., Hsu, C.W., Bonet-Ponce, L., Wu, W.H., Tsai, Y.T., Wu, W.P., Jia, Y., Duong, J.K. et al. (2016) Reprogramming metabolism by targeting sirtuin 6 attenuates retinal degeneration. J. Clin. Invest., 126, 4659–4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xue, Y., Wang, S.K., Rana, P., West, E.R., Hong, C.M., Feng, H., Wu, D.M. and Cepko, C.L. (2021) AAV-Txnip prolongs cone survival and vision in mouse models of retinitis pigmentosa. elife, 10, e66240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Perkins, G.A., Ellisman, M.H. and Fox, D.A. (2003) Three-dimensional analysis of mouse rod and cone mitochondrial cristae architecture: bioenergetic and functional implications. Mol. Vis., 9, 60–73. [PubMed] [Google Scholar]
- 56. Vlachantoni, D., Bramall, A.N., Murphy, M.P., Taylor, R.W., Shu, X., Tulloch, B., Van Veen, T., Turnbull, D.M., McInnes, R.R. and Wright, A.F. (2011) Evidence of severe mitochondrial oxidative stress and a protective effect of low oxygen in mouse models of inherited photoreceptor degeneration. Hum. Mol. Genet., 20, 322–335. [DOI] [PubMed] [Google Scholar]
- 57. Nicholls, D.G. (2010) Mitochondrial ion circuits. Essays Biochem., 47, 25–35. [DOI] [PubMed] [Google Scholar]
- 58. Klaus, S. and Ost, M. (2020) Mitochondrial uncoupling and longevity - a role for mitokines? Exp. Gerontol., 130, 110796. [DOI] [PubMed] [Google Scholar]
- 59. Perron, N.R., Beeson, C. and Rohrer, B. (2013) Early alterations in mitochondrial reserve capacity; a means to predict subsequent photoreceptor cell death. J. Bioenerg. Biomembr., 45, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fox, D.A., Poblenz, A.T. and He, L. (1999) Calcium overload triggers rod photoreceptor apoptotic cell death in chemical-induced and inherited retinal degenerations. Ann. N. Y. Acad. Sci., 893, 282–285. [DOI] [PubMed] [Google Scholar]
- 61. Glancy, B. and Balaban, R.S. (2012) Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry, 51, 2959–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. He, L., Poblenz, A.T., Medrano, C.J. and Fox, D.A. (2000) Lead and calcium produce rod photoreceptor cell apoptosis by opening the mitochondrial permeability transition pore. J. Biol. Chem., 275, 12175–12184. [DOI] [PubMed] [Google Scholar]
- 63. Petit, L., Ma, S., Cipi, J., Cheng, S.Y., Zieger, M., Hay, N. and Punzo, C. (2018) Aerobic glycolysis is essential for normal rod function and controls secondary cone death in retinitis Pigmentosa. Cell Rep., 23, 2629–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pilkis, S.J., Claus, T.H., Kurland, I.J. and Lange, A.J. (1995) 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: a metabolic signaling enzyme. Annu. Rev. Biochem., 64, 799–835. [DOI] [PubMed] [Google Scholar]
- 65. Randle, P.J. (1986) Fuel selection in animals. Biochem. Soc. Trans., 14, 799–806. [DOI] [PubMed] [Google Scholar]
- 66. Rowe, A.A., Patel, P.D., Gordillo, R. and Wert, K.J. (2021) Replenishment of TCA cycle intermediates provides photoreceptor resilience against neurodegeneration during progression of retinitis pigmentosa. JCI Insight, 6, e150898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yuan, T., Walsh, M.P., Sutherland, C., Fabian, H. and Vogel, H.J. (1999) Calcium-dependent and -independent interactions of the calmodulin-binding domain of cyclic nucleotide phosphodiesterase with calmodulin. Biochemistry, 38, 1446–1455. [DOI] [PubMed] [Google Scholar]
- 68. Hsu, Y.T. and Molday, R.S. (1994) Interaction of calmodulin with the cyclic GMP-gated channel of rod photoreceptor cells. Modulation of activity, affinity purification, and localization. J. Biol. Chem., 269, 29765–29770. [PubMed] [Google Scholar]
- 69. Godoy, J.A., Rios, J.A., Picon-Pages, P., Herrera-Fernandez, V., Swaby, B., Crepin, G., Vicente, R., Fernandez-Fernandez, J.M. and Munoz, F.J. (2021) Mitostasis, calcium and free radicals in health, aging and neurodegeneration. Biomol. Ther., 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Area-Gomez, E., Guardia-Laguarta, C., Schon, E.A. and Przedborski, S. (2019) Mitochondria, OxPhos, and neurodegeneration: cells are not just running out of gas. J. Clin. Invest., 129, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Brooks, M.J., Chen, H.Y., Kelley, R.A., Mondal, A.K., Nagashima, K., De Val, N., Li, T., Chaitankar, V. and Swaroop, A. (2019) Improved retinal organoid differentiation by modulating signaling pathways revealed by comparative transcriptome analyses with development in vivo. Stem Cell Reports, 13, 891–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yang, F., Shen, Y., Camp, D.G., 2nd and Smith, R.D. (2012) High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev Proteomics, 9, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ting, L., Rad, R., Gygi, S.P. and Haas, W. (2011) MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods, 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Brosch, M., Yu, L., Hubbard, T. and Choudhary, J. (2009) Accurate and sensitive peptide identification with Mascot percolator. J. Proteome Res., 8, 3176–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Spivak, M., Weston, J., Bottou, L., Kall, L. and Noble, W.S. (2009) Improvements to the percolator algorithm for peptide identification from shotgun proteomics data sets. J. Proteome Res., 8, 3737–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Janssen, A.J., Trijbels, F.J., Sengers, R.C., Smeitink, J.A., van den Heuvel, L.P., Wintjes, L.T., Stoltenborg-Hogenkamp, B.J. and Rodenburg, R.J. (2007) Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin. Chem., 53, 729–734. [DOI] [PubMed] [Google Scholar]
- 77. Jiang, K., Nellissery, J. and Swaroop, A. (2021) Determination of mitochondrial respiration and glycolysis in ex vivo retinal tissue samples. J. Vis. Exp., 174. 10.3791/62914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ogilvy, A.J., Shen, D., Wang, Y., Chan, C.-C. and Abu-Asab, M.S. (2014) Implications of DNA leakage in eyes of mutant mice. Ultrastruct. Pathol., 38, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.