

Abstract
Human immunodeficiency virus type 1 (HIV-1) invades the central nervous system (CNS) shortly after infection and becomes localized in varying concentrations in different brain regions, the most vulnerable is the basal ganglia (BG). It is hypothesized that HIV-1mediated neuropathogenesis involves degeneration of dopaminergic neurons in the substantia nigra and the loss of dopaminergic terminals in the BG, leading to deficits in the central dopaminergic activity, resulting in progressive impairment of neurocognitive and motor functions. In the era of highly active antiretroviral therapy (HAART), although the incidence of HIV-associated dementia (HAD) has decreased, the neurocognitive and neuropsychological deficits continue to persist after HAART. In this study, We investigated the impact of HIV-1 on dopaminergic activity with respect to concentrations of dopamine (DA) and homovanillic acid (HVA) in different regions of postmortem human brains of HIV-1–negative and HIV-1+ individuals and their relationship to neurocognitive impairment. We found that in HIV-1+ as well as HIV-negative cases, dopamine and HVA concentrationsin ranged widely in different brain regions. In HIV-negative brain regions, the highest concentration of DA was found in putamen, caudate, substantia nigra, and the basal ganglia. In HIV-1+ cases, there was a significant decrease in DA levels in caudate nucleus, putamen, globus pallidus, and substantia nigra compared to that in HIV-negative cases. In HIV-1+ cases, a strong correlation was found between DA levels in substantia nigra and other brain regions. Concentration of HVA in HIV-negative cases was also highest in the regions containing high dopamine levels. However, no significant decrease in regional HVA levels was found in HIV-1+ cases. HIV-1 RNA load (nondetectable [ND] to log10 6.9 copies/g tissue) also ranged widely in the same brain regions of HIV-1+ cases. Interestingly, the brain regions having the highest HIV-1 RNA had the maximum decrease in DA levels. Age, gender, ethnicity, and postmortem interval were not correlated with decrease in DA levels. Profile of DA, HVA, and HIV-1 RNA levels in the brain regions of HIV-1+ individuals treated with HAART was similar to those not treated with HAART. A majority of HIV-1+ individuals had variable degrees of neurocognitive impairments, but no specific relationship was found between the regional DA content and severity of neurocognitive deficits. These findings suggest widespread deficits in dopamine in different brain regions of HIV-1–infected cases, and that these deficits may be the results of HIV-1–induced neurodegeneration in the subcortical regions of human brain.
Keywords: CNS, dopamine, HIV-1, HVA
Introduction
Human immunodeficiency virus type 1 (HIV-1) has affected millions of people worldwide, and despite multipronged international efforts to contain this pandemic, the disease continues to spread unabated. According to the recent updated reports by WHO/UNAIDS (AIDS Epidemic Update, 2007), the current prevalence of HIV-1 infection around the world remains a major concern, as there are 33.2 million people living with HIV infection, 2.5 million are newly infected, and 2.1 million died of HIV-1–related complications. Irrespective of the regional and demographic diversity of people, mode of infection and health consequences, including progressive immune suppression, opportunistic infections, and neurocognitive deficits, remain common among all the people who are infected with HIV-1.
In the very early phase of the epidemic, the central nervous system (CNS) was considered as an important target of HIV-1 infection that could lead to neurodegeneration (Ho et al, 1985). It is now well recognized that within days of primary systemic infection, the HIV-1 virus invades the CNS and is localized in varying concentrations in different brain regions (Resnick et al, 1988; Masliah et al, 1992; Wiley et al, 1986; Kumar et al, 2007). Although the mode of entry of HIV-1 into the brain remains debatable, one of the most favored proposed models is that the virus enters the CNS through trafficking of HIV-1–infected monocytes, or possibly lymphocytes, by using the “Trojan horse mechanism” and penetrating the blood-brain barrier (Nottet et al, 1996; Persidsky et al, 1997; Persidsky and Gendelman, 2003). The other models may include the virus’ entry through the infected vascular endothelium (Maslin et al, 2005), and/or through direct infiltration of cell free viral particles from circulation in to the CNS (Banks et al, 2004; Bobardt et al, 2004). Once in the brain, HIV-1 has the propensity to become localized in the frontostriatal pathways and other subcortical regions, particularly in the basal ganglia, causing progressive neurodegenerative changes, and impairment in neuromotor and neurocognitive functions regulated by the neuronal activities in these regions (Kure et al, 1990). Although progression of HIV-1 infection to symptomatic acquired immunodeficiency syndrome (AIDS) occurs after a number of years, studies have demonstrated that even in the early stage of infection the basal ganglia (BG) are highly vulnerable to HIV-1 assault. Using quantitative magnetic resonance imaging (MRI), and positron emission tomography (PET), a number of reports show a selective reduction in the volume of caudate nucleus and white matter, as well as the cortical and subcortical gray matter, and this loss continues to progress over the course of the infection (Jernigan et al, 1993; Kieburtz et al, 1996; Aylward et al, 1993). Furthermore, PET studies with fluorodeoxyglucose (18F-FDG) also found changes in the metabolic activity of the basal ganglia, showing hypermetabolism in the early stages of HIV-1 infection, and hypometabolism in more advanced stages of the disease (Rottenberg et al, 1996). These observations of disturbance in brain metabolic activity are further supported by a recent study that used voxel based 18F-FDG PET brain imaging in individuals with HIV-1 infection who were also injecting drug users (IDUs), and found that HIV-1 had a synergistic effect with IDU on hypermetabolic activity in the subcortical deep white matter, the basal ganglia, the thalami, and the brain stem, and on the decreased activity in the cortical structures (Georgiou et al, 2008).
The neuropathology of HIV-1 infection is driven by many complex processes involving the impact of neurotoxic virus-related products, as well as virus-host interaction–related neurotoxic factors contributing to HIV encephalitis, CNS injury, neurodegeneration, and cognitive impairment (Masliah et al, 1992; Brew, 1993). Although HIV-1 does not infect neurons directly because of the absence of receptors (Broder and Dimitrov, 1996), experimental studies have found that two HIV-1 proteins, Tat and gp120, are highly neurotoxic and can cause neuronal injury and ultimately the neuronal cell death (Nath, 2002). Additionally, a number of neurotoxic factors are generated by the brain perivascular macrophages, resident microglia, as well as astrocytes in response to entry of M-tropic HIV-1 into these cells by binding to their CD4 receptors and CCR5 coreceptors (Choe et al, 1996; Kaul et al, 2007). These infected brain cells produce an array of neurotoxins, including proinflammatory cytokines such as tumor necrosis factor alpha (TNFα), and other neurotoxic factors, including β2-microglobulins, neopterins, kynurenine, excitotoxins such as quinolinic acid, and chemokines such as monocyte chemotactic protein-1 (MCP-1), and proteases (Ranki et al, 1995; Mattson et al, 2005; Heyes et al, 1992). These neurotoxic factors play an important role in the degenerating processes of neurons and their supporting structures. Neuronal degeneration in different brain regions, specifically that of the dopaminergic system in the basal ganglia, has been found to relate to mild to severe cognitive impairment and HIV-associated dementia (HAD) found in 20% to 30% of HIV-1–infected individuals (Navia and Price, 1987; Berger and Arendt 2000).
The introduction of highly active antiretroviral therapy (HAART) over the past several years has resulted in the improvement of the immune status with increased CD4+ cells, and decrease in plasma viral load, but has changed HIV infection in to a chronic disease with increasing prevalence and extended life expectancy (Dore et al, 2003). There is a large body of evidence suggesting that although HAART has minimized the incidence of severe form of HAD, the prevalence rate of neurological problems, cognitive impairment, and HAD continue to increase among HIV-1–infected individuals (Sacktor et al, 2002), because some of the drugs in HAART, such as protease inhibitors, have poor oral absorption, variable tissue distribution, and poor penetration of the blood-brain barrier to reach the brain in therapeutic levels to be effective (Kim et al, 1998). Thus, despite intervention with HAART, the brain continues to remain a sanctuary for HIV-1, causing persistent neuropathogenesis in different areas of the brain (Bagasra et al, 1996; Schrager and D’Souza, 1998).
The concept that HIV-1–induced neurodegeneration of the basal ganglia may result in dopaminergic deficits was based on early clinical observations that some of the HIV-1–infected patients with AIDS developed the Parkinson’s disease symptoms of cognitive and motor disorders when treated with dopamine receptor–blocking drugs, such as haloperidol, chlorpromazine, or metoclopropamide. The severity of parkinsonian symptoms observed in these patients was attributed to HIV-1–induced hypersensitivity of dopamine receptors to antipsychotic agents (Kieburtz et al, 1991; Mirsattari et al, 1998; Koutsilieri et al, 2002). Further indirect evidence that HIV-1–induced damage to basal ganglia may involve dopaminergic system dysfunction came from the treatment strategies for alleviating parkinsonian symptomatology by augmenting dopaminergic activity in HIV-1–infected individuals. These treatment strategies designed to enhance dopaminergic activity included substances such as l-DOPA (the precursor of dopamine synthesis), selegeline (deprenyl), an inhibitor of monoamine oxidase (MAO) A and B activities, and methylphenidate (a psychostimulant) to improve the availability of dopamine. For example, a controlled clinical trial of selegiline was found to improve cognitive functions in patients with HAD (Danna Consortium, 1998; Sacktor et al, 2000). Selegeline acts by slowing down the catabolism of dopamine in the CNS by inhibiting MAO, thus increasing the availability of dopamine (Magyar, 1993). Similarly, methylphenidate, a dopaminergic agonist and a psychostimulant, was also found to have beneficial effects on some of the cognitive functions in HIV-1–infected patients (Hinkin et al, 2001). So far, the evidence that HIV infection may lead to deficits in the CNS dopmaminergic activity has come from a few studies involving cerebrospinal fluid (CSF) and finding a decrease in the levels of dopamine and homovanillic acid (HVA) in the CSF of patients who were at different stages of disease progression (Berger, et al, 1994; Larsson et al, 1991; Ellis et al, 2004). However, CSF does not represent any specific region of the CNS, therefore the levels of dopamine or HVA in the CSF may not reflect the actual deficits in the regional dopaminergic activity in the brains of HIV-1–infected individuals.
The evidence thus far suggests that the basal ganglia are the main site of HIV-1 assault, and that the neurodegeneration, as well as metabolic abnormalities in the basal ganglia, and dopaminergic system dysfunction may be associated with neurocognitive deficits (Nath et al, 2000; von Gisslen et al, 2001). Although dopamine levels have been found to decrease in the CSF of HIV-1–infected individuals, and dopaminergic activity–enhancing drugs have been reported to improve cognitive functions, investigations have remained scarce on the evaluation of direct impact of HIV-1 on dopamine activity in the CNS. Because the regional analysis of dopaminergic activity could allow identification of the brain areas most affected by HIV-1 that may be contributing to neurocognitive deficits in patients with HIV/AIDS, we investigated tissue samples from different areas of postmortem human brains to determine the regional status of dopamine in the CNS. The samples of tissues were obtained from the frontal cortex and basal ganglia (BG) of a few cases, and from the nuclei of BG (caudate nucleus, putamen, globus pallidus, and substantia nigra) from the other cases, as well as the CSF of individuals who were assessed during life for HIV-associated cognitive deficits, irrespective of HAART treatment, and died of HIV/AIDS. The tissues from well-characterized brain regions were obtained from the National NeuroAIDS Tissue Consortium (NNTC), and included tissues from individuals irrespective of their HAART during life. The CNS regional dopaminergic activity in HIV-1–infected cases was compared with that in the same brain regions of HIV-negative cases.
Results
The demographic characteristics, including age, gender, ethnicity, as well as years of infection and postmortem interval (PMI) of HIV-1+ (N=38) and HIV-negative (N=11) cases are presented in Table 1. The other specific characteristics pertaining to HIV-1+ cases presented in Table 1 include neurocognitive status, MSK rating (0–4) used for the diagnosis of neurocognitive impairment, presence of opportunistic infections (OIs) at the time of death, and the number of individuals (n=33) who used highly active antiretroviral therapy (HAART) and those who did not use (n=5).
Table 1.
Demographic and other characteristics of HIV-1+ and HIV− cases
| Specific characteristics | HIV-1+ cases | HIV-negative cases |
|---|---|---|
|
| ||
| n | 38 | 11 |
| Year diagnosed with HIV-1 infection | 1978–2001 | No infection |
| Known duration of HIV-1 disease | 1–21 years | N/A |
| Age at death | 31–58 Years | 35–66 years |
| Year of death | 1998–2005 | 1998–2005 |
| Gender | 32 men, 6 women | 6 men, 5 women |
| Ethnicity | 29 Caucasian, 4 Hispanics, 2 African American (AA), 3 Native American | 9 Caucasians, 2 AA |
| MSK rating, AIDS dementia | 0–4 | N/A |
| Neurocognitive status | 12 probable HAD, 8 possible HAD, 5 probable MCMD, 2 possible MCMD, 2 MCMD, 5 Impaired, 2 NPI, 2 normal | N/A |
| Opportunistic infections | No opportunistic infections at the time of death | No infections |
| Antiretroviral therapy | Majority on HAART (N = 33), No HAART (N = 5) | N/A |
| Postmortem interval (PMI) | 2–24 h (mean ± SD, 9.41 ± 5.74 h) | 3–22 h (mean ± SD, 7.28 ± 5.77 h) |
The concentrations of dopamine (log10 pg/g tissue) and HVA (log10 ng/g tissue) are presented as mean ± SD in different brain regions of HIV-1+ and HIV-negative cases (Table 2). A wide variation was found in DA concentration in all regions of the autopsied brains of HIV-1+ and HIV-negative individuals (Figure 1). In HIV-1+ cases, there was a significant decrease in the overall concentration of DA in the basal ganglia (P<.005), as well as in the individual nuclei, caudate (P<.005), putamen (P<.000), and substantia nigra (P<.005), and no significant decrease in DA levels was found in FC, FC4, FC6, and GP of HIV-1+ cases compared to that in the same brain regions of HIV- negative cases. Strong correlations (rs) were found between concentration of DA in SN and DA in other brain regions (FC, BG, CAUD, PUTA, GP), rs=.679−.779, P <0.001 to 0.052 (Table 3), as well as between DA and HVA in FC4 and GP (rs=.697, P<.025), and FC4 and putamen (rs=.867, P<.001). The concentration of HVA was also found to vary widely among different brain regions of HIV-1+ and HIV-negative cases (Figure 2). However, HVA levels in different brain regions of HIV-1+ cases did not differ significantly from those in HIV-negative cases (Table 2). Furthermore, HIV-1 RNA levels (Table 2; mean ± SD, log10 copies/g tissue) were higher in the brain regions having maximum decrease in dopamine concentration. However, this relationship was not observed between the regional HVA levels and HIV-1 RNA load. There was no significant correlation (rs) between the levels of DA and viral load within the same brain region (Table 4). However, HVA concentration in SN and CAUD correlated negatively with VL in the same brain regions (“rs” −.782, P<.008, P<=−.771, P<.072, respectively) (Table 4).
Table 2.
Dopamine and HVA concentrations (log10) and HIV-1 viral RNA levels in postmortem brains of HIV-1+ and HIV− cases
| Dopamine concentration (log10 pg/g tissue) |
% change | P value | HVA concentration (log10 ng/g tissue) |
% change | P value | HIV-1 viral RNA load (log10 copies/g tissue) | |||
|---|---|---|---|---|---|---|---|---|---|
| HIV− | HIV-1+ | HIV− | HIV-1+ | ||||||
|
| |||||||||
| Frontal cortex (FC) | 2.186 ± 0.46 (5) | 1.90 ± 0.75 (24) | −13% | NS | 1.624 ± 0.615 (6) | 1.24 ± 0.61 (24) | −24% | NS | 2.68 ± 1.32 FC (9) |
| FC4 | 2.072 ± 0.495 (6) | 1.957 ± 0.637 (13) | −5% | NS | 1.85 ± 1.055 (5) | 1.06 ± 0.43 (13) | 43% | NS | 3.80 ± 0.96 FC4 (5) |
| FC6 | 1.911 ± 0.70 (6) | 1.952 ± 0.646 (13) | −2% | NS | .73 ± 0.685 (3) | 1.08 ± 0.34 (12) | +48% | NS | 3.55 ± 1.89 Basal ganglia (10) |
| Basal ganglia | 2.915 ± 0.20 (4) | 2.102 ± 0.928 (15) | −28% | <.005 | .75 ± 0.683 (4) | 2.13 ± 0.76 (15) | −22.5% | NS | 3.29 ± 1.49 FC6 (7) |
| Caudate | 3.90 ± 1.143 (6) | 2.086 ± 0.68 (14) | −47% | <.005 | 1.913 ± 0.519 (5) | 2.23 ± 0.34 (14) | +16% | NS | 4.63 ± 1.38 Caudate (6) |
| Putamen | 4.563 ± 0.305 (5) | 2.163 ± 0.435 (14) | −53% | <.000 | 2.506 ± 0.65 (5) | 2.56 ± 0.258 (14) | +2% | NS | 3.05 ± 1.52 Putamen (9) |
| Globus pallidus | 2.91 ± 1.182 (5) | 2.198 ± 0.51 (13) | −24% | NS | 1.872 ± 0.65 (5) | 2.198 ± 0.696 (13) | +17% | NS | 3.42 ± 1.10 Globus pallidus (5) |
| Substantia nigra | 3.202 ± 0.863 (6) | 1.747 ± 0.742 (23) | −45% | <.000 | 1.837 ± 0.68 (7) | 1.844 ± 0.62 (23) | +0.4% | NS | 3.44 ± 1.77 substantia nigra (10) |
| Cerebrospinal fluid | No sample | 2.137 ± 1.47 (9) | ? | No sample | 1.572 ± 0.70 (7) | ? | 2.66 ± 0.92 cerebrospinal fluid (8) | ||
Note. Dopamine and HVA concentrations are expressed as mean ± SD. FC4 = frontal cortex 4; FC6 = frontal cortex 6. Numbers in parentheses = samples of each brain region of HIV-1+ and HIV− cases. NS =not significant. Dopamine and HVA concentrations in tissues were measured using the CoulAray HPLC-ECD system. HIV-1 RNA load in the same brain regions was determined by real-time RT-PCR system.
Figure 1.
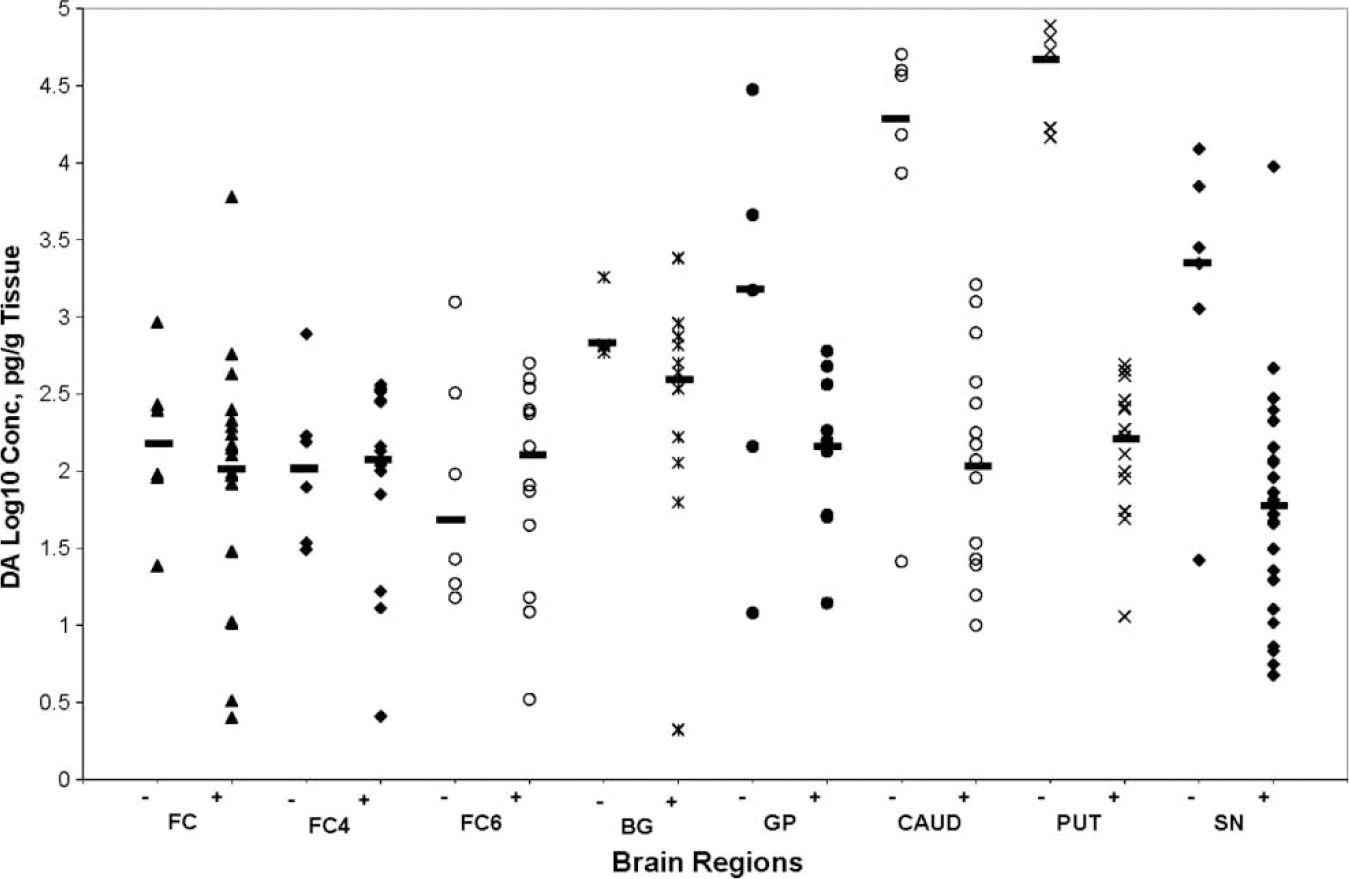
Scatter plot showing dopamine concentration (y-axis, log10 pg/g tissue) in each brain region (x-axis) of HIV-1+ and HIV-negative individuals. The horizontal bars represent the median value in each region. Dopamine concentration decreased significantly in the basal ganglia, caudate, putamen, and substantia nigra. No significant change was found in the levels of DA in FC, FC4, FC6, and globus pallidus of HIV-1+ cases compared to those in the brain regions of HIV-negative cases. The values of DA in CSF are not shown because CSF samples from HIV-1 negative cases were not available.
Table 3.
Correlations between DA levels in SN and other brain regions in HIV-1+ cases
| Brain regions | Correlations, “rs” | P value |
|---|---|---|
|
| ||
| SN vs FC | .779 | .001 |
| SN vs BG | .767 | .016 |
| SN vs CAUD | .750 | .052 |
| SN vs PUTA | .679 | .094 |
| SN vs GP | .714 | .071 |
| FC4 vs FC6 | .747 | .003 |
| BG vs FC | .907 | .000 |
| CAUD vs PUTA | .819 | .001 |
| CAUD vs GP | .636 | .026 |
| GP vs PUTA | .755 | .007 |
| DA FC4 vs HVA GP | .697 | .025 |
| DA FC4 vs HVA PUTA | .867 | .001 |
| HVA FC4 vs HVA FC6 | .846 | .001 |
| HVA BG vs HVA SN | .867 | .002 |
Note. DA levels in substantia nigra were significantly correlated with DA levels in FC, BG, and CAUD and weekly correlated with that in putamen and globus pallidus. Significant correlations were also found in DA levels between other regions.
Figure 2.
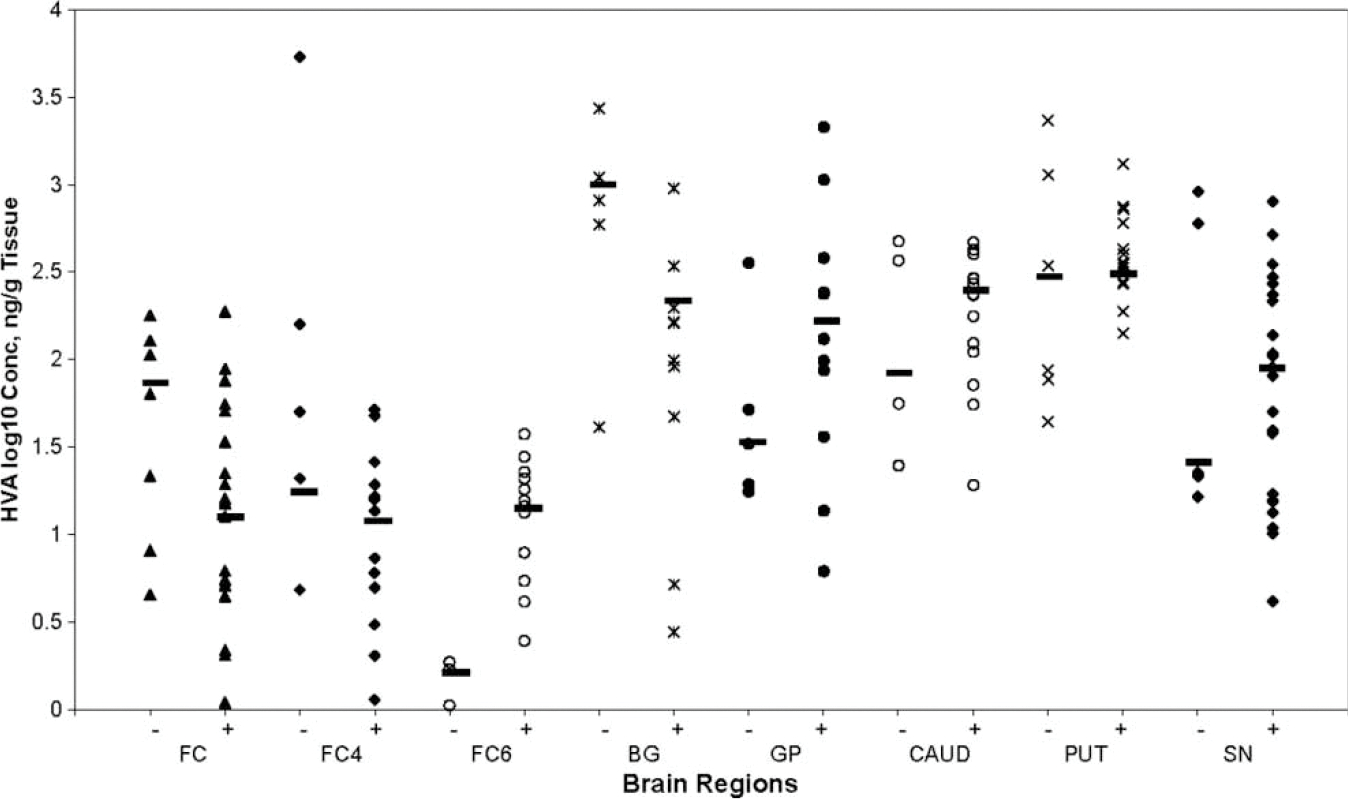
Scatter plot showing HVA concentration (y-axis, log10 ng/g tissue) in each brain region (x-axis) of HIV-1+ and HIV-negative individuals. The horizontal lines represent the median value in each region. There was no significant change found in the values of HVA in each region of HIV-1+ brain compared to that in HIV-negative brain.
Table 4.
Correlations between regional DA and HVA levels and VL
| DA vs VL |
HVA vs VL |
|||
|---|---|---|---|---|
| Correlations, “rs” | P value | Correlations, “rs” | P value | |
|
| ||||
| Brain region | ||||
| FC | .417 (24) | .265 | −.517 (25) | .154 |
| FC4 | −.700 (13) | .188 | .300 (13) | .624 |
| FC6 | −.536 (13) | .215 | −.086 (12) | .535 |
| BG | .430 (15) | .214 | .091 (15) | .813 |
| Caudate | −.314 (14) | .544 | −.771 (14) | .072* |
| Putamen | −.217 (14) | .576 | −.217 (14) | .576 |
| Globus pallidus | .200 (13) | .747 | .500 (13) | .391 |
| Substantia nigra | .236 (23) | .511 | −.782 (23) | .008** |
| Cerebrospinal fluid | 2.137 (7) | 1.417 | 1.88 (7) | −0.41 |
Note. Correlations were determined using Spearman’s rank correlations coefficient.
The level of significance, P, is denoted with * and **. There was no significant correlation between DA or HVA and HIV-1 RNA in different brain regions. Only significant negative correlation was found between HVA and HIV-1 RNA in the caudate and substantia nigra. Numbers in parenthesis represent tissue sample.
There was no statistically significant correlation, “rs” (P>.1), observed between age, gender, ethnicity, as well as PMI (HIV-1+, PMI range 3 to 22 h, mean ± SD, 7.28 ± 5.77 h; HIV-1–negative, 2 to 24 h, mean ± SD, 9.41 ± 5.74 h) and DA or HVA levels in different brain regions of HIV-1+ and HIV-negative cases.
Although a majority (87%) of HIV-1+ individuals had used highly active antiretroviral treatment (HAART) during life, there was no significant difference in the values of DA or HVA between the brain regions of those who used HAART (n=33) versus those who did not use HAART (13%), except in putamen where a significant difference was found in DA levels between HAART+ and HAART− individuals (n=11HAART+ versus n=3HAART−; P<.01) (Table 5).
Table 5.
Dopamine and HVA concentration in different regions of brain of HIV-1+ with and without HAART
| Brain region | DA concentration |
t | P value | HVA concentration |
t | P value | ||
|---|---|---|---|---|---|---|---|---|
| With HAART | No HAART | With HAART | No HAART | |||||
|
| ||||||||
| FC | 1.907 ± 0.75 (24) | — | 1.191 ± 0.65 (24) | — | ||||
| FC4 | 1.90 ± 0.705 (8) | 2.03 ± 0.40 (5) | 0.42 | NS | 1,038 ± 0.45 (8) | 0.90 ± 0.541 (5) | 0.47 | NS |
| FC6 | 2.06 ± 0.57 (8) | 1.78 ± 0.721 (5) | 0.73 | NS | 1.077 ± 0.32 (8) | 1.095 ± 0.36 (4) | 0.08 | NS |
| Basal ganglia | 2.04 ± 0.98 (14) | 2.22 ± xx (1) | — | — | 2.23 ± 0.68 (14) | 0.7126 ± xx (1) | — | — |
| Caudate | 2.026 ± 0.62 (10) | 2.24 ± 0.81 (4) | 0.47 | NS | 2.16 ± 0.35 (10) | 2.41 ± 0.386 (4) | 1.124 | NS |
| Putamen | 2.068 ± 0.436 (11) | 2.514 ± 0.17 (3) | 2.719 | <.01* | 2.56 ± 0.26 (11) | 2.584 ± 0.25 (3) | 0.146 | NS |
| Globus pallidus | 2.133 ± 0.53 (9) | 2.34 ± 0.42 (4) | 0.75 | NS | 2.30 ± 0.67 (9) | 1.964 ± 0.698 (4) | 0.81 | NS |
| Substantia nigra | 1.79 ± 0.77 (20) | 1.514 ± 0.47 (3) | 0.85 | NS | 1.819 ± 0.56 (20) | 2.013 ± 0.72 (3) | 0.45 | NS |
| CSF | 1.747 ± 1.56 (8) | 3.143 ± xx (1) | 1.498 ± 0.73 (6) | 2.0703 ± xx (1) | — | — | ||
Note. Dopamine and HVA concentration (log10 ng/g tissue) are presented as mean ± SD in different brain regions. — = no sample; Students’ t test was used to calculate the significance of difference (P values) in DA and HVA levels between HAART-treated and non–HAART-treated individuals. There was no significant difference in the levels of DA and HVA in different brain regions between HAART+ and HAART− individuals, excepting a significant difference (*P<.01) in DA levels was found in putamen. Interestingly, HAART-treated individuals had lower DA concentration (log10 pg/g tissue) in putamen compared to that in non–HAART-treated individuals. Numbers in parenthesis are the tissue samples from the number of cases. Samples of brain regions of individuals not using HAART varied between 1 and 5.
Surprisingly, among HIV-1+ cases, the decrease in DA levels in different brain regions (basal ganglia, caudate, putamen, and substantia nigra) did not significantly correlate (rs) with specific neurocognitive status (diagnosed as probable or possible HAD, probable or possible MCMD, or MCMD). Moreover, even when data from all HIV-1+ individuals were categorized into groups with diagnosis of HAD or MCMD, or unimpaired, no significant correlation (rs) was found between DA levels and HAD or MCMD, and there was no significant difference (Mann-Whitney test) in the regional DA levels between the two groups (HAD and MCMD), except that a trend towards decrease in DA levels was found in CAUD, PUTA, and GP of those with MCMD compared to those with HAD. There was no apparent difference in the regional levels of HVA among the three groups (Table 6).
Table 6.
Dopamine and HVA distribution in different brain regions with respect to neurocognitive status in HIV-1–infected individuals
| DA concentration (pg/g tissue) |
HVA concentration (ng/g tissue) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HAD vs MCMD |
HAD vs UnImp |
MCMD vs UnImp |
||||||||||
| Brain regions | HAD | MCMD | Unimpaired | t | P | t | P | t | P | HAD | MCMD | UnImp |
|
| ||||||||||||
| FC | 1.98 ± 0.75 (16) | 1.74 ± 0.83 (8) | – | 0.73 | .5 | – | – | – | 1.43 ± 0.64 (14) | 0.94 ± 0.54 (9) | – | |
| FC4 | 2.26 ± 0.28 (4) | 1.49 ± 0.86 (5) | 2.23 ± 0.31 (4) | 1.88 | .12 | 0.13 | NS | 1.63 | .15 | 1.09 ± 1.45 (4) | 0.88 ± 0.45 (5) | 1.33 ± 0.43 (3) |
| FC6 | 2.20 ± 0.39 (4) | 1.51 ± 0.83 (5) | 2.27 ± 0.44 (4) | 1.48 | <.18 | 0.21 | NS | 1.60 | .15 | 1.17 ± 0.26 (4) | 0.98 ± 0.41 (5) | 1.08 ± 0.42 (3) |
| BG | 2.14 ± 0.83 (10) | 2.02 ± 1.28 (5) | – | 0.23 | .83 | – | – | – | 2.20 ± 0.71 (4) | 1.96 ± 0.94 (5) | – | |
| CAUD | 2.36 ± 0.81 (6) | 1.39 ± 0.41 (4) | 2.36 ± 0.18 (4) | 2.20 | .06* | N/A | NS | 4.27 | <.005 | 2.22 ± 0.48 (10) | 2.32 ± 0.31 (4) | 2.16 ± 0.42 (4) |
| PUTA | 2.31 ± 0.34 (7) | 1.60 ± 0.49 (3) | 2.32 ± 0.28 (4) | 2.68 | .028* | N/A | NS | 2.51 | <.05 | 2.56 ± 0.22 (7) | 2.50 ± 0.32 (3) | 2.63 ± 0.36 (4) |
| GP | 2.35 ± 0.46 (6) | 1.67 ± 0.51 (3) | 2.35 ± 0.49 (4) | 2.03 | .08* | N/A | NS | 1.80 | <.13 | 2.22 ± 0.79 (7) | 2.25 ± 0.22 (3) | 1.97 ± 0.92 (4) |
| SN | 1.91 ± 0.83 (11) | 1.59 ± 0.73 (11) | 1.7 (1) | 0.95 | .36 | N/A | NS | – | – | 1.76 ± 0.73 (11) | 1.88 ± 0.63 (11) | 2.37 ± xx (1) |
| CSF | 2.08 ± 1.35 (3) | 1.81 ± 1.86 (4) | – | 0.21 | .84 | No sample | – | – | 0.59 (1) | 1.62 ± 0.71 | – | |
Note. Dopamine concentrations (mean ± SD) are presented in different brain regions of HIV-1+ cases. Numbers in parenthesis are the number of samples from each region.
HAD = HIV-associated dementia; MCMD = minor cognitive motor disorders; UnImp = unimpaired. The individuals with neurocognitive deficits were grouped into those diagnosed with HAD or MCMD. A limited number of individuals were also found to have no impairment, and were included as the comparison group. Using Mann-Whitney test, no significant (NS) difference in the regional DA levels was found between individuals with HAD and MCMD, excepting that there was a trend towards significant decrease in CAUD, PUTA, and GP in MCMD group compared to that in the same regions of HAD group. There was a significant decrease in the DA levels in CAUD and PUTA of individuals with MCMD compared to those who were unimpaired, but no difference was found in the DA levels in any region between the HAD group and the unimpaired group. From the unimpaired individuals, only one sample of SN but no sample of CSF was available. Levels of HVA in different brain regions did not appear significantly different among the three groups (statistical significance not presented).
Discussion
The major findings of this study demonstrate that the concentration of dopamine was very variable in different regions of post mortem brains of HIV-1+ and HIV-negative cases, and that there was a significant decrease in the regional DA concentration in individuals who died of HIV/AIDS (and were free of opportunistic infections at the time of death), compared with that in the same brain regions of HIV-1–negative cases. We further found that among HIV-negative cases, the heterogeneous distribution of DA was marked by the highest concentration present in the putamen and caudate nucleus, a finding consonant with that reported earlier in noninfected postmortem human brains (Arai et al, 1984; Herregodts et al, 1991). On the other hand, in HIV-1+ individuals, DA concentration decreased significantly in putamen, substantia nigra, caudate nucleus, and the basal ganglia, but no significant decrease was found in DA levels in globus pallidus and the three frontocortical areas (Table 2, Figure 1). However, DA levels in SN correlated significantly with DA levels in the other brain regions, including FC, BG, PUTA, and GP, as well as between different brain regions (Table 3), suggesting that DA content and its availability in different brain regions may be regulated by its production in SN, and that the decreased DA concentration in these regions may be due to HIV-1–associated degeneration of dopamine producing neurons in the SN. Although the brain regions having maximum decrease in dopamine concentration had the highest concentration of HIV-1 RNA, no correlation was found between DA concentration and HIV-1 viral load in any brain region (Table 4). The concentration of HVA in HIV-negative cases was also high in the same brain regions containing high DA levels (Table 2), and were consistent with those reported earlier (Mackay et al, 1986). However, changes in HVA levels in HIV-1+ cases were not as remarkable as those of dopamine, and correlated negatively with viral load (VL) only in substantia nigra and in caudate nucleus, but not with VL in the other brain regions (Table 4).
Previous studies carried out on CSF of HIV-1–infected asymptomatic and symptomatic patients during life found decrease in biogenic amines, including dopamine and HVA, as well as indolea-mines (Larsson et al, 1991; Berger et al, 1994; Kumar et al, 2001), implicating that damage to biogenic amine neurons may also occur in early stages of the disease. However, the relationship between DA deficits in CSF and clinical symptomatology of neurological and neurocognitive problems in HIV/AIDS has not been clearly understood. Moreover, because CSF does not represent any specific region of the CNS, these findings were taken as the overall reflection of deficits in the brain dopaminergic activity caused by HIV-1 infection. Early evidence that damage to DA neurons occurs in HIV-1 infection came from studies on postmortem brain tissues, showing a significant decrease in DA concentration in the caudate nucleus of patients with AIDS (Sardar et al, 1996), and in putamen of simian immunodeficiency virus (SIV)-infected rhesus monkeys with and without symptoms of AIDS (Scheller et al, 2005). However, investigations in these studies were limited to one or two brain regions to assess the dopaminergic activity in postmortem brains of HIV-1− or SIV-infected cases, and, moreover, the viral load in the same brain regions was not known. The present study, to our knowledge, reports for the first time the widespread depletion in dopamineregic activity and a widely variable distribution of HIV-1 RNA in the same brain regions of individuals who died of HIV/AIDS.
Other studies carried out in HIV-1–infected men during life have indirectly revealed deficits in the brain dopaminergic activity, compared to that in age-matched, noninfected individuals, and these deficits were reflected in the hypothalamic dysregulation of prolactin secretion in response to intravenous administration of metoclopramide, a neuroleptic and a dopamine-blocking agent (Parra et al, 2001). Because the hypothalamus receives dopaminergic innervations from the basal ganglia, these findings may also implicate deficits in dopaminergic input from the basal ganglia to the hypothalamus, suggesting that the decreased dopaminergic tone in hypothalamus may be a consequence of HIV-1–induced neurodegenerative changes in the basal ganglia (Aylward et al, 1993; Kure et al, 1990), resulting in DA deficits and thus limiting the DA-hypothalamic interaction and a decreased serum prolactin response to metoclopramide in HIV-1–infected men.
Furthermore, the decreased DA concentration found in putamen, substantia nigra, and other brain regions of HIV-1+ individuals in this study (Table 2) is also in accord with the decreased presynaptic dopamine transporter (DAT) availability when determined during life with positron emission tomography (PET) using 11C-cocaine as ligand for DAT, and 11C-raclopride for D2 receptors in putamen and ventral striatum in HIV-1+ patients with dementia and HIV-negative controls (Wang et al, 2004; Volkow et al, 1996). Although in the above study, DAT availability in putamen and ventral striatum was significantly decreased in patients with HAD, in the caudate nucleus of these patients, DAT was not significantly different from that in seronegative controls, and D2 receptor availability in all three regions did not differ between HIV-1+ and HIV-negative patients. Although this discrepancy in the relationship between regional DAT activity and HAD is not clearly understood at present, it is well recognized that during neurotransmission, the DA transporter functions are regulated mostly by the neuronal and synaptic DA concentration. Moreover, after its synthesis in the presynaptic dopaminergic neurons, DA is transferred to different sites within the neuron, including in the presynaptic storage vesicles, is released into the synapse in response to stimuli (excess DA in the synapse is transported back to presynaptic neuron by binding to DAT), and binds to D2 receptors at the postsynaptic neuron terminals. Because all DA-related functions, including neurocognitive, motor, and behavioral functions resulting from the interactions between DA and other neuronal components require an adequate availability of DA, the decreased DAT in the brain regions of HIV-1+ patients during life as shown by Wang et al (2004) may be a consequence of HIV-1–induced degeneration of dopaminergic neurons, and decreased dopamine availability, in these regions as found in the present study. In fact, studies carried out on human brains using serological and morphometric techniques found significant neuronal loss in SN of individuals with HIV/AIDS, suggesting degeneration of neurons in the pars compacta of SN (Reyes et al, 1991; Itoh et al, 2000). Because neurons in the pars compacta of the nigral complex are implicated as the site of dopamine synthesis, it may be speculated that the decrease in DA concentration in SN found in this study would be due to the presence of high levels of HIV-1 RNA in SN as well as in the other brain regions associated with dopaminergic activity (Table 2). However, a lack of direct correlation between dopamine concentration and HIV-1 RNA, although surprising, supports the concept that neurodegenerative changes in dopaminergic regions may not be caused by the direct interaction between HIV-1 and dopaminergic neurons, and that high levels of HIV-1 RNA in these regions may not be the determining factor for causing dopamine deficits. In fact, a number of studies suggest that different neurotoxic factors (cytokines and tryptophan metabolites) secreted in large quantities during interaction between HIV-1 and non-neuronal brain cells, including macrophages, microglia, and astrocytes, are involved in causing damage to dopaminergic neurons and frontostriatal circuitries (Itoh et al, 2000; Reyes et al, 1991). Recent studies by Taylor et al (2007) evaluated the damage to neuronal integrity induced by HIV-1 in the CNS, using MR spectroscopy, and found that in the basal ganglia and frontal lobe white matter, there was a 4% to 5% reduction in the cerebral metabolite N-acetylaspartate (NAA), the neuronal integrity marker, in individuals with HIV-1 infection compared to that in HIV-negative groups, a finding consistent with the earlier reports by Chang et al (2004), suggesting that the poor neural integrity may be the predictor of severity of illness.
Dopamine is synthesized both in the periphery and in the CNS from the dietary amino acid l-tyrosine. In the CNS, dopamine is synthesized in the dopaminergic neurons, localized mainly in the substantia nigra. Because dopamine does not cross the blood-brain barrier, it is not transported from the peripheral system to the CNS. However, the amino acid l-tyrosine is taken up from circulation and is converted to l-dihydroxyphenyl-alanine (l-DOPA) by the rate-limiting enzyme, l-tyrosine hydroxylase (TH) present in the neurons. l-DOPA is then converted to dopamine by the enzyme, L-aromatic aminoacid dopadecarboxylase (L-AADD). After synthesis, dopamine is stored within the cytoplasmic storage vesicles where it is protected from oxidation by the mitochondrial monamine oxidase (MAO). Dopamine is released into the synapse in response to action potential generating stimuli, and the slow steady-state concentration of dopamine and its release is regulated primarily by its reuptake into the presynaptic neurons by binding to dopamine transporter (DAT) (Garris et al, 1994). The metabolism of dopamine to HVA, a two-step process, involving the enzymes MAO-B and catechol-o-methyl transferase (COMT), also contributes to maintaining a steady-state concentration of dopamine in the neurons. Although some studies have found HIV-1 Tat protein mediated inhibition of tyrosine hydroxylase gene expression in dopaminergic neurons, which may result in decreased dopamine synthesis (Zauli et al, 2000), specific mechanisms involving various sequence of events leading to HIV-1–induced neurodegenerative changes resulting in dopaminergic deficits in different brain regions are not clearly understood.
Investigations of biogenic amine neurotransmitters, including dopamine in the postmortem human brain tissues, are quite challenging. Because dopamine is present in ultramicro concentration in different brain regions, and we anticipated it to further decrease in the postmortem brains of HIV-1–infected cases, we used the highly sensitive CoulArray HPLC-ECD technology to quantify dopamine concentration in small quantity of tissue from different brain regions. In addition, all procedures were carefully conducted in order to prevent the influence of relevant extrinsic factors that may affect the final results. Some of these factors included (1) the postmortem interval (PMI) variability; (2) handling of tissue at autopsy—snap freezing after excision; (3) preventing extra thawing of the tissue between initial freezing and homogenization before analysis; (4) standardization of the methodology for HPLC-ECD CoulArray technology for high recovery, sensitivity, and specificity for quantification of dopamine and its metabolite (HVA) in small samples of postmortem brain tissues.
The procedures used in this study for handling the postmortem human brain tissues and quantification of dopamine have been described in details in the earlier reports by us (Kumar et al, 2006) as well as by other investigators (Mackay et al, 1986; Herregdots et al, 1991). These studies have provided important evidence for obtaining reliable and meaningful results regarding the status of dopamine in different regions of postmortem brains of normal individuals as well as of those with neurodegenerative disease, including Alzheimer’s disease and HIV/AIDS (Adolfsson et al, 1979; Arai et al, 1984; Mackay et al, 1986; Herregdots et al, 1991; Sardar et al, 1996).
With regards to PMI and its potential influence on the stability of dopamine and HVA in the postmortem brain tissues, it has been shown that PMI between 4 and 28 h did not affect the results of dopamine and HVA concentration in different brain regions as well as in CSF (Carlsson et al, 1976; Mackay et al, 1986; Wester et al, 1990). For example, Sardar et al (1996) did not find any difference in dopamine and HVA concentration in the caudate nucleus of patients with HIV/AIDS and normal controls when tissues were obtained after 24 or 72 h of death. In the present study, statistical evaluation of our data (by multivariate analysis after Bonferroni correction) did not show influence of PMI on DA and HVA levels in different brain regions. Moreover, all tissues were obtained from the NNTC and were collected under the guidelines established at NNTC (Morgello et al, 2001), and PMI was maintained within the recommended range, varying between 2 and 24 h (mean ± SD, 9.41 ± 5.74 h) for HIV-1+ cases, and 3 and 22 h (7.28 ± 5.77 h) for HIV-negative cases.
Also, after dissection, all tissues were immediately snap frozen at −80°C at NNTC, and were transported to us in the frozen state in dry ice, and were kept frozen at −80°C (without intermittent thawing) until used for the assays, the conditions that have been described to preserve the stability of CNS dopamine and HVA in the autopsied brain samples of HIV-negative cases (Carlsson et al, 1976; Mackay et al, 1986; Wester et al, 1990). Furthermore, we used the procedures standardized by us earlier (Kumar et al, 2006) for high sensitivity and specificity for ultramicro quantification of dopamine in small quantities of CSF and brain tissues using the CoulArray HPLC-ECD multielectrode system. Thus, our values of dopamine and HVA in different brain regions may represent as closely as possible the concentrations present at the time when tissues were excised within the above-mentioned PMI range. Additionally, because the tissues at the NNTC centers are well characterized for authenticity of the specific region, the values of DA and HVA obtained in this study may represent the content present in the specific region.
The levels of DA and HVA in different brain regions of HIV-1+ or HIV-negative cases were not influenced by gender differences in our sample (multivariate analysis after Bonferroni correction). Moreover, the majority of HIV-1+ cases in this study were males (n=32) and only a few were females (n=6), and among HIV – cases, there were 6 males and 5 females. Because the number of individuals in each gender group was limited, values of DA and HVA from all individuals were pooled to represent one group. Earlier studies carried out on postmortem human brains of noninfected individuals have also reported that the gender differences did not influence the DA content in different brain regions (Spokes, 1979).
Age or ethnic differences were also not found to contribute to changes in dopamine content in different brain regions. The age of HIV-1+ and HIV – individuals investigated in this study ranged from 31 to 58 and 38 to 66 years, respectively, and the number of individuals in a specific age group was limited only to one to three per group. Because age range was a continuous variable, there were no specific age-related differences found in DA or HVA levels in the brain regions of HIV-1+ or HIV-negative cases (multivariate analysis after Bonferroni correction). Although in noninfected postmortem human brains, DA concentration in some of the brain regions have been found to change with age (Carlsson et al, 1976; Gottfreis, 1990), these studies were carried out in larger number of individuals belonging to specific age groups (young versus the older adults). Regarding ethnicity, because the majority of HIV-1+ as well as HIV-negative individuals were Caucasians, and only a limited number of individuals (two to four) belonged to the other three ethnic groups (Hispanics, AA, native Americans), ethnic differences were not found to contribute to difference in the content of dopamine and HVA in different brain regions.
The content of DA and HVA in the brain regions of HIV-1+ cases who used HAART (n=33) did not differ from those who did not use HAART (n=5). Surprisingly, the individuals who used HAART (87%) had a significant decrease in DA content in putamen (P<.01), an observation contrary to the expected neuroprotective role of HAART. The between-group differences (HAART+ and HAART−) in the values of DA in different regions could not be delineated (Table 5), because the number of HAART-negative individuals (n=5), as well as the number of samples available for each region (n=1–5) were limited. However, these findings may suggest that despite HAART, HIV-1–infected individuals continue to have dopaminergic deficits in the basal ganglia nuclei, including putamen, a constituent of nigrostriatal dopaminergic pathway involved in motor regulation. These findings concur with the earlier observations suggesting that antiretroviral therapy may not be effective in improving DA levels in CSF or Parkinson-like symptoms in HIV-1–infected individuals (Gisslen et al, 1994; Mirsattari et al, 1998). Moreover, some studies have also shown that HIV-1–associated neurodegenerative changes in the subcortical regions continue to prevail in individuals using HAART, including atrophy of the basal ganglia in the late stages of the disease (Stout et al, 1998), hypertrophy of putamen in early stages and hypotrophy in the late stages (Jernigan et al, 1993; Castello et al, 2007), and reduction in blood flow as well as in the volume of the caudate nucleus (Ances et al, 2006, 2007). Recent studies carried out by AIDS Clinical Trial Group (ACTG) (Robertson et al, 2007), as well as reports by Nath and Sacktor (2006), also suggest that despite the success with HAART in making significant impact on severity of dementia and in some cases reversing some of the HIV-related neurocognitive impairment, persistence of HIV in the CNS and incidence of neuropsychological deficits continue to remain, possibly by the nonreversible neural injury that may be related with the history of disease progression. Although the direct impact of HAART on the central dopaminergic activity in HIV-1 infection remains poorly understood, in the present study, the number of individuals who did not use HAART was too limited (n=5) for delineating the impact of HAART on the central regional dopaminergic status, and further investigations are warranted with a larger number of HAART-naïve individuals in order to unravel the differences in dopaminergic activity between the two groups.
With respect to relationship between deficits in brain dopamine concentration and clinical manifestation of neurocognitive impairment, we found that despite the overall deficits in DA levels in the brain of HIV-1+ cases, there was a lack of correlation (rs) between decrease in DA concentration in each brain region and severity of neurocognitive impairment, diagnosed as HAD or MCMD. The neurocognitive impairment in HIV-1+ individuals was assessed at the NNTC centers between 1 and 19 months before their death by the criteria of Memorial Sloan-Kettering (MSK) rating scale (0 to 4) (Price and Brew, 1988), and 53% of individuals in our sample were diagnosed with HIV-associated dementia (HAD, probable or possible; score=2–4), 31% with minor cognitive motor disorders (MCMD, probable or possible; score1–2), 5% had some impairment (score=1), and 11% had no impairment (score=0). Whereas HAD is the major manifestation of HIV-1 within the central nervous system, and is characterized by cognitive, motor, and psychiatric syndrome that usually develops in the last stages HIV-1 infection, MCMD is characterized by movement disorders, similar to those seen in Parkinson’s disease. However, both HAD and MCMD have been associated with dysfunctions of dopaminergic system in the basal ganglia with core symptoms similar to those seen in subcortical dementia of Parkinson’s and Huntington diseases (Lopez et al, 1999; Nath et al, 2000).
In this study, we found that although an overall depletion of DA in different brain regions of HIV-1+ cases was significant, when we dichotomized our sample into groups of those who had HAD versus those who had MCMD, there was no significant difference in DA levels between the two groups (Table 6), and there was no correlation (rs) between DA content in different brain regions and HAD or MCMD, a finding in agreement with the earlier suggestions that pathological changes may not always correlate with the clinical manifestations (Berger and Nath, 1997). Although one of the important limiting factors in this study has been the inadequate availability of number of tissue samples of all regions from each individual case, our findings of discordance between decrease in DA levels in the basal ganglia regions and neurocognitive deficits concur with those reported earlier by Wang et al (2004), who used PET scanning during life to assess DA activity in the brain as reflected by DA transporter availability showing that although DAT was decreased in patients with HIV-dementia, HIV-dementia scores (HDS) did not correlated with DAT availability. Thus our findings of a lack of correlation between neurocognitive deficits and DA levels, and the findings on decreased DAT activity in different brain regions reported earlier (Wang et al, 2004), may suggest that, despite HAART intervention (majority of individuals in this sample were treated with HAART during life), HIV-related neurologic damage continues to remain. These findings may suggest that all neurocognitive deficits in HIV-1 infection may not be exclusively related to the dopaminergic system dysfunction, and that the interaction between DA and the other neurotransmitter systems present in the basal ganglia, including γ-aminobutyric acid (GABA), substance P, norepinephrine, glutamate, and enkephalin, may be contributing to different mechanisms involved in the performance of neurocognitive functions (Morgensen and Yang, 1991). However, impact of HIV-1 on these neurotransmitters systems and their relationship to DA and neurocognitive functions are not presently understood and may be the subject of future investigations.
Regarding the relationship between HIV-1 viral load and dementia ratings, we have reported earlier a wide variability in the distribution HIV-1 RNA levels in different brain regions. Although we found high HIV-1 RNA levels in substantia nigra, caudate, putamen, and other nuclei of the basal ganglia, there was no significant correlation between the viral RNA levels and severity of dementia in HIV-1+ cases (Kumar et al, 2007), a finding similar to that reported by Johnson et al (1996). Such discordance between the severity of dementia and the underlying neuropathology of AIDS has also been reported by other investigators (Gray et al, 2001). Our findings of variable HIV-1 RNA levels in different brain regions and lack of correlation with HAD also concurs with the earlier reports showing a lack of correlation between HAD and the viral protein gp41 (the immunocytochemical marker for HIV-1) in the basal ganglia and frontal lobes of patients with AIDS, although HAD was found to correlate better with presence of macrophages and microglia (Glass et al, 1995). These findings suggest that the presence of virus, even in high concentration in the brain, may not be directly related to the neuronal damage, but the neuron may be injured and die by apoptosis induced by the indirect mechanisms involving neurotoxic cytokines (TNFα, interleukin [IL]-1) and tryptophan metabolites, such as quinolinic acid, generated by the microglia, resident macrophages, and astrocytes in response to HIV-1 infection (Merril and Chen, 1991, Heyes et al, 1992). In fact, high concentrations of TNFα and quinolinic acid have been found in the basal ganglia of patients with HIV encephalitis (Achim et al, 1993). The other neurotoxins found to cause neuronal damage include the HIV-1 proteins gp120 and Tat, which bind to the cell surface and alter the metabolic activities of the neurons, and may then undergo degenerative changes in patients with HAD (Bennet et al, 1995; Nath, 2002).
Moreover, a majority of HIV-1+ cases included in this study had been treated with HAART, and some of the antiretroviral drugs in HAART, such as protease inhibitors, do not cross the blood-brain barrier, whereas others may be present in suboptimal and variable concentrations in different brain regions, and may exert variable impact on HIV-1 viral load as well on the regional dopaminergic neuronal loss. These events may also contribute to the lack of direct relationship between dopamine concentration, as well as HIV-1 RNA levels, and severity of neurocognitive deficits. Although recent evidence also suggests that in some patients, symptoms of HIV dementia may be reversible by HAART (Fillipi et al, 1998), these effects may not be long lasting due to development of drug resistance, and that despite HAART, in some individuals HIV-1 RNA may continue to replicate in the CNS, and may continue to produce increasing quantity of neurotoxic products and cause progressive neurocognitive and motor dysfunctions (Tozzi et al, 2004, 2007; Anthony et al, 2005; Eden et al, 2007).
The findings of the present study are of considerable significance, because these findings provide, to our knowledge for the first time, direct evidence that the levels of dopamine are depleted in the basal ganglia nuclei of individuals who died of HIV/AIDS and during life had suffered from HAD or MCMD. Because there were no significant changes in HVA levels in different brain regions of HIV-1+ cases, our focus of discussion remained primarily with respect to changes in dopamine. However, our findings also support other reports from various pathological, neurological, immunohistochemical, and physiological studies showing abnormalities of dopaminergic system in the basal ganglia nuclei regions and frontocortical structures of human brain. Although HAD and MCMD have been associated primarily with dopaminergic system dysfunctions, further studies are warranted in a larger sample to delineate the complex mechanisms involving interaction of DA with other neurotransmitters systems that may contribute to HIV-associated CNS disease.
In summary, the findings of the present study demonstrate the deleterious effect of HIV-1 infection on dopaminergic activity, with a significant decrease in dopamine content in different regions of postmortem brains of individuals who died of HIV/AIDS. The decrease in dopamine concentration was widely variable in different brain regions, and was not influenced by the differences in age, gender, ethnicity, and PMI. These findings support the earlier reports of decreased dopamine levels in CSF of HIV-1–infected patients, as well as the indirect evidence of dopaminergic deficits, when manifested as Parkinson-like symptoms of cognitive, motor, and behavioral disorders that develop in HIV-1–infected patients. Furthermore, these findings show, to our knowledge for the first time, a widespread impact of HIV-1 on dopamine concentration in the brain regions, including the frontocortical areas, basal ganglia, caudate, putamen, globus pallidus, that are actively involved in dopaminergic functions and in substantia nigra, the main site of dopamine synthesis in the brain. Moreover, the concentration of HIV-1 RNA was also highest in these same brain regions, indicating the vulnerability of these regions to HIV-1 assault, although there was no significant direct correlation found between the regional dopamine concentration and HIV-1 RNA levels, suggesting that HIV-1 may not be directly involved in the degeneration of dopamine-producing neurons or the regions innervated by dopaminergic projections. Our findings concur with the previous studies suggesting that the neurodegeneration may be the result of a number of neurotoxic viral proteins, such as gp120 and Tat, as well as the neurotoxic products, including various cytokines (TNFα, IL-1, IL-6, IL-1β) and metabolites, such as quinolinic acid, generated by HIV-1–infected non-neuronal cells, including resident macrophages, microglia, and astrocytes. Furthermore, dopamine levels in different brain regions of individuals who used HAART did not differ from those who did not use HAART, and severity of HAD and MCMD did not show correspondence to decrease in dopamine levels in specific regions of individuals who died of HIV/AIDS.
Material and methods
Human postmortem brain tissues
Postmortem human brain tissues from different regions of HIV-1–seropositive and HIV-negative control individuals used in this study were procured from the four centers of the National Institutes of Health (NIH)-supported National NeuroAIDS Tissue Consortium (NNTC). Each of these centers obtained the informed consent of the family and approval of their Institutional Review Board for human ethics for enrollment, evaluation, and follow up of these patients every 6 months during life, and collection of data on various aspects of their health including physical, physiological, mental, cognitive, and neuropsychological functions, as well as for autopsy and for donation of organ and body fluids as gifts after their death (Anatomical Gift Act, 1990). At the NNTC Centers, the individuals who are invited for enrollment are evaluated for their HIV-1 serostatus and are followed for progression of disease. After death, their brain and other body tissues are harvested at autopsy for research investigations related with HIV/AIDS. The NNTC guidelines for postmortem interval (PMI) are within 24 h at all centers. All procedures for tissue extraction, labeling, and preservation are carried out according to the NNTC protocol (Morgello et al, 2001). Briefly, tissues after careful harvesting are immediately transferred to separate containers, appropriately labeled for specific tissues, and are snap frozen in liquid nitrogen and transferred to −80°C freezers for storage until used. Postmortem cerebrospinal fluid (CSF) is collected from the lateral ventricles or basilar cistern and stored at −80°C.
This study was approved by the Institutional Review Board of the Miller School of Medicine, University of Miami. The tissues obtained from NNTC were shipped in dry ice according to the guidelines for overnight shipping and handling established by the NIH/NNTC (US Federal Regulations 49 CFR 172 subpart H). Brain tissues were excluded from those with neuropsychiatric illness, such as schizophrenia, long-term use of antipsychotic medication, history of stimulant drug dependency, and CNS cancer, and from those with opportunistic infection of the CNS or CNS complications due to severe head injury with loss of consciousness for more than 30 min. The brain tissues of 49 individuals included 38 HIV-1 positives and 11 HIV negatives, with known demographic characteristics and belonging to different ethnic categories, and had no evidence of active opportunistic infection of the CNS at the time of death. The HIV-1+ patients died during 1998 to 2005. Tissues of HIV-1+ (N=38) and HIV-negative cases (N=11) were obtained from the National Neurological Brain Bank (NNAB; Los Angeles, CA), Texas Repository for AIDS Neuropathogenesis Research (TRANR: Galveston, TX), the Manhattan Brain Bank (MHBB; New York, NY), and HIV Neurobehavioral Research Center (HNRC) brain bank at the University of California at San Diego. The age of HIV-1+ and HIV-negative cases ranged from 31 to 58 years and 35 to 66 years, respectively. Among the 38 HIV-1+ cases investigated, 32 (84%) were males and 6 (16%) were females, 24 (63%) were white, 5 (13.3%) were African American (AA), 4 (10.5%) were Hispanic whites, 4 (10.5%) were native Americans, and 1 (2.7%) was Asian. Among the 11 HIV-negative cases, 6 individuals (54.5%) were males and 5 (45.4%) were females. Nine of them (81.8%) were white, 1 (9.1%) was AA, and 1 (9.1%) was Hispanic black. The assessment of neurocognitive status of HIV-1+ individuals carried out within 1 to 19 months before their death revealed the presence of probable or possible HAD, probable or possible minor cognitive motor disorder (MCMD), and impairment to a degree that they could carry out the activities of daily living. All HIV-negative individuals were cognitively normal except one, who had some impairment not related to HIV-1 or any other infection. The HIV-negative cases included in this study also died during the same span of time (1998 to 2005) due to different causes. The PMI of all HIV-1+ and HIV-negative cases ranged from 2 to 24 h, except one HIV-1+ case had PMI of 29 h.
The majority of HIV-1+ cases (n=33) had used highly active antiretroviral therapy (HAART) regimen prescribed as standard of care for each individual and included combinations of protease inhibitors, single or multiple nucleoside reverse transcriptase inhibitors (NRTIs), plus one non-nucleoside reverse transcriptase inhibitor (NNRTI). Among the five individuals who did not use antiretroviral medication, one had no medication before death, four did not use HAART, and information was not available for one person.
The neurocognitive deficits were assessed by the criteria defined by Memorial Sloan-Kettering (MSK) rating scale used at NNTC. There was a wide variation in the severity of dementia among the 38 HIV-1+ cases; 12 individuals (32%) had diagnosis of probable HIV-associated dementia (HAD; score=2–4); 8 (21%) had possible HAD (score=2–3); 5 (13%) had probable minor cognitive motor disorders (MCMDs; score=2); 2 (5%) had possible MCMD (score=1–2); 2 (5%) had MCMD (score=1); 4 (10.5%) were impaired (score=1); 1 (2.5%) had no impairment (NPI; score=0); 4 (10.5%) were cognitively normal (score=0); and 1 (2.5%) did not get diagnosis.
The brain tissues of different regions of each individual used in this study were characterized and labeled by the respective centers at the time of autopsy and included the frontal cortex (FC), frontal cortex areas 4 (FC4) and 6 (FC6), and basal ganglia from a few cases, and individual nuclei, including caudate nucleus (CAUD), putamen (PUTA), globus pallidus (GP), and substantia nigra (SN) from the other cases, as well as cerebrospinal fluid (CSF). All brain regions of each autopsied case were not available for this study; the tissues available from HIV-1+ cases included the FC from 25 cases, areas FC4 and FC6 were each from 13 cases. The basal ganglia as one region was obtained from 15 HIV-1+ cases, and CAUD, PUTA, and GP were from 14, 13, and 12 cases, respectively. The tissue samples from SN were from 22 cases, and CSF from 14 cases. The brain tissues of HIV-1− individuals (N=11) included the same regions as those of HIV-1+ cases. Dopamine and HVA concentrations were measured in each brain region using highly sensitive CoulArray high-performance liquid chromatography equipped with multielectrode chemical detector (HPLC-ECD) system. The levels of HIV-1 RNA in each region were determined as described earlier by using the real-time reverse transcriptase–polymerase chain reaction (Kumar et al, 2007).
Dopamine and HVA extraction and quantification
The brain tissues and CSF were received from each center through the overnight courier service on dry ice and were kept frozen at −80°C until the day of analysis for DA and HVA. Dopamine and HVA were extracted from the brain tissues, and separation, detection, and quantification were carried out using the highly sensitive CoulArray HPLC-ECD system as described previously (Kumar et al, 2006). Briefly, for extraction of dopamine and HVA, the tissue from each region was weighed and homogenized in chilled phosphate-buffered saline (PBS), pH 7.4, in an Eppendorf tube using disposable plastic mini-homogenizer, to a concentration of 200 mg tissue/ml. The homogenate was centrifuged at 3000 rpm at 4°C for 15 min, and the supernatant was transferred to chilled tube kept in ice. For determination of dopamine, the internal standard (IS), dihydroxybenzylamine (DHBA; 100 pg), was added to an 1.0-ml aliquot of the supernatant and the mixture was treated with activated alumina for extraction of dopamine and the IS, as described earlier (Kumar et al, 2003; Kumar et al, 2006). The extract was filtered through 0.2-micron syringe filters, and 40 μl of filtrate was injected in to the reverse-phase, stainless steel (3.9 × 150-mm), 5-μ, C18 resolve column (Water’s, Milford, MA, USA) for separation of dopamine and DHBA using the mobile phase at a flow rate of 1.0 ml/min, and containing 50 mM sodium acetate, 50 mM citric acid, 0.5 mM Sodium Octyl Sulfate (SOS), 0.15 mM disodium-EDTA, 1.0 mM dibutylamine, and 14% methanol, and the mixture was adjusted to pH 4.0. The peaks of DA and IS were identified by their retention times obtained with the standard solutions during the calibration procedure.
For extraction of HVA, the homogenate was treated with 200 μl of 1.0 M Perchloric acid (PCA) for precipitation of proteins, centrifuged, and the supernatant was separated and processed as described previously (Kumar et al, 2006). Briefly, the IS (isoproterinol [ISOP]) was added to 1.0 ml of supernatant, and HVA and IS were extracted with diethyl ether. The ether extract was evaporated in vacuum, and the residue was reconstituted in 200μl of sodium acetate, pH 5.0, and filtered through 0.2-micron syringe filters before injection of 40μl into the CoulArray HPLC-ECD system. The separation of HVA and IS was carried out using mobile phase with composition similar to that used for DA, but containing different concentrations of sodium acetate (0.1 M) and citric acid (0.1 M), and methanol 20%, at a flow rate of 0.7 ml/min. All solutions including standards and mobile phase were prepared in HPLC-grade water. The concentrations of DA as well as HVA were calculated based on the ratio of the peak area of unknown to that of IS, and were expressed as pg/g wet weight and ng/g wet weight, respectively. The sensitivity was adjusted to detect DA between 5 and 500 pg /ml and HVA between 0.1 and 20.0 ng/ml. The intra- and interassay coefficient of variance (% CV) for DA was 5.35% and 7.05%, respectively; and for HVA, it was 10.9% and 11.5%, respectively.
Statistical analysis
Statistical analyses were carried out using the Statistical Package for the Social Sciences (SPSS, version 14). The concentration of both dopamine and HVA in different brain regions of each individual case (HIV-1+ and HIV-negative) were log10 transformed, and all values from each brain region were expressed as mean ± SD. A paired t test was used to determine the between group differences in the levels of dopamine as well as HVA in different brain regions of HIV-1+ and HIV-negative cases, and difference of P<.05 was considered as significant. The correlation (rs) of DA and HVA with demographic characteristics, including age, gender, and ethnicity as well as PMI, were determined after Bonferroni correction for multiple comparisons. Correlations for the levels of DA, HVA, as well as HIV-1 RNA (log10 copies/g tissue) in different brain regions were determined using Spearman’s rank correlations coefficient (rs).
Acknowledgments
This study was supported by the NIH grants R01 NS43982, R01 NS41205, and R01 DA 13550. The authors thank the NNTC for providing the postmortem brain tissues for this project under the request #77. The authors would also like to express their gratitude to the individuals who made commitment during life to donate their brains as gift for the advancement of scientific knowledge.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Achim CL, Heyes MP, Wiley C (1993). Quantitation of human immunodeficiency virus immune activation factors, and quinolininc acid in AIDS brains. J Clin Invest 91: 2769–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adolfsson R, Gottfries CG, Roos BE, Winblad B (1979). Post-mortem distribution of dopamine and homovanillic acid in human brain, variations related with age, and a review of literature. J Neurol Transm 45: 81–105. [DOI] [PubMed] [Google Scholar]
- Ances BM, Roc AC, Wang J, Korczkowski M, Okawa J, Stern J, Kim J, Wolf R, Lawler K, Kolson DL, Detre JA (2006). Caudate blood flow and volume are reduced in HIV+ neurocognitively impaired patients. Neurology 66: 862–866. [DOI] [PubMed] [Google Scholar]
- Ances BM, Ellis RJ (2007). Dementia and neurocognitive disorders due to HIV-1 infection. Semin Neurol 27: 86–92. [DOI] [PubMed] [Google Scholar]
- Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE (2005). Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol 64: 529–536. [DOI] [PubMed] [Google Scholar]
- Arai H, Kosaka K, Lizuka R (1984). Changes in biogenic amines and their metabolites in postmortem brains from patients with Alzheimer-type dementia. J Neurochem 43: 388–393. [DOI] [PubMed] [Google Scholar]
- Aylward EH, Henderer JD, McArthur JC, Brettschneider PD, Harris GJ, Barta PE, et al. (1993). Reduced basal ganglia volume in HIV-1-associated dementia: results from quantitative neuroimaging. Neurology 43: 2099–2104. [DOI] [PubMed] [Google Scholar]
- Bagasra O, Levi E, Bobroski L (1996). Cellular reservoirs of HIV-1 in the central nervous system of infected individuals: identification by the combination of in situ polymerase chain reaction and immunohistochemistry. AIDS 10: 573–585. [DOI] [PubMed] [Google Scholar]
- Banks WA, Robinson SM, Wolf KM, Bess JW Jr, Arthur LO (2004). Binding, internalization, and membrane incorporation of human immunodeficiency virus-1 at the blood-brain barrier is differentially regulated. Neuroscience 128: 143–153. [DOI] [PubMed] [Google Scholar]
- Bennet BA, Rusyniak DE, Hollingsworth CK (1995). HIV-1 gp120-induced neurotoxicity to midbrain dopamine cultures. Brain Res 705: 168–176. [DOI] [PubMed] [Google Scholar]
- Berger JR, Kumar M, Kumar A, Fernandez JB, Levine B (1994). Cerebrospinal fluid dopamine in HIV-1 infection. AIDS. 8: 67–71. [DOI] [PubMed] [Google Scholar]
- Berger JR, Nath A (1997). HIV dementia and the basal ganglia. Intervirology 40: 122–131. [DOI] [PubMed] [Google Scholar]
- Berger JR, Arendt G (2000). HIV-dementia: the role of the basal ganglia and dopaminergic systems. J Psychopharmacol 14: 214–221. [DOI] [PubMed] [Google Scholar]
- Bobardt MD, Salmon P, Wang L, Esko JD, Gabzuda D, Fiala M, Trono D, Van der Schueren B, David G, Gallay PA (2004). Contribution of proteoglycans to human immunodeficiency virus type 1 brain invasion. J Virol 78: 6567–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew BJ (1993). HIV-1 related neurological disease. J Acquired Immune Defic Syndr 6 (Suppl 1): S10–S15. [PubMed] [Google Scholar]
- Broder CC, Dimitrov DS (1996). HIV and the 7-transmembrane domain receptors. Pathobiology 64: 171–179. [DOI] [PubMed] [Google Scholar]
- Castello JMB, Courtney MG, Melrose RJ, Stern CE (2007). Putamen hypertrophy in nondemented patients with human immunodeficiency virus infection and cognitive compromise. Arch Neurol 64: 1275–1280. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Winblad B (1976). Influence of age and time interval between death and autopsy on dopamine and 3methoxytyramine levels in human basal ganglia. J Neural Transm 38: 271–276. [DOI] [PubMed] [Google Scholar]
- Chang L, Ernst T, Leonido-Yee M, Witt M, Speck O, Walot I, Miller EN (1999). Highly active antiretroviral therapy reverses brain metabolite abnormalities in mild HIV dementia. Neurology 53: 782–789. [DOI] [PubMed] [Google Scholar]
- Chang L, Lee PL, Yiannoutsos CT, Ernst T, Marra CM, Richards T, Kolson D, Schifitti G, Jarvik JG, Miller EN, Lenkinski R, Gonzalez G, Navia BA (2004). A multicenter in vivo protons-MRS study of HIV-associated dementia and its relationship to age. Neuroimage 23: 1336–1347. [DOI] [PubMed] [Google Scholar]
- Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J (1996). The β-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85: 1135–1148. [DOI] [PubMed] [Google Scholar]
- Consortium Danna (1998). A randomized double blind placebo controlled trial of deprenyl and thioctic acid in human immunodeficiency virus-associated cognitive impairment. Danna Consortium on the therapy of HIV-dementia and related cognitive disorders. Neurology 50: 645–651. [DOI] [PubMed] [Google Scholar]
- Dore GJ, McDonald A, Li Y, Kaldor JM, Brew BJ (2003). Marked improvement in survival following AIDS dementia complex in the era of highly active antiretroviral therapy. AIDS 17: 1539–1545. [DOI] [PubMed] [Google Scholar]
- Eden A, Price RW, Spudich S, Fuchs L, Hargberg L, Gisslen M (2007). Immune activation of the central nervous system is still present after >4 years of effective highly active antiretroviral therapy. J Infect Dis 196: 1779–1783. [DOI] [PubMed] [Google Scholar]
- Ellis R Childers ME Kumar AM Kumar M Goetz T Caligiuri M(2004). Decreased cerebrospinal fluid dopamine correlates with impaired motor skills in HIV-1 infection and methamphetamine dependence. Presented at the Society for Neuroscience, 34th Annual Meeting, San Diego, Oct 23–27. [Google Scholar]
- Fillipi CG, Sze G, Farber SJ, Shahmanesh M, Selwyn PA (1998). Regression of HIV encelopathy and basal ganglia signal intensity abnormality at MR imaging in patients with AIDS after the initiation of protease inhibitor therapy. Radiology 206: 491–498. [DOI] [PubMed] [Google Scholar]
- Ensoli F, Wang H, Fiorelli V, Zeichner SL, Cristofero De M R, Luzi G, Thiele CJ (1997). HIV-1 infection and the developing nervous system: lineage-specific regulation of viral gene expression and replication in distinct neuronal precursors. J NeuroVirol 3: 290–298. [DOI] [PubMed] [Google Scholar]
- Garris PA, Ciolkowski EL, Pastore P, Weightman RM (1994). Efflux of dopamine from the synaptic cleft in the nucleus accumbence of the rat brain. J Neurosci 14: 6084–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou FM, Gonenc A, Waldrop-Valverde D, Kuker RA, Ezzuddin SH, Sfakianakis GN, Kumar M (2008). Analysis of the effect of injecting drug use and HIV-1 infection on 18F-FDG PET brain metabolism. J Nucl Med 49: 1999–2005. [DOI] [PubMed] [Google Scholar]
- Gisslen M, Larsson M, Norkrans G, Fuchs D, Wachter H, Hagberg L (1994). Tryptophan concentrations increase in cerebrospinal fluid and blood after zidvudine treatment in patients with HIV type 1 infection. AIDS Res Hum Retroviruses 10: 947–951. [DOI] [PubMed] [Google Scholar]
- Glass JD, Fedor H, Wesselingh SL, McArthur JC (1995). Immunocytochemical quantification of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol 38: 755–762. [DOI] [PubMed] [Google Scholar]
- Gray F, Adle-Biassette H, Chreien F, Lorin de la Grandmaison G, Force G, Keohane C (2001). Neuropatholoy and neurodegeneration in human immunodeficiency virus infection. Pathogenesis of HIV-induced lesion of the brain, correlations with HIV-associated disorders and modifications according to treatments. Clin Neuropathol 20: 146–155. [PubMed] [Google Scholar]
- Harrison MIJ, Newman SP, Hall-Craggs MA, et al. (1998). Evidence of CNS impairment in HIV-1 infection: clinical, neuropsychological, EEG, and MRI/MNRS study. J Neurosurg Psychiatr 5: 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herregodts P, Ebinger G, Michotte Y (1991). Distribution of monoamines in human brain: evidence for neurochemical heterogeneity in subcortical as well as cortical areas. Brain Res 542: 300–306. [DOI] [PubMed] [Google Scholar]
- Heyes MP, Saito K, Markey SP (1992). Human macrophages convert l-tryptophan in to the neurotoxin quinolininc acid. Biochem J 283: 633–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkin CH, Castellon SA, Hardy DJ, Farinpour R, Newton J, Singer E (2001). Methylphenidate improves HIV-associated cognitive slowing. J Neuropsychiatry Clin Neurosci 13: 248–254. [DOI] [PubMed] [Google Scholar]
- Ho D, Rota TR, Schooley RT, Kaplan JC, Allan JD, Groopman JE, et al. (1985). Isolation of HTLV-III from cerebrospinal fluid and neural tissues of patients with neurologic syndromes related to the acquired immunodeficiency syndrome. N Eng J Med 313: 1493–1497. [DOI] [PubMed] [Google Scholar]
- Hriso E, Kuhn T, Masdeu JC, Grundman M (1991). Extrapyramidal symptoms due to dopamine-blocking agents in patients with AIDS encephalopathy. Am J Psychiatry 11: 1558–156. [DOI] [PubMed] [Google Scholar]
- Itoh K, Mehraein P, Weis S (2000). Neuronal damage of the substantia nigra in HIV-infected brains. Acta Neuropathol 99: 376–384. [DOI] [PubMed] [Google Scholar]
- Jernigan TL, Archibald S, Hesselink JR (1993). Magnetic resonance imaging morphometric analysis of cerebral volume loss in human deficiency virus infection. Arch Neurol 50: 250–255. [DOI] [PubMed] [Google Scholar]
- Johnson RT, Glass JD, McArthur JC, Chesebro BW (1996). Quantitation of human deficiency virus in brains of demented and non-demented patients with acquired immunodeficiency syndrome. Ann Neurol 39: 392–395. [DOI] [PubMed] [Google Scholar]
- Kaul M, Ma Q, Medders KE, Desai MK, Lipton SA (2007). HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ 14: 296–305. [DOI] [PubMed] [Google Scholar]
- Kieburtz KD, Epstein LG, Gelbard HA, Greenamyre JT (1991). Excitotoxicity and dopaminergic dysfunction in the acquired immunodeficiency syndrome dementia complex. Therapeutic implications. Arch Neurol 48: 1281–1284. [DOI] [PubMed] [Google Scholar]
- Kieburtz KD, Ketonen L, Cox C, Grossman H, Holloway R, Booth H, Hickey C, Feigin A, Caine D (1996). Cognitive performance and regional brain volume in human immunodeficiency virus type 1 infection. Arch Neurol 53: 155–158. [DOI] [PubMed] [Google Scholar]
- Kim RB, Fromm MF, Wandel C, Leake B, Wood AJJ, Roden DM (1998). The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest 101: 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsillieri E, Sopper S, Scheller C, ter Meulen V, Riederer P (2002). Parkinson in HIV dementia. J Neural Transm 109: 767–775. [DOI] [PubMed] [Google Scholar]
- Kumar AM, Berger JR, Eisdorfer C, Fernandez JB, Goodkin K, Kumar M (2001). Cerebrospinal fluid 5-hydroxytryptamine and 5-hydroxyindoleacetic acid in HIV-1 infection. Neuropsychobiology 44: 13–18. [DOI] [PubMed] [Google Scholar]
- Kumar AM, Borodowsky I, Fernandez JB, Gonzalez L, Kumar M (2007). Human immunodeficiency virus type 1 RNA levels in different brain regions of human brain: quanyification using real-time reverse transcriptasepolymerase chain reaction. J NeuroVirol 13: 210–224. [DOI] [PubMed] [Google Scholar]
- Kumar AM, Fernandez JB, Antoni MH, Eisdorfer S, Kumar M (2003). Catecholamines quantification in body fluids using isocratic, reverse phase HPLC-CoulArray multielectrode chemical detector system: Investigation of sensitivity, stability, and reproducibility. J Liquid Chromatogr Relat Technol 26: 3433–3451. [Google Scholar]
- Kumar AM, Fernandez JB, Gonzalez L, Kumar M (2006). Ultramicro quantification of dopamine and homovanillic acid in human brain tissue: quest for higher recovery and sensitivity with CoulArray HPLC-ECD system. J Liquid Chromatogr Relat Technol 29: 777–799. [Google Scholar]
- Kure K, Weidenheim KM, Lyman WD, Dickson DW (1990). Morphology and distribution of HIV-1 gp41-positive microgila in subacute AIDS encephalitis: pattern of involvement resembling a multisystem degeneration. Acta Neuropahol (Berl) 80: 393–400. [DOI] [PubMed] [Google Scholar]
- Larsson M, Hagberg L, Forsman A, Norkrans G (1991). Cerebrospinal fluid catecholamine metabolites in HIV-1 infected patients. J Neurosci Res 28: 406–409. [DOI] [PubMed] [Google Scholar]
- Lopez OL, Smith G, Meltzer CC, Becker JT (1999). Dopamine systems in human immunodeficiency virus-associated dementia [review]. Neuropsychiatry Neuropsychol Behav Neurol 12: 184–192. [PubMed] [Google Scholar]
- Mackay AVP, Yates CM, Wright A, Hamilton P, Davies P (1986). Regional distribution of monoamines and their metabolites in the human brain. J Neurochem 65: 135–153. [DOI] [PubMed] [Google Scholar]
- Magyar R (1993). Pharmacology of monoamine oxidase type B inhibitors. In: Inhibitors of monoamine oxidase B, pharmacology and clinical use in neurodegenerative disorders. Basel Szelenyi I., Switzerland: Birkhauser, pp 125–143. [Google Scholar]
- Masliah E, Achim CL, Ge N, DeTeresa R, Terry D, Wiley CA (1992). Spectrum of HIV associated neocortical damage. Ann Neurol 32: 85–93. [DOI] [PubMed] [Google Scholar]
- Maslin CLV, Kedzierska K, Webseter N, Muller W, Crowe S (2005). Transendothelial migration of monocytes: the underlying molecular mechanisms and consequences of HIV-1 infection. Curr HIV Res 3: 303–317. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Haughey NJ, Nath A (2005). Cell death in HIV dementia [review]. Cell Death Differ 12 (Suppl 1): 893–904. [DOI] [PubMed] [Google Scholar]
- Merril JE, Chen ISV (1991). HIV-1, macrophages, glial cells, and cytokines in AIDS nervous system disease. FASEB J 5: 2391–2397. [DOI] [PubMed] [Google Scholar]
- Mirsattari SM, Power C, Nath A (1998). Parkinsonism with HIV infection. Movement Disord 13: 684–689. [DOI] [PubMed] [Google Scholar]
- Morgello S, Gelman BB, Kozlowski PB, Vinters HV, Masliah E, Cornford M, Cavert W, Marra C, Grant I, Singer EJ (2001). The National NeuroAIDS Tissue Consortium: a new paradigm in brain banking with an emphasis on infectious diseases. Neuropathol Appl Neurobiol 27: 326–335. [DOI] [PubMed] [Google Scholar]
- Morgenson GJ, Yang CR (1991). The contribution of basal forebrain to limbic-motor integration and the mediation of motivation to action. Adv Exp Med Biol 295: 267–273. [DOI] [PubMed] [Google Scholar]
- Nath A (2002). Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis 186 (Suppl 2): S193–S198. [DOI] [PubMed] [Google Scholar]
- Nath A, Anderson C, Jones M, Maragose W, Booz R, Mactutus C, Bell J, Hauser K, Mattson M (2000). Neurotoxicity and dysfunction of dopaminergic systems associated with AIDS dementia. J Psychopharmacol 14: 222–227. [DOI] [PubMed] [Google Scholar]
- Nath A, Sacktor N (2006). Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Curr Opin Neurol 19: 358–361. [DOI] [PubMed] [Google Scholar]
- Navia BA, Price RW (1987). The acquired immune deficiency syndrome dementia complex as the presenting or sole manifestation of human immune deficiency virus infection. Arch Neurol 44: 65–69. [DOI] [PubMed] [Google Scholar]
- Nottet HS, Persidsky Y, Sasseville VG, Nukuna AN, Bock P, Zhai QH, Sharer LR, McComb RD, Swindells S, Soderland C, Gendelman HE (1996). Mechanisms for the transendothelial migration of HIV-1 infected monocytes in to the brain. J Immunol 156: 1284–1295. [PubMed] [Google Scholar]
- Parra A, Ramirez-Peredo J, Larrea F, Cabrera V, Coutino B, Torres I, Angeles A, Perez-Romano B, Ruiz-Arguelles G, Ruiz-Arguelles A (2001). Decreased dopaminergic tone and increased basal bioactive prolactin in men with human immunodeficiency virus infection. Clin Endocrinol 54: 731–738. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Gendelman HE (2003). Mononuclear phagocyte immunity and the neuropathogenesis of HIV-1 infection. J Leuk Biol 74: 691–701. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Stins M, Way D, Witte MH, Weinand M, Kim KS, Bock P, Gendelman HE, Fiala M (1997). A model for monocyte migration through the blood brain barrier during HIV-1 encephalitis. J Immunol 158: 3499–3510. [PubMed] [Google Scholar]
- Price RW, Brew BJ (1988). The AIDS dementia complex. J Infect Dis 158: 1079–1083. [DOI] [PubMed] [Google Scholar]
- Ranki A, Nyberg M, Ovod V, Haltia M, Elovaara I, Raininko R, Haapasalo H, Krohn K (1995). Abundant expression of HIV Nef and Rev proteins in brain astrocytes in vivo is associated with dementia. AIDS 9: 1001–1008. [DOI] [PubMed] [Google Scholar]
- Resnick L, Berger JR, Shapshak P, Tourtellotte WW (1988). Early penetration of the blood brain barrier by HIV. Neurology 38: 9–14. [DOI] [PubMed] [Google Scholar]
- Reyes MG, Faraldi F, Senseng CS, Flowers C, Fariello R (1991). Nigral degeneration in acquired immunodeficiency syndrome (AIDS). Acta Neuropathol 82: 39–44. [DOI] [PubMed] [Google Scholar]
- Robertson KR, Smurzynski M, Parsons TD, Wu K, Bosch RJ, Wu J, McArthur JC, Collier AC, Evans SR, Ellis RJ (2007). The prevalence of incidence of neurocognitive impairment in the HAART era. AIDS 21: 1915–1921. [DOI] [PubMed] [Google Scholar]
- Rottenberg DA, Sidtis JJ, Strother SC, Schaper KA, Anderson JR, Nelson MJ, et al. (1996). Abnormal cerebral glucose metabolism in HIV-1 seropositive subjects with and without dementia. J Nucl Med 37: 1133–1141. [PubMed] [Google Scholar]
- Sacktor N, McDermott MP, Marder K, Schiffito G, Selnes OA, McArthur JC, Stern Y, Albert S, Palumbo D, Kieburtz K, De Marcaida JA, Cohen B, Epstein L (2002). HIV-associated cognitive impairment before and after the advent of combination therapy. J NeuroVirol 8: 136–142. [DOI] [PubMed] [Google Scholar]
- Sacktor N, Schiffito G, McDermott MP, Marder K, McArthur JC, Kieburtz K (2000). Transdermal selegeline in HIV-associated cognitive impairment. Pilot, placebo controlled study. Neurology 54: 233–235. [DOI] [PubMed] [Google Scholar]
- Sardar AM, Czudek C, Reynolds GP (1996). Dopamine deficits in the brain: The neurochemical basis of Parkinsonian symptoms in AIDS. Neuroreport 7: 910–912. [DOI] [PubMed] [Google Scholar]
- Scheller C, Sopper S, Jenuwein M, Neuen-Jacob E, Tatschner T, Grunblatt E, ter Meulen V, Riederer P, Koutsilieri E (2005). Early impairment in dopaminergic neurotransmission in brains of SIV-infected rhesus monkey due to microglia activation. J Neurochem 95: 377–387. [DOI] [PubMed] [Google Scholar]
- Schrager LK, D’Souza MP (1998). Cellular and anatomical reservoirs of HIV-1 in patients receiving potent antiretroviral combination therapy. JAMA 280: 67–71. [DOI] [PubMed] [Google Scholar]
- Spokes EGS (1979). An analysis of factors influencing measurements of dopamine, noradrenaline, glutamate decarboxylase and cholineacetylase in human postmortem brain tissue. Brain 102: 333–346. [DOI] [PubMed] [Google Scholar]
- Stout JC, Ellis RJ, Jernigan TL, et al. (1998). HIV Neurobehavioral Research Center Group. Progressive cerebral volume loss in human deficiency virus infection: a longitudinal volumetric magnetic resonance imaging study. Arch Neurol 55: 161–168. [DOI] [PubMed] [Google Scholar]
- Taylor M Schweinsberg BC Alhassoon OM Gangavatana A Brown GG Young-Casey C, Letendre SL Grant I the HNRC group (2007). Effects of human immunodeficiency virus and methamphetamine on cerebral metabolites measured with magnetic resonance spectroscopy. J NeuroVirol 13: 150–159. [DOI] [PubMed] [Google Scholar]
- Tozzi V, Balestra P, Murri R, Galgani S, Bellagamba R, Narciso P, Antonori A, Giulianelli M, Tosi G, Fantoni M, Sampaolesi A, Noto P, Ippolito G, Wu AW (2004). Neurocognitive impairment influences quality of life in HIV-infected patients receiving HAART. Int J STD AIDS 15: 254–259. [DOI] [PubMed] [Google Scholar]
- Tozzi V, Balestra P, Bellagamba R, Corpolongo A, Salvatori MF, Visco-Comandini U, Vlassi C, Giulianelli M, Galgani S, Antinore A, Narciso P (2007). Persistence of neuropsychological deficits despite long-term highly active antiretroviral therapy in patients with HIV-related neurocognitive impairment: prevalence and risk factors. J Acquir Immune Defic Syndr 45: 174–182. [DOI] [PubMed] [Google Scholar]
- Volkow N, Fowler J, Gately S, Logan J, Wang G, Ding Y et al. (1996). PET evaluation of the dopamine system of the human brain. J Nucl Med 37: 1242–1256. [PubMed] [Google Scholar]
- Von Giesen HJ, ittsack HJ, Wenserski F, Koller H, Hefter H, Arendt G (2001). Basal ganglia metabolite abnormalities in minor motor disorders associated with human immunodeficiency virus type 1. Arch Neurol 58: 1281–1286. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Chang L, Volkow ND, Telang F, Logan J, Ernst T, Fowler JS (2004). Decreased brain dopaminergic transporters in HIV-1 associated dementia patients. Brain 127: 2452–2458. [DOI] [PubMed] [Google Scholar]
- Wester P, Bergstrom U, Erikson A, Gezelius C, Hardy J, Winblad B (1990). Ventricular cerebrospinal fluid monoamine transmitters and metabolite concentrations reflect human brain neurochemistry in autopsy cases. J Neurochem 54: 1148–1156. [DOI] [PubMed] [Google Scholar]
- WHO/UNAIDS Agency (2007). AIDS Epidemic Update, November, 2007.
- Wiley CA, Soontornniyomkij V, Radhakrishnan L, Masliah E, Mellors J, Herman SA, Dalley P, Achim CL (1998). Distribution of brain HIV load in AIDS. Brain Pathol 8: 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CA, Shrie RD, Nelson JA, Lampert PW, Oldstone MB (1986). Cellular localization of human immunodeficiency virus infection within the brains of acquired immunodeficiency syndrome patients. Proc Natl Acad Sci USA 83: 7089–7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zauli G, Secchiero P, Luigi R, Gibellini D, Mirandola P, Mazzoni M, Milani MD, Dowd DR, Capitani S, Vitale M (2000). HIV-1 Tat-mediated inhibition of the tyrosine hydroxylase gene expression in dopaminergic neuronal cells. J Biol Chem 275: 4159–416. [DOI] [PubMed] [Google Scholar]