

Abstract
Directed evolution (DE) is a widely used method for improving the function of biomolecules via multiple rounds of mutation and selection. Microfluidic droplets have emerged as an important means to screen the large libraries needed for DE, but this approach was so far partially limited by the need to lyse cells, recover DNA, and retransform into cells for the next round, necessitating the use of a high‐copy number plasmid or oversampling. The recently developed live cell recovery avoids some of these limitations by directly regrowing selected cells after sorting. However, repeated sorting cycles used to further enrich the most active variants ultimately resulted in unfavourable recovery of empty plasmid vector‐containing cells over those expressing the protein of interest. In this study, we found that engineering of the original expression vector solved the problem of false positives (i.e. plasmids lacking an insert) cells containing empty vectors. Five approaches to measure activity of cell‐displayed enzymes in microdroplets were compared. By comparing various cell treatment methods prior to droplet sorting two things were found. Substrate encapsulation from the start, that is prior to expression of enzyme, showed no disadvantage to post‐induction substrate addition by pico‐injection with respect to recovery of true positive variants. Furthermore in‐droplet cell growth prior to induction of enzyme production improves the total amount of cells retrieved (recovery) and proportion of true positive variants (enrichment) after droplet sorting.
In this study five methods for to measure activity of cell‐displayed enzymes were compared. By comparing various cell treatment methods it was found that substrate encapsulation prior to enzyme expression in a one‐pot reaction showed no disadvantage in cell growth, and cell growth in‐droplet increased both the total amount of cells recovered, as well as the proportion of positives recovered.
INTRODUCTION
Directed evolution mimics the process of natural evolution in the laboratory and has been used to engineer bio‐ molecules with improved functionalities via multiple rounds of mutation followed by selection of variants that show improvement of the desired properties (Arnold, 1998, 2018; Renata et al., 2015; Zeymer & Hilvert, 2018). Specifically, enzymes have been evolved that show increased activity, increased thermostability (Giver et al., 1998; Qu et al., 2020), stability in an alternative medium resembling human blood (Mate et al., 2013), enantioselectivity (Prier et al., 2017), novel reactions (Molina‐Espeja et al., 2016; Renata et al., 2015), or a combination of above‐mentioned properties (Li et al., 2016).
Smart library design addresses the problem of library size by targeting mutations to small regions of interest in a sequence, thus achieving a high coverage even with a small library size (Qu et al., 2020). Smart library design can reduce the amount of sequence space needed to explore to find variants with improved functions. However, the design of smart libraries relies on existing knowledge of enzyme structure and function to identify and select regions to mutate. Furthermore, small library design restricts mutations to a small part of the sequence. Thus, smart libraries negate a main advantage of directed evolution. As smart library design focuses on mutational hotspots, variants with mutations in unexpected locations will be missed, as such locations are excluded from the library designed (Romero & Arnold, 2009). Furthermore, a larger library size allows screening of double or even triple mutations allowing ‘jumps’ through sequence space, bypassing dead ends and epistatic ratchets in which a double mutation is beneficial but neither of the single mutants are (Bridgham et al., 2009; Starr & Thornton, 2016).
Many methods have been used for high‐throughput screening, but they have various limitations. For example, colorimetric screening of colonies on agar plate gives only a yes or no answer regarding catalytic activity (Pikkemaat & Janssen, 2002; van Loo et al., 2004). Other methods are only able to detect shifts in catalytic activity in a narrow range. Antibiotic selection allows for screening of very large libraries, but narrows the substrate scope to those with antibiotic properties (MacBeath et al., 1998; Orencia et al., 2001). Fluorescence‐cctivated cell sorting (FACS) is useful for sorting large libraries but is unsuited for enzymatic activity, as substrates and reaction products can move freely in solution and thus do not stay linked to the cell genotype (Leemhuis et al., 2005; Yang & Withers, 2009). Microtiter plate screening is quantitative, but throughput is limited and requires complex robotic setups to achieve throughputs above 103.
Microfluidics has recently emerged as an technique with which to screen the large libraries needed for DE (Neun et al., 2020). By encapsulation of single cells inside microdroplets, a high‐throughput can be achieved with a wide range of enzymatic reactions, as each microdroplet functions as a mini‐reactor with one genotype and phenotype (Agresti et al., 2010; Fallah‐Araghi et al., 2012; Gielen et al., 2016; Mair et al., 2017). Neun et al. (2019) is an example of encapsulation of single cells in a droplet. In droplet‐compartmentalised in vitro expression systems (Courtois et al., 2018; Holstein et al., 2021), there is no barrier to the encounter of enzyme and substrate. The use of microfluidics to generate water‐in‐oil‐in‐water double emulsions allows turnover screening using FACS (Tauzin et al., 2020; Zinchenko et al., 2014). A disadvantage of many cell‐based protein expression systems is that they require at least a partial or temporary lysis to allow a reaction prior to product detection in microfluidic droplet sorting. Cell lysis releases the expressed enzyme into the solution and allows interaction between enzyme and substrate. Partial cell lysis using carefully adjusted reaction conditions (Tauzin et al., 2020) or a kill switch (Wong et al., 2021) allows recovery of the DNA of selected hits after regrowing cells from droplet hits. When cells cannot be regrown a retransformation step is required to recover the DNA.
DNA is usually recovered from high‐copy plasmids with or without an extra step of PCR amplification. As a consequence, in order to recover the plasmids of selected variants, obtained plasmid DNA needs to be retransformed into fresh E. coli cells (Colin et al., 2015; Kintses et al., 2012; Neun et al., 2022; Zinchenko et al., 2014). The retransformation is very efficient (>85%) (Kintses et al., 2012) when a high‐copy plasmid is used, but a PCR step is necessary when such a plasmid is not suitable, leading to a potential loss of sorted diversity (Obexer et al., 2017). Furthermore, the addition of lysis agents containing biosurfactants soluble in aqueous phase, such as Triton or Tween, might lead to reduced stability of the droplet emulsion, which has to be prevented by increasing the concentration of expensive fluorsurfactants in the oil phase.
The advantage of cell growth in droplets in E. coli for recovery of variants has been shown recently, to effectively increase recovery, and obtain much higher diversity after transformation (Zurek et al., 2021). All these methods so far employ cell lysis as a part of the protocol to enable the access of the substrate to the expressed enzymes for enzymatic turnover to occur. (Colin et al., 2015; Kintses et al., 2012; Zinchenko et al., 2014). To circumvent the need for cell lysis protein can be displayed in the periplasm or on the surface of intact cells (Agresti et al., 2010). Periplasmic expression of L‐asparaginase in E. coli was recently shown by Karamitros et al. (2020). By expressing enzyme in the periplasm, substrate turnover can be performed by intact cells. Another promising method for surface display is the E. coli autodisplay system, which uses the AIDA autotransporter system to present proteins, including enzymes, on the surface of E. coli (Jose, 2006; Jose et al., 2012). Autodisplay has been used to display various proteins on the surface of E. coli (Jose, 2006; Jose et al., 2012; van Loo, Bayer, et al., 2019) and applied before for high‐throughput screening towards substrates covalently bound to the cell surface (Becker et al., 2007, 2008) recently used in high‐throughput screening for direct enzymatic turnover, identifying a 30‐fold improved variant in activity towards fluorescein disulfate after only one round of evolution. Furthermore, fluorescence‐activated droplet sorting (FADS) has already been successfully used to sort DE libraries of arylsulfatase SpAS1, presented on the outer membrane of E. coli (van Loo, Heberlein, et al., 2019). However, in that study, only 12% of the cells selected and sorted were recovered. Whilst this is a substantial fraction of the diversity, this means that recovery could potentially be up to around eightfold more efficient.
In the study by van Loo et al., expression was induced in bulk in a flask prior to encapsulation of single cells into microdroplets (hereafter described as bulk induction). A downside of bulk induction is the risk of formation of a biofilm which can interfere with droplet encapsulation, this biofilm is suspected to be the result of dimerization of SpAS1 on the cells surface, although this was not confirmed. Thus, oversampling during the droplet sorting is required to fully recover diversity, that is to obtain each unique variant found in the assay. By performing both growth and induction after encapsulation inside a microdroplet, biofilm formation is prevented. Recovering at least one selected variant from each droplet reduces the amount number of droplets that need to be screened, as each theoretical variant needs to be selected only once. This in turn reduces the time needed and the amount of reagents consumed, such as substrates and fluorosurfactants. Consequently, a library could be screened faster, and a larger diversity could be covered in the same time with complete recovery of all variants. The advantage of screening microdroplets containing a single cell is the linkage of genotype/phenotype as each droplet contains only one distinct variant. However, the presence of only one cell per variant limits the maximal recovery of variants. To recover 100% of sorted diversity (i.e. every variant that gives a positive signal is retrieved), every sorted cell has to grow into a full‐sized colony, allowing zero room for mechanical or biological losses. By inoculating droplets with a single cell and including a growth step in the protocol genotype/phenotype linkage is also retained as all cells inside each droplet are clones of the encapsulated cell (Tauzin et al., 2020; Wong et al., 2021; Zurek et al., 2021). At the same time by including a growth step, several shortcomings of working with single cells can be avoided though following advantages: (i) multiple copies of each variant can be recovered, thus allowing recovery of full library diversity even if not all sorted cells grow into a colony (ii) more cells per volume, and thus more enzymatic turnover, can increase sensitivity of the method (iii) effect of false positives may be less pronounced, by preventing the dilution of positive variants in iterative screening rounds.
Although live cell recovery sidesteps some of the issues of conventional lysis protocols. This approach can still be improved. In this study, we seek to address the issue of optimal recovery of diversity after live cell screening with FADS. To this end, two things should be optimised: (i) to recover a higher percentage of sorted diversity and (ii) to obtain a higher rate of true positive variants. Therefore, we compare screening of single cells in microdroplet to in‐droplet cell growth. We also investigate the effect of utilising pico‐injection to add solutions to microdroplets as opposed to a single‐step process in which all reagents are added during the initial encapsulation in microdroplets, after which the microdroplets are not further manipulated prior activity measurement and sorting. Through combinations of single‐cell vs. cell growth and one‐pot vs. multistep assay, we compare five different strategies for (Figure 1) live cell recovery after FADS. The previously used method as described in van Loo, Bayer, et al., (2019) is methodically compared with a novel method for growing E. coli cells inside microdroplets to further increase the total amount of cell retrieved after sorting (recovery) and proportion of true positive cells containing the plasmid encoding the active enzyme (enrichment) achievable. The aim is to improve on the current method by: (i) vector redesign, eliminating the ‘empty plasmid’ variants in the sorting procedure (ii) growing an E. coli culture inside microdroplets starting from a single‐cell and (iii) using pico‐injection (Abate et al., 2010) to add inducer and/or substrate after single‐cell encapsulation.
FIGURE 1.
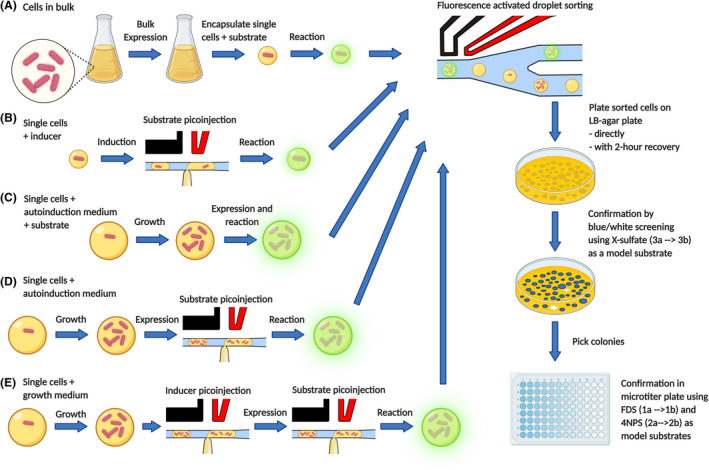
Overview of sorting protocols compared. Single cells were sorted after induction in bulk (A) or induction in droplet by pico‐injection (B). Cells were grown inside droplets using autoinduction, where substrate was added during encapsulation (C) or pico‐injected after induction (D). Finally, cells were incubated inside droplet with two separate pico‐injections to (1) induce and (2) add substrate (E). This figure was prepared with BioRender.com.
RESULTS AND DISCUSSION
The effects of different methods on the recovery (amount of variants recovered) and enrichment (high proportion of true positives) of an enzyme expressed on the surface of E. coli through an autotransporter system were investigated for the specific example of SpAS1. SpAS1 is part of the Aryl Sulfatase clade of the Alkaline Phosphatase (AP) superfamily (Mohamed & Hollfelder, 2013; van Loo, Bayer, et al., 2019; van Loo, Heberlein, et al., 2019). During initial droplet sorting experiments the amount of blue (active) colonies increased after each subsequent sort, and the amounts of (inactive) variants increased (Table S2), resulting in nearly all variants being negative after four rounds (Table S1). PCR testing confirmed all negative variants contained the pBAD‐AT plasmid with no SpAS1 insert. To verify the reason for negative enrichment, a mock recovery after bulk expression with known amounts of positives (pBAD‐AT‐His6‐SpAS1WT) and negatives (pBAD‐AT without insert) was performed. It was shown that the negative plasmid was enriched at three‐ to fivefold after recovery from bulk‐expression (Table S2). As the amount of colonies recovered is proportional to the amount of cells plated, the negative enrichment hinges on lower than expected recovery of active (blue) variants (Table S3).
To address negative enrichment, a twofold approach was taken. The initial vector was modified to move the auto‐ transporter sequence in the empty plasmid in frame with the start codon. Expression of the autotransporter in the empty plasmid should reduce negative enrichment by bringing recovery of ‘empty vectors’ on par with expressing variants. Second, glucose and fucose were added during the recovery process to aid in recovery of the cells after the droplet sorting procedure.
None of the additives (Glucose or Fucose) substantially altered the enrichment of pBAD‐AT or pBAD‐AT‐shift (Table S4). Vector pBAD‐AT‐shift led to substantially reduced negative enrichment during competition experiments (Figure S2) compared with vector pBAD‐AT. In pBAD‐AT‐shift the autotransporter construct is in frame with the constructs start codon even if vector is lacking the insert (Figure 2). Whilst use of pBAD‐AT‐shift instead of the pBAD‐AT plasmid (Figure 3) may reduce negative enrichment, it did not improve the recovery of active variants. Next, the effects on enrichment in microdroplet format were investigated. Whilst encapsulated single cells have been successfully used for high‐throughput lysis‐free screening (van Loo, Heberlein, et al., 2019), full recovery of selected variants was not realised, requiring oversampling to recover full diversity. By combining encapsulation of single cells in microdroplet with subsequent cell growth in droplets spatial genotype/phenotype linkage is maintained whilst increasing the amounts of copies per variant recovered.
FIGURE 2.
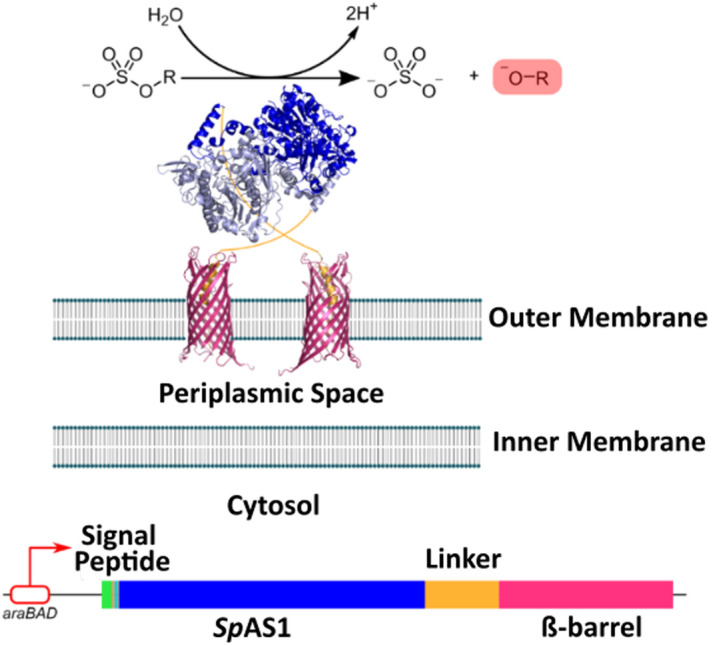
SpAS1 expressed on the surface of E. coli. Aryl sulfatase SpAS1 is linked to a B‐barrel that transports the protein to the outer membrane and displays the enzyme on the surface of the cell. A signal peptide directs the protein to the inner membrane. The plasmid is induced upon adding arabinose by the araBAD promotor.
FIGURE 3.

Plasmid sequence of pBAD‐AT (top) and pBAD‐AT‐shift (bottom). The pBAD‐AT vector contains the autotransporter sequence, an ampicillin resistance gene (AmpR), pBR322 origin of replication, AraC and AraBAD cassete. The pBAD‐AT‐shift is a modified version of pBAD‐AT in which the autotransporter construct has been brought in frame with the start codon. This means any ‘empty plasmid’ without sequence insert will express the Aida autotransporter construct.
Due to the overwhelming effect of the pBAD‐AT‐shift vector on negative variants, only pBAD‐AT‐shift (Figure 3) was used for all experiments in microdroplet. The single‐cell protocol (A) was repeated and compared with additional protocol using pico‐injection to induce individual cells in droplet rather than in bulk (B).
In‐droplet cell growth was implemented in three protocols: encapsulating cells with autoinduction medium and substrate (1a) (Figure 1C, Figure S1), encapsulation of cells with autoinduction medium, followed by pico‐injection of substrate (1a) (Figure 1D, Figure S1) and encapsulation of cells with growth medium followed by pico‐injection of inducer (l‐arabinose) and subsequent second pico‐injection of substrate (1a) (Figure 1E, Figure S1).
The new workflows seek to address two different issues. (1) Increasing the amount of recovered cells, for which in‐droplet cell growth is used (method C–E) and (2) improving the degree of control over the system through inclusion of pico‐injections. A downside of expression in droplet over flask expression in the difficulty of further manipulating the cell(culture) after cells and medium have been encapsulated. Using pico‐injection, the contents of the droplet can be modified after encapsulation, allowing manipulations such as induction or addition of substrate whilst maintaining the genotype/phenotype link in the droplet. Sorted droplets were plated after a two‐hour incubation in Lucigen recovery medium (Lucigen). The amount of cells recovered per droplet varied per competition but was below 1 in all cases (Figure 4). In the case of in‐droplet growth, 3–5 colonies were recovered when recovered droplets were plated directly after sorting (Figure 4). Although recovery was improved after a two‐hour incubation, omitting this step still resulted in recovery of multiple colonies per sorted droplet. As such, cells can be plated directly after sorting rather than after an incubation step that could introduce additional bias. Recovery values were substantially higher in the populations sorted using cell‐growth protocols as opposed to single‐cell protocols (Figure 4).
FIGURE 4.
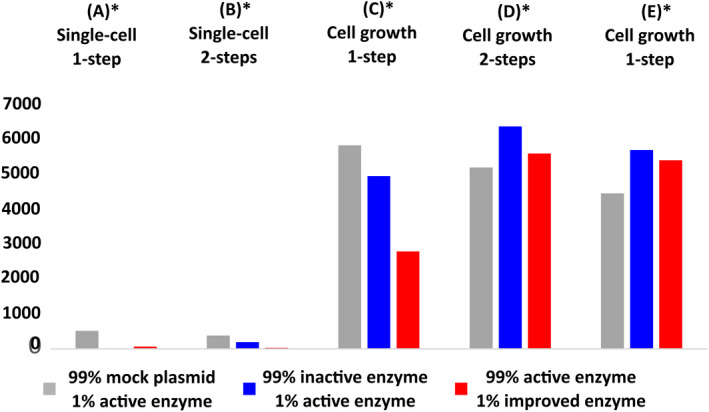
Cells recovered on agar plate per 1000 sorted droplets for single‐cell methods. Cells recovered per sorted droplet for each competition are shown side by side: 99% mock plasmid +1% active enzyme, 99% inactive enzyme +1% active enzyme and 99% active enzyme +1% improved enzyme. Results for methods (A) and (B) are based upon the screening of 1,000,000 droplets and sorting the 0.1% fraction (1000) of droplets showing highest activity during fluorescence activated droplet sorting (FADS). Results for methods (C), (D) and (E) are based upon screening of 300,000 droplets and sorting and recovery of the 0.1% fraction (300) of droplets showing highest activity during FADS. The sorted droplet emulsion was broken and the released cells spread on agar plate. Cells were plated across three agar plates and total amount of recovered colonies was counted. *Sorting was performed according to the methods described in Figure 1A–E, respectively.
Recovered colonies were counted, scraped from agar plate and resuspended into LB medium +15% (v/v) glycerol. Next, cells were transferred to nitrocellulose plates on LB‐medium containing sulfate ester (3a) (Figure S1). White (inactive) and blue (active) colonies were counted (Table S5).
For each five protocols (A–E), three competition experiments were performed: mock plasmid vs. active enzyme, inactive enzyme (C53A) vs. active enzyme (WT), and active enzyme (WT) vs. improved enzyme (R419Q). Cells recovered from protocols (A) and (B) (Figure 1) showed recovery below 100% in all cases and substantially lower recovery for the C53A vs. WT competition. Cells recovered from growth methods (C), (D) and (E) (Figure 1) show recovery of multiple copies each variant per collected droplet (Figure 4, Figures S3–S7). Recovery was increased approximately threefold after a 2‐h incubation (Figure S8) in Lucigen recovery medium (Lucigen). In contrast to the single‐cell protocol, the incubation step is not necessary to recover cells, whilst it quantitatively increases recovery. Recovery of active variants was markedly highest for protocol (C), in which cells were grown in autoinduction medium with the presence of substrate from the beginning of droplet culture. Addition of pico‐injection steps in protocol (D) and (E) (Figure 1) appear to reduce the enrichment of active variants (Figure 4).
In this study, we have compared five different methods to screen and recover live E. coli using FADS. We have shown that using in‐droplet cell growth increases both the total amount of cells retrieved (recovery) and the proportion of true positives retrieved (enrichment) obtained through live cell FADS. Three lines of inquiry were pursued to improve the efficiency of live cell recovery after microfluidic droplet sorting: (i) engineering of the expression vector (ii) Incorporating cell‐growth in droplet. (iii) Incorporating pico‐injection to add inducer and/or substrate.
It was found that an empty plasmid not expressing any protein (Figure 3) had a substantial selective advantage over a vector expressing SpAS1 as a passenger (gene of interest linked to autotransporter construct). Whilst an empty plasmid that expresses only the autotransporter/linker protein without passenger has no substantial selective advantage over a plasmid carrying SpAS1 as passenger (Figure S1), this may be explained by the added burden on the cell expressing the autotransporter.
Incorporating a growth step prior to sorting has a substantial effect on recovery of cells per droplet, whilst a fraction of variants were recovered on plate after sorting for the single‐cell methods (Figure 4). Several copies of each variant per droplet were recovered after sorting using cell‐growth methods. As each cell recovered from the same droplet is a descendent of the initially encapsulated cell, full recovery of sorted diversity is possible, even when not every cell is recovered.
Although single‐cell sorting leads to enrichment of active variants, the amount of cells recovered after sorting remains relatively low. In addition, enrichment is still far below theoretically possible numbers, and many variants remain unrecovered. The main issue lies with the negative enrichment that occur during incubation after sorting. Addition of fucose or glucose during the incubation or recovery had no substantial effect on negative enrichment during mock encapsulation or recovery (Figure S1). Recovery of live cells removes the need to amplify DNA and retransform cells plasmid DNA after selection. Whilst small plasmids can achieve high efficiency in retransformation, the recovery of larger plasmids requires a DNA amplification step for sufficient recovery of diversity (van Loo, Heberlein, et al., 2019). After sorting, intact cells can be directly plated on agar medium to obtain colonies, preventing the loss of diversity and introduction of bias. The initial protocol based on screening single cells in droplets has been successfully used in a directed evolution protocol resulting in up to 30‐fold improved catalytic efficiency (kcat/Km) in variants of aryl sulfatase SpAS1 (van Loo, Heberlein, et al., 2019). However, the number of recovered variants and the enrichment of active variants were limited. In this work, the original method is compared with an updated method with several changes in protocol to improve both the recovery and enrichment of improved variants after fluorescence‐activated droplet sorting (FADS).
Incorporating a growth step increased the enrichment of positive variants after one round of sorting (Table 1). Incorporation of pico‐injection steps did not affect the total recovery (Figure 4). However, it led to a lower proportion of true positives, reducing, rather than increasing the enrichment of positive variants after one round of sorting (Table 1). Thus, it is concluded that using cell growth in droplet without addition of pico‐injection steps results in the greatest improvement in recovery and enrichment. By using in‐droplet cell growth sufficient recovery efficiency to retrieve every unique variant in a sorting population can be achieved. Due to redundancy (multiple copies of each variant), this can be achieved even if less than 100% of sorted cells are recovered. Recovering all the variety multiple cells per droplet can reduces sorting time needed to oversample, thus allowing larger libraries to be screened faster. Additionally, it makes multiple successive rounds of screening more feasible, as no unique variants are lost each round. The success of the single‐step cell growth procedure (C) with similar efficiency to more involved methods involving methods involving pico‐injections shows that it is desirable to keep the protocol as simple as possible whilst more complicated methods can improve experimental procedures, as was found for cell growth in‐droplet, keeping the protocol simple is a consideration. Despite the tunability that pico‐injection provides to the reaction process, a relatively straightforward protocol in which all reagents were added in a simple step ultimately provided better results. Whilst improving flexibility, every additional microfluidic step introduces an additional chance for experimental variation to affect the whole protocol. It should be kept in mind that whilst striving to improve experimental protocols, a return to simplicity is a worthy consideration in experimental design.
TABLE 1.
Enrichment of active variants after droplet sorting.
| Method | Competition | Enrichment fold |
|---|---|---|
| (A) | Mock vs. active | No recovery |
| (A) | Inactive vs. active | No recovery |
| (B) | Mock vs. active | 6 |
| (B) | Inactive vs. active | 1 |
| (C) | Mock vs. active | 97 |
| (C) | Inactive vs. active | 96 |
| (D) | Mock vs. active | 1 |
| (D) | Inactive vs. active | 1 |
| (E) | Mock vs. active | 24 |
| (E) | Inactive vs. active | 81 |
Note: Enrichment is determined by dividing the proportion of active variants after droplet sorting a mixture of 99% mock plasmid +1% active enzyme and 99% inactive enzyme +1% active enzyme, respectively a . Cells were plated across two agar plates and enrichment was calculated based on the average cell count of the plates.
Sorting was performed according to the methods described in Figure 1A–E, respectively.
FUNDING INFORMATION
This project has received funding from the EU under the Horizon 2020 Research and Innovation Framework Programme No 722610 to FH and EBB through which BDGE was supported. FH and EBB acknowledge support from HFSP grant RGP006/2013. TSK was supported as an EU H2020 Marie Skłodowska‐Curie Fellow No 750772. FH is an ERC Advanced Investigator (695669).
CONFLICT OF INTEREST
None declared.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
We thank the members of the Hollfelder lab (Department of Biochemistry, UCAM) for help and assistance, in particular Paul Zurek for insightful discussions and experimental tips, Josephin M. Holstein for aid in synthesis of fluorescein disulphate and Anna Kersting (Institute for Evolution and Biodiversity, WWU) for aid in the proofreading process. BDGE thanks Ulrich Ernst (Institute for Evolution and Biodiversity, WWU), for guidance during the writing process. This project has received funding from the EU under the Horizon 2020 Research and Innovation Framework Programme No 722610 to FH and EBB through which BDGE was supported. FH and EBB acknowledge support from HFSP grant RGP006/2013. TSK was supported as an EU H2020 Marie Skłodowska‐Curie Fellow No 750772. FH is an ERC Advanced Investigator (695669).
Eenink, B.D.G. , Kaminski, T.S. , Bornberg‐Bauer, E. , Jose, J. , Hollfelder, F. & van Loo, B. (2022) Vector redesign and in‐droplet cell‐growth improves enrichment and recovery in live Escherichia coli . Microbial Biotechnology, 15, 2845–2853. Available from: 10.1111/1751-7915.14144
REFERENCES
- Abate, A. , Hung, T. , Mary, P. , Agresti, J. & Weitz, D. (2010) High‐throughput injection with microfluidics using picoinjectors. Proceedings of the National Academy of Sciences of the United States of America, 107(45), 19163–19166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agresti, J. , Antipov, E. , Abate, A. , Ahn, K. , Rowat, A. , Baret, J.C. et al. (2010) Ultrahigh‐throughput screening in drop‐based microfluidics for directed evolution. Proceedings of the National Academy of Sciences of the United States of America, 9, 4004–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold, F. (1998) Design by directed evolution some directed evolution experiments. Accounts of Chemical Research, 31(3), 125–131. [Google Scholar]
- Arnold, F. (2018) Directed evolution: bringing new chemistry to life. Angewandte Chemie, 57(16), 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, S. , Michalczyk, A. , Wilhelm, S. , Jaeger, K. & Kolmar, H. (2007) Ultrahigh‐ throughput screening to Identify E. coli cells expressing functionally active enzymes on their surface. Chembiochem, 8(8), 943–949. [DOI] [PubMed] [Google Scholar]
- Becker, S. , Höbenreich, H. , Vogel, A. , Knorr, J. , Wilhelm, S. , Rosenau, F. et al. (2008) Single‐cell high‐throughput screening to identify enantioselective hydrolytic enzymes. Angewandte Chemie, 47(27), 5085–5088. [DOI] [PubMed] [Google Scholar]
- Bridgham, J. , Ortlund, E.A. & Thornton, J. (2009) An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature, 461(7263), 515–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin, P. , Kintses, B. , Gielen, F. , Miton, M. , Fischer, G. , Mohamed, M. et al. (2015) Ultrahigh‐throughput discovery of promiscuous enzymes by picodroplet functional metagenomics. Nature Communications, 1, 10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtois, F. , Olguin, L.F. , Whyte, G. , Bratton, D. , Huck, W. , Abell, C. et al. (2018) An integrated device for monitoring time‐dependent in vitro expression from single genes in picolitre droplets. Chembiochem, 9(3), 439–446. [DOI] [PubMed] [Google Scholar]
- Fallah‐Araghi, A. , Baret, J.‐C. , Ryckelynck, M. & Griffiths, A. (2012) A completely in vitro ultrahigh‐throughput droplet‐based microfluidic screening system for protein engineering and directed evolution. Lab on a Chip, 12(5), 882–891. [DOI] [PubMed] [Google Scholar]
- Gielen, F. , Hours, R. , Emond, S. , Fischlechner, M. , Schell, U. & Hollfelder, F. (2016) Ultrahigh‐ throughput‐directed enzyme evolution by absorbance‐activated droplet sorting (AADS). Proceedings of the National Academy of Sciences of the United States of America, 113(47), 7383–7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giver, L. , Gershenson, A. , Freskgard, P. & Arnold, F. (1998) Directed evolution of a thermostable esterase. Proceedings of the National Academy of Sciences of the United States of America, 95(22), 12809–12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstein, J. , Gylstorff, C. & Hollfelder, F. (2021) Cell‐free directed evolution of a protease in microdroplets at ultrahigh‐throughput. ACS Synthetic Biology, 10(2), 252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose, J. (2006) Autodisplay: efficient bacterial surface display of recombinant proteins. Applied Microbiology and Biotechnology, 69(6), 607–614. [DOI] [PubMed] [Google Scholar]
- Jose, J. , Maas, R. & Teese, M. (2012) Autodisplay of enzymes‐molecular basis and perspectives. Journal of Biotechnology, 161(2), 92–103. [DOI] [PubMed] [Google Scholar]
- Karamitros, C. , Morvan, M. , Vigne, A. , Lim, J. , Gruner, P. , Beneyton, T. et al. (2020) Bacterial expression systems for enzymatic activity in droplet‐based microfluidics. Analytical Chemistry, 92(7), 4908–4916. [DOI] [PubMed] [Google Scholar]
- Kintses, B. , Hein, C. , Mohamed, M. , Fischlechner, M. , Courtois, F. , Lainé, C. et al. (2012) Picoliter cell lysate assays in microfluidic droplet compartments for directed enzyme evolution. Chemistry & Biology, 19(8), 1001–1009. [DOI] [PubMed] [Google Scholar]
- Leemhuis, H. , Stein, V. , Griffiths, A. & Hollfelder, F. (2005) New genotype‐phenotype linkages for directed evolution of functional proteins. Current Opinion in Structural Biology, 15(4), 472–478. [DOI] [PubMed] [Google Scholar]
- Li, G. , Zhang, H. , Sun, Z. , Liu, X. & Reetz, M. (2016) Multiparameter optimization in directed evolution: engineering thermostability, enantioselectivity, and activity of an epoxide hydrolase. ACS Catalysis, 6(6), 3679–3687. [Google Scholar]
- MacBeath, G. , Kast, P. & Hilvert, D. (1998) Redesigning enzyme topology by directed evolution. Science, 279(5358), 1958–1961. [DOI] [PubMed] [Google Scholar]
- Mair, P. , Gielen, F. & Hollfelder, F. (2017) Exploring sequence space in search of functional enzymes using microhuidic droplets. Current Opinion in Chemical Biology, 37, 137–144. [DOI] [PubMed] [Google Scholar]
- Mate, D. , Gonzalez‐Perez, D. , Falk, M. , Kittl, R. , Pita, M. , De Lacey, A. et al. (2013) Blood tolerant laccase by directed evolution. Chemistry & Biology, 20(2), 223–231. [DOI] [PubMed] [Google Scholar]
- Mohamed, M. & Hollfelder, F. (2013) Efficient, crosswise catalytic promiscuity among enzymes that catalyze phosphoryl transfer. Biochimica et Biophysica Acta, 1834(1), 417–424. [DOI] [PubMed] [Google Scholar]
- Molina‐Espeja, P. , Viña‐Gonzalez, J. , Gomez‐Fernandez, B.J. , Martin‐Diaz, J. , Garcia‐Ruiz, E. & Alcalde, M. (2016) Beyond the outer limits of nature by directed evolution. Biotechnology Advances, 34(5), 754–767. [DOI] [PubMed] [Google Scholar]
- Neun, S. , Kaminski, T. & Hollfelder, F. (2019) Single‐cell activity screening in microfluidic droplets. Methods in Enzymology, 628, 95–112. [DOI] [PubMed] [Google Scholar]
- Neun, S. , Zurek, P. , Kaminski, T. & Hollfelder, F. (2020) Ultrahigh‐throughput screening for enzyme function in droplets. Methods in Enzymology, 643, 317–343. [DOI] [PubMed] [Google Scholar]
- Neun, S. , Brear, P. , Campbell, E. , Tryfona, T. , El Omani, K. , Wagner, A. et al. (2022) Functional metagenomic screening identifies an unexpected β‐glucuronidase. Nature Chemical Biology. Online ahead of print. Available from: 10.1038/s41589-022-01071-x [DOI] [PubMed] [Google Scholar]
- Obexer, R. , Godina, A. , Garrabou, X. , Mittl, P. , Baker, D. , Griffiths, A. et al. (2017) Emergence of a catalytic tetrad during evolution of a highly active artificial aldolase. Nature Chem, 9, 50–56. [DOI] [PubMed] [Google Scholar]
- Orencia, M. , Yoon, J. , Ness, J. , Stemmer, W. & Stevens, R. (2001) Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Nature Structural Biology, 8(3), 238–242. [DOI] [PubMed] [Google Scholar]
- Pikkemaat, M. & Janssen, D. (2002) Generating segmental mutations in haloalkane dehalogenase: a novel part in the directed evolution toolbox. Nucleic Acids Research, 30(8), e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prier, C. , Zhang, R. , Buller, A. , Brinkmann‐Chen, S. & Arnold, F. (2017) Enantioselective, intermolecular benzylic C–H amination catalysed by an engineered iron‐haem enzyme. Nature Chemistry, 9(7), 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu, G. , Li, A. , Acevedo‐Rocha, C. , Sun, Z. & Reetz, M. (2020) The crucial role of methodology development in directed evolution of selective enzymes. Angewandte Chemie, International Edition, 59(32), 13204–13231. [DOI] [PubMed] [Google Scholar]
- Renata, H. , Wang, Z. & Arnold, F. (2015) Expanding the enzyme universe: accessing non‐natural reactions by mechanism‐guided directed evolution. Angewandte Chemie, International Edition, 54(11), 3351–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero, P. & Arnold, F. (2009) Exploring protein fitness landscapes by directed evolution. Nature Reviews. Molecular Cell Biology, 10(12), 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr, T. & Thornton, J. (2016) Epistasis in protein evolution. Protein Science, 25(7), 1204–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauzin, A. , Pereira, M. , Van Vliet, L. , Colin, P. , Laville, E. , Esque, J. et al. (2020) Investigating host‐microbiome interactions by droplet based microfluidics. Microbiome, 8(1), 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loo, B. , Spelberg, J. , Kingma, J. , Sonke, T. , Wubbolts, M. & Janssen, D. (2004) Directed evolution of epoxide hydrolase from a. radiobacter toward higher enantioselectivity by error‐prone PCR and DNA shuffling. Chemistry & Biology, 11(7), 981–990. [DOI] [PubMed] [Google Scholar]
- van Loo, B. , Bayer, C. , Fischer, G. , Jonas, S. , Valkov, E. , Mohamed, M. et al. (2019) Balancing specificity and promiscuity in enzyme evolution: multidimensional activity transitions in the alkaline phosphatase superfamily. Journal of the American Chemical Society, 141(1), 370–387. [DOI] [PubMed] [Google Scholar]
- van Loo, B. , Heberlein, M. , Mair, P. , Zinchenko, A. , Schüürmann, J. , Eenink, B.D.G. et al. (2019) High‐throughput, lysis‐free screening for sulfatase activity using Escherichia coli autodisplay in micro‐ droplets. ACS Synthetic Biology, 8(12), 2690–2700. [DOI] [PubMed] [Google Scholar]
- Wong, C. , van Vliet, L. , Bhujbal, S. , Guo, V. , Sletmoen, M. , Stokke, B. et al. (2021) Titratable cell lysis‐on‐demand system for droplet‐compartmentalized ultrahigh‐throughput screening in functional metagenomics and directed evolution. ACS Synthetic Biology, 10(8), 1882–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, G. & Withers, S. (2009) Ultrahigh‐throughput FACS‐based screening for directed enzyme evolution. Chembiochem, 10(17), 2704–2715. [DOI] [PubMed] [Google Scholar]
- Zeymer, C. & Hilvert, D. (2018) Directed evolution of protein catalysts. Annual Review of Biochemistry, 87(1), 131–157. [DOI] [PubMed] [Google Scholar]
- Zinchenko, A. , Devenish, S. , Kintses, B. , Colin, P. , Fischlechner, M. & Hollfelder, F. (2014) One in a million: flow cytometric sorting of single‐cell‐lysate assays in monodisperse picolitre double emulsion droplets for directed evolution. Analytical Chemistry, 86(5), 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurek, P. , Hours, R. , Schell, U. , Pushpanath, A. & Hollfelder, F. (2021) Growth amplification in ultrahigh‐throughput microdroplet screening increases sensitivity of clonal enzyme assays and minimizes phenotypic variation. Lab on a Chip, 21(1), 163–173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1