

Abstract
Soluble oligomers populating early amyloid aggregation can be regarded as nanodroplets of liquid-liquid phase separation (LLPS). Amyloid peptides typically contain hydrophobic aggregation-prone regions connected by hydrophilic linkers and flanking sequences, and such sequence hydropathy pattern drives the formation of supramolecular structures in the nanodroplets and modulates subsequent fibrillization. Here, we studied LLPS and fibrillization of coarse-grained amyloid peptides with increasing flanking sequences. Nanodroplets assumed lamellar, cylindrical micellar and spherical micellar structures with increasing peptide hydrophilic/hydrophobic ratios, and such morphologies governed subsequent fibrillization processes. Adding glycine-serine repeats as flanking sequences to Aβ16-22, the amyloidogenic core of amyloid-β, our computational predictions of morphological transitions were corroborated experimentally. The uncovered inter-relationships between peptide sequence pattern, oligomer/nanodroplet morphology, and fibrillization pathway, kinetics and structure may contribute to our understanding of pathogenic amyloidosis in ageing, facilitate future efforts ameliorating amyloidosis through peptide engineering, and aid in the design of novel amyloid-based functional nano-biomaterials and nanocomposites.
Keywords: liquid-liquid phase separation (LLPS), amyloid oligomer, nanodroplet, fibrillization, sequence hydropathy pattern
Graphical Abstract
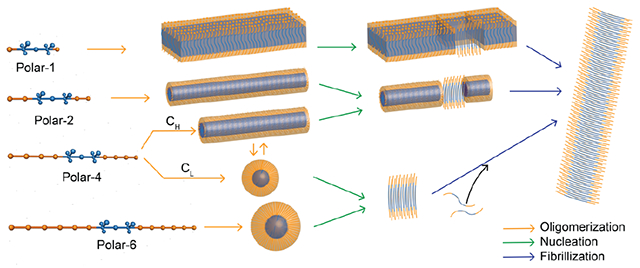
Introduction
Amyloid aggregation, the biophysical process of misfolded or intrinsically disordered amyloidogenic proteins self-assembling into fibrils, is associated with a long list of human diseases such as Alzheimer’s and Parkinson’s disease as well as type 2 diabetes.1 Amyloid fibrillization also plays important functional roles in biology, including catalysis,2 protein storage,3,4 intercellular communication and host defense.5,6 With recent advances, especially with cryo-electron microscopy (cryo-EM) and solid-state nuclear magnetic resonance (NMR) in structural biology, the fibrillar structures of many amyloid proteins have been solved, showing a ubiquitous cross-β core.7–10 Majority of the resolved fibrils are comprised of parallel in-register β-sheets, but fibrils formed by the same amyloid proteins may display distinct morphologies a.k.a. polymorphism.8,11–13 The fibrillization process of a given amyloid protein is usually driven by one or more amyloidogenic core fragments, or aggregation-prone regions (APRs), within which the sequence displays a high β-sheet propensity and, often, a high hydrophobicity (to entail toxicity). APRs may also be identified in non-amyloidogenic proteins but are usually buried inside,14,15 and have been exploited as building blocks for nano-biotechnology and novel biomaterials.16,17 Proteins with APR repeats connected by flexible linker sequences, such as spidroin in spider silk and suckerin in squid sucker ring teeth where APRs form nanofibrils within an amorphous matrix comprising linkers,18,19 have been explored for novel biomedical applications.20,21 On the other hand, flanking sequences around the APRs, naturally-occurring or engineered, may contribute to the peptide aggregation kinetics, pathway, aggregate structure, fibril function, and interaction with membranes as a result of the changing energetics of the peptides.22 Mechanistic insights into the role of the flanking sequences in amyloid aggregation are, therefore, essential for understanding amyloid function, disease pathology and mitigation, as well as the design of functional amyloid-based novel nano-biomaterials and nanocomposites.
Increasing experiments suggest that many amyloidogenic proteins, such as tau,23 islet amyloid polypeptide (IAPP),24 and α-synuclein25 undergo liquid-liquid phase separation (LLPS) en route to amyloid fibrillization. LLPS is a ubiquitous phenomenon in biology,26,27 where biomolecules including proteins, nucleotides, and metabolites28 coalesce into meta-stable or stable liquid droplets in equilibrium with isolated monomers.29 These liquid biomolecular condensates are termed membraneless organelles which play important roles in physiology and pathology, including amyloidogenesis.29–31 LLPS can be modulated by salt concentration, temperature, pH, and driven by different interaction types such as cation-π, electrostatic attraction, and hydrophobic interactions.32–35 Using coarse-grained (CG) computer simulations complemented with experiments, we previously showed that LLPS could describe the early aggregation of amyloid proteins over a wide-range of concentrations.36 Amyloid oligomers that are highly dynamic and undergoing frequent exchanges with soluble peptide monomers (i.e., the low-density liquid phase)37 can be regarded as nanodroplets of the high-density liquid phase.38 At low peptide concentrations, these nanodroplets are meta-stable and transient. As the peptide concentration increases, protein condensates become stable and more peptides are partitioned into the high-density phase.36 Out of the high-density liquid phase, the nucleated conformational conversion to fibril seeds occurs, followed by fibril elongation and/or fibril-dependent secondary nucleation.39
In the high-density liquid phase, where composite molecules are highly dynamic, amyloidogenic proteins containing APRs and flanking sequences that have distinct physiochemical properties in terms of hydrophobicity, secondary structure, and conformational flexibility may behave like block copolymers, rendering the formation of supramolecular assemblies.40 We therefore postulated that these supramolecular patterns and structures in the high-density liquid phase might in turn modulate their subsequent amyloid fibrillization.
A myriad of studies focusing on block copolymer self-assembly have yielded much insights about the assembly kinetics and versatile structural motifs of these polymeric materials.41–44 Depending on their composition,40,45 amphiphilic block copolymers can form a variety of self-assembled structures. For example, ABA block copolymers with a constant hydrophobic B block and increasingly longer hydrophilic A blocks can self-assemble into lamellar, cylindrical micellar, and spherical micellar conformations, respectively (Fig. 1). Most of the APRs of amyloidogenic proteins are hydrophobic, such as the well-studied sequences of 16KLVFFAE22 in amyloid-β17 and 22NFGAIL27 in IAPP.46 Inspired by the block copolymer self-assembly and the crucial need of elucidating the molecular dynamics of protein aggregation underpinning amyloid diseases, here we proposed to develop a set of CG peptide models mimicking ABA block copolymers with varying hydrophilic/hydrophobic ratios and study their corresponding LLPS and amyloid aggregation in silico. The peptide condensates could indeed form lamellar, cylindrical micellar and spherical micellar structures with increasing hydrophilic/hydrophobic ratios. Such high-density liquid phase morphologies played an important role in the subsequent fibrillization pathway, kinetics and fibril structures. Experimentally, we utilized the amyloidogenic core of amyloid-β, 16KLVFFAE22, and designed a set of new sequences by adding hydrophilic GS (glycine-serine) repeats to one or both ends of the peptide termini. Glycine-rich sequences are commonly found in nature as linkers or spacers flanking APRs in the block copolymer-like proteins of spidroin and suckerin.18,19 Our computational predictions, corroborated by experimental characterizations of the designed peptides, established the inter-relationships between peptide sequence hydropathy pattern, the high-density liquid phase morphology, and subsequent fibrillization pathway and kinetics. These new insights are central to our understanding of amyloidosis as well as efforts on ameliorating amyloid diseases through peptide engineering, and could prove beneficial for future design of amyloid-based functional nano-biomaterials and nanocomposites.
Figure 1.
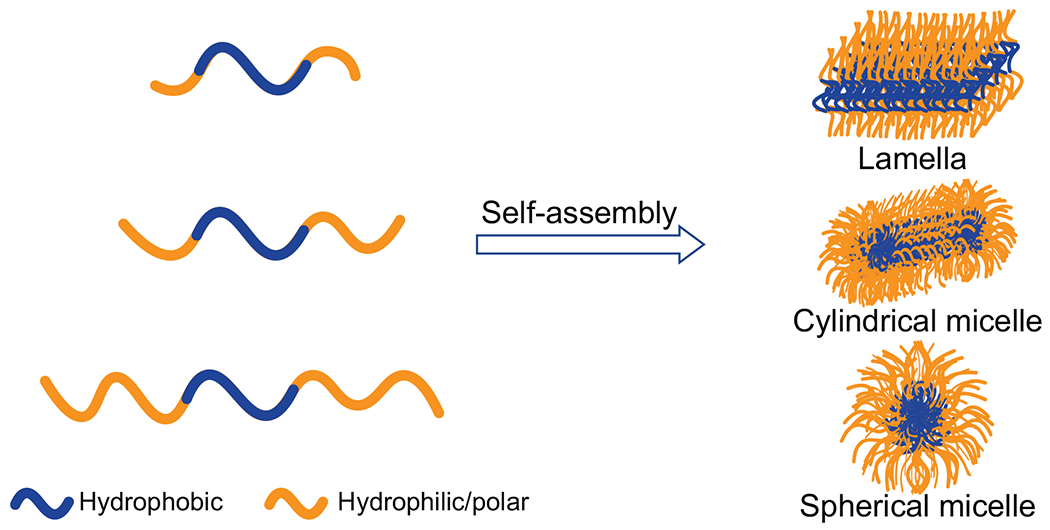
Schematic diagram demonstrating the ABA block-copolymer (BCP) self-assembly. With increasing hydrophilic/hydrophobic ratio, the self-assembly morphology changes from lamella to cylindrical and to spherical micelles.
Materials and Methods
Discrete Molecular Dynamics (DMD) simulations
DMD is a type of molecular dynamics algorithm where conventional continuous interaction potentials are replaced by optimized step-wise potential functions. Comprehensive description of the DMD algorithm has been published elsewhere.47 Briefly, the evolution of the system in DMD is driven by a set of interatomic collisions, where two atoms met at an energy step and their velocities were updated according to conservation laws. Thus, the system’s dynamics is dictated by iteratively updating only the two colliding atoms, predicting their new collisions with corresponding neighbors, and finding the next collision via quick sort algorithms. Compared with classic molecular dynamics, the sampling efficiency of DMD is significantly enhanced and the method has been widely used to study protein folding, amyloid aggregation, and interactions with nanoparticles.36,47–49 The DMD simulation engine can be obtained at Molecules In Action, LLC. (www.moleculesinaction.com).
Coarse-grained amyloid peptide model
A CG amyloid peptide model was developed in the DMD simulations to study the formation of amyloid fibrils. The detailed peptide model can be found elsewhere.48 Specifically, each CG peptide was composed by 11 beads, namely, two donors, two acceptors, two carbons, two hydrophobic atoms, and three hydrophilic atoms. Two donors and two acceptors were able to form interchain peptide hydrogen bonds. The angular and distance-dependent hydrogen bond formed between C–donor and C–acceptor, where C denoted two carbon atoms covalently linked to either the donor or the acceptor with a bond length of 1.5 Å, was modeled by a reaction-like algorithm. A hydrogen bond with the lowest potential energy had a linear alignment between C–donor and C–accepter and a donor-acceptor distance of ≈2 Å. Two hydrophobic beads–atoms attached to two C with a bond length of 3 Å, respectively–were introduced to model side-chain interactions between different peptides. Each peptide also included three hydrophilic beads, which were collinear with two C separated by 5 Å forming the CG peptide “backbone.” Both two acceptor–C–donor in a peptide were collinear and perpendicular to the backbone. Hydrophobic–C was perpendicular to both the backbone and acceptor–C–donor. The conformation of a peptide was, thus, determined by the dihedral angle between hydrophobic–C–C–hydrophobic, modeled by a multiple-well step function. Motivated by a previous CG peptide model,50–52 we allowed the peptide to adopt two conformations: aggregation-prone (β) and aggregation-incompetent (π) states. In the β-state, the dihedral angle with a minimum free energy was ≈15°, and thus, both donors and acceptors in the peptide were approximately parallel to each other, compatible with a long fibrillar state. On the other hand, the π-state had the dihedral angle ≈90° and was incompatible with the linear fibril. In this study, the π-state of an isolated peptide was more favorable than the β-state with a lower free energy, ΔEπβ ≈3.08 kBT, where kB denotes the Boltzmann constant and T the simulation temperature. We adopted an HP-like interaction potential model for non-bonded interactions – an attractive potential, ~2 kBT, was assigned among hydrophobic atoms, and a hard-core only interaction potential was assigned among hydrophilic atoms and between hydrophobic and hydrophilic atoms. The net energy gain for a hydrogen bond was ~3.5 kBT. For the non-fibrillizing model, the π state was unfavoured by assigning ΔEπβ=21.54 kBT so that the peptide could only occupy the aggregation-incompetent state most of simulation time. All the CG peptide model simulation details can be found at https://dlab.clemson.edu/research/LLPS-HDLP-Mophology/.
Coarse-grained simulation setup
We systematically investigated the LLPS and amyloid aggregation of the polar-2, polar-4, and polar-6 peptides by utilizing non-fibrillizing and fibrillizing models. For the non-fibrillizing models, we fixed the number of peptides at 100 in boxes with different sizes to manipulate LLPS (Table S1). For the fibrillizing models, we gave 60 peptides in different boxes to study fibrillization pathways under different concentrations (Table S2). At each concentration, 50 independent self-assembly simulations were performed starting from randomized monomers.
Analysis methods
For the analysis of the CG aggregation simulations, the number of peptides in each fibril was monitored. A peptide belonged to a β-sheet only if it was in the β-conformation and was stabilized by at least two interpeptide hydrogen bonds. As for analyzing the number of peptides in each oligomer, a single-linkage clustering analysis of snapshot structures along the simulation trajectories was performed: two peptides formed an oligomer if they were in contact (i.e., making at least one intermolecular contact) and a peptide belonged to an oligomer if it was in contact with any of the member peptides.
The two-dimensional potential mean force (2D PMF) was computed according to PMF = − kBT ln P(Nagg, Nfibril), where P(Nagg, Nfibril) denoted the probability of finding Nfibril peptides forming β-sheet structures in an aggregate with the size Nagg. We also plotted PMF = − kBT ln P(Nagg, Qfibril), where P(Nagg, Qfibril) denoted the probability of finding peptides in an aggregate with the size Nagg and having a fraction Qfibril of peptides forming fibrils. Briefly, for each snapshot, we performed the single-linkage clustering analysis. For each of the aggregate, its Nfibril and Qfibril values were obtained by computing the number of composite peptides forming fibrils – i.e., stable β-sheets with at least 4 peptides. By analyzing all independent simulations starting form time zero, the mass-weighted histogram was computed.
Materials
Peptides Ac-KLVFFAE-NH2, Ac-GSKLVFFAE-NH2, Ac-GSKLVFFAEGS-NH2, Ac-GSGSKLVFFAEGSGS-NH2 were custom synthesized by Mimotopes (Australia). Ac- stands for acetyl at the N-terminus and -NH2 stands for amide at the C-terminus. The purity of the peptides was determined by HPLC in water/acetonitrile as >95%. Liquid chromatography mass spectrometry (LC-MS) provided by the peptide supplier (Perkin-Elmer Sciex API 100) indicated a MW of 839.9 Da, consistent with the theoretical MW of the parent, 840.1 Da, for Ac-KLVFFAE-NH2. LC-MS results for the other peptides indicated MWs of 1038.1, 1182.2 and 1470.4 Da, accordingly with a theoretical MW of 1038.2, 1182.3, 1470.6 Da for Ac-GSKLVFFAE-NH2, Ac-GSKLVFFAEGS-NH2, and Ac-GSGSKLVFFAEGSGS-NH2, respectively. All peptides were readily dissolved in ultrapure H2O, when necessary, and incubated at 37 °C inside of a water bath.
Transmission electron microscopy (TEM)
TEM image acquisition was performed with a FEI Tecnai F20 transmission electron microscope (200 kV). All peptide samples were readily dissolved in ultrapure H2O at 1mM concentration and incubated at 37 °C inside of a water bath. Sample-grid preparation included the addition of each sample (10 μL) at the top of a glow-discharged formvar/carbon-coated copper grid (400 mesh, ProSciTech) for 60 s. Thereafter, grids were dried by simply drawing off the excess of sample with Whatmann filter paper. Grids were then negatively stained with uranyl acetate (UA, 1%) by placing a 5 μL liquid drop at the top of the grid for 30 s. Uranyl acetate 1% solution was initially centrifuged and the volume used for staining originated from its supernatant. Grids were dried similarly by using Whatmann filter papers and were finally let to air-dry for 120 s. Image acquisition was initiated upon mounting the samples into a single tilt specimen holder.
Fibril morphology analysis
Fibril tracking and mesoscopic analysis (i.e., internal contour length, persistence length, local curvature) were performed with FiberApp software.53 Semi-automatic fibril tracking was performed utilizing two complementary plugin tracking algorithms, A* pathfinding algorithm for preliminary tracking and active contour models energy minimization algorithm to obtain a sub-pixel level accuracy (Fig. S8). Internal contour length (L, nm) values were determined by the plugin algorithm log-normal probability density function: , in which L is the total length, μ is the mean value and σ the standard deviation of the length natural logarithm and A is a normalizing constant. In order to quantify the rigidity of the fibrillar aggregates, persistence length (λ) values of the fibrils were calculated and expressed as mean values determined by three different methods, bond correlation function (BCF), mean-squared end-to-end distance (MSED), and mean-squared midpoint displacement (MSMD).54 BCF method represents a more generic approach to evaluate the persistence length of polymers in 3D or 2D as the length over which tangent angle decreases by a factor of e. λBCF values in 2D were determined by the function: 〈cos θ〉 = e−l/2λ where θ is the tangent angle of any two segments along a fibril contour separated by an arc length of l.53 λMSED values in 2D derived from the function: 〈R2〈 = 4λ[l − 2λ(1 − e−l/2λ)] and were dependent from R, which is the direct distance between any pair of segments along a contour separated by an arc length of l. Evaluation of λMSMD values in 2D was performed according to the following equation, , which describes the displacement of a mean-squared midpoint between any two segments along a contour, separated by an arc length of l. Fibril width (W, nm) and size (nm) distribution values were obtained after deactivating the fiber tracking parameters and manually selecting the distances of interest through FiberApp.
Attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectroscopy
ATR-FTIR spectra (1610-1690 cm−1) were acquired from an IRTracer-100 (Shimadzu) equipped with a single-reflection ATR accessory (GladiATR 10-diamond prism), a He-Ne laser and an MCT detector (Hg-Cd-Te) under liquid nitrogen cooling. Initial background spectrum was acquired prior to each sample’s spectrum acquisition at the operating data acquisition parameters (1610-1690 cm−1, 512 number of scans at resolution of 4 cm−1) and was automatically subtracted from the input software (LabSolutions IR). After each spectrum acquisition, sample reservoirs had been washed initially with ultrapure H2O and afterwards with ethanol. 0 h spectra were acquired from samples placed on the sample reservoir in powder form. Sample preparation for the incubated samples included the addition of 5 μL of each sample on the prism reservoir (incidence angle 45°) as well as the latter use of an air-dryer gun operating at room temperature. Once solvent was totally evaporated, spectrum acquisition was initiated. Data acquisition included an absorbance measurement mode of 512 number of scans at resolution of 4 cm−1 accompanied with an Happ-Genzel apodization function. Peptides were instantly dissolved in ultrapure H2O at 1 mM working concentrations.
Cell culture
SH-SY5Y human neuroblastoma cells were cultured in Dulbecco’s modified Eagle’s medium: Nutrient Mixture F-12 (DMEM/F12, ATCC) containing 10% fetal bovine serum (FBS), 1% penicillin/streptomycin and passaging was performed every 5-6 days. Passages did not exceed the number of 20. Regarding the cell viability assays, 96-well black plate (Costar) wells were initially coated with 70 μL of attachment factor solution (37 °C, 30 min incubation). SH-SY5Y cells were seeded at a density of 30,000 per well and were further cultured for 24 h under ~80% cell confluency. Optimized peptide concentration solutions in H2O were added into propidium iodine (PI) contained DMEM/F12 medium solutions to form 1 μM PI mixtures. Each 300 μL mixture was afterwards split into triplicate wells (3×100 μL) and well plate incubation (37 °C, 30 min) then followed. PI-positive cell quantification and mapping was acquired every hour using nine reads per well by an Operetta CLS High-Content Analyzer (PerkinElmer) at 37 °C (5% CO2). Peptides were instantly dissolved in ultrapure H2O at 300 μM working concentrations prior to each assay. DMEM/F-12 medium solutions containing ultrapure H2O were used as controls.
Results and Discussion
Changing morphology of self-assembled high-density liquid phase of CG amyloid peptides with increasing hydrophilic/hydrophobic ratios.
The 11-bead CG peptide model (denoted as the polar-1 model, Fig. 2A) developed in the prior study of LLPS and amyloid aggregation36,48,49 resembled an ABA block copolymer with a minimal A block. The A block (orange) was hydrophilic and had only hard-core interactions with each other and other CG atoms. The B block (blue) contained both hydrophobic atoms and hydrogen bond donors and acceptors, driving the peptide self-assembly. The peptide adopted either a fibrillization-incompetent π-state representing random coil or helical conformations, or a fibrillization-prone β-state representing β-sheet conformation (Methods). Only in the β-state, the could continuously grow into long left-handed fibrils by forming parallel in-registered β-sheets stabilized by hydrogen bonds. The relative energy between the π- and β-states as well as the energy barriers could be adjusted to control the amyloid aggregation kinetics in discrete molecular dynamics (DMD) simulations.36 For example, we assigned the π-state with a lower energy than the β-state (ΔEβ < ΔEπ) to account for the observation that amyloid peptides have to undergo conformational changes to form amyloid fibrils and the high-energy conformation of an individual peptide in fibrils is stabilized by extensive backbone hydrogen bonds and side-chain contacts with neighboring peptides. The oligomerization/LLPS processes could also be decoupled from the fibrillization process by assigning ΔEπ ≫ ΔEβ and ΔEπ ≫ kBT, where kB is the Boltzmann constant and T is the system temperature, such that the fibrillization-prone β-state is energetically unfavored and the peptide becomes nonamyloidogenic. Before fibrillization, the self-assembled aggregates of polar-1 peptides adopted the lamellar morphology (middle panel, Fig. 2A). Hence, we added hydrophilic and flexible flanking sequences with various lengths (2, 4, 6 hydrophilic A blocks) to both the N- and C-termini of the hydrophobic B block based on polar-1 model with shortest A block (polar-2, 4 and 6 in right panel, Fig. 2A) to increase the hydrophilic/hydrophobic ratio.
Figure 2.
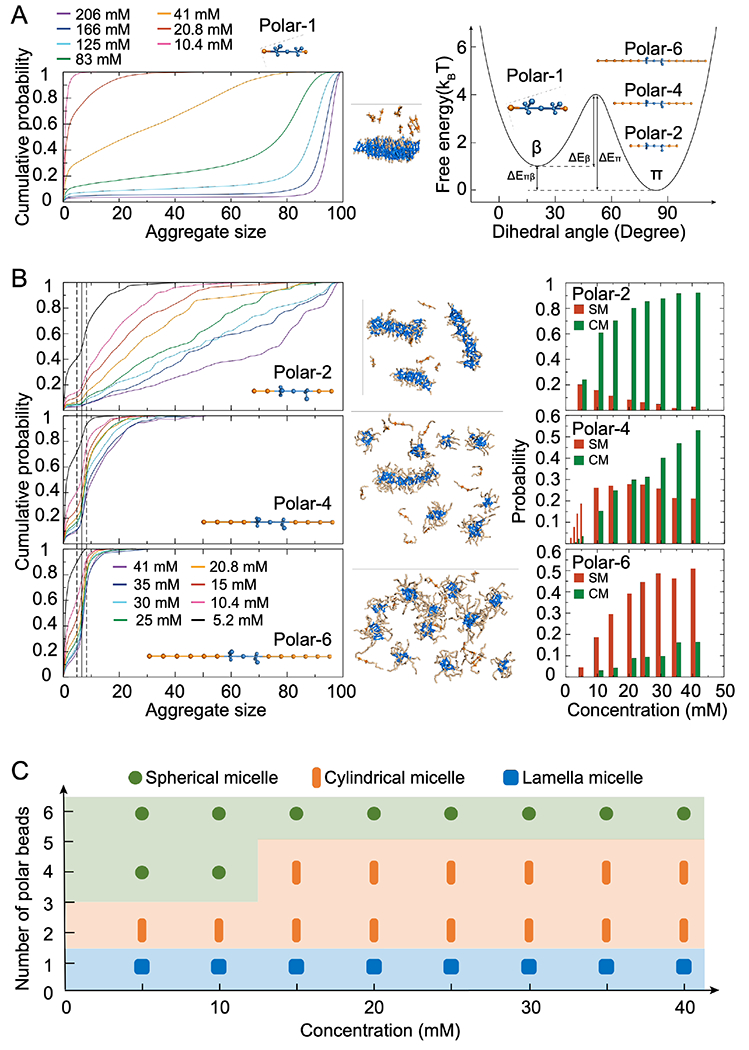
(A) Cumulative peptide population as a function of aggregate size for different concentrations for polar-1. The mass-weighted size distribution of aggregates corresponds to the first-order derivative of the presented cumulative probability distribution. The coarse-grained peptide model with two lowest energy states of the peptide monomer, corresponding to the fibrillization-prone state β and fibrillization-incompetent state π. Polar-2, polar-4, and polar-6 with increasing flanking sequences are shown in the inset. (B) Cumulative peptide populations as functions of aggregate size for different concentrations of non-fibrillizing systems, with the CG peptide models of polar-2, polar-4 and polar-6, are shown in the left column. The representative structures formed by different models with peptide concentration at 20.8 mM are shown in the middle, where the hydrophobic and hydrophilic moieties are colored in blue and orange, respectively. Partitions of peptides in spherical micelle (SM) and cylindrical micelle (CM) formed by Polar-2, Polar-4, and Polar-6 peptides as a function of peptide concentration are shown in the right column. (C) Phase diagrams of polar-1, polar-2, polar-4, and polar-6 as a function of the peptide concentration.
Non-amyloidogenic CG peptide models were first used to decouple LLPS from fibrillization in silico. For each CG peptide, a wide range of concentrations were investigated in DMD simulations. The main advantage of DMD is its superior sampling efficiency, both spatially and temporally, compared with conventional MD.47,55,56 At each concentration, 50 independent self-assembly simulations were performed starting from randomized monomers. After reaching equilibrium, the cumulative peptide fraction as a function of aggregate size was calculated with time and ensemble averages (left panel, Fig. 2B). A common inflection point around 7 was observed for the polar-2, 4, and 6 models under different concentrations, but not for the polar-1 model (left panel, Fig. 2A). These stable or metastable oligomers formed by 5-9 (i.e., 7±2) identical peptides corresponded to spherical micelles (middle panel in Fig. 2B, equilibrium conformations). We chose the cutoff aggregate size of ~5, corresponding to the free energy barrier separating the monomer and micelle states and thus the local minima in the aggregate size distribution or the inflection points in-between in the cumulative probability distribution in Fig. 2B. The aggregates with sizes smaller than 5 were unstable and tended to dissociate into monomers, and thus, constituted the low-density liquid phase (LDLP) together with monomers. Stable and metastable aggregates with at least five peptides were counted as the high-density liquid phase (HDLP) in our simulations. Larger aggregates adopting cylindrical micellar structures were observed for the polar-2 and 4 peptides. Polar-2 had spherical micelles as transient self-assembly intermediates. Polar-4, on the other hand, mainly formed spherical micelles at lower peptide concentrations (e.g., 5.2 mM) and self-assembled into cylindrical micelles at higher peptide concentrations (e.g., 41 mM), displaying a concentration-dependent morphology of HDLP. Polar-6, with the longest flanking sequence, only formed spherical micelles under a wide range of concentrations. Based on the population of various aggregate morphologies in terms of spherical and cylindrical micelles at different peptide concentrations (right panel, Fig. 2B), a phase diagram for different peptides at varied concentration can thus be obtained (Fig. 2C). Taken together, longer flanking sequence peptides displayed higher probabilities to assume the smaller sized structures, consistent with the block copolymer self-assembly.
Distinct fibrillization kinetics and pathways for designed amyloid peptides with flanking sequences of different lengths in silico.
Next, we studied the fibrillization of different amyloidogenic CG peptides under a wide range of peptide concentrations in DMD simulations. Averaged over 50 independent simulations, the fraction of peptides forming fibrils — defined as parallel in-register β-sheets with at least four strands — was calculated as a function of simulation time for each CG model at different concentrations (Fig. 3A). For the polar-2 model, saturation of fibrillization was observed at all concentrations within ~10 μs of CG simulations. Variations in terms of aggregation kinetics among independent simulations at different peptide concentrations were small. The aggregation kinetics of polar-4, on the other hand, displayed a strong concentration dependence. For example, saturation of fibrillization was observed at and above 15 mM, but the fibrillization did not saturate at lower concentrations within 120 μs of simulation time. The fibrillization of polar-6 did not saturate even at the highest peptide concentration of 41 mM within the simulation time of 120 μs.
Figure 3.
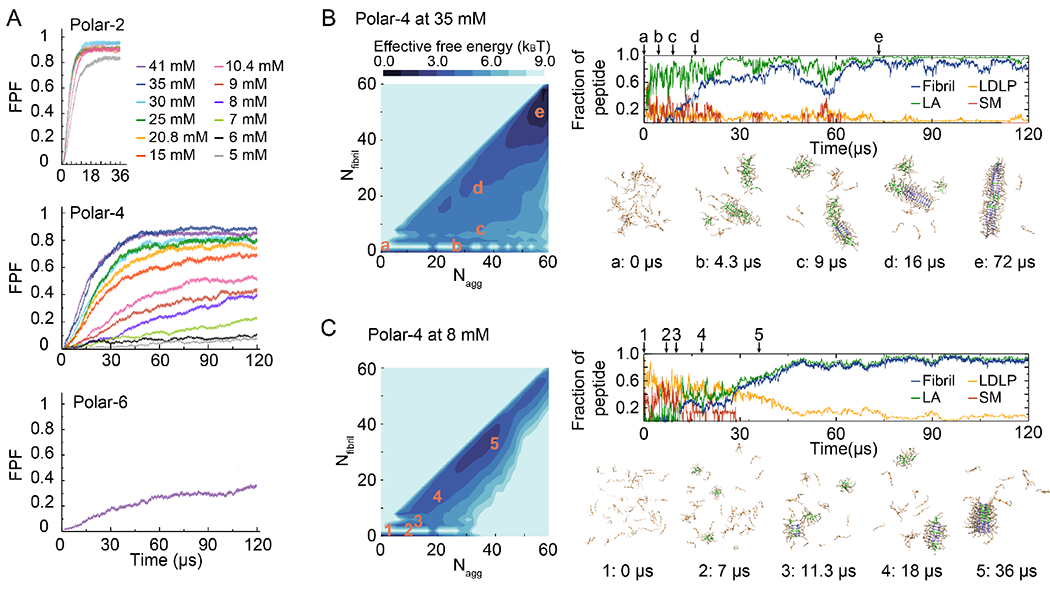
(A) Averaged-time course of the fraction of peptides in fibrils (FPF) for different concentrations of the polar-2, polar-4, and polar-6 model system, respectively. 2D PMF as a function of the aggregate size (Nagg) and corresponding number of peptides forming fibrils (Nfibril) for polar-4 at 35 mM and 8 mM are shown in the left of (B) and (C). Fraction of peptides in different components – i.e., low-density liquid phase (LDLP), spherical micelle (SM), large aggregates (LA) with Nagg > 9 in which the spherical micelles were excluded but nucleated fibril structures were included, and fibrils – as a function of simulation time are shown on the top right, and representative snapshots at different time points in the right bottom at 35 mM (B) and 8 mM (C). (Nagg, Nfibril) of each snapshot is marked as orange in the 2D PMF.
To further characterize the phase separation and amyloid aggregation, we also computed the two dimensional potential of mean force (2D PMF, or the effective free energy), −kBTlnP(Nagg, Qfibril), where P(Nagg, Qfibril) corresponded to the probability of finding peptides in an aggregate with the size Nagg and having a fraction Qfibril of peptides forming fibrils (Fig. 3B&C, Figs. S2, S4). We included all simulation trajectories starting from time zero in order to capture the dynamics of initial phase separations of the peptides. Since polar-4 at high concentrations formed stable cylindrical micelles as polar-2 (Fig. 2C), the fibrillization of both peptides displayed a similar pathway (Fig. 3B and Fig. S1). At a relatively high peptide concentration of ~35 mM (Fig. 3B), polar-4 peptides first underwent LLPS rapidly and majority of the peptides self-assembled to cylindrical micelles (e.g., two aggregates were formed by 19 and 27 peptides at ~4.3 μs in a presentative trajectory, Fig. 3B) partitioned in the cylindrical micelles of the high-density liquid phase (Fig. 2C), the nucleated conformational conversion to fibril seeds took place in these cylindrical micelles and the fibril growth was also dominated by the rapid addition of peptides within cylindrical micelles.
In contrast, polar-4 at low concentrations had spherical micelles as stable species of the HDLP similar to polar-6 (Fig. 2C). At a low concentration of 8 mM (Fig. 3C), polar-4 peptide formed spherical micelles instead of cylindrical micelles (e.g., at ~7 μs in a presentative trajectory, Fig. 3C). These spherical micelles exchanged frequently with monomers in the low-density liquid phase, and occasionally underwent conformational conversions to proto-fibril or fibril seeds (e.g., ~11.3 μs, Fig. 3C). The growth of fibrils was mainly through a gradual elongation via an addition of monomers in the solution of the LDLP (e.g., the decrease of monomers along with the increase of fibril population from ~18 to 36 μs, Fig. 3C), and thus, the low equilibrium peptide concentration in the LDLP resulted in slow aggregation kinetics. The fibrillization kinetics of polar-4 at the low concentration also displayed a greater heterogeneity (Fig. S2). Polar-6 followed a similar aggregation pathway (Fig. S3) via the formation of spherical micelles over a wide range of concentrations. It was noticed that the conformational conversion of micelles was intermittent during the simulation. For example, another micelle converted to a protofibril and rendered a new fibril at ~60 μs (e.g., Fig. S3B). Interestingly, the formed fibrils of the polar-6 peptides could also disaggregate in some simulations (e.g., Fig. S3C).
Together, our systematic computational study suggested that the sequence composition of amyloid peptides determined the morphologies of the HDLP, which in turn governed the fibrillization pathway and kinetics of the peptides. Both lamellar and cylindrical micelle-shaped condensates as the HDLP had at least one infinitely extendable dimension, which should enable rapid fibril growth via elongation and/or secondary nucleation by the high concentration of peptides in proximity already in the condensate. Although the zero-dimensioned spherical micelles could facilitate the nucleation of fibrils seeds, a slower fibril growth was determined by the low concentration of monomers in the LDLP which was in equilibrium with other spherical micelles in the HDLP. For peptides with an intermediate hydrophilic/hydrophobic ratio that featured distinct HDLP morphologies at low and high concentrations (e.g., polar-4), different fibrillization pathways and kinetics could be observed correspondingly.
Experimental characterizes revealed fibril morphologies could be modulated by extending hydrophilic amino acids to the amyloidogenic hydrophobic core.
To further corroborate our simulation results, we utilized the central hydrophobic core of amyloid-β (16-22), Ac-KLVFFAE-NH2, as a reference for our experiments. At one or both termini of the peptide core, single or multiple GS (glycine-serine) residue repeats were incorporated, containing overall a positive net charge at pH 7. Herein, Ac-KLVFFAE-NH2 at pH 7 self-assembled rapidly within 15 min, into either long or shortened amyloid-like fibrils (Fig. 4A-i), originated from some intermediate protofibrillar aggregates observed (Fig. 4A-i, right panel). Statistical analysis showed that the fibrils possessed a mean contour length of 186.4 nm (σL = 198.1 nm) (Fig. 4A-iii) and a mean width of 8.8 nm (σw = 3.2 nm) (Fig. 4A-v). The ability of the amyloidogenic core motif to self-assemble was due to its high hydrophobic attraction and aromatic π-π stacking between the FF residues.57,58 After 5 h of incubation, mature twisted fibrils formed (Fig. 4A-ii), accompanied by an increased contour length (<L> = 889.0 nm, σL = 538.9 nm) (Fig. 4A-iv) and a slightly increased width (<W> =10.8 nm, σw = 2.9 nm) (Fig. 4A-vi).
Figure 4.

(A) In vitro aggregation analysis of AC-KLVFFAE-NH2 over time (15 min and 5 h). (i) TEM revealed the formation of pre-fibrillar and fibrillar species over the first 15 min of incubation. (ii) Mature fibrils with increased twistedness were observed over 5h of incubation. (iii), (iv) The internal contour length (L, nm) of the analyzed fibrils (n=450) for the two timepoints revealed a 376.9% increase over time. (v), (vi) Fibril width (W, nm) analysis (n=300) for the respective fibrils analyzed in panels iii and iv. (B) In vitro aggregation analysis of Ac-GSKLVFFAE-NH2 over 15 min and 5 h reveals how terminal hydrophilic capping on a hydrophobic amyloidogenic core can manipulate self-assembly structures into lamellar ones, respectively. (i), (ii) TEM images of 15 min and 5 h incubated Ac-GSKLVFFAE-NH2 indicating the formation of lamellar nanotape structures. (iii), (iv) Number (%) of the internal contour length (L, nm) values over 15 min and 5 h indicate a 147.53 nm increase over time for n=80 full-length analyzed nanotapes. (v), (vi) Nanotape width analysis (W, nm) (n=300) for the respective nanotapes analyzed in panels (iii) and (iv). Ac-KLVFFAE-NH2 and Ac-GSKLVFFAE-NH2 concentration: 1 mM in MilliQ H2O, incubation temperature: 37 °C. Internal contour length and fibril width values were calculated through FiberApp.
Several morphology studies of KLVFF-based peptides have been performed and reviewed elsewhere,59–61 revealing the influence of pH62,63 and terminal capping on the role of electrostatic interactions that drive the formation of lamellar morphologies. Herein, the incorporation of a GS residue into the N terminus (Ac-GSKLVFFAE-NH2) led to an early onset (15 min) of peptide aggregation into long flexible nanotapes mesoscopically (Fig. 4B-i), with a mean contour length of 623.5 nm (σL = 323.9 nm) (Fig. 4B-iii) and a mean width of 57.1 nm (σw = 27.2 nm) (Fig. 4B-v). Such phenomenon may be attributed to the salt bridge affinity weakening through electrostatic charge reduction by the non-polar glycine (G) between the polar residues K16 and E22 and electrostatic repulsion between K16 and S15. The specific salt bridge has been found to mediate fibrillar assembly and was initially reported by Mehta et al..64 Moreover, aromatic stacking between F19/F19 and F20/F20 both contributed to the lamination and creation of a Phe zipper in the resulted β-sheet conformation.64 On the other hand, a longer incubation time (5 h) (Fig. 4B-ii) did not alter significantly the mean internal contour length (<L> = 771.1 nm, σL = 362.9 nm) (Fig. 4B-iv) and width of the rendered morphologies (<W> =59.2 nm, σw = 28.9 nm) (Fig. 4A-vi).
Upon incorporation of GS residues to both termini (Ac-GSKLVFFAEGS-NH2) and corresponding increases of the hydrophilic/hydrophobic ratio, the peptides self-assembled into rigid cylindrical micellar morphologies (Fig. 5A-i) shortly upon the onset of incubation (15 min). Such cylindrical micelles possessed a mean internal contour length of 319.1 nm (σL = 205.1 nm) (Fig. 5A-iii) and a mean width of 15.7 nm (σw = 6.6 nm) (Fig. 5A-v). However, upon a longer incubation time of 5 h, the peptides tended to assume lamellar morphologies (<W> =38.8 nm, σw = 17.3 nm) (Fig. 5A-vi) while losing their rigidity. As a longer peptide motif, two repeating GS units were incorporated into both termini of the amyloidogenic core (Ac-GSGSKLVFFAEGSGS-NH2) resulting in a significant increase of the hydrophilic/hydrophobic ratio (10:5 residues). Such primary structure motif led to the initial (15 min) formation of ~8 nm spherical micelles (Fig. 5B-i) consisting of an inner hydrophobic LVFFA unit and outer GSGS-containing compartments. Nevertheless, based on the amyloid nucleation theory,65–67 which posits the formation of metastable oligomeric species – spherical micelles in our case – from monomers involving primary and secondary steps unlike in nucleation-conversion-polymerization, spherical micelles upon a critical nucleus size further self-assembled into short rigid protofibrils as larger aggregate assembly became a much more energetically favorable process. Their mean internal contour length value was 200.9 nm (σL = 267.7 nm) (Fig. 5B-vii) and their mean width was 16.1 nm (σw = 4.7 nm) (Fig. 5B-ix). Interestingly, the protofibrillar species co-existed with some small (~ 20 nm) (Fig. 5B-iv bottom row) and large (50-80 nm) spherical aggregates (Fig. 5B-ii, iii, iv-top, middle row), which tended to be similar in morphology to our previous reports regarding the co-existence of HDLP droplets with IAPP amyloid protofibrils.36 Longer peptide motif further assembled into mature rigid fibrils upon 5 h of incubation characterized by the absence of such spherical aggregates. The regarding mature fibrils possessed a mean internal contour length of 557.1 nm (σL = 326.4 nm) (Fig. 5B-viii) and a slightly larger mean width of 19.6 nm (σw = 13.2 nm) (Fig. 5B-ix) compared to the aforementioned intermediate species. As a following step, additional mesoscopic analysis based on the persistence length (λ) and local curvature (1/Rc) for all peptides-derived fibrillar/lamellar/cylindrical aggregates was performed (Fig. 5C–E). In order to quantify the rigidity of the fibrillar aggregates, persistence length values were calculated and determined as mean values by three different methods, bond correlation function (BCF), mean-squared end-to-end distance (MSED), and mean-squared midpoint displacement (MSMD), through FiberApp.53 Among all peptides, Ac-GSGSKLVFFAEGSGS-NH2 (5 h) possessed the highest bending stiffness (λ = 6227.2 ± 2287.5 nm) (Fig. 5C) as reflected by the absence of bending and twisting observed through transmission electron microscopy (TEM). On the other hand, lamellar morphologies observed for Ac-GSKLVFFAE-NH2 at both 15 min and 5 h were characterized by soft looping fibrils with a persistence length of ~1000 nm (Fig. 5C). For Ac-KLVFFAE-NH2, a combination of long and short fibrils at 15 min led to a reduced persistence length (λ = 3568.9 ± 1277.5 nm) and high variations on local curvature compared to the persistence length (λ =3901.9 ± 885.2 nm) and local curvature distribution for the 5 h mature elongated fibrils (Fig. 5C–E). Moreover, the initial 15 min cylindrical micellar morphologies for Ac-GSKLVFFAEGS-NH2 were more rigid with an increased persistence length of λ = 4502.3 ± 1379.1 nm in comparison with the soft lamellar morphologies observed at 5 h with a persistence length of λ = 3767.0 ± 990.8 nm (Fig. 5C). In addition to the intermediate (30 min) short fibrils of the longer peptide motif that exhibited low local curvature values, both morphologies at the two different timepoints shared significantly low local curvature values (Fig. 5D–E). Therefore, our experimental characterizations of Aβ16-22 with designed flanking sequences confirmed our computational predictions regarding the morphology of the peptide assemblies.
Figure 5.

(A) In vitro aggregation analysis of AC-GSKLVFFAEGS-NH2 (i), (ii) TEM characterization of AC-GSKLVFFAEGS-NH2 over 15 min showing the initial formation of narrow lamellar aggregates, which tend to wider lamellar aggregates over 5 h of incubation. (iii), (iv) Number (%) of the internal contour length (L, nm) values over 15 min and 5 h for n=80 full-length analyzed lamellar aggregates. (v), (vi) Number (%) fibril width (W, nm) analysis (n=300) indicates a significant 23.05 nm increase over time. (B) In vitro aggregation analysis of Ac-GSGSKLVFFAEGSGS-NH2 reveals the spontaneous formation of micellar morphologies over 15 min of incubation followed by their morphological evolution into short protofibrilar species accompanied by 10-40 nm spherical aggregates over 30 min of incubation. Mature rigid fibrils were finally formed over 5 h of incubation. (i-iii, v) TEM images of the 15 min, 30 min and 5 h incubated Ac-GSGSKLVFFAEGSGS-NH2 peptide depicting the morphology transition from micelles to rigid fibrils. (iv) Small (~ 20 nm) and large nanoscale aggregates (50-80 nm) in diameter after 30 min of incubation. Scale bars: 25 nm, except for bottom right-100 nm. (vi) Internal contour length (L, nm) of the micelles determined by number (%) count. (vii) Number (%) determination of the internal contour fibril length (L, nm) upon 30 min of incubation (n=390). (viii) Number (%) determination of the internal contour fibril length (L, nm) upon 5 h of incubation (n=80). (ix) Fibril width (W, nm) analysis (n=300) of the respective protofibrils and mature fibrils analyzed in panels vii and viii. L (nm), W (nm) values were estimated similarly as for the other peptides. (C) Persistence length (λ, nm) bar chart indicating the bending rigidity of the four respective peptides (Ac-KLVFFAE-NH2, Ac-GSKLVFFAE-NH2, Ac-GSKLVFFAEGS-NH2, Ac-GSGSKLVFFAEGSGS-NH2) over time (< 1 h-orange and 5 h-green). Data points shown are the mean values (n=3) ± (SEM). Statistical analysis performed through unpaired t-test determining two tailed p-values (APA): < 0.12 (ns), < 0.033 (*), < 0.002 (**), < 0.001 (***). (D), (E) Number (%) vs local curvature (1/Rc, nm) graphs of the four respective peptides over time. Peptides’ concentration: 1 mM in MilliQ H2O, incubation temperature: 37 °C. Internal contour length and fibril width values were calculated through FiberApp. λ values were estimated using the bond correlation function (BCF), mean squared end-to-end distance (MSED), and mean-squared midpoint displacement (MSMD) methods and are presented as mean values determined by the three methods. λ and 1/Rc values were obtained through FiberApp from the corresponding contour length fibril analysis.
We also characterized the secondary structures of all four different peptides over time using attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectroscopy. Specifically, we studied bond vibrations in the amide I region (1610-1690 cm−1), which is mainly (70-85%) governed by the stretching vibrations of the C=O bond. In a qualitative manner, all peptides over time exhibited a major peak in the wavenumber region of 1623-1641 cm−1, which was associated with β-sheet structures. In particular, the strong amide I band at the lower frequencies 1623-1641 cm−1 combined with the weaker amide I band at the higher frequencies 1674-1695 cm−1 suggested an antiparallel β-sheet conformation of the peptide chains (Fig. 6A–D). Such interpretation was in agreement with previous secondary structure studies on either amyloid fibrillar Aβ16-22 or lamellar Aβ16-22 contained but modified peptides.62,64 Interestingly, low frequency β-sheet region (1610-1640 cm−1) was accompanied by a shift of the maxima to lower wavenumbers upon the addition of terminal capping GS residues, which was likely linked with an increase in the number of strands per β-sheets followed by reduced twist angles δ (°) (Fig. 6A–D).68
Figure 6.
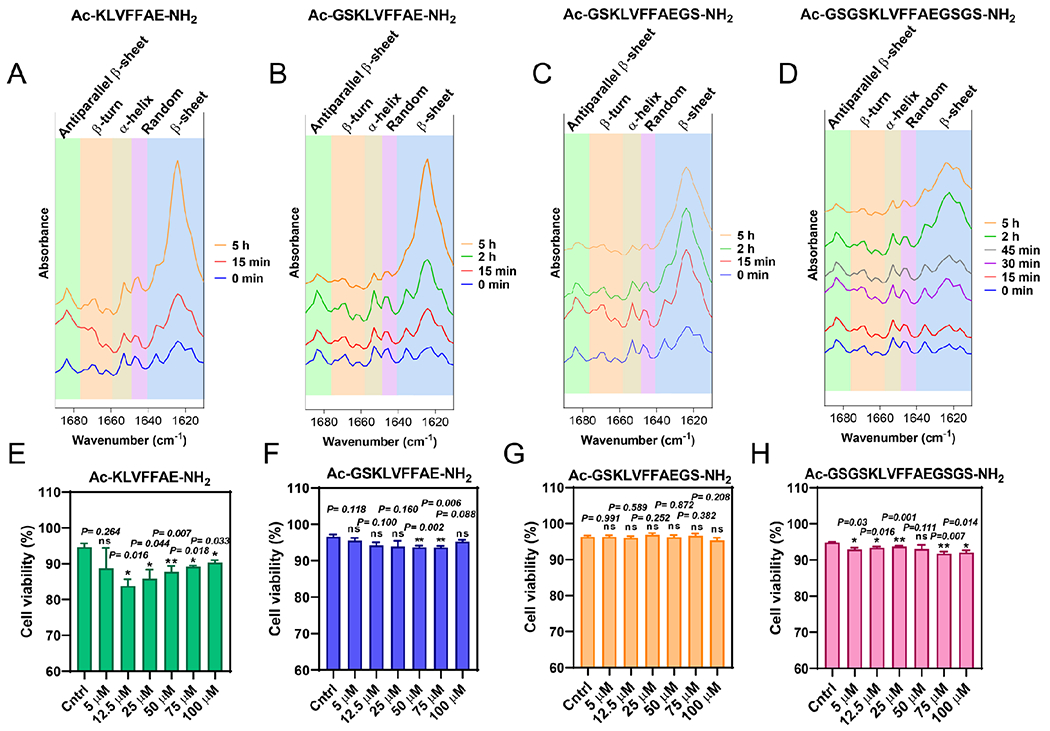
ATR-FTIR amide I region spectra (1610-1690 cm−1) of AC-KLVFFAE-NH2 (A), Ac-GSKLVFFAE-NH2 (B), Ac-GSKLVFFAEGS-NH2 (C) and Ac-GSGSKLVFFAEGSGS-NH2 (D) for different timepoints (0 min-5 h) revealing the universal adoption of anti-parallel β-sheet conformations for all peptides over time. Each type of conformation (anti-parallel β-sheet, β-turn, α-helix, random and β-sheet) is highlighted accordingly with their wavenumber (cm−1) region. (E)-(H) In vitro neuronal cell viability induced by Ac-KLVFFAE-NH2, Ac-GSKLVFFAE-NH2, Ac-GSKLVFFAEGS-NH2 and Ac-GSGSKLVFFAEGSGS-NH2 in a range of concentrations (5-100 μM) at SH-SY5Y neuroblastoma cells after 15 h of incubation. DMEM/F-12 medium solutions with MilliQ H2O were used as controls. Data points shown are the mean values (n=3) ± (SEM). Statistical analysis performed through paired t-test determining two tailed p-values (APA): < 0.12 (ns), < 0.033 (*), < 0.002 (**), < 0.001 (***).
All four peptides were tested in a range of concentrations (5-100 μM) whether they could induce significant neuronal cell death in human neuroblastoma cells (SH-SY5Y) after 15 h of incubation (Fig. 6E–H, Fig. S4–7). Cell viability (%) for SH-SHY5 cells incubated with GS-modified peptides did not go below 90% indicating their non-cytotoxic profile prior and post LLPS (Fig. 6F–H, Fig. S5–7). On the other hand, SH-SY5Y cells exposed to 5-100 μM Ac-KLVFFAE-NH2 displayed up to 20% of cell mortality, especially for the lower concentrations 12.5 and 25 μM (Fig. 6E, Fig. S4). Such result may be attributed to the slower nucleation rates of fibrillization, which resulted in longer cell exposure to pre-fibrillar (oligomeric/protofibrillar) amyloid aggregates, the most toxic amyloid species acknowledged.69,70 On the other hand, the long GS-modified peptides did not affect cell viability to such extent at low concentrations, despite their even slower nucleation rates of fibrillization compared to Ac-KLVFFAE-NH2. The specific result is likely attributed to the hydrophilic moiety on the outer sphere of the early micellar assemblies that prevented the aberrant interactions of peptides with cellular membranes. However, none of the peptide samples induced significant cytotoxicity in SH-SY5Y cells.
Conclusions
In sum, our combined computational and experimental study was inspired by recent discoveries that many intrinsically disordered amyloid proteins undergo LLPS before fibrillization25,30,36,71 and the knowledge derived from polymer physics that protein condensates in the HDLP may adopt supramolecular patterns and structures depending on their sequence hydropathy pattern. The coarse-grained simulations in this study, despite lacking specific sequence details, effectively modeled hydrophobicity, hydrogen bonding, nucleated conformational conversion, and twisted β-sheet formation, and thus captured the general properties of LLPS and fibrillization of amyloid peptides driven by these interactions and behaviors. The excellent agreement between computational predictions and in vitro characterizations of correspondingly designed peptides revealed that the peptide condensates in the HDLP could indeed form lamellar, cylindrical micellar and spherical micellar structures with increasing hydrophilic/hydrophobic ratios. Such HDLP morphologies played an important role in the subsequent fibrillization pathway and kinetics. Specifically, with the nucleation of fibril seeds in the HDLP condensates due to a high local concentration of peptides, the readily available peptides in the two-dimensional lamellae and one-dimensional cylindrical micelles allowed rapid fibril growth. For peptides forming the zero-dimensional spherical micelles as the HDLP, fibril growth was determined by peptide monomers in the solution – i.e., the LDLP, thereby resulting in slow fibrillization kinetics. Fragment derivatives of amyloid peptides, including amyloidogenic cores, are often used to inhibit aggregation of full-length proteins by themselves or as parts of designed composites. Our results suggest that introducing hydrophilic and flexible flanking sequences may help avoid or delay the potential self-aggregation of these fragments.
Our results also shed a new light on the diverse morphologies of soluble aggregates or oligomers reported in the literature for various amyloid proteins and peptides. While remaining to be verified in future work, those curly aggregates observed prior to, or coexisting with the formation of rigid fibrils could originate from cyclical micelles.72–74 Since CG peptides in this study contained only one B block (i.e., hydrophobic APRs), the self-assembled HDLP aggregates with microphase separation of A and B blocks had at least one of the dimensions limited by the sequence length – e.g., lamellae, cylindrical and spherical micelles have constraints in one, two, and tree dimensions, respectively. For block copolymers with more than one hydrophobic B blocks as in many amyloidogenic peptides or proteins containing multiple APRs connected by linkers and flanking sequences, microphase separated domains could be inter-connected by the linkers, resulting into large coalesces without limits in any dimensions. Such large aggregates or oligomers with dimensions much large than that of the fully extend monomers have been commonly observed during the early aggregation of many amyloid peptides and proteins.36,72,75
In addition, by adding one to two copies of a hydrophilic moiety we demonstrated with simplicity the key roles of amphiphilicity, sequence and chain length on peptide amyloid aggregation. These insights pointed to the potential of engineering peptide sequence and structure for the purposes of biotechnology and anti-amyloidosis medicine. Moreover, our work also underscored a translational value between the field of amyloidosis and (amphiphilic) block copolymer self-assembly as both are governed, in principle, by hydrophobic interaction, hydrogen bonding and the laws of thermodynamics and polymer physics. Together, our results established the inter-relationships between peptide sequence hydropathy pattern, HDLP morphology upon LLPS, and subsequent fibrillization pathway and kinetics for amyloid aggregation. These new insights advanced our fundamental understanding of amyloidosis, a foundation to our combat of amyloid diseases and the design of functional amyloid-based novel nano-biomaterials and nanocomposites.
Supplementary Material
Acknowledgements
This work was supported in part by National Key R&D Program of China (2021YFA1200900) (Ke), NSF CAREER CBET-1553945 (Ding), NIH MIRA R35GM119691 (Ding), and the National Natural Science Foundation of China 11904189 (Sun). Computer simulations were supported by the multi-scale computational modeling core of NIH P20GM121342. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NSFC, NIH, and NSF.
Footnotes
Simulation setup details (Table S1–S2); Polar-2 fibrillization at 5 mM (Fig. S1); Polar-4 fibrillization at 8 and 35 mM (Fig. S2); Polar-6 aggregation at 40 mM (Fig. S3); Propidium iodide channel map of SH-SY5Y cells upon incubation with different peptides at various concentrations (Fig. S4–S7); TIFF images before and after fiber tracking analysis (Fig. S8).
References
- (1).Ke PC; Sani M-A; Ding F; Kakinen A; Javed I; Separovic F; P. Davis T; Mezzenga R Implications of Peptide Assemblies in Amyloid Diseases. Chem. Soc. Rev 2017, 46 (21), 6492–6531. 10.1039/C7CS00372B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Fowler DM; Koulov AV; Alory-Jost C; Marks MS; Balch WE; Kelly JW Functional Amyloid Formation within Mammalian Tissue. PLoS Biol. 2006, 4 (1), e6. 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Seuring C; Verasdonck J; Gath J; Ghosh D; Nespovitaya N; Wälti MA; Maji SK; Cadalbert R; Güntert P; Meier BH; Riek R The Three-Dimensional Structure of Human β-Endorphin Amyloid Fibrils. Nat. Struct. Mol. Biol 2020, 27 (12), 1178–1184. 10.1038/s41594-020-00515-z. [DOI] [PubMed] [Google Scholar]
- (4).Antonets KS; Belousov MV; Sulatskaya AI; Belousova ME; Kosolapova AO; Sulatsky MI; Andreeva EA; Zykin PA; Malovichko YV; Shtark OY; Lykholay AN; Volkov KV; Kuznetsova IM; Turoverov KK; Kochetkova EY; Bobylev AG; Usachev KS; Demidov ON; Tikhonovich IA; Nizhnikov AA Accumulation of Storage Proteins in Plant Seeds Is Mediated by Amyloid Formation. PLOS Biol. 2020, 18 (7), e3000564. 10.1371/journal.pbio.3000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Van Gerven N; Van der Verren SE; Reiter DM; Remaut H The Role of Functional Amyloids in Bacterial Virulence. J. Mol. Biol 2018, 430 (20), 3657–3684. 10.1016/jjmb.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fowler DM; Koulov AV; Balch WE; Kelly JW Functional Amyloid–from Bacteria to Humans. Trends Biochem. Sci 2007, 32 (5), 217–224. 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- (7).Li Y; Zhao C; Luo F; Liu Z; Gui X; Luo Z; Zhang X; Li D; Liu C; Li X Amyloid Fibril Structure of α-Synuclein Determined by Cryo-Electron Microscopy. Cell Res. 2018, 28 (9), 897–903. 10.1038/s41422-018-0075-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cao Q; Boyer DR; Sawaya MR; Ge P; Eisenberg DS Cryo-EM Structure and Inhibitor Design of Human IAPP (Amylin) Fibrils. Nat. Struct. Mol. Biol 2020, 27 (7), 653–659. 10.1038/s41594-020-0435-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kollmer M; Close W; Funk L; Rasmussen J; Bsoul A; Schierhorn A; Schmidt M; Sigurdson CJ; Jucker M; Fändrich M Cryo-EM Structure and Polymorphism of Aβ Amyloid Fibrils Purified from Alzheimer’s Brain Tissue. Nat. Commun 2019, 10 (1), 4760. 10.1038/s41467-019-12683-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang L-Q; Zhao K; Yuan H-Y; Wang Q; Guan Z; Tao J; Li X-N; Sun Y; Yi C-W; Chen J; Li D; Zhang D; Yin P; Liu C; Liang Y Cryo-EM Structure of an Amyloid Fibril Formed by Full-Length Human Prion Protein. Nat. Struct. Mol. Biol 2020, 27 (6), 598–602. 10.1038/s41594-020-0441-5. [DOI] [PubMed] [Google Scholar]
- (11).Röder C; Kupreichyk T; Gremer L; Schäfer LU; Pothula KR; Ravelli RBG; Willbold D; Hoyer W; SchRöder GF Cryo-EM Structure of Islet Amyloid Polypeptide Fibrils Reveals Similarities with Amyloid-β Fibrils. Nat. Struct. Mol. Biol 2020, 27 (7), 660–667. 10.1038/s41594-020-0442-4. [DOI] [PubMed] [Google Scholar]
- (12).Gallardo R; Iadanza MG; Xu Y; Heath GR; Foster R; Radford SE; Ranson NA Fibril Structures of Diabetes-Related Amylin Variants Reveal a Basis for Surface-Templated Assembly. Nat. Struct. Mol. Biol 2020, 27 (11), 1048–1056. 10.1038/s41594-020-0496-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kapurniotu A Enlightening Amyloid Fibrils Linked to Type 2 Diabetes and Cross-Interactions with Aβ. Nat. Struct. Mol. Biol 2020, 27 (11), 1006–1008. 10.1038/s41594-020-00523-z. [DOI] [PubMed] [Google Scholar]
- (14).Fernandez-Escamilla A-M; Rousseau F; Schymkowitz J; Serrano L Prediction of Sequence-Dependent and Mutational Effects on the Aggregation of Peptides and Proteins. Nat. Biotechnol 2004, 22 (10), 1302–1306. 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- (15).Tartaglia GG; Pawar AP; Campioni S; Dobson CM; Chiti F; Vendruscolo M Prediction of Aggregation-Prone Regions in Structured Proteins. J. Mol. Biol 2008, 380 (2), 425–436. 10.1016/j.jmb.2008.05.013. [DOI] [PubMed] [Google Scholar]
- (16).Jacob RS; Ghosh D; Singh PK; Basu SK; Jha NN; Das S; Sukul PK; Patil S; Sathaye S; Kumar A; Chowdhury A; Malik S; Sen S; Maji SK Self Healing Hydrogels Composed of Amyloid Nano Fibrils for Cell Culture and Stem Cell Differentiation. Biomaterials 2015, 54, 97–105. 10.1016/j.biomaterials.2015.03.002. [DOI] [PubMed] [Google Scholar]
- (17).Xing Y; Sun Y; Wang B; Ding F Morphological Determinants of Carbon Nanomaterial-Induced Amyloid Peptide Self-Assembly. Front. Chem 2020, 8, 160. 10.3389/fchem.2020.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Guerette PA; Hoon S; Ding D; Amini S; Masic A; Ravi V; Venkatesh B; Weaver JC; Miserez A Nanoconfined β-Sheets Mechanically Reinforce the Supra-Biomolecular Network of Robust Squid Sucker Ring Teeth. ACS Nano 2014, 8 (7), 7170–7179. 10.1021/nn502149u. [DOI] [PubMed] [Google Scholar]
- (19).Ding D; Guerette PA; Hoon S; Kong KW; Cornvik T; Nilsson M; Kumar A; Lescar J; Miserez A Biomimetic Production of Silk-like Recombinant Squid Sucker Ring Teeth Proteins. Biomacromolecules 2014, 15 (9), 3278–3289. 10.1021/bm500670r. [DOI] [PubMed] [Google Scholar]
- (20).Ding D; Pan J; Lim SH; Amini S; Kang L; Miserez A Squid Suckerin Microneedle Arrays for Tunable Drug Release. J. Mater. Chem. B 2017, 5 (43), 8467–8478. 10.1039/c7tb01507k. [DOI] [PubMed] [Google Scholar]
- (21).Ping Y; Ding D; Ramos RANS; Mohanram H; Deepankumar K; Gao J; Tang G; Miserez A Supramolecular β-Sheets Stabilized Protein Nanocarriers for Drug Delivery and Gene Transfection. ACS Nano 2017, 11 (5), 4528–4541. 10.1021/acsnano.6b08393. [DOI] [PubMed] [Google Scholar]
- (22).Ulamec SM; Brockwell DJ; Radford SE Looking Beyond the Core: The Role of Flanking Regions in the Aggregation of Amyloidogenic Peptides and Proteins. Front. Neurosci 2020, 14, 1216. 10.3389/fnins.2020.611285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wegmann S; Eftekharzadeh B; Tepper K; Zoltowska KM; Bennett RE; Dujardin S; Laskowski PR; MacKenzie D; Kamath T; Commins C; Vanderburg C; Roe AD; Fan Z; Molliex AM; Hernandez-Vega A; Muller D; Hyman AA; Mandelkow E; Taylor JP; Hyman BT Tau Protein Liquid–Liquid Phase Separation Can Initiate Tau Aggregation. EMBO J 2018, 37 (7), e98049. 10.15252/embj.201798049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pytowski L; Lee CF; Foley AC; Vaux DJ; Jean L Liquid-Liquid Phase Separation of Type II Diabetes-Associated IAPP Initiates Hydrogelation and Aggregation. Proc. Natl. Acad. Sci. U. S. A 2020. 117 (22), 12050–12061. 10.1073/pnas.1916716117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ray S; Singh N; Kumar R; Patel K; Pandey S; Datta D; Mahato J; Panigrahi R; Navalkar A; Mehra S; Gadhe L; Chatterjee D; Sawner AS; Maiti S; Bhatia S; Gerez JA; Chowdhury A; Kumar A; Padinhateeri R; Riek R; Krishnamoorthy G; Maji SK α-Synuclein Aggregation Nucleates through Liquid–Liquid Phase Separation. Nat. Chem 2020, 12 (8), 705–716. 10.1038/s41557-020-0465-9. [DOI] [PubMed] [Google Scholar]
- (26).Dignon GL; Best RB; Mittal J Biomolecular Phase Separation: From Molecular Driving Forces to Macroscopic Properties. Annu. Rev. Phys. Chem 2020, 71 (1), 53–75. 10.1146/annurev-physchem-071819-113553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Alberti S; Gladfelter A; Mittag T Considerations and Challenges in Studying Liquid–Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176 (3), 419–434. 10.1016/j.cell.2018.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yuan C; Levin A; Chen W; Xing R; Zou Q; Herling TW; Challa PK; Knowles TPJ; Yan X Nucleation and Growth of Amino Acid and Peptide Supramolecular Polymers through Liquid–Liquid Phase Separation. Angew. Chem 2019, 131 (50), 18284–18291. 10.1002/ange.201911782. [DOI] [PubMed] [Google Scholar]
- (29).Shin Y; Brangwynne CP Liquid Phase Condensation in Cell Physiology and Disease. Science 2017, 357 (6357), aaf4382. 10.1126/science.aaf4382. [DOI] [PubMed] [Google Scholar]
- (30).Babinchak WM; Surewicz WK Liquid–Liquid Phase Separation and Its Mechanistic Role in Pathological Protein Aggregation. J. Mol. Biol 2020, 432 (7), 1910–1925. 10.1016/j.jmb.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kostylev MA; Tuttle MD; Lee S; Klein LE; Takahashi H; Cox TO; Gunther EC; Zilm KW; Strittmatter SM Liquid and Hydrogel Phases of PrPC Linked to Conformation Shifts and Triggered by Alzheimer’s Amyloid-β Oligomers. Mol. Cell 2018, 72 (3), 426–443.e12. 10.1016/j.molcel.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Singh V; Xu L; Boyko S; Surewicz K; Surewicz WK Zinc Promotes Liquid–Liquid Phase Separation of Tau Protein. J. Biol. Chem 2020, 295 (18), 5850–5856. 10.1074/jbc.AC120.013166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dignon GL; Zheng W; Kim YC; Mittal J Temperature-Controlled Liquid–Liquid Phase Separation of Disordered Proteins. ACS Cent. Sci 2019, 5 (5), 821–830. 10.1021/acscentsci.9b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Vega IE; Umstead A; Kanaan NM EFhd2 Affects Tau Liquid-Liquid Phase Separation. Front. Neurosci 2019, 13, 845. 10.3389/fnins.2019.00845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Qamar S; Wang G; Randle SJ; Ruggeri FS; Varela JA; Lin JQ; Phillips EC; Miyashita A; Williams D; Ströhl F; Meadows W; Ferry R; Dardov VJ; Tartaglia GG; Farrer LA; Kaminski Schierle GS; Kaminski CF; Holt CE; Fraser PE; Schmitt-Ulms G; Klenerman D; Knowles T; Vendruscolo M; St George-Hyslop P FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-π Interactions. Cell 2018, 173 (3), 720–734.e15. 10.1016/j.cell.2018.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Xing Y; Nandakumar A; Kakinen A; Sun Y; Davis TP; Ke PC; Ding F Amyloid Aggregation under the Lens of Liquid–Liquid Phase Separation. J. Phys. Chem. Lett 2020. 12 (1), 368–378. 10.1021/acs.jpclett.0c02567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Michaels TCT; šarić A; Curk S; Bernfur K; Arosio P; Meisl G; Dear AJ; Cohen SIA; Dobson CM; Vendruscolo M; Linse S; Knowles TPJ Dynamics of Oligomer Populations Formed during the Aggregation of Alzheimer’s Aβ42 Peptide. Nat. Chem 2020, 12 (5), 445–451. 10.1038/s41557-020-0452-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Pittman JM; Srivastava AK; Boughter CT; Venkata BS; Zerweck J; Moore PC; Smok I; Tonelli M; Sachleben JR; Meredith SC Nanodroplet Oligomers (NanDOs) of Aβ40. Biochemistry 2021, 60 (36), 2691–2703. 10.1021/acs.biochem.1c00147. [DOI] [PubMed] [Google Scholar]
- (39).Cohen SIA; Linse S; Luheshi LM; Hellstrand E; White DA; Rajah L; Otzen DE; Vendruscolo M; Dobson CM; Knowles TPJ Proliferation of Amyloid-B42 Aggregates Occurs through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci 2013, 110 (24), 9758–9763. 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Klok H-A; Lecommandoux S Supramolecular Materials via Block Copolymer Self-Assembly. Adv. Mater 2001, 13 (16), 1217–1229. . [DOI] [Google Scholar]
- (41).Cui S; Yu L; Ding J Thermogelling of Amphiphilic Block Copolymers in Water: ABA Type versus AB or BAB Type. Macromolecules 2019, 52 (10), 3697–3715. 10.1021/acs.macromol.9b00534. [DOI] [Google Scholar]
- (42).Lyubimov I; Beltran-Villegas DJ; Jayaraman A PRISM Theory Study of Amphiphilic Block Copolymer Solutions with Varying Copolymer Sequence and Composition. Macromolecules 2017, 50 (18), 7419–7431. 10.1021/acs.macromol.7b01419. [DOI] [Google Scholar]
- (43).Beltran-Villegas DJ; Jayaraman A Assembly of Amphiphilic Block Copolymers and Nanoparticles in Solution: Coarse-Grained Molecular Simulation Study. J. Chem. Eng. Data 2018, 63 (7), 2351–2367. 10.1021/acs.jced.7b00925. [DOI] [Google Scholar]
- (44).J. Holder S; Sommerdijk M, N. A. J. New Micellar Morphologies from Amphiphilic Block Copolymers : Disks, Toroids and Bicontinuous Micelles. Polym. Chem 2011, 2 (5), 1018–1028. 10.1039/C0PY00379D. [DOI] [Google Scholar]
- (45).Alexandridis P; Lindman B Amphiphilic Block Copolymers: Self-Assembly and Applications; Elsevier, 2000. [Google Scholar]
- (46).Sun Y; Wang B; Ge X; Ding F Distinct Oligomerization and Fibrillization Dynamics of Amyloid Core Sequences of Amyloid-Beta and Islet Amyloid Polypeptide. Phys. Chem. Chem. Phys 2017, 19 (41), 28414–28423. 10.1039/C7CP05695H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ding F; Dokholyan NV Discrete Molecular Dynamics Simulation of Biomolecules. In Computational Modeling of Biological Systems: From Molecules to Pathways; Dokholyan NV, Ed.; Biological and Medical Physics, Biomedical Engineering; Springer US: Boston, MA, 2012; pp 55–73. 10.1007/978-1-4614-2146-7_3. [DOI] [Google Scholar]
- (48).Faridi A; Sun Y; Okazaki Y; Peng G; Gao J; Kakinen A; Faridi P; Zhao M; Javed I; Purcell AW; Davis TP; Lin S; Oda R; Ding F; Ke PC Mitigating Human IAPP Amyloidogenesis In Vivo with Chiral Silica Nanoribbons. Small 2018, 14 (47), 1802825. 10.1002/smll.201802825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kakinen A; Xing Y; Hegoda Arachchi N; Javed I; Feng L; Faridi A; Douek AM; Sun Y; Kaslin J; Davis TP; Higgins MJ; Ding F; Ke PC Single-Molecular Heteroamyloidosis of Human Islet Amyloid Polypeptide. Nano Lett. 2019, 19 (9), 6535–6546. 10.1021/acs.nanolett.9b02771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Pellarin R; Schuetz P; Guarnera E; Caflisch A Amyloid Fibril Polymorphism Is under Kinetic Control. J. Am. Chem. Soc 2010, 132 (42), 14960–14970. 10.1021/ja106044u. [DOI] [PubMed] [Google Scholar]
- (51).Pellarin R; Caflisch A Interpreting the Aggregation Kinetics of Amyloid Peptides. J. Mol. Biol 2006, 360 (4), 882–892. 10.1016/j.jmb.2006.05.033. [DOI] [PubMed] [Google Scholar]
- (52).Pellarin R; Guarnera E; Caflisch A Pathways and Intermediates of Amyloid Fibril Formation. J. Mol. Biol 2007, 374 (4), 917–924. 10.1016/j.jmb.2007.09.090. [DOI] [PubMed] [Google Scholar]
- (53).Usov I; Mezzenga R FiberApp: An Open-Source Software for Tracking and Analyzing Polymers, Filaments, Biomacromolecules, and Fibrous Objects. Macromolecules 2015, 48 (5), 1269–1280. 10.1021/ma502264c. [DOI] [Google Scholar]
- (54).Pilkington EH; Lai M; Ge X; Stanley WJ; Wang B; Wang M; Kakinen A; Sani M-A; Whittaker MR; Gurzov EN; Ding F; Quinn JF; Davis TP; Ke PC Star Polymers Reduce Islet Amyloid Polypeptide Toxicity via Accelerated Amyloid Aggregation. Biomacromolecules 2017, 18 (12), 4249–4260. 10.1021/acs.biomac.7b01301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Proctor EA; Dokholyan NV Applications of Discrete Molecular Dynamics in Biology and Medicine. Curr. Opin. Struct. Biol 2016, 37, 9–13. 10.1016/j.sbi.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Shirvanyants D; Ding F; Tsao D; Ramachandran S; Dokholyan NV Discrete Molecular Dynamics: An Efficient And Versatile Simulation Method For Fine Protein Characterization. J. Phys. Chem. B 2012, 116 (29), 8375–8382. 10.1021/jp2114576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Favrin G; Irbäck A; Mohanty S Oligomerization of Amyloid Aβ16–22 Peptides Using Hydrogen Bonds and Hydrophobicity Forces. Biophys. J 2004, 87 (6), 3657–3664. 10.1529/biophysj.104.046839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Gazit E A Possible Role for π-Stacking in the Self-Assembly of Amyloid Fibrils. FASEB J. 2002, 16 (1), 77–83. 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- (59).Dehsorkhi A; Castelletto V; Hamley IW Self-Assembling Amphiphilic Peptides. J. Pept. Sci 2014, 20 (7), 453–467. 10.1002/psc.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Hamley IW Peptide Nanotubes. Angew. Chem. Int. Ed 2014, 53 (27), 6866–6881. 10.1002/anie.201310006. [DOI] [PubMed] [Google Scholar]
- (61).Valéry C; Artzner F; Paternostre M Peptide Nanotubes: Molecular Organisations, Self-Assembly Mechanisms and Applications. Soft Matter 2011, 7 (20), 9583–9594. 10.1039/C1SM05698K. [DOI] [Google Scholar]
- (62).Lu K; Jacob J; Thiyagarajan P; Conticello VP; Lynn DG Exploiting Amyloid Fibril Lamination for Nanotube Self-Assembly. J. Am. Chem. Soc 2003, 125 (21), 6391–6393. 10.1021/ja0341642. [DOI] [PubMed] [Google Scholar]
- (63).Tao K; Wang J; Zhou P; Wang C; Xu H; Zhao X; Lu JR Self-Assembly of Short Aβ(16–22) Peptides: Effect of Terminal Capping and the Role of Electrostatic Interaction. Langmuir 2011, 27 (6), 2723–2730. 10.1021/la1034273. [DOI] [PubMed] [Google Scholar]
- (64).Mehta AK; Lu K; Childers WS; Liang Y; Dublin SN; Dong J; Snyder JP; Pingali SV; Thiyagarajan P; Lynn DG Facial Symmetry in Protein Self-Assembly. J. Am. Chem. Soc 2008, 130 (30), 9829–9835. 10.1021/ja801511n. [DOI] [PubMed] [Google Scholar]
- (65).šarić A; Chebaro YC; Knowles TPJ; Frenkel D Crucial Role of Nonspecific Interactions in Amyloid Nucleation. Proc. Natl. Acad. Sci. U. S. A 2014, 111 (50), 17869–17874. 10.1073/pnas.1410159m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Knowles TPJ; Vendruscolo M; Dobson CM The Amyloid State and Its Association with Protein Misfolding Diseases. Nat. Rev. Mol. Cell Biol 2014, 15 (6), 384–396. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- (67).Hills RD; Brooks CL Hydrophobic Cooperativity as a Mechanism for Amyloid Nucleation. J. Mol. Biol 2007, 368 (3), 894–901. 10.1016/j.jmb.2007.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Zandomeneghi G; Krebs MRH; McCammon MG; Fändrich M FTIR Reveals Structural Differences between Native β-Sheet Proteins and Amyloid Fibrils. Protein Sci. 2004, 13 (12), 3314–3321. 10.1110/ps.041024904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Hayden EY; Teplow DB Amyloid β-Protein Oligomers and Alzheimer’s Disease. Alzheimers Res. Ther 2013, 5 (6), 60. 10.1186/alzrt226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med 2016, 8 (6), 595–608. 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Wen J; Hong L; Krainer G; Yao Q-Q; Knowles TPJ; Wu S; Perrett S Conformational Expansion of Tau in Condensates Promotes Irreversible Aggregation. J. Am. Chem. Soc 2021, 143 (33), 13056–13064. 10.1021/jacs.1c03078. [DOI] [PubMed] [Google Scholar]
- (72).Watanabe-Nakayama T; Ono K; Itami M; Takahashi R; Teplow DB; Yamada M High-Speed Atomic Force Microscopy Reveals Structural Dynamics of Amyloid B1–42 Aggregates. Proc. Natl. Acad. Sci 2016, 113 (21), 5835–5840. 10.1073/pnas.1524807113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Nakajima K; Yamazaki T; Kimura Y; So M; Goto Y; Ogi H Time-Resolved Observation of Evolution of Amyloid-β Oligomer with Temporary Salt Crystals. J. Phys. Chem. Lett 2020, 11 (15), 6176–6184. 10.1021/acs.jpclett.0c01487. [DOI] [PubMed] [Google Scholar]
- (74).Ogi H; Fukukshima M; Hamada H; Noi K; Hirao M; Yagi H; Goto Y Ultrafast Propagation of β-Amyloid Fibrils in Oligomeric Cloud. Sci. Rep 2014, 4 (1), 6960. 10.1038/srep06960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Serrano AL; Lomont JP; Tu L-H; Raleigh DP; Zanni MT A Free Energy Barrier Caused by the Refolding of an Oligomeric Intermediate Controls the Lag Time of Amyloid Formation by HIAPP. J. Am. Chem. Soc 2017, 139 (46), 16748–16758. 10.1021/jacs.7b08830. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.