

Summary
Homozygous familial hypercholesterolemia (HoFH) is an extremely rare metabolism disorder usually caused by low-density lipoprotein receptor (LDLR) mutations. LDLR genotype is commonly known to determine blood concentrations of LDL cholesterol. However, effects of LDLR genotype on holistic metabolome remain unclear. Herein, we present metabolomic, genetic, and clinical datasets from a large multi-center panel of 142 patients with LDLR-mutated HoFH. We found that true homozygotes and compound heterozygotes showed few differences in clinical and metabolomic phenotypes. Compared with defective/defective mutation carriers, patients carrying one or two null mutation showed profound alterations in clinical laboratory lipids and serum cholesterol esters, lysophosphocholines, bile acids, and amino acids. Importantly, these altered metabolites are implicated in multiple biochemical reactions and associated with LDL cholesterol. This study extends the first map of different LDLR genotypes influencing the metabolome and suggests that the small-molecule metabolites serve as potential targets to mitigate the deleterious impact of LDLR mutations on HoFH.
Subject areas: Clinical genetics, Genetics, Health sciences, Human genetics, Human metabolism
Graphical abstract
Highlights
-
•
Patients with TH-HoFH and DH-HoFH show few differences in sera metabolome
-
•
LDLR genotype-related metabolic network correlates with clinical phenotypes
-
•
Null and defective LDLR mutation carriers exhibit distinct metabolite profiles
-
•
Null LDLR mutations are associated with oxidative stress and cholesterol metabolism
Clinical genetics; Genetics; Health sciences; Human genetics; Human metabolism
Introduction
Homozygous familial hypercholesterolemia (HoFH) is an extremely rare, but devastating inborn errors of metabolism disorder (Thompson et al., 2018). HoFH is commonly caused by biallelic mutations in genes involved in the catabolism of low-density lipoprotein cholesterol (LDL-C) (Cuchel et al., 2014). These mutations lead to decreased clearance of LDL-C from blood, and consequently to extremely elevated LDL-C levels and increased risks for premature cardiovascular diseases and death (Cuchel et al., 2013, 2014). Currently, its global prevalence is estimated to be between 1:160,000 and 1:400,000 (Beheshti et al., 2020; Tromp et al., 2022). In China, it was estimated that there are between 2,000 and 5,000 individuals with HoFH, in line with a prevalence of 1:300,000 to 1:600,000 (Chen et al., 2019; Jiang et al., 2022).
More than 90% of patients with HoFH have two pathogenic variants in gene encoding low-density lipoprotein receptor (LDLR). Patients may carry two same mutated LDLR alleles (true homozygotes) or two different variants (compound heterozygotes) in either LDLR allele (Bertolini et al., 2020; Sjouke et al., 2015). The cell-surface LDLR is the key protein that maintains the level of cholesterol in hepatocytes by controlling the rate of LDL-C uptake from circulation. Different LDLR mutations might cause different impacts on LDLR protein expression and LDL-C uptake cycle. Regarding the type of LDLR pathogenic variants, two major classes based on phenotypic effects have been proposed, including null mutations and defective mutations (Cuchel et al., 2014; Santos et al., 2016). Generally, patients carrying of null LDLR mutations exhibit a more aggressively elevated LDL-C than those carrying of two defective variants (Alves et al., 2020; Raal et al., 2020).
Metabolomics has been broadly acknowledged to be the omics discipline that is closest to the phenotype (Guijas et al., 2018). High-throughput metabolomic analysis provides an unbiased and holistic view on the metabolism changes between DNA and expressed phenotypes by quantitative assessments of small-molecule metabolites (molecular weight <1500 Da) in biofluids (Argmann et al., 2016; Goncalves and Frezza, 2021). Metabolome senses DNA variations and causes perturbations in individual biochemical phenotypes that ultimately influence the phenotypic presentation in inborn errors of metabolism disorders (Argmann et al., 2016; Vernon, 2015).
Recent metabolomic studies revealed that patients with heterozygous familial hypercholesterolemia (HeFH) and LDLR−/− mice that mimic human HoFH with two LDLR null variants did not just simply form an LDLR-specific lipoprotein spectrum, but also presented a variety of significantly changed small-molecule metabolites. Of note, most of these altered metabolites are not the direct production of LDLR pathways. More importantly, these metabolic alterations were implicated in multiple biological processes and closely associated with the development of atherosclerotic cardiovascular diseases (Lauterbach et al., 2020; Li et al., 2015; Olkowicz et al., 2021; Saulnier-Blache et al., 2018). However, current knowledge of LDLR mutations-caused HoFH is limited in the datasets of clinical laboratory and genetic tests; information on metabolomic effects of LDLR genotypes are still lacking.
We hypothesized that the different LDLR genotypes that result in heterogeneous clinical phenotypes might also render an altered metabolomic phenotypes in patients with HoFH. Therefore, in the present study, we performed a comprehensive metabolome analysis to examine the serum metabolome map of 142 patients with HoFH with different LDLR genotypes. We sought to investigate the potential links between LDLR genotypes and metabolomic phenotypes and infer potential molecular mechanisms underlying the association between LDLR genotypes and clinical phenotypes of unknown origin in HoFH.
Results
Demographic and clinical characteristics of study subjects
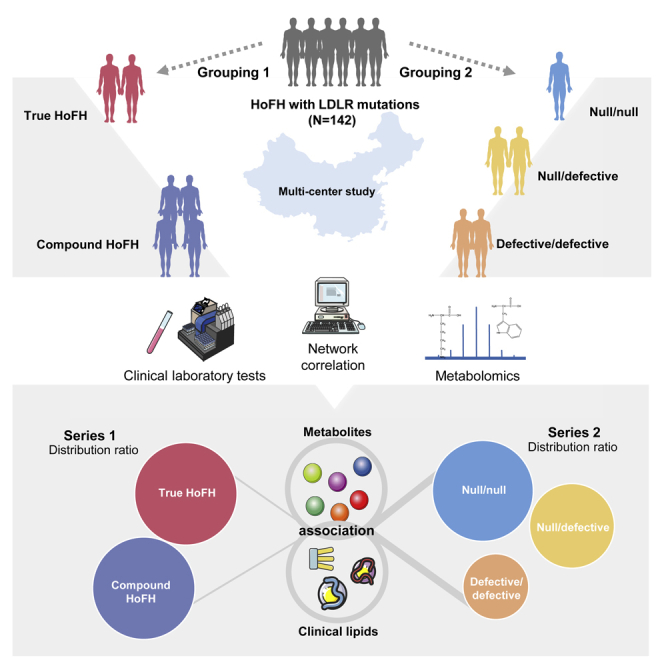
A total of 142 patients with HoFH with genetically identified LDLR gene mutations participated in the study. The detailed characteristics of LDLR mutations were summarized in Table S1. According to the classical categories in HoFH population, all patients were firstly subdivided into true homozygous group (TH: n = 33; ages = 23.8 ± 8.1; male, 48.5%) and compound heterozygous group (CH: n = 109; ages = 20.2 ± 13.1; male, 51.4%). Then, we grouped the participants into three groups according to the LDLR status as follows: defective/defective group (n = 58; ages = 21.4 ± 12.4; male, 51.7%), null/defective group (n = 61; ages = 20.5 ± 12.7; male, 51.0%), and null/null group (n = 23; ages = 22.1 ± 9.6; male, 47.8%). The detailed grouping categories and clinical characteristics of study patients were depicted in Figure 1A and summarized in Table 1.
Figure 1.

Study design and multivariate statistical pattern analysis of metabolomic data
(A) Grouping design of the study patients.
(B) Treemap overview for the category distribution of identified metabolites.
(C and D) PCA and PLS-DA scores plot of the metabolomic data from serum samples of patients with TH and CH; each point represents an individual serum sample.
(E) PLS-DA permutations plot of metabolomic data of TH and CH groups; each green point and black point represents R2-value and Q2-value, respectively. The criteria for PLS-DA model validity: All Q2-values to the left are lower than the original points to the right; The left regression line of the Q2-points is below zero; all R2-values to the left are lower than the original point to the right.
(F–H) PCA scores plot, PLS-DA scores plot, PLS-DA permutations plot of the metabolomic data from defective/defective, null/defective, and null/null groups.
Table 1.
Demographic and clinical characteristics of all subjects
| Total (n = 142) | True homozygosity (n = 33) | Compound heterozygosity (n = 109) | P | Defective/Defective (n = 58) | Null/Defective (n = 61) | Null/Null (n = 23) | P | |
|---|---|---|---|---|---|---|---|---|
| Ages | 21.1 ± 12.1 | 23.8 ± 8.1 | 20.2 ± 13.1 | 0.14 | 21.4 ± 12.4 | 20.5 ± 12.7 | 22.1 ± 9.6 | 0.85 |
| Male sex, n (%) | 72, (50.1%) | 16, (48.5%) | 56, (51.4%) | 0.77 | 30, (51.7%) | 31, (51.0%) | 11, (47.8%) | 0.95 |
| Hypertension, n (%) | 4, (2.8%) | 1, (3.0%) | 3, (2.8%) | 0.93 | 2, (3.4%) | 2, (3.3%) | 0, (0.0%) | 0.67 |
| CVD, n (%) | 42, (30.3%) | 8, (24.2%) | 35, (32.1%) | 0.39 | 15, (26.9%) | 18, (29.5%) | 10, (43.5%) | 0.29 |
| LDL-C, mmol/L | 10.55 ± 3.63 | 10.35 ± 2.82 | 10.61 ± 3.86 | 0.72 | 8.75 ± 3.39 | 11.36 ± 3.43 | 12.96 ± 2.52 | <0.0001 |
| TC, mmol/L | 12.47 ± 3.67 | 12.21 ± 3.07 | 12.55 ± 3.84 | 0.65 | 10.68 ± 3.49 | 13.20 ± 3.31 | 15.06 ± 2.84 | <0.0001 |
| TG, mmol/L | 0.97 [0.69, 1.41] | 1.11 [0.59, 1.48] | 0.95 [0.71, 1.40] | 0.78 | 0.90 [0.61, 1.24] | 0.82 [0.69, 1.59] | 1.30[1.11, 1.48] | 0.0025 |
| HDL-C, mmol/L | 0.85 ± 0.38 | 0.91 ± 0.36 | 0.83 ± 0.34 | 0.31 | 0.93 ± 0.31 | 0.81 ± 0.41 | 0.74 ± 0.45 | 0.077 |
| APOB, g/L | 2.47 ± 0.64 | 2.47 ± 0.56 | 2.48 ± 0.67 | 0.94 | 2.20 ± 0.64 | 2.60 ± 0.54 | 2.89 ± 0.43 | <0.0001 |
| LP(a), mg/dL | 41.0 [21.0, 75.0] | 40.0 [24.1, 57.0] | 43.1 [18.0, 77.2] | 0.44 | 44.9 [17.6, 65.8] | 41.0 [23.0, 76.3] | 33.4 [16.0, 72.3] | 0.56 |
| Statin alone | 9, (6.3%) | 2, (6.1%) | 7, (6.4%) | 0.94 | 6, (10.3%) | 2, (3.3%) | 1, (4.3%) | 0.26 |
| Statin + Probucol | 22, (15.5%) | 6, (18.1%) | 16, (14.7%) | 0.63 | 11, (19.0%) | 8, (13.1%) | 3, (13.0%) | 0.64 |
| Statin + Ezetimibe | 111, (78.2%) | 25, (75.8%) | 86, (78.9%) | 0.70 | 41, (70.7%) | 51, (83.6%) | 19, (82.7%) | 0.20 |
Continuous data are presented as mean ± SD or median [interquartile range], categorical variables are presented as %. Two-tailed Student’s t test or Mann Whitney U test were used for continuous data in comparison of true homozygosity and compound heterozygosity. One-way ANOVA or Kruskal Wallis test was used for continuous data in comparison of defective/defective, null/defective, and null/null groups. The Chi-square test was used for categorical data. CVD, cardiovascular disease; LLT, lipid-lowering therapy; LDL-C, low-density lipoprotein cholesterol; TC, total cholesterol; TG, triglycerides; HDL-C, high-density lipoprotein cholesterol; APOB, apolipoprotein B; Lp(a), lipoprotein (a).
Hypertension and cardiovascular disease (CVD) were recorded in four and forty-two subjects, respectively. Although no significant difference was observed in the prevalence of CVD in each subgroup comparison, the results demonstrated that patients with LDLR null/null mutations (43.5%) exhibited a higher trend in the prevalence of CVD than those with null/defective (29.5%) and defective/defective (26.9%) mutations. No significant difference was observed in the lipid-lowering treatments (LLT) and the concentrations of clinical laboratory lipids between TH and CH groups. In addition, there was also no significant difference in LLT status among the defective/defective, null/defective, and null/null groups. Null/null group had the highest levels of LDL-C, total cholesterol (TC), triglycerides (TG), and apolipoprotein B (APOB). This is followed by null/defective and defective/defective groups (Table 1).
Metabolomic phenotypes of true homozygotes and compound heterozygotes
Using UPLC-QTOF/MS, we identified 808 circulating metabolites in the serum of study patients, with 245 metabolites being annotated by chemical standards. The relative standard derivations (RSD) of the distribution for the identified metabolites in the quality control (QC) samples are shown in Figure S1A. A total of 696 metabolites with less than 20% RSD of peak intensity were included for unsupervised principal component analysis (PCA). The resultant PCA scores plot demonstrated that the QC samples were clustered closely (Figure S1B), indicating that the proposed metabolomic approach was robust and reproducible for further analysis. The treemap-based distribution of identified metabolites into each metabolic category was as follows (Figure 1B): phospholipids (17.4%), fatty acyls (9.3%), lysophospholipids (9.1%), carbohydrates (9.5%), amino acid and its derivatives (7.8%), organic acids (6.9%), nucleotides (6.3%), triacylglycerols (4.7%), sphingolipids (3.4%), bile acids (4.0%), carnitines (2.6%), cholesterol esters (1.9%), benzenoids (1.7%), and indoles (1.6%).
To investigate the potential differences in the sera metabolome phenotypes of patients with HoFH with TH and CH, both unsupervised and supervised multivariate statistical analysis (MVA) models were performed. By using the datasets of all identified 696 serum metabolites, the unsupervised PCA score plot showed unsatisfactory classifications between TH and CH groups (Figure 1C). Furthermore, the supervised partial least squares discriminate analysis (PLS-DA) scores plot and permutations plot (Figures 1D and 1E) also demonstrated a poor separation between TH and CH groups and indicated a lower explanatory and predictive ability (R2 = 0.137; Q2 = 0.105; CV-ANOVA p value = 0.0789). These results suggested that the overall metabolite profiles could not discriminate patients with HoFH with TH and DH.
LDLR null mutations associated with significant metabolomic changes
We further investigated whether different LDLR functional status (carriers of null mutations or defective mutations) could cause prominent changes in the sera metabolome of patients with HoFH. By using the overall datasets of 696 metabolites, unsupervised PCA of was firstly performed to obtain an overview of metabolomic phenotypes among the three groups. As shown in Figure 1F, PCA score plots showed a partial discrimination between the null/null, null/defective, and defective/defective groups. A distinct group separation was achieved by supervised PLS-DA scores plot (Figure 1G) with an excellent fitness, as evidenced by higher values of R2, Q2 (R2 = 0.432; Q2 = 0.484), and significant CV-ANOVA p value (<0.0001). A satisfactory predictability without overfitting was also highlighted in the permutations plot (all Q2 and R2 values are lower than the original points, Figure 1H). These results indicated that patients with HoFH carrying different functional LDLR mutations (null or defective) exhibited a significant heterogeneity in the serum metabolome.
To determine which metabolomic changes are mainly associated with LDLR functional status, we employed multivariate variable importance projection (VIP) plot and univariate volcano plot analyses to identify the discriminatory metabolites contributing to the separations among null/null, null/defective, and defective/defective groups (Figures 2A and S1C). A total of 41 differentially expressed metabolites among three groups were identified and summarized in Table S2. Notably, most of the differentially expressed metabolites between null/null and defective/defective groups were also significantly altered between null/defective and defective/defective groups.
Figure 2.

Metabolomic signatures associated with LDLR functional status
(A) VIP plots of each metabolite variation in pairwise comparisons of defective/defective, null/defective, and null/null groups; the dashed lines indicate the threshold for the significant differences.
(B) Hierarchical clustering heatmap of LDLR status-associated metabolite intensities in different groups.
(C) PCA scores plot of the differentiated metabolites; each point represents an individual sample. CE = cholesterol esters, TAG = triacylglycerides, LysoPC = lysophosphocholines, LysoPC O = alkyl LysoPC, LysoPE O = alkyl lysophosphoethanolamines, LysoPA = lysophosphatidic acid, LPI = lysophosphatidylinositol.
Using the normalized levels of these differentiated metabolites, the cluster trends between groups were visualized by hierarchical clustering heatmap and PCA score plot (Figures 2B and 2C). From the resultant graphs, it was evident that the null/null and null/defective groups were clustered nearby and were distinct from defective/defective group. Patients carrying null/null variants exhibited the most aggressive phenotype in the perturbed sera levels of free cholesterol, cholesterol esters (CE), bile acids, triacylglycerides (TAG), lysophosphocholines (LysoPC), fatty acids, long-chain carnitine, purines, and amino acids (as shown in Figure 2B and Table S2). The differential metabolites obtained from the comparison of null/null group versus defective/defective group and null/defective group and defective/defective group exhibited significant enrichments in various identical metabolic pathways, mainly including fatty acid metabolism, betaine metabolism, retinol metabolism, steroid metabolism, multiple phospholipids metabolism, and amino acids metabolism (Figure 3A).
Figure 3.

Metabolic pathway enrichment and regression analyses
(A) Pathway enrichment analysis of the differentiated metabolites that distinguished the study groups; the bubble size refers to the enrichment factor of the pathway and the color represents the natural log of the pathway p value.
(B) Correlation plot depicting the relation of each metabolite and LDLR status; positive or negative β-coefficient values indicate a positive or negative correlation between metabolites and LDLR status (at least LDLR null mutation: null/null or null/defective), FDR-adjusted p < 0.05 was considered as significant threshold, n.s. represents no significance; Model 1: Adjustments for ages, gender, and CVD history; Model 2: Adjustments for all variables in model 1 and clinical lipids (TC, TG, LDL-C, HDL-C, Lp(a), and APOB). NN = null/null, DD = defective/defective, ND = null/defective. Other abbreviations are seen in Figure 2.
Regression analyses of LDLR null mutations-associated metabolite signatures
Our results indicated that carriers of one or two null LDLR variants might cause profound sera metabolome perturbations than two defective LDLR variants carriers (Figure 2B and Table S2). To further validate and confirm the associations between metabolomic signatures and null LDLR mutations, regression analyses were calculated using multiple covariates adjustments.
We firstly constructed the regression analyses adjusted for the well-known important modifiers in metabolome changes, including ages, gender, and CVD (Model 1, Figure 3B). The results demonstrated that 40 in 41 analytes (exception of TAG 52:2) demonstrated significant associations with at least one LDLR null mutation (p < 0.05), indicating that the associations between the identified metabolite signatures and null LDLR mutations were independent of the commonly known metabolic factors. After stepwise adjustment for all tested clinical lipids (Model 2, Figure 3B), a broad array of metabolites (25 in 41 metabolites) still remained significantly associated with LDLR null mutations, mainly including histidine, hypoxanthine, lysine, purine, serine, taurine, phenylalanine, methionine, glutamine, betaine, retinoic acid, glycocholic acid, linoleic acid, palmitic acid, stearoyl-carnitine, two CE species, two TAG species, and several lysophospholipids. These results indicated that LDLR-null mutations might also cause a variety of metabolome alterations, in addition to the well-known LDLR pathway-altered lipoproteins, such as LDL-C and APOB.
Metabolomic signatures network correlates with phenotypic alterations in clinical lipids
As described above, patients carrying one or two LDLR null mutations showed remarkable differences in the circulating levels of clinical lipids and sera metabolome compared with two defective LDLR mutations carriers. To further investigate the association between altered metabolites and clinical lipid features, correlation analysis was calculated using Spearman’s rank correlation coefficients and debiased sparse partial correlation (DSPC) network. The resultant Spearman’s rank correlogram (Figure 4A) demonstrated that the free cholesterol, retinoic acid, palmitic acid, palmitoyl-carnitine, stearoyl-carnitine, betaine, phenylalanine, bile acids, CE species, and LysoPC species were positively associated with LDL-C, TC, and APOB, whereas lysine and histidine were negatively associated with these clinical lipid markers.
Figure 4.

Differential metabolites correlated with clinical lipids
(A) Spearman’s rank correlation heatmap depicting the relationship of each LDLR status-associated metabolites with clinical lipids. Positive correlations are displayed in red and negative correlations in blue. Color intensity and size of the circle are proportional to the Spearman correlation coefficients. In this correlogram, correlations with p < 0.05∗ or p < 0.01∗∗ are considered significant.
(B) Debiased sparse partial correlation network plot visualizing the major relationships of clinical lipids and key metabolites; circles represent metabolites, squares represent clinical lipids, the solid lines indicate positive correlation, the dashed lines indicate negative correlation, the thickness of the edges represents the strength of the partial correlation coefficients (|coefficients| ranges from 0.5 to 1.0). Abbreviations are seen in Figure 2.
As metabolites act in a coordinated manner and not in isolation, an association network between metabolites and the clinical lipids could provide valuable insights into the underlying processions. As shown in Figure 4B, the DSPC network was dominated mainly by retinoic acid, palmitoyl-carnitine, stearoyl-carnitine, cholesterol, betaine, phenylalanine, lysine, histidine, CE species, LysoPC species, and 12α-hydroxylated primary bile acids (cholic acid, taurocholic acid, and glycocholic acid). They were significantly correlated with each other. In line with the Spearman’s rank coefficients (Figure 4A), most of these altered metabolites were also positively correlated with TC, LDL-C, and APOB.
Discussion
LDLR genotype is the strongest prevalent genetic risk factor for HoFH and plays determined roles in the sera concentrations of LDL-C (Banerjee et al., 2019; Zhao et al., 2020). Different pathogenic mutations in LDLR gene may affect LDLR protein expression and activity, resulting different uptake rates of LDL-C from circulation (Santos et al., 2016; Zhao et al., 2020). Besides the limited information on the circulating levels of LDL-C, the impact of LDLR genotype on holistic metabolism in human HoFH remains largely unknown. To our knowledge, the current study marks the first metabolomic analysis that compares sera metabolome of patients with HoFH with different LDLR genotypes and identifies previously unknown LDLR genotypes-associated metabolomic signatures and pathways. The associations of metabolome alterations with LDLR genotypes and clinical laboratory alterations provide potential molecular targets and may help in giving future directions for discovering additional therapeutical strategies in patients with HoFH, specifically null LDLR variants carriers.
As previously described mostly in European, Iberoamerica, and North American populations (Alves et al., 2020; Luirink et al., 2019; Raal et al., 2016; Sanchez-Hernandez et al., 2016), patients with TH carrying two identical LDLR mutations showed more severe hypercholesterolemia than patients with DH carrying two differential LDLR mutations. Recent studies within a smaller number of Chinese patients with HoF also revealed that individuals with TH showed higher untreated-LDL-C levels than individuals with CH (Jiang et al., 2022). Interestingly, the present revealed that the patients with TH and patients with CH who had received similar LLT exhibited few differences in the levels of all tested clinical lipids and sera metabolome phenotype (Table 1 and Figures 1C and 1D). The pathogenic status of LDLR mutations plays critical roles in determining the circulating cholesterol levels (Sniderman et al., 2022). Patients with TH may carry two LDLR defective mutations (residual LDLR function ranging from 2% to 25%), whereas subjects that are CH can bear one or two LDLR null mutations (residual LDLR function <2%) (Alves et al., 2020; Raal et al., 2020). Patients with HoFH carrying one or two LDLR null mutations may show a higher level of circulating LDL-C and TC than those carrying two LDLR defective mutations (Drouin-Chartier et al., 2017). Thus, the observed phenotypes of clinical lipids between the TH and CH that differed from previous reports may be explained by the LDLR mutation types and LLT status of the study subjects.
Another important finding of our study was that different LDLR functional mutations could lead to remarkable differences in the clinical and metabolomic phenotypes of patients with HoFH. Patients carrying two LDLR null mutations had the highest serum levels of LDL-C, TC, APOB, and TG and highest prevalence of CVD. This is followed by those with LDLR null/defective mutations and those with two LDLR defective mutations. In the view of metabolome, carriers of one or two LDLR null mutations also exhibited more aggressive phenotype in the elevation of free cholesterol, retinoic acid, primary bile acids, long-chain carnitines, CE species, and LysoPC species than those with two defective LDLR mutations. Importantly, these metabolomic signatures were mapped to multiple metabolic pathways and significantly correlated with the levels of HoFH-related clinical lipids, including LDL-C, TC, and APOB.
CE and LysoPC are the major basic elements of LDL-C (Luo et al., 2020; Zhou et al., 2019). Consistent with the discriminating capabilities of LDL-C in distinguishing patients with null/null, null/defective, and defective/defective mutations (Table 1), a variety of CE and LysoPC species in serum samples also showed significant discriminating power (Table S2). In addition, the correlation and network analyses demonstrated that these altered small-molecular lipids were positively correlated with the direct metabolic markers in the defected LDLR pathway, including LDL-C, TC, and APOB (Figure 4). Previous evidence indicated an intricate connection between CE biosynthetic pathways and LysoPC metabolism. CE can be produced by transfer of a fatty acid from the phosphatidylcholines to cholesterol, resulting in the formation of LysoPC (Claria et al., 2021; Huang et al., 2020). Furthermore, previous studies also revealed that LysoPC administration could also induce a significant elevation of cholesterol in LDLR−/− mice, and induce cholesterol biosynthesis in macrophages (Cha et al., 2018; Mukherjee et al., 2018). Thus, the increased circulating LysoPC in the sera of patients with HoFH with LDLR null mutations might also promote cholesterol-related small-molecular lipids and lipoprotein accumulation.
LysoPC is also known as the major element and hydrolyzed production of LDL-C and oxidized LDL-C, which is a useful marker of oxidative stress in hypercholesterolemic subjects (Kim et al., 2020). In addition, this study also demonstrated several oxidative stress-associated hydrophilic metabolites were associated with LDLR genotypes. We observed patients with HoFH with one or two null mutations expressed lower sera levels of methionine, taurine, histidine, and glutamine than those carrying two defective mutations. These metabolites have been reported to act as the endogenous free radical scavenger for reactive oxygen species (Afolabi et al., 2022; Chen et al., 2012; Watanabe et al., 2008). Interestingly, the decreased plasma level of taurine, methionine, and glutamine was also observed in the comparisons of LDLR null/null mice versus wild-type mice and patients with HeFH versus normolipidemic subjects (Olkowicz et al., 2021). Furthermore, we also identified the elevation of serum hypoxanthine and purine was positively with null LDLR mutations. Hypoxanthine is a downstream product in the purine degradation pathway. The elevated hypoxanthine has been demonstrated to be a marker of oxidative stress (Irvine et al., 2022). These evidences suggest that declined metabolic antioxidants and elevated purines reflect a more severe oxidative stress condition in null LDLR mutations carriers comparing to subjects carrying defective mutations.
It is well documented that LDLR gene and its regulated pathway play a critical role in maintaining cholesterol homeostasis in hepatocytes by controlling the rate of cholesterol uptake from circulating LDL-C (Santos et al., 2016; Zhao et al., 2020). Interestingly, the present study demonstrated that several LDLR genotype-associated metabolites were also implicated in the routes of cholesterol absorption and excretion. In patients with one or two LDLR null mutations, the sera levels of retinoic acid and three 12α-hydroxylated bile acid species (including cholic acid, glycocholic acid, and taurocholic acid) were significantly higher than those with two LDLR defective mutations. Retinoic acid is a functional derivative of retinol which plays a vital role in regulating the cholesterol absorption genes expression and 12α-hydroxylated bile acid synthesis (Hong et al., 2021; Pathak et al., 2013). 12α-hydroxylated bile acid, the major products in the cholesterol excretion route, can also promote cholesterol absorption and increase hepatic and plasma cholesterol. Inhibition of 12α-hydroxylated bile acid synthesis could significantly prevent hypercholesterolemia in mice (Semova et al., 2022; Zurkinden et al., 2020). These evidences suggested new insights into the additional signaling routes of LDLR in regulating cholesterol homeostasis.
Limitations of the study
Some limitations warrant discussion. It is an observational cross-sectional clinical study that cannot accurately establish causality of the observed associations. Functional assessments were not available for all LDLR mutations identified in the present study. Future studies on determining the activity of LDLR mutations and investigating the metabolite signatures in purified LDL-C will offer great promise for a more comprehensive understanding of the effects of LDLR genotypes on HoFH to establish mechanism-based precise therapy. Nevertheless, it is a large-scale and multi-center study investigating over 140 individuals with LDLR-mutated HoFH with advanced metabolomic approaches, providing new metabolism information which is beyond existing knowledge of LDLR genetic effects on this orphan disease. The collective evidence here suggests a possible role of small-molecular metabolites as intervention point to mitigate the impact of LDLR pathogenic mutations in the pathophysiology progression of HoFH.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Serum samples | Beijing Anzhen Hospital | https://www.anzhen.org/ |
| Chemicals, peptides, and recombinant proteins | ||
| LC-MS grade acetonitrile | Fisher Scientific | Cat # A955-4 |
| LC-MS grade methanol | Fisher Scientific | Cat # A456-4 |
| LC-MS grade formic acid | Fisher Scientific | Cat # A117-50 |
| L-phenyl-d5-alanine | Sigma-Aldrich | Cat # 615870 |
| L-leucine-5, 5, 5-d3 | Sigma-Aldrich | Cat # 486825 |
| Stearic acid-18, 18, 18-d3 | Sigma-Aldrich | Cat # 490393 |
| Cholic acid-2, 2, 4, 4-d4 | Sigma-Aldrich | Cat # 903809 |
| L-arginine-d7 | Cayman Chemical | Cat # 34834 |
| PC (18:0/20:4)-d11 | Cayman Chemical | Cat # 27928 |
| Stearoyl-L-carnitine-d3 | Cayman Chemical | Cat # 26580 |
| Cholesterol-d7 | Cayman Chemical | Cat # 25265 |
| LysoPC (19:0)-d5 | Avanti Polar Lipids, Inc. | Cat # 855778 |
| Critical commercial assays | ||
| TC | Tellgen Corporation. | BH016Z |
| TG | Tellgen Corporation. | BH017Z |
| APOB | Tellgen Corporation. | BH021Z |
| HDL | Tellgen Corporation. | BH018Z |
| LDL | Beijing Strong Biotechnologies, Inc. | EGS141Z |
| Lp(a) | Beijing Strong Biotechnologies, Inc. | GM9151Z |
| Deposited data | ||
| LC-MS raw data | This paper | https://service.most.gov.cn/ |
| Software and algorithms | ||
| Progenesis QI software | Waters, Manchester, U.K. | https://www.waters.com/waters/en_US/Progenesis-QI-Software |
| SIMCA-P software | Umetric, Umeå, Sweden | https://www.sartorius.com/ |
| SPSS Statistics | IBM Corp, New York, USA | N/A |
| MetaboAnalyst | https://www.metaboanalyst.ca | N/A |
| Bioinformatics platform | http://www.bioinformatics.com.cn/login/ | N/A |
Resource availability
Lead contact
Further information and requests for reagent and resource may be directed to and will be fulfilled by the lead contact, Prof. Yanwen Qin (qinyanwen@vip.126.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human subjects
A total of 142 subjects (ages 21.1 ± 12.1, male sex 50.1%) were enrolled from Beijing Anzhen Hospital, Peking University First Hospital, and Suzhou Municipal Hospital between June 2015 and December 2021. This study complies with the Declaration of Helsinki and was approved by the Ethics Committee of Beijing Anzhen Hospital of the Capital University of Medical Sciences and Peking University First Hospital. Verbal and written consent was obtained from all subjects.
Method details
HoFH diagnosis and study design
Criteria for the diagnosis of HoFH were as follows: An untreated LDL-C ≥ 13 mmol/L or a treated LDL-C ≥ 8 mmol/L, either cutaneous or tendon xanthomas before the age of 10 years, together with two mutant alleles at LDLR, APOB, PCSK9, or LDLRAP1 genes(Wu et al., 2021). Genotyping was obtained from patients if it had been performed or tested according to our previously reported methods (Jiang et al., 2017, 2022). Only subjects with two mutant alleles at LDLR gene were included in this study.
All study patients were further divided into true homozygous (carriers of two identical pathogenic variants of LDLR) and compound heterozygous (carriers of two different pathogenic variants in either LDLR allele). Assignment of functional status of LDLR variants were determined as previously described and classified as either null or defective according to previous in vitro functional experiments, the National Center for Biotechnology Information ClinVar database, LDLR-specific Leiden Open Variation database, and predicted dysfunctional effects of variants in silico analysis (Alves et al., 2020; Jiang et al., 2017; Thompson et al., 2018): All LDLR variants with less than 2% activity verified by in vitro functional assays, nonsense, large deletions, and frameshift variants were considered to be null variants. All other variants were classified as defective. The pathogenicity classification of variants was annotated by using ClinVar database, LDLR-specific Leiden Open Variation database, and MLb-LDLr software (Larrea-Sebal et al., 2021).
CVD history for the subjects were defined as a composite of myocardial infarction, coronary and carotid revascularization, ischemic or atherothrombotic stroke, premature carotid artery disease, peripheral artery disease, and supra-aortic valve disease. Hypertension was defined as a condition in individuals with mean office systolic blood pressure (SBP) ≥ 140 mmHg or diastolic blood pressure (DBP) ≥ 90 mmHg (three consecutive measurements at 5-min intervals), or a condition in those who underwent antihypertensive agent therapy with a defined history of hypertension.
Blood sample collection, transportation, and preservation
Blood sample was collected by trained technologists following a uniform standard guideline: After signing the informed consent, we requested that the participants to record the time of dinner and maintain an overnight fasting state of 10–12 h before blood collection in the next day. Blood sample was collected from the antecubital vein of patients. All blood samples were collected into BD vacutainer serum separator tube containing micronized silica particles (clot activators) and a polymer gel separator to obtain serum. All vacutainer were filled to capacity and were inverted five times immediately after blood drawing to mix the clot activators with the whole blood. Serum was obtained after clotting for 30 min at room temperature followed by centrifugation at 1300 ×g for 20 min. Finally, three serum aliquots of 200 μL were transferred into new plastic test tubes with no additives and stored at − 80°C. The processing time, collection location, sample size, storage temperature, and technologist names for the blood collection were clearly recorded in the three hospitals. All serum samples were transported to Metabolomics Analysis Platform, State Key Laboratory of Natural and Biomimetic Drugs, Peking University by using ultra-low temperature cold chain. Prior to metabolomic analyses, all samples were separated by centrifugation at 3000×g for 20 min. The serum levels of LDL-C, TC, TG, APOB, Lp(a), and HDL-C were determined using an automatic biochemistry analyzer (Beckman AU 5400, Brea, USA).
Metabolomic analyses
Metabolomics analysis was performed as described previously (Du et al., 2021). Briefly, the metabolites were extracted as follows: 50 μL serum was extracted by 4-fold volume of ice-cold acetonitrile: water (1:1, v/v) containing internal standards (0.012 mg/mL L-phenyl-d5-alanine, 0.014 mg/mL L-arginine-d7, 0.10 mg/mL stearic acid-18, 18, 18-d3, 0.09 mg/mL L-leucine-5, 5, 5-d3, 0.10 mg/mL LysoPC (19:0)-d5, 0.04 mg/mL PC(18:0/20:4)-d11, 0.04 mg/mL cholesterol-d7, 0.1 mg/mL stearoyl-L-carnitine-d3, and 0.05 mg/mL cholic acid-2, 2, 4, 4-d4). The mixture was vortexed and centrifuged at 14,500 rpm for 10 min at 4°C. Then, 150 μL supernatant was transferred into a clean dry tube and evaporated to dryness. The dried residue was stored at −80°C. Following the above protocol, QC samples were prepared by mixing equal aliquots from each sample.
Metabolomic profiling was performed on a high-resolution UPLC-SYNAPT Xevo-G2 XS Q-TOF/MS system (Waters Corporation, Milford, USA). The dried residue was reconstituted in 100 μL of water: methanol (1:1, v/v) solution. Metabolites separations were performed on an Acquity UPLC BEH C18 column (2.1 mm × 100 mm, 1.7 μm, Waters Corp., Milford, USA). The column temperature was maintained at 35°C and the flow rate remained constant at 0.4 mL/min. The optimal mobile phase consisted of a linear gradient system of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B): 0–4.0 min, 2–20% B; 4.0–7.0 min, 20–60% B; 7.0–9.0 min, 60% B; 9.0–12.5 min, 60–90% B; 12.5–14.0 min, 90–100% B; 14.0–16.0 min, 100% B; 16.0–19.0 min, 0% B. Mass acquisition was performed on positive and negative ion modes. The capillary voltage was 3.0 kV for positive mode and 2.3 kV for negative model. The source temperature was set at 110°C. The sampling cone voltage and cone gas rate were set at 35V and 50 L/h, respectively. The desolvation gas temperature and desolvation gas flow were 450°C and 600 L/h, respectively. All analyses were acquired using a LockSpray interface to ensure the accuracy and reproducibility. Data was collected in centroid mode from 50 to 1100 Da. The collision energy parameters were ranging from 5 to 60 eV. The QC sample was injected every 6 real samples throughout the entire experiment.
Quantification and statistical analysis
Metabolomic data processing
All the LC-MS acquired data was transformed to Progenesis QI software (Waters, Manchester, U.K.) for deconvolution, alignment, and data reduction by using the QC samples. The MS1 and MS2 spectra-based metabolite identification was performed by using QI MetaScope database, METLIN database, HMDB database, LIPIDMAPS database, and in-house metabolite library. The semi-quantitative intensity was calculated by using the isotope-labeled internal standards. The identified metabolites that were absent in more than 10% of all QC samples together with those with more than 20% relative SD in peak intensity across QC samples were removed. Subsequently, a list of normalized data matrix including information of identified metabolite names, precursor ions, fragment ions, neutral molecules, retention time, and normalized intensity was generated for further analysis. The MVA for the data matrix was performed using SIMCA-P software (v14.1, Umetric, Umeå, Sweden). Unsupervised PCA was employed to explore the clustering trends among different groups. The quality of PCA models was controlled by evaluating the R2 and Q2 values. Supervised PLS-DA was performed to maximally discriminate groups and to compute the variable importance projection (VIP) score plot for each metabolite. The quality of PLS-DA model was controlled by R2, Q2, permutation plot (200 permutations), and cross validation ANOVA(CV-ANOVA; significance threshold ≤0.05). VIP value >2.0 and Mann Whitney U test p < 0.05 after false discovery rate (FDR) were used as the thresholds to identify differentially expressed metabolites between groups. Hierarchical clustering heatmap analyses of metabolite features were performed by using TBtools software v1.082.
Statistics and pathway enrichment analysis
Categorical clinical variables were summarized by frequency (n) and percentages (%) and compared using Chi-square test or Fisher exact test. Continuous and non-normally distributed variables were presented by mean and SD(means ± SD) and expressed by medians and interquartile ranges [IQR], respectively. The unpaired two-tailed Student’s t test and Mann Whitney U test were used for the comparisons of normally distributed data and non-normally distributed data, respectively. The associations between LDLR status and metabolites were performed by regression analyses using the sum of standardized metabolite values (z-scores) weighted according to the value of their corresponding β-coefficients(Lin et al., 2020). Relationships between clinical lipids and metabolic variables were calculated using Spearman’s rank correlation coefficient. All analyses were performed by using SPSS Statistics 26 (IBM Corp, New York, USA) and bioinformatics platform (http://www.bioinformatics.com.cn/login/). Debiased Sparse Partial Correlation algorithm was used for estimating partial correlation networks of metabolites and clinical lipids by using MetScape (Basu et al., 2017). The quantitative metabolic pathway enrichment analysis was derived by using MetaboAnalyst (http://www.metaboanalyst.ca/).
Additional resources
This study is registered with www.chictr.org.cn/index.aspx, number: ChiCTR1900022156.
Acknowledgments
This work was supported by National Key Research and Development Program of China (Grant Nos. 2021YFC2500600, 2021YFC2500603, 2021YFC2500602, 2021YFC2500601), National Natural Science Foundation of China (Grant Nos. 82100295, 81970224), and State Key Laboratory of Natural and Biomimetic Drugs, Peking University (Grant Nos. K202227).
Author contributions
Y.W.Q. and L.Y.W. conceived and designed the study. Y.W.Q., L.Y.W., Y.Y., and J.P.L. made clinical diagnosis, recruited subjects. Z.Y.D., Y.W., and F.L. collected samples and clinical phenotypes. L.J. performed genetic phenotypes. Z.Y.D. and L.Y.L. performed metabolomics and data analysis. Z.Y.D., L.Y.W., and Y.W.Q. wrote and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: November 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105334.
Contributor Information
Luya Wang, Email: wangluya@126.com.
Yanwen Qin, Email: qinyanwen@vip.126.com.
Supplemental information
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. Raw metabolomic data has been deposited at https://service.most.gov.cn/ and is available from the lead contact and National Key Research and Development Program of China upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Afolabi O.A., Anyogu D.C., Hamed M.A., Odetayo A.F., Adeyemi D.H., Akhigbe R.E. Glutamine prevents upregulation of NF-kB signaling and caspase 3 activation in ischaemia/reperfusion-induced testicular damage: an animal model. Biomed. Pharmacother. 2022;150:113056. doi: 10.1016/j.biopha.2022.113056. [DOI] [PubMed] [Google Scholar]
- Alves A.C., Alonso R., Diaz-Diaz J.L., Medeiros A.M., Jannes C.E., Merchan A., Vasques-Cardenas N.A., Cuevas A., Chacra A.P., Krieger J.E., et al. Phenotypical, clinical, and molecular aspects of adults and children with homozygous familial hypercholesterolemia in Iberoamerica. Arterioscler. Thromb. Vasc. Biol. 2020;40:2508–2515. doi: 10.1161/ATVBAHA.120.313722. [DOI] [PubMed] [Google Scholar]
- Argmann C.A., Houten S.M., Zhu J., Schadt E.E. A next generation multiscale view of inborn errors of metabolism. Cell Metab. 2016;23:13–26. doi: 10.1016/j.cmet.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P., Chan K.C., Tarabocchia M., Benito-Vicente A., Alves A.C., Uribe K.B., Bourbon M., Skiba P.J., Pordy R., Gipe D.A., et al. Functional Analysis of LDLR (low-density lipoprotein receptor) variants in patient lymphocytes to assess the effect of Evinacumab in homozygous familial hypercholesterolemia patients with a spectrum of LDLR activity. Arterioscler. Thromb. Vasc. Biol. 2019;39:2248–2260. doi: 10.1161/ATVBAHA.119.313051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S., Duren W., Evans C.R., Burant C.F., Michailidis G., Karnovsky A. Sparse network modeling and metscape-based visualization methods for the analysis of large-scale metabolomics data. Bioinformatics. 2017;33:1545–1553. doi: 10.1093/bioinformatics/btx012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beheshti S.O., Madsen C.M., Varbo A., Nordestgaard B.G. Worldwide prevalence of familial hypercholesterolemia: meta-analyses of 11 million subjects. J. Am. Coll. Cardiol. 2020;75:2553–2566. doi: 10.1016/j.jacc.2020.03.057. [DOI] [PubMed] [Google Scholar]
- Bertolini S., Calandra S., Arca M., Averna M., Catapano A.L., Tarugi P., Italian Study Group of Homozygous Familial Hypercholesterolemia Homozygous familial hypercholesterolemia in Italy: clinical and molecular features. Atherosclerosis. 2020;312:72–78. doi: 10.1016/j.atherosclerosis.2020.08.027. [DOI] [PubMed] [Google Scholar]
- Cha M.H., Lee S.M., Jung J. Lysophosphatidylcholine induces expression of genes involved in cholesterol biosynthesis in THP-1 derived macrophages. Steroids. 2018;139:28–34. doi: 10.1016/j.steroids.2018.09.003. [DOI] [PubMed] [Google Scholar]
- Chen G., Nan C., Tian J., Jean-Charles P., Li Y., Weissbach H., Huang X.P. Protective effects of taurine against oxidative stress in the heart of MsrA knockout mice. J. Cell. Biochem. 2012;113:3559–3566. doi: 10.1002/jcb.24233. [DOI] [PubMed] [Google Scholar]
- Chen P., Chen X., Zhang S. Current status of familial hypercholesterolemia in China: a need for patient FH registry systems. Front. Physiol. 2019;10:280. doi: 10.3389/fphys.2019.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clària J., Curto A., Moreau R., Colsch B., López-Vicario C., Lozano J.J., Aguilar F., Castelli F.A., Fenaille F., Junot C., et al. Untargeted lipidomics uncovers lipid signatures that distinguish severe from moderate forms of acutely decompensated cirrhosis. J. Hepatol. 2021;75:1116–1127. doi: 10.1016/j.jhep.2021.06.043. [DOI] [PubMed] [Google Scholar]
- Cuchel M., Bruckert E., Ginsberg H.N., Raal F.J., Santos R.D., Hegele R.A., Kuivenhoven J.A., Nordestgaard B.G., Descamps O.S., Steinhagen-Thiessen E., et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 2014;35:2146–2157. doi: 10.1093/eurheartj/ehu274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchel M., Meagher E.A., du Toit Theron H., Blom D.J., Marais A.D., Hegele R.A., Averna M.R., Sirtori C.R., Shah P.K., Gaudet D., et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40–46. doi: 10.1016/S0140-6736(12)61731-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin-Chartier J.P., Tremblay A.J., Bergeron J., Lamarche B., Couture P. The low-density lipoprotein receptor genotype is a significant determinant of the rebound in low-density lipoprotein cholesterol concentration after lipoprotein apheresis among patients with homozygous familial hypercholesterolemia. Circulation. 2017;136:880–882. doi: 10.1161/CIRCULATIONAHA.117.029435. [DOI] [PubMed] [Google Scholar]
- Du Z., Lu Y., Sun J., Chang K., Lu M., Fang M., Zeng X., Zhang W., Song J., Guo X., et al. Pharmacokinetics/pharmacometabolomics-pharmacodynamics reveals the synergistic mechanism of a multicomponent herbal formula, Baoyuan decoction against cardiac hypertrophy. Biomed. Pharmacother. 2021;139:111665. doi: 10.1016/j.biopha.2021.111665. [DOI] [PubMed] [Google Scholar]
- Gonçalves E., Frezza C. Genome and metabolome: chance and necessity. Genome Biol. 2021;22:276. doi: 10.1186/s13059-021-02501-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guijas C., Montenegro-Burke J.R., Warth B., Spilker M.E., Siuzdak G. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat. Biotechnol. 2018;36:316–320. doi: 10.1038/nbt.4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W., Xu D., Song X., Niu B., Zhuang Z., Lu Y., Lei X., Ma R., Lu C., Sun N., et al. Vitamin A and retinoic acid accelerate the attenuation of intestinal adaptability upon feeding induced by high-fat diet in mice. J. Nutr. Biochem. 2021;97:108803. doi: 10.1016/j.jnutbio.2021.108803. [DOI] [PubMed] [Google Scholar]
- Huang J.P., Cheng M.L., Wang C.H., Huang S.S., Hsieh P.S., Chang C.C., Kuo C.Y., Chen K.H., Hung L.M. Therapeutic potential of cPLA2 inhibitor to counteract dilated-cardiomyopathy in cholesterol-treated H9C2 cardiomyocyte and MUNO rat. Pharmacol. Res. 2020;160:105201. doi: 10.1016/j.phrs.2020.105201. [DOI] [PubMed] [Google Scholar]
- Irvine H.J., Acharjee A., Wolcott Z., Ament Z., Hinson H.E., Molyneaux B.J., Simard J.M., Sheth K.N., Kimberly W.T. Hypoxanthine is a pharmacodynamic marker of ischemic brain edema modified by glibenclamide. Cell Rep. Med. 2022;3:100654. doi: 10.1016/j.xcrm.2022.100654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L., Benito-Vicente A., Tang L., Etxebarria A., Cui W., Uribe K.B., Pan X.D., Ostolaza H., Yang S.W., Zhou Y.J., et al. Analysis of LDLR variants from homozygous FH patients carrying multiple mutations in the LDLR gene. Atherosclerosis. 2017;263:163–170. doi: 10.1016/j.atherosclerosis.2017.06.014. [DOI] [PubMed] [Google Scholar]
- Jiang L., Stoekenbroek R.M., Zhang F., Wang Q., Yu W., Yuan H., Cai G., Chen Y., Li G., Yang Y., et al. Homozygous familial hypercholesterolemia in China: genetic and clinical characteristics from a real-world, multi-center, cohort study. J. Clin. Lipidol. 2022;16:306–314. doi: 10.1016/j.jacl.2022.03.003. [DOI] [PubMed] [Google Scholar]
- Kim M., Yoo H.J., Lee D., Lee J.H. Oxidized LDL induces procoagulant profiles by increasing lysophosphatidylcholine levels, lysophosphatidylethanolamine levels, and Lp-PLA2 activity in borderline hypercholesterolemia. Nutr. Metab. Cardiovasc. Dis. 2020;30:1137–1146. doi: 10.1016/j.numecd.2020.03.015. [DOI] [PubMed] [Google Scholar]
- Larrea-Sebal A., Benito-Vicente A., Fernandez-Higuero J.A., Jebari-Benslaiman S., Galicia-Garcia U., Uribe K.B., Cenarro A., Ostolaza H., Civeira F., Arrasate S., et al. MLb-LDLr: a machine learning model for predicting the pathogenicity of LDLr missense variants. JACC. Basic Transl. Sci. 2021;6:815–827. doi: 10.1016/j.jacbts.2021.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterbach M.A., Latz E., Christ A. Metabolomic profiling reveals distinct and mutual effects of diet and inflammation in shaping systemic metabolism in Ldlr(-/-) Mice. Metabolites. 2020;10:E336. doi: 10.3390/metabo10090336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Zhang L., Dong F., Liu Y., Li N., Li H., Lei H., Hao F., Wang Y., Zhu Y., Tang H. Metabonomic changes associated with atherosclerosis progression for LDLR(-/-) Mice. J. Proteome Res. 2015;14:2237–2254. doi: 10.1021/acs.jproteome.5b00032. [DOI] [PubMed] [Google Scholar]
- Lin Y.T., Salihovic S., Fall T., Hammar U., Ingelsson E., Ärnlöv J., Lind L., Sundström J. Global plasma metabolomics to identify potential biomarkers of blood pressure progression. Arterioscler. Thromb. Vasc. Biol. 2020;40:e227–e237. doi: 10.1161/ATVBAHA.120.314356. [DOI] [PubMed] [Google Scholar]
- Luirink I.K., Braamskamp M.J.A.M., Wiegman A., Hartgers M.L., Sjouke B., Defesche J.C., Hovingh G.K. The clinical and molecular diversity of homozygous familial hypercholesterolemia in children: results from the GeneTics of clinical homozygous hypercholesterolemia (GoTCHA) study. J. Clin. Lipidol. 2019;13:272–278. doi: 10.1016/j.jacl.2018.12.003. [DOI] [PubMed] [Google Scholar]
- Luo J., Yang H., Song B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020;21:225–245. doi: 10.1038/s41580-019-0190-7. [DOI] [PubMed] [Google Scholar]
- Mukherjee P., Hough G., Chattopadhyay A., Grijalva V., O'Connor E.I., Meriwether D., Wagner A., Ntambi J.M., Navab M., Reddy S.T., Fogelman A.M. Role of enterocyte stearoyl-Co-A desaturase-1 in LDLR-null mice. J. Lipid Res. 2018;59:1818–1840. doi: 10.1194/jlr.M083527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olkowicz M., Czyzynska-Cichon I., Szupryczynska N., Kostogrys R.B., Kochan Z., Debski J., Dadlez M., Chlopicki S., Smolenski R.T. Multi-omic signatures of atherogenic dyslipidaemia: pre-clinical target identification and validation in humans. J. Transl. Med. 2021;19:6. doi: 10.1186/s12967-020-02663-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak P., Li T., Chiang J.Y.L. Retinoic acid-related orphan receptor alpha regulates diurnal rhythm and fasting induction of sterol 12alpha-hydroxylase in bile acid synthesis. J. Biol. Chem. 2013;288:37154–37165. doi: 10.1074/jbc.M113.485987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raal F.J., Rosenson R.S., Reeskamp L.F., Hovingh G.K., Kastelein J.J.P., Rubba P., Ali S., Banerjee P., Chan K.C., Gipe D.A., et al. Evinacumab for homozygous familial hypercholesterolemia. N. Engl. J. Med. 2020;383:711–720. doi: 10.1056/NEJMoa2004215. [DOI] [PubMed] [Google Scholar]
- Raal F.J., Sjouke B., Hovingh G.K., Isaac B.F. Phenotype diversity among patients with homozygous familial hypercholesterolemia: a cohort study. Atherosclerosis. 2016;248:238–244. doi: 10.1016/j.atherosclerosis.2016.03.009. [DOI] [PubMed] [Google Scholar]
- Sánchez-Hernández R.M., Civeira F., Stef M., Perez-Calahorra S., Almagro F., Plana N., Novoa F.J., Sáenz-Aranzubía P., Mosquera D., Soler C., et al. Homozygous familial hypercholesterolemia in Spain: prevalence and phenotype-genotype relationship. Circ. Cardiovasc. Genet. 2016;9:504–510. doi: 10.1161/CIRCGENETICS.116.001545. [DOI] [PubMed] [Google Scholar]
- Santos R.D., Gidding S.S., Hegele R.A., Cuchel M.A., Barter P.J., Watts G.F., Baum S.J., Catapano A.L., Chapman M.J., Defesche J.C., et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 2016;4:850–861. doi: 10.1016/S2213-8587(16)30041-9. [DOI] [PubMed] [Google Scholar]
- Saulnier-Blache J.S., Wilson R., Klavins K., Graham D., Alesutan I., Kastenmüller G., Wang-Sattler R., Adamski J., Roden M., Rathmann W., et al. Ldlr-/- and ApoE-/- mice better mimic the human metabolite signature of increased carotid intima media thickness compared to other animal models of cardiovascular disease. Atherosclerosis. 2018;276:140–147. doi: 10.1016/j.atherosclerosis.2018.07.024. [DOI] [PubMed] [Google Scholar]
- Semova I., Levenson A.E., Krawczyk J., Bullock K., Gearing M.E., Ling A.V., Williams K.A., Miao J., Adamson S.S., Shin D.J., et al. Insulin prevents hypercholesterolemia by suppressing 12 alpha-hydroxylated bile acids. Circulation. 2022;145:969–982. doi: 10.1161/CIRCULATIONAHA.120.045373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjouke B., Kusters D.M., Kindt I., Besseling J., Defesche J.C., Sijbrands E.J.G., Roeters van Lennep J.E., Stalenhoef A.F.H., Wiegman A., de Graaf J., et al. Homozygous autosomal dominant hypercholesterolaemia in The Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur. Heart J. 2015;36:560–565. doi: 10.1093/eurheartj/ehu058. [DOI] [PubMed] [Google Scholar]
- Sniderman A.D., Glavinovic T., Thanassoulis G. Key questions about familial hypercholesterolemia: JACC review topic of the week. J. Am. Coll. Cardiol. 2022;79:1023–1031. doi: 10.1016/j.jacc.2022.01.010. [DOI] [PubMed] [Google Scholar]
- Thompson G.R., Blom D.J., Marais A.D., Seed M., Pilcher G.J., Raal F.J. Survival in homozygous familial hypercholesterolaemia is determined by the on-treatment level of serum cholesterol. Eur. Heart J. 2018;39:1162–1168. doi: 10.1093/eurheartj/ehx317. [DOI] [PubMed] [Google Scholar]
- Tromp T.R., Hartgers M.L., Hovingh G.K., Vallejo-Vaz A.J., Ray K.K., Soran H., Freiberger T., Bertolini S., Harada-Shiba M., Blom D.J., et al. Worldwide experience of homozygous familial hypercholesterolaemia: retrospective cohort study. Lancet. 2022;399:719–728. doi: 10.1016/S0140-6736(21)02001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon H.J. Inborn errors of metabolism: advances in diagnosis and therapy. JAMA Pediatr. 2015;169:778–782. doi: 10.1001/jamapediatrics.2015.0754. [DOI] [PubMed] [Google Scholar]
- Watanabe M., Suliman M.E., Qureshi A.R., Garcia-Lopez E., Bárány P., Heimbürger O., Stenvinkel P., Lindholm B. Consequences of low plasma histidine in chronic kidney disease patients: associations with inflammation, oxidative stress, and mortality. Am. J. Clin. Nutr. 2008;87:1860–1866. doi: 10.1093/ajcn/87.6.1860. [DOI] [PubMed] [Google Scholar]
- Wu Y., Jiang L., Zhang H., Cheng S., Wen W., Xu L., Zhang F., Yang Y., Wang L., Chen J. Integrated analysis of microRNA and mRNA expression profiles in homozygous familial hypercholesterolemia patients and validation of atherosclerosis associated critical regulatory network. Genomics. 2021;113:2572–2582. doi: 10.1016/j.ygeno.2021.05.036. [DOI] [PubMed] [Google Scholar]
- Zhao H., Li Y., He L., Pu W., Yu W., Li Y., Wu Y.T., Xu C., Wei Y., Ding Q., et al. In vivo AAV-CRISPR/Cas9-mediated gene editing ameliorates atherosclerosis in familial hypercholesterolemia. Circulation. 2020;141:67–79. doi: 10.1161/CIRCULATIONAHA.119.042476. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Little P.J., Ta H.T., Xu S., Kamato D. Lysophosphatidic acid and its receptors: pharmacology and therapeutic potential in atherosclerosis and vascular disease. Pharmacol. Ther. 2019;204:107404. doi: 10.1016/j.pharmthera.2019.107404. [DOI] [PubMed] [Google Scholar]
- Zurkinden L., Sviridov D., Vogt B., Escher G. Downregulation of Cyp7a1 by cholic acid and chenodeoxycholic acid in Cyp27a1/ApoE double knockout mice: differential cardiovascular outcome. Front. Endocrinol. 2020;11:586980. doi: 10.3389/fendo.2020.586980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. Raw metabolomic data has been deposited at https://service.most.gov.cn/ and is available from the lead contact and National Key Research and Development Program of China upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.