

Abstract
Tumor vaccination is a promising approach for tumor immunotherapy because it presents high specificity and few side effects. However, tumor vaccines that contain only a single tumor antigen can allow immune system evasion by tumor variants. Tumor antigens are complex and heterogeneous, and identifying a single antigen that is uniformly expressed by tumor cells is challenging. Whole tumor cells can produce comprehensive antigens that trigger extensive tumor-specific immune responses. Therefore, tumor cells are an ideal source of antigens for tumor vaccines. A better understanding of tumor cell-derived vaccines and their characteristics, along with the development of new technologies for antigen delivery, can help improve vaccine design. In this review, we summarize the recent advances in tumor cell-derived vaccines in cancer immunotherapy and highlight the different types of engineered approaches, mechanisms, administration methods, and future perspectives. We discuss tumor
cell-derived vaccines, including whole tumor cell components, extracellular vesicles, and cell membrane-encapsulated nanoparticles. Tumor cell-derived vaccines contain multiple tumor antigens and can induce extensive and potent tumor immune responses. However, they should be engineered to overcome limitations such as insufficient immunogenicity and weak targeting. The genetic and chemical engineering of tumor cell-derived vaccines can greatly enhance their targeting, intelligence, and functionality, thereby realizing stronger tumor immunotherapy effects. Further advances in materials science, biomedicine, and oncology can facilitate the clinical translation of tumor cell-derived vaccines.
Keywords: Vaccine, Cancer, Immunotherapy, Immunogenicity, Engineering, Nanotechnology, Biomaterials
Graphical abstract
Highlights
-
•
Summarize versatile engineering strategies of tumor cell-derived vaccines.
-
•
Elucidate the anticancer mechanisms of engineered tumor cell-derived vaccines.
-
•
Discuss clinical applications, challenges and perspectives of engineered tumor cell-derived vaccines.
Abbreviations
- (TSA)
Tumor-specific antigen
- (APC)
Antigen-presenting cell
- (FDA)
Food and Drug Administration
- (GM-CSF)
Granulocyte-macrophage colony-stimulating factor
- (TAA)
Tumor-associated antigen
- (TME)
Tumor microenvironment
- (CAF)
Cancer-associated fibroblast
- (DC)
Dendritic cell
- (CTL)
Cytotoxic T cell
- (GPI)
Glycosylphosphatidylinositol
- (PTT)
Photothermal therapy
- (Treg)
Regulatory T cell
- (FAP)
Fibroblast activation protein-α
- (ICD)
Immunogenic cell death
- (MHC)
Major histocompatibility complex
- (TCL)
Tumor cell lysate
- (CSC)
Cancer stem cell
- (NK)
Natural killer
- (ICI)
Immune checkpoint inhibitor
- (EV)
Extracellular vesicle
- (TEX)
Tumor-derived exosome
- (TMV)
umor-derived microvesicle
- (CCM)
Cancer cell membrane
1. Introduction
Tumor immunotherapy is aimed at activating the immune system to effectively inhibit the occurrence, development, and recurrence of tumors by mounting an antitumor immune response [1,2]. Currently used tumor immunotherapy strategies include monoclonal antibodies3, immune checkpoint blockers [4,5], lytic virus therapy [6,7], and tumor vaccines [8]. Tumor vaccines can effectively deliver tumor-specific antigens (TSAs) to antigen-presenting cells (APCs) and activate tumor-specific T cells. They establish long-lasting antitumor memory and reduce nonspecific killing and adverse reactions, ultimately inducing local tumor regression and eradicating metastatic lesions [9,10]. Tumor vaccines are generally divided into preventive and therapeutic vaccines [11,12]. Therapeutic tumor vaccines (hereafter called tumor vaccines) are used to deliver tumor cell-specific antigens to stimulate the immune system to produce specific immune cells or antibodies and eliminate tumor cells. Provenge® is a tumor vaccine used to treat asymptomatic or mildly symptomatic metastatic prostate cancer that is resistant to hormone therapy. In 2010, it became the first tumor vaccine approved for marketing by the U.S. Food and Drug Administration (FDA). This vaccine is derived from patient-derived APCs. The APCs are cocultured and activated using prostate acid phosphatase and granulocyte-macrophage colony-stimulating factor (GM-CSF) binding protein in vitro and then transferred back to the patients for tumor treatment. Phase III clinical trials of the vaccine confirmed that it can reduce the mortality risk of patients and prolong their survival time by an average of 4.1 months [13]. However, no new tumor vaccine has been recently approved by the FDA. Over 500 clinical trials (clinicaltrials.gov) related to tumor vaccines are in progress or have been completed. In 2021, OSE Immunotherapeutics announced favorable results for their cancer vaccine Tedopi® to treat advanced non-small cell lung cancer patients who did not respond to treatment with immune checkpoint inhibitors (PD-1/PD-L1) (NCT02654587). This vaccine screens 10 optimal neo-antigenic epitopes from five tumor-associated antigens (TAAs) that are commonly expressed in lung cancer cells and combines them to stimulate T lymphocytes to recognize and attack cancer cells. Tedopi® has been patented and granted orphan drug status in the United States for HLA-A2-positive non-small cell lung cancer. Although tumor vaccines have shown therapeutic effects in clinical trials, their overall clinical efficacy has been unsatisfactory [14], and the expected immune response was observed in only a small proportion of patients.
The failure of tumor vaccines can be attributed to insufficient tumor immunogenicity, which prevents the generation of sufficient robust T cells that are required to induce long-term immunity. Furthermore, when immunosuppressive cells, such as tumor-associated macrophages, regulatory T cells, cancer-associated fibroblasts (CAFs), and myeloid-derived suppressor cells, accumulate at tumor sites, they can create an immunosuppressive microenvironment by expressing suppressor receptors and immunosuppressive cytokines that impede antitumor immune responses [[15], [16], [17], [18]]. To activate tumor immune responses using tumor vaccines, APCs, especially dendritic cells (DCs), are required to absorb a wide range of TSAs for optimal T cell activation [19,20]. Therefore, a wider range of tumor antigens and more effective delivery methods would contribute to the development of tumor vaccines [21], and they should be optimized to maximize tumor immunogenicity. Owing to the heterogeneity and complexity of tumor antigens [22,23], few antigens can be used to prepare universal vaccines [24]. Furthermore, the predominant mutation-inducing antigens in tumors can vary according to cancer type and even among different patients. Therefore, most tumor patients have few common antigens elsewhere in the body, and actual tumor cells contain ideal and comprehensive antigens to induce tumor-specific immune responses; thus, tumor cells are the best source of antigens to prepare tumor vaccines [[25], [26], [27]]. Although tumor cell-derived vaccines contain a wide range of antigens, their ability to stimulate tumor immunogenicity needs substantial improvement, and genetic and chemical engineering methods could be utilized to improve the immunogenicity of tumor vaccines [28,29]. In the field of tumor vaccine therapy, researchers need to address three major challenges: enhancing the immunogenicity of antigens, countering the immune escape mechanism of tumors, and achieving effective delivery of tumor vaccines. Engineering strategies can offer the following great opportunities for improving the efficacy of tumor-derived vaccines [[30], [31], [32], [33]]: 1) codelivery of antigens and novel adjuvants such as natural, synthetic, or genetically engineered immune-stimulatory molecules; 2) modulation of the tumor microenvironment (TME) in such a manner that cytotoxic T lymphocytes (CTLs) are more effective in killing; 3) methods of enhancing traditional treatment modalities such as chemotherapy, radiotherapy, and phototherapy with tumor vaccines; and 4) building more efficient and accurate delivery systems.
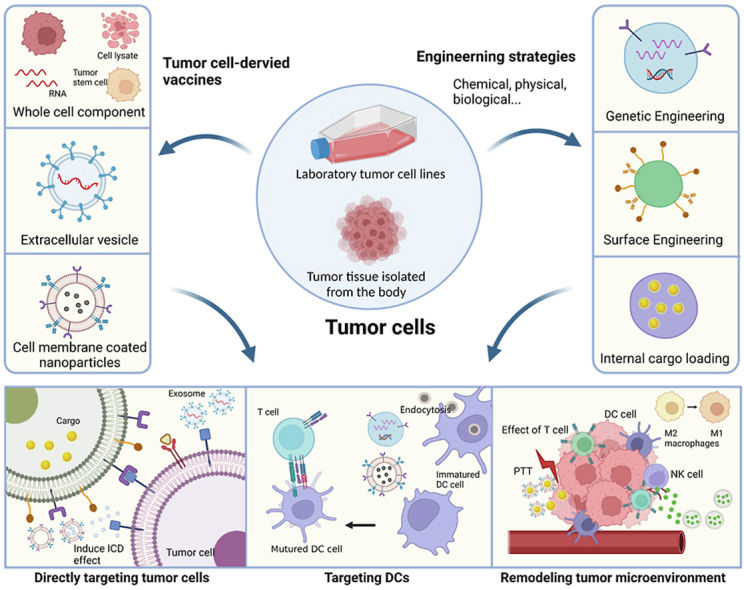
Over the past decade, tumor cell-derived vaccines have been considered to improve the long-term immune memory effect against cancer, which has attracted great attention from researchers, and several research groups have reported encouraging preclinical results [21,25]. Many reviews have discussed the potential and significance of a single or several kinds of tumor-derived vaccines (e.g., tumor cell membranes, extracellular vesicles) [20,[34], [35], [36]]. However, to the best of our knowledge, no review has provided a comprehensive summary of engineered tumor cell-derived vaccines. Based on the literature, we summarized common engineering strategies and recent research progress (Scheme 1.). Finally, we discuss the advantages and disadvantages of different types of therapeutic vaccines for oncological treatment and clinical translation; current challenges in this regard have also been presented, revealing the state of progress and highlighting the potential for future research.
Scheme 1.

Illustration of novel strategies for engineered tumor cell-derived vaccines. The cellular components used as tumor cell-derived vaccines include tumor whole cell fractions, extracellular vesicles, and cell membrane-coated nanoparticles. Three main engineering strategies (comprising genetic engineering, surface engineering, and internal cargo loading) are used to enhance the therapeutic efficacy of tumor vaccines. The antitumor mechanisms of tumor cell-derived vaccines include: 1) direct killing of tumor cells through surface-specific receptor targeting, the release of cytokines at the tumor site, or drug-induced tumor immunogenic cell death (ICD); 2) targeting dendritic cells (DCs) by delivering large amounts of antigen; the mature DCs then present the antigen as an antigenic peptide on the cell surface and secrete a variety of cytokines to induce T cell activation and kill the tumor; and 3) remodeling the tumor microenvironment (TME) by eliciting local inflammation, polarization of macrophages, natural killer cell (NK) recruitment, etc. (Created with BioRender.com).
2. Versatile engineering strategies
Modified strategies to engineer vaccines can lead to improved delivery efficiency, targeting ability, and therapeutic efficacy [29,37,38]. Different engineering modifications can be used to flexibly increase the therapeutic efficacy of tumor cell-derived vaccines. The current engineering approaches are: (1) genetically engineered modification of cell surface protein expression by introducing genes to increase the immunogenicity of tumor vaccines; (2) surface engineering modification by attaching functionalized groups to the cell surface to increase the targeting specificity and immunogenicity of tumor vaccines; and (3) internal cargo loading by loading the desired cargo, such as antigens and adjuvants, into tumor cell-derived vaccines (Scheme 1). These engineering strategies can considerably enhance the immunogenicity of tumor vaccines.
2.1. Genetic engineering
Genetic engineering can be used to modify the genetic information of almost any living cell; exogenous genes are introduced into recipient cells through specific gene editing to alter the cellular phenotype [39]. Therefore, genetic engineering can be used to develop more effective tumor vaccines based on different requirements. The knockout, insertion, or replacement of nucleic acid sequences can be performed using gene editing techniques or direct construction of gene sequences of target expression molecules in vitro, followed by transfer into the cytoplasm. Physical transfer methods and viral and nonviral vectors are used for genetic engineering [40]. Viral vectors, including adenovirus and lentivirus, are commonly used for the transfection of genes, and they show superior transfection efficiency. However, viral transduction may be subject to insertional mutations because the random integration of a virus into the host genome may disrupt the original gene sequence, thereby leading to cancer or loss of original function [41]. To improve safety, nonviral carriers such as cationic lipid or polymer-based nanoparticles can be used for gene delivery. A distinctive feature of nucleic acid molecules is that they are negatively charged; therefore, they can be adsorbed to cationic carriers and delivered into cells. However, nonviral carriers can potentially be toxic and show lower cellular uptake than viral vectors [42]. Finally, some physical methods, such as gene guns, electroporation, and laser irradiation, deliver naked nucleic acids into the cells. Electroporation, acoustic pore effects, and laser irradiation are based on external physical actions that can alter the permeability of the cell membrane, thereby allowing nucleic acids to enter the cells [43,44]. However, these methods do not show high transfection efficiency, and the outcomes are dependent on external conditions [45]. In contrast, gene guns and microinjection, which use mechanical means to introduce genes directly into cells, are effective physical methods. In this context, genetic engineering (e.g., CRISPR/Cas9 technology), which is a more flexible modification method, has been widely used [46]. Compared to DNA, mRNA does not need to be integrated into the host genome, is expressed more rapidly and can be introduced into the cell at any amount to avoid overexpression [40]. For example, Huang et al. [47] used lentivirus to transfect α-lactalbumin (α-LA) mRNA into breast cancer tumor cells to enrich their secreted exosomes with large amounts of α-LA, which enhanced the targeting ability of exosomes. With genetic engineering, either DNA plasmid transfection can be utilized or mRNA can be introduced directly into host cells, and the expression of cell-specific molecules can be controlled and enhanced; thus, it is considered a flexible and selective method of modification, and it can effectively increase the expression of specific functional molecules in large quantities to improve antitumor immunity.
2.2. Surface engineering
Biological membranes such as cell membranes or the lipid outer layer of exosomes can be modified to increase cell or tissue targeting and interaction and in vivo delivery, thereby improving the therapeutic efficacy of vaccines [48,49]. Biological membranes are composed of complex biological substances such as polysaccharides, proteins, and lipids, which contain a range of functional groups that allow chemical and physical modification of their surfaces using various synthetic materials. Numerous reactive groups, such as amines, can be chemically coupled to the surface of cell membranes through specific reactions. For example, amination and click reactions are chemical reactions that are commonly used for cell or vesicle surface coupling. Amination usually requires 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) as condensation agents to link the carboxyl and amine groups to form amide bonds. In click chemistry, a cycloaddition reaction of azide and alkyne catalyzed by copper produces nontoxic byproducts under mild conditions [50]. The click chemistry-based coupling approach requires cell membrane and surface modifiers with azide and alkyne groups on either end. Therefore, targeting peptides can be coupled to the membrane surface. For instance, EDC-NHS condensation can be used to produce surface alkyne-based exosomes, followed by a click reaction with a neuropilin-1-targeted peptide (RGERPPR, RGE)-targeting peptide and azide group to successfully enable alkyne-based exosomes to target gliomas [51]. Most of these chemical reactions are time consuming and can potentially damage biological materials because multiple chemical reactions require the use of organic reagents [52]. In contrast, physical methods are usually driven by electrostatic and/or hydrophobic interactions [53]. Based on the inherent nature of biological membranes, hydrophobic molecules or positively charged nanoparticles can be adsorbed on their surface. For example, small molecules (e.g., mannose) and large antibodies have been successfully inserted on cell membrane surfaces with the aid of hydrophobic ligands to improve targeting [[54], [55], [56]]. In this context, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] is a commonly used anchoring ligand that has a long hydrocarbon chain structure and, thus, can be easily inserted into cell membranes [53,57]. Some proteins can be anchored to cell membranes using glycosylphosphatidylinositol (GPI) anchors and glycolipids [58]. Similarly, immunoadjuvants can also be directly inserted into lipid bilayer membranes using physical methods [3]. Compared to chemical reactions, physical methods are milder and faster; the original physical and chemical properties of the cell membrane are less damaged [48]. However, physical methods present the disadvantage of modification instability. Other specific interactions, such as avidin-biotin, have also been explored as simple and mild conjugation methods owing to the high binding affinity between two functional groups [[59], [60], [61]], such as the modification of cell surfaces from GM-CSF to polymeric nanoparticles by streptavidin–biotin cross-linking [29,62]. Overall, researchers have developed a series of engineering methods to build composites based on biological components using the inherent properties of biofilms, including chemical coupling based on carboxyl, amine, sulfhydryl, and biotin groups, as well as physical adsorption via electrostatic or hydrophobic interactions. However, chemical coupling relies heavily on appropriate chemical modification of the inherent biological factors on the extracellular surface; therefore, the choice of ligands is limited. This may also lead to unavoidable cell damage. In contrast, physical adsorption, especially the most commonly used electrostatic adsorption method, tends to facilitate the integration of two different components.
2.3. Internal cargo loading
Internal cargo loading refers to the encapsulation of exogenous substances (such as drug molecules, photosensitizers, and inorganic/organic biomaterials) within tumor cell-derived carriers or to the addition of tumor cell-derived vaccines to the interior of other carriers (non-tumor origin biomaterials such as hydrogels, organic/inorganic nanoparticles). For the former method, use of extracellular vesicles and cell membrane vesicles is suitable, due to the interior capacity of these vesicles. The latter method is more suitable for the delivery of cell lysates, RNA vaccines because they cannot be used as a vehicle themselves. The incorporation of exogenous substances can confer superior properties to tumor cell-derived vesicles, such as better immunogenicity and photothermal properties, which can lead to better therapeutic efficacy against cancer cells [32]. To load internal cargo into tumor cells, diffusion, endocytosis, and electroporation can be used depending on the cell type [39]. Small molecules can diffuse into cells, leading to different concentrations inside and outside the cell. All eukaryotic cells can absorb large molecules or nanoparticles by endocytosis, which characterizes their internal cargo loading [63,64]. Nonphagocytic cells have limited uptake capacity, whereas macrophages have superior phagocytic ability [65]. However, endocytosis might promote the degradation of biodegradable nanoparticles, which leads to uncontrolled drug leakage and adverse side effects on cells. When a cell is exposed to a sufficiently high electric field, the permeability of the cell membrane rapidly increases. Therefore, electroporation is an efficient approach for internal cargo loading. Gold nanorods encapsulated into platelets by electroporation have been successfully delivered to tumor tissue for enhanced photothermal therapy (PTT) [66]. For internal cargo loading of cell-derived vesicles, membrane extrusion and ultrasonication are commonly used. Membrane extrusion uses coextrusion of the cell membrane or exosomes and nanoparticles through a porous membrane, and it can reassemble the lipid layer around the nanoparticles during membrane extrusion [67]. However, large-scale application of this method is difficult. Therefore, an ultrasonic method was proposed in which membrane-encapsulated nanoparticles were produced by sonicating vesicles or cell membranes with nanoparticles [68]. Despite the simplicity of the approach, the time and power of the ultrasound should be optimized to obtain maximum fusion efficiency with minimal damage to the membrane-coated nanoparticles.
Natural tumor cells are thought to provide both an abundance of TAAs and the ability to act as a covert coating to evade clearance by the immune system. Engineering strategies for tumor cell-derived vaccines could provide smart modules by enabling complex modifications, such as protein expression and antibody cross-linking, thereby compensating for the shortcomings of traditional tumor vaccines and providing more definite therapeutic effects.
3. Tumor cell-derived vaccines
3.1. Whole tumor cell vaccines
Whole tumor cell vaccines have attracted attention because they contain the complete antigen spectrum of tumor cells [27,69]. The first attempt at a cancer vaccine in the early 20th century involved the injection of whole autologous tumor cells [70]. A complete set of autologous antigens includes epitopes recognized by CD8+ CTLs and by CD4+ helper T cells, inducing powerful T cell activation [71]. Whole-cell vaccines can reduce the chances of tumor escape at a higher rate than single-epitope vaccines [72]. These vaccines can be classified into autologous and allogeneic sources, depending on the origin of the tumor cells [73]. Autologous vaccines use a patient's own tumor cells as the source of antigen, thereby facilitating personalized therapy [74]. Allogenic vaccines use tumor cell lines derived from different individuals of the same race; these have the advantages of large-scale production and easy standardized quality control. However, these unmodified tumor cells do not produce a strong immune response [75].
Enhanced expression of immunostimulators in tumor vaccines is crucial to promote the effectiveness of whole-cell tumor vaccines; using genetic engineering techniques to modify cells to secrete more immunostimulatory factors is a convenient way to achieve this. GVAX is a genetically engineered vaccine that uses tumor cells modified to express GM-CSF, which are used as immune adjuvants and can increase the antigen-presenting capacity of DCs and promote DC survival [[76], [77], [78]]. One approach for immunotherapy of pancreatic cancer combined GM–CSF–secreting pancreatic cancer cell vaccine (GVAX Pancreas) with live attenuated Listeria monocytogenes modified to express mesothelin (CRS-207) [79]. The GVAX pancreas induced specific T cell immunity against tumor antigens and mesothelin, and in combination with low-dose cyclosporine, it achieved regulatory T cell (Treg) suppression. GVAX Pancreas combined with CRS-207 prolonged the survival of patients with metastatic pancreatic cancer in clinical trials [79]. GVAX has also been combined with other treatments, but the clinical efficacy did not meet expectations [70,76,80]. As tumor stromal cells also contribute to the progression and metastasis of tumors, they should be eliminated to achieve better therapeutic efficacy against tumors [81]. Noncancerous stromal cells in the TME represent promising and genetically stable therapeutic targets [82]. More than 90% of CAFs of malignant epithelial carcinomas express fibroblast activation protein-α (FAP). CAFs are the major type of cells within the TME, and they deliver nutrition and protection to tumors and regulate immunosuppression. To eliminate CAFs, tumor vaccines targeting FAP can be used with a heterologous prime-boost strategy to enhance FAP-specific cellular immunity. Chen et al. [83] prepared an autologous tumor whole-cell vaccine with high FAP expression. The vaccine targeted both tumor cells and CAFs, leading to a significant reduction in CAFs, lifting of the immunosuppressive TME, and enhanced recruitment of effector T cells to improve antitumor success. In addition to the enhanced expression of immunostimulators, enriching the antigen spectrum also improved the immunogenicity of tumor vaccines. Vaccines prepared by crossing tumor cells of the same gene with heterozygous cells can significantly enhance the original immunogenicity, promote APC activation, and inhibit tumor-induced immune tolerance [84]. Gattoni-Celli and Young constructed a semiallogeneic glioma vaccine by mixing H-2b GL261 glioma cells and H-2d RAG-neo cells [85]. The vaccine increased immunoregulatory cytokine production, leading to a high profile of antitumor cytokines after inoculation in tumor-bearing mice. This suggests that the modification of an autologous whole-cell tumor vaccine to form a semiallogeneic whole-cell tumor vaccine can effectively utilize the allogeneic response to enhance the antitumor immune effect.
The effect of downregulated gene expression in tumor vaccines has also been used to enhance immunogenicity. Modulation of MYC oncogenes has been reported to affect immunogenicity against cancer cells [86]. High MYC expression decreases tumor cell immunogenicity and is associated with immunosuppression in the TME [87]. Wu et al. [88] inhibited MYC expression in neuroblastoma and melanoma cells, thereby enhancing their immunogenicity, using a whole-cell tumor vaccine. In a different study, Srinivasan et al. [89] observed that knockout of the inhibitor of differentiation protein 2 in murine neuroblastoma cells enhanced immunogenicity and served as a whole-cell tumor vaccine (Fig. 1A and B). Immunized mice resisted subsequent wild-type neuroblastoma cells, and tumor growth was inhibited even in established neuroblastomas. CTLA-4 checkpoint inhibitors synergistically caused an increase in the numbers of CD8+ T cells in the TME and increased interferon-γ (IFN-γ) production. Moreover, after IFN-γ treatment or T cell tumor infiltration, PD-L1 expression in mice and human neuroblastomas was upregulated for adaptive immune resistance.
Fig. 1.

Whole tumor cell vaccine. (A) Therapeutic effects of knockout Id2 mouse neuroblastoma cells (Id2kd-N2a) as a vaccine in combination with checkpoint inhibitors α-PD-L1 and α-CTLA-4 in tumor-bearing mice. (B) Tumor imaging in mice after α-PD-L1+α-CTLA-4+Id2kd N2a treatment. Copyright © 2018 Srinivasan et al. [89] (C) Schematic illustration of the design of walking dead triple-negative breast cancer cells for suppressing lung metastasis with temporal chemoimmunotherapy. (D) After intravenous injection of 4T1-tumor-bearing mice, cell-PD-1/Liposomes (DiR-labeled) accumulated in major organs over time. (E) Inhibitory effect on subcutaneous 4T1 tumor mouse model. Copyright © 2022 Wiley-VCH GmbH [90].
Numerous available reactive groups on the cell membrane surface have been used for surface chemical modification. Recently, Zhao et al. [90] coupled DOX-loaded liposomes and anti-PD-1 antibodies to the surface of inactivated tumor cells (Cell-PD-1/Liposome) (Fig. 1C–E). Inactivated tumor cells facilitate the targeting of drugs to lung metastases and the induction of an antitumor immune response by providing TAAs. DOX-induced immunogenic cell death (ICD) and the immune checkpoint blocking effect of PD-1 antibody synergistically activated the antitumor immune response and lifted immunosuppression, improving the immune microenvironment in the lung. Cell-PD-1/liposomes significantly improved the in vivo antitumor and antimetastatic effects in a 4T1 lung metastasis-bearing mouse model of triple-negative breast cancer; the mouse model exhibited delayed tumor growth, reduced lung metastases, and prolonged overall survival. In this way, the design of cells as modifiable carriers to deliver drugs and active molecules enriches the functionality of tumor vaccines.
Tumors contain a unique subpopulation of cells similar to stem cells, called cancer stem cells (CSCs), which possess tumor properties such as drug resistance, metastasis, and recurrence. CSC-based vaccines can produce effective antitumor immunity [91]. Contrary to unsorted tumor cells, CSC-initiated antibodies and T cells can selectively target CSCs [92]. To further increase the efficacy of CSC vaccines, a variety of engineering strategies have been developed (Table 1). Tumor-associated molecular mucin 1 (MUC1) is a transmembrane mucin glycoprotein that is overexpressed in colorectal cancer (CRC); it has been identified as a therapeutic target for immunotherapy [93]. Colorectal CSC vaccines overexpressing epithelial cell MUC1 have potential as a new preventive vaccine for CRC, and they have exhibited inhibitory effects on the CD4+CD25+ Treg cell subpopulation, significantly increased the cytotoxicity of natural killer (NK) cells and splenocytes, and exhibited targeted colorectal CSC killing [94]. In another study, vaccines with GM-CSF surface modification of CSCs induced a higher number of CD8+ T cells to infiltrate tumor tissues, but some CD8+ T cells expressed PD-1. In addition, CSC vaccines upregulate PD-L1 expression in tumor cells, leading to immune resistance. Therefore, it is a reasonable hypothesis that CSC vaccines combined with PD-1 blockers can be used to treat tumors more effectively. Streptavidin-GM-CSF surface-modified vaccine for bladder CSCs was used in combination with PD-1 blockers to treat bladder cancer. This treatment improved the function of specific T lymphocytes but did not increase the number of CD4+ Foxp3+ T cells [62]. Nevertheless, it increased the cure rate in mice and effectively protected mice from a second CSC attack.
Table 1.
Tumor cell-derived vaccines.
| Vaccine type | Modification strategy | Feature | Effect of vaccine | Ref. |
|---|---|---|---|---|
| Whole tumor cell vaccine | Genetic engineering | Expresses GM-CSF | Increases DC antigen presentation ability, promotes DC survival | [79] |
| Expresses high levels of FAP | Targets tumor cells and tumor-associated fibroblasts | [83] | ||
| Inhibits MYC expression | Appropriate targets to induce and improve tumor cell immunogenicity | [88] | ||
| Knocks out Id2 gene in mouse neuroblastoma cells | Imparts immunogenicity to tumor cells in hosts with normal immunity, acting synergistically with costimulatory CTLA-4 checkpoint inhibitors | [89] | ||
| Surface engineering | Inactivates tumor cells surface couple with DOX-loaded liposomes and anti-PD-1 antibodies | Delays tumor growth, reduces lung metastases, increases overall survival time | [90] | |
| Cell hybridization | H-2b GL261 glioma cells fuse with H-2d RAG-neo cells | Increases cytokine production by immune cells, produces high levels of antitumor cytokines | [85] | |
| Tumor cell-derived nanovaccines | Genetic engineering | Transduces FAP cDNA into tumor cells | Targets tumor parenchyma and stroma, promotes tumor iron droop | [101] |
| Tumor cell lysate | Genetic engineering | Blocks STAT3 signaling pathway | Inhibits tumor cell proliferation, promotes tumor cell apoptosis, promotes the generation of immune memory for HCC, prolongs the survival period of mice | [106] |
| Surface engineering | Tumor cell lysates are covalently attached to polydopamine nanoparticles | Enhances antigen uptake and maturation of BMDCs, as well as the expression of surface molecules and cytokine secretion associated with Th1 | [105] | |
| Internal cargo loading | Tumor cell lysates are loaded inside and on PLGA nanoparticle surfaces | Maximizes the delivery load of tumor antigens, stimulates a broader cancer-specific immune response | [21] | |
| Oxidized tumor cell lysates are loaded inside PLGA nanoparticles | Heightens immunity stimulation | [102] | ||
| Tumor cell lysates are loaded in chitosan nanoparticles of which, the surface has been modified with mannose | Activates DCs in vitro and in vivo, and prevents tumor growth | [108] | ||
| CpG and tumor cell lysates are loaded into temperature-sensitive PLEL hydrogels | Inhibits CT26 colon cancer in mice, forms immune memory, reduces tumor recurrence rate, inhibits distant tumors. | [111] | ||
| Tumor cell lysates are loaded into multiarmed poly(ethylene glycol) (8-arm PEG)/oxidized dextran dynamically cross-linked hydrogels | Recruits DCs, releases antigens gradually, induces a tumor-specific immune response, prevents tumor recurrence after surgery in mouse models | [112] | ||
| Self-assembled poly(l-valine) hydrogels | Recruits, activates, and matures DCs in vitro and in vivo | [113] | ||
| PEG-b-poly(l-alanine) hydrogels are loaded with a dual checkpoint inhibitor, tumor antigen that is continuously released, and GM-CSF | Increases the proportion of activated effector CD8+ T cells in spleens and tumors of immunized mice, and decreases the proportion of Tregs | [114] | ||
| Cancer stem cell vaccine | Surface engineering | SA-GM-CSF surface-modified bladder CSCs | Increases the number of CD8+ T cells by the activation of tumor specific T cells | [62] |
| Genetic engineering | Overexpression of epithelial cell molecule mucin 1 | Enhances innate and adaptive immune responses and immune memory | [94] | |
| Tumor whole RNA vaccine | Encapsulate into cationic liposomes | Total tumor-derived RNAs are encapsulated by DOTAP nanoparticles | Nonspecifically targets the lung, heart, liver, and lymphatic organs, activates systemic and intratumoral immunity within 24 h | [123] |
| Total tumor-derived RNAs are loaded into lipid nanoparticles | Promotes DC maturation, induces T lymphocytes to kill HEPA1-6 cells, prevents and inhibited the growth of HCC in vivo | [124] |
Although CSC vaccines have shown therapeutic potential in tumor therapy, the small fraction of CSCs in tumor tissues complicates the preparation and application of these vaccines. The number of CSCs isolated from a tumor during preparation is limited. Moreover, the small number of CSCs in tumor tissues hinders the precise targeting of CSCs by vaccines. Furthermore, some surface markers detected on CSCs are also found on normal stem cells [95], which may result in potential damage to healthy cells. Considering that the main goal of CSC vaccine therapy is to target residual CSCs, it could be used as an adjuvant therapy with surgery, chemotherapy, or radiotherapy.
Whole tumor cell vaccines can provide a wide range of personalized tumor antigens (including TAAs and neoantigens) and epitopes of CD4+ and CD8+ T cells [96], thereby offering advantages for tumor treatment (Table 1). As we have described above, engineering can increase immune factor release, enhance tumor immunogenicity, and develop carriers for the delivery of chemotherapeutic drugs, nanoparticles, and antibodies. Despite progress in laboratory studies, clinical translation has been difficult, with most vaccine developments being discontinued when clinical trials stop after Phases I and II. On one hand, the reasons for the suboptimal treatment outcomes may include low vaccine immunogenicity, in vivo immune escape, and the immunosuppressive nature of the TME [10]; combination therapy may overcome these obstacles, for instance, by using a combination of immune checkpoint inhibitors. On the other hand, tumor cells derived from patients may not be sufficient to prepare an adequate dose of whole-cell vaccine, and in vitro expansion of tumor cells is required. The bioactivity of laboratory-expanded or modified cells may differ from that of cell lines grown in vivo. Based on such risks and negative clinical trial results, whole-cell vaccines may not represent ideal tumor vaccines on their own; as a result, research in recent years has focused on tumor cell-derived vaccines. In the diversification of immunotherapy research, there is much scope for the development of whole tumor cell vaccines, with the potential-strengthening strategies of combination therapies and nanomaterials. Photodynamic therapy [97] and photothermal therapy-induced ICD can convert tumor cells into in situ vaccines, thereby avoiding complex cell preparation. In addition, the use of intact cells as a physical platform to efficiently deliver adjuvants or nanomaterials into the body may be used to enhance cellular vaccines synergistically.
3.2. Tumor cell-derived nanovaccines
Unlike intact tumor cells that are introduced into the body as a vaccine, tumor cell-derived nanovaccines are produced by extruding intact tumor cells directly into nanovesicles. The nanovaccine not only retains all the antigens of the tumor cells but can also overcome cell or tissue barriers and exhibit longer circulation times [98]. The nanovesicles used here have similar characteristics to exosomes, such as surface-labeled proteins and morphological size; however, they produce a 100-fold higher yield than do exosomes [99]. Compared to whole tumor cells, nanovesicles not only retain TAAs and cytoplasmic contents of tumor cells, but also have smaller particle sizes, which enable them to overcome biological barriers and circulate longer. Notably, APCs prefer to phagocytose nanoparticles. Therefore, nanovaccines are more likely to be phagocytosed by APCs, which increases lymph node retention, thereby activating lymphocyte-mediated antitumor immune responses [100]. Genetic engineering techniques can enable tumor cell-derived nanovesicles to carry specific molecules. For example, CAFs are the most abundant stromal cells in TMEs, and they promote tumor cell proliferation and invasion. Therefore, they have been selected as targets for tumor immunotherapy. Tumor cells have been genetically engineered to express FAP [101] and then extruded through polycarbonate porous membranes to obtain a large number of nanovesicular vaccines. These vaccines contained both tumor self-specific antigens and FAP antigens, thereby targeting both tumor parenchymal and mesenchymal cells and achieving significant antitumor effects in tumor-bearing mouse models (Table 1). Nanoengineering is therefore an economically viable and sensible strategy compared to the isolation of exosomes. Stable production lines and easy access to the relevant technology are essential for clinical translation and large-scale application. In the field of material design, less means more.
3.3. Tumor cell lysate vaccines
A wide variety of TAAs that can improve antitumor immunity are induced by the production of homologous tumor lysates. Tumor lysate antigen-based vaccines appear to have a greater clinical benefit than peptide-based approaches in cancer immunotherapy [102,103]. Moreover, the simultaneous use of major histocompatibility complex (MHC) class I- and II-restricted antigens from tumor cell lysates (TCLs) would likely lead to a more robust antitumor response and longer-lasting memory of T cells [104]. Despite this, few TSAs are present in TCLs, suggesting their weak immunogenicity. Moreover, TCLs have a rapid degradation rate when entering the body, and APCs do not efficiently take up these lysates [105]. Therefore, engineering strategies have focused on modifying TCLs, aiming to enhance their immunogenicity (Table 1).
Tumor cells have been genetically engineered by regulating crucial molecules in the tumor immune response to enhance their immunogenicity. Blocking the STAT3 signaling pathway in hepatocellular carcinoma (HCC) cells could inhibit the proliferation and promote the apoptosis of tumor cells. The efficacy of STAT3-blocked whole-cell lysate as a vaccine against HCC has been verified [106], as the vaccine promoted the formation of immune memory against HCC in vivo. In contrast, upon being injected with HCC cells, mice developed a secondary immune response that inhibited tumor growth and tumorigenesis while prolonging survival. The combination of a whole-cell vaccine with a checkpoint inhibitor produced broad tumor-specific cellular immunity against neuroblastoma and melanoma tumors in mice.
Effective antigen delivery is a critical step in cancer vaccination, and nanoparticle-based delivery is expected to improve this aspect. Wang et al. [105] covalently attached TCLs to polydopamine (PDA) nanoparticles (TCL@PDA) (Fig. 2B and C). The lysate antigen was enriched in free amine and sulfhydryl groups, which were covalently attached to the catechol group in PDA by Michael addition and Schiff base reactions under simple mixing. The TCL@PDA nanoparticles facilitated antigen uptake by DCs and enhanced surface molecule expression and cytokine secretion, delaying tumor development. Whole-cell lysis fractions contain water-soluble and water-insoluble antigens. If a water-insoluble component is added to a tumor vaccine, a greater variety of tumor antigens are delivered to the body. A higher load of antigens in tumor vaccines would lead to a more extensive cancer cell-specific immune response, representing a more effective vaccine. Therefore, polymeric nanoparticles can be used to maximize the loading of tumor antigens. Previously [21], whole-cell lysis fractions were simultaneously loaded in the interior and on the surface of poly(lactic-co-glycolic acid) (PLGA) nanoparticles (Fig. 2A). Meanwhile, immune adjuvants were coloaded into nanovaccines to increase their efficacy. These nanovaccines prevented lung cancer and melanoma in 100% and 70% of mice, respectively. The nanovaccines were effective in treating melanoma and triple-negative breast cancer in mice and cured 25% of them. Upon combination with the immune checkpoint inhibitor PD-1 antibody, the cure rate of the nanovaccine for melanoma-bearing mice increased to 40%. Another study showed that treatment with hypochlorite enhanced the immunogenicity of TCLs [107]. To stimulate greater immunogenicity, Berti et al. [102] used PLGA nanoparticles loaded with hypochlorite-oxidized tumor lysates, which could be efficiently internalized by DC uptake and induce efficient DC maturation. Accordingly, animals in the PLGA nanoparticle formulation group survived longer than those in the free oxidized tumor lysate vaccine group. Another example of the delivery of TCLs with nanoparticles is the use of chitosan nanoparticles with mannose modified on the surface as a carrier (Man-CTS NPs) [108]. Mannose receptors are expressed by immature DCs, enabling DCs to detect Man-CTS NPs and thereby increasing the uptake of antigens associated with the nanoparticles. In addition, chitosan has potential proinflammatory properties, and Man-CTS NPs trigger significant adjuvant effects by stimulating the intrinsic immune response [109]. Man-CTS NPs encapsulated with TCL effectively enhance both cellular and humoral antitumor immunity, providing a viable therapeutic approach by increasing the efficacy of the antitumor immune response.
Fig. 2.

Tumor lysate vaccines delivered by carriers for cancer immunotherapy. (A) The preparation of nanovaccines and the tumor-specific immune responses induced by nanovaccines. Copyright © 2021, Wiley–VCH GmbH [21]. (B) Polydopamine nanoparticles loaded with whole tumor cell lysates (TCL@PDA) as a therapeutic vaccine for colorectal cancer. (C) Anticancer preventive effect of TCL@PDA in vivo at day 20. Copyright © The Royal Society of Chemistry 2019 [105]. (D) The PLEL-based combination strategy to amplify cancer immunotherapy. Copyright © 2021 Yang et al. [111].
Injectable hydrogels are sustained-release drug delivery systems that can be used as chemotherapy drug delivery systems and as good carriers for tumor vaccines [110]. Injectable hydrogels are soluble in vitro (which allows tumor antigens to be loaded internally) but transforms into gel upon injection into the body, thereby achieving a slow release of antigens in situ and potentially triggering a systemic antitumor immune response with local administration. Previously, a temperature-sensitive poly(D,l-lactide)-poly(ethylene glycol)-poly(D,l-lactide) (PLEL) hydrogel vaccine loaded with TCLs was combined with a PLEL hydrogel loaded with cyclophosphamide (Fig. 2D) [111]. Tumor cells killed by cyclophosphamide released personalized TAAs and immunostimulatory danger signals to trigger antitumor immunity, while the TCL vaccine promoted further activation and maturation of dendritic cells following uptake and recognition of tumor antigens. This combination therapy strategy enhanced the antitumor effects of TCL-based tumor vaccines and reduced the toxic side effects of conventional chemotherapy. In the same year, Yu et al. [112]reported that cell lysates extracted from excised tumors were used as antigens and loaded into a multiarmed poly(ethylene glycol) (8-armPEG)/oxidized dextran dynamically cross-linked hydrogel together with adjuvants. The results showed that DCs can be recruited into the in situ stroma by subcutaneous injection of hydrogels and trigger a strong tumor-specific immune response. The treatment effectively inhibited the postoperative growth of residual tumors in several mouse models, thus providing a personalized protocol for cancer prevention and treatment in postoperative patients. Moreover, TCLs loaded with self-assembled peptide hydrogels can induce strong T cell responses in vivo [108,[113], [114], [115]]. Injectable and self-assembled poly(l-valine) hydrogels have been employed as delivery vehicles for cargoes, including antigens and immune enhancers, to effect DC modulation. Their sustained release of TCLs and immune adjuvants efficiently recruits, activates, and matures DCs in vitro and in vivo [114]. Similarly, a PEG-b-poly(l-alanine) hydrogel loaded with dual checkpoint inhibitors displayed a continuous release of tumor antigens and GM-CSF, maintaining the recruitment and activation of DC while inducing a strong T cell response in vivo. This response was enhanced by dual immune checkpoint treatment, and the immunotherapy was superior to vaccination alone or immune checkpoint blockade alone. The proportion of activated effector CD8+ T cells was significantly increased in the spleens and tumors of immunized mice, whereas the proportion of Tregs decreased [113]. Therefore, injectable hydrogels provide a unique method for targeting the immunosuppressive TME with desirable features such as convenient synthesis, a high drug loading capacity, controllable release, and low toxicity. The peptide hydrogel-based TCL delivery system has great potential as a treatment modality for a variety of cancers.
Overall, TCLs have been used as an abundant antigen source to stimulate the immune system against cancer cells. TCL vaccines not only efficiently present multiple antigens to activate T cell responses but also provide immunomodulatory cytokines to DCs, thus inducing tolerogenic transformation [116]. However, soluble TCLs containing antigens and cytokines are inherently unstable, which often results in poor uptake by DCs, inefficient antigen cross-presentation, and limited induction of CTL responses. The development of suitable delivery vectors can overcome these limitations to improve the efficiency of antigen presentation. Furthermore, TCLs contain complex components, including mutated and unmutated tumor antigens, immunosuppressive cytokines, and nucleic acids. These inexact factors may be detrimental to the activation and delivery of APCs; therefore, purification methods must urgently be developed.
3.4. Whole tumor RNA vaccines
Tumor mRNA vaccines bear mRNA that simultaneously expresses multiple antigens, with the vaccine containing all antigenic sequences of the tumor, including those of TAAs and true TSAs. Tumor mRNA vaccines have several distinct advantages over conventional tumor vaccines. First, tumor mRNA can simultaneously encode multiple tumor antigens [117]. Second, mRNA can bypass the MHC classification restrictions and obtain immunogenicity without an adjuvant [118]. Third, mRNA vaccines are safer than DNA and live attenuated vaccines because they have no insertional mutations and do not integrate into the host genome. As the heterogeneity of tumor cells makes it difficult to identify specific antigens that are expressed by all tumor cells [119], whole tumor RNA vaccines (derived from the whole transcriptome) could transcribe whole tumor cell antigen components, thereby causing the extensive activation of specific tumor immune responses.
A significant drawback of RNA vaccines is their inherent instability. Naked RNA is rapidly degraded by RNases outside the cell, resulting in the inactivation of naked RNA before it can be localized to APCs. Furthermore, the negative charge of naked RNA could also significantly impede its internalization in APCs [120]. Therefore, more efficient drug delivery systems should be selected to facilitate the efficient delivery of RNA (Table 1). Nucleic acids such as RNA have numerous negatively charged phosphates, which can bind electrostatically to cationic substances. Cationic lipid nanoparticles (LNPs), such as cationic liposomes, are lipid vesicles that are widely used for the delivery of small molecules and nucleic acid drugs. In preclinical and clinical trials, the first generation of carriers for RNA delivery are cationic liposomes, which contain ionizing lipids. These lipids maintain a positive charge at all physiological pH values and readily condense anionic RNA. In particular, 1,2-dioleyl-3-trimethylammonium propane (DOTAP) is one of the most widely used cationic lipids that can efficiently encapsulate RNA [121]. Cationic liposomes with DOTAP-encapsulated mRNA increase the expression of MHC I/II B7 costimulatory molecules and maturation markers on splenic APCs, successfully inducing systemic immune activation [122]. Furthermore, a personalized RNA nanoparticle vaccine was produced by extracting whole tumor RNA from cells obtained from patient tumor biopsies, followed by encapsulation into lipid DOTAP nanoparticles [123]. The vaccine activated systemic and intratumoral immunity within 24 h, but the proportion of PD-L1+ CD86+ myeloid cells in systemic organs (e.g., spleen, bone marrow, liver) and the TME increased significantly. The concomitant administration of an anti-PD-L1 monoclonal antibody amplified the therapeutic efficacy of the vaccine. We have also successfully constructed a DC-targeted RNA LNP tumor vaccine using total RNA extracted from liver cancer cells [124].
LNP improves the RNA stability and transfection efficiency and promotes phagocytosis and antitumor immunity of DCs, thereby triggering specific antitumor immune responses.
Compared to the above specific tumor RNA antigens, whole-cell tumor RNA antigens offer a broad spectrum approach, eliminating the need to identify specific TAAs or neoantigens. Whole tumor RNA vaccines induce PD-L1 expression, and therefore, the combination of immune checkpoint inhibitors with whole tumor RNA vaccines may be required to produce synergistic antitumor efficacy. Furthermore, a variety of novel RNA carriers, such as pH-responsive ionizable lipids and polymeric materials, may be used to efficiently deliver whole tumor RNA.
4. Tumor-derived extracellular vesicles (TEVs)
Almost all types of cells secrete extracellular vesicles (EVs), which are crucial for intercellular communication. As a new type of cell-free therapy, extracellular vesicles are widely used in the treatment of various diseases, including cancer [37]. EVs are complex mixtures composed of multiple lipids, nucleic acids, and membrane proteins, which have tissue targeting ability. Therefore, they are often used as efficient carriers for various drugs [36,125,126]. EVs are divided into four subgroups: exosomes (30–150 nm in diameter), microvesicles (100–1000 nm in diameter), apoptotic bodies (100–5000 nm in diameter), and oncosomes (1–10 μm in diameter). TEVs are promising tumor vaccines due to the existence of immunogenic molecules, such as nucleic acids, TAAs, and damage-associated molecular patterns, which can stimulate the maturation of immune cells to initiate an antitumor response [[127], [128], [129]]. Current studies on the application of TEVs in vaccines have mainly focused on exosomes and microvesicles.
4.1. Tumor cell-derived exosome vaccines
Exosomes are EVs with a diameter of 30–150 nm composed of a lipid bilayer structure containing abundant proteins [130]. Tumor-derived exosomes (TEXs) from several cancers (including that of the kidney, blood, breast, and skin) carry a variety of functional molecular cargoes from cell membranes and nuclear endosomes of primary tumor cells that can be transferred to recipient cells [131,132]. The abundant proteins on the exosome membrane have efficient cellular uptake and targeted homing capabilities so that they can bring the internal cargo to the tumor site and promote immune responses [133]. The suppressed TME hinders immunotherapy, and pharmacological intervention in the metabolic circuit of the TME can potentially improve the therapeutic outcomes of immunotherapy. Antigens derived from donor tumor cells and found in TEXs are presented to MHCs after uptake by DCs, stimulating naïve T cells to generate antitumor responses. Immunostimulatory components, which can induce antitumor immune responses, are more abundant in TEXs than in cells [134,135]. The transmembrane CD47 on TEXs provides them with the ability to escape immune system attack [136]. In addition, the amount of TEXs released by cancer cells is generally 10 times higher than that released by normal cell-derived exosomes [95]. Wolfers et al. [137] highlighted the potential of TEXs in cancer immunotherapy for the first time in 2001. Since then, many studies have reported their ability as effective tumor vaccines to trigger T cell-mediated antitumor immune responses [[138], [139], [140], [141]]. However, the limited immunogenicity of TEXs often results in poor in vivo antitumor immunity. Therefore, TEXs have been engineered using various strategies to increase their immunogenicity (Table 2).
Table 2.
Tumor cell-derived exosome-based vaccines.
| Type of engineering | Engineering strategy | Effect of modification | Effect of vaccine | Ref. |
|---|---|---|---|---|
| Genetic engineering | Mouse melanoma cell lines transduced with CIITA-inserted retrovirus | Highly enriched MHC Class II on exosomes | Increases the expression of MHC class II molecule CD86 in DCs, higher mRNA levels of inflammatory cytokine TNF-α and maturation marker chemokine receptor 7 | [142] |
| Mouse melanoma cell lines transduced with CIITA-inserted retrovirus | Highly enriched MHC Class II on exosomes | Increases the expression of MHC class II molecules CD86 and CD80 in DCs, increases Th1 immune response | [143] | |
| Mda-mb-231 cells were transfected with lentivirus containing α-La RNA encoding sequence | α-LA is overexpressed in exosomes | Promotes in situ type 1 conventional DC activation, improves tumor-responsive CD8+T cell response | [47] | |
| Wild-type MC38 cell lines were transduced with lentiviral vectors encoding IL-12 gene and/or shRNA against TGF-β1 | Il-12 and TGF-β1 shRNA are overexpressed in exosomes | Reshapes TME to increase tumor-infiltrating CLT and NK cells to aid the therapeutic effect of DC vaccine | [145] | |
| HEK293 cells were transfected with pDisplay encoding GE11 | GE11-positive exosomes containing let-7a miRNA | Downregulates the expression of HMGA2 or RAS family members, inhibits breast cancer tumor growth | [146] | |
| Surface engineering | Combined adjuvant polyinosinic-polycytidylic acid | Inserted adjuvant polyinosinic-polycytidylic acid on exosomes surface | Stimulate DC maturation and activation, activate natural killer cells. Mediates Th1 immune response, delays tumor growth, inhibits B16 lung tumor nodules | [141] |
| Protein transfer | Staphylococcal enterotoxin A is anchored to the exosomal membrane | Enhances CTL response in vivo, stimulates antitumor effects of CD4+ T cells and NK cells | [147] | |
| Internal cargo loading | Electroporation | Exosomes contain sodium polytungstate and metformin | Inhibits distant metastasis of tumor, induces long-term immune memory | [150] |
| Cultured cells with medium containing HAuCl4 solution | Cells release exosomes containing Au nanoparticles | Homologous targeting and radiosensitization, strong immune response to cancer | [151] |
Direct genetic manipulation of tumor cells that secrete TEXs can increase the enrichment of their target molecules and enhance their immunostimulatory effect, which is a practical alternative to increasing immunogenicity. Lee et al. [142] transduced melanoma B16F10 cells with the MHC II transactivator protein CIITA (Class II transactivator) gene, and TEXs were produced by this species enriched with MHC class II molecules. The exosome vaccine increased the expression of MHC class II and CD86 on the surface of DCs and increased the mRNA levels of the inflammatory cytokines TNF-α and chemokine receptor 7. Therefore, CIITA-TEXs induced a significantly enhanced immune response. Fan et al. [143] reported similar results using CIITA-transduced exosomes from a mouse colon cancer CT-26 cell line. CT26-CIITA-derived TEXs increased Th1 immune responses, as evidenced by the significant increases in TNF-α, IFN-γ, and interleukin (IL)-12 and a significant decrease in IL-10 expression. In addition to the direct genetic manipulation of tumor cells that secrete TEXs, there is extensive research interest in combining engineered exosomes and immunostimulators. Huang et al. [47] engineered exosome HELA-Exos with a combination of TLR3 agonists and an ICD inducer. α-LA is a breast-specific immunodominant protein expressed in most human breast cancers [144]. After α-LA was overexpressed in triple-negative breast cancers, which are poorly immunogenic tumors, α-LA was further enriched as a specific tumor homing protein on the surface of exosomes to enhance targeting and immunogenicity. The HELA-Exos activated DCs in situ and specifically induced ICD in breast cancer cells. Moreover, inhibiting the expression of immunosuppressive factors in exosomes can further amplify the antitumor immune response. Rossowska et al. developed exosomes secreted from genetically engineered MC38 tumor cells overexpressing IL-12, and TGF-β1 shRNA, which further enhanced the antitumor activity of DC-based immunotherapy and inhibited tumor growth [145]. Similarly, the GE11 peptide efficiently transfected the gene into cells expressing high levels of EGFR or into tumor xenografts. Delivery of let-7a (a tumor suppressor miRNA) to EGFR-expressing cancer tissues using genetically modified surface GE11-positive exosomal vaccines significantly inhibited breast cancer tumor growth by downregulating the expression of HMGA2 or RAS family members [146].
The lipid membrane structure of exosomes allows them to carry multiple molecules for better antigen delivery, inhibiting immunosuppression and increasing circulating time. Through protein transfer, Xiu et al. [147] produced TEXs with staphylococcal enterotoxin A (SEA), including a SEA with a highly hydrophobic transmembrane structural domain at its tail (SEA-TM) on the exosome surface. Immunization of mice with modified TEXs resulted in increased IFN-γ and IL-2 secretion from T cells, enhanced CTL responses in vivo, and stimulated the antitumor effects of CD4+ T cells and NK cells. Treatment with SEA-TEX inhibited tumor growth and prolonged survival, with stronger effects than those of untreated TEXs or mixtures of TEXs and SEA. These effects might have occurred because SEA promotes the binding of TEXs to DCs while decreasing the immunosuppressive activity of Tregs.
Owing to their lipid bilayer structure, TEXs are also promising drug delivery systems for hydrophilic or hydrophobic drugs or even nanoparticles [148,149]. A CD39 inhibitor, sodium polytungstate, and the AMP-activated protein kinase agonist metformin (C-PMet) were electroporated into the interior of B16F10 tumor cell-derived exosomes (Fig. 3A and B) [150]. Therapeutic drugs are carried around the tumor by TEXs to synergistically inhibit tumor progression, prevent distant metastases, and provide long-term immune memory protection while improving antitumor efficiency. In addition to delivering small molecule drugs, TEXs can be manipulated to load gold nanoparticles. For example, tumor cells can be cultured in a medium containing HAuCl4 solution, and the cells can be used to synthesize gold nanoparticles and release them directly as TEX-encapsulated gold nanoparticles (Au@MC38) [151] (Fig. 3C and D). Au@MC38 was found to preserve the biological integrity of the original cancer cells and amplify radiation-induced DNA damage, generating reactive oxygen species (ROS); this exacerbated the ICD of tumor cells, thereby resulting in an improved immune response.
Fig. 3.

Exosomes with internal cargo as therapeutic tumor vaccines. (A) Antitumor immune responses induced by C-PMet-based immunometabolic therapy. (B) Therapeutic effect of inhibiting lung metastasis of tumors. Copyright © 2022 Wu et al. [150]. (C) The preparation of Au@MC38 and in vivo radiosensitization for cancer therapy. (D) In vivo homologous targeting and in vitro transcytosis. Copyright © 2021, Wiley–VCH GmbH [151].
Loading TEXs into DCs can also increase the ability of TEXs to activate immune responses, because T cell activation depends on MHC presentation of antigenic peptides. Moreover, TEX-loaded DCs can facilitate the processing of TEX-derived tumor antigens and the presentation of processed tumor antigenic peptides in MHC slots [138]. Wolfers et al. [137] demonstrated that human DCs loaded with melanoma TEXs induced the in vitro production of IFN-γ from CTL clones and increased the immune response against cancer. Rao et al. [152] also reported that TEX-DC-stimulated T cells presented higher cytotoxic capacity than tumor lysate-DCs [153], and they were more effective in inducing immune responses, as reflected by their higher expression of IFN-γ and lower expression of immunosuppressive IL-10 and TGF-β in HCC mouse models [154].
4.2. Tumor-derived microvesicle vaccines
Tumor-derived microvesicles (TMVs) are EVs ranging between 100 and 1000 nm in diameter secreted by tumor or other cells in the TME [155]. They are shed from the plasma membrane and carry a variety of tumor-associated nucleic acids, proteins, and additional bioactive substances that modulate the characteristics and activities of tumors, such as metastasis, invasion, angiogenesis, and immune response [156,157]. Several factors can increase the amount of TMVs shed from cells, including hypoxia [158] and irradiation [159]. For example, tumor cells induced by hypoxic conditions (1% O2) can release large amounts of TMVs into the peripheral circulation [160]. Pineda et al. used TMVs produced by C6 glioma cells after ionizing radiation as a therapeutic tumor vaccine [161]. This vaccine was found to release tumor antigens upon irradiation, contributing to T cell infiltration in immunized rat tumors and promoting tumor cell death, thereby significantly reducing tumor volume.
TMVs can contain the tumor antigen profile and carry innate DNA signals [162,163]. These DNA components are involved in the cGAS/STING pathway to induce type I IFN, which effectively promotes DC maturation, T cell activation, and tumor rejection. Furthermore, TMVs can be absorbed by DCs and enter lysosomes, thereby increasing the pH within lysosomes, which facilitates the processing and presentation of TMVs carrying tumor antigens [164]. Moreover, TMVs can also lead to the release of calcium ions in lysosomes, promote the dephosphorylation of the transcription factor EB into the nucleus, regulate the expression of CD80 and CD86, and promote the activation and maturation of DCs. These studies demonstrate the great potential of TMVs as tumor vaccines.
Although systemic administration by injection is the most common mode of vaccine administration, the oral route has considerable advantages owing to its simplicity, safety, and induction of mucosal and systemic immune responses [165,166]. Dong et al. administered a tumor-derived TMV vaccine to mice orally and it was absorbed by ileal epithelial cells (IECs) [167]. Through NOD2 signaling, TMV activates the ileal epithelium, causing IECs to release chemokines that attract CD103+ CD11c+ DCs. The TMV vaccine was transported to the basolateral side by IECs, and DCs captured and effectively cross-presented TMV-derived antigens to activate CD8+ T cells. In malignant tumors, Tregs aid in the immune escape of tumor cells so that the patient's immune system is considerably less effective in fighting the tumor [168]. Parenky et al. also extracted TMV from mouse prostate cancer cells as an oral tumor vaccine [169]. When combined with cyclophosphamide and GM-CSF, oral vaccines can significantly reduce Treg numbers in vivo.
EVs play a crucial role in immune regulation, intercellular communication, and inflammatory responses, with TEVs carrying tumor-specific antigens, cytokines, and nucleic acids that can activate immune cells, representing an ideal tumor vaccine for cancer therapy. Nevertheless, research on TEXs and TMVs are still at the initial stages, and the therapeutic applications of TEVs are mainly explored in preclinical animal studies. A deeper understanding of the interactions between cell-specific molecules on exosomes and immune cells, as well as tumor cells, will help provide the foundation for the development of new exosome-based biomarkers and effective drugs for cancer immunotherapy. In addition, TEVs face several challenges with respect to characterization, purification, fabrication, and upscaled production [170]; among them, the differentiation and isolation of EVs are the main challenges. TMVs overlap with TEXs in size (100–1000 nm and 10–150 nm diameters, respectively), and there is still no universal biomarker to distinguish the various isoforms of TEVs. Therefore, their separation is a barrier to the clinical quality grading of their vaccines. Only after accurate isolation of extracellular vesicles can we gain insight into the characteristics and labeling of each subpopulation and elucidate the role of each subset in cellular communication and physiological processes. In addition, the mass production of exosomes is challenging. Quality classification and standards for biopharmaceuticals should be addressed before EV vaccines can be formally used in the clinic, and specific Good Manufacturing Practice regulations need to be developed by the FDA at the earliest to ensure the safety of exosome treatments. Efforts have already been made to establish quality control of extracellular vesicles [171], and various studies have reported strategies that can increase the yield of exosomes (including genetic manipulation and adjustment of cell culture media) [172,173]. In the future, novel engineering tools could effect more powerful antitumor immune responses to TEVs, and integration of clinical data and emerging technologies presents the foundation for the clinical application and production of TEVs.
5. Tumor cell membrane-derived vaccines
Although whole-cell vaccines have been studied in clinical trials for many years, they have not produced significant long-term therapeutic effects, possibly because of the excess of nontumor-associated antigenic materials [174,175]. Tumor cell membranes possess several unique characteristics as an ideal tumor vaccine. First, tumor cells are easily cultured in vitro on a large scale, representing an abundant source. Second, tumor cell membranes are rich in tumor antigens [56,176] because they possess membrane proteins and polysaccharides containing TAAs or TSAs [177]. Third, tumor cell membranes can be modified or reconfigured, providing flexibility for the membrane coating platform. The development of more personalized and novel therapeutic approaches using primary tumor cell membranes might represent a promising strategy for the development of cancer cell membrane (CCM) systems. The engineering methods and therapeutic effects of tumor cell membrane-derived vaccines are listed in Table 3.
Table 3.
Tumor cell membrane-derived vaccines.
| Type of engineering | Engineering strategy | Effect of vaccine | Ref. |
|---|---|---|---|
| Genetic engineering | KillerRed (KR)-expressing plasmid is transfected into tumor cells to express KR protein on the cell membrane surface | KR proteins produces cytotoxic reactive oxygens species under laser irradiation at 510–600 nm | [184] |
| Surface engineering | Cholesterol-modified CpG and DC-SIGN aptamer are inserted onto tumor cell membrane vesicles | Targets DCs, improves delivery to lymph nodes, increases the nanovaccine efficacy | [179] |
| B7-1 and IL-12 bind to glycosylphosphatidylinositol, which anchors them to tumor cell membrane vesicles | Induces antitumor protective immunity, inhibits tumor growth, improves the survival rate of mice with squamous cell carcinoma | [180] | |
| 4T1 (breast cancer) cell membranes expressing photosensitizer was fused with recombinant liposome containing monophosphoryl lipid A adjuvant | Promotes generation of cytotoxic T lymphocytes after DC maturation and photodynamic therapy, eradicates primary tumors, inhibits metastasis | [184] | |
| Internal cargo loading | 4T1 (breast cancer) cell membranes were coated with liquid metal nanoparticles | Promotes maturation and activation of antigen-presenting cells, inhibits tumor growth in mouse breast tumor model | [32] |
| 4T1 (breast cancer) cell membrane were coated with PSiNPs@Au | Activates antitumor immune response in vivo, reverses the immunosuppressive microenvironment to eliminate the established solid tumor and inhibits its metastasis | [192] | |
| B16–F10 cell membranes were coated with PLGA nanoparticles containing CpG sequences | Activates antigen-presenting cells to secrete pro-inflammatory cytokines IL-6 and IL-12, promotes DC maturation to provide a strong antitumor response | [174] | |
| 4T1 (breast cancer) cell membrane were coated with PLGA nanoparticles containing imiquimod | Increases CD8+ T cells, decreases regulatory T cells, increases effector memory T cells in the spleen | [67] | |
| MCF-7 cell membranes were coated with PLGA nanoparticles containing indocyanine green | Displays homologous targeting in vitro and tumor targeting in vivo, with high spatial resolution and penetration depth | [187] |
5.1. Tumor cell membrane vaccines
In 1974, Hollinshead et al. [178] proposed that malignant melanoma could be treated by vaccination using the soluble membrane antigen fraction of tumor cells. Compared to cellular vaccines, tumor cell membrane vesicles do not contain genetic material and have the advantages of better biosafety, easier mass manufacturing, longer storage time, and good tumor-targeting ability owing to the homology of the outer membrane. To increase the effectiveness of the vaccine, adjuvant components can be modified directly on the tumor cell membrane surface. Liu et al. [179] successfully constructed a tumor cell-derived DC-targeted tumor vaccine (CMV-CpG/Apt) by inserting cholesterol-modified CpG and cholesterol-modified DC-SIGN aptamer as adjuvants into tumor cell membrane vesicles (Fig. 4A and B). CMV-CpG/Apt could specifically target DCs for rapid accumulation in lymph nodes, which improved their delivery efficiency and triggered stronger antitumor immune responses. Notably, CMV-CpG/Apt effectively inhibited tumor growth and produced long-term immune memory. In addition to the conjugation of ligands to increase targeting to immune cells, researchers have attempted to incorporate immunostimulatory molecules into tumor cell membrane vesicles. Previously, vaccines were produced by anchoring two immunostimulatory molecules (B7-1 and IL-12) to GPI and then binding them to tumor membrane vesicles (Fig. 4C and D) [180]. The expression of GPI-B7-1 and GPI-IL-12 on tumor cells induces potent antitumor protective immunity [181]. When such a vaccine was administered to mice bearing head and neck squamous cell carcinoma tumors, tumor growth was inhibited and survival rate was increased. This application has shown similar results in both oral and breast cancer tumors [182]. In addition, immunostimulatory adjuvants are often incorporated into vaccines to promote their immune response initiation activity. For example, the lipid adjuvant monophosphoryl lipid A was efficiently embedded in lipid membranes and induced DC maturation by targeting Toll-like receptor 4 [183]. Kim et al. introduced a KillerRed (KR) protein-expressing plasmid into 4T1 breast cancer cells to obtain KR-containing tumor cell membranes; liposomes containing monophosphate lipid A were hybridized with KR-embedded CCMs, and a composite vaccine with KR-CCM and adjuvant functions was synthesized (Lp-KR-CCM-A) (Fig. 4E) [184]. The KR protein is a red fluorescent photosensitizer that effectively produces cytotoxic ROS when irradiated with laser light between 510 and 600 nm [185]. The plasmid contained genes encoding membrane localization signal peptides; therefore, the KR protein was firmly anchored in the lipid bilayer of the CCM. Lp-KR-CCM-A produced cytotoxic ROS after photodynamic therapy, which fully stimulated tumor immune activation and inhibited primary tumor growth and lung metastasis. Using such purified tumor cell membranes with appropriate modifications as antigen sources can improve effective immune responses.
Fig. 4.

Cancer cell membranes combined with immune adjuvants as a nanovaccine for cancer immunotherapy. (A) Preparation of a DC-targeted tumor vaccine with cholesterol-modified CpG and cholesterol-modified DC-SIGN aptamer inserted on the surface of the tumor cell membrane (CMV-CpG/Apt). (B) The therapeutic effect of CMV-CpG/Apt in B16-OVA tumor-bearing mice. Copyright © 2021, American Chemical Society [179]. (C–D) Therapeutic efficacy of a tumor cell membrane vaccine with surface modifications of glycolipid-anchored immune stimulatory molecules GPI-B7-1 and GPI-IL-12 combined with anti-PD-1 mAb, inhibiting MOC1 tumor growth (C) and MOC2 tumor growth (D). Copyright © 2020 Bommireddy et al. [180]. (E) Cancer therapy with lipocomplexes (Lp-KR-CCM-A). When laser irradiated, Lp-KR-CCM-A produces reactive oxygen species and kills cancer cells [184]. Copyright © 2019, American Chemical Society.
5.2. Tumor cell membrane-coated nanoparticle vaccines
With the development of nanomaterials, nanoparticles have increasingly been used in medical research, especially in the field of drug delivery. However, the human body is a complex environment that is adept at recognizing and excluding foreign elements; therefore, when these nanoparticles enter the body, they become highly susceptible to uptake and clearance by the reticuloendothelial system. To address this issue, cell membrane-based nanoparticles have been developed. Many cell membranes, including erythrocyte, platelet, macrophage, bacterial, neutrophil, and tumor cell membranes, can be used for nanoparticle coating [39]. The properties of tumor cells, such as immune activation, immune escape, and the ability for “self-homing,” are primarily mediated by membrane proteins on their surfaces [185]. Therefore, this feature can be exploited to replicate these surface antigen structures from cells to nanoparticles using tumor cell membrane encapsulation, which confers long circulation capability to nanoparticles and transports them to self-targeting homologous tumors [177]. Therefore, nanoparticles coated with tumor cell membranes may enhance the development of tumor vaccines by broadening their design possibilities and application potential. Some studies have reported the encapsulation of nanoparticles (such as metal-organic framework nanoparticles [33], gold nanoparticles [151,186], and PLGA nanoparticles [187]) with CCMs for cancer therapy.
Tumor cell membrane-coated inorganic nanoparticles represent a promising treatment modality against cancer. The physicochemical properties of inorganic nanoparticles, including photothermal properties, are useful in improving the therapeutic efficacy of tumor vaccines. A typical example is that using photothermal materials can cause local inflammation in situ, promoting the recruitment of APCs and stimulating cellular uptake [188]. A tumor cell membrane-coated liquid gold metal nanoparticle (LMNP) has been developed as a tumor preventive vaccine [32]. Photothermal conversion was induced by LMNP injection when it was irradiated with near infrared light within the 808 nm wavelength, and APCs were recruited due to the photothermal effect. The APCs internalized LMNP present in the antigens, thereby inducing an antitumor immune response and inhibiting tumor growth in a mouse mammary tumor model. Furthermore, inorganic nanoparticles encapsulated in cell membranes can act as drug carriers to deliver therapeutic or adjuvant molecules. For example, porous silicon (PSi) has an important function in drug delivery owing to its porous structure, biodegradability, and compatibility [[189], [190], [191]]. In addition, PSi microparticles can promote antigen cross-presentation and IFN-γ secretion to achieve antitumor effects [153]. Researchers constructed a novel nanovesicular vaccine, defined as CCM@(PSiNPs@Au), by combining Psi nanoparticles, gold elements, and tumor cell membranes, which showed superior immunostimulatory and photothermal effects [192]. CCM@(PSiNPs@Au) exhibited good biosafety, acted as a photothermal agent to eliminate established solid tumors, and inhibited their metastasis by initiating antitumor immune responses in vivo and reversing the immunosuppressive microenvironment.
The application of inorganic nanoparticles is occasionally constrained by concerns regarding their potential toxicity. In contrast, organic polymeric materials such as PLGA have been approved by the FDA as safe for clinical applications [127], and they are commonly used as biomaterials for drug delivery because of their good biocompatibility, degradability, resistance to degradation of carried substances, and slow release [185,193,194]. In tumor cell membrane-derived nanovaccines, PLGA loaded with immune adjuvant allows stable delivery to the target site and activates the immune response. Kroll et al. [174] encapsulated CpG sequences into biodegradable PLGA nanoparticles, coating them with membranes obtained from mouse B16F10 melanoma cells to form adjuvant-loaded tumor cell membrane-coated nanoparticles. The produced nanoparticles were more readily endocytosed by DCs, activated the secretion of the proinflammatory cytokines IL-6 and IL-12, and promoted the maturation of DCs. Researchers have also developed a vaccine consisting of PLGA nanoparticles containing the TLR7 agonist imiquimod encapsulated by CCM [67]. The vaccine could promote the maturation of DCs and increase the antitumor response against breast cancer 4T1 cells in vitro. Mice with established immune memory exhibited tumor growth inhibition and longer survival (following vaccination, 75% of mice survived longer than 50 d after tumor formation). In addition, photosensitizers have been coloaded in PLGA nanoparticles to further increase their therapeutic efficacy. Chen et al. encapsulated a tumor membrane with indocyanine green (ICG)-containing PLGA nanoparticles (ICNPs) to simultaneously recognize and eradicate tumors [187]. The cell membrane surface was modified with polyethylene glycol (PEG) to avoid aggregation and nonspecific phagocytosis of ICNPs in vivo. The ICNPs showed homologous and specific targeting with deep tumor penetration. With laser treatment and a single dose of ICNPs, complete tumor ablation was achieved, showing promising synergistic antitumor efficacy in vivo.
In summary, cell membrane coating methods that transfer the original tumor cell membrane to the surface of nanoparticles have been applied successfully to design nanovaccines for immunotherapy. Numerous cargoes can be loaded into the inner core of these nanoparticles, while they can be coated with a variety of cell membranes, conferring a broad spectrum of therapeutic potential for cancer immunotherapy. Reconstruction or modification of the outer membrane layer to add functionality provides further flexibility to the membrane coating platform.
5.3. Hybrid membrane-coated nanoparticle vaccines
Although tumor cell membrane-coated nanoparticles show great potential for tumor therapy, limitations such as short circulation and low immunogenicity impede their application. Contrary to monotypic cell membranes, the nanoparticles of hybrid membranes can inherit the prominent features of the tumor-targeting and immunostimulatory capabilities of tumor cell membranes while acquiring additional features (such as long circulation) from other cell membranes. These hybrid membranes can be extracted from hybrid cells by fusing two cells or by fusing two cell membranes extracted from separate cells [195].
Erythrocytes are the most predominant blood cells; because of their inherent biocompatibility and long circulation ability, the erythrocyte membrane was the first to be used for biomimetic nanoparticles [196]. Furthermore, CD47 molecules (expressed at high levels on erythrocyte membranes) reduce phagocytosis by reticuloendothelial cells and inhibit in vivo clearance. In particular, incomplete erythrocytes are transported to the spleen by macrophages and DCs. Several immune cells reside in the spleen, an important secondary lymphoid organ. Therefore, the erythrocyte membrane can be fused with tumor cells, deliver tumor antigens to immune cells in the spleen, and activate immune responses. Han et al. [197] designed an erythrocyte-tumor cell membrane-coated nanosilver vaccine. The system exhibited a significant ability to target splenic APCs, thereby activating various immune cells in the spleen and providing a more active T cell immune response. Use of this vaccine suppressed tumor metastasis in B16F10-Luc murine melanoma and 4T1-Luc murine mammary carcinoma models. Wang et al. [198] fused membranes from erythrocytes and melanoma cells to prepare drug-laden hollow copper sulfide nanoparticles coated with a hybrid membrane (DCuS@[RBC-B16] NPs) for treating melanoma. The modified nanoparticles showed a significantly increased circulating lifetime and homogeneous targeting in vivo.
Tumor antigens (both known and unknown) are highly expressed on tumor cell membranes, but the lack of immune costimulatory molecules leads to inefficient delivery of tumor antigens to APCs, thereby limiting the effectiveness of immune stimulation [36]. In contrast, DCs express high levels of the B7 family of immune costimulatory molecules, and they also recognize antigens and present them to the cell membrane as MHCs. Therefore, DC-tumor fusion cells express high levels of antigens and costimulatory molecules while enhancing T cell activation [199]. Lymph nodes and spleens accumulate significantly higher levels of membrane-encapsulated nanoparticles from DC-tumor fusion cells [200]. In previous studies [33,201], hybrid membranes derived from fused cells formed by DCs and 4T1 tumor cells were wrapped around the surface of PCN-224 nanoparticles (NP@FMs). The developed vaccine inherited and even amplified most interface properties of the two parental cells, including specific targeting of homologous tumors and lymph node homing ability, and the proportion of CD8+ CTLs induced by NP@FMs was substantially higher than that of single membrane-coated nanoparticles (Fig. 5).
Fig. 5.

Hybrid cell membrane nanovaccines based on DCs and tumor cells for cancer immunotherapy. (A) Schematic of fused cells membrane-coated PCN-224 (PCN@FM) for combined tumor therapy and (B) evaluation of homotypic targeting, immune activation, and PDT efficacy in vitro [33]. Copyright © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (C) Schematic of fused cells membrane-coated PCN-224 nanoparticles (NP@FM) for tumor prevention and the mechanisms by which NP@FM induces immune responses. (D) The effect of fused cells membrane-coated PCN-224 MOF (MOF@FM) as a preventive vaccine against tumor suppression [201]. Copyright © 2019, Liu et al.
Immune cells such as macrophages are capable of targeting metastatic cancer cells by mechanisms such as convergent targeting of lesion sites, penetration of blood vessels, and interaction through adhesion molecules such as integrins. Both M1 macrophages and their derived exosomes have good tumor targeting and deep tumor penetration abilities [202]. Therefore, the extraction of macrophage membranes and their encapsulation onto nanoparticles can effectively increase the targeted accumulation of nanoparticles in tumors [203]. This active targeting ability is mainly attributed to the presence of specific proteins on the macrophage membrane surface, such as Toll-like receptors, which recognize the tumor endothelium [204]. Rao et al. [205] extracted the cell membranes of M1 macrophages, platelets, and cancer cells with high affinity for the signal regulatory protein α variant to produce vaccines of mixed cell membrane vesicles. The vaccine accumulated reasonably at the wound site after surgery and interacted effectively with tumor cells circulating in the blood. Furthermore, it promoted macrophage phagocytosis and enhanced antitumor immunity by repolarizing the TME into the M1 phenotype and blocking the CD47-signal regulatory protein α interaction.
In addition to the aforementioned intrinsic cell membrane components, bacterial outer membranes can also be combined with tumor cell membranes to produce hybrid membrane vaccines. Bacterial outer membranes are composed of various immunostimulatory components that recruit immune cells in vivo; they can be used as immunostimulants or natural adjuvants to enhance immune performance. Previously, tumor cell membranes and the outer membranes of Salmonella were fused to construct a eukaryotic–prokaryotic vesicle (EPV) nanoplatform [125] (Fig. 6). EPVs inherited the benefits of the original composition, and melanoma antigens were integrated with this natural adjuvant to stimulate a tumor-specific immune response. In vivo prophylactic trials have shown that EPVs act as a prophylactic vaccine, in addition to providing high levels of tumor infiltration (16.4%) by CD8+ CTLs. On this basis, EPV can be combined with ICG-based PTT and immunotherapy. PLGA-ICG nanoparticles (PI) have been implanted into EPV (PI@EPV), and the therapeutic vaccine demonstrated a synergistic antitumor effect combining local elimination and persistent inhibition.
Fig. 6.

Fusion of bacterial outer membranes/tumor cell membrane-based nanovaccines for immunotherapy. (A) The fabrication of eukaryotic-prokaryotic vesicle-coated PI@EPV nanovaccines. (B) Fusion membrane vesicle endocytosis by DC2.4 murine dendritic cells (C) Tumor growth curves in a melanoma breast cancer model after immunization with the nanovaccine. (D) Percentage of tumor-free mice after tumor challenge. (E–F) The proportions of splenic lymphocytes expressing different T cells after immunization. Copyright © 2020, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim [125].
Overall, cell membrane-based bionanotechnology is widely studied and is often employed to develop tumor vaccines based on the presence of bioactive surface molecules, facilitating synergy with other therapeutic modalities. Tumor vaccines fused with cell membranes have shown promising therapeutic efficacy against cancer cells because they can inherit the prominent features of tumor cell membranes and other cell membranes. More membrane types (such as those from bacteria and algae) and application areas remain to be explored. Currently, the development of cell-membrane nanovaccines is still in its infancy, and certain issues must be resolved to optimize the effectiveness of their use. Cell membrane production technology lacks a common process or quality control. It must be established how acceptable levels of batch variation can be achieved during mass production. Tumor cell lines cultured in the laboratory are different to those cultured in vivo, and different tumor cell membrane surface molecules have varying biological properties. The extraction method should be carefully selected because complex procedures can affect the integrity of membrane proteins. Moreover, the biosafety of the hybrid membrane cell method cannot be guaranteed from the few existing studies. In particular, as the application range of hybrid membrane vaccines is steadily increasing, there is a growing concern regarding the safety and storage stability of materials for clinical applications. As the exploration of this bionic strategy continues, its true potential will be revealed, and researchers will attempt to translate it into clinical applications that could substantially impact human healthcare.
6. Treatment strategies using tumor cell-derived vaccines
To develop tumor cell vaccines and effective immunotherapies for cancer, an understanding of the mechanisms responsible for the immunity against tumors is required (Fig. 7). These treatment strategies can be summarized as follows. For tumor vaccines to effectively stimulate antitumor immunity, APCs should absorb antigens and be activated to induce strong and sustained helper T cell and CTL responses. Efficient antigen uptake by APCs is particularly important in this process. Therefore, tumor-derived vaccines have been designed to target DCs and deliver large amounts of high-quality antigens. Specific targeted molecular modifications of the vaccine facilitate the accumulation of DCs in lymph nodes [179,206]. Thereafter, antigens and adjuvants in the vaccine stimulate the maturation of DCs, and the mature DCs then present the antigens to activate T cells, which release cytotoxic effectors. In addition, the homologous targeting of tumor-derived vaccines allows them to effectively target tumor tissue in vivo, thereby releasing tumor antigens that attract immune cells to the tumor site. Utilizing the homologous targeting of tumor vaccines, some chemotherapeutic drugs can be effectively delivered to the tumor site by tumor-derived vaccines [193,207]. After efficient delivery, chemotherapeutic drugs cause apoptosis of tumor cells while changing from nonimmunogenic cells to immunogenic cells, thereby causing a certain degree of tumor-specific immune response, i.e., ICD [36,110]. Pattern recognition receptors expressed on DCs are capable of recognizing damage-associated molecular patterns released during ICD. The DCs that acquire antigens around tumor cells transfer the antigens to lymph node-resident DCs to activate T cells, which return to the tumor site and impede tumor growth by directly killing tumor cells and releasing type II interferon [208,209]. In addition, ICD promotes the release of type I interferons and chemokines and the recruitment of APCs and T cells, further activating immunity. Tumor-derived vaccine-delivered ICD inducers, photodynamic therapy, and PTT can provide ICD with a tumor killing ability [20].
Fig. 7.

Therapeutic strategies of engineered tumor cell-derived vaccines. These strategies include the delivery of adjuvants, chemotherapy drugs, photosensitizers, radioactive molecules, antigens, MHC molecules, and immunomodulatory molecules, which can activate the immune system and reverse the suppressive tumor microenvironment.
Tumor cells tend to evade immune attacks through multiple mechanisms. For instance, hypoxia, acidification, immune checkpoint overexpression, accumulation of regulatory immune cells, and infiltration of tumor-associated macrophages in tumor tissues create an immunosuppressive microenvironment that can severely impede immunotherapy. Conventional protein vaccines that rely on the delivery of antigen alone cannot overcome this immunosuppressive TME. Therefore, to achieve a more effective tumor therapy, vaccine strategies have been developed based on the characteristics of the TME [150]. For example, immune checkpoint inhibitors and tumor vaccine-induced T cell responses can address the lack of sufficient endogenous viable T cells infiltrating the immunosuppressive TME. Vaccines can be combined with immune checkpoint inhibitors to suppress immune resistance [210]. Modulating the expression of immune-stimulating genes in whole tumor cell vaccines can also improve immunosuppression in TMEs [88]. For example, a hybrid membrane nanovaccine composed of tumor cells and macrophages promoted the conversion of macrophages to the M1 phenotype in the TME [205]. FAP genetically engineered tumor vaccines target CAFs in tumor tissue by reprogramming the immunosuppressive TME to inhibit tumor growth and by activating CTLs to release IFN-γ and consume FAP+ CAFs to promote iron death in tumors [101].
Integration of inorganic or organic nanoparticles with tumor-derived vaccines is also an effective strategy to increase the immunogenicity of tumor vaccines. Inorganic nanoparticles, such as gold nanoparticles, can produce mild thermal effects in situ. This causes local inflammation, which in turn stimulates the active recruitment of APCs, initiates an antitumor immune response in vivo, and modifies the TME [32,192]. Moreover, gold is associated with a good X-ray absorption coefficient, and radiation-induced DNA damage and ROS generation can be amplified, thereby exacerbating apoptosis and necrosis. Local radiation-induced ICD also initiates immune responses [151]. ZnO can serve as a site for redox reactions that produce ROS and increase oxidative stress [211]. Organic nanoparticles, such as biodegradable PLGA nanoparticles, are widely used in tumor vaccines and have promising applications. For example, PLGA nanoparticles can enhance the delivery of vaccine components, protect antigens and adjuvants from degradation, and control antigen release, thereby prolonging antigen presentation and allowing simultaneous delivery of both antigens and adjuvants to the same APC [212]. The uptake of PLGA microspheres loaded with antigen and photosensitizer by APCs also promoted the active translocation of APCs to draining lymph nodes after light exposure, which resulted in a strong CD8+ T cell response [213].
7. Tumor cell-derived vaccines in clinical trials
Tumor cell-derived vaccines (such as whole tumor cell vaccines, tumor cell-derived nanovaccines, and tumor cell lysate vaccines derived from intact tumor cells) have obvious tumor targeting properties and are rich in specific antigens that can avoid the immune escape caused by tumor antigen loss [214]. However, intact tumor cell vaccines contain an excess of nontumor-associated antigenic material [175], and the inherent instability of soluble tumor lysates often results in poor DC uptake, inefficient antigen cross-presentation, and limited induction of CTL responses [215]. Engineering techniques can be applied to address these challenges. For example, tumor lysates can be loaded into PLGA microspheres and hydrogels to promote antigen uptake by APCs and increase T cell responses. However, whole-cell vaccine production remains complex and time consuming. EVs, as a third mechanism of intercellular communication [216], have a wide range of bioactive molecules both on the membrane surface and within the membrane and are natural intercellular carriers of protein, mRNA, and miRNA, thus presenting great potential in the field of immunotherapy and drug delivery. EVs have inherent bionic properties and the ability to fuse with specific target cells to transfer membrane proteins and internal components. Exosomes, in particular, are suitable for long-term cryopreservation, which is highly conducive to the production and storage of large amounts of tumor vaccines for subsequent drug delivery. However, even though tumor cells secrete 10 times more exosomes than normal cells do, the exosomes yield is still very low and their accurate isolation is challenging, rendering current technology unsuitable for large-scale production. Artificial membrane vesicles can be obtained by extrusion of broken tumor cell membranes. They are similar to EVs, in that, they possess many tumor antigens on their surface but no excess nontumor antigen-related material, which is ideal for triggering tumor-specific immune responses [217,218]. Compared with exosomes, the extraction and preparation of cell membranes formed by membrane vesicles is straightforward, the vaccine preparation process is established, and the ultimate yield is much higher. Nevertheless, many cells still need to be cultured to obtain enough membranes. The membrane extraction process may cause loss of proteins, and targeting ability may subsequently be impaired (Table 4).
Table 4.
Advantages and limitations of tumor cell-derived vaccines.
| Whole cell component | Extracellular vesicle | Cell membrane-coated nanoparticles | |
|---|---|---|---|
| Extraction and isolation | Easy isolation in the lab | Difficult to isolate efficiently, with limited scalability | Easy to separate and obtain using established processes, but with limited scalability |
| Engineering | Easy to be genetically engineered | Sensitive to physical and chemical modifications | Withstands moderate physical and chemical changes and multiple engineering strategies |
| Biosafety and immunogenicity | A source of all potential specific antigens, but with an excess of nontumor-associated antigenic substances that limits immunogenicity | Good biocompatibility with a wide range of bioactive molecules (e.g., antigens, nucleic acids) both on the membrane surface and inside the membrane | Good biocompatibility, inheriting the complex surface protein structure of the source cell, and has a large number of tumor antigens without excess nontumor antigen-related substances |
| Quality control and production | Production is complex and time consuming: large numbers of cells still need to be cultured under cGMP conditions, and the reproducibility is affected by the condition of the source cells | Sensitive to parental cell conditions and isolation methods, but suitable for long-term storage; standardized production and quality control methods remain to be established | Reproducibility is affected by the state of the source cell; standardized production and quality control methods remain to be established |
Among the three main bioengineering sectors, genetic engineering has been developed over a long period and, with the rapid evolution of technology, gene editing has become economical and faster. Gene modification is often used to enhance protein or cytokine expression to increase immunogenicity. It is a more flexible engineering approach than surface modification methods, which are limited by the inherent functional groups present on the cell membrane surface. Clinical trials testing whole-cell vaccines for tumors as developed via genetic modification are very common and show promising clinical applications. In contrast, the biosafety of vaccines produced by surface engineering (both modification by chemical means or internal loading) has not been fully demonstrated in humans. Physical adsorption is convenient and fast, but it is difficult to control the adsorption strength and release in vivo. Such biomedical applications still need to be explored in vivo.
To date, only sipuleucel-T (Provenge®) has been approved for marketing by the FDA to treat metastatic prostate cancer. Nevertheless, the clinical benefits of sipuleucel-T are limited and several engineered tumor cell-derived vaccines have entered FDA clinical trials. Incomplete statistics show that more than 20 tumor vaccine programs are undergoing or have completed phase III clinical studies, and nearly 80 tumor cell-derived vaccines are in phases I or II of clinical studies (clinicaltrials.gov). Most of them focus on DC or tumor whole-cell vaccines. For the latter, trials have focused on unmodified autologous or allogeneic tumor immunization, often combined with other immune-modulating agents. However, the vast majority of whole tumor-cell vaccines remain in phase I or II trials owing to unsatisfactory efficacy, and few have entered phase III trials (Table 5). The immune mechanism of whole-cell vaccines still remains to be investigated, and more effective whole-cell vaccines in the context of individual patients should also be explored. In addition, for tumor EVs, tumor cell membrane-coated nanoparticles used as vaccines are rarely seen in clinical trial programs owing to limitations such as safety, stability issues, and difficulty in large-scale production. Future studies investigating these challenges can assist in the development of more effective tumor vaccines with good potential for clinical translation.
Table 5.
Clinical trials of tumor cell-derived vaccines.
| Vaccine Type | Strategy | Condition or disease | Treatment(s) | Trial phase | NCT number | Dose and schedules |
|---|---|---|---|---|---|---|
| Whole tumor cell vaccine | Autologous or allogeneic tumor | |||||
| Mesothelioma | Drug: Celebrex | I | NCT01258868 | 1 × 107 to 1 × 108 patient tumor cells every four weeks for six injections; if subject shows immune response, vaccine will be given every three months from months 9–24 | ||
| Esophageal cancer | ||||||
| Lung cancer | ||||||
| Thoracic sarcomas | ||||||
| Thymoma | ||||||
| Metastatic solid tumors | Biological: Tumor cell vaccine | I, II | NCT00148993 | – | ||
| Malignant mesothelioma | Biological: PA-1-STK ovarian carcinoma vaccine | I | NCT00006216 | – | ||
| Drug: Ganciclovir | ||||||
| Melanoma | Biological: autologous, DNP-modified vaccine (M-Vax) | I, II | NCT00257465 | Four doses of 5.0 × 106, 2.5 × 106, 0.5 106, or 0 cells; eight injections of the vaccine will be administered as an injection into the skin of the arm over a 6-month period; all participants receive low dose cyclophosphamide and BCG | ||
| Biological: autologous, DNP-modified melanoma vaccine | ||||||
| Biological: autologous, DNP-modified vaccine | ||||||
| Non-small cell lung cancer | Drug: 1650-G Vaccine | II | NCT00654030 | Patients receive 2 injections of 1650-G Vaccine given 4 weeks apart, for a total of 52 weeks on study. | ||
| Lung cancer | Drug: autologous tumor cell vaccine | I | NCT00098917 | Patients receive autologous DC loaded with irradiated autologous tumor cells intradermally on approximately days 1, 30, and 60 in the absence of unacceptable toxicity. | ||
| Drug: therapeutic autologous dendritic cells | ||||||
| Colorectal neoplasms | Biological: autologous tumor cell + CpG vaccine | I | NCT00780988 | Patients will receive subcutaneous vaccination at weeks 1 and 2 after resection. If needed patients will receive chemotherapy for tumor reduction. When disease is controlled off chemotherapy, patients will receive a conditioning regimen of fludarabine (30 mg/m2 daily × 3 days) followed by intensive fractionated total body irradiation. The dose of fTBI will be escalated using a 3 + 3 design to ensure safety and will range from 400 to 800 Gy. The patient will then undergo hematopoietic and immune cell rescue. They will undergo a third vaccination within 7–14 days after transplant. | ||
| Multiple myeloma | Biological: autologous tumor cell vaccine | I | NCT00469820 | – | ||
| Plasma cell neoplasm | Biological: peripheral blood lymphocyte therapy | |||||
| Skin melanoma | Biological: bystander-based autologous tumor cell vaccine | II | NCT00101166 | Intradermal vaccine injections at 28-day intervals for a total of three immunizations (performed on days 1, 29, and 57) | ||
| Chronic myeloid leukemia | Biological: Allogeneic tumor cell vaccine | I, II | NCT00162513 | Six vaccine doses, 2 weeks apart. | ||
| Breast cancer | Biological: allogeneic tumor cell vaccine, autologous tumor cell vaccine, recombinant interferon alfa, recombinant interferon gamma, sargramostim | II | NCT00002475 | Treatment repeats every 2 weeks for three courses. | ||
| Colorectal cancer | ||||||
| Kidney cancer | ||||||
| Lung cancer | ||||||
| Malignant mesothelioma | ||||||
| Pancreatic cancer | ||||||
| Genetically modified tumor cell | ||||||
| Ewing's sarcoma | Biological: Vigil™ | I | NCT01061840 | Once a month and up to 12 doses. | ||
| Non-small cell lung cancer | ||||||
| Liver cancer | ||||||
| Ovarian cancer | Biological: Vigil™ | II | NCT01309230 | Intradermal autologous Vigil™ (1.0 × 107 cells/injection, maximum of 12 vaccinations) | ||
| Stage III/IV ovarian cancer | Biological: Vigil™ vaccine | II | NCT01551745 | Intradermal injection of Vigil™ (1.0 × 107 cells once a month, maximum of 12 doses); Bevacizumab 10 mg/kg intravenously (prior to Vigil™ administration) every 2 weeks | ||
| Drug: Bevacizumab | ||||||
| Advanced melanoma | Biological: Vigil™ vaccine | II | NCT01453361 | Intradermal injection (1 × 107 cells once a month, maximum of 12 doses) | ||
| Breast cancer | Biological: autologous, lethally irradiated breast cancer cells | I | NCT00880464 | Doses of approximately 1 × 107, 4 × 106, 1 × 106, or 1 × 105 cells (depending on the final cell yield) administered on days 1, 8, 15, 29, and then every 2 weeks until the vaccine supply is exhausted or the patient is removed from study | ||
| Breast cancer | Biological: autologous, lethally irradiated breast cancer cells | I | NCT00317603 | Administered on days 1, 8, 15, 29, and then every 2 weeks until the vaccine supply is exhausted | ||
| Lung neoplasm | Biological: Lucanix | II | NCT01058785 | Patients are randomized to receive either 1.25 × 107, 2.5 × 107, or 50 × 107 cells per injection once a month (maximum of 16 injections) | ||
| Bronchogenic carcinoma | ||||||
| Colon cancer | Biological: Vigil™ vaccine<Drug: placebo | II | NCT01505166 | Patients will receive 1 × 107 cells (Group A) or placebo (Group B) via 5–12 doses of intradermal injections starting 4–8 weeks post-surgery (C1W1D1) and continuing C1W3D1, C2W3D1, then every 28 days. Starting C1W4D1, all patients will receive oxaliplatin 85 mg/m2, l-leucovorin 200 mg/m2, fluorouracil 400 mg/m2 IV bolus, and fluorouracil 2400 mg/m2 for 46 h continuous infusion every 14 days for six cycles (one cycle = 4 weeks) | ||
| Acute myeloid leukemia | Biological: AML cell vaccine | I | NCT02493829 | – | ||
| Myelodysplastic syndromes | ||||||
| Lung neoplasms | Biological: Hyperacute lung cancer cell vaccine | I | NCT00073398 | Intradermal injection every four weeks for four cycles. Dosage will vary from 3 × 106 to 100 × 106 HyperAcute lung cancer vaccine cells | ||
| Pancreatic cancer | Biological: PANC 10.05 pcDNA-1/GM-Neo and PANC 6.03 pcDNA-1 neo vaccine | II | NCT01088789 | Administered every 6 months | ||
| Metastatic breast cancer | Biological: KS24.22 cells | I | NCT01127074 | The first four vaccinations, which were given every two weeks, were followed by four monthly vaccinations. | ||
| Kidney cancer | Biological: IL-2 | II | NCT00031564 | At 3–6 weeks after surgery, patients receive B7-1 gene-modified autologous tumor cell vaccine subcutaneously once on days 1, 29, and 57; 6 weeks after the first vaccination, patients receive IL-2 subcutaneously for 5 days a week for 6 weeks | ||
| Biological: B7-1 | ||||||
| Metastatic colorectal cancer | Drug: CYBiological: GVAXDrug: SGI-110 | I | NCT01966289 | Cyclophosphamide is administered intravenously at 200 mg/m2; GVAX is administered intradermally at 5E8 colon cancer cells + 5E7 GM-CSF secreting cells; SGI-110 is administered subcutaneously at 60 mg/m2; each cycle is 28 days | ||
| Prostate cancer | Biological: Immunotherapy allogeneic GM-CSF secreting cellular vaccine | I, II | NCT00140374 | – | ||
| Prostate cancer | Biological: Immunotherapy allogeneic GM-CSF secreting cellular vaccine | I, II | NCT00140387 | – | ||
| Colorectal cancerMetastatic cancer | Biological: colon GVAXDrug: cyclophosphamide | I | NCT00656123 | Dose escalation of 1.4 × 108 to 7 × 108 cells, administered in up to 15 intradermal injections on day two of cycles 1–4; cyclophosphamideis administered intravenously at 200 mg/m2 on day 1 of cycles 1–4 | ||
| Prostate cancer | Biological: GM-CSF gene transduced allogeneic vaccine GVAX | I, II | NCT00122005 | – | ||
| Melanoma | Biological: Tag-7 gene modified inactivated tumor cells | I, II | NCT04180774 | Vaccinations every 3 weeks, dosage of 10 million transfected and inactivated tumor cells | ||
| Kidney cancer | ||||||
| Pancreatic cancer | Biological: GVAX pancreatic cancer vaccine | II | NCT00084383 | First vaccination 6–8 weeks after surgery; eligible patients receive three additional vaccinations at one-month intervals; patients who remain disease-free receive a fifth booster at 6 months following the fourth vaccination | ||
| Lung cancer | Biological: GVAX lung cancer vaccine | II | NCT00074295 | Patients receive GVAX lung cancer vaccine intradermally (ID) (6–7 injections per vaccination) on weeks 1, 3, 5, 7, and 9 for a total of 5 vaccinations. Treatment continues in the absence of disease progression or unacceptable toxicity. | ||
| Surface modified tumor cells | ||||||
| Colorectal cancer | Biological: Autologous or Allogeneic tumor cells | I, II | NCT00722228 | Five vaccine doses, 3 weeks apart, injected subcutaneously | ||
| Ovarian cancer | ||||||
| Gastric cancer | ||||||
| Breast cancer | ||||||
| Lung cancer | ||||||
| Kidney cancer | ||||||
| Melanoma | ||||||
| Dendritic cells loaded with pulsed tumor cell lysate-based vaccine | Autologous dendritic cells & autologous/allogeneic tumor lysate | |||||
| Metastatic pancreatic cancer | Biological: KLH-pulsed autologous dendritic cell vaccine | II | NCT00868114 | Weekly dose of 5 × 107 KLH-pulsed autologous dendritic cells for 3 weeks | ||
| Non-Hodgkin's lymphoma | Biological: tumor-pulsed dendritic cells | III | NCT00006434 | – | ||
| Stage IV colorectal cancer | Biological: autologous dendritic cells loaded with autologous tumour homogenate | II | NCT02919644 | Intradermal injection of autologous dendritic cells loaded with autologous tumor homogenate (day 1); subcutaneous injection of IL-2 for five days (days 3–7) | ||
| Curative resection | Drug: IL-2 | |||||
| Glioblastoma | Biological: autologous dendritic cells pulsed with tumor lysate antigen vaccine | II | NCT03014804 | Vaccination on days 0, 7, and 14, and at weeks 4, 6, 8, 11, 14, 17, and 20 | ||
| Drug: Nivolumab | ||||||
| Giant cell glioblastoma | Biological: malignant glioma tumor lysate-pulsed autologous dendritic cell vaccineDrug: Temozolomide | I | NCT01957956 | Intradermal vaccinations with malignant glioma tumor lysate-pulsed autologous dendritic cell vaccine on day 1 or on days 1, 3, and 5 | ||
| Glioblastoma | ||||||
| Gliosarcoma | ||||||
| Metastatic melanoma | Drug: GM-CSF | II | NCT00948480 | A series of eight vaccinations administered over 6 months | ||
| Renal cell carcinoma | Biological: allogeneic DCs and autologous RCC tumor derived cells | I | NCT00050323 | – | ||
| Ovarian neoplasms | Biological: CSC-DC | I, II | NCT02178670 | – | ||
| Sarcoma | Hematopoietic stem cell transplantation | II | NCT00405327 | – | ||
| Neuroblastoma | ||||||
| Wilm's Tumor | ||||||
| Kidney cancer | Biological: autologous tumor cell vaccine, keyhole limpet hemocyanin, therapeutic autologous dendritic cells | II | NCT00093522 | Arm I: 3 weeks after leukapheresis, patients receive intradermal vaccination comprising DCs loaded with autologous tumor lysate and KLH once every 14 days for a total of four injections; Arm II: 2 weeks after leukapheresis, patients receive fludarabine intravenously for 15–30 min once daily for 3 days; at 5 weeks after leukapheresis, these patients also receive DC vaccine as in arm I | ||
| Drug: fludarabine phosphate | ||||||
| Procedure: conventional surgery | ||||||
| Lung cancer | Biological: autologous tumor cell vaccine, therapeutic autologous dendritic cells | I | NCT00023985 | Patients receive autologous tumor lysate-pulsed dendritic cell vaccine subcutaneously twice, 4 weeks apart | ||
| Procedure: conventional surgery | ||||||
| Mesothelioma | Drug: MesoPher | II, III | NCT03610360 | Starting 9–13 weeks after the last chemotherapy dose, three biweekly injections with MesoPher; if stable, an additional two injections at weeks 18 and 30 (a maximum of five doses) | ||
| Fusion cell-based vaccine | DC-tumor fusion cell | |||||
| Breast cancer | Biological: Dendritic Cell/Tumor Fusion Vaccine | I, II | NCT00622401 | A subcutaneous injection of tumor cells fused with dendritic cells every 3 weeks for a total of 9 weeks | ||
| Drug: Interleukin-12 | ||||||
| Acute myelogenous leukemia | Drug: decitabine | I | NCT03679650 | Two vaccines, 3 weeks apart, with potential for a booster vaccine | ||
| Biological: DC/AML fusion cells | ||||||
| Renal cancer | Biological: Dendritic Cell Tumor Fusion Vaccine | I, II | NCT00458536 | The treatment will consist of 3 vaccinations of fused cells given by an injection under your skin at 3-week intervals. The first six participants will receive only the study vaccine. The remaining participants will receive the study vaccine combined with GM-CSF. | ||
| Drug: GM-CSF | ||||||
| Multiple myeloma | Biological: Dendritic Cell Tumor Fusion Vaccine | I | NCT00459069 | Administered on weeks 0, 3, and 6 | ||
| Ovarian cancer | Drug: imiquimod, GM-CSFBiological: dendritic cell tumor fusion vaccine | II | NCT00799110 | Subcutaneous vaccinations once every 3 weeks for a total of three vaccines | ||
| Primary peritoneal cancer | ||||||
| Fallopian tube cancer | ||||||
| Multiple myeloma | Single agent | I | NCT00458653 | Subcutaneous administration every 4 weeks for three doses | ||
| Metastatic melanoma | Single agent | I | NCT00626860 | Vaccinations administered at 3-week intervals for 2–3 doses | ||
| Glioblastoma | Biological: dendritic cell/tumor fusion vaccineDrug: IL-12, Temozolomide (TMZ) | I, II | NCT04388033 | Surgery is followed by concomitant radiation and TMZ-chemotherapy (75 mg/m2 per day for 42 days); maintenance chemotherapy with TMZ is administered at 150–200 mg/m2/day for 5 days in each 28-day cycle; fusion cells are injected intradermally; 6 μg of IL-12 is injected twice subcutaneously over an interval of 1 h | ||
| Glioma | ||||||
| Neuroectodermal tumors | ||||||
| Leukemia | Drug: autologous tumor cell vaccine | I | NCT00100971 | After completing chemotherapy, patients receive vaccine subcutaneously every 2 weeks for a total of four doses | ||
| Drug: therapeutic autologous dendritic cells | ||||||
8. Summary and perspectives
In this paper, we extensively reviewed the progress of engineered tumor cell-derived vaccines (including whole tumor cells, exosomes, and cell membrane-encapsulated nanoparticles), which can deliver tumor antigens abundantly and activate a broad range of antitumor immune responses (Table 6). Although efficacy and safety have prevented large-scale clinical translation of tumor vaccines, genetic engineering may overcome this obstacle by modifying tumor cells to produce molecules containing specific immune stimuli that enhance immunogenicity. Tumor cell membrane vaccines have superior homologous tumor targeting properties, and an introduced nanoparticle core can further enhance their immunostimulatory properties. Briefly, bioengineering has enhanced the intelligence, targeting, and functionality of tumor cell-derived vaccines. Following modifications, these vaccines are either loaded with cargo, combined with chemotherapy or light/radiation (leading to the release of TAAs from ICDs to enhanced immune activation), or possess an increased content of tumor antigens, immunomodulatory molecules, MHC molecules, and immune adjuvants, thus improving their preventive and therapeutic efficacy (Fig. 8).
Table 6.
Characteristics of tumor cell-derived vaccines.
| Vaccine type | Cell-derived components | Feature | Reference |
|---|---|---|---|
| Tumor cell-derived vaccines | Whole tumor cells | Contain intact autoantigens, reducing the chance of tumor escape | [79,83,85,[88], [89], [90]] |
| Tumor cell-derived nanovesicles | Contain the full range of tumor cell antigens; nanoscale particle sizes can improve pharmacokinetics by overcoming cell or tissue barriers and prolonging circulation times | [101] | |
| Tumor lysates | Contain a wide range of tumor-associated epitopes, but the low number of tumor-specific antigens may induce a transformation of DC tolerance | [21,105,106,108,[111], [112], [113], [114]] | |
| Tumor stem cells | Contain CSC vaccine-initiated antibodies and T cells that selectively target tumor stem cells to inhibit tumor growth, reduce the development of primary tumor lung metastasis, and prolong survival | [62,94] | |
| Whole tumor RNAs | Contain all the antigen sequences of the tumor; can induce stronger and more durable antitumor immunity; has self-adjuvant properties; do not integrate mutations into the gene sequence; pose no risk of infection | [119,123,125] | |
| Tumor extracellular vesicles | Exosomes | Contain a variety of functional molecular cargoes from primitive tumor cell membranes and endosomes; avoid immune rejection; release TEXs in large numbers; are easy to obtain | [47,137,142,143,145,147,[150], [151], [152]] |
| Microvesicles | Contain a variety of tumor-related nucleic acid proteins and other bioactive substances; can regulate the characteristics and activities of tumors, including tumor metastasis, invasion angiogenesis, and immune regulation | [161,167,169] | |
| Tumor cell membrane-driven vaccines | Tumor cell membranes | Contain tumor-associated antigens or tumor-specific antigens; do not contain genetic material; have increased biological safety; pose easier mass manufacturing and longer storage time; have targeting ability | [179,180,182,184] |
| Tumor cell membrane-coated nanoparticles | Combine with nanoparticles loaded inside to start immunotherapy | [32,67,174,187,192] | |
| Fusion membranes | Possess tumor targeting, stronger immune stimulation, longer circulation time, and enhances growth of specific organs (such as liver and spleen) | [33,36,197,198,201,205] |
Fig. 8.

Summary of tumor-derived nanovaccines. Tumor tissue/cells removed from the patient or cultured in the laboratory can be extracted to obtain the desired tumor cell components. After engineered modifications, they can deliver a variety of active substances to the tumor originator and exert antitumor effects through different strategies. In the future, we will need to pay attention to standardized quality specifications during production to ensure a pure, safe, and economically viable vaccine, which is a challenge (Created with BioRender.com).
Tumor cells can play different roles in vaccines. When used as a coating, the tumor cell membrane can act as camouflage to avoid the clearance of nanoparticles by the reticuloendothelial system, thus prolonging the circulation time. Furthermore, it can increase the accumulation of therapeutic nanoparticles in the tumor tissue by homologous targeting of tumor cells. When tumor cell exosomes and microvesicles are used as delivery vehicles, the reduced particle size increases stability and prolongs circulation time while increasing lymph node targeting, allowing immune-activating antigens to reach the lymph nodes directly to activate lymphocytes. The engineering strategies that allow greater flexibility in the implementation of these specific delivery functions are best reflected in genetic engineering, for which the clinical safety and efficacy of gene modification have been tested thoroughly; genetically modified properties can also be inherited by tumor cell-derived vaccines. This convenient modification strategy showcases a major advantage of tumor cell-derived tumor vaccines over other vaccines.
The immunosuppressive microenvironment is a major barrier to the therapeutic effect of tumor cell-derived vaccines. Engineered vaccines allow the immunosuppressive microenvironment to evolve toward supporting tumor immunity by releasing various immunomodulatory factors, targeting tumor-associated stromal cells, and reducing tumor suppressor immune cells. The expression of these immune factors can, on one hand, be genetically engineered to express specific cytokines and, on the other hand, be stimulated by several exogenous toxins. The level of immunosuppression in the TME directly influences the responsiveness of tumor cells to tumor vaccine stimulation and the resultant clinical benefits. Although immune checkpoint inhibitors have been widely used in tumor immunotherapy in recent years, their side effects have become more prominent with an increase in clinical trials. Since the immune system operates in a Janus fashion, systemic inhibition in one direction may cause unintended side effects in another direction. Compared to checkpoint inhibitors, most tumor vaccines achieve immunotherapeutic sensitization through local modulation of the immune microenvironment while reducing systemic side effects.
Although many tumor cell-derived vaccines have shown significant immune responses and antitumor efficacy in animals, only a few whole-cell vaccines have been evaluated in clinical trials, which suggests that numerous methodological problems regarding tumor cell-derived vaccines remain to be overcome. Few antitumor vaccines have achieved the same clinical translation as anti-infection vaccines, and a major challenge is the low immunogenicity of tumors and associated antigens, which, unlike pathogens such as bacteria, originate from a patient's own healthy tissue and are thus difficult for the autoimmune system to distinguish. Advances in engineering strategies can partially address this problem, but there is still a long way to go before tumors can be cured. One reason for the low immunogenicity of tumor cell-derived nanovaccines is the insensitivity of the immune system to tumor components from autologous sources. Bacteria, as foreign substances in vitro, are more likely to elicit an immune response from the body. Combining bacterial components (such as bacterial membranes) with tumor cell-derived components may be an effective approach, and researchers are already producing fused cell membrane vaccines from bacterial and tumor cell membranes. As we have summarized in a recent review, microalgae and their extracts can serve as immunomodulators, photosensitizers, and oxygen generators, thereby representing an ideal and natural weapon against cancer [183]. The combination of tumor cells and microalgae may exert a synergistic antitumor effect. On one hand, tumor cells can use homologous targeting to deliver microalgae to the tumor site for effective accumulation within the tumor. On the other hand, microalgae can act as oxygen generators, alleviating the tumor hypoxia and sensitizing other tumor treatment strategies. Another impediment to the clinical translation of tumor vaccines is that the current preparation process leads to uncontrollable stability and low quality of tumor cell-derived vaccines, and the yield is relatively low for clinical use, so higher quality large-scale preparation methods need to be explored. In addition, the long-term safety of tumor cell-derived vaccines has not been evaluated in depth. Moreover, most biomaterials are not licensed for clinical use, which is a major obstacle for the clinical application of tumor cell-derived vaccines engineered using biomaterials. Among the biomaterials commonly employed in tumor vaccines, polyalpha-hydroxy acids (such as PLGA) were the first to be approved by the FDA for use in humans and are currently used in a variety of applications [219]. The degradation products of these biomaterials are lactic and hydroxyacetic acid, which are eventually excreted as carbon dioxide, water, and hydroxyacetic acid which can participate in the tricarboxylic acid cycle or be excreted in the form of urine. Therefore PLGA is a promising carrier material. Among the inorganic biomaterials, Prussian blue is an FDA-approved antidote against radioactive poisoning by elements such as thallium [[220], [221], [222]]. Prussian blue-based tumor vaccines enable simultaneous photothermal, photodynamic, and chemodynamic therapy while acting as an ICD inducer to synergistically increase antitumor immunity [[223], [224], [225]].
The goal of therapeutic tumor vaccines is to achieve clinical translation that ultimately induces tumor regression, eradicates minor tumors, and establishes a durable immune memory with minimal side effects. We offer the following views on the future of therapeutic tumor vaccine development: (1) Novel tumor vaccines must be based on a more comprehensive and in-depth understanding of the tumor immune system. Advances in basic research will provide a theoretical basis for the evolution of therapeutic strategies. (2) Tumor vaccines that underperformed in clinical trials nevertheless increased our knowledge base, allowing for continuous improvements by adapting engineering strategies to reap more significant vaccine efficacy. (3) Breakthroughs in computer algorithm design and high-throughput sequencing have already elevated the accuracy of antigen prediction, making personalized analyses possible [9,10,226]. Personalized tumor vaccines will eventually provide more effective and higher quality antigenic epitopes to achieve definitive tumor cures. We are confident that these step-by-step improvements will eventually lead to tumor elimination.
Declaration of competing interest
The authors do not have any conflicts of interest to declare.
Funding information
This study was supported by the National Natural Science Foundation of China (Nos. 82072051 and 81771964 ).
Author contributions
Conceptualization, Xinyi Zhang, Hengqing Cui, Wenjun Zhang; writing-original draft preparation, Xinyi Zhang; review and editing, Jie Gao, Hengqing Cui, Wenjun Zhang; supervision, Jie Gao, Zhaoshen Li. All authors have read and agreed to the published version of the manuscript.
Ethical statement
Hereby, I Prof. Jie Gao, consciously assure that for the manuscript entitled “Engineered tumor cell-derived vaccines against cancer: the art of combating poison with poison” the following is fulfilled:
1)This material is the authors' own original work, which has not been previously published elsewhere.
2)The paper is not currently being considered for publication elsewhere.
3)The paper reflects the authors' own research and analysis in a truthful and complete manner.
4)The paper properly credits the meaningful contributions of co-authors and co-researchers.
5)The results are appropriately placed in the context of prior and existing research.
6)All sources used are properly disclosed (correct citation). Literally copying of text must be indicated as such by using quotation marks and giving proper reference.
7)All authors have been personally and actively involved in substantial work leading to the paper, and will take public responsibility for its content.
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Contributor Information
Zhaoshen Li, Email: zhsl@vip.163.com.
Jie Gao, Email: gaojiehighclea@163.com.
References
- 1.Wolchok J. Putting the immunologic brakes on cancer. Cell. 2018;175(6):1452–1454. doi: 10.1016/j.cell.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Riley R.S., June C.H., Langer R., Mitchell M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019;18(3):175–196. doi: 10.1038/s41573-018-0006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Me A., C X., Jj M. Engineered nanoparticles for cancer vaccination and immunotherapy. Acc. Chem. Res. 2020;53(10) doi: 10.1021/acs.accounts.0c00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020;11(1):3801. doi: 10.1038/s41467-020-17670-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akinleye A., Rasool Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019;12(1):92. doi: 10.1186/s13045-019-0779-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abd-Aziz N., Poh C.L. Development of oncolytic viruses for cancer therapy. Transl. Res. 2021;237:98–123. doi: 10.1016/j.trsl.2021.04.008. [DOI] [PubMed] [Google Scholar]
- 7.Lawler S.E., Speranza M.C., Cho C.F., Chiocca E.A. Oncolytic viruses in cancer treatment: a review. JAMA Oncol. 2017;3(6):841–849. doi: 10.1001/jamaoncol.2016.2064. [DOI] [PubMed] [Google Scholar]
- 8.Qin L., Zhang H., Zhou Y., Umeshappa C.S., Gao H. Nanovaccine-based strategies to overcome challenges in the whole vaccination cascade for tumor immunotherapy. Small. 2021;17(28) doi: 10.1002/smll.202006000. [DOI] [PubMed] [Google Scholar]
- 9.Sahin U., Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359(6382):1355–1360. doi: 10.1126/science.aar7112. [DOI] [PubMed] [Google Scholar]
- 10.Saxena M., van der Burg S.H., Melief C.J.M., Bhardwaj N. Therapeutic cancer vaccines. Nat. Rev. Cancer. 2021;21(6):360–378. doi: 10.1038/s41568-021-00346-0. [DOI] [PubMed] [Google Scholar]
- 11.Blass E., Ott P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021;18(4):215–229. doi: 10.1038/s41571-020-00460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finn O.J. Cancer vaccines: between the idea and the reality. Nat. Rev. Immunol. 2003;3(8):630–641. doi: 10.1038/nri1150. [DOI] [PubMed] [Google Scholar]
- 13.Buonerba C., Ferro M., Di Lorenzo G. Sipuleucel-T for prostate cancer: the immunotherapy era has commenced. Expert Rev. Anticancer Ther. 2011;11(1):25–28. doi: 10.1586/era.10.180. [DOI] [PubMed] [Google Scholar]
- 14.Bouzid R., Peppelenbosch M., Buschow S.I. Opportunities for conventional and in situ cancer vaccine strategies and combination with immunotherapy for gastrointestinal cancers, A review. Cancers. 2020;12(5):E1121. doi: 10.3390/cancers12051121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welters M.J., van der Sluis T.C., van Meir H., et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci. Transl. Med. 2016;8(334) doi: 10.1126/scitranslmed.aad8307. [DOI] [PubMed] [Google Scholar]
- 16.Mazzarella L., Duso B.A., Trapani D., et al. The evolving landscape of “next-generation” immune checkpoint inhibitors: a review. Eur. J. Cancer. 2019;117:14–31. doi: 10.1016/j.ejca.2019.04.035. [DOI] [PubMed] [Google Scholar]
- 17.Mariathasan S., Turley S.J., Nickles D., et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saxena M., Bhardwaj N. Re-emergence of dendritic cell vaccines for cancer treatment. Trends Cancer. 2018;4(2):119–137. doi: 10.1016/j.trecan.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y., Slone N., Chrisikos T.T., et al. Vaccine efficacy against primary and metastatic cancer with in vitro-generated CD103+ conventional dendritic cells. J Immunother Cancer. 2020;8(1) doi: 10.1136/jitc-2019-000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korbelik M. Optimization of whole tumor cell vaccines by interaction with phagocytic receptors. Vaccines (Basel). 2021;9(8):904. doi: 10.3390/vaccines9080904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma L., Diao L., Peng Z., et al. Immunotherapy and prevention of cancer by nanovaccines loaded with whole-cell components of tumor tissues or cells. Adv. Mater. 2021;33(43) doi: 10.1002/adma.202104849. [DOI] [PubMed] [Google Scholar]
- 22.Qiu S., Wei Q., Liang Z., et al. Biodegradable polylactide microspheres enhance specific immune response induced by Hepatitis B surface antigen. Hum. Vaccines Immunother. 2014;10(8):2350–2356. doi: 10.4161/hv.29559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pavot V., Berthet M., Rességuier J., et al. Poly(lactic acid) and poly(lactic-co-glycolic acid) particles as versatile carrier platforms for vaccine delivery. Nanomedicine (Lond) 2014;9(17):2703–2718. doi: 10.2217/nnm.14.156. [DOI] [PubMed] [Google Scholar]
- 24.You G., Kim Y., Lee J.H., Song J., Mok H. Exosome-modified PLGA microspheres for improved internalization into dendritic cells and macrophages. Biotechnol Bioproc E. 2020;25(4):521–527. doi: 10.1007/s12257-020-0008-7. [DOI] [Google Scholar]
- 25.Qi S., Lu L., Zhou F., et al. Neutrophil infiltration and whole-cell vaccine elicited by N-dihydrogalactochitosan combined with NIR phototherapy to enhance antitumor immune response and T cell immune memory. Theranostics. 2020;10(4):1814–1832. doi: 10.7150/thno.38515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanyi J.L., Bobisse S., Ophir E., et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018;10(436) doi: 10.1126/scitranslmed.aao5931. [DOI] [PubMed] [Google Scholar]
- 27.Chiang C.L.L., Coukos G., Kandalaft L.E. Whole tumor antigen vaccines: where are we? Vaccines (Basel) 2015;3(2):344–372. doi: 10.3390/vaccines3020344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y., Guo Y., Tang L. Engineering cancer vaccines using stimuli-responsive biomaterials. Nano Res. 2018;11(10):5355–5371. doi: 10.1007/s12274-018-2162-1. [DOI] [Google Scholar]
- 29.Ahmed K.K., Geary S.M., Salem A.K. Surface engineering tumor cells with adjuvant-loaded particles for use as cancer vaccines. J. Contr. Release. 2017;248:1–9. doi: 10.1016/j.jconrel.2016.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J., Li Z., Huang H., et al. Improved antigen cross-presentation by polyethyleneimine-based nanoparticles. Int. J. Nanomed. 2011;6 doi: 10.2147/IJN.S15457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J., Wang H., Xu L., et al. Nanovaccine based on a protein-delivering dendrimer for effective antigen cross-presentation and cancer immunotherapy. Biomaterials. 2019;207:1–9. doi: 10.1016/j.biomaterials.2019.03.037. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y., Liu M., Li C., Li B., Zhang X. Tumor cell membrane-coated liquid metal nanovaccine for tumor prevention. Chin. J. Chem. 2020;38(6):595–600. doi: 10.1002/cjoc.201900557. [DOI] [Google Scholar]
- 33.Liu W.L., Zou M.Z., Liu T., et al. Expandable immunotherapeutic nanoplatforms engineered from cytomembranes of hybrid cells derived from cancer and dendritic cells. Adv. Mater. 2019;31(18) doi: 10.1002/adma.201900499. [DOI] [PubMed] [Google Scholar]
- 34.Li A., Zhao Y., Li Y., Jiang L., Gu Y., Liu J. Cell-derived biomimetic nanocarriers for targeted cancer therapy: cell membranes and extracellular vesicles. Drug Deliv. 2021;28(1):1237–1255. doi: 10.1080/10717544.2021.1938757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raza F., Zafar H., Zhang S., et al. Recent advances in cell membrane‐derived biomimetic nanotechnology for cancer immunotherapy. Adv Healthcare Mater. 2021;10(6) doi: 10.1002/adhm.202002081. [DOI] [PubMed] [Google Scholar]
- 36.Meng Z., Zhang Y., Zhou X., Ji J., Liu Z. Nanovaccines with cell-derived components for cancer immunotherapy. Adv. Drug Deliv. Rev. 2022;182 doi: 10.1016/j.addr.2021.114107. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X., Zhang H., Gu J., et al. Engineered extracellular vesicles for cancer therapy. Adv. Mater. 2021;33(14) doi: 10.1002/adma.202005709. [DOI] [PubMed] [Google Scholar]
- 38.Kooijmans S.A.A., Aleza C.G., Roffler S.R., van Solinge W.W., Vader P., Schiffelers R.M. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J. Extracell. Vesicles. 2016;5 doi: 10.3402/jev.v5.31053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo G.F., Chen W.H., Zeng X., Zhang X.Z. Cell primitive-based biomimetic functional materials for enhanced cancer therapy. Chem. Soc. Rev. 2021;50(2):945–985. doi: 10.1039/d0cs00152j. [DOI] [PubMed] [Google Scholar]
- 40.Kim T.K., Eberwine J.H. Mammalian cell transfection: the present and the future. Anal. Bioanal. Chem. 2010;397(8):3173–3178. doi: 10.1007/s00216-010-3821-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woods N.B., Muessig A., Schmidt M., et al. Lentiviral vector transduction of NOD/SCID repopulating cells results in multiple vector integrations per transduced cell: risk of insertional mutagenesis. Blood. 2003;101(4):1284–1289. doi: 10.1182/blood-2002-07-2238. [DOI] [PubMed] [Google Scholar]
- 42.Yan Y., Liu X.Y., Lu A., Wang X.Y., Jiang L.X., Wang J.C. Non-viral vectors for RNA delivery. J. Contr. Release. 2022;342:241–279. doi: 10.1016/j.jconrel.2022.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaroszeski M.J., Heller L.C., Gilbert R., Heller R. Electrically mediated plasmid DNA delivery to solid tumors in vivo. Methods Mol. Biol. 2004;245:237–244. doi: 10.1385/1-59259-649-5:237. [DOI] [PubMed] [Google Scholar]
- 44.Miller D.L., Song J. Tumor growth reduction and DNA transfer by cavitation-enhanced high-intensity focused ultrasound in vivo. Ultrasound Med. Biol. 2003;29(6):887–893. doi: 10.1016/s0301-5629(03)00031-0. [DOI] [PubMed] [Google Scholar]
- 45.Mehier-Humbert S., Guy R.H. Physical methods for gene transfer: improving the kinetics of gene delivery into cells. Adv. Drug Deliv. Rev. 2005;57(5):733–753. doi: 10.1016/j.addr.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 46.Wang H., La Russa M., Qi L.S. CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem. 2016;85:227–264. doi: 10.1146/annurev-biochem-060815-014607. [DOI] [PubMed] [Google Scholar]
- 47.Huang L., Rong Y., Tang X., et al. Engineered exosomes as an in situ DC-primed vaccine to boost antitumor immunity in breast cancer. Mol. Cancer. 2022;21(1):45. doi: 10.1186/s12943-022-01515-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Q., Cheng H., Peng H., Zhou H., Li P.Y., Langer R. Non-genetic engineering of cells for drug delivery and cell-based therapy. Adv. Drug Deliv. Rev. 2015;91:125–140. doi: 10.1016/j.addr.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 49.Yoo J.W., Irvine D.J., Discher D.E., Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat. Rev. Drug Discov. 2011;10(7):521–535. doi: 10.1038/nrd3499. [DOI] [PubMed] [Google Scholar]
- 50.Smyth T., Petrova K., Payton N.M., et al. Surface functionalization of exosomes using click chemistry. Bioconjugate Chem. 2014;25(10):1777–1784. doi: 10.1021/bc500291r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jia G., Han Y., An Y., et al. NRP-1 targeted and cargo-loaded exosomes facilitate simultaneous imaging and therapy of glioma in vitro and in vivo. Biomaterials. 2018;178:302–316. doi: 10.1016/j.biomaterials.2018.06.029. [DOI] [PubMed] [Google Scholar]
- 52.Spicer C.D., Pashuck E.T., Stevens M.M. Achieving controlled biomolecule-biomaterial conjugation. Chem Rev. 2018;118(16):7702–7743. doi: 10.1021/acs.chemrev.8b00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ai X., Wang S., Duan Y., et al. Emerging approaches to functionalizing cell membrane-coated nanoparticles. Biochemistry. 2021;60(13):941–955. doi: 10.1021/acs.biochem.0c00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong X., Mu L.L., Liu X.L., et al. Biomimetic, hypoxia-responsive nanoparticles overcome residual chemoresistant Leukemic cells with Co-targeting of therapy-induced bone marrow niches. Adv. Funct. Mater. 2020;30(12) doi: 10.1002/adfm.202000309. [DOI] [Google Scholar]
- 55.Chen H., Sha H., Zhang L., et al. Lipid insertion enables targeted functionalization of paclitaxel-loaded erythrocyte membrane nanosystem by tumor-penetrating bispecific recombinant protein. Int. J. Nanomed. 2018;13:5347–5359. doi: 10.2147/IJN.S165109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang R., Xu J., Xu L., et al. Cancer cell membrane-coated adjuvant nanoparticles with mannose modification for effective anticancer vaccination. ACS Nano. 2018;12(6):5121–5129. doi: 10.1021/acsnano.7b09041. [DOI] [PubMed] [Google Scholar]
- 57.Miao L., Zhang Y., Huang L. mRNA vaccine for cancer immunotherapy. Mol. Cancer. 2021;20(1):41. doi: 10.1186/s12943-021-01335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Medof M.E., Nagarajan S., Tykocinski M.L. Cell-surface engineering with GPI-anchored proteins. FASEB J. 1996;10(5):574–586. doi: 10.1096/fasebj.10.5.8621057. [DOI] [PubMed] [Google Scholar]
- 59.Luo C.H., Huang C.T., Su C.H., Yeh C.S. Bacteria-mediated hypoxia-specific delivery of nanoparticles for tumors imaging and therapy. Nano Lett. 2016;16(6):3493–3499. doi: 10.1021/acs.nanolett.6b00262. [DOI] [PubMed] [Google Scholar]
- 60.Cheng H., Kastrup C.J., Ramanathan R., et al. Nanoparticulate cellular patches for cell-mediated tumoritropic delivery. ACS Nano. 2010;4(2):625–631. doi: 10.1021/nn901319y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morishita M., Takahashi Y., Matsumoto A., Nishikawa M., Takakura Y. Exosome-based tumor antigens-adjuvant co-delivery utilizing genetically engineered tumor cell-derived exosomes with immunostimulatory CpG DNA. Biomaterials. 2016;111:55–65. doi: 10.1016/j.biomaterials.2016.09.031. [DOI] [PubMed] [Google Scholar]
- 62.Shi X., Zhang X., Li J., et al. PD-1 blockade enhances the antitumor efficacy of GM-CSF surface-modified bladder cancer stem cells vaccine. Int. J. Cancer. 2018;142(10):2106–2117. doi: 10.1002/ijc.31219. [DOI] [PubMed] [Google Scholar]
- 63.Yong T., Zhang X., Bie N., et al. Tumor exosome-based nanoparticles are efficient drug carriers for chemotherapy. Nat. Commun. 2019;10(1):3838. doi: 10.1038/s41467-019-11718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mooney R., Roma L., Zhao D., et al. Neural stem cell-mediated intratumoral delivery of gold nanorods improves photothermal therapy. ACS Nano. 2014;8(12):12450–12460. doi: 10.1021/nn505147w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brynskikh A.M., Zhao Y., Mosley R.L., et al. Macrophage delivery of therapeutic nanozymes in a murine model of Parkinson's disease. Nanomedicine (Lond). 2010;5(3):379–396. doi: 10.2217/nnm.10.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rao L., Bu L.L., Ma L., et al. Platelet-facilitated photothermal therapy of head and neck squamous cell carcinoma. Angew Chem. Int. Ed. Engl. 2018;57(4):986–991. doi: 10.1002/anie.201709457. [DOI] [PubMed] [Google Scholar]
- 67.Xiao L., Huang Y., Yang Y., et al. Biomimetic cytomembrane nanovaccines prevent breast cancer development in the long term. Nanoscale. 2021;13(6):3594–3601. doi: 10.1039/D0NR08978H. [DOI] [PubMed] [Google Scholar]
- 68.Wu T., Liu Y., Cao Y., Liu Z. Engineering macrophage exosome disguised biodegradable nanoplatform for enhanced sonodynamic therapy of glioblastoma. Adv. Mater. 2022;34(15) doi: 10.1002/adma.202110364. [DOI] [PubMed] [Google Scholar]
- 69.de Gruijl T.D., van den Eertwegh A.J.M., Pinedo H.M., Scheper R.J. Whole-cell cancer vaccination: from autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol. Immunother. 2008;57(10):1569–1577. doi: 10.1007/s00262-008-0536-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Soiffer R.J., Kooshesh K.A., Ho V. Whole tumor cell vaccines engineered to secrete GM-CSF (GVAX) ImmunoMedicine. 2021;1(1) doi: 10.1002/imed.1025. [DOI] [Google Scholar]
- 71.Gibney G.T., Kudchadkar R.R., DeConti R.C., et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin. Cancer Res. 2015;21(4):712–720. doi: 10.1158/1078-0432.CCR-14-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu J., Shao B., Luo M., et al. Irradiated lactic acid-stimulated tumour cells promote the antitumour immunity as a therapeutic vaccine. Cancer Lett. 2020;469:367–379. doi: 10.1016/j.canlet.2019.11.018. [DOI] [PubMed] [Google Scholar]
- 73.Parmiani G., Pilla L., Maccalli C., Russo V. Autologous versus allogeneic cell-based vaccines? Cancer J. 2011;17(5):331–336. doi: 10.1097/PPO.0b013e3182337a76. [DOI] [PubMed] [Google Scholar]
- 74.Kurtz S.L., Ravindranathan S., Zaharoff D.A. Current status of autologous breast tumor cell-based vaccines. Expert Rev. Vaccines. 2014;13(12):1439–1445. doi: 10.1586/14760584.2014.969714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wong K.K., Li W.A., Mooney D.J., Dranoff G. Advances in therapeutic cancer vaccines. Adv. Immunol. 2016;130:191–249. doi: 10.1016/bs.ai.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 76.Tsujikawa T., Crocenzi T., Durham J.N., et al. Evaluation of cyclophosphamide/GVAX pancreas followed by listeria-mesothelin (CRS-207) with or without nivolumab in patients with pancreatic cancer. Clin. Cancer Res. 2020;26(14):3578–3588. doi: 10.1158/1078-0432.CCR-19-3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alson D., Schuyler S.C., Yan B.X., Samimuthu K., Qiu J.T. Combination vaccination with tetanus toxoid and enhanced tumor-cell based vaccine against cervical cancer in a mouse model. Front. Immunol. 2020;11:927. doi: 10.3389/fimmu.2020.00927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim V.M., Pan X., Soares K.C., et al. Neoantigen-based EpiGVAX vaccine initiates antitumor immunity in colorectal cancer. JCI Insight. 2020;5(9) doi: 10.1172/jci.insight.136368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Le D.T., Wang-Gillam A., Picozzi V., et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015;33(12):1325–1333. doi: 10.1200/JCO.2014.57.4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Salgia R., Lynch T., Skarin A., et al. Vaccination with irradiated autologous tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor augments antitumor immunity in some patients with metastatic non-small-cell lung carcinoma. J. Clin. Oncol. 2003;21(4):624–630. doi: 10.1200/JCO.2003.03.091. [DOI] [PubMed] [Google Scholar]
- 81.Berg T.J., Pietras A. Radiotherapy-induced remodeling of the tumor microenvironment by stromal cells. Semin Cancer Biol. Published online February. 2022;8(22) doi: 10.1016/j.semcancer.2022.02.011. S1044-579X. 00033-5. [DOI] [PubMed] [Google Scholar]
- 82.Pyonteck S.M., Akkari L., Schuhmacher A.J., et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen M., Xiang R., Wen Y., et al. A whole-cell tumor vaccine modified to express fibroblast activation protein induces antitumor immunity against both tumor cells and cancer-associated fibroblasts. Sci. Rep. 2015;5 doi: 10.1038/srep14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu J., Kindy M.S., Gattoni-Celli S. Semi-allogeneic vaccines and tumor-induced immune tolerance. J. Transl. Med. 2009;7(1):3. doi: 10.1186/1479-5876-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gattoni-Celli S., Young M.R.I. Restoration of immune responsiveness to glioma by vaccination of mice with established brain gliomas with a semi-allogeneic vaccine. Int. J. Mol. Sci. 2016;17(9):E1465. doi: 10.3390/ijms17091465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang P., Wu X., Basu M., et al. MYCN amplification is associated with repressed cellular immunity in neuroblastoma: an in silico immunological analysis of TARGET database. Front. Immunol. 2017;8:1473. doi: 10.3389/fimmu.2017.01473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li Y., Casey S.C., Felsher D.W. Inactivation of MYC reverses tumorigenesis. J. Intern. Med. 2014;276(1):52–60. doi: 10.1111/joim.12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu X., Nelson M., Basu M., et al. MYC oncogene is associated with suppression of tumor immunity and targeting Myc induces tumor cell immunogenicity for therapeutic whole cell vaccination. J Immunother Cancer. 2021;9(3) doi: 10.1136/jitc-2020-001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Srinivasan P., Wu X., Basu M., Rossi C., Sandler A.D. PD-L1 checkpoint inhibition and anti-CTLA-4 whole tumor cell vaccination counter adaptive immune resistance: a mouse neuroblastoma model that mimics human disease. PLoS Med. 2018;15(1) doi: 10.1371/journal.pmed.1002497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao Z., Fang L., Xiao P., et al. Walking dead tumor cells for targeted drug delivery against lung metastasis of triple-negative breast cancer. Adv Mater. Published online June. 2022;27 doi: 10.1002/adma.202205462. [DOI] [PubMed] [Google Scholar]
- 91.Ning N., Pan Q., Zheng F., et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res. 2012;72(7):1853–1864. doi: 10.1158/0008-5472.CAN-11-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dashti A., Ebrahimi M., Hadjati J., Memarnejadian A., Moazzeni S.M. Dendritic cell based immunotherapy using tumor stem cells mediates potent antitumor immune responses. Cancer Lett. 2016;374(1):175–185. doi: 10.1016/j.canlet.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 93.Guo M., You C., Dou J. Role of transmembrane glycoprotein mucin 1 (MUC1) in various types of colorectal cancer and therapies: current research status and updates. Biomed. Pharmacother. 2018;107:1318–1325. doi: 10.1016/j.biopha.2018.08.109. [DOI] [PubMed] [Google Scholar]
- 94.Guo M., Luo B., Pan M., et al. Colorectal cancer stem cell vaccine with high expression of MUC1 serves as a novel prophylactic vaccine for colorectal cancer. Int. Immunopharm. 2020;88 doi: 10.1016/j.intimp.2020.106850. [DOI] [PubMed] [Google Scholar]
- 95.Huang B., Yan X., Li Y. Cancer stem cell for tumor therapy. Cancers. 2021;13(19):4814. doi: 10.3390/cancers13194814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Field C.S., Hermans I.F., Hunn M.K. Whole tumor cell vaccines for glioma immunotherapy. Immunotherapy. 2016;8(4):387–389. doi: 10.2217/imt-2015-0022. [DOI] [PubMed] [Google Scholar]
- 97.Meng Z., Zhou X., Xu J., et al. Light-triggered in situ gelation to enable robust photodynamic-immunotherapy by repeated stimulations. Adv. Mater. 2019;31(24) doi: 10.1002/adma.201900927. [DOI] [PubMed] [Google Scholar]
- 98.Klochkov S.G., Neganova M.E., Nikolenko V.N., et al. Implications of nanotechnology for the treatment of cancer: recent advances. Semin. Cancer Biol. 2021;69:190–199. doi: 10.1016/j.semcancer.2019.08.028. [DOI] [PubMed] [Google Scholar]
- 99.Jang S.C., Kim O.Y., Yoon C.M., et al. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano. 2013;7(9):7698–7710. doi: 10.1021/nn402232g. [DOI] [PubMed] [Google Scholar]
- 100.Dewangan H.K., Tomar S. Nanovaccine for transdermal delivery system. J. Drug Deliv. Sci. Technol. 2022;67 doi: 10.1016/j.jddst.2021.102988. [DOI] [Google Scholar]
- 101.Hu S., Ma J., Su C., et al. Engineered exosome-like nanovesicles suppress tumor growth by reprogramming tumor microenvironment and promoting tumor ferroptosis. Acta Biomater. 2021;135:567–581. doi: 10.1016/j.actbio.2021.09.003. [DOI] [PubMed] [Google Scholar]
- 102.Berti C., Graciotti M., Boarino A., Yakkala C., Kandalaft L.E., Klok H.A. Polymer nanoparticle-mediated delivery of oxidized tumor lysate-based cancer vaccines. Macromol. Biosci. 2022;22(2) doi: 10.1002/mabi.202100356. [DOI] [PubMed] [Google Scholar]
- 103.Neller M.A., López J.A., Schmidt C.W. Antigens for cancer immunotherapy. Semin. Immunol. 2008;20(5):286–295. doi: 10.1016/j.smim.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 104.Ochyl L.J., Bazzill J.D., Park C., Xu Y., Kuai R., Moon J.J. PEGylated tumor cell membrane vesicles as a new vaccine platform for cancer immunotherapy. Biomaterials. 2018;182:157–166. doi: 10.1016/j.biomaterials.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang X., Wang N., Yang Y., et al. Polydopamine nanoparticles carrying tumor cell lysate as a potential vaccine for colorectal cancer immunotherapy. Biomater. Sci. 2019;7(7):3062–3075. doi: 10.1039/c9bm00010k. [DOI] [PubMed] [Google Scholar]
- 106.Han Q., Wang Y., Pang M., Zhang J. STAT3-blocked whole-cell hepatoma vaccine induces cellular and humoral immune response against HCC. J. Exp. Clin. Cancer Res. 2017;36(1):156. doi: 10.1186/s13046-017-0623-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chiang C.L.L., Kandalaft L.E., Tanyi J., et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: from bench to bedside. Clin. Cancer Res. 2013;19(17):4801–4815. doi: 10.1158/1078-0432.CCR-13-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shi G.N., Zhang C.N., Xu R., et al. Enhanced antitumor immunity by targeting dendritic cells with tumor cell lysate-loaded chitosan nanoparticles vaccine. Biomaterials. 2017;113:191–202. doi: 10.1016/j.biomaterials.2016.10.047. [DOI] [PubMed] [Google Scholar]
- 109.Carroll E.C., Jin L., Mori A., et al. The vaccine adjuvant chitosan promotes cellular immunity via DNA sensor cGAS-STING-dependent induction of type I interferons. Immunity. 2016;44(3):597–608. doi: 10.1016/j.immuni.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Adu-Berchie K., Mooney D.J. Biomaterials as local niches for immunomodulation. Acc. Chem. Res. 2020;53(9):1749–1760. doi: 10.1021/acs.accounts.0c00341. [DOI] [PubMed] [Google Scholar]
- 111.Yang F., Shi K., Hao Y., et al. Cyclophosphamide loaded thermo-responsive hydrogel system synergize with a hydrogel cancer vaccine to amplify cancer immunotherapy in a prime-boost manner. Bioact. Mater. 2021;6(10):3036–3048. doi: 10.1016/j.bioactmat.2021.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yu Z., Xu Y., Yao H., et al. A simple and general strategy for postsurgical personalized cancer vaccine therapy based on an injectable dynamic covalent hydrogel. Biomater. Sci. 2021;9(20):6879–6888. doi: 10.1039/d1bm01000j. [DOI] [PubMed] [Google Scholar]
- 113.Song H., Yang P., Huang P., Zhang C., Kong D., Wang W. Injectable polypeptide hydrogel-based co-delivery of vaccine and immune checkpoint inhibitors improves tumor immunotherapy. Theranostics. 2019;9(8):2299–2314. doi: 10.7150/thno.30577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Song H., Huang P., Niu J., et al. Injectable polypeptide hydrogel for dual-delivery of antigen and TLR3 agonist to modulate dendritic cells in vivo and enhance potent cytotoxic T-lymphocyte response against melanoma. Biomaterials. 2018;159:119–129. doi: 10.1016/j.biomaterials.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 115.Björklund M., Pettersson S., Coutinho A. Ontogenic development of “natural” and induced plaque-forming cell isotypes in normal mice. Eur. J. Immunol. 1985;15(10):1003–1007. doi: 10.1002/eji.1830151008. [DOI] [PubMed] [Google Scholar]
- 116.González F.E., Gleisner A., Falcón-Beas F., Osorio F., López M.N., Salazar-Onfray F. Tumor cell lysates as immunogenic sources for cancer vaccine design. Hum. Vaccines Immunother. 2014;10(11):3261–3269. doi: 10.4161/21645515.2014.982996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Beck J.D., Reidenbach D., Salomon N., et al. mRNA therapeutics in cancer immunotherapy. Mol. Cancer. 2021;20(1):69. doi: 10.1186/s12943-021-01348-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen N., Xia P., Li S., Zhang T., Wang T.T., Zhu J. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life. 2017;69(5):297–304. doi: 10.1002/iub.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Miao L., Li L., Huang Y., et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 2019;37(10):1174–1185. doi: 10.1038/s41587-019-0247-3. [DOI] [PubMed] [Google Scholar]
- 120.Naka T., Iwahashi M., Nakamura M., et al. Tumor vaccine therapy against recrudescent tumor using dendritic cells simultaneously transfected with tumor RNA and granulocyte macrophage colony-stimulating factor RNA. Cancer Sci. 2008;99(2):407–413. doi: 10.1111/j.1349-7006.2007.00698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zeng C., Zhang C., Walker P.G., Dong Y. Formulation and delivery technologies for mRNA vaccines. Curr Top Microbiol Immunol. Published online June. 2020;2 doi: 10.1007/82_2020_217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sayour E.J., De Leon G., Pham C., et al. Systemic activation of antigen-presenting cells via RNA-loaded nanoparticles. OncoImmunology. 2017;6(1) doi: 10.1080/2162402X.2016.1256527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sayour E.J., Grippin A., De Leon G., et al. Personalized tumor RNA loaded lipid-nanoparticles prime the systemic and intratumoral Milieu for response to cancer immunotherapy. Nano Lett. 2018;18(10):6195–6206. doi: 10.1021/acs.nanolett.8b02179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang Y., Xie F., Yin Y., et al. Immunotherapy of tumor RNA-loaded lipid nanoparticles against hepatocellular carcinoma. Int. J. Nanomed. 2021;16:1553–1564. doi: 10.2147/IJN.S291421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen Q., Huang G., Wu W., et al. A hybrid eukaryotic-prokaryotic nanoplatform with photothermal modality for enhanced antitumor vaccination. Adv. Mater. 2020;32(16) doi: 10.1002/adma.201908185. [DOI] [PubMed] [Google Scholar]
- 126.Gao J., Chu D., Wang Z. Cell membrane-formed nanovesicles for disease-targeted delivery. J. Contr. Release. 2016;224:208–216. doi: 10.1016/j.jconrel.2016.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Feltrin F. da S., Agner T., Sayer C., Lona L.M.F. Curcumin encapsulation in functional PLGA nanoparticles: a promising strategy for cancer therapies. Adv. Colloid Interface Sci. 2022;300 doi: 10.1016/j.cis.2021.102582. [DOI] [PubMed] [Google Scholar]
- 128.Astete C.E., Sabliov C.M. Synthesis and characterization of PLGA nanoparticles. J. Biomater. Sci. Polym. Ed. 2006;17(3):247–289. doi: 10.1163/156856206775997322. [DOI] [PubMed] [Google Scholar]
- 129.Swider E., Koshkina O., Tel J., Cruz L.J., de Vries I.J.M., Srinivas M. Customizing poly(lactic-co-glycolic acid) particles for biomedical applications. Acta Biomater. 2018;73:38–51. doi: 10.1016/j.actbio.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 130.Théry C., Ostrowski M., Segura E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009;9(8):581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 131.Li Q., Cai S., Li M., et al. Tumor-derived extracellular vesicles: their role in immune cells and immunotherapy. Int. J. Nanomed. 2021;16:5395–5409. doi: 10.2147/IJN.S313912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Syn N.L., Wang L., Chow E.K.H., Lim C.T., Goh B.C. Exosomes in cancer nanomedicine and immunotherapy: prospects and challenges. Trends Biotechnol. 2017;35(7):665–676. doi: 10.1016/j.tibtech.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 133.Zhu L., Sun H.T., Wang S., et al. Isolation and characterization of exosomes for cancer research. J. Hematol. Oncol. 2020;13(1):152. doi: 10.1186/s13045-020-00987-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Naseri M., Bozorgmehr M., Zöller M., Ranaei Pirmardan E., Madjd Z. Tumor-derived exosomes: the next generation of promising cell-free vaccines in cancer immunotherapy. OncoImmunology. 2020;9(1) doi: 10.1080/2162402X.2020.1779991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chen T., Guo J., Yang M., Zhu X., Cao X. Chemokine-containing exosomes are released from heat-stressed tumor cells via lipid raft-dependent pathway and act as efficient tumor vaccine. J. Immunol. 2011;186(4):2219–2228. doi: 10.4049/jimmunol.1002991. [DOI] [PubMed] [Google Scholar]
- 136.Zhang W., Huang Q., Xiao W., et al. Advances in anti-tumor treatments targeting the CD47/SIRPα Axis. Front. Immunol. 2020;11:18. doi: 10.3389/fimmu.2020.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wolfers J., Lozier A., Raposo G., et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;7(3):297–303. doi: 10.1038/85438. [DOI] [PubMed] [Google Scholar]
- 138.Ren G., Wang Y., Yuan S., Wang B. Dendritic cells loaded with HeLa-derived exosomes simulate an antitumor immune response. Oncol. Lett. 2018;15(5):6636–6640. doi: 10.3892/ol.2018.8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang Y., Luo C.L., He B.C., Zhang J.M., Cheng G., Wu X.H. Exosomes derived from IL-12-anchored renal cancer cells increase induction of specific antitumor response in vitro: a novel vaccine for renal cell carcinoma. Int. J. Oncol. 2010;36(1):133–140. [PubMed] [Google Scholar]
- 140.Gu X., Erb U., Büchler M.W., Zöller M. Improved vaccine efficacy of tumor exosome compared to tumor lysate loaded dendritic cells in mice. Int. J. Cancer. 2015;136(4):E74–E84. doi: 10.1002/ijc.29100. [DOI] [PubMed] [Google Scholar]
- 141.Lee E.Y., Park K.S., Yoon Y.J., et al. Therapeutic effects of autologous tumor-derived nanovesicles on melanoma growth and metastasis. PLoS One. 2012;7(3) doi: 10.1371/journal.pone.0033330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lee Y.S., Kim S.H., Cho J.A., Kim C.W. Introduction of the CIITA gene into tumor cells produces exosomes with enhanced anti-tumor effects. Exp. Mol. Med. 2011;43(5):281–290. doi: 10.3858/emm.2011.43.5.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fan W., Tian X.D., Huang E., Zhang J.J. Exosomes from CIITA-transfected CT26 cells enhance anti- tumor effects. Asian Pac. J. Cancer Prev. APJCP. 2013;14(2):987–991. doi: 10.7314/apjcp.2013.14.2.987. [DOI] [PubMed] [Google Scholar]
- 144.Jaini R., Kesaraju P., Johnson J.M., Altuntas C.Z., Jane-Wit D., Tuohy V.K. An autoimmune-mediated strategy for prophylactic breast cancer vaccination. Nat Med. 2010;16(7):799–803. doi: 10.1038/nm.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rossowska J., Anger N., Wegierek K., et al. Antitumor potential of extracellular vesicles released by genetically modified murine colon carcinoma cells with overexpression of interleukin-12 and shRNA for TGF-β1. Front. Immunol. 2019;10:211. doi: 10.3389/fimmu.2019.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.ichiro Ohno S., Takanashi M., Sudo K., et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013;21(1):185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Xiu F., Cai Z., Yang Y., Wang X., Wang J., Cao X. Surface anchorage of superantigen SEA promotes induction of specific antitumor immune response by tumor-derived exosomes. J. Mol. Med. (Berl.) 2007;85(5):511–521. doi: 10.1007/s00109-006-0154-1. [DOI] [PubMed] [Google Scholar]
- 148.Vader P., Mol E.A., Pasterkamp G., Schiffelers R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016;106(Pt A):148–156. doi: 10.1016/j.addr.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 149.van den Boorn J.G., Dassler J., Coch C., Schlee M., Hartmann G. Exosomes as nucleic acid nanocarriers. Adv. Drug Deliv. Rev. 2013;65(3):331–335. doi: 10.1016/j.addr.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 150.Wu L., Xie W., Li Y., et al. Biomimetic nanocarriers guide extracellular ATP homeostasis to remodel energy metabolism for activating innate and adaptive immunity system. Adv Sci (Weinh). Published online April. 2022;9 doi: 10.1002/advs.202105376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Qin X., Yang C., Xu H., et al. Cell-derived biogenetic gold nanoparticles for sensitizing radiotherapy and boosting immune response against cancer. Small. 2021;17(50) doi: 10.1002/smll.202103984. [DOI] [PubMed] [Google Scholar]
- 152.Rao Q., Zuo B., Lu Z., et al. Tumor-derived exosomes elicit tumor suppression in murine hepatocellular carcinoma models and humans in vitro. Hepatology. 2016;64(2):456–472. doi: 10.1002/hep.28549. [DOI] [PubMed] [Google Scholar]
- 153.Xia X., Mai J., Xu R., et al. Porous silicon microparticle potentiates anti-tumor immunity by enhancing cross-presentation and inducing type I interferon response. Cell Rep. 2015;11(6):957–966. doi: 10.1016/j.celrep.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Conniot J., Scomparin A., Peres C., et al. Immunization with mannosylated nanovaccines and inhibition of the immune-suppressing microenvironment sensitizes melanoma to immune checkpoint modulators. Nat. Nanotechnol. 2019;14(9):891–901. doi: 10.1038/s41565-019-0512-0. [DOI] [PubMed] [Google Scholar]
- 155.Zhu S., Li S., Yi M., Li N., Wu K. Roles of microvesicles in tumor progression and clinical applications. Int. J. Nanomed. 2021;16:7071–7090. doi: 10.2147/IJN.S325448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bian X., Xiao Y.T., Wu T., et al. Microvesicles and chemokines in tumor microenvironment: mediators of intercellular communications in tumor progression. Mol. Cancer. 2019;18(1):50. doi: 10.1186/s12943-019-0973-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Morel O., Jesel L., Freyssinet J.M., Toti F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler. Thromb. Vasc. Biol. 2011;31(1):15–26. doi: 10.1161/ATVBAHA.109.200956. [DOI] [PubMed] [Google Scholar]
- 158.Wang T., Gilkes D.M., Takano N., et al. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. U. S. A. 2014;111(31):E3234–E3242. doi: 10.1073/pnas.1410041111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lin W., Xu Y., Chen X., et al. Radiation-induced small extracellular vesicles as “carriages” promote tumor antigen release and trigger antitumor immunity. Theranostics. 2020;10(11):4871–4884. doi: 10.7150/thno.43539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhang H., Yu Y., Zhou L., et al. Circulating tumor microparticles promote lung metastasis by reprogramming inflammatory and mechanical niches via a macrophage-dependent pathway. Cancer Immunol Res. 2018;6(9):1046–1056. doi: 10.1158/2326-6066.CIR-17-0574. [DOI] [PubMed] [Google Scholar]
- 161.Pineda B., Sánchez García F.J., Olascoaga N.K., et al. Malignant glioma therapy by vaccination with irradiated C6 cell-derived microvesicles promotes an antitumoral immune response. Mol. Ther. 2019;27(9):1612–1620. doi: 10.1016/j.ymthe.2019.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang H., Tang K., Zhang Y., et al. Cell-free tumor microparticle vaccines stimulate dendritic cells via cGAS/STING signaling. Cancer Immunol Res. 2015;3(2):196–205. doi: 10.1158/2326-6066.CIR-14-0177. [DOI] [PubMed] [Google Scholar]
- 163.Zhang H., Huang B. Tumor cell-derived microparticles: a new form of cancer vaccine. OncoImmunology. 2015;4(8) doi: 10.1080/2162402X.2015.1017704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ma J., Wei K., Zhang H., et al. Mechanisms by which dendritic cells present tumor microparticle antigens to CD8+ T cells. Cancer Immunol Res. 2018;6(9):1057–1068. doi: 10.1158/2326-6066.CIR-17-0716. [DOI] [PubMed] [Google Scholar]
- 165.Mitragotri S. Immunization without needles. Nat. Rev. Immunol. 2005;5(12):905–916. doi: 10.1038/nri1728. [DOI] [PubMed] [Google Scholar]
- 166.Lycke N. Recent progress in mucosal vaccine development: potential and limitations. Nat. Rev. Immunol. 2012;12(8):592–605. doi: 10.1038/nri3251. [DOI] [PubMed] [Google Scholar]
- 167.Dong W., Zhang H., Yin X., et al. Oral delivery of tumor microparticle vaccines activates NOD2 signaling pathway in ileac epithelium rendering potent antitumor T cell immunity. OncoImmunology. 2017;6(3) doi: 10.1080/2162402X.2017.1282589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chen B.J., Zhao J.W., Zhang D.H., Zheng A.H., Wu G.Q. Immunotherapy of cancer by targeting regulatory T cells. Int. Immunopharm. 2022;104 doi: 10.1016/j.intimp.2021.108469. [DOI] [PubMed] [Google Scholar]
- 169.Parenky A.C., Akalkotkar A., Mulla N.S., D'Souza M.J. Harnessing T-cell activity against prostate cancer: a therapeutic microparticulate oral cancer vaccine. Vaccine. 2019;37(41):6085–6092. doi: 10.1016/j.vaccine.2019.08.033. [DOI] [PubMed] [Google Scholar]
- 170.Xu Z., Zeng S., Gong Z., Yan Y. Exosome-based immunotherapy: a promising approach for cancer treatment. Mol. Cancer. 2020;19(1):160. doi: 10.1186/s12943-020-01278-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mehanny M., Lehr C.M., Fuhrmann G. Extracellular vesicles as antigen carriers for novel vaccination avenues. Adv. Drug Deliv. Rev. 2021;173:164–180. doi: 10.1016/j.addr.2021.03.016. [DOI] [PubMed] [Google Scholar]
- 172.Huda M.N., Nurunnabi M. Potential application of exosomes in vaccine development and delivery. Pharm Res. Published online January. 2022;13 doi: 10.1007/s11095-021-03143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jafari D., Malih S., Eini M., et al. Improvement, scaling-up, and downstream analysis of exosome production. Crit. Rev. Biotechnol. 2020;40(8):1098–1112. doi: 10.1080/07388551.2020.1805406. [DOI] [PubMed] [Google Scholar]
- 174.Kroll A.V., Fang R.H., Jiang Y., et al. Nanoparticulate delivery of cancer cell membrane elicits multiantigenic antitumor immunity. Adv. Mater. 2017;29(47) doi: 10.1002/adma.201703969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lokhov P.G., Balashova E.E. Cellular cancer vaccines: an update on the development of vaccines generated from cell surface antigens. J. Cancer. 2010;1:230–241. doi: 10.7150/jca.1.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rao L., Yu G.T., Meng Q.F., Bu L.L. Cancer cell membrane‐coated nanoparticles for personalized therapy in patient‐derived xenograft models - rao - 2019 - advanced functional materials - Wiley online library. https://onlinelibrary.wiley.com/doi/full/10.1002/adfm.201905671 Accessed.
- 177.Fang R.H., Hu C.M.J., Luk B.T., et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14(4):2181–2188. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hollinshead A.C., Herberman R.B., Jaffurs W.J., Alpert L.K., Minton J.P., Harris J.E. Soluble membrane antigens of human malignant melanoma cells. Cancer. 1974;34(4):1235–1243. doi: 10.1002/1097-0142(197410)34:4<1235:aid-cncr2820340433>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 179.Liu B., Yang Y., Chao Y., et al. Equipping cancer cell membrane vesicles with functional DNA as a targeted vaccine for cancer immunotherapy. Nano Lett. 2021;21(22):9410–9418. doi: 10.1021/acs.nanolett.1c02582. [DOI] [PubMed] [Google Scholar]
- 180.Bommireddy R., Munoz L.E., Kumari A., et al. Tumor membrane vesicle vaccine augments the efficacy of anti-PD1 antibody in immune checkpoint inhibitor-resistant squamous cell carcinoma models of head and neck cancer. Vaccines (Basel) 2020;8(2):E182. doi: 10.3390/vaccines8020182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bozeman E.N., He S., Shafizadeh Y., Selvaraj P. Therapeutic efficacy of PD-L1 blockade in a breast cancer model is enhanced by cellular vaccines expressing B7-1 and glycolipid-anchored IL-12. Hum. Vaccines Immunother. 2016;12(2):421–430. doi: 10.1080/21645515.2015.1076953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Pack C.D., Bommireddy R., Munoz L.E., et al. Tumor membrane-based vaccine immunotherapy in combination with anti-CTLA-4 antibody confers protection against immune checkpoint resistant murine triple-negative breast cancer. Hum. Vaccines Immunother. 2020;16(12):3184–3193. doi: 10.1080/21645515.2020.1754691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Agrahari V., Agrahari V., Burnouf P.A., Chew C.H., Burnouf T. Extracellular microvesicles as new industrial therapeutic frontiers. Trends Biotechnol. 2019;37(7):707–729. doi: 10.1016/j.tibtech.2018.11.012. [DOI] [PubMed] [Google Scholar]
- 184.Kim H.Y., Kang M., Choo Y.W., et al. Immunomodulatory lipocomplex functionalized with photosensitizer-embedded cancer cell membrane inhibits tumor growth and metastasis. Nano Lett. 2019;19(8):5185–5193. doi: 10.1021/acs.nanolett.9b01571. [DOI] [PubMed] [Google Scholar]
- 185.Xu C., Liu W., Hu Y., Li W., Di W. Bioinspired tumor-homing nanoplatform for co-delivery of paclitaxel and siRNA-E7 to HPV-related cervical malignancies for synergistic therapy. Theranostics. 2020;10(7):3325–3339. doi: 10.7150/thno.41228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sun Q., Wu J., Jin L., et al. Cancer cell membrane-coated gold nanorods for photothermal therapy and radiotherapy on oral squamous cancer. J. Mater. Chem. B. 2020;8(32):7253–7263. doi: 10.1039/d0tb01063d. [DOI] [PubMed] [Google Scholar]
- 187.Chen Z., Zhao P., Luo Z., et al. Cancer cell membrane-biomimetic nanoparticles for homologous-targeting dual-modal imaging and photothermal therapy. ACS Nano. 2016;10(11):10049–10057. doi: 10.1021/acsnano.6b04695. [DOI] [PubMed] [Google Scholar]
- 188.Zhang L., Zhang Y., Xue Y., et al. Transforming weakness into strength: photothermal-therapy-induced inflammation enhanced cytopharmaceutical chemotherapy as a combination anticancer treatment. Adv. Mater. 2019;31(5) doi: 10.1002/adma.201805936. [DOI] [PubMed] [Google Scholar]
- 189.Zhang H., Liu D., Shahbazi M.A., et al. Fabrication of a multifunctional nano-in-micro drug delivery platform by microfluidic templated encapsulation of porous silicon in polymer matrix. Adv. Mater. 2014;26(26):4497–4503. doi: 10.1002/adma.201400953. [DOI] [PubMed] [Google Scholar]
- 190.Savage D.J., Liu X., Curley S.A., Ferrari M., Serda R.E. Porous silicon advances in drug delivery and immunotherapy. Curr. Opin. Pharmacol. 2013;13(5):834–841. doi: 10.1016/j.coph.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tasciotti E., Liu X., Bhavane R., et al. Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications. Nat. Nanotechnol. 2008;3(3):151–157. doi: 10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]
- 192.Li J., Huang D., Cheng R., et al. Multifunctional biomimetic nanovaccines based on photothermal and weak-immunostimulatory nanoparticulate cores for the immunotherapy of solid tumors. Adv Mater. Published online December. 2021;7 doi: 10.1002/adma.202108012. [DOI] [PubMed] [Google Scholar]
- 193.Gong C., Yu X., You B., et al. Macrophage-cancer hybrid membrane-coated nanoparticles for targeting lung metastasis in breast cancer therapy. J. Nanobiotechnol. 2020;18(1):92. doi: 10.1186/s12951-020-00649-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Keshavarz Shahbaz S., Foroughi F., Soltaninezhad E., Jamialahmadi T., Penson P.E., Sahebkar A. Application of PLGA nano/microparticle delivery systems for immunomodulation and prevention of allotransplant rejection. Expet Opin. Drug Deliv. 2020;17(6):767–780. doi: 10.1080/17425247.2020.1748006. [DOI] [PubMed] [Google Scholar]
- 195.Liao Y., Zhang Y., Blum N.T., Lin J., Huang P. Biomimetic hybrid membrane-based nanoplatforms: synthesis, properties and biomedical applications. Nanoscale Horiz. 2020;5(9):1293–1302. doi: 10.1039/d0nh00267d. [DOI] [PubMed] [Google Scholar]
- 196.Li R., He Y., Zhang S., Qin J., Wang J. Cell membrane-based nanoparticles: a new biomimetic platform for tumor diagnosis and treatment. Acta Pharm. Sin. B. 2018;8(1):14–22. doi: 10.1016/j.apsb.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Han X., Shen S., Fan Q., et al. Red blood cell-derived nanoerythrosome for antigen delivery with enhanced cancer immunotherapy. Sci. Adv. 2019;5(10) doi: 10.1126/sciadv.aaw6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wang D., Dong H., Li M., et al. Erythrocyte-cancer hybrid membrane camouflaged hollow copper sulfide nanoparticles for prolonged circulation life and homotypic-targeting photothermal/chemotherapy of melanoma. ACS Nano. 2018;12(6):5241–5252. doi: 10.1021/acsnano.7b08355. [DOI] [PubMed] [Google Scholar]
- 199.Koido S., Homma S., Okamoto M., et al. Fusions between dendritic cells and whole tumor cells as anticancer vaccines. OncoImmunology. 2013;2(5) doi: 10.4161/onci.24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ma J., Liu F., Sheu W.C., et al. Copresentation of tumor antigens and costimulatory molecules via biomimetic nanoparticles for effective cancer immunotherapy. Nano Lett. 2020;20(6):4084–4094. doi: 10.1021/acs.nanolett.9b05171. [DOI] [PubMed] [Google Scholar]
- 201.Liu W.L., Zou M.Z., Liu T., et al. Cytomembrane nanovaccines show therapeutic effects by mimicking tumor cells and antigen presenting cells. Nat. Commun. 2019;10(1):3199. doi: 10.1038/s41467-019-11157-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Guerra A.D., Yeung O.W.H., Qi X., Kao W.J., Man K. The anti-tumor effects of M1 macrophage-loaded poly (ethylene glycol) and gelatin-based hydrogels on hepatocellular carcinoma. Theranostics. 2017;7(15):3732–3744. doi: 10.7150/thno.20251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Madsen S.J., Hirschberg H. Macrophages as delivery vehicles for anticancer agents. Ther. Deliv. 2019;10(3):189–201. doi: 10.4155/tde-2019-0004. [DOI] [PubMed] [Google Scholar]
- 204.Xuan M., Shao J., Dai L., Li J., He Q. Macrophage cell membrane camouflaged Au nanoshells for in vivo prolonged circulation life and enhanced cancer photothermal therapy. ACS Appl. Mater. Interfaces. 2016;8(15):9610–9618. doi: 10.1021/acsami.6b00853. [DOI] [PubMed] [Google Scholar]
- 205.Rao L., Wu L., Liu Z., et al. Hybrid cellular membrane nanovesicles amplify macrophage immune responses against cancer recurrence and metastasis. Nat. Commun. 2020;11(1):4909. doi: 10.1038/s41467-020-18626-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.He L.Z., Weidlick J., Sisson C., Marsh H.C., Keler T. Toll-like receptor agonists shape the immune responses to a mannose receptor-targeted cancer vaccine. Cell. Mol. Immunol. 2015;12(6):719–728. doi: 10.1038/cmi.2014.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chen D., Cai L., Guo Y., et al. Cancer cell membrane-decorated zeolitic-imidazolate frameworks codelivering cisplatin and oleanolic acid induce apoptosis and reversed multidrug resistance on bladder carcinoma cells. ACS Omega. 2020;5(2):995–1002. doi: 10.1021/acsomega.9b02261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yewdall A.W., Drutman S.B., Jinwala F., Bahjat K.S., Bhardwaj N. CD8+ T cell priming by dendritic cell vaccines requires antigen transfer to endogenous antigen presenting cells. PLoS One. 2010;5(6) doi: 10.1371/journal.pone.0011144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Roberts E.W., Broz M.L., Binnewies M., et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. 2016;30(2):324–336. doi: 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Field C.S., Hunn M.K., Ferguson P.M., Ruedl C., Ancelet L.R., Hermans I.F. Blocking CTLA-4 while priming with a whole cell vaccine reshapes the oligoclonal T cell infiltrate and eradicates tumors in an orthotopic glioma model. OncoImmunology. 2017;7(1) doi: 10.1080/2162402X.2017.1376154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Dumontel B., Susa F., Limongi T., et al. ZnO nanocrystals shuttled by extracellular vesicles as effective Trojan nano-horses against cancer cells. Nanomedicine (Lond) 2019;14(21):2815–2833. doi: 10.2217/nnm-2019-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Han L., Peng K., Qiu L.Y., et al. Hitchhiking on controlled-release drug delivery systems: opportunities and challenges for cancer vaccines. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.679602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Schineis P., Kotkowska Z.K., Vogel-Kindgen S., et al. Photochemical internalization (PCI)-mediated activation of CD8 T cells involves antigen uptake and CCR7-mediated transport by migratory dendritic cells to draining lymph nodes. J. Contr. Release. 2021;332:96–108. doi: 10.1016/j.jconrel.2021.02.014. [DOI] [PubMed] [Google Scholar]
- 214.Keenan B.P., Jaffee E.M. Whole cell vaccines-past progress and future strategies. Semin. Oncol. 2012;39(3):276–286. doi: 10.1053/j.seminoncol.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Solbrig C.M., Saucier-Sawyer J.K., Cody V., Saltzman W.M., Hanlon D.J. Polymer nanoparticles for immunotherapy from encapsulated tumor-associated antigens and whole tumor cells. Mol. Pharm. 2007;4(1):47–57. doi: 10.1021/mp060107e. [DOI] [PubMed] [Google Scholar]
- 216.Crooke S.T., Wang S., Vickers T.A., Shen W., Liang X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017;35(3):230–237. doi: 10.1038/nbt.3779. [DOI] [PubMed] [Google Scholar]
- 217.Zhang P., Liu G., Chen X. Nanobiotechnology: cell membrane-based delivery systems. Nano Today. 2017;13:7–9. doi: 10.1016/j.nantod.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Luk B.T., Zhang L. Cell membrane-camouflaged nanoparticles for drug delivery. J. Contr. Release. 2015;220(Pt B):600–607. doi: 10.1016/j.jconrel.2015.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Li X., Jiang X. Microfluidics for producing poly (lactic-co-glycolic acid)-based pharmaceutical nanoparticles. Adv. Drug Deliv. Rev. 2018;128:101–114. doi: 10.1016/j.addr.2017.12.015. [DOI] [PubMed] [Google Scholar]
- 220.Jing L., Liang X., Deng Z., et al. Prussian blue coated gold nanoparticles for simultaneous photoacoustic/CT bimodal imaging and photothermal ablation of cancer. Biomaterials. 2014;35(22):5814–5821. doi: 10.1016/j.biomaterials.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 221.Zhang W., Hu S., Yin J.J., et al. Prussian blue nanoparticles as multienzyme mimetics and reactive oxygen species scavengers. J. Am. Chem. Soc. 2016;138(18):5860–5865. doi: 10.1021/jacs.5b12070. [DOI] [PubMed] [Google Scholar]
- 222.Li J., Zhang F., Hu Z., et al. Drug “pent-up” in hollow magnetic prussian blue nanoparticles for NIR-induced chemo-photothermal tumor therapy with trimodal imaging. Adv Healthc Mater. 2017;6(14) doi: 10.1002/adhm.201700005. [DOI] [PubMed] [Google Scholar]
- 223.Xu Z., Liu Y., Ma R., et al. Thermosensitive hydrogel incorporating prussian blue nanoparticles promotes diabetic wound healing via ROS scavenging and mitochondrial function restoration. ACS Appl. Mater. Interfaces. 2022;14(12):14059–14071. doi: 10.1021/acsami.1c24569. [DOI] [PubMed] [Google Scholar]
- 224.Guan S., Liu X., Fu Y., et al. A biodegradable “Nano-donut” for magnetic resonance imaging and enhanced chemo/photothermal/chemodynamic therapy through responsive catalysis in tumor microenvironment. J. Colloid Interface Sci. 2022;608(Pt 1):344–354. doi: 10.1016/j.jcis.2021.09.186. [DOI] [PubMed] [Google Scholar]
- 225.Hou L., Gong X., Yang J., Zhang H., Yang W., Chen X. Hybrid-membrane-decorated prussian blue for effective cancer immunotherapy via tumor-associated macrophages polarization and hypoxia relief. Adv. Mater. 2022;34(14) doi: 10.1002/adma.202200389. [DOI] [PubMed] [Google Scholar]
- 226.Abbasi J. Personalized cancer vaccine approach safe in early trial. JAMA. 2021;325(18):1825. doi: 10.1001/jama.2021.6828. [DOI] [PubMed] [Google Scholar]