

Summary
Alternative splicing is crucial for a wide range of biological processes. However, limited by the availability of reference genomes, genome-wide patterns of alternative splicing remain unknown in most nonmodel organisms. We present an attention-based convolutional neural network model, DeepASmRNA, for predicting alternative splicing events using only transcriptomic data. DeepASmRNA consists of two parts: identification of alternatively spliced transcripts and classification of alternative splicing events, which outperformed the state-of-the-art method, AStrap, and other deep learning models. Then, we utilize transfer learning to increase the performance in species with limited training data and use an interpretation method to decipher splicing codes. Finally, applying Amborella, DeepASmRNA can identify more AS events than AStrap while maintaining the same level of precision, suggesting that DeepASmRNA has superior sensitivity to identify alternative splicing events. In summary, DeepASmRNA is scalable and interpretable for detecting genome-wide patterns of alternative splicing in species without a reference genome.
Subject areas: Biological sciences, Molecular biology, Molecular biology experimental approach, Artificial intelligence, Artificial intelligence applications
Graphical abstract
Highlights
-
•
DeepASmRNA uses only the transcriptome to predict alternative splicing events
-
•
DeepASmRNA identifies adjacent HSPs to greatly improve the recall
-
•
DeepASmRNA uses attention-based convolutional neural network to classify AS events
-
•
Transfer learning is used to increase the predictive power of a target species
Biological sciences; Molecular biology; Molecular biology experimental approach; Artificial intelligence; Artificial intelligence applications
Introduction
Alternative splicing (AS), generating multiple isoforms from a single gene (Lee and Rio, 2015), has a crucial role in increasing the biological complexity and diversity of the transcriptome and the encoded proteome (Barash et al., 2010). AS is involved in many biological processes, including development, growth, and stress responses (Staiger and Brown, 2013). It also amplifies evolutionary differences between vertebrates (Gueroussov et al., 2015), and mistakes in splicing may be associated with various diseases, such as cancers (Cherry and Lynch, 2020; Oka et al., 2021; Xiong et al., 2015).
RNA sequencing (RNA-Seq) (Wang et al., 2009) and single-molecule real-time (SMRT) long-read isoform sequencing (Iso-Seq) (Sharon et al., 2013) have greatly extended our knowledge of AS at the genome-wide scale. To date, most genome-wide AS detection methods are based on a reference genome, such as SUPPA2 (Trincado et al., 2018) and AStool (Qi et al., 2022). Applying these methods, researchers found that AS is ubiquitous and tissue specific in eukaryotes (Chen and Manley, 2009; Marquez et al., 2012; Pan et al., 2008; Thatcher et al., 2014; Wang et al., 2008). These studies significantly expanded knowledge of AS in model organisms with a reference genome, and multiple databases of AS events have been set up (Sun et al., 2020). However, it is challenging to detect AS events when a reference genome is not available, which limits our understanding of AS in nonmodel organisms lacking a reference genome.
To explore AS in species without a reference genome, several algorithms have been designed for detecting AS at the genome-wide scale. The pattern of AS transcripts is that these transcripts share common coding regions but differ in the alternatively spliced regions. Thus, Liu et al. (2017) utilized the insertions and deletions (indels) flanking perfectly aligned regions from all-versus-all BLAST to identify AS transcripts from transcriptomic data without a reference genome. IsoSplitter used SIM4 (Florea et al., 1998) to align the sequences and identify AS transcripts from the transcriptome (Wang et al., 2021). However, the sensitivities of the methods were not high, and the types of AS events were not further classified. Subsequently, Ji et al. developed AStrap (Ji et al., 2019) to identify AS isoforms using CD-HIT and GMAP and to predict the types of AS events using a machine learning framework. However, the recall of AStrap was only 34.50% (553/1603) in Amborella, suggesting that it has limited sensitivity to detect AS events.
Compared with classical machine learning, deep learning eliminates the requirement of manual feature engineering by automatically extracting useful information from sequences (Eraslan et al., 2019). Importantly, deep neural networks can discover a hierarchy of vectors to dramatically improve prediction accuracy (Minar and Naher, 2018). Therefore, various applications of deep learning have been reported in genomics (Eraslan et al., 2019). For example, a sequence-based deep learning model was used to predict the RNA splicing branchpoint (Paggi and Bejerano, 2018). Splice junctions were also accurately predicted from primary pre-mRNA sequences by deep learning (Jaganathan et al., 2019). Despite the success of deep learning in genomics, previous studies have not used deep learning to classify the types of AS events from transcriptomic data.
Here, we introduce an approach called DeepASmRNA to identify AS transcripts and classify AS events from isoform-level transcriptomic data without a reference genome. Our method outperformed the state-of-the-art methods in both AS transcript detection and AS event classification. In addition, our method is interpretable and makes use of transfer learning to improve AS event prediction in species with limited training data.
Results
A framework for reference-free prediction of AS events
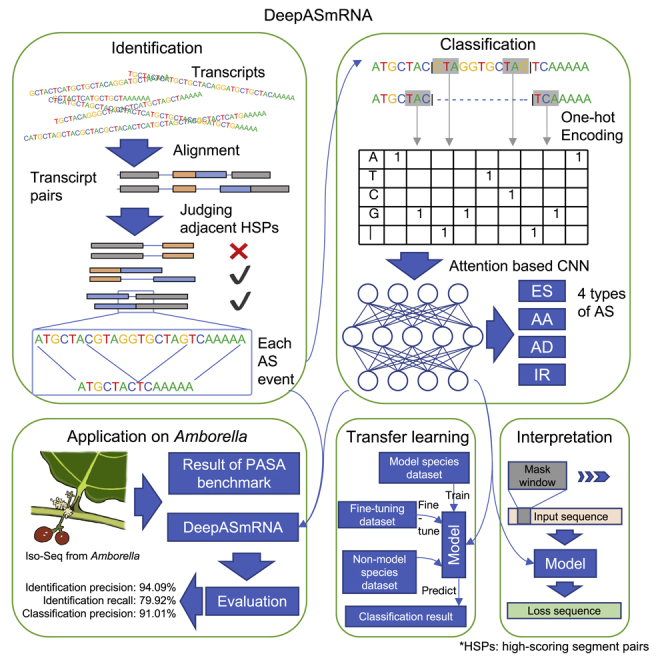
To explore AS in species without a reference genome, we proposed DeepASmRNA to predict AS from only full-length transcripts. DeepASmRNA only takes transcript sequences as input and is composed of two parts (Figure 1). For the first part, we used the results of all-versus-all BLASTN (Altschul et al., 1990) for recognition of AS patterns for identifying alternatively spliced transcripts. For the second part, we used an attention-based CNN model to classify four basic types of AS: exon skipping (ES), alternative acceptor site (AA), alternative donor site (AD), and intron retention (IR). Therefore, DeepASmRNA outputs AS transcript pairs and the corresponding probabilities of ES, AA, AD, and IR for each AS event of a transcript pair. As the sum of four probabilities is one, the final prediction is the AS type with the maximum probability. To assess DeepASmRNA, we trained three models for human, Arabidopsis thaliana, and rice. Moreover, to apply our model to other nonmodel species with limited training data, we provided a transfer learning method based on our pretrained model.
Figure 1.

The workflow of DeepASmRNA
DeepASmRNA is composed of two parts: identification of alternatively spliced transcripts and classification of AS events. For identification, identifying AS patterns was based on the results of all-versus-all alignment transcripts obtained by BLASTN. For classification, an attention-based CNN model was used to classify the AS events into 4 types. HSP: high-scoring segment pairs in BLAST, which indicated constitutive region. Indels: insertion and deletion, which indicated the alternative region.
Accurate identification of alternatively spliced transcripts from transcriptomes
To verify the flexibility of DeepASmRNA in different species, the transcripts of human, A. thaliana, and rice, respectively, were used as the input of DeepASmRNA. To obtain the transcripts and the labeled AS events of the three species, we downloaded the full-length transcripts and gene annotation files of human (GRCh38.p13, v37) (Frankish et al., 2021), Arabidopsis thaliana (Araport11) (Cheng et al., 2017; Arabidopsis Genome Initiative et al., 2000), and rice (MSU7) (Kawahara et al., 2013) (Table S1). Then, 159,505; 48,358; and 66,338 transcripts were obtained for human, Arabidopsis thaliana, and rice, respectively. Next, SUPPA2 (Trincado et al., 2018) guided by gene structures was applied to obtain AS events and AS transcript pairs. For human, 79,320 AS events, including 40,055 ES, 16,727 AA, 15,209 AD, and 7,329 IR, were identified in 232,908 AS transcript pairs; for Arabidopsis thaliana, 14,415 AS events, including 1,051 ES, 4,211 AA, 3,161 AD, and 5,992 IR, were identified in 24,964 transcript pairs; and for rice, 7,542 AS events, including 788 ES, 2,472 AA, 1,336 AD, and 2,946 IR, were identified in 10,389 transcript pairs. These datasets were used as labeled data (Table 1).
Table 1.
Labeled datasets obtained by SUPPA2
| Features | Human | Arabidopsis thaliana | Rice |
|---|---|---|---|
| ES | 40,055 | 1,051 | 788 |
| AA | 16,727 | 4,211 | 2,472 |
| AD | 15,209 | 3,161 | 1,336 |
| IR | 7,329 | 5,992 | 2,946 |
| Total AS events | 79,320 | 14,415 | 7,542 |
| AS transcript pairs | 232,908 | 24,964 | 10,389 |
Applying the first part of DeepASmRNA, the identification of alternatively spliced transcripts (see method details), we predicted 204,910; 21,747; and 8,873 AS transcript pairs for human, Arabidopsis thaliana, and rice, respectively (Table S2). Using the AS transcript pairs obtained by SUPPA2 (Table 1) as the ground truth, we found that the precisions of DeepASmRNA were 91.30% (187,083/204,910), 94.70% (20,595/21,747), and 93.19% (8,269/8,873) in human, Arabidopsis thaliana, and rice, respectively, whereas the recalls were 80.32% (187,083/232,908), 82.50% (20,595/24,964), and 79.59% (8,269/10,389) (Figure 2 and Table S2). Moreover, we examined existing mRNAs from the gene locus to verify the AS events, and the accuracies were 98.33%, 99.63%, and 99.23% in human, Arabidopsis thaliana, and rice, respectively. In addition, we compared the performance of DeepASmRNA with those of AStrap and IsoSplitter by applying them to the same datasets. The results (Figure 2) suggest that the recalls of DeepASmRNA are significantly higher than those of AStrap and IsoSplitter, whereas precision remains comparable.
Figure 2.

Performance of DeepASmRNA in the identification of AS transcript pairs in human, Arabidopsis thaliana, and rice
(A) Precision.
(B) Recall. The x axis presents species, and the y axis shows the value of precision or recall. See also Table S2.
A highly accurate landscape of AS classification
Applying the first part of DeepASmRNA, the identification of alternatively spliced transcripts from transcriptomes, we can obtain all the AS events of each AS transcript pair. To further predict whether an AS event is ES, AA, AD, or IR between the AS transcript pair without a reference genome, in the second part of DeepASmRNA, we further constructed an attention-based CNN classification model (Yin et al., 2015), using only the mRNA transcript sequences as input (Figure 3). The one-hot encoding of transcript sequences from four fragments was put into the model (see method details) (Figure 3A). Then, high-level sequence information was extracted by two attention-based CNN layers followed by a global CNN layer. Finally, the model mapped learned key information to final outputs (i.e., ES, AA, AD, or IR) using two fully connected layers (Figure 3B). The model outputs the probabilities of ES, AA, AD, and IR for each AS event. The sum over all four AS types of each AS event was always one, thus, the max probability was taken as the final prediction. In contrast to the previous machine learning method, AStrap, which requires manual feature engineering, our CNN automatically learns predictive key information from raw transcript sequences. In addition, our method utilizes a self-attention mechanism to learn long-range interactions between vectors and perform word sense disambiguation (Tang et al., 2018).
Figure 3.

The workflow of the attention-based convolutional neural network for the classification of alternative splicing events
(A) One-hot encoding of the mRNA transcript sequence, taking intron retained events as an example. The blue parts on the sequence represent constitutive regions, and the gray parts represent alternatively spliced regions. The yellow parts are the sequence we selected to encode the AS event.
(B) The architecture of the attention-CNN model.
(C) Transfer learning workflow.
Our model was trained on the three labeled datasets from human, Arabidopsis thaliana, and rice, which were obtained from SUPPA2. To examine whether the length of the input sequence impacts the accuracy of our model, we compared the performance of models trained on N bp (N ranges from 10 to 60, steps by 10). As shown in Figure 4A, with the increase in input sequence length, the accuracy of the model increases gradually, suggesting that a long sequence context can capture more information than a shorter context. However, as the length reached 50 bp, the accuracy tended to be stable (Table S3). Based on the accuracies of different input lengths, we chose DeepASmRNA-50bp as the final model (Table 2). In addition, the area under the curve (AUC) of our method was consistently higher than 0.97 in each dataset (Figures 4B, S1A, and S1B), highlighting that our model has a strong ability to distinguish between the positive and negative datasets in a variety of species.
Figure 4.

Performance of DeepASmRNA in AS classification
(A) The accuracy of training on different input lengths. The line plot illustrates the accuracy of DeepASmRNA with the different input segment lengths.
(B) The receiver operating characteristic (ROC) curves of DeepASmRNA in human. AUCs are listed at the bottom right of each subplot. Solid lines of different colors represent four types of AS events, whereas dotted lines represent micro- and macro-average ROC curves. Micro-average regards each instance equally, whereas macro-average considers each class equally.
(C) The accuracy compared with AStrap. The x axis shows the different datasets, which consist of two species from two dataset sources. Human_AStrap: human from AStrap’s dataset; Rice_AStrap: rice from AStrap’s dataset; Human_ DeepASmRNA: human from DeepASmRNA’s dataset; Rice_DeepASmRNA: rice from DeepASmRNA’s dataset. The different colors represent the different models. Blue represents the performance of AStrap, whereas orange represents DeepASmRNA.
(D) The accuracy compared with the other deep learning model; each color represents a model. See also Figure S1 and Tables S3–S5.
Table 2.
Performance of DeepASmRNA-50bp in AS classification
| Metrics | Human | Arabidopsis thaliana | Rice | |
|---|---|---|---|---|
| Overall | Accuracy (%) | 88.49 | 90.73 | 90.02 |
| ES | Precision (%) | 90.61 | 91.99 | 88.22 |
| Recall (%) | 90.52 | 87.68 | 88.48 | |
| F1-score | 0.91 | 0.90 | 0.88 | |
| AUPRC | 0.97 | 0.96 | 0.96 | |
| AA | Precision (%) | 83.93 | 90.99 | 93.07 |
| Recall (%) | 83.01 | 91.31 | 88.07 | |
| F1-score | 0.83 | 0.91 | 0.91 | |
| AUPRC | 0.91 | 0.97 | 0.97 | |
| AD | Precision (%) | 85.45 | 92.16 | 90.70 |
| Recall (%) | 87.29 | 82.53 | 78.00 | |
| F1-score | 0.86 | 0.87 | 0.84 | |
| AUPRC | 0.94 | 0.94 | 0.91 | |
| IR | Precision (%) | 91.16 | 89.53 | 88.50 |
| Recall (%) | 90.54 | 95.79 | 97.63 | |
| F1-score | 0.91 | 0.93 | 0.93 | |
| AUPRC | 0.97 | 0.96 | 0.97 |
Because AStrap, a machine learning method based on hand-crafted features (Ji et al., 2019), is the only method performing the classification of AS events, we only compared the performance of our models with those of AStrap. Without the model of Arabidopsis thaliana in AStrap, we only compared the human and rice models. We used two datasets to compare our models with those of AStrap. First, we tested DeepASmRNA based on AStrap’s dataset. DeepASmRNA outperformed AStrap in both human and rice (87.8% versus 86.5% and 91.2% versus 88.3%, respectively) (Figure 4C and Table S4). Second, we tested AStrap based on our dataset. Similar performance was observed in rice (90.02% versus 90.11%), whereas significant improvement was observed in human (88.49% versus 81.44%), indicating that an attention-based CNN model is more effective than machine learning for AS event classification without a reference genome.
Comparison with other deep learning models
We also compared the performance of our model with those of other widely used deep learning models, including LeNet (Lecun et al., 1998), ResNet (Kaiming et al., 2016), and LSTM (Greff et al., 2017). We used the same training, validation, and test sets for these models.
As shown in Figure 4D, the accuracy of our model was 88.49% in human, which was the highest among the four models. The accuracies of DeepASmRNA were 90.73% and 90.02% in Arabidopsis thaliana and rice, respectively, which were also higher than those of other deep learning models (Table S5). These findings show that DeepASmRNA generally outperformed the other deep learning models regarding the three metrics (accuracy, precision, and recall) when using similar hyperparameter settings. These results suggest that deep learning models, especially our proposed method, are powerful in the classification of AS events.
Transfer learning further improves the performance of DeepASmRNA
In the previous section, we demonstrated the high performance of DeepASmRNA in three model organisms, human, Arabidopsis thaliana, and rice. Here, we explored how to apply our model to other nonmodel species with limited training data. To this end, an increase in the generalizability of the model is needed, i.e., DeepASmRNA must be able to be applied efficiently to unseen data from other species. Transfer learning, which attempts to transfer the knowledge from related tasks to a target task when the latter has little or no labeled training data (Sinno Jialin and Qiang, 2010), can solve the task by training a base model (pretrained model), copying its first n layers to the first n layers of a target model (Yosinski et al., 2014), and training the remaining layers of the target model on a small number of labeled data for the target task.
We used the datasets and models of the three species to verify the transfer learning effects of our models. We first used Arabidopsis thaliana and rice to verify the efficacy of transfer learning between relatively close species. When we tested the Arabidopsis thaliana model on the rice dataset, the overall accuracy of the model reached 89.67%, and after fine-tuning learning, the accuracy of the model was increased to 90.44% (Table S6). When the rice model was applied to the Arabidopsis thaliana dataset, the accuracy of the model reached 88.50%; after fine-tuning learning, the accuracy of the model increased to 88.69% (Table S6). The results suggest that in relatively closely related species such as rice and Arabidopsis thaliana, a model without fine-tuning learning has achieved good performance, in which fine-tuning plays a small role.
For distantly related species for transfer learning, such as human and Arabidopsis thaliana, when we transferred the Arabidopsis thaliana dataset to the human model, the overall accuracy of the model was 72.13% (Table S6). After fine-tuning learning, the accuracy of the model reached 84.96%, significantly improving the performance of the model. Thus, fine-tuning learning may be critical for the application of our model to distant species. However, when the Arabidopsis thaliana model was applied to the human dataset, the accuracy was only 66.50%, and it moderately increased to 66.82% after transfer learning or fine-tuning. This might be because human AS events are more complex, and key sequence information in human is absent in Arabidopsis thaliana. According to the above performance, we suggested using our model directly without fine-tuning when applying it to relatively close species. However, when applying it to distantly related species, we suggest utilizing fine-tuning to achieve better performance based on a human model whose AS event is more complex.
Interpretation of DeepASmRNA
Because we directly used exon sequences as model inputs, we sought to examine key information the model learned to predict each type of AS event. We chose human and Arabidopsis thaliana as representatives of animals and plants, respectively. In the model for each species, we selected the top ten samples with the smallest prediction loss in each type of AS event, which may best represent key information learned by the model. To search for the key information in the DNA sequence, we masked different parts of the sequence to observe the change in the loss function. The larger the change is, the more important the masked part is. Specifically, we used a sliding window with a window size of 12 bp and a step size of 1 bp to mask transcript sequences, where the window size matched the kernel size of the first convolution layer. We changed all the one-hot encoding to either 0 or 1 in a window; thus, we generated a series of values along the input sequence, in which each value represented the change in loss function after masking a sliding window. Similar patterns were observed for the human and Arabidopsis thaliana models (Figures 5A and 5B). Specifically, the loss value significantly increased near 50 bp and 150 bp, which are proximal to exon junctions. For AD events, in both 0-masked and 1-masked data, models focused more on the former exon junctions, whereas the AA events focused on the later exon junctions. For ES and IR events, both exon junctions were the focus. Interestingly, there is a difference between the Arabidopsis thaliana and human data in ES and IR, implying the two species may have focused on different positions. To further explore which positions and which bases are critical for distinguishing different types of AS events, a more precise mask of each base type at each position was carried out, and TF-MoDISco (Shrikumar et al., 2018) was used to determine the pattern of sequence based on the importance score of the precise mask. Unfortunately, we did not find out any patterns in the sequences. However, using the visualization tools in TF-MoDISco, we visualized the mean importance score of the top 10 sequences with the lowest loss in each type of AS event. The ‘GT’-like pattern after the AD event donor and ‘AG’-like pattern before the acceptor were highlighted in the 0-mask approach (Figure S2), which is consistent with the sequence patterns around splice sites for four types visualized by sequence logo (Omar, 2017) (Figure S3). In addition, compared with AStrap, our interpretation can not only find that the pattern has a positive contribution to a specific type of AS but also determine some patterns that significantly reject a certain type of AS. For example, the donor of AD should not be ‘CG’ or ‘AG’ in Arabidopsis thaliana.
Figure 5.

Visualization of the mRNA transcript sequence interpreting DeepASmRNA
The results on the left are from the human model, whereas the results on the right are from Arabidopsis thaliana.
(A) Cross entropy loss values of DNA sequences using 0 masked windows.
(B) Cross entropy loss values of DNA sequences using 1 masked window. The y axis indicates the top ten sequences with the smallest prediction loss in each class of AS events. The x axes represent the start position of the masked window, and the brightness levels show the loss values. See also Figures S2 and S3.
Implementation of DeepASmRNA model in the Iso-Seq data from Amborella
To observe the robustness of our method, we used Iso-Seq data (Sharon et al., 2013) from Amborella to identify AS transcripts and classify AS events (Amborella Genome Project et al., 2013). Overall, we obtained 8,918 full-length transcripts (Table S7). Next, considering the high errors of Iso-Seq, we used the same parameters as rice except for 90% identity for each high-scoring segment pair (HSP) when identifying alternatively spliced transcripts, which resulted in 1,676 identified transcript pairs. Then, we extracted the sequences involved in AS events and encoded them into one-hot coding. Finally, we inputted them into the rice-based DeepASmRNA model because rice is the most closely related species of Amborella among the three species with trained models. In all, we obtained 3,058 AS events classified into 372 ES, 1,014 AA, 484 AD, and 1,188 IR (Table S8).
To observe the performance of our method in Amborella, we assessed our results at two levels: alternatively spliced transcripts and AS events. PASA (Campbell et al., 2006) was applied to obtain the evaluation set of AS events. Overall, PASA generated 1,973 alternatively spliced transcript pairs and 3,312 AS events, including 325 ES, 1,236 AA, 444 AD, and 1,307 IR (Table S8).
To compare our model with AStrap, the 8,918 full-length transcripts were also used as inputs to apply AStrap based on the rice model with default parameters. A total of 760 AS events, including 139 ES, 66 AA, 90 AD, and 465 IR events in 688 transcript pairs were detected by AStrap (Table S8).
The performance of DeepASmRNA and AStrap in Amborella is compared at two levels. At the transcript-pair level, in the 1,676 transcript pairs identified by DeepASmRNA, 1,577 pairs overlapped with the results of PASA, suggesting that the precision of the identification of alternatively spliced transcripts was 94.09% and the recall was 79.92%. For AStrap, the precision was 98.17%, whereas the recall was 34.21%. At the AS event level, in the transcript pairs shared by PASA, we predicted 2,813 AS events in which 2,628 AS events were the same as the results of PASA, suggesting that the precision was 91.01% (2,613/2,871), which was higher than that of AStrap (87.67% = 654/746).
Combining the results of AS transcript identification and AS event classification, the precision of our method reached 85.74% (Figure 6), which is similar to the 86.05% precision of AStrap (Ji et al., 2019), whereas the recall of our method was higher than that of AStrap (79.17% versus 22.95%), suggesting that our model can identify significantly more AS events while maintaining the accuracy. We observed that DeepASmRNA could find four types of events, and the proportions of the four types were similar to those of the four types in PASA, whereas AStrap only found more IR events and only small amounts of the other three types (ES, AA, and AD) (Figure S4 and Table S9). The Venn diagram and confusion matrix of prediction results from DeepASmRNA and AStrap are shown in Figure S4 and Table S9. In the first part, AStrap used GMAP for sequence alignment. GMAP is a program for aligning cDNA sequences to a genome by searching for GT and AG pairs in the 5′ and-3′ end surrounding the intron (Wu and Watanabe, 2005). Only the IR event contains GT-AG at both ends. On contrary, for most of the ES, AA, and AD events, there is no complete intron and thus no GT-AG boundary (Figure S3). Therefore, many ES, AA, and AD events were missed by AStrap.
Figure 6.

The distribution of each type of AS event in Amborella
(A) Precisions of AS events predicted by DeepASmRNA and AStrap. Overlapping: the AS events identified by DeepASmRNA or AStrap are also in PASA; nonoverlapping: the AS events identified by DeepASmRNA or AStrap are not in PASA.
(B) Recalls of AS events predicted by DeepASmRNA and AStrap. Overlapping: the AS events identified by DeepASmRNA or AStrap are also in PASA; nonoverlapping: the AS events are included in PASA but are not identified by DeepASmRNA or AStrap. See also Table S8.
We also compared the runtime and memory among DeepASmRNA, AStrap, and IsoSplitter, using the full-length transcripts of Amborella. Lacking the AS classification of AS events in IsoSplitter, we only listed its runtime and memory in the identification of AS transcript pairs. As shown in Table 3, both runtime and the memory of DeepASmRNA outperformed than AStrap and IsoSplitter.
Table 3.
Runtime and memory comparison between DeepASmRNA, AStrap, and IsoSplitter in Amborella
| Methods | Parts | Runtime | Memory |
|---|---|---|---|
| DeepASmRNA | Identification | 32 s | 282 M |
| Classification | 9 s | 378 M | |
| AStrap | Identification | 3 min 16 s | 460 M |
| Classification | 31 s | 1240 M | |
| IsoSplitter | Identification | 67 minuts | 2662 M |
Discussion
In the current study, we developed DeepASmRNA, a computational approach to identify AS transcripts and classify AS events from an isoform-level transcriptome without a reference genome. Our method outperformed the state-of-the-art method, AStrap, in both AS transcript identification and AS event classification. In the identification of AS transcripts, we found that the precisions of our method were 91.30%, 94.70%, and 93.19% for human, Arabidopsis thaliana, and rice, respectively, higher than those of AStrap. In the classification of AS events, the overall accuracies of our method for human, Arabidopsis thaliana, and rice were 88.49%, 90.73%, and 90.02%, respectively, which are higher than those of AStrap and other deep learning models. In addition, DeepASmRNA could utilize transfer learning to improve its performance in nonmodel organisms and was interpretable. Applying DeepASmRNA to Iso-Seq data from Amborella, the performance of our method was also better than that of AStrap.
The main factor affecting the identification precision of alternatively spliced transcripts is the ability to distinguish between alternatively spliced transcripts and paralogous transcripts. In general, variants resulting from AS are indels, whereas variants resulting from gene duplication are indels and single nucleotide variants. In this study, a variety of parameters were designed to capture the differences between transcripts, but limited by the local alignment of BLASTN, the global difference between two transcripts may not be fully illuminated. In particular, the uncertainty of the boundaries of HSPs may affect the extraction of AS regions, which may reduce the accuracy of the classification of AS events to a certain degree. It is tempting to address these problems in future studies.
In AS event classification, due to the extreme imbalance of the classes of AS in labeled data, the training may decrease the performance of rare classes to improve the overall performance of DeepASmRNA. However, because DeepASmRNA inputs are exon sequences, we cannot use the oversampling method to balance data. Inspired by the image data augmentation method (Cao and Zhang, 2019), we use the reverse complement (RC) augmentation trick as they did in DNA sequence, which adds the reverse complementary strand of the least common class to mitigate label imbalance. We augmented the ES class in Arabidopsis thaliana and rice as well as IR in the human dataset. For ES and IR events, such data augmentation is biologically logical; that is, these events are expected to occur in both the original sequence and its reverse complement. Furthermore, we augmented and balanced the dataset after it was divided into training, validation, and test sets to avoid data leakage. After data augmentation, although the overall performance of DeepASmRNA did not improve much, DeepASmRNA has a significantly better power to predict the augmentation class (Table S10). For example, in Arabidopsis thaliana, before data augmentation, the precision and recall of ES events were only 83.33% and 71.82%, respectively, whereas after data augmentation, they were 91.99% and 87.68%, respectively. As the performance of the augmented class was enhanced and the performance of the remaining classes remained constant, our data augmentation strategy might be a solution for dealing with the unbalanced labels in AS classification.
Using the masked input methods, we explored what useful information has been learned from mRNA transcript sequences. We observed that DeepASmRNA has learned different sequences of useful information for different AS event classes, and this key information is in line with domain knowledge in biology. The interpretability of DeepASmRNA shows that deep learning is not merely a black box.
The rapid increase in the number of sequenced transcriptomes without reference genomes has empowered the investigation of AS events in nonmodel organisms. Here, we present DeepASmRNA, an accurate, scalable, and biologically interpretable tool for predicting AS events using the transcriptome alone, which is a step toward investigating AS genome-wide in species without a reference genome. We expect that DeepASmRNA will greatly empower the studies of alternative splicing in nonmodel species.
Limitations of the study
We note that DeepASmRNA still has some limitations. First, DeepASmRNA can only classify the 4 basic classes of AS events considered in this study. Second, the performance of DeepASmRNA depends heavily on accurately identifying exon boundaries. Because BLAST may not be able to accurately identify exon boundaries, we used Chambon’s (GU-AG) rule to adjust exon boundaries in HSPs. In the future, we will explore novel methods to improve the identification of exon boundaries.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests should be directed to the Lead Contact, Dr. Erli Pang (pangerli@bnu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Method details
Overview
To detect AS from full-length transcripts without a reference genome, we developed DeepASmRNA including the identification of alternatively spliced transcripts by alignments between two transcripts and the classification of AS events from primary transcript sequences by an attention-based CNN model (Figure 1). For the identification of alternatively spliced transcripts, there are two steps needed: sequence alignment and recognition of AS patterns. For the classification of AS events, two steps are also needed: AS flanking sequence encoding and deep learning model training. The four basic types of AS events are included in our model: ES, AA, AD, and IR.
Data preparation
To train and verify DeepASmRNA, full-length transcripts and structural annotations of the three well-annotated species including human (GRCh38.p13, v37) (Frankish et al., 2021), Arabidopsis thaliana (Araport11) (Cheng et al., 2017; Arabidopsis Genome Initiative et al., 2000), and rice (MSU7) (Kawahara et al., 2013) were downloaded from GENECODE (https://www.gencodegenes.org/), Araport (https://www.araport.org/), and the Rice Genome Annotation Project website (http://rice.plantbiology.msu.edu/), respectively (Table S1). Additionally, the Iso-Seq data (Sharon et al., 2013) from Amborella obtained by Pacific BioSciences (PacBio) single-molecule real-time (SMRT) Iso-Seq and the genome sequence (AMTR1.0) of Amborella (Amborella Genome Project et al., 2013) were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/, accession: PRJNA374048), including three samples from leaves and three samples from flowers. Full-length transcripts of Amborella were detected by PacBio SMRT analysis (https://www.pacb.com/support/software-downloads/) with default parameters (Table S7). To compare with AStrap (Ji et al., 2019), we also obtained the datasets used in AStrap.
Identification of alternatively spliced transcripts
To detect potentially alternatively spliced transcripts, we first performed all-versus-all alignments between full-length transcripts using BLASTN (v2.10.1) (Altschul et al., 1990) with the arguments “-evalue 1E-10” and “-ungapped”. The transcript pairs with more than 70% reciprocal overlap and all HSP identities greater than 99% were retained for downstream analysis. The criteria were chosen by the grid search approach (Table S11). The transcript pairs were considered alternatively spliced transcripts, and only the HSPs and indels simultaneously met the following criteria: 1) the number of HSPs was more than one, 2) the number of mismatched bases in all HSPs was less than 3, and 3) the length of indels between two adjacent HSPs used a different strategy; for human data, it was longer than 1 and simultaneously shorter than 2000 bp, and for Arabidopsis thaliana and rice data, it was longer than 1 and simultaneously shorter than 400 bp, based on the distribution of lengths of introns in the respective species.
Evaluation of alternatively spliced transcripts
To verify the performance of detecting alternatively spliced transcripts, four basic types of AS events of human, Arabidopsis thaliana, and rice were obtained by SUPPA2 with the default parameters (Trincado et al., 2018) guided by their structural annotation of the genome. Meanwhile, we examined existing mRNAs from the gene locus to verify the AS events. For Amborella, the AS events were obtained by ALT_SPLICE function in PASA (Campbell et al., 2006) with default parameters. A pair of transcripts were considered alternatively spliced transcripts if there was an AS event between them. The alternatively spliced transcripts obtained by SUPPA2 and PASA were considered positive datasets. Our prediction was compared with the ground truth. The precision and recall were measured by the following formulas.
where TP is true positive, FP is false positive, and FN is false negative.
Extracting and encoding AS flanking sequences
DeepASmRNA predicted AS events from mRNA transcript sequences, so we first obtained the mRNA transcript sequences involved in each AS event. For each type of AS event from the above SUPPA2 results, we extracted the sequences of the alternatively spliced region and N (ranging from 10 to 60, steps by 10) bp flanking exon sequences in constitutive regions (Figure S5). Thus, an AS event was represented by alternative region sequences and a pair of flanking sequences in constitutive regions.
Next, we encoded the alternative region sequences and a pair of flanking sequences in constitutive regions into a tensor that can be used for the deep learning model. For the alternatively spliced regions with varying lengths, only the sequences near the AS sites had a great influence on the AS event classification, so we retained N bp sequences upstream and downstream of splicing sites and combined them. For sequences less than N bp, we used 0 to pad the positions far from AS sites to ensure equal length. Thus, an AS event is represented by a 4N+2 bp sequence, which is composed of four sequence fragments, and the character ‘|’ was used to indicate the splicing sites (Figure 3A). Then, we utilized the most common method for encoding mRNA sequences, one-hot encoding (Jing et al., 2020). A, T, C, G, and ‘|’ were represented by a five-dimensional binary vector. The four bases and ‘|’ were fixed in a specific order, and then the ith (i ∈ 1,..,4N+2}) position was represented by five binary bits with the jth (j ∈ 1,..,5}) bit set to “1” and others to “0” (Figure 3A) (Eraslan et al., 2019). After the encoding, each AS event was transformed into a tensor with the shape of (4N+2)∗5.
DeepASmRNA architecture
Recently, attention mechanisms developed to improve the performance of encoder-decoder models have achieved successful applications in many fields (Vaswani et al., 2017). This approach overcomes the word sense disambiguation limitation, which may help CNN focus on positions where sequences are similar (Tang et al., 2018). Considering its efficiency and precision in machine translation, we also attempted to use the self-attention mechanism to enhance the CNN performance.
The complete architecture of DeepASmRNA (Figure 3B) is composed of the following parts: a multi-layer, self-attention-based CNN for extracting efficient information from raw data, a global convolution layer for summarizing the key information and generating vectors, and two layers of fully connected neural network layers with a dropout layer as the output layer.
The basic element of the DeepASmRNA architecture is a self-attention layer (Figure 3B). First, the one-hot encoding of a transcript sequence was inputted, and three one-dimensional convolution layers scanned information in the sequence to generate query (Q), key (K), and value (V) vectors for each base. Next, we used the Q, K, and V vectors to generate dot-product attention; thus, each base of the sequence obtained global information of the sequence in the first layer. Then, we concatenated the vectors extracted by attention and the Q tensor, which was regarded as the output of self-attention (Yin et al., 2015). Finally, a max-pooling layer was used to downsample and shorten the sequence.
Overall, we designed multiple architectures, namely, DeepASmRNA-Nbp, which used N (N ranges from 10 to 60, steps by 10) nucleotides on each side of splicing sites as input. Our model took one-hot coded transcript sequences of an AS event as input. After the multiple layer neural network, the output of the model consisted of four scores that sum to one, corresponding to the probabilities of different types of AS events (ES, AA, AD, and IR).
Classification model training and tuning
We used TensorFlow 2.1 (Abadi et al., 2016) as the basic framework to build and train our models. All models were trained on servers with NVIDIA Tesla P100 dual GPUs. The AS events obtained by SUPPA2 from human, Arabidopsis thaliana, and rice data were used to train and test the models. Because the numbers of different types of AS events were very different, which may significantly affect the multiclass classification performance, we balanced the AS events. We used the reverse complementary strand of ES events in the Arabidopsis thaliana and rice as well as IR events in the human dataset to augment the unbalanced data (Cao and Zhang, 2019). The datasets were divided into training, validation, and testing sets at a ratio of 6:2:2, and then each set was balanced according to the reverse complementary metrics.
For each model, we tuned multiple hyperparameters, including the number of convolution kernels, the length of convolution kernels, the number of hidden layers, and the dropout rate. Finally, the hyperparameters of each model were determined according to model performance in the validation sets (Table S12). Since our study is a multiclass classification problem, we used softmax as the activation function of the output layer, ReLU (Nair and Hinton, 2010) as the activation function of hidden layers, cross entropy as the loss function, and Adam (Kingma and Ba, 2014) as the optimizer with a learning rate of 0.001. To avoid overfitting, we adopted early stopping and dropout strategies. A dropout rate of 0.6 was settled after a few attempts.
Evaluation of AS event classification
We used several evaluation metrics to assess the performance of the DeepASmRNA models. For each model, we computed the precision, recall, accuracy, and area under the curve (AUC). For each type of AS event, we used precision, recall, F1-score, and area under precision-recall curve (AUPRC) as evaluation metrics. The receiver operating characteristic (ROC) curves of the models for classification were plotted, and the AUCs were calculated. Accuracy was used to assess the overall performance of the DeepASmRNA model. The accuracy and F1-score formulas are:
where TP is true positive, TN is true negative, FP is false-positive, and FN is false negative.
Comparison to other universal deep learning models
To find the most suitable model for our problems and goals, we also tried several models that are widely used in deep learning, including LeNet (Lecun et al., 1998), ResNet (Kaiming et al., 2016), and long short-term memory (LSTM) (Greff et al., 2017). The input layer, output layer, and final fully connected layer of all models were consistent with the DeepASmRNA architecture, but the difference was the structure of the information extraction layer. For LeNet, a one-dimensional convolution layer and a maximum pooling layer were used as convolution units. After two convolution units, a global convolution layer summarizes the information. For ResNet, we used a one-layer convolution and two-layer residual blocks to extract information. For LSTM, we used one-layer convolution, one-layer pooling, and three layers LSTM. All models used the same training, validation, and test datasets, and the training environment was the same as that of DeepASmRNA.
Transfer learning
Scalability applied efficiently to external data is another model requirement in addition to accuracy and generalizability. Transfer learning is a technique for solving tasks with a small labeled dataset by using a pre-trained model for another task, which breaks the assumption that the test set and training set have the same distribution to some extent (Sinno Jialin and Qiang, 2010). One of the most common methods of transfer learning is fine-tuning learning, which uses a very small number of training sets to train only a few layers of the model to fine-tune the model for better performance on new problems (Sinno Jialin and Qiang, 2010). In the process of fine-tuning learning, the information extracted from the first few layers is usually more basic, while the later layers are more likely to be task-related (Yosinski et al., 2014). Therefore, in fine-tuning learning, we freeze all the convolution layers and train only the last two fully connected layers (Figure 3C). However, because the fine-tuning set was very small and seriously biased, the overfitting of the model was serious. With the increase in training epochs, the performance of the validation set decreased rapidly (Yosinski et al., 2014). Considering that there is no good validation set in practical application, we only trained two fully connected layers for 20 epochs to ensure the fine-tuning effect. In addition, we found that the epochs of re-train influence the performance of fine-tune learning, Here, we used 20 epochs to calculate the result while a smaller value should carry out when applied to closely related species.
We chose human and Arabidopsis thaliana as well as Arabidopsis thaliana and rice to represent distant species and closely related species, respectively. A fine-tuning set was composed of 2 samples for each type of AS event which was 8 samples for each species, while the test set was the above test set.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31571361).
Our deepest gratitude goes to Prof. Xiaohui Wu and Wenbin Ye from Xiamen University for providing the dataset used in AStrap.
Author contributions
E.L.P. designed research. L.C. Q.B.Z., and H.T.S. performed research. E.L.P., L.C., Q.B.Z., and K.L. wrote the original draft of the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: November 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105345.
Supplemental information
Data and code availability
-
•
All original code has been deposited at https://github.com/CMB-BNU/DeepASmRNA or http://cmb.bnu.edu.cn/DeepASmRNA/index.php/download and is publicly available as the data of publication.
-
•
All processed data to reproduct the results in DeepASmRNA has been deposited at https://data.mendeley.com/datasets/r5ccs67jhd/draft?a=03c273e2-55b6-44d6-8557-297f77a2a1bb.
-
•
Any additional required to reanalyze the data report in this paper is available from the lead contact upon request.
References
- Abadi M., Agarwal A., Barham P., Brevdo E., Chen Z., Citro C., Corrado G.S., Davis A., Dean J., Devin M., et al. TensorFlow: large-scale machine learning on heterogeneous distributed systems. arXiv. 2016 doi: 10.48550/arXiv.1603.04467. Preprint at. [DOI] [Google Scholar]
- Amborella Genome Project. Barbazuk W., dePamphilis C.W., Der J.P., Leebens-Mack J., Ma H., Palmer J.D., Rounsley S., Sankoff D., Schuster S.C., et al. The Amborella genome and the evolution of flowering plants. Science. 2013;342:1241089. doi: 10.1126/science.1241089. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative. Koo H.L., Jenkins J., Rizzo M., Rooney T., Tallon L.J., Feldblyum T., Nierman W., Benito M.I., Lin X., Stiekema W.J. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Barash Y., Calarco J.A., Gao W., Pan Q., Wang X., Shai O., Blencowe B.J., Frey B.J. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- Campbell M.A., Haas B.J., Hamilton J.P., Mount S.M., Buell C.R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genom. 2006;7:327. doi: 10.1186/1471-2164-7-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z., Zhang S. Simple tricks of convolutional neural network architectures improve DNA-protein binding prediction. Bioinformatics. 2019;35:1837–1843. doi: 10.1093/bioinformatics/bty893. [DOI] [PubMed] [Google Scholar]
- Chen M., Manley J.L. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C.Y., Krishnakumar V., Chan A.P., Thibaud-Nissen F., Schobel S., Town C.D. Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 2017;89:789–804. doi: 10.1111/tpj.13415. [DOI] [PubMed] [Google Scholar]
- Cherry S., Lynch K.W. Alternative splicing and cancer: insights, opportunities, and challenges from an expanding view of the transcriptome. Genes Dev. 2020;34:1005–1016. doi: 10.1101/gad.338962.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eraslan G., Avsec Ž., Gagneur J., Theis F.J. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 2019;20:389–403. doi: 10.1038/s41576-019-0122-6. [DOI] [PubMed] [Google Scholar]
- Florea L., Hartzell G., Zhang Z., Rubin G.M., Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A., Diekhans M., Jungreis I., Lagarde J., Loveland J.E., Mudge J.M., Sisu C., Wright J.C., Armstrong J., Barnes I., et al. Gencode 2021. Nucleic Acids Res. 2021;49:D916–D923. doi: 10.1093/nar/gkaa1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greff K., Srivastava R.K., Koutnik J., Steunebrink B.R., Schmidhuber J. LSTM: a search space odyssey. IEEE Trans. Neural Netw. Learn. Syst. 2017;28:2222–2232. doi: 10.1109/TNNLS.2016.2582924. [DOI] [PubMed] [Google Scholar]
- Gueroussov S., Gonatopoulos-Pournatzis T., Irimia M., Raj B., Lin Z.Y., Gingras A.C., Blencowe B.J. An alternative splicing event amplifies evolutionary differences between vertebrates. Science. 2015;349:868–873. doi: 10.1126/science.aaa8381. [DOI] [PubMed] [Google Scholar]
- Jaganathan K., Kyriazopoulou Panagiotopoulou S., McRae J.F., Darbandi S.F., Knowles D., Li Y.I., Kosmicki J.A., Arbelaez J., Cui W., Schwartz G.B., et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176:535–548.e24. doi: 10.1016/j.cell.2018.12.015. [DOI] [PubMed] [Google Scholar]
- Ji G., Ye W., Su Y., Chen M., Huang G., Wu X. AStrap: identification of alternative splicing from transcript sequences without a reference genome. Bioinformatics. 2019;35:2654–2656. doi: 10.1093/bioinformatics/bty1008. [DOI] [PubMed] [Google Scholar]
- Jing X., Dong Q., Hong D., Lu R. Amino acid encoding methods for protein sequences: a comprehensive review and assessment. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020;17:1918–1931. doi: 10.1109/TCBB.2019.2911677. [DOI] [PubMed] [Google Scholar]
- Kaiming H., Xiangyu Z., Shaoqing R., Jian S. IEEE; 2016. Deep Residual Learning for Image Recognition. [Google Scholar]
- Kawahara Y., de la Bastide M., Hamilton J.P., Kanamori H., McCombie W.R., Ouyang S., Schwartz D.C., Tanaka T., Wu J., Zhou S., et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013;6:4. doi: 10.1186/1939-8433-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma D.P., Ba J. Adam: a method for stochastic optimization. arXiv. 2014 doi: 10.48550/arXiv.1412.6980. Preprint at. [DOI] [Google Scholar]
- Lecun Y., Bottou L., Bengio Y., Haffner P. Gradient-based learning applied to document recognition. Proc. IEEE. 1998;86:2278–2324. doi: 10.1109/5.726791. [DOI] [Google Scholar]
- Lee Y., Rio D.C. Mechanisms and regulation of alternative Pre-mRNA splicing. Annu. Rev. Biochem. 2015;84:291–323. doi: 10.1146/annurev-biochem-060614-034316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Mei W., Soltis P.S., Soltis D.E., Barbazuk W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017;17:1243–1256. doi: 10.1111/1755-0998.12670. [DOI] [PubMed] [Google Scholar]
- Marquez Y., Brown J.W.S., Simpson C., Barta A., Kalyna M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012;22:1184–1195. doi: 10.1101/gr.134106.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minar M.R., Naher J. Recent advances in deep learning: an overview. arXiv. 2018 doi: 10.13140/RG.2.2.24831.10403. Preprint at. [DOI] [Google Scholar]
- Nair V., Hinton G.E. Proceedings of the 27th International Conference on International Conference on Machine Learning. 2010. Rectified linear units improve restricted Boltzmann machines vinod nair; pp. 807–814. [Google Scholar]
- Oka M., Xu L., Suzuki T., Yoshikawa T., Sakamoto H., Uemura H., Yoshizawa A.C., Suzuki Y., Nakatsura T., Ishihama Y., et al. Aberrant splicing isoforms detected by full-length transcriptome sequencing as transcripts of potential neoantigens in non-small cell lung cancer. Genome Biol. 2021;22:9. doi: 10.1186/s13059-020-02240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagih O. ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics. 2017;33:3645–3647. doi: 10.1093/bioinformatics/btx469. [DOI] [PubMed] [Google Scholar]
- Paggi J.M., Bejerano G. A sequence-based, deep learning model accurately predicts RNA splicing branchpoints. RNA. 2018;24:1647–1658. doi: 10.1261/rna.066290.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Qi H., Guo X., Wang T., Zhang Z. ASTool: an easy-to-use tool to accurately identify alternative splicing events from plant RNA-seq data. Int. J. Mol. Sci. 2022;23:4079. doi: 10.3390/ijms23084079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon D., Tilgner H., Grubert F., Snyder M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013;31:1009–1014. doi: 10.1038/nbt.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrikumar A., Tian K., Avsec Ž., Shcherbina A., Banerjee A., Sharmin M., Nair S., Kundaje A. Technical note on transcription factor motif discovery from importance scores (TF-MoDISco) version 0.5.6.5. arXiv. 2018 doi: 10.48550/arXiv.1811.00416. Preprint at. [DOI] [Google Scholar]
- Pan S.J., Yang Q. A survey on transfer learning. IEEE Trans. Knowl. Data Eng. 2010;22:1345–1359. doi: 10.1109/TKDE.2009.191. [DOI] [Google Scholar]
- Staiger D., Brown J.W.S. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell. 2013;25:3640–3656. doi: 10.1105/tpc.113.113803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Zhang Q., Liu B., Lin K., Zhang Z., Pang E. CuAS: a database of annotated transcripts generated by alternative splicing in cucumbers. BMC Plant Biol. 2020;20:119. doi: 10.1186/s12870-020-2312-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G., Müller M., Rios A., Sennrich R. EMNLP; 2018. Why Self-Attention? a Targeted Evaluation of Neural Machine Translation Architectures. [Google Scholar]
- Thatcher S.R., Zhou W., Leonard A., Wang B.B., Beatty M., Zastrow-Hayes G., Zhao X., Baumgarten A., Li B. Genome-wide analysis of alternative splicing in Zea mays: landscape and genetic regulation. Plant Cell. 2014;26:3472–3487. doi: 10.1105/tpc.114.130773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trincado J.L., Entizne J.C., Hysenaj G., Singh B., Skalic M., Elliott D.J., Eyras E. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018;19:40. doi: 10.1186/s13059-018-1417-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser L., Polosukhin I. Attention is all you need. arXiv. 2017 doi: 10.48550/arXiv.1706.03762. Preprint at. [DOI] [Google Scholar]
- Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Hu Z., Ye N., Yin H. IsoSplitter: identification and characterization of alternative splicing sites without a reference genome. RNA. 2021;27:868–875. doi: 10.1261/rna.077834.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.D., Watanabe C.K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- Xiong H.Y., Alipanahi B., Lee L.J., Bretschneider H., Merico D., Yuen R.K.C., Hua Y., Gueroussov S., Najafabadi H.S., Hughes T.R., et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347 doi: 10.1126/science.1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W., Schütze H., Xiang B., Zhou B. ABCNN: attention-based convolutional neural network for modeling sentence pairs. Trans. Assoc. Comput. Linguist. 2016;4:259–272. [Google Scholar]
- Yosinski J., Clune J., Bengio Y., Lipson H. How transferable are features in deep neural networks? arXiv. 2014 doi: 10.48550/arXiv.1411.1792. Preprint at. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All original code has been deposited at https://github.com/CMB-BNU/DeepASmRNA or http://cmb.bnu.edu.cn/DeepASmRNA/index.php/download and is publicly available as the data of publication.
-
•
All processed data to reproduct the results in DeepASmRNA has been deposited at https://data.mendeley.com/datasets/r5ccs67jhd/draft?a=03c273e2-55b6-44d6-8557-297f77a2a1bb.
-
•
Any additional required to reanalyze the data report in this paper is available from the lead contact upon request.