

Abstract
Glutathione S-transferase alpha 4 (GSTA4) is a phase II detoxifying enzyme that metabolizes electrophiles and carcinogens including 4-hydroxynonenal (4-HNE), an endogenous carcinogen that contributes to colorectal carcinogenesis. In this study, we investigated GSTA4 expression and regulation in murine primary colonic epithelial cells, microbiome-driven murine colitis, and human carcinomas. Exposure of YAMC cells to 4-HNE induced Gsta4 expression. Using an inflammation-associated model of colorectal cancer (CRC), Gsta4 expression increased in vivo in colon macrophages and serum after 2 weeks of colonization of IL-10 deficient (Il10−/−) mice with Enterococcus faecalis. Increased expression was noted after 9 months of colonization in colon macrophages and epithelia in areas of inflammation. In human colon biopsies immunohistochemistry showed no GSTA4 expression in normal epithelial cells whereas GSTA4 was strongly expressed in the neoplastic epithelia of invasive carcinomas. For tubular adenomas increased expression was primarily noted in stromal macrophages. Increased GSTA4 was confirmed in established human CRC cell lines and associated with 4-HNE-protein adducts in human colon adenomas and CRC. Next, we showed that 4-HNE induced activation of c-Jun and Nrf2, two components of the oncogenic transcription factor AP-1. AP-1 inhibitors and gene specific small interfering RNAs partially suppressed GSTA4 expression. Co-immunoprecipitation confirmed interactions between c-Jun and Nrf2 supporting a role for AP-1 in regulating 4-HNE-induced GSTA4 expression. These findings demonstrate GSTA4 activation during 4-HNE-induced neoplastic transformation in colorectal carcinogenesis. GSTA4 is a potential surrogate biomarker for CRC screening and should provide novel approaches for chemoprevention.
Keywords: Glutathione S-transferase alpha 4, c-Jun, Nrf2, colorectal cancer, biomarker
INTRODUCTION
Glutathione S-transferases (GSTs) are phase II enzymes that metabolize a range of electrophilic toxic xenobiotics and carcinogens.1 The mammalian GST family comprises seven major classes based on primary structure and metabolized substrates. Polymorphisms and mutations in GSTs are associated with an increased susceptibility to human cancer including lung,2 bladder,3 breast,4 and colon.5,6 Animal models show that mutations in GSTs can increase susceptibility to cancer. For example, deleting glutathione S-transferase Pi (Gstp) in ApcMin/+ mice significantly increases colon adenoma multiplicity and decreases survival compared to controls.7 Several GSTs are overexpressed in cancer cells compared to corresponding normal tissues.8–10 This phenotype is also associated with increased resistance to cisplatin.11 Finally, in selected human cancers, GST expression is associated with oncogene activation and may aid in early diagnosis or prognosis.8,12
Glutathione S-transferase alpha 4 (GSTA4) catalyzes the conjugation of glutathione with trans-4-hydroxy-2-nonenal (4-HNE) and thereby detoxifies 4-HNE.13 4-HNE is a highly reactive breakdown product of ω-6 polyunsaturated fatty acids that forms mutagenic adducts with DNA14 and generates Michael adducts with proteins that adversely affect function.15 This aldehyde plays a diverse role in carcinogenesis by inhibiting DNA repair,16 regulating the cell cycle,17,18 modulating p53-mediated apoptosis, and activating mitogen-activated protein kinases, nuclear factor κB, cyclooxygenase-2, and stress response genes.19–22
Limited data shows that GSTA4 is normally expressed in human liver, testis, skin, and colon.23,24 In conventionally housed mice, Gsta4 expression has been observed in colon epithelial cells.25 Expression of this GST along with other alpha class GSTs has been noted during inflammation and in selected human cancers.8,25 Notably, in a rat model of chemically induced hepatocarcinogenesis Gsta4, but not Gstp, correlated with nuclear factor-erythroid 2-related factor 2 (Nrf2) activation.9 This transcription factor regulates multiple antioxidant and cellular protective genes involved in oxidative stress including GSTs. Nonetheless, mechanisms for GSTA4 regulation during inflammation and cancer initiation remain unclear.
Recent studies have shown that 4-HNE is a potent endogenous mutagen that causes double-strand DNA breaks, spindle dysfunction, tetraploidy, and chromosomal instability.26,27 Furthermore, repeated in vitro exposure to sublethal doses of 4-HNE can transform primary colonic epithelial cells into poorly differentiated carcinomas.27 4-HNE is a mediator for microbiome-driven bystander effects.26,27 This mechanism for cancer initiation occurs through endogenous mutagenesis via COX-2-dependent production of 4-HNE by polarized macrophages.28 These findings strongly implicate 4-HNE in malignant transformation and suggest GSTA4, as a primary detoxifying enzyme for 4-HNE, is an important defense against cancer initiation in inflammation-associated CRC. Using a dual-chamber system that mimics cancer-inducing bystander effects, we previously showed that silencing Gsta4 in murine colonic epithelial cells significantly increased the susceptibility of cells to 4-HNE-induced genotoxicity.26 Other studies using Gsta4 null mice have shown an accumulation of 4-HNE-protein adducts in multiple organs. These mice are more susceptible to bacterial infection but not cancer.29,30 In contrast, Gsta4 was recently demonstrated to be a tumor suppressor for skin cancer in mice.31 Polymorphisms in a corresponding human gene have been shown to significantly affect the risk for nonmelanoma skin cancer.
In this study we investigated the expression of Gsta4 in human colon cancer cell lines and human adenoma and carcinoma biopsies as well as in microbiome-driven CRC using E. faecalis colonized interleukin-10 knockout mice (Il10−/−). GSTA4 was noted in premalignant human adenomas with maximal staining found in epithelial cells from CRC biopsies. We found that Gsta4 was strongly expressed by macrophages during the early stages of colon inflammation and later in colon epithelial cells as CRC developed. Furthermore, we demonstrated that oncogenic transcription factor activator protein 1 (AP-1) regulated GSTA4 induction by 4-HNE. c-Jun, a critical component of AP-1, was rapidly activated in primary murine colonic epithelial cells after exposure to 4-HNE. Finally, we linked Nrf2 to GSTA4 expression and confirmed its interaction with c-Jun. These results demonstrate that GSTA4 is activated during the initiation phase of colorectal carcinogenesis and has the potential to serve as a novel biomarker in CRC screening.
RESULTS
4-HNE induces Gsta4
We initially determined expression of Gsta4 in YAMC murine primary colonic epithelial cells following exposure to purified 4-HNE. Untreated YAMC cells expressed Gsta4 but that expression was further increased upon 4-HNE treatment (Figures 1a and b), suggesting an adaptive response to this aldehyde. The total Gst activity significantly increased 24 h following 4-HNE treatment and reverted back to control levels by 48 h (Figure 1c). This may reflect 4-HNE-induced gene suppression of other Gst sub-classes such as Gstp1 and Gstk1.27,32 To investigate Gsta4 expression in vivo, we stained for Gsta4 in colon biopsies from Il10−/− mice colonized with E. faecalis that had developed colitis and colon cancer and from sham-colonized control mice.26 After 2 weeks of colonization with E. faecalis, Gsta4-positive cells were found scattered in the colon mucosal stroma of Il10−/− mice compared to sham-colonized mice (Figures 2a and b). In addition, Gsta4 increased in serum of these mice at 2 weeks compared to controls. Increased serum Gsta4 was not noted in wild-type mice (Figure 2c). After 9 months colonization of Il10−/− mice with E. faecalis Gsta4 increased in areas of inflammation compared to sham-colonized mice (Figures 2d and e). Colonic epithelial cells stained for Gsta4 in areas of inflammation when compared to normal crypts (Figures 2d and e). Finally, Gsta4 induction was largely localized to macrophages by immunofluorescent imaging (Figures 2f–i), suggesting increased expression in these myeloid cells.
Figure 1.
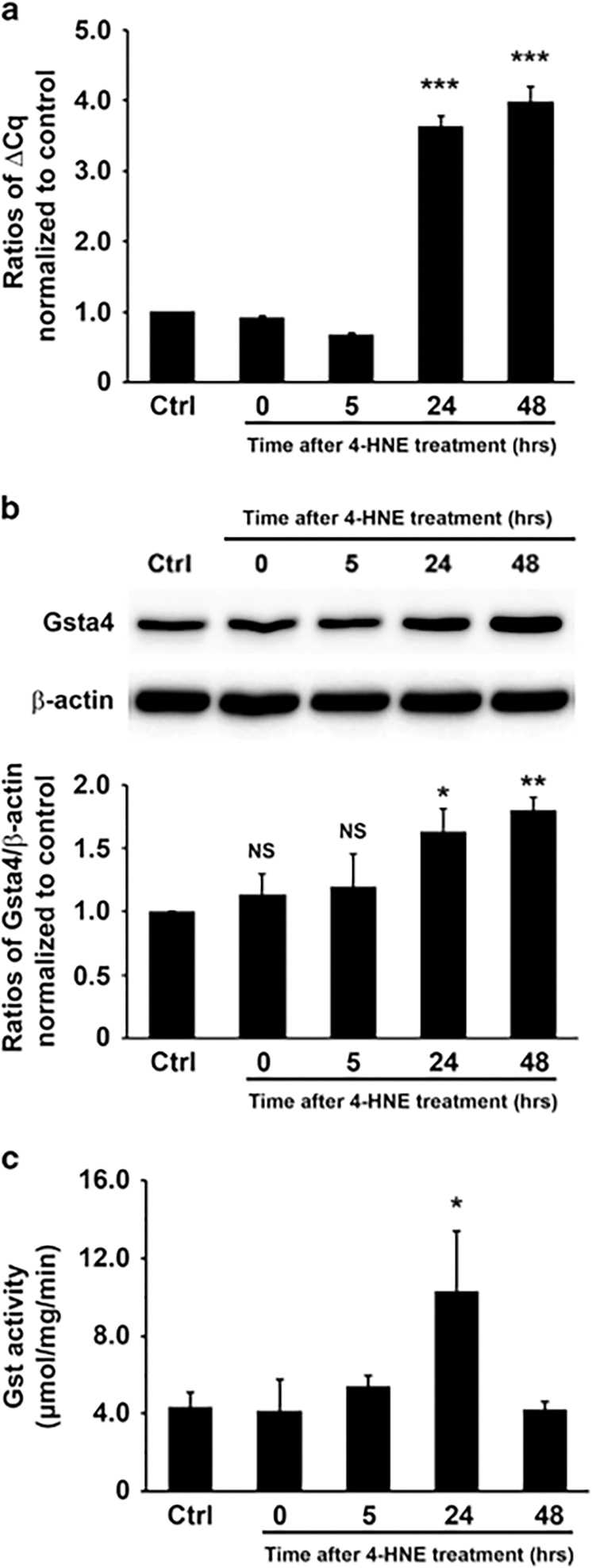
4-HNE induces Gsta4 expression. (a) qRT-PCR shows significantly increased Gsta4 expression in YAMC cells 24 to 48 h following 4-HNE treatment (***P < 0.001; ctrl, untreated YAMC cells). (b) Western blot shows increased Gsta4 in YAMC cells 24 and 48 h following 1 h treatment with purified 4-HNE (upper). Bar graph shows ratios of Gsta4/β-actin after being normalized to untreated control (lower). NS, not significant, *P < 0.05, and **P < 0.01 compared to ctrl. (c) Gst activity increases 24 h following 4-HNE treatment (*P < 0.05; ctrl, untreated YAMC cells). All data represent mean ± s.d. for three independent experiments.
Figure 2.
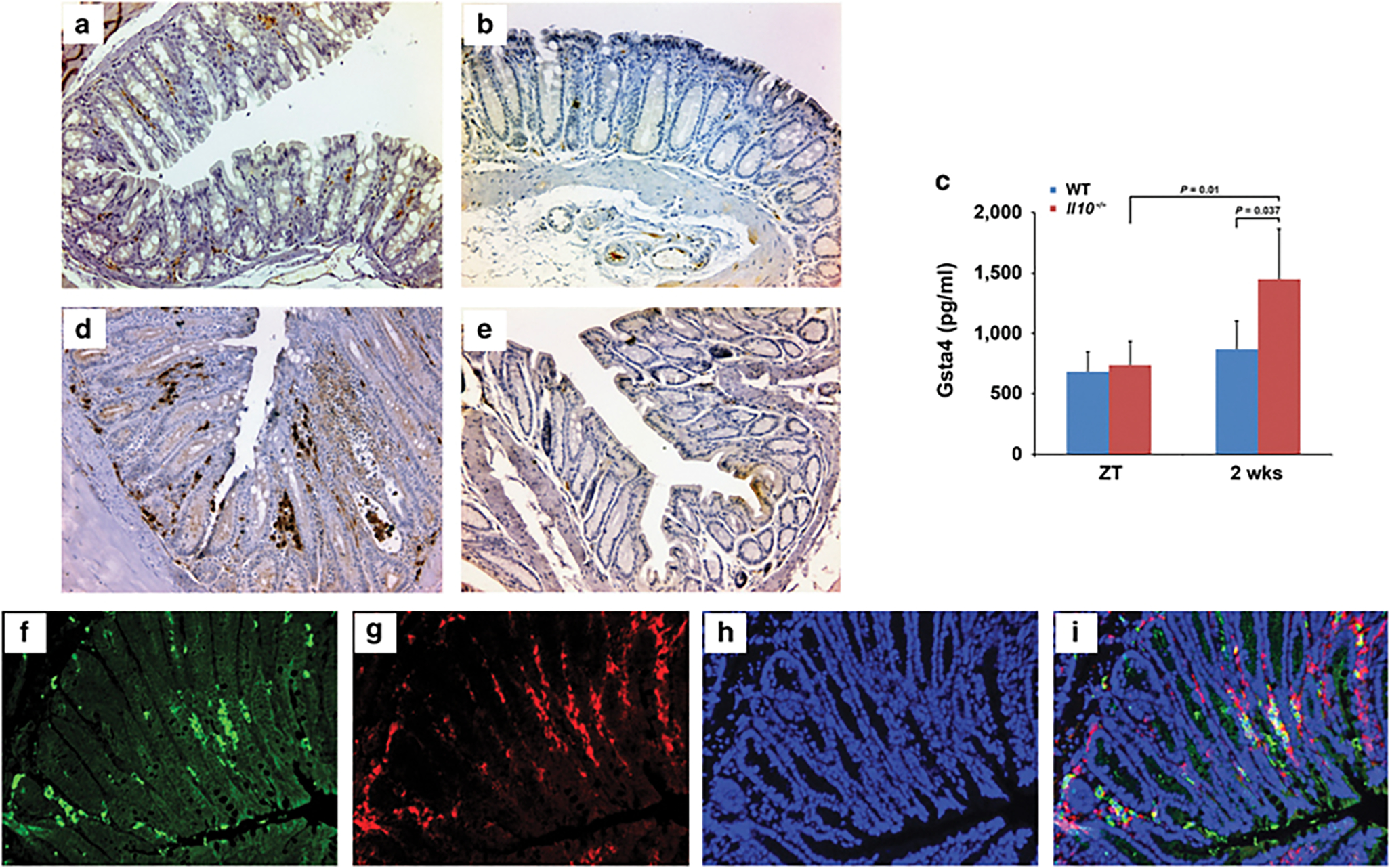
Gsta4 is produced in microbiome-driven colitis and cancer. (a) Immunohistochemical staining shows Gsta4 expression in the lamina propria of colon biopsies from Il10−/− mice colonized with E. faecalis for 2 weeks. (b) No Gsta4 staining is seen in biopsies from sham-colonized mice. (c) Gsta4 significantly increases in serum from Il10−/− mice (n = 5, red bars) compared to wild-type mice (n = 5, blue bars) after 2 weeks colonization with E. faecalis. ZT, zero time prior to colonization. (d) Intense staining for Gsta4 is seen in lamina propria and epithelial cells in areas of inflammation and neoplasia in colon sections from Il10−/− mice colonized with E. faecalis after 9 months. (e) No positive staining for Gsta4 is noted in colon sections from sham-colonized Il10−/− mice. (f-i), Immunofluorescent staining for Gsta4 (f, green) and F4/80 (g, red) confirms Gsta4 expression in macrophages (i, yellow). Nuclei are counterstained using 4’,6-diamidino-2-phenylindole (h and i, blue).
GSTA4 in human colon adenomas and carcinomas
To confirm whether GSTA4 expression was upregulated during human colorectal carcinogenesis, we analyzed expression in established CRC cell lines compared to a fetal human colon epithelial cell line. All human CRC cell lines strongly expressed GSTA4 compared with a fetal human colon epithelial cell line (Figure 3a). For human colon biopsies, no GSTA4 expression was found in epithelial cells for tumor-adjacent normal colon (TANC) biopsies, except for one sample, or in hyperplastic polyps (Table 1). In the stroma, moderate staining was noted in 17 of 45 (38%) biopsies for TANCs and in 11 of 30 (37%) biopsies for hyperplastic polyps (Table 1, Figures 3b, c, and d). GSTA4 was more strongly expressed in the stroma of tubular adenomas than in colon epithelial cells (P < 0.001; Table 1 and Figures 3b and e). In contrast, for invasive CRCs, GSTA4 was more strongly expressed in epithelial cells than the stroma (P < 0.001; Table 1 and Figures 3b and f). In addition, immunofluorescent imaging showed that most GSTA4-positive cells in the stroma stained for F4/80 indicating the cells were macrophages (Figures 3g–j). Finally, increased GSTA4 expression was associated with an accumulation of 4-HNE-protein adducts in tubular adenomas and invasive CRCs compared to rare positive cells in biopsies from TANCs or hyperplastic polyps (Figures 4a–d). These findings indicate increased GSTA4 expression in premalignant human adenomas with maximal staining in colon epithelial cells from CRC.
Figure 3.
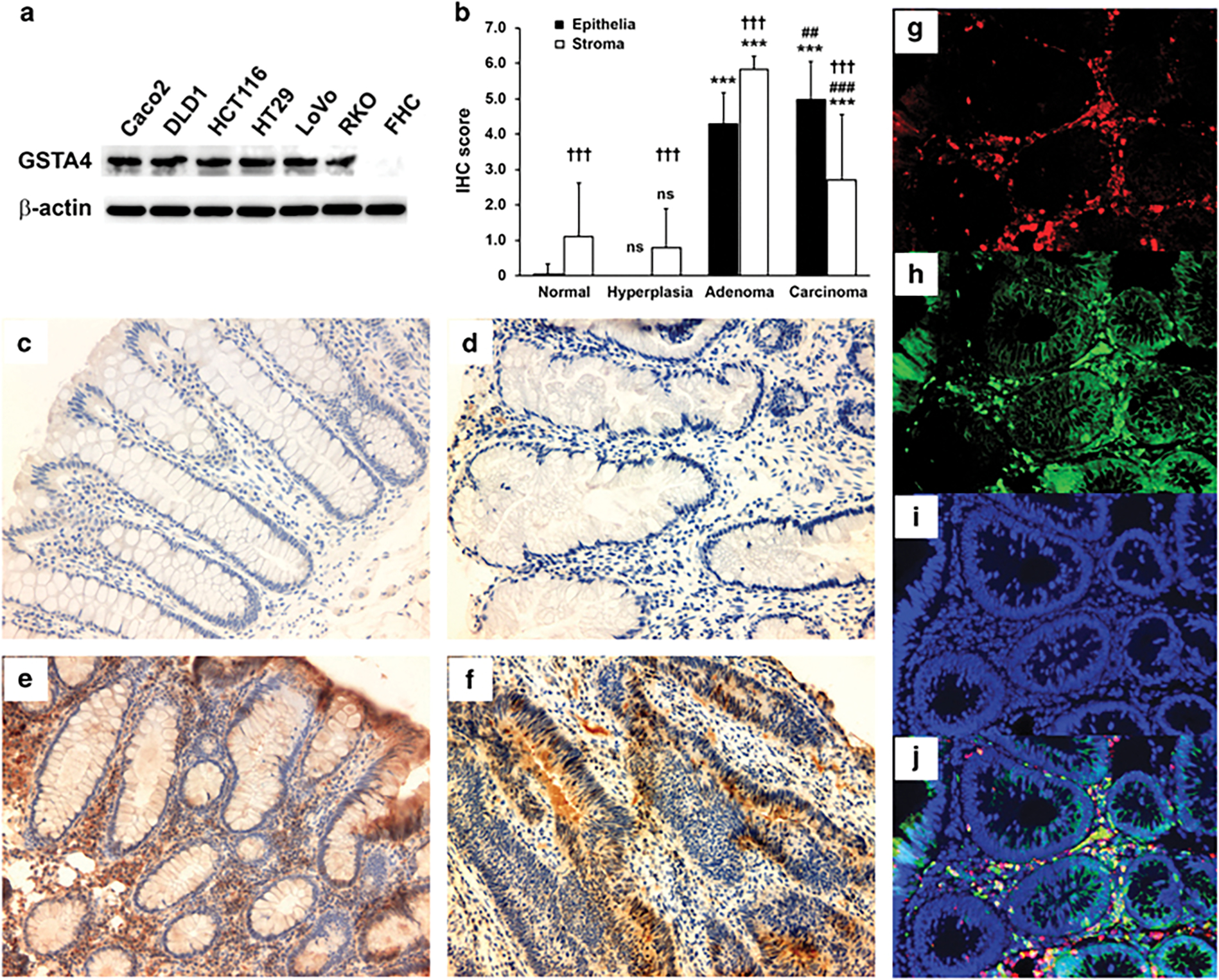
GSTA4 is expressed in human colon adenomas and carcinomas. (a) Western blots show increased expression of GSTA4 in colon cancer cell lines compared to a fetal human colonic epithelial cell line (FHC). (b) Immunohistochemical staining scores for GSTA4 in human colon tissues (NS, not significant, ***P < 0.001 compared to TANC biopsies; ##P < 0.01 and ###P < 0.001 compared to tubular adenomas; †††P < 0.001 compared to epithelia). (c-f) Representative immunohistochemical staining of colon biopsies for GSTA4 in TANCs (c), hyperplastic polyps (d), tubular adenomas (e), and invasive carcinomas (f); intense staining is evident for GSTA4 in tubular adenomas and invasive carcinomas compared to normal tissue and hyperplastic polyps. (g-j) Immunofluorescent staining for F4/80 (g, red) and GSTA4 (h, green) shows co-localization of GSTA4 and macrophages (j, yellow) in a tubular adenoma. Nuclei are counterstained with DAPI (i and j, blue).
Table 1.
Summary of IHC staining for GSTA4 in human colon biopsies
| Tissue type | Scorea |
Total | |||||
|---|---|---|---|---|---|---|---|
| 0 | 2 | 3 | 4 | 5 | 6 | ||
|
| |||||||
| Epithelia | |||||||
| TANC | 44b | 1 | 0 | 0 | 0 | 0 | 45 |
| Hyperplastic colon | 30 | 0 | 0 | 0 | 0 | 0 | 30 |
| Tubular adenoma | 0 | 1 | 3 | 14 | 10 | 2 | 30 |
| Invasive carcinoma | 0 | 1 | 3 | 5 | 13 | 13 | 35 |
| Stroma | |||||||
| TANC | 28 | 5 | 8 | 4 | 0 | 0 | 45 |
| Hyperplastic colon | 19 | 9 | 2 | 0 | 0 | 0 | 30 |
| Tubular adenoma | 0 | 0 | 0 | 0 | 5 | 25 | 30 |
| Invasive carcinoma | 7 | 11 | 4 | 6 | 5 | 2 | 35 |
Abbreviations: ICH, immunohistochemistry; TANC, tumor-adjacent normal colon.
Score was determined by the combination of overall number of GSTA4-positive cells and IHC intensity (see Materials and Methods section for detailed categories).
No. of samples with the score listed above the numbers.
Figure 4.
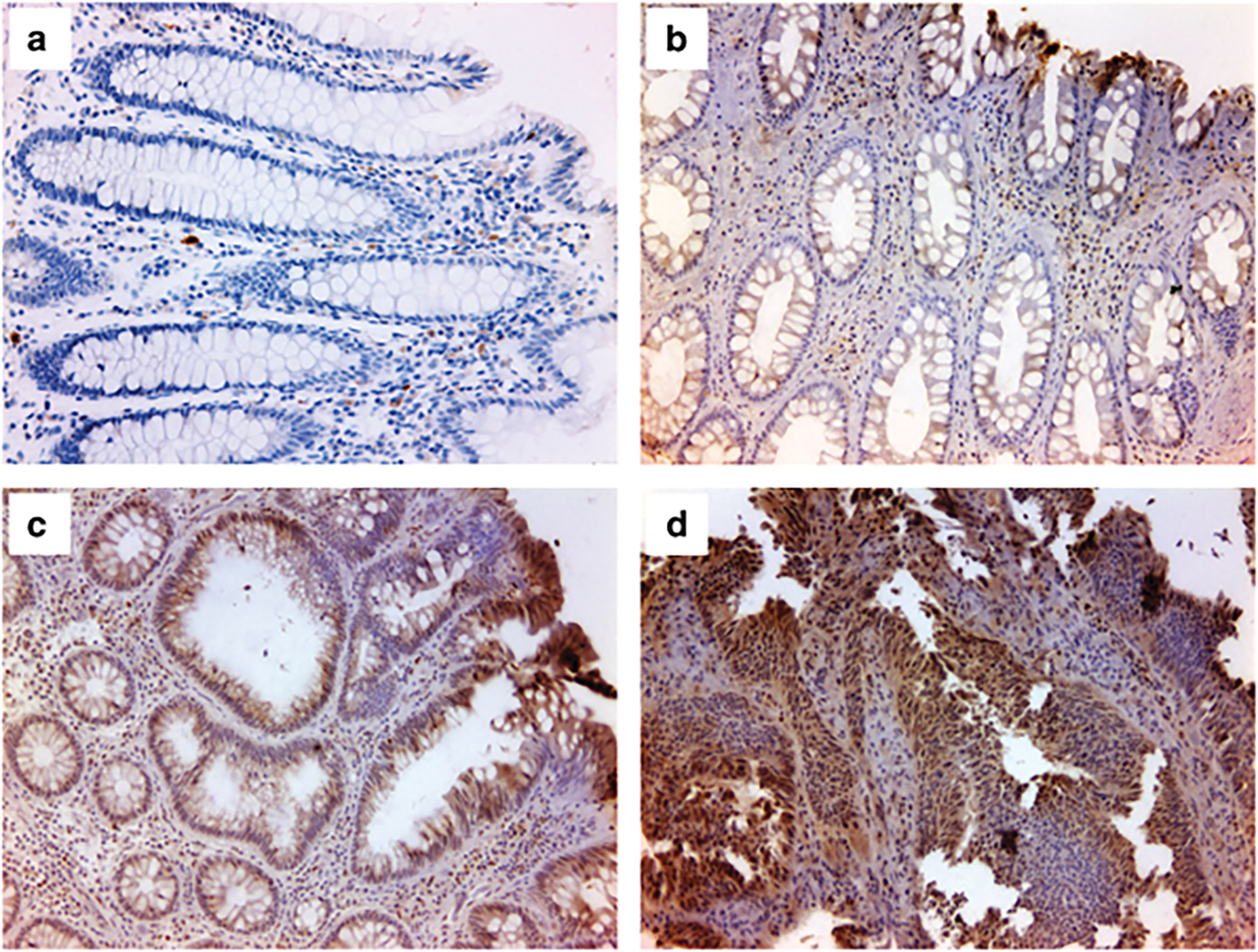
4-HNE-protein adducts in human colon adenomas and colorectal cancer. (a and b) Immunohistochemical staining for 4-HNE-protein adducts shows rare 4-HNE-positive cells in lamina propria of biopsies from normal human colon (a) or hyperplastic polyps (b). (c and d) 4-HNE-protein adducts are increased in the lamina propria and epithelia for biopsies from tubular adenomas (c) and invasive CRCs (d).
4-HNE activates AP-1 transcription factor
GSTA4 consists of 7 exons in both human and mouse genomes, but upstream gene structures vary between these species (Figure 5a). In the human genome, upstream GSTA4 is a 239 bp non-coding sequence followed by the RN7SK gene. In the mouse genome, however, there are >13,000 bp of non-coding sequence and a non-coding RNA gene (C920006O11Rik) between Gsta4 and Rn7sk (Figure 5a). To investigate the regulation of GSTA4 during CRC development, we performed an in silico analysis for potential transcription factors that might induce GSTA4. We found several potential binding sites for proto-oncogene activator protein 1 (AP-1) within RN7SK (Figure 5b, red boxes). For Gsta4, two sites with this motif were identified approximately −500 bp upstream of Gsta4 (Supplementary Figure 1). To test the binding capacity of these motifs, we performed electrophoretic mobility shift assay (EMSA) using 3 probes (Figure 5b). EMSA indicated that Probe 3, but not others, bound nuclear extracts from HCT116 and YAMC cells (Figure 5c), confirming an AP-1 binding site for GSTA4 induction.
Figure 5.
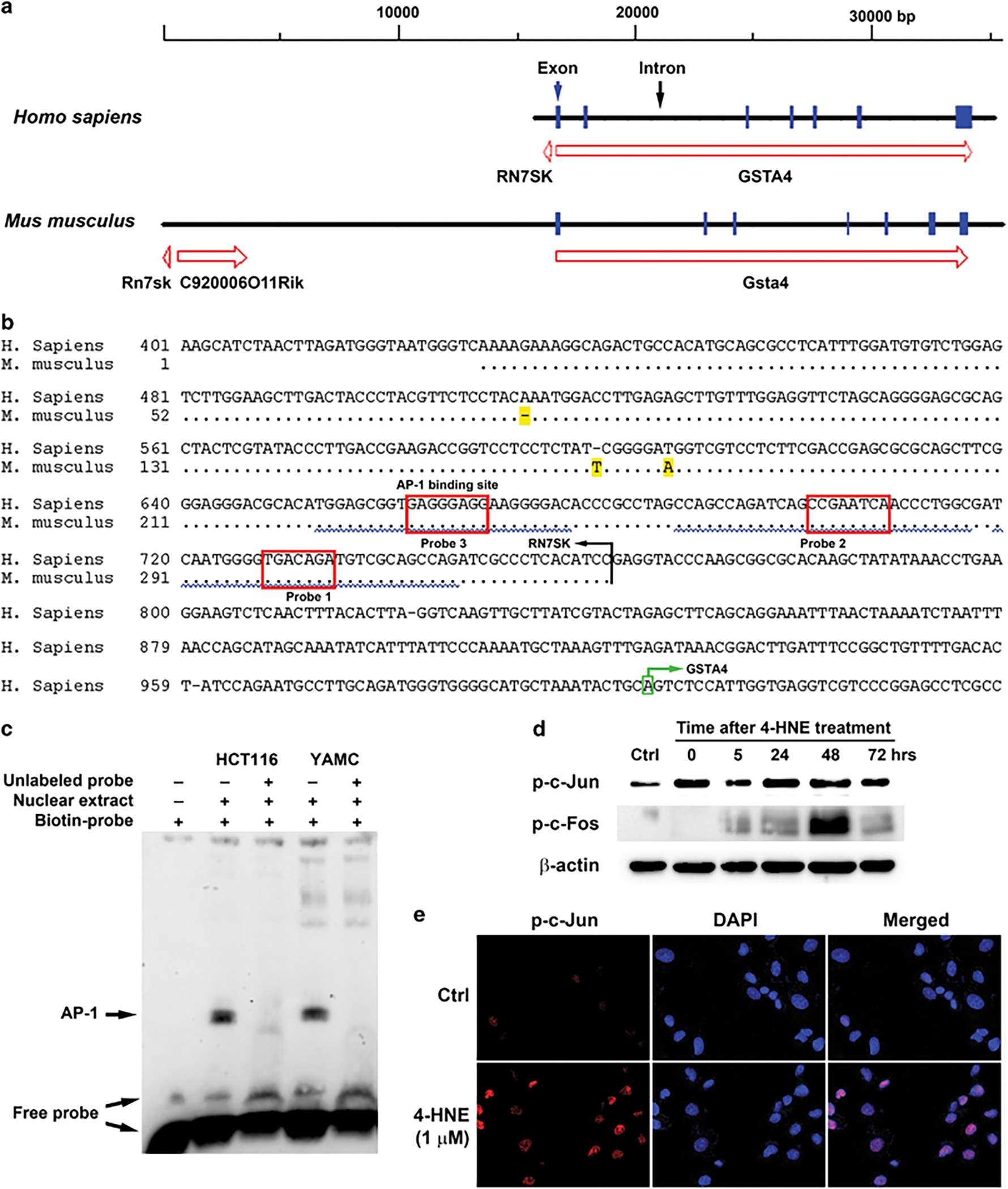
Proto-oncogene AP-1 regulates GSTA4 expression. (a) GSTA4 and upstream genes for human (upper) and mouse (lower). (b) Sequence alignments of putative promoter for human GSTA4 and murine Rn7sk. Non-matching bases are highlighted in yellow. Probes for EMSA assay are underlined by squiggled blue lines. Predicted AP-1 binding sites are shown in red boxes. The transcription start site for GSTA4 is highlighted in green box. (c) EMSA assay shows mobility shift of biotin-labeled probe after incubating with nuclear extracts from HCT116 and YAMC cells. This shift is eliminated by adding 100-fold excess unlabeled competitors. (d) Phospho-c-Jun (Ser73) increases in YAMC cells following 1 h treatment with 1 μM 4-HNE and persists throughout the experimental period. Phospho-c-Fos (Ser32) only transiently increases at 48 h post-treatment. (e) Immunofluorescent imaging shows nuclear translocation of phospho-c-Jun (Ser73) following 1 h treatment with 4-HNE (lower panels) compared to negative staining for untreated controls (upper panels).
AP-1 is a transcription factor that activates target genes through homo- or heterodimers of proteins belonging to JUN, FOS, ATF, and MAF families.33 Of these, c-Jun regulates proliferation, apoptosis, inflammation, and tumorigenesis.33 To test AP-1 induction by 4-HNE, we treated YAMC cells with 1 μM 4-HNE for 1 h and assayed for phosphorylated (active) c-Jun (Ser73). p-c-Jun (Ser73) immediately increased and persisted following 4-HNE treatment (Figure 5d). As c-Fos protein may interact with c-Jun to form AP-1,33 we found that phosphorylated c-Fos (Ser32) only transiently increased following 4-HNE treatment at 48 h (Figure 5d and Supplementary Figure 2), suggesting it may not be associated with p-c-Jun for induction of Gsta4 following 4-HNE treatment. Immunofluorescent imaging confirmed nuclear translocation of p-c-Jun immediately following 4-HNE treatment (Figure 5e).
GSTA4 is induced by c-Jun
To determine whether c-Jun contributes to GSTA4 gene expression, we examined the effect of SR11302, an AP-1 inhibitor,34 on GSTA4 expression in macrophages, primary epithelial cells, and CRC cells. Treatment of RAW264.7 macrophages with 10 μM SR11302 reduced Gsta4 gene product by 44% and 52% after 24 and 48 h, respectively, compared to untreated controls (Figure 6a). We next exposed HCT116 cells to 20 μM SR11302 and found no cellular toxicity. After 24 h, no significantly reduced GSTA4 gene product was seen for cells treated with SR11302 compared to untreated control. By 48 h post-treatment GSTA4 was reduced by 30% compared to untreated controls (Figure 6b). Similarly, treatment of YAMC cells (10 μM) showed no effect on Gsta4 at 24 h but decreased expression by 30% after 48 h compared to untreated controls (Figure 6c). The induction of Gsta4 in these cells by 4-HNE was inhibited at 24 and 48 h (Figure 6d). Finally, to eliminate potential off-target effects of SR11302, c-Jun was silenced in HCT116 cells using human c-Jun-specific siRNA and GSTA4 expression measured. A 30% reduction was noted in c-Jun and GSTA4 mRNA 24 h post-transfection compared to cells treated with non-targeting siRNA (Figures 6e). A 30% reduction in GSTA4 protein was observed after 72 h for cells treated with c-Jun siRNA compared to controls (Figure 6f). These results suggest GSTA4 activation through c-Jun.
Figure 6.
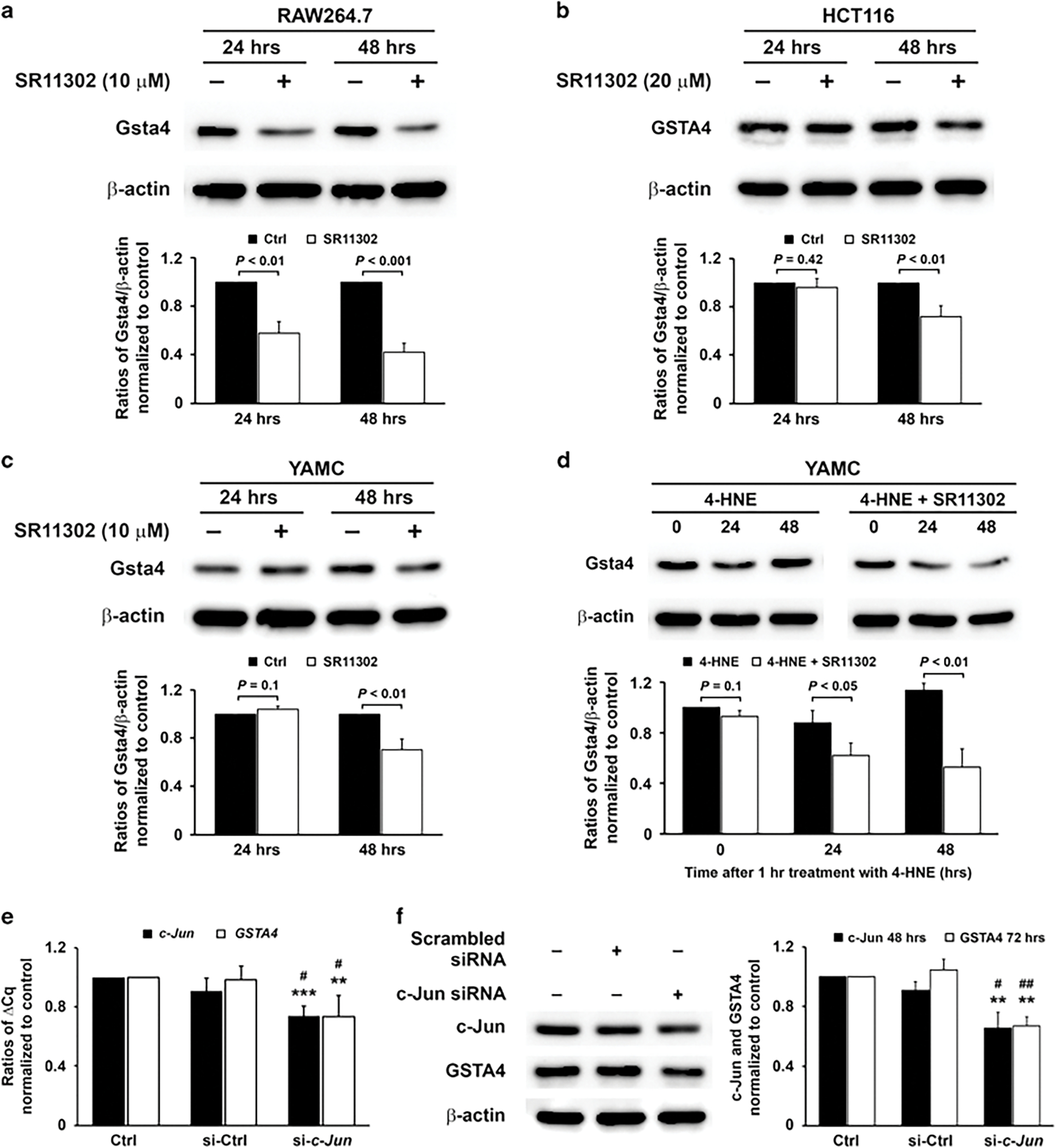
4-HNE induces Gsta4 via AP-1 activation. (a) Western blots (upper) show decreases in Gsta4 gene product in RAW264.7 murine macrophages at 24 and 48 h post-treatment using AP-1 inhibitor SR11302 (10 μM) after normalized to controls (lower). (b) In HCT116 colon cancer cells, GSTA4 gene product decreases after 48 h using 20 μM SR11302. (c) In YAMC murine colonic epithelial cells, Gsta4 gene product decreases after 48 h treatment with 10 μM SR11302. (d) Gsta4 gene product increases in YAMC cells following 1 h treatment with 4-HNE (upper left) after normalized to zero time (lower). This effect is diminished by 10 μM SR11302 (upper right and lower). (e) qRT-PCR shows decreased c-Jun and GSTA4 expression for HCT116 cells transfected with c-Jun-specific siRNA 24 h after transfection. (f) Western blots show decreased c-Jun and GSTA4 gene products at 48 and 72 h, respectively. All data represent mean ± s.d. for three independent experiments.
Nrf2 activates GSTA4
Nrf2 is a transcription factor that regulates gene response and detoxification to oxidative stress.35 Nrf2 interacts with c-Jun to form the AP-1 complex and binds to antioxidant-response elements in promoter or enhancer sequences leading to gene activation.33,35 To assess the role of Nrf2 in 4-HNE-induced Gsta4 activation, YAMC cells were treated with 4-HNE for 1 h and Nrf2 expression measured. Nrf2 gene product increased 48 h in 4-HNE-treated cells compared to untreated controls (Figure 7a). Immunofluorescent imaging showed nuclear translocation of Nrf2 following 4-HNE treatment (Figure 7b, lower) compared to untreated controls (Figure 7b, upper). In addition, Nrf2 expression increased in 6 CRC cell lines compared to fetal human colon cells (Figure 7c). Furthermore, we treated HCT116 cells with trigonelline, an Nrf2 inhibitor,36 and measured Nrf2 and GSTA4 gene products. We found Nrf2 and GSTA4 slightly increased at 10 μM, but was inhibited at 50 μM compared to untreated controls (Figure 7d). To eliminate off-target effects of trigonelline, Nrf2 was silenced using Nrf2-specific siRNA. Nrf2 mRNA was decreased 85% by siRNA-mediated knockdown (Figure 7e) that resulted in a 44% and 35% reduction of Nrf2 and GSTA4 gene products, respectively (Figure 7f). These results suggest that induction of GSTA4 is regulated, in part, through Nrf2.
Figure 7.
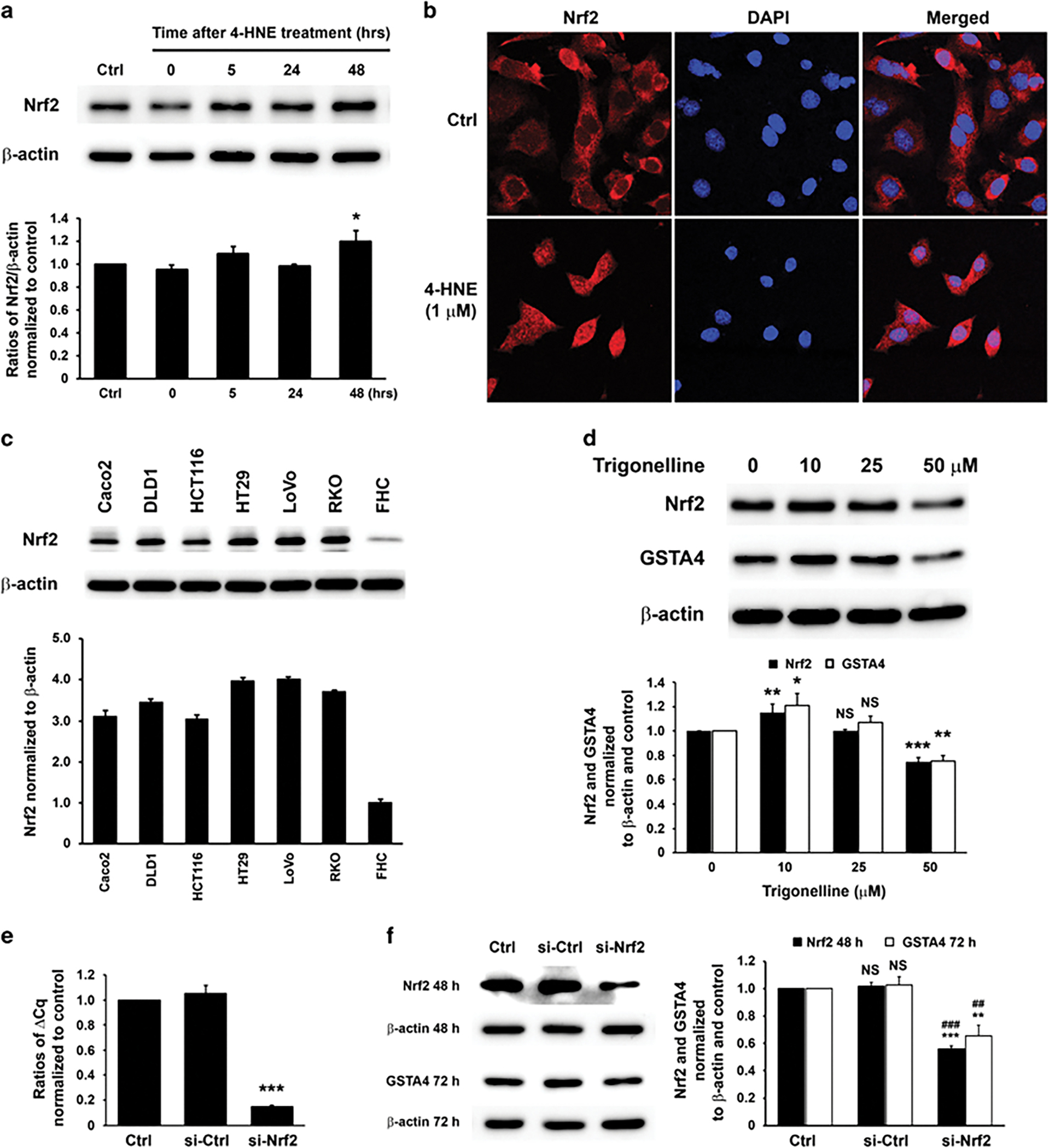
Nrf2 is involved in Gsta4 activation. (a) Western blots (upper) show increased Nrf2 in YAMC cells 48 h following 1 h treatment with 4-HNE after normalized to β-actin (lower). (b) Immunofluorescent imaging shows cytoplasmic localization of Nrf2 (red) in untreated YAMC cells (upper) and nuclear translocation at 5 h following 4-HNE-treatment (lower). (c) Nrf2 is strongly expressed in colon cancer cell lines compared to a fetal human colonic epithelial cell line (FHC; upper). Bar graph shows relative intensity of Nrf2 by normalizing to β-actin (lower). P < 0.01 for all CRC cell lines compared to FHC cells. (d) Western blots (upper) show slightly increased Nrf2 and GSTA4 in HCT116 cells treated with 10 μM of Nrf2 inhibitor trigonelline compared to untreated control after normalized to β-actin (lower). However, Nrf2 and GSTA4 gene products are suppressed by trigonelline at 50 μM. NS, not significant, *P < 0.05, **P < 0.01, and ***P < 0.001 compared to untreated controls. (e) qRT-PCR shows significantly decreased Nrf2 expression for HCT116 cells 24 h transfected with Nrf2-specific siRNA compared with controls (***P < 0.001). (f) Western blots (left) show decreased Nrf2 and GSTA4 gene products 48 and 72 h, respectively, following transfection with Nrf2-specific siRNA after normalized to β-actin (right). NS, not significant, **P < 0.01, and ***P < 0.001 compared to ctrl; ##P < 0.01 and ###P < 0.001 compared to cells transfected with non-targeting siRNA (si-ctrl). All data represent mean ± s.d. for three independent experiments.
Gsta4 induction by c-Jun and Nrf2
To determine whether Nrf2 interacts with c-Jun to induce Gsta4, YAMC cell lysates were co-immunoprecipitated following 4-HNE treatment. p-c-Jun was pulled down by anti-Nrf2 antibody and Nrf2 by anti-phospho-c-Jun antibody (Figure 8a and Supplementary Figure 3). In complexes recovered by anti-Nrf2 antibody, Nrf2 increased following 4-HNE treatment compared to untreated controls (Figure 8a, left). Similarly, anti-phospho-c-Jun antibody pulled down nuclear-localized Nrf2/p-c-Jun complexes and both Nrf2 and p-c-Jun increased for cells treated with 4-HNE compared to controls (Figure 8a, middle). Finally, immunofluorescent imaging confirmed co-localization of p-c-Jun and Nrf2 in nuclei of cells treated with 4-HNE (Figure 8b), supporting activation of the c-Jun/Nrf2 complex by 4-HNE and thereby increasing expression of Gsta4.
Figure 8.
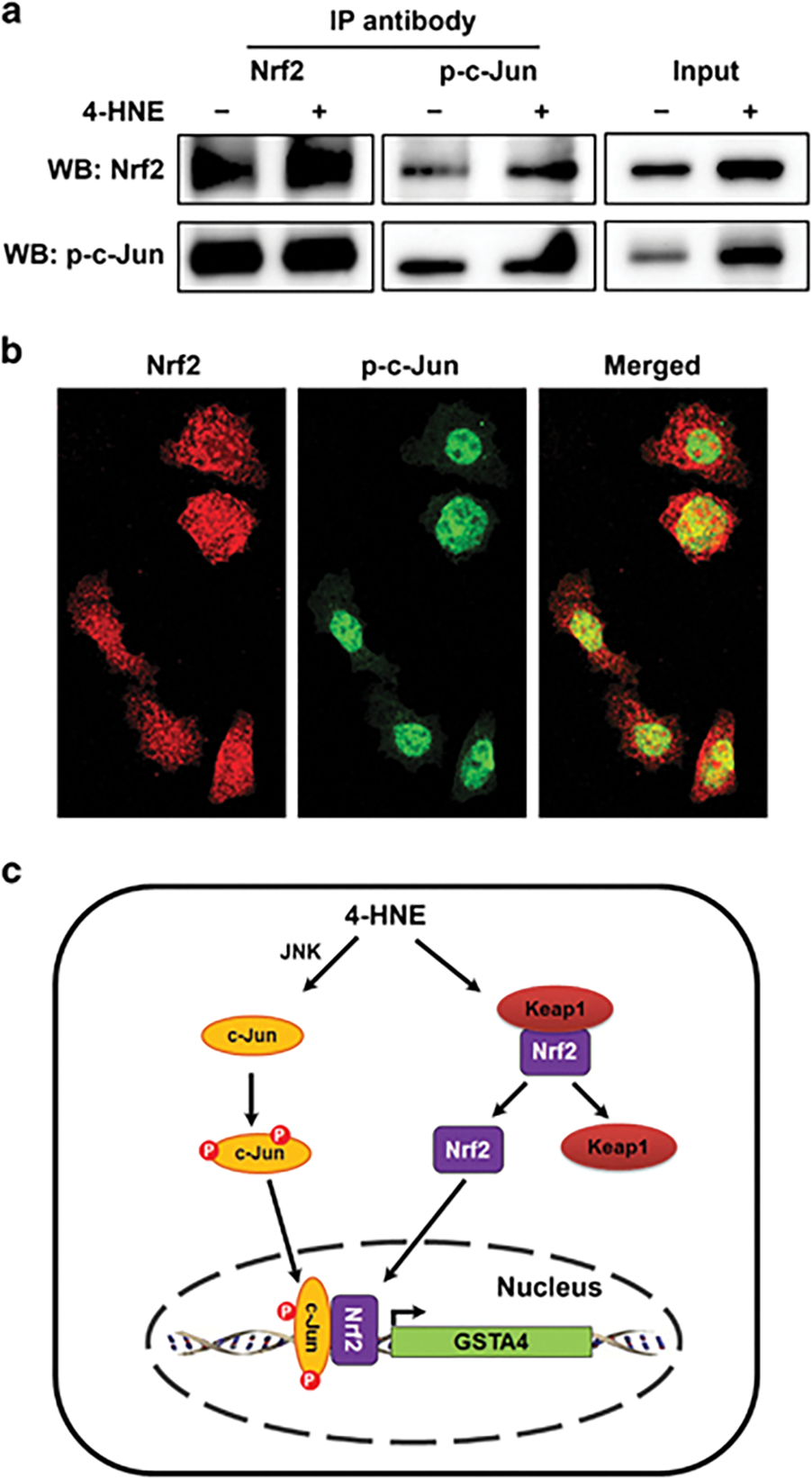
Interaction of c-Jun and Nrf2 regulates Gsta4 expression. (a) For 4-HNE-treated YAMC cells, and compared to untreated controls, Western blots show increased Nrf2 (upper left) in complexes co-immunoprecipitated using anti-Nrf2 antibody. An increase is not apparent for p-c-Jun (lower left). In contrast, when using an anti-phospho-c-Jun antibody for co-immunoprecipitation (middle panels), both Nrf2 (upper middle) and p-c-Jun (lower middle) were increased for 4-HNE-treated YAMC cells compared to untreated controls. Input controls show increased Nrf2 and p-c-Jun in 4-HNE-treated YAMC cells compared with untreated controls (right panels). (b) Immunofluorescent staining for Nrf2 (red) and p-c-Jun (green) confirms co-localization of p-c-Jun and Nrf2 to nuclei (yellow) following 4-HNE treatment. (c) A proposed model for GSTA4 activation during colorectal carcinogenesis: 4-HNE induces phosphorylation of c-Jun and releases Nrf2 from Kelch-like ECH-associated protein 1 (Keap1); both translocate into nuclei to form AP-1 complexes that bind antioxidant-response elements and activate GSTA4.
Discussion
In this study we report that GSTA4, a detoxifying enzyme for 4-HNE,13 is strongly expressed in macrophages and epithelial cells in microbiome-driven murine colitis as well as in biopsies from human colon adenomas and carcinomas. These findings add support for a bystander model of inflammation-associated CRC.26,27 Furthermore, we showed that oncogenic transcription factor AP-1 containing c-Jun/Nrf2 regulated 4-HNE-induced GSTA4 during colorectal carcinogenesis.
Previous studies have identified GSTs in multiple human cancers.12,37 Several transcription factors can regulate GST expression although those that control GSTA4 expression in colon epithelial cells are unclear.1,38 One recent study found that the GSTA family of genes in steroidogenic cells were controlled by steroidogenic factor 1 (SF-1), a key regulator of steroidogenesis.39 However, SF-1 did not appear to effectively bind the promoter of GSTA4.39 In another report KRAS-induced AP-1 activation was linked to GSTP1 expression in colorectal adenomas and carcinomas.12 We similarly found that AP-1 regulated the induction of GSTA4 by 4-HNE during colorectal carcinogenesis. These results are consistent with observations made using human bronchial epithelial and hepatoma cells wherein 4-HNE induces phase II detoxifying genes through c-Jun.38
c-Jun, a component of AP-1 transcription factors, regulates cellular proliferation, apoptosis, inflammation, and tumorigenesis through homodimerization or by forming heterodimers with other AP-1 components.33 Phosphorylation of c-Jun at positive regulatory sites within the N-terminal transactivation domain (Ser63 and Ser73) activates c-Jun.40 In colorectal carcinogenesis c-Jun is activated through β-catenin signaling and forms a ternary complex that contains c-Jun, TCF4, and β-catenin.41,42 β-catenin can activate GSTs in mouse liver.37 Although macrophage-induced bystander effects lead to the activation of β-catenin in colon epithelial cells,43 silencing β-catenin in HCT116 cells had no effect on GSTA4 expression (data not shown). This suggests that 4-HNE does not induce GSTA4 through β-catenin. In contrast, the induction of GSTA4 by 4-HNE through c-Jun positively links bystander effects to AP-1 activation and likely represents an adaptive response to 4-HNE overloading.26 Data from human colon adenomas and CRC confirm a causal link between 4-HNE and increased GSTA4 expression as was previously reported by us in a model of microbiome-driven murine colitis.26
Several studies have shown that 4-HNE can activate c-Jun N-terminal kinase (JNK) and mitogen-activated protein (MAP) kinases.19 JNK (but not MAP kinases) specifically phosphorylates c-Jun N-terminal amino acids, including Ser63 and Ser73, and is required for Gsta4 activation in mouse liver following oxidative stress.40,44 Based on these observations, we postulated that 4-HNE-induced c-Jun phosphorylation in colonic epithelial cells was driven by JNK (Figure 8c). Indeed c-Jun was immediately activated after 4-HNE treatment. In contrast, p-c-Fos (Ser32), an active form of c-Fos, was noted only at 48 h following 4-HNE treatment. This time gap suggested that p-c-Fos was unlikely to bind with p-c-Jun to induce Gsta4 following 4-HNE exposure. As an alternate mechanism, we considered binding of c-Jun to transcription factor Nrf2 in the regulation of phase II detoxifying genes such as GSAT4.33,35 Notably, Nrf2 promotes tumorigenesis when activated by oncogenes including KRAS, BRAF, and MYC.45,46 Using co-immunoprecipitation, we confirmed interactions between p-c-Jun and Nrf2 following 4-HNE exposure. Interestingly, using anti-Nrf2 antibody, p-c-Jun was unchanged for 4-HNE-treated cells compared to controls, suggesting binding of Nrf2 at basal levels and during induced states.46 This binding has not been observed following 4-HNE treatment in human bronchial epithelial and hepatoma cells and may reflect tissue specific responses.38 In colonic epithelial cells, however, 4-HNE appears to activate Nrf2. This likely results in diverse cellular effects that can modulate colorectal carcinogenesis. Furthermore, we noted that 50 μM of trigonelline, a dose similar to that used for reducing reactive oxygen species in HT29 and Caco2 CRC cells (≥ 30 μM)47 but >50-fold for other cell types,36,48 was required to effectively inhibit expression of Nrf2 and GSTA4 in HCT116 cells. This might be due to the strong expression of Nrf2 at basal levels in these CRC cells (Figure 7c). Although trigonelline-induced reduction of GSTA4 may occur through other Nrf2-independent pathways, silencing of Nrf2 by Nrf2-specific siRNA confirmed Nrf2 as an important regulator of GSTA4 expression.
The induction of GSTA4 occurs soon after macrophage polarization and is consistent with our previous findings showing increased production of 4-HNE by microbiome-polarized macrophages.26,28 Although AP-1 can regulate Gsta4 in macrophages, as supported by our data using AP-1 inhibitor, it remains unclear whether Nrf2 binds p-c-Jun to regulate Gsta4 expression. One study showed JUNB was an important regulator for macrophage activation.49 Further investigation, however, is needed to clarify the role of other AP-1 components such as ATF, JUNB, and MAF in GSTA4 activation.
Searching for human GSTA4 promoters using several promoter databases including Ensembl (http://uswest.ensembl.org/index.html), Eukaryotic Promoter Database (http://epd.vital-it.ch/), and Mammalian Promoter/Enhancer Database (http://promoter.cdb.riken.jp/) resulted in putative promoter sequences contain RN7SK. This gene is located 239 bp upstream of GSTA4 and encodes an abundant small nuclear RNA that negatively regulates gene expression by binding the positive transcription elongation factor b.50 Several potential AP-1 binding sites were identified within the murine Rn7sk gene, but because this gene was far upstream of Gsta4, an AP-1 binding site within Rn7sk may not effectively regulate Gsta4. However, 2 motifs were identified 586 and 506 bp upstream Gsta4 and may serve as binding sites for AP-1-mediated Gsta4 expression. Interestingly, in the murine genome C920006O11Rik was located between Rn7sk and Gsta4 and represented a large intergenic non-coding RNA (lincRNA) that may act over long distances to activate transcription at distal promoters.51 Whether these closely adjacent genes regulate GSTA4 (or Gsta4) is unclear. Investigation of these genes using promoter bashing and luciferase reporter assays would help elucidate their roles in GSTA4 expression but was beyond the scope of the current study.
Prior reports show that GSTP1 is highly expressed in colorectal carcinomas and ovarian tumors, associated with multidrug resistance, and confers a worsened clinical prognosis.12,52,53 Deletion of GSTP1 in HCT116 CRC cells results in decreased proliferation under growth-limiting conditions, increases apoptosis, and abrogates formation of xenografts in immunodeficient mice.10 Similarly, we speculate that GSTA4 induction by c-Jun/Nrf2 in CRC cells may contribute to cancer cell survival in otherwise oxidative microenvironments and help generate resistance to anti-cancer agents. Because many anti-cancer agents, including radiation therapy, produce tumor cell killing by generating cytotoxic reactive oxygen species, overexpression of antioxidant enzymes such as GSTA4 may contribute to cellular resistance to therapeutic agents. Thus, inhibition of GSTA4 could conceivably sensitize tumor cells to oxidative stress and/or apoptosis and enhance the efficacy of cancer treatments. The role of GSTA4 in protecting CRC cells against anti-cancer agents is an area of active investigation in our laboratory.
Previous studies found GSTA4 expression in a small number of normal human colon tissues (4 biopsies) using a home-made polyclonal antibody.24 In the current study we investigated GSTA4 expression in a relatively large number of human colon biopsies using tissue microarray. We found that all TANC biopsies, except for one sample, and all hyperplastic polyp biopsies, were negative for GSTA4 staining in epithelial cells. In contrast, GSTA4 stained strongly in epithelial cells for biopsies of adenomas and carcinomas. These observations are consistent with reports of increased total GST activity in the plasma from CRC patients compared to cancer negative controls.54 Further investigation of GSTA4 levels in blood for human subjects with a normal colonoscopy, or who have biopsy confirmed adenomas or CRC, will help clarify whether this particular GST is a potential biomarker for CRC screening.
In conclusion, GSTA4, a detoxifying enzyme for 4-HNE, was induced in macrophages during the early stages of inflammation and strongly expressed in neoplastic epithelial cells in early and advanced human CRC. Activation of the oncogenic transcription factor c-Jun/Nrf2 complex was associated with 4-HNE-induced GSTA4 expression. These results provide new insights into mechanisms by which GSTA4 is expressed during colorectal carcinogenesis. Finally, these results suggest that GSTA4 may be a potential biomarker for CRC screening and lead to novel strategies for CRC chemoprevention.
MATERIALS AND METHODS
Cell culture
YAMC murine primary colonic epithelial cells (Ludwig Institute for Cancer Research, New York, NY, USA), murine macrophage RAW264.7 cells, and human colon cancer cell lines (American Type Culture Collection, Manassas, VA, USA) were grown as previously described.55 SR11302 was purchased from Santa Cruz Biotechnology (Dallas, TX, USA) and trigonelline hydrochloride from Sigma (St. Louis, MO, USA).
Animal model and human tissues
Murine studies were approved by the University of Oklahoma Health Sciences Center and Oklahoma City VA Health Care System animal care and use committees. Specific pathogen-free Il10−/− mice (The Jackson Laboratory, Bar Harbor, ME, USA) were colonized with E. faecalis OG1RFSS or phosphate buffered saline (PBS) sham as previously described (n = 10 per group).26 Mice were necropsied after 2 weeks and 9 months of colonization and colons fixed in 10% formalin. Human colon tissue arrays including tumor-adjacent normal colon (TANC, 45 cores) and invasive colorectal carcinoma (35 cores) were purchased from US Biomax, Inc. (Rockville, MD, USA). Tissue arrays for hyperplastic colons (30 cores) and tubular adenomas (30 cores) were made by de-identified tissue blocks collected from the Oklahoma City VA Health Care System under a study approved by the Institutional Review Board at the University of Oklahoma Health Sciences Center.
Immunohistochemistry
Epitope retrieval and immunohistochemical (IHC) staining were performed as previously described.26 A polyclonal rabbit antibody against full-length human GSTA4 with cross-reactivity to murine Gsta4 (Abnova, Walnut, CA, USA; H00002941-D01P, 1:100) and anti-4-HNE serum (Alpha Diagnostic International Inc, San Antonio, TX, USA; HNE11-S, 1:1,000) were used as primary antibodies. Goat anti-rabbit IgG-HRP conjugate (Life Technologies, Grand Island, NY, USA) was used as secondary antibody. Chromogenic color development was performed using 3,3′-diaminobenzidine enhanced liquid substrate (Sigma) and nuclei counterstained by Mayer’s hematoxylin (Sigma). IHC staining for GSTA4 in human tissues was scored using a combination of stain intensity (0, negative; 1, weak; 2, moderate; 3, strong) and overall number of positive cells per 20X field (0, none; 1, < 10%; 2, 10 – 50%; 3, >50% positive cells).
Immunofluorescence
For YAMC staining, cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with pre-cold methanol for 10 min at −20 °C. Slides were blocked using Image-iT® FX Signal Enhancer (Life Technologies) for 30 min and then 5% normal goat serum for 1 h at room temperature. Mouse anti-GSTA4 (Abnova; H00002941-B01P, 1:100) and rabbit anti-Nrf2 (Santa Cruz Biotechnology; sc-722, 1:400) polyclonal antibodies, rabbit anti-F4/80 (Thermo Scientific, Waltham, MA, USA; MA5-16363, 1:100) and mouse anti-phospho-c-Jun (Ser73) (Santa Cruz Biotechnology; sc-822, 1:400) monoclonal antibodies were incubated at 4 °C overnight. Goat anti-mouse IgG-Alexa Fluor® 488 conjugate and goat anti-rabbit IgG-Alexa Fluor® 647 conjugate (Life technologies; A11029 and A21244, 1:1,000) were used as secondary antibodies. DNA was counterstained with 4’,6-diamidino-2-phenylindole (DAPI). To stain F4/80 and GSTA4 in formalin-fixed, paraffin-embedded tissues, epitope retrieval was conducted as described in the IHC staining followed by blocking and staining protocols as for YAMC staining. Images were analyzed by laser scanning confocal microscopy (Leica Microsystems, Buffalo Grove, IL, USA).
Gsta4
Gsta4 in mouse serum was quantified by enzyme-linked immunosorbent assay (ELISA) using a commercial kit (Cloud-Clone Corp, Houston, TX, USA). Serum was diluted in 1:10 in PBS and 100 μl used per manufacturer’s instructions. Absorbance was measured at 450nm on a microplate reader (BioTek, Winooski, VT, USA).
Gst activity
Gst activity was analyzed using Glutathione S-transferase Assay kit (Sigma) per manufacturer’s instructions with minor modification. YAMC cells treated with 4-HNE or sham were harvested in PBS and sonicated using a Sonic Dismembrator (Fisher Scientific, Pittsburgh, PA, USA). Cell lysates were centrifuged at 10,000 × g for 5 min and supernatants used for analysis. The reaction solution consisted of 490 μl Dulbecco’s PBS, 5 μl of 200 mM L-glutathione reduced, 5 μl of 100 mM 1-chloro-2,4-dinitrobenzene as substrate, and 1 μl of cell lysate. Absorbance was measured at 340 nm at 37 °C for 5 min using a Cary 300 Bio spectrometer (Varian, Santa Clara, CA, USA). Gst activity was normalized to milligram total protein.
EMSA
Nuclear protein was extracted from HCT116 and YAMC cells using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Life Technologies). Single strand probes were synthesized by Integrated DNA Technologies (Coralville, IA, USA) and labeled using a Pierce™ Biotin 3’ End DNA Labeling Kit (Life Technologies). After labeling, single strand probes were annealed at 60 °C for 30 min followed by 30 min at room temperature. EMSA was performed using a LightShift™ Chemiluminescent EMSA Kit (Life Technologies) per manufacturer’s instructions.
c-Jun and Nrf2 silencing
Silencing of c-Jun and Nrf2 in HCT116 cells was carried out using gene specific small interfering RNA (siRNA) for c-Jun (Cell Signaling Technology, Danvers, MA, USA) and Nrf2 (Santa Cruz Biotechnology) as per instructions. HCT116 cells were seeded at 5 × 105 in 6-well plates and incubated at 37 °C overnight. Transfection was performed using DharmaFECT 4 transfection reagent (GE Healthcare, Pittsburgh, PA, USA) as previously described.28 Cells were lysed 24–72 h after transfection for RNA and protein extraction.
Real-time quantitative reverse transcriptase PCR (qRT-PCR)
Total RNA was isolated from YAMC and HCT116 cells using the NucleoSpin RNA II kit (Clontech Laboratories, Mountain View, CA, USA). PrimeTime® qPCR primers for murine Gsta4, human GSTA4, JUN, Nrf2, and β-actin were purchased from Integrated DNA Technologies. qRT-PCR was performed using iTaq™ universal SYBR® Green one-step kit (Bio-Rad, Hercules, CA, USA) per manufacturer’s instruction. cDNA synthesis and target gene amplification were carried out on the CFX96 Real-Time System (Bio-Rad) at 50 °C for 10 min followed by 40 cycles of at 95 °C for 10 sec and 60 °C for 30 sec.
Western blot
Whole-cell extracts were prepared using lysis buffer (Cell Signaling Technology). Cell lysates (20 μg) were separated by SDS-PAGE, and blotting performed as previously described.26 Rabbit anti-GSTA4 (Abnova; H00002941-D01P, 1:2,000), anti-Nrf2 (Cell Signaling Technology; #12721, 1:1,000), anti-phospho-c-Jun (Ser73) (Cell Signaling Technology; #3270, 1:1,000), and anti-phospho-c-Fos (Ser32) (Cell Signaling Technology; #5348, 1:1,000) were used as primary antibodies. Murine anti-β-actin loading control was purchased from Santa Cruz Biotechnology (sc-81178, 1:2,000). Goat anti-rabbit IgG-horseradish peroxidase (HRP) conjugate (Life Technologies; #65–6120, 1:10,000) and horse anti-mouse IgG-HRP conjugate (Cell Signaling Technology; #7076, 1:5,000) were used as secondary antibodies. Signals were generated by Clarity™ Western ECL Substrate (Bio-Rad). Images were captured by ChemiDoc XRS+ system and analyzed using Image Lab software (Bio-Rad).
Co-immunoprecipitation
Co-immunoprecipitation was carried out using Catch & Release v2.0 Reversible Immunoprecipitation System (EMD Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. In brief, 250 μg of cell lysate was incubated with 2 μg of antibody to Nrf2 or p-c-Jun (Santa Cruz Biotechnology) in a resin column at 4 °C overnight. Nrf2/p-c-Jun complexes were eluted with denaturing elution buffer for Western blotting using rabbit anti-Nrf2 and anti-phospho-c-Jun (Cell Signaling Technology) antibodies as previously described.
Statistical analysis
Data are expressed as means with standard deviations. Nonparametric t tests (Mann Whitney test) were used for comparisons between groups of human colon tissues. Student t tests were used for all other comparisons. P values < 0.05 were considered statistically significant.
Supplementary Material
Supplementary Figure 2. Immunofluorescent imaging shows nuclear localization of phospho-c-Fos (Ser32) (red) 48 h after 4-HNE treatment (lower panels) compared to negative staining for untreated controls (upper panels). Nuclei are counterstained using 4’,6-diamidino-2-phenylindole (DAPI) (blue).
{kind=link}
Supplementary Figure 1. The proximal promoter sequence upstream of mouse Gsta4. Sequence alignment shows two potential binding sites for AP-1 at −586 and −506 bp upstream of transcription start site for Gsta4.
{kind=link}
Supplementary Figure 3. Western blots for supernatants of co-immunoprecipitation. Trace Nrf2 and p-c-Jun are detected in the supernatants after co-immunoprecipitation.
{kind=link}
ACKNOWLEDGEMENTS
We thank Jim Henthorn in the Flow Cytometry and Imaging Laboratory, the Histology and Immunohistochemistry Core in the Peggy and Charles Stephenson Cancer Center (NIH GM103639) at the University of Oklahoma Health Sciences Center for technical assistance. Supported in part by the Oklahoma Center for the Advancement of Science and Technology grant HR10-032 (XW) and Francis Duffy Endowment (MH).
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol 2005; 45: 51–88. [DOI] [PubMed] [Google Scholar]
- 2.Miller DP, Neuberg D, de Vivo I, Wain JC, Lynch TJ, Su L et al. Smoking and the risk of lung cancer: susceptibility with GSTP1 polymorphisms. Epidemiology 2003; 14: 545–551. [DOI] [PubMed] [Google Scholar]
- 3.Johns LE, Houlston RS. Glutathione S-transferase mu1 (GSTM1) status and bladder cancer risk: a meta-analysis. Mutagenesis 2000; 15: 399–404. [DOI] [PubMed] [Google Scholar]
- 4.Mitrunen K, Jourenkova N, Kataja V, Eskelinen M, Kosma VM, Benhamou S et al. Glutathione S-transferase M1, M3, P1, and T1 genetic polymorphisms and susceptibility to breast cancer. Cancer Epidemiol Biomarkers Prev 2001; 10: 229–236. [PubMed] [Google Scholar]
- 5.Sweeney C, Coles BF, Nowell S, Lang NP, Kadlubar FF. Novel markers of susceptibility to carcinogens in diet: associations with colorectal cancer. Toxicology 2002; 181–182: 83–87. [DOI] [PubMed] [Google Scholar]
- 6.Zhong S, Wyllie AH, Barnes D, Wolf CR, Spurr NK. Relationship between the GSTM1 genetic polymorphism and susceptibility to bladder, breast and colon cancer. Carcinogenesis 1993; 14: 1821–1824. [DOI] [PubMed] [Google Scholar]
- 7.Ritchie KJ, Walsh S, Sansom OJ, Henderson CJ, Wolf CR. Markedly enhanced colon tumorigenesis in ApcMin mice lacking glutathione S-transferase Pi. Proc Natl Acad Sci USA 2009; 106: 20859–20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Searchfield L, Price SA, Betton G, Jasani B, Riccardi D, Griffiths DF. Glutathione S-transferases as molecular markers of tumour progression and prognosis in renal cell carcinoma. Histopathology 2011; 58: 180–190. [DOI] [PubMed] [Google Scholar]
- 9.Shimizu T, Fan Y, Yamana D, Miura T, Nanashima N, Yamada T et al. Glutathione S-transferase A4 is a positive marker for rat hepatic foci induced by clofibrate and genotoxic carcinogens. Cancer Sci 2010; 101: 1093–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dang DT, Chen F, Kohli M, Rago C, Cummins JM, Dang LH. Glutathione S-transferase pi1 promotes tumorigenicity in HCT116 human colon cancer cells. Cancer Res 2005; 65: 9485–9494. [DOI] [PubMed] [Google Scholar]
- 11.Kalinina EV, Berozov TT, Shtil AA, Chernov NN, Glasunova VA, Novichkova MD et al. Expression of genes of glutathione transferase isoforms GSTP1–1, GSTA4–4, and GSTK1–1 in tumor cells during the formation of drug resistance to cisplatin. Bulletin of experimental biology and medicine 2012; 154: 64–67. [DOI] [PubMed] [Google Scholar]
- 12.Miyanishi K, Takayama T, Ohi M, Hayashi T, Nobuoka A, Nakajima T et al. Glutathione S-transferase-pi overexpression is closely associated with K-ras mutation during human colon carcinogenesis. Gastroenterology 2001; 121: 865–874. [DOI] [PubMed] [Google Scholar]
- 13.Coles BF, Kadlubar FF. Human alpha class glutathione S-transferases: genetic polymorphism, expression, and susceptibility to disease. Methods Enzymol 2005; 401: 9–42. [DOI] [PubMed] [Google Scholar]
- 14.Schaur RJ. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol Aspects Med 2003; 24: 149–159. [DOI] [PubMed] [Google Scholar]
- 15.Uchida K 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res 2003; 42: 318–343. [DOI] [PubMed] [Google Scholar]
- 16.Feng Z, Hu W, Tang MS. Trans-4-hydroxy-2-nonenal inhibits nucleotide excision repair in human cells: a possible mechanism for lipid peroxidation-induced carcinogenesis. Proc Natl Acad Sci USA 2004; 101: 8598–8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrera G, Pizzimenti S, Dianzani MU. 4-hydroxynonenal and regulation of cell cycle: effects on the pRb/E2F pathway. Free Radic Biol Med 2004; 37: 597–606. [DOI] [PubMed] [Google Scholar]
- 18.Chaudhary P, Sharma R, Sahu M, Vishwanatha JK, Awasthi S, Awasthi YC. 4-Hydroxynonenal induces G2/M phase cell cycle arrest by activation of the ataxia telangiectasia mutated and Rad3-related protein (ATR)/checkpoint kinase 1 (Chk1) signaling pathway. J Biol Chem 2013; 288: 20532–20546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zarrouki B, Soares AF, Guichardant M, Lagarde M, Geloen A. The lipid peroxidation end-product 4-HNE induces COX-2 expression through p38MAPK activation in 3T3-L1 adipose cell. FEBS Lett 2007; 581: 2394–2400. [DOI] [PubMed] [Google Scholar]
- 20.Sharma A, Sharma R, Chaudhary P, Vatsyayan R, Pearce V, Jeyabal PV et al. 4-Hydroxynonenal induces p53-mediated apoptosis in retinal pigment epithelial cells. Arch Biochem Biophys 2008; 480: 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.West JD, Marnett LJ. Alterations in gene expression induced by the lipid peroxidation product, 4-hydroxy-2-nonenal. Chem Res Toxicol 2005; 18: 1642–1653. [DOI] [PubMed] [Google Scholar]
- 22.Kumagai T, Matsukawa N, Kaneko Y, Kusumi Y, Mitsumata M, Uchida K. A lipid peroxidation-derived inflammatory mediator: identification of 4-hydroxy-2-nonenal as a potential inducer of cyclooxygenase-2 in macrophages. J Biol Chem 2004; 279: 48389–48396. [DOI] [PubMed] [Google Scholar]
- 23.Cheng JZ, Yang Y, Singh SP, Singhal SS, Awasthi S, Pan SS et al. Two distinct 4-hydroxynonenal metabolizing glutathione S-transferase isozymes are differentially expressed in human tissues. Biochem Biophys Res Commun 2001; 282: 1268–1274. [DOI] [PubMed] [Google Scholar]
- 24.Desmots F, Rissel M, Loyer P, Turlin B, Guillouzo A. Immunohistological analysis of glutathione transferase A4 distribution in several human tissues using a specific polyclonal antibody. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society 2001; 49: 1573–1580. [DOI] [PubMed] [Google Scholar]
- 25.Edalat M, Mannervik B, Axelsson LG. Selective expression of detoxifying glutathione transferases in mouse colon: effect of experimental colitis and the presence of bacteria. Histochem Cell Biol 2004; 122: 151–159. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Yang Y, Moore DR, Nimmo SL, Lightfoot SA, Huycke MM. 4-Hydroxy-2-nonenal mediates genotoxicity and bystander effects caused by Enterococcus faecalis-infected macrophages. Gastroenterology 2012; 142: 543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Yang Y, Huycke MM. Commensal bacteria drive endogenous transformation and tumour stem cell marker expression through a bystander effect. Gut 2015; 64: 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Allen TD, Yang Y, Moore DR, Huycke MM. Cyclooxygenase-2 generates the endogenous mutagen trans-4-hydroxy-2-nonenal in Enterococcus faecalis-infected macrophages. Cancer Prev Res (Phila) 2013; 6: 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engle MR, Singh SP, Czernik PJ, Gaddy D, Montague DC, Ceci JD et al. Physiological role of mGSTA4–4, a glutathione S-transferase metabolizing 4-hydroxynonenal: generation and analysis of mGsta4 null mouse. Toxicol Appl Pharmacol 2004; 194: 296–308. [DOI] [PubMed] [Google Scholar]
- 30.Apidianakis Y, Que YA, Xu W, Tegos GP, Zimniak P, Hamblin MR et al. Down-regulation of glutatione S-transferase alpha 4 (hGSTA4) in the muscle of thermally injured patients is indicative of susceptibility to bacterial infection. FASEB J 2012; 26: 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abel EL, Angel JM, Riggs PK, Langfield L, Lo HH, Person MD et al. Evidence that Gsta4 modifies susceptibility to skin tumor development in mice and humans. J Natl Cancer Inst 2010; 102: 1663–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shireman LM, Kripps KA, Balogh LM, Conner KP, Whittington D, Atkins WM. Glutathione transferase A4–4 resists adduction by 4-hydroxynonenal. Arch Biochem Biophys 2010; 504: 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 2003; 3: 859–868. [DOI] [PubMed] [Google Scholar]
- 34.Huang C, Ma WY, Dawson MI, Rincon M, Flavell RA, Dong Z. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc Natl Acad Sci USA 1997; 94: 5826–5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 2009; 284: 13291–13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arlt A, Sebens S, Krebs S, Geismann C, Grossmann M, Kruse ML et al. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 2013; 32: 4825–4835. [DOI] [PubMed] [Google Scholar]
- 37.Giera S, Braeuning A, Kohle C, Bursch W, Metzger U, Buchmann A et al. Wnt/beta-catenin signaling activates and determines hepatic zonal expression of glutathione S-transferases in mouse liver. Toxicol Sci 2010; 115: 22–33. [DOI] [PubMed] [Google Scholar]
- 38.Levy S, Jaiswal AK, Forman HJ. The role of c-Jun phosphorylation in EpRE activation of phase II genes. Free Radic Biol Med 2009; 47: 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsumura T, Imamichi Y, Mizutani T, Ju Y, Yazawa T, Kawabe S et al. Human glutathione S-transferase A (GSTA) family genes are regulated by steroidogenic factor 1 (SF-1) and are involved in steroidogenesis. FASEB J 2013; 27: 3198–3208. [DOI] [PubMed] [Google Scholar]
- 40.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev 1993; 7: 2135–2148. [DOI] [PubMed] [Google Scholar]
- 41.Hasselblatt P, Gresh L, Kudo H, Guinea-Viniegra J, Wagner EF. The role of the transcription factor AP-1 in colitis-associated and beta-catenin-dependent intestinal tumorigenesis in mice. Oncogene 2008; 27: 6102–6109. [DOI] [PubMed] [Google Scholar]
- 42.Nateri AS, Spencer-Dene B, Behrens A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 2005; 437: 281–285. [DOI] [PubMed] [Google Scholar]
- 43.Yang Y, Wang X, Huycke T, Moore DR, Lightfoot SA, Huycke MM. Colon macrophages polarized by commensal bacteria cause colitis and cancer through the bystander effect. Transl Oncol 2013; 6: 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desmots F, Loyer P, Rissel M, Guillouzo A, Morel F. Activation of C-Jun N-terminal kinase is required for glutathione transferase A4 induction during oxidative stress, not during cell proliferation, in mouse hepatocytes. FEBS Lett 2005; 579: 5691–5696. [DOI] [PubMed] [Google Scholar]
- 45.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011; 475: 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shelton P, Jaiswal AK. The transcription factor NF-E2-related factor 2 (Nrf2): a protooncogene? FASEB J 2013; 27: 414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bakuradze T, Lang R, Hofmann T, Stiebitz H, Bytof G, Lantz I et al. Antioxidant effectiveness of coffee extracts and selected constituents in cell-free systems and human colon cell lines. Mol Nutr Food Res 2010; 54: 1734–1743. [DOI] [PubMed] [Google Scholar]
- 48.Sirota R, Gibson D, Kohen R. The role of the catecholic and the electrophilic moieties of caffeic acid in Nrf2/Keap1 pathway activation in ovarian carcinoma cell lines. Redox Biol 2015; 4: 48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fontana MF, Baccarella A, Pancholi N, Pufall MA, Herbert DR, Kim CC. JUNB is a key transcriptional modulator of macrophage activation. J Immunol 2015; 194: 177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Diribarne G, Bensaude O. 7SK RNA, a non-coding RNA regulating P-TEFb, a general transcription factor. RNA Biol 2009; 6: 122–128. [DOI] [PubMed] [Google Scholar]
- 51.Orom UA, Shiekhattar R. Long noncoding RNAs usher in a new era in the biology of enhancers. Cell 2013; 154: 1190–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nobuoka A, Takayama T, Miyanishi K, Sato T, Takanashi K, Hayashi T et al. Glutathione-S-transferase P1–1 protects aberrant crypt foci from apoptosis induced by deoxycholic acid. Gastroenterology 2004; 127: 428–443. [DOI] [PubMed] [Google Scholar]
- 53.Sawers L, Ferguson MJ, Ihrig BR, Young HC, Chakravarty P, Wolf CR et al. Glutathione S-transferase P1 (GSTP1) directly influences platinum drug chemosensitivity in ovarian tumour cell lines. Br J Cancer 2014; 111: 1150–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nomani H, Ghobadloo SM, Yaghmaei B, Rezvanie NA, Yaghmaei K. Glutathione S-transferases activity in patients with colorectal cancer. Clinical biochemistry 2005; 38: 621–624. [DOI] [PubMed] [Google Scholar]
- 55.Wang X, Allen TD, May RJ, Lightfoot S, Houchen CW, Huycke MM. Enterococcus faecalis induces aneuploidy and tetraploidy in colonic epithelial cells through a bystander effect. Cancer Res 2008; 68: 9909–9917. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 2. Immunofluorescent imaging shows nuclear localization of phospho-c-Fos (Ser32) (red) 48 h after 4-HNE treatment (lower panels) compared to negative staining for untreated controls (upper panels). Nuclei are counterstained using 4’,6-diamidino-2-phenylindole (DAPI) (blue).
Supplementary Figure 1. The proximal promoter sequence upstream of mouse Gsta4. Sequence alignment shows two potential binding sites for AP-1 at −586 and −506 bp upstream of transcription start site for Gsta4.
Supplementary Figure 3. Western blots for supernatants of co-immunoprecipitation. Trace Nrf2 and p-c-Jun are detected in the supernatants after co-immunoprecipitation.