

Summary
Hematological cancers such as leukemia, lymphoma, and multiple myeloma have traditionally been treated with chemo and radiotherapy approaches. Introduction of immunotherapies for treatment of these diseases has led to patient remissions that would not have been possible with traditional approaches. In this critical review we identify main disease characteristics, symptoms, and current treatment options. Five common immunotherapies, namely checkpoint inhibitors, vaccines, cell-based therapies, antibodies, and oncolytic viruses, are described, and their applications in hematological cancers are critically discussed.
Subject areas: Health sciences, Immunology, Oncology, Biological sciences, Cancer
Graphical abstract
Health sciences; Immunology; Oncology; Biological sciences; Cancer
Introduction
Immunotherapies harness the immune system to fight diseases. Thus, they have potential to revolutionize cancer treatment and have been applied to improve current outlooks in hematological cancers specifically. Immunotherapy first emerged as a new treatment for cancer in the 1890s when William Coley began injecting cancer patients with bacteria to trigger immune responses against cancer. Throughout the 20th century, knowledge of the immune system was gained, including discoveries of cytokines and immune cells, and immunotherapy re-emerged in the 1980s when the hepatitis B vaccine based on single cell surface antigens was developed (Allison, 2014). Since that time, the field has seen an evolution in its application against cancer and in 2013 was Science’s breakthrough of the year (Couzin-Frankel, 2013).
There are cell-mediated mechanisms in place that are used by the immune system to respond to pathogens and damage, and to differentiate between self and non-self antigens. For example, immune cells of the innate immune system have membrane-associated or cytosolic pattern recognition receptors that can recognize pathogen-associated molecular patterns (PAMPs) expressed by microbes, and subsequently activate an acute inflammatory response (Kogut et al., 2020). In addition, major molecules belonging to the histocompatibility complex (MHC) class I and II display peptide fragments of intracellular proteins on cell surfaces for presentation to T-cell receptors, which can then recognize if they are non-self-antigens and respond (Cooper and Alder, 2006).
However, cancerous cells can mutate to evade recognition by the immune system through numerous mechanisms. Malignant cells can express fewer antigens on their surface, can lose the MHC Class I expression, and can express immune checkpoint molecules (Oiseth and Aziz, 2017). Not only cancerous cells lead to the immune evasion, but the tumor microenvironment that includes cellular and extracellular materials surrounding the cancer cells expresses properties that lead to improved growth of the cancer cells and makes it difficult for the immune system and drugs to kill the cancer cells (Thakkar et al., 2020). This microenvironment is characterized by metabolic reprogramming such as overexpression of growth factors and enhanced glycolysis, hypoxia, and acidic conditions (Pérez-Herrero and Fernández-Medarde, 2021). Therefore, immunotherapy seeks to restore the function of the immune system to attack the cancer cells by altering the tumor microenvironment or blocking the cancer’s immune suppression tactics.
In recent years, immunotherapy has gained importance in the treatment of leukemia, lymphoma, and multiple myeloma. In fact, it is suggested that the progression from monoclonal gammopathy of undetermined significance and latent multiple myeloma to multiple myeloma is because of an immune imbalance (Minnie and Hill, 2020). Immune system escape plays an important role in the progression of multiple myeloma, which may be because of several reasons: T cell exhaustion or senescence, variations in cytokine secretion, tolerance of dysfunctional antigen-presenting cells, and accumulation of tumor-associated suppressor macrophages and myeloid-derived suppressor cells (Minnie and Hill, 2020).
Cancer immunotherapies have been included in current hematological cancer treatments and are integrated in standard regimens. This is the case for immunomodulatory drugs and monoclonal antibodies for which new combinations or new molecules are being found. Moreover, as discussed by Minnie and Hill (2020) current research on hematological cancer therapies is focused on some immunotherapy-based strategies and many clinical trials are being reported in this regard (Minnie and Hill, 2020). Undoubtedly, the future for treatment of hematological cancers that currently evade remission will require a combination of immunotherapy and chemotherapy approaches.
Here we address the main hematological cancers along with their standard current treatments, and to address the application of specific types of immunotherapies, including checkpoint inhibitors, therapeutic vaccines, antibodies, cell-based therapies, and oncolytic viruses (OVs), in improving the treatment of hematological cancers.
Hematological cancers, a general description
Leukemia
Leukemias, the cancers of the blood and bone marrow, can be divided into four main types, including acute lymphocytic leukemia (ALL), chronic lymphocytic leukemia (CLL), acute myeloid leukemia (AML), and chronic myeloid leukemia (CML). They can be differentiated from each other based on morphology during maturation and linage commitment, and they can be subdivided based on genetic aberrations (Kampen, 2012). Chronic leukemia develops slowly in more mature cells whereas acute leukemia develops quickly in immature cells and can be classified by cytomorphology and immunophenotyping (Szczepański et al., 2003). Of the leukemia types, CLL and AML are the most diagnosed forms of leukemia.
Lymphocytic leukemias
Disease overview and diagnosis
Acute lymphocytic or acute lymphoblastic leukemias (ALL) are estimated to affect 6,660 people for 2022 in the United States, making it one of the rare forms of leukemia among adults. However, this form of leukemia is quite prevalent among children diagnosed with leukemia, being the most common form of pediatric cancer and accounting for most pediatric cancer deaths (Teachey and Pui, 2019). Among the ALLs, there are a few different subtypes, based on the World Health Organization (WHO) system: B-cell ALL and T cell ALL.
B-cell ALL occurs much more frequently than T cell ALL in children 9 years old and younger (Teachey and Pui, 2019). T cell ALL is known to be more predominant in adult cases of ALL than pediatric cases, accounting for roughly 25% of adult ALL cases (Van Vlierberghe and Ferrando, 2012), and occurs 2 to 3 times more frequently in males than females (Teachey and Pui, 2019). T cell ALL malignancy most frequently occurs by constitutive action of NOTCH1 signaling. In addition, deletions of the CDKN2A locus encompassing the p16/INK4A and p14/ARF suppressor genes in chromosome band 9p21 are found in over 70% of T cell ALL cases (Van Vlierberghe and Ferrando, 2012). In the past, T cell ALL in children gave much poorer prognoses than B-cell ALL, but with more recent research and treatment strategies it has become nearly as treatable as B- cell ALL (Teachey and O’Connor, 2020).
Chronic lymphocytic leukemia (CLL) is one of the most common leukemias in adults, and is most frequent among older individuals, with the average age of diagnosis at 71 years old (Desantis et al., 2014). In addition, only 10–15% of patients receive a CLL diagnosis before age 50. CLL is almost always because of B-cell malignancies, with malignant T cell phenotypes constituting roughly 2–5% of CLL patients of which have significantly less successful outcomes compared to B-CLL patients (Shahjahani et al., 2015). This malignancy of B lymphocytes is often characterized by an accumulation of high CD5−expressing B-cells in the blood. Mutations and chromosomal alterations including the deletion of chromosome 13q, chromosome 11q, and/or 17p are associated with a more aggressive and treatment-resistive leukemia (Hallek et al., 2018).
Current treatments
B-cell ALL has historically had higher success rates compared to T cell ALL following various treatments because of several reasons. Because patients with T-ALL are typically older than patients with B-ALL, chemotherapeutic treatment in T-ALL cases results in much weaker tolerance and presents higher chances of relapse. For these reasons, recent studies indicate treatment of B-ALL sees overall survival rates above 90%, whereas T-ALL trails behind with around 80–85% overall survival (Teachey and Pui, 2019). Though prognosis is relatively positive for individuals diagnosed with ALL, an estimated 1 in 5 children will relapse following an initially successful treatment, in which the prognosis is much poorer. In addition, 30–40% of ALL relapse cases are associated with complications involving the central nervous system (CNS) that arise because of infiltration of leukemia cells in the cerebrospinal fluid (CSF), providing an extra barrier for successfully treating ALL relapse patients (Lenk et al., 2020).
Contemporary therapies for children with ALL have been developed consisting of remission induction therapy followed by consolidation therapy for 8 weeks and maintenance therapy for 18 to 30 months. Remission induction therapy using a glucocorticoid, vincristine, an asparaginase preparation, optional use of an anthracycline, and intrathecal chemotherapy leads to remission in almost all patients. To consolidate remission and prevent the development of overt CNS leukemia, an 8-week delayed intensification protocol is administered using methotrexate and folinic acid. The final maintenance phase involves daily oral mercaptopurine or thioguanine and weekly oral methotrexate for 18 to 30 months. Low patient adherence to maintenance therapy is associated with a 4 times higher risk of relapse (Hunger and Mullighan, 2015).
Advances in ALL genomic profiling has facilitated the development of targeted-therapy strategies for specific subtypes of ALL. For example, patients diagnosed with ALL with the BCR-ABL1 fusion oncoprotein are prime candidates for targeted treatments using tyrosine kinase inhibitors (TKIs) (Kantarjian et al., 2002). Further, patients diagnosed with B-cell ALL are ideal candidates for cell-based therapy approaches to treatment because of the high density of CD19 present on the surface of most B-cell ALL cells (Turtle et al., 2016).
Because of its slow growth rate, CLL requires monitoring after treatment, which traditionally included combination therapy using fludarabine and phosphadamine and immunotherapy using the anti-CD20 monoclonal antibody rituximab (Hallek et al., 2018). However, more recent approaches have led to a decline in the use of chemoimmunotherapy to treat CLL, including venetoclax and several different kinase inhibitors. These kinase inhibitors work to transport CLL cells from the tissues to the peripheral blood, increasing peripheral-blood CLL-cell counts in a phenomenon referred to as “redistribution lymphocytosis” and resulting in decreased enlargement of lymph nodes (Faderl and Keating, 2005). Targeting Bruton’s tyrosine kinase (BTK) with ibrutinib, one of several BTK inhibitors studied for improved treatment of CLL, results in direct cytotoxicity, inhibition of CLL-cell proliferation, the disruption of cytokine/chemokine signaling, and inhibition of cell migration (Bond and Woyach, 2019). BTK inhibitors and other similar advances in CLL treatment will be discussed further in the sections on checkpoint inhibitors.
Myeloid leukemias
Disease overview and diagnosis
According to the American Cancer Society, AML is the most common among the myeloid leukemias, and it is the most common acute type among adults (Bray et al., 2018). It has a cure rate of 35–40% in patients aged 60 years or younger (Döhner et al., 2010). This form, one of the most heavily researched, is classified according to specific factors that impact patient prognosis. According to the WHO classification system, AML can be split into 4 major categories: AML with certain genetic abnormalities, AML with myelodysplasia-related changes, AML related to previous chemotherapy or radiation, and AML not otherwise specified (Döhner et al., 2015).
CML, the more slowly developed form of myeloid leukemia, impacted approximately 34,200 in 2017 globally (Dong et al., 2020). This form of leukemia arises when the Philadelphia chromosome, caused by a BCR-ABL1 fusion gene formed by t(9;22)(q34;q11.2), is generated. The BCR-ABL1 fusion gene promotes unregulated cell proliferation by encoding an active BCR-ABL1 tyrosine kinase (Loghavi et al., 2015).
Current treatments
Standard treatment for AML involves a two-phase approach, beginning with remission induction therapy using cytarabine paired with an anthracycline, such as daunorubicin or idarubicin, followed by post-remission therapy using higher doses of cytarabine to eliminate any remaining leukemia cells (Döhner et al., 2015). This treatment can lead to cures in 30–40% of younger patients (Kantarjian et al., 2021). For the highly aggressive acute promyelocytic leukemia, the frontline therapeutic strategy is all-trans retinoic acid and arsenic trioxide (Sanz et al., 2019). In recent years, research has helped to identify targetable abnormalities for AML. Newer treatments of interest for AML include combining epigenetic therapy with hypomethylating agents such as azacitidine and decitabine, adding fms-like tyrosine kinase 3 inhibitors to intensive chemotherapy, adding IDH inhibitors in AML with IDH1/2 mutations, using anti-CD47 antibodies in treating TP53-mutated AML, using memin inhibitors for treatment of mixed-lineage leukemia-rearranged acute leukemia, using combinations of small-molecule target therapies with intensive chemotherapy, and many immunotherapy approaches that are covered in later section (Kantarjian et al., 2021).
In the case of CML, standard treatment involves targeted therapies using TKIs. One commonly used inhibitor is imatinib, and more recently, asciminib (Goldman and Melo, 2003; Hughes et al., 2019). Clinical trial data is also available for newer inhibitors such as nilotinib, dasatinib, and bosutinib, showing superior response rates compared to imatinib (Ferdinand et al., 2012). In the past, interferon-α therapy was commonly used for treating CML, however it is associated with undesired side effects (Goldman and Melo, 2003). In addition, patients with CML can be cured using allogenic stem cell transplantation, which is covered further in the cell-based therapies section.
Lymphoma
Lymphomas affect lymphocytes that are located in the lymph system. Lymphocytic leukemias can affect the same cell types as lymphomas but are characterized by location in mainly the bone marrow and blood. Lymphomas can be categorized into many subtypes, mainly Hodgkin’s and non-Hodgkin’s (Küppers, 2008; Shankland et al., 2012).
Hodgkin’s lymphomas
Disease overview and diagnosis
Hodgkin’s lymphoma (HL) is a type of cancer originating from B lymphocytes in the lymphatic system (Küppers et al., 2012). Of all lymphomas, HL consists of 11%, whereas the remaining can be attributed to non-Hodgkin’s lymphoma (NHL) (Shankland et al., 2012).
HL consists of two histological disease entities: classical HL and nodular lymphocyte predominant HL (Ansell, 2015). Classical HL comprises 90% of all HL cases. This disease is characterized by the presence of Reed-Sternberg cells, which are mutated B lymphocytes that are multinucleated and cancerous, proliferating in the lymph nodes and tissues. The Reed-Sternberg cells express CD30 and CD15 antigens that are not expressed by normal B cells. The diagnosis for classical HL typically requires a biopsy sample for lymph nodes to confirm the presence of Reed-Sternberg cells (Yung and Linch, 2003). Within classical Hodgkin’s lymphoma, several subgroups of the disease exist: nodular sclerosis, mixed cellularity, lymphocyte depletion, and lymphocyte-rich HL (Ansell, 2014). Nodular sclerosis comprises a majority (75%–80%) of all classical Hodgkin’s disease cases, characterized by the presence of nodules.
The other subtype of HL is nodular lymphocyte predominant Hodgkin’s lymphoma (NLPHL). NLPHL is significantly rarer than the classical HL, only making up around 5% of all classical HL cases (Fanale et al., 2010). In addition, unlike classical HL, NLPHL is not characterized by a presence of Reed-Sternberg cells; instead, large “popcorn cells”, or lymphocyte predominant cells, are present. These lymphocyte predominant cells can be detected in tumor cells and are variants of the Reed-Sternberg cells that express CD19+ and CD20 antigens instead of CD30 and CD15 (Wahed et al., 2015). Diagnosis of NLPHL consists of a primary analysis of the lymph nodes of a patient for swelling. A more specific diagnosis can be conducted by an excision surgical biopsy of the lymph node, in which the neoplastic cells can be screened for (Mak and Saunders, 2006).
Current treatments of Hodgkin’s lymphoma
Treatments for HL have been shown to have significant efficiency, with the five-year survival rate being 87%. Common treatment options include chemotherapy, radiation therapy, target therapy, and immunotherapy. The specific treatment options are unique to each patient and are highly dependent on the subtype and stage of the HL, as well as the patient’s background. For patients with favorable-risk disease, three rounds of a hybrid MOPP (chlormethine-vincristine-procarbazine-prednisolone)/ABV (doxorubicin-bleomycin-vinblastine) treatment as well as involved-field radiotherapy is preferred over complete nodal irradiation (Yung and Linch, 2003). Depending on the patient response, the chemotherapy and radiation may be followed up with a stem cell transplant or immunotherapy. With unfavorable-risk disease patients, typically a higher intensity chemotherapy regimen followed by radiation therapy is recommended, such as high-dose ABVD (doxorubicin-bleomycin-vinblastine-dacarbazine) or subtotal nodal irradiation (Yung and Linch, 2003). In every case, a stem cell transplant and immunotherapy are options if the patient does not respond to initial treatment (Ansell, 2015).
Non-Hodgkin’s lymphoma (NHL)
Disease overview and diagnosis
Non-Hodgkin’s Lymphoma is a tumor that arises from a geneticmutation in a lymphocyte and lacks key markers of HL, Reed Sternberg cells. This disease develops when a B-cell or T cell differentiates uncontrollably instead of undergoing programmed cell-death or apoptosis. At this stage, these lymphocytes become neoplastic cells which form lymphomas. Nodal lymphomas are those that develop in the lymph nodes, and extranodal lymphomas are those which develop in other areas, such as the stomach and skin. Lymphoma cells also have the ability to travel through the bloodstream and cause complications in other organs. In the gastrointestinal tract, this can lead to bowel obstructions (Greiner et al., 1995; Shankland et al., 2012).
NHLs can be classified into B-cell and T cell lymphomas. Non-Hodgkin B-cell lymphomas are more common than Non-Hodgkin T cell lymphomas and involve the typical expression of CD20 on neoplastic B-cell surfaces. Various classes of B-cell lymphomas range from indolent or slow growing to highly aggressive. Diffuse, large B- cell lymphoma is the most common form of B-cell lymphoma and acts aggressively in its development. In contrast, follicular B-cell lymphoma is indolent and is known to arise from a chromosomal translocation between chromosome 14 and 18. In this translocation, large chromosomal segments are exchanged, and the BCL2 gene from chromosome 18 is placed after the immunoglobulin heavy chain promoter on chromosome 14, resulting in overexpression of BCL2 which normally blocks apoptosis (Greiner et al., 1995; Shankland et al., 2012). A third type of B-cell lymphoma is Burkitt-Lymphoma (Nogai et al., 2011), a highly aggressive class which can also result from a chromosomal translocation where the MYC gene from chromosome 8 becomes adjacent to the IgH promoter of chromosome 14 and upregulates MYC gene expression, stimulating cell growth and metabolism and leading to further cell proliferation. A fourth type of B-cell lymphoma is Mantle Cell Lymphoma (Nogai et al., 2011), which is aggressive and also involves a chromosomal translocation of the BCL1 gene of chromosome 11 becoming adjacent to the Ig promoter on chromosome 14. The BCL1 gene encodes the protein, Cyclin D1, promoting cell growth and division. Additional types of B-cell Lymphomas include Marginal Zone Lymphoma (nodal and MALT) and Lymphoplasmacytic Lymphoma.
Within the class of T cell lymphomas, there is Adult T cell lymphoma. This type of lymphoma is believed to be caused by Human T-Lymphotropic Virus (HTLV) which travels through body fluids to infect T-cells, by incorporating viral DNA into the genetic sequence of T-cells. Mycosis fungoides is another type of T cellLymphoma and involves the skin. In this form, the neoplastic cell is a CD4+ helper T-cells that can be identified by its cerebriform nucleus. As these cells circulate the blood, patients experience Sezary Syndrome (erythroderma and pruritus) (Hristov et al., 2019; Oka and Miyagaki, 2019).
General symptoms of NHL include painless lymphadenopathy, and cytokine release related fever, night sweats, and weight loss. With extranodal involvement of the GI tract, patients can experience bowel obstructions. If the bone marrow is involved, additional symptoms include fatigue, easy bruising, and recurrent infections. Diagnostic techniques for identifying NHL includes imaging studies such as computed tomography (CT) scans to help establish the stage of lymphoma (extent of nodal and extranodal involvement), and lymph node biopsies to confirm tissue malignancy (Singh et al., 2020).
Current treatments of Non-Hodgkin lymphoma
Current gold standard treatments (Avanzi and Brentjens, 2017) for both B-cell NHL and T cell NHL involve several chemotherapy and radiotherapy cycles and have shown high success in patient remission, despite the presence of disseminated disease at time of diagnosis. However, many NHL patients still experience relapse and systemic resistance to therapies, preventing the goal of curing the disease. Many different chemotherapies are currently used for various subtypes, levels of aggression and extent of spreading and often are combined for higher efficacy. Alkylating agents, corticosteroids, platinum drugs, purine analogs, anti-metabolites, and anthracyclines are some classes of chemotherapeutics for treatment of NHL.
The most common combinational chemotherapy for both B-cell NHL and T cell NHL is usually abbreviated as CHOP, a treatment including cyclophosphamide, hydroxydaunorubicin (doxorubicin), oncovin (vincristine), and prednisone. With NHL that exhibit CD20+ B-cells, the monoclonal antibody Rituximab (Advani et al., 2018) can be used to bind CD20+ and induce complement mediated lysis, as well as cytotoxicity and apoptosis abilities. This regimen of R-CHOP is especially prescribed to patients of diffuse large B-cell lymphoma and is prescribed in cycles 3 weeks apart from one another (Flinn et al., 2014). The specific regimen for T cell lymphoma is more intensive chemotherapy cycles, up to 2 years, with one or more of the following accepted chemical therapeutics (Rodriguez et al., 2001): cyclophosphamide, doxorubicin (Adriamycin), vincristine, L-asparaginase, methotrexate, prednisone, and, sometimes, cytarabine (ara-C). Sometimes this high-dose, aggressive chemotherapy is followed by stem cell transplants (Corradini et al., 2006). Although the chance of chemotherapeutic remission is high in T cell lymphoma which has not spread to the bone marrow, the chances of effective treatment vary by compounding factors and can be very difficult once metastasis to the bone marrow has occurred (Singh et al., 2020).
Multiple myeloma
Disease overview and diagnosis
Multiple myeloma is a disease that exhibits an excess of plasma cells in the bone marrow. Although it was originally thought that the origin was a single tumor stem cell, it has been shown to be composed of clonally different subgroups of tumor cells (Sirohi and Powles, 2004; Röllig et al., 2015). The disease is characterized by a range of symptoms including osteolytic bone lesions, kidney damage, hypercalcemia and anemia. The presence of these symptoms is because of the tumor itself and the response of the immune system to the tumor. In addition, the presence of monoclonal protein (paraprotein/light chain/”M″ component) in blood or urine is common (Sirohi and Powles, 2004; Raab et al., 2009) as well as a lack of immunoglobulin M (Ig M) (Röllig et al., 2015).
Within the pathogenesis of multiple myeloma, there is an important genetic component associated with the disease as B lymphocytes undergo DNA breakage processes to transform into plasma cells, which makes them more susceptible to mutations. Many of these mutations are located in the region corresponding to the Ig heavy chain (IgH) at 14q32 and are essentially related to seven chromosomes, having identified PRAD1, BCL1, cyclin D1 (11q13), cyclin D3 (6p21), C-MAF (16q23), FGFR3-MMSET (4p16.3) and MAFB (20q11) as specific non-random chromosomal translocations. Deletions can also occur on chromosomes 13 and 17p (p53) as well as t(4;14) translocations. Other important mutations occur in NRAS, KRAS, FAM46C, BRAF and TP53 (Figure 1) (Sirohi and Powles, 2004).
Figure 1.

Multiple myeloma pathogenesis
Created with BioRender.com.
Furthermore, the microenvironment of multiple myeloma cells has been shown to play a key role in the development of the disease. It consists of extracellular matrix proteins (such as collagen, fibronectin, laminin and vitronectin), bone marrow stromal cells (hematopoietic and non-hematopoietic), osteoclasts, osteoblasts, vascular endothelial cells, lymphocytes and fluid composed of cytokines and growth factors, like interleukin 6 and 10 (IL-6 and 10), vascular endothelial growth factor (VEGF), insulin-like growth factor 1 (IGF1), transforming growth factor β 1 (TGFB1), tumor necrosis factor (TNF)- superfamily members, chemokine ligand CCL3, stem cell factor (SCF) and hepatocyte growth factor (HGF) (Sirohi and Powles, 2004; Raab et al., 2009). Interactions between myeloma cells and bone marrow stromal cells or between matrix proteins, whichtake place through cell surface receptors (such as integrins, syndecans, cadherins and selectins) or through the immunoglobulin superfamily of adhesion molecules, trigger different signaling pathways (KRAS, RAF1, MAP2K1, MAP2K1, PIK3, and AKT; JAK and STAT3; PRKC; NFKB; and WNT) that generate growth and progression of the cancerous cells. Moreover, adhesion of myeloma cells to matrix proteins leads to the secretion of molecules such as urokinase-type plasminogen activator and metalloproteinases 2 and 9 (Raab et al., 2009). The following facts are known: interaction of myeloma cells with fibronectin confers protection against apoptosis, IL-6 (the most potent growth factor in multiple myeloma) increases VEGF production in myeloma cells and vice versa, and interaction of myeloma cells with hemopoietic and non-hemopoietic cells results in suppression of the immune system, as well as lytic bone lesions (Sirohi and Powles, 2004; Raab et al., 2009).
Within the pathology there are different stages. First, the disease appears as a plasmacytoma in a premyelomatous stage, characterized by the presence of a monoclonal paraprotein in serum. This stage is known as monoclonal gammopathy of unknown significance (MGUS) and may progress to smoldering multiple myeloma, where there is no end-organ damage but it is a more advanced stage than MGUS (Sirohi and Powles, 2004; Röllig et al., 2015; Ho et al., 2020). From here on, 5–7% of MGUS patients and approximately 50% of patients with smoldering multiple myeloma will develop clinical multiple myeloma (van de Donk et al., 2021a). Because most of the somatic mutations found in symptomatic multiple myeloma are also found in MGUS and latent multiple myeloma, there must be other causes related to the development of symptomatic multiple myeloma (Dutta et al., 2019; Minnie and Hill, 2020). In this regard, from asymptomatic stages onwards, intraclonal heterogeneity exists, and clonal stability drives the multiple myeloma progression (Dutta et al., 2019).
Although the disease can be diagnosed accidentally when it is asymptomatic because of a persistent back pain or an atypical anemia, certain clear symptoms, like bone pain and fractures, anemia, infections and renal failure, among others, may suggest the presence of multiple myeloma and are reasons to test for the presence of the disease. Concerning this, there are numerous techniques used to diagnose multiple myeloma, including paraprotein detection in urine or serum by immunofixation, serum and urine protein electrophoresis, imaging techniques, bone marrow analysis and cytogenetic analysis, among others (Röllig et al., 2015; van de Donk et al., 2021a). In addition to the previous diagnostic methods, it is important to assess the bone lesions using imaging techniques. In particular, functional imaging techniques, such as 18F-fluorodeoxyglucose positron emission tomography combined with computed tomography (18F-FDG-PET/CT), or diffusion-weighted MRI, are recommended (van de Donk et al., 2021a).
Once the disease has been diagnosed, patients need to be evaluated for prognosis of the disease that depends on their stage. The monoclonal protein marker is the most commonly used marker for monitoring and staging the disease as well as assessing the efficacy of treatment, according to the traditional Durie-Salmon System. However, the combination of beta2-microglobulin with albumin levels represents the new standard staging system (International Staging System or ISS) because of the reproducibility, simplicity and effectiveness of these prognosis factors, which was revised in 2015 to include chromosomal abnormalities and serum lactate dehydrogenase level as prognosis factors (Tables 1 and 2) (Greipp et al., 2005; Palumbo et al., 2015; Röllig et al., 2015).
Table 1.
New staging systems for multiple myeloma (Palumbo et al., 2015): International Staging System (ISS)
| ISS stages | Parameters of diagnosis | OSa rate |
|---|---|---|
| Stage I | Serum β2-microglobulin level <3.5 mg/L and serum albumin level ≥3.5 g/dL |
62 months |
| Stage II | Those patients who are not in either of the other two states | 44 months |
| Stage III | Serum β2-microglobulin level ≥5.5 mg/L, regardless of serum albumin level | 29 months |
Risk stratification by International Staging System (ISS), result of the combination of serum β2-microglobulin level and serum albumin level.
OS, overall survival.
Table 2.
New staging systems for multiple myeloma (Palumbo et al., 2015): Revised International Staging System (R-ISS)
| R-ISS stages | Parameters of diagnosis | 5-year OS rate |
|---|---|---|
| Stage I | ISS stage I, normal LDHa and no high-risk CA | 82% |
| Stage II | Those conditions which are not covered by either stage I or stage III of R-ISS | 62% |
| Stage III | Stage III ISS with high LDH levels or high-risk CA | 40% |
Risk stratification by revised ISS (R-ISS), result of the combination of ISS, Chromosomal abnormalities (CA) and Serum lactate dehydrogenase (LDH).
Normal LDH levels ranged from 140 units per liter (U/L) to 280 U/L or 2.34 μkat/L to 4.68 μkat/L. Values taken from HealthLinkBC, British Columbia.
Current treatments of multiple myeloma
Treatment of multiple myeloma should start when the patient presents end-organ damage, which is defined by the CRAB criteria (i.e., hypercalcemia, renal failure, anemia and bone lesions), or a biomarker of malignancy (Röllig et al., 2015; van de Donk et al., 2021a). Despite new treatments, none have been found to cure the disease (van de Donk et al., 2021a).
For people younger than 70 years, the current first-line treatment (Figure 2A) is based on induction therapy followed by high-dose melphalan and autologous stem cell transplantation. It has been observed that the use of high-dose myeloablative therapy with autologous stem cell transplantation prolongs patient survival compared to conventional cytostatic treatments (Röllig et al., 2015; van de Donk et al., 2021a). Tandem stem cell transplantation (two sequential transplants) is being utilized, although its usefulness is controversial, because better progression-free survival has been found, but with no significant differences in overall survival (Sirohi and Powles, 2004; Raab et al., 2009; Röllig et al., 2015; van de Donk et al., 2021a). In addition, allogeneic stem cell transplantation can be considered after the use of high-dose chemotherapy or an autologous stem cell transplantation. This treatment is an option for relapsed/refractory patients (Shah and Mailankody, 2020).
Figure 2.

Current treatments for Multiple myeloma
(A) First-line therapy for patients under 70 years.
(B) Regimens for patients not eligible for ASCT (65–75 years).
(C) Established treatments for patient relapse. BTZ, Bortezomib; CFZ, Carfilzomib; IXZ, ixazomib; DEX, Dexamethasone; CP, cyclophosphamide; MEL, melphalan; IMiD, immunomodulatory drugs; THAL, thalidomide; LD, lenalidomide; POM, pomalidomide; PRD, prednisone; mAb, monoclonal antibodies; DARA, daratumumab; ELO, elotuzumab; ISA, isatuximab; SEL, selinexor; ASCT, autologous stem cell transplantation
The treatment of choice in induction therapy consists of a regimen of bortezomib with either dexamethasone and an immunomodulatory drug (thalidomide or lenalidomide) or bortezomib-cyclophosphamide-dexamethasone. Although carfilzomib is not approved for use in first-line therapy, the use of this second-generation proteasome inhibitor together with lenalidomide and dexamethasone is achieving high response rates in some clinical trials. In addition, the FDA and the EMA have recently cleared (September 2019 and January 2020) therapy with four drugs, bortezomib, thalidomide, dexamethasone and the CD38-directed antibody daratumumab for newly diagnosed transplant eligible patients. This four-drug regimen is being studied in a phase II clinical trial, using lenalidomide instead of thalidomide, with promising results (van deDonk et al., 2021a; Clinical Trials, 2022).
Although the median overall survival is higher for patients receiving autologous transplantation, 10 against 4–5 years for non-eligible patients, many patients are not eligible for it. Within this group, in which older patients are usually included, triple regimens are being used (Figure 2B), such as bortezomib-cyclophosphamide-prednisone, bortezomib-lenalidomide-dexamethasone, and bertezomib-melphalan-prednisone. Moreover, FDA approved triplet and quadruplet regimens that include daratumumab in the following combinations: lenalidomide-dexamethasone and bortezomib-melphalan- prednisolone, respectively (van de Donk et al., 2021a).
After completion of the autologous transplantation, consolidation treatment of limited duration is necessary to enhance progression-free and overall survivals in patients. This usually involves the same treatment used for induction or a second transplant, three months after the first one (tandem autologous stem cell transplantation), in the case of high-risk disease (van de Donk et al., 2021a). The emergence of drugs with better safety profiles has led to the consideration of continuous treatments to maintain response (Röllig et al., 2015). In this regard, lenalidomide has been approved as maintenance therapy based on several phase III clinical trials with increased durations of progression-free and overall survivals. Also, bertezomib and ixazomib, as single agents, are under study as maintenance therapy in different clinical trials with promising results (van de Donk et al., 2021a; Clinical Trials, 2022).
When a patient relapses (Figure 2C) and meets the CRAB criteria, or a paraprotein (M-protein) increase is detected, treatment should be initiated immediately (Röllig et al., 2015; van de Donk et al., 2021a). Combination of dexamethasone with lenalidomide, bertezomib or carfilzomib is the established treatment for frail relapsed or refractory multiple myeloma patients, being the doublet carfilzomib-dexamethasone the regimen with better clinical results. In the case of non-frail patients, four lenalidomide-based and three bortezomib-based triplet regimens, which are applied to lenalidomide-refractory patients, have been approved, and two carfilzomib-based and two pomalidomide-based triplets are being used (van de Donk et al., 2021a). Monoclonal antibodies, such as elotuzumab, daratumumab and isatuximab, have also been efficiently used for relapsed or refractory multiple myeloma in different combination therapies that have been reviewed in detail in the section monoclonal antibodies for hematological cancers.
In the case of second and subsequent relapses (Figure 2C), a change in drug type is usually effective. In this regard, for patients who are resistant to both lenalidomide and bortezomib, treatments containing pomalidomide, daratumumab, or carfilzomib can be used. Thus, the FDA and the EMA have approved the use of pomalidomide-dexamethasone with daratumumab, elotuzumab or isatuximab. For patients resistant to proteasome inhibitors, immunomodulatory drugs and antibodies against CD38, the FDA has approved the combined use of selinexor with dexamethasone. Similarly, the FDA and EMA have approved the use of an antibody-drug conjugate against BCMA, belantamab mafodotin, to this triple-class refractory patients (van de Donk et al., 2021a).
Nevertheless, despite the great advances achieved so far in the treatment of multiple myeloma, there are certain groups of patients in which the development of new drugs based on innovative mechanisms of action are required. This is the case of the multiple myeloma patients that are resistant to all approved drugs or with a high-risk disease. In this regard, immunotherapy is gaining prominence with numerous innovative treatments that will be discussed next.
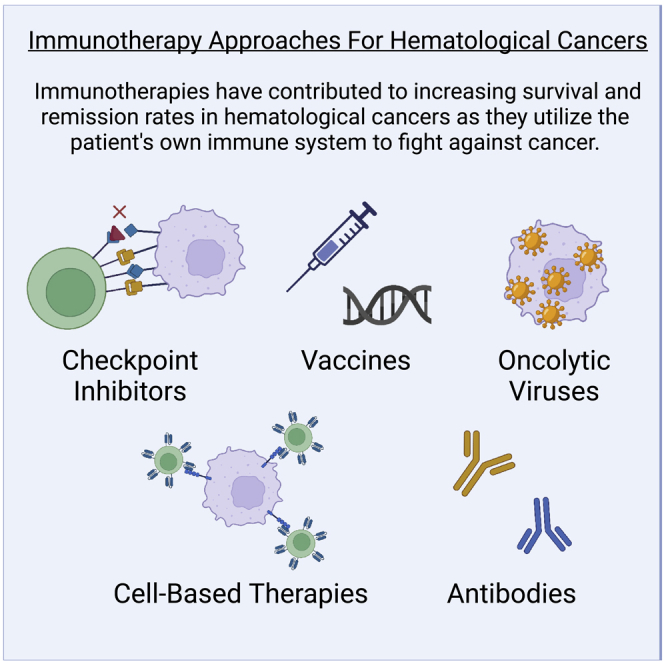
Immunotherapy strategies for hematological cancer treatment
The application of immunotherapy to hematological cancer has used advanced approaches including checkpoint inhibitors, vaccines, antibodies, cell-based therapies, and OVs (Figure 3). This section will briefly review these approaches and their successful applications toward cancer, with a particular focus on their use in hematological cancers. Challenges still exist with immunotherapies regarding their limited success rates and their high market price. Pros and cons of each immunotherapy approach are discussed throughout the manuscript and summarized in Table 3.
Figure 3.

Main immunotherapies used for hematological cancers: checkpoint inhibitors, vaccines, cell-based, antibodies, and oncolytic viruses
Table 3.
Pros and Cons of the reviewed immunotherapies
| TherapyType | Pros | Cons |
|---|---|---|
| Checkpoint Inhibitors | Can achieve long-term remission | Can cause autoimmunity/chronic inflammation side effects |
| Can increase patient survival time | Response rate varies in patient populations | |
| Several FDA approved treatments | Resistance or relapse can occur | |
| Can be used in many types of cancer | Expensive | |
| Responses can be predicted with biomarkers | Can be limited by the tumor microenvironment | |
| Targeted against a single antigen | ||
| Vaccines | Can achieve long-term remission | Can be easily degraded by the body |
| Can increase patient survival time | Limited shelf-life | |
| Easy and cost-effective synthesis of nucleic acid and peptide vaccines | Diverse tumor neoepitopes require personalized vaccines | |
| Can be used in many types of cancer | Cell-based vaccines require arduous isolation processes | |
| Low carcinogenic potential for peptide-based vaccines | mRNA is a large, negatively charged molecule, with poor uptake without a carrier |
|
| High chemical stability for peptide vaccines | Carriers for mRNA or DNA vaccines can cause immunogenicity | |
| Naked nucleic acid vaccines require frequent administration because of degradation by nucleases | ||
| Peptide vaccines have short half-lives | ||
| Antibodies | Highly reproducible and uniform | Short half-lives |
| Can achieve long-term patient survival time | Requires high doses that lead to increased toxicity | |
| Several FDA approved treatments | Can cause uncomfortable side effects such as fever, nausea, weakness | |
| Can be used in many types of cancer | Expensive | |
| Responses can be predicted prior to treatment | ||
| Cell-Based Therapies | Can achieve long-term patient remission | Relapse common because of developed resistance |
| Can increase patient survival time | Cytokine release syndrome leads to off-target side effects and toxicity | |
| Several FDA approved treatments and ongoing clinical trials | Difficulty in selective targeting | |
| Can be used in many types of cancer | CAR T cell therapy can cause cytokine release syndrome | |
| Have high utility for hematological cancers | CAR T cell therapy is directed against a single antigen | |
| CAR NK-cells have higher safety profiles than CAR T-cells | Arduous production process for CAR T-cells | |
| Expensive | ||
| Allogenic transplants can cause graft-vs-host responses | ||
| Oncolytic Viruses | High patient tolerability | Can be destroyed by the immune system |
| Can achieve long-term patient remission | Limited replication and spreading | |
| Can increase patient survival time | Some cancer cells are resistant | |
| 4 FDA approved treatments and several ongoing clinical trials | ||
| Can be used in many types of cancer | ||
| Can be engineered to eliminate pathogenic effects | ||
| Can add immunomodulatory transgenes to the viruses |
CAR, chimeric antigen receptor; NK, natural killer; FDA, Food and Drug Administration.
Checkpoint inhibitors
Overview
One of the most common types of immunotherapies are checkpoint inhibitors that work by blocking immune suppression techniques employed by the cancer cells. Cytotoxic T-cells conventionally target tumor cells using T-cell receptors or co-signaling receptors (Huse, 2009; Chen and Flies, 2013; Vaddepally et al., 2020). Cancer cells can activate immune checkpoints that suppress the immune system, and specifically the cytotoxic T-cells, from attacking them. Checkpoint inhibitors are used to block this immune evasion technique by cancer cells so that the cytotoxic T-cells can attack the cancer as customary.
Progress has been made rapidly in the field since the year 2018, when two scientists, Dr. Tasuku Honjo and Dr. James Allison of MD Anderson, won the Nobel Prize in Physiology or Medicine for their work on programmed death 1 (PD-1) and cytotoxic T-lymphocyte antigen (CTLA-4) checkpoint proteins (Ledford et al., 2018). PD-1 is an inhibitory transmembrane protein expressed on multiple cell types and an immune checkpoint pathway that can be used to inhibit T cell activation and proliferation (Vaddepally et al., 2020). The mechanism can be seen in Figure 4. Tumor cells can promote ligands of this pathway, B7-H1/PD-L1 and B7-DC/PD-L2, as an immune evasion technique. CTLA-4 checkpoint proteins are part of the B7/CD28 family, where CD28 is a co-stimulatory receptor for T cell responses. CTLA-4 prevents dendritic cells from priming T-cells to recognize tumors (Ledford et al., 2018).
Figure 4.

Mechanism of PD-1 checkpoint inhibitors
PD-1/PD-L1 checkpoint inhibitors are the most used checkpoint inhibitors today in Phase III/IV clinical trials (Darvin et al., 2018). Inhibitors of CTLA-4 are the other checkpoint inhibitor in Phase III/IV trials (Darvin et al., 2018). The first approved checkpoint inhibitor was anti- CTLA-4, called Ipilimumab (Yervoy), in 2011, for the treatment of advanced melanoma (Robert, 2020). Nivolumab and pembrolizumab are the first two FDA approved checkpoint inhibitors targeting PD-1. The binding of PD-1 and PD-L1 on tumor cells and antigen-presenting cells blocks T-cell receptor signaling and response leading to cell exhaustion. Nivolumab and pembrolizumab block the binding interaction of PD-1 and PD-L1 which improves the activity of antitumoral T-cells (Strati et al., 2018). There are seven total FDA approved checkpoint inhibitors on the market, with two that have indications for HL (Vaddepally et al., 2020).
Responses to checkpoint inhibitors vary across patient populations, with many patients never experiencing response and further patients undergoing relapse (Hargadon et al., 2018). Predictors for response to checkpoint inhibitors are being studied to determine patient outcome and guide approaches in terms of pursuing single-agent or combination therapy and when the therapy is administered. A key predictor is the PD-L1 presence in the tumor; however, some patients without PD-L1 expression can still respond positively to anti-PD-1 or anti-PD-L1 therapies (Darvin et al., 2018). This could be because of the transient nature of the PD-L1 expression in the tumor microenvironment rather than the tumor itself, or the lack of uniformity in PD-L1 immunohistochemistry antibodies (Darvin et al., 2018). Other biomarker strategies are reviewed in reference (Gibney et al., 2016) and include tumor-infiltrating lymphocytes, T-cell receptor clonality, mutational burden, neoantigen burden, immune gene signatures, and multiplex immunohistochemistry. Recently it was shown that chromosome arm aneuploidy (CAA) score can predict response to immune-checkpoint inhibition, where a lower CAA score has better prognosis to immune-checkpoint inhibition. Undoubtedly, the availability of a standard predictive model for efficacy in this immunotherapy approach would greatly reduce costs and minimize toxicities.
The future of immune checkpoint inhibitors lies in increasing the small percentage of patients who can benefit from them because of primary and acquired resistances. In this regard, numerous efforts are being made through combinational therapies, as evidenced by the impressive number of clinical trials that are currently underway (952 combinational studies including pembrolizumab, 706 with nivolumab or 330 involving ipilimumab) (Clinical Trials, 2022). Clearly, there is a need for a better understanding of the genomic, epigenomic and transcriptomic features involved in tumor resistance to achieve more personalized combination therapies involving checkpoint inhibitors that improve their clinical outcomes.
Immune checkpoint inhibitors can result in adverse effects on the immune system, leading to autoimmune diseases such as hypothyroidism or inflammatory bowel diseases (Robert, 2020). In rare cases, they can even lead to death, so their administration needs to be carefully monitored. However, this may be difficult as immune checkpoint inhibitor toxicity is not correlated with the dose, and signs of toxicity often require discontinuation of the treatment (deMiguel and Calvo, 2020).
Checkpoint inhibitors for hematological cancers
Although immune checkpoint inhibitors have significantly improved the clinical outcomes in solid tumors, this immunotherapy approach has only shown success in limited types of hematological tumors with high infiltration of immune cells. A better understanding of the heterogeneity of the immune microenvironments in hematological cancers that are disease-specific can dramatically improve the efficacy of checkpoint inhibitors to treat these malignancies.
Certain biomarkers can be applied for hematological cancers to predict the patient’s response to therapy with checkpoint inhibitors. Higher percentages of CD3+ and CD8+T-cells present in the bone marrow was used as a response predictor for anti-PD-1 therapy for patients with AML (Daver et al., 2019). Some preclinical studies have suggested that Reed-Sternberg cells use the PD-1 checkpoint to evade immune detection, so anti-PD-1 checkpoint inhibitors have been employed for patients with HL.
In patients with relapsed/refractory HL, after receiving ASCT and brentuximab vedotin (BV), treatment with anti-PD-1 was employed and had an 87% overall response rate (ORR) (Ansell et al., 2015). Moreover, PD-L1 is overexpressed in many subtypes of NHL and anti-PD-1 has been applied as a promising treatment in early clinical phases (Pianko et al., 2018). In initial phase I studies, it was seen that HL patients showed greater response to antibody studies because of higher expressions of PD-L1 as compared to more common NHL subtypes with lower expressions of PD-L1. The use of nivolumab and BV in patients with relapsed refractory cHL showed an ORR of 82% and complete response of 61%. Based on these results, elderly patients with chemotherapy ineligible HL are being studied for treatment with this combinatorial treatment of nivolumab and BV.
For patients of NHL, the most promising anti-PD-1 therapy has been identified as pembrolizumab for higher PD-L1 expression subtypes (relapsed primary mediastinal B- cell lymphoma showed an ORR of 41%) as well as nivolumab for patients with primary CNS lymphoma.
For aggressive T cell lymphomas, overall response rates range from 15% to 40% with nivolumab and pembrolizumab. Other clinical trials are in progress for the treatment of multiple lymphoma subtypes, HL, indolent B-cell NHL and T cell NHL with promising results (Strati et al., 2018).
However, many lymphoma patients are resistant to treatment with checkpoint inhibitors, and more studies are needed to elucidate and predict these responses. One marker that has been associated with increased response rates to anti-PD-1 therapy is an amplification of the 9p24.1 locus that encodes the PD-1 ligands, but more research into reliable biomarkers for response prediction is needed (Pianko et al., 2018).
In addition, the combination of PD-1 inhibitors with chemotherapies has been under investigation because higher expression of PD-L1 has correlated with poor outcomes with cHL chemotherapy patients. Cohort D of the CheckMate 205 study, which included patients with advanced cHL after frontline treatment with nivolumab (four cycles), showed that the combination of nivolumab with doxorubicin, vinblastine and dacarbazine resulted in an ORR of 86% and complete response of 80%. The use of pembrolizumab in conjunction with anti-CD19 CAR T cell therapy is also under phase I/II clinical trials for relapsed B-NHL (Strati et al., 2018).
Anti-PD-1 drugs, used as monotherapy, have only been found to be useful in myeloma when administered after stem cell transplantation. The combined use of different checkpoint inhibitors has also been reported, with a phase I/II clinical trial using ipilimumab with nivolumab shortly after ASCT in high-risk patients or with recurrent multiple myeloma, showing a promising progression-free survival after 18 months in the 71 and 67% of patients, respectively. However, this combination triggered events that required the use of systemic steroids (Minnie and Hill, 2020; Clinical Trials, 2022).
Moreover, the use of PD-1 inhibitors seems to be necessary as adjuvant treatment in the use of CD137 receptor agonist antibodies, which, in preclinical studies, have been shown to maintain control over myeloma by enhancing CD8+T cell action in both transplant and non-transplant settings. Because PD-1 is overexpressed after CD137 agonist therapy, the addition of anti-PD-1 permit to increase the disease control (Minnie and Hill, 2020; Yang et al., 2020).
CTLA-4 inhibitors have not been used as often as PD-1 and PD-L1 in regard to hematological cancer treatment. However, there is a phase I clinical trial, where the CTLA-4 inhibitor is being studied in hematological cancers with promising results (Yang et al., 2020). It has also been shown to obtain promising results in combination therapy with anti-PD-1. Impressively, the use of nivolumab, ipilimumab (an anti-CTLA-4 monoclonal antibody) and BV in a parallel patient population to show an ORR of 100% and complete response rate of 63%, as well as mild toxicity response (Diefenbach et al., 2016). The mechanism of action of CTLA-4 is parallel to PD-1. Yet another phase 1b study using dual PD-1 and CTLA-4 or KIR blockade in patients with HL, NHL, and multiple myeloma, suggests that dual therapy does not significantly improve therapeutic outcomes compared to anti-PD-1 therapy alone. Therefore, more studies need to be performed to determine if dual therapy improves patient outcomes for hematological cancers in the same way as solid tumors (Armand et al., 2021).
Apart from the checkpoint inhibitors that target PD-1 and CTLA-4 proteins in T-cells, the blockage of TIGIT (T cell immunoreceptor with Ig and ITIM domains), which is overexpressed in multiple myeloma, by novel checkpoint inhibitors have significantly improved CD8+ effector T cell activity and survival after ASCT in mice. In addition, this approach prevented dendritic cell immunosuppression and T cell exhaustion and prevented multiple myeloma progression in preclinical studies without transplantation, so new myeloma therapies based on checkpoint inhibitors are moving in this direction (Minnie and Hill, 2020).
In the case of CLL, checkpoint inhibitors that target B cell receptor signaling are at the forefront. One such inhibitor for CLL is Bruton’s tyrosine kinase (BTK). The first-generation BTK inhibitor approved for clinical use, ibrutinib, has been shown to improve T cell number and function in CLL patients (Long et al., 2017). However, incidences of ibrutinib resistance in CLL patients have led to the development of several next-generation BTK inhibitors for leukemia treatment. Those include the FDA-approved acalabrutinib and zanubrutinib (Wen et al., 2021). Although these checkpoint inhibitor-based therapies are considered breakthroughs for CLL treatment, more research is needed to identify treatment strategies to minimize toxicity, off-target effects, and the onset of inhibitor resistance (Faderl and Keating, 2005).
Although immune checkpoint inhibitors such as nivolumab have shown great promise in the treatment of hematological malignancies, particularly HL, recent studies indicate that the use of nivolumab for adult T cell leukemia/lymphoma leads to rapid disease progression (Ratner et al., 2018). This phenomenon highlights the importance of proper disease diagnosis for effective treatment.
Therapeutic vaccines
General observations
Therapeutic vaccines for the use of cancer treatment are engineered to trigger immune responses against target tumor-associated or tumor-specific antigens. These vaccines can be comprised of DNA, mRNA, peptide/protein, and cell-based components (Zhang et al., 2018). Cancer vaccines work in the same function by presenting some form of the cancer to the immune system so that it can better recognize and attack it. They can consist of antigens (DNA, mRNA, protein) injected into the patient to stimulate an immune response or consist of immune cells that have been trained ex vivo with tumor isolates to recognize the tumor, as shown in Figure 5.
Figure 5.

Different types of cancer vaccines and mechanisms of delivery
Vaccines can consist of antigens of DNA, mRNA, protein/peptide, or cell-based. They can be introduced into the patient directly to recruit immune cells or immune cells can be trained ex vivo to respond to antigen and then introduced back to the patient. Created with BioRender.com.
DNA vaccines use synthetic DNA sequences to be transcribed and translated in vivo, producing proteins to be presented to immune cells (Gary and Weiner, 2020). Non-replicating mRNA vaccines contain the sequence of the antigen of interest surrounded by two untranslated regions at both ends of the sequence. This results in fully mature mRNA with a 5′ cap and a poly(A) tail that can translate proteins of interest (Cappellano et al., 2021). There are also self-replicating mRNAs that are based on the alphavirus genome and can replicate to amplify itself using the replication machinery of the alphavirus; however, the genes encoding the structural proteins of the alphavirus are replaced with the sequence of the antigen of interest. For long sequences, it is possible to combine the two approaches to encode the alphavirus replication genes and the gene of interest.
Peptide and protein-based vaccines mainly consist of epitope peptides that stimulate CD8+T cells or CD4+ T helper cells to target tumor-associated or tumor-specific antigens. They typically consist of 8–12 amino acid peptides from a tumor antigen coding sequence (Liu et al., 2021). Protein-based vaccines for cancer include formulations based on anti-idiotype antibodies and heat shock proteins (Hu et al., 2018). Anti-idiotype vaccines use antibodies that target idiotypes of another antibody that targets a tumor antigen. Heat shock proteins can be used to activate anti-tumor T-cells through binding to and presenting antigens to antigen-presenting cells using MHC class I and II molecules (Hu et al., 2018).
Cell-based vaccines are based on the use of immune cells such as dendritic cells, NK cells, and T-cells, or stem cells. Dendritic cells, which are cells involved with antigen processing and presentation, have been utilized often in the form of vaccines for cancer therapies (Shang et al., 2017; Huber et al., 2018). These therapeutic vaccines consist of autologous dendritic cells with tumor-specific antigens that aim to increase the T cell response against tumor cells in the patient (Huber et al., 2018). Dendritic cell vaccines can be prepared in vivo by activating the dendritic cells that are already present or ex vivo by loading them with tumor-associated antigens (Shang et al., 2017). Dendritic cells loaded with tumor associated mRNA ex vivo is a commonly used approach in current clinical trials (Jahanafrooz et al., 2020). Anti-cancer vaccines can also be produced by induced pluripotent stem cells iPSC-based whole cells (Li et al., 2009; Iriguchi and Kaneko, 2019; Chu et al., 2020).
Variations of vaccine type (DNA, mRNA, peptide, or cell) affect their efficacy, and a short list of more pros and cons of these therapies can be found in Tables 1 and 2. For example, peptide and nucleic acid vaccines have fast and easy synthesis methods. However, they can be easily degraded by the body and have limited shelf-lives. The different types of vaccines all have different delivery targets which can affect their efficacy; peptides can function from the bloodstream, but mRNA must enter the cell, and DNA must go into the nucleus. Thereto, mRNA vaccines have slight better immunogenicity than DNA vaccines when compared directly; however, it should be noted that mRNA and DNA can be loaded into their target cells ex vivo. The appropriate type of vaccine to use is dependent on the cancer of interest, and in the case of hematological cancers the various available vaccines are discussed in the next section.
Therapeutic vaccines for hematological cancers
The ability of therapeutic vaccines to effectively fight hematological cancers is demonstrated by the fact that allogeneic stem cell transplantation can induce remission in a significant number of patients. A better understanding of tumor microenvironment and the use of combination therapies involving checkpoint inhibitors are the main ways that are being explored to improve the efficacy of therapeutic vaccines in hematological malignancies. In the same way, the use of vaccine approaches after hematopoietic cell transplantation is being studied.
Therapeutic vaccines are promising in the case of adult T cell leukemia and CLL because of the increased PD-1 expression on cytomegalovirus (CMV)- and Epstein-Barr virus (EBV)-specific cytotoxic T-lymphocytes (Ahmad et al., 2014). For adult T cell leukemia, Tax-specific cytotoxic T lymphocytes have been identified as a selective target for a vaccine. In a 2015 study, Suehiro et al. analyzed the clinical outcomes of a therapeutic vaccine with Tax peptide-pulsed dendritic cells for adult T cell leukemia and found that Tax-specific dendritic cell vaccine therapy resulted in partial remission or stable disease 8 weeks after initiation (Suehiro et al., 2015).
Therapeutic vaccines are being explored for AML, as well. Currently, Stroopinsky et al. are developing a personalized vaccine for AML using patient-derived dendritic cell-tumor cell fusions. This vaccine, in conjunction with PD-1 checkpoint inhibition, resulted in prolonged survival and an increase in tumor-specific immunity in a murine AML model (Stroopinsky et al., 2021). Further, the peptide vaccine, galinpepimut- S, is being explored as an immunotherapeutic for AML. Results of a phase 2 trial using galinpepimut indicated that the vaccine was well-tolerated and induced specific immunological responses in AML patients (Maslak et al., 2018).
Early vaccine trials for lymphoma were started by extracting native idiotype from a tumor biopsy and producing the vaccines through rescue fusion hybridization from myeloma cell lines or cloning the idiotype through recombinant DNA techniques. Often the idiotype must be coupled to a carrier protein such as keyhole limpet hemocyanin so the immune system can recognize it as foreign, and immunological adjuvants that stimulate dendritic cells are often given as a follow-up therapeutic (Metzger and Mauz- Körholz, 2019). Numerous phase 2 trials have used antigen-based vaccine approaches targeting idiotype for patients with lymphoma. Results in clinical trials using this approach have had mixed results (Avigan and Rosenblatt, 2018).
One phase I clinical trial of idiotypic DNA vaccine administered as a complex with cationic polymer polyethyleneimine (DEI) to patients with B-cell NHL led to stabilization of first remission. Linear PEI can be used to deliver the DNA into the cell (Meleshko et al., 2017). Ongoing studies on safety, tolerability, and immunogenicity of vaccine are being performed, and preclinical studies show increases in immunogenicity and reduction of toxicity with lower molecular weight conjugates. Another study showed that follicular NHL patients who received a recombinant idiotype vaccine (MyVax) did not improve progression-free survival unless patients mounted an anti-idiotype immune response (Levy et al., 2014). Moreover, another trial of idiotype vaccination, BioVaxID, was used in follicular lymphoma patients and shown to improve disease-free survival (Schuster et al., 2011).
Dendritic cells with immunological adjuvants have also been used. In a phase I clinical trial in patients with relapsed/refractory follicular NHL, intranodal injections of IFN-α cultured dendritic cells in combination with rituximab showed clinical and anti-tumor immune responses. IFN-α dendritic cells exhibited a partially mature phenotype, high migratory behavior, and immunostimulatory ability (Cox et al., 2019).
Patients with multiple myeloma have not seen expected results from idiotype protein-based vaccines because of their deficient expression on the surface of plasma cells and their insufficient immunogenicity. However, McCann et al. presented a phase I clinical study of an idiotypic DNA vaccine with promising results in multiple myeloma patients that received the vaccine after a treatment with high dose chemotherapy and an ASCT (McCann et al., 2015; Avigan and Rosenblatt, 2018). Moreover, a vaccine based on a peptide derived from B-cell lymphoma 2 (Bcl-2) family has been reported in a phase I clinical study with relapsed multiple myeloma patients, resulting in promising immune responses in combination with bertezomib with no added toxicity (Jørgensen et al., 2016; Avigan and Rosenblatt, 2018). Another study reported a vaccine based on a synthetic long peptide that targets the MUC1 protein (mucin 1, cell surface associated). This peptide-based vaccine was evaluated in combination with the human granulocyte-macrophage colony stimulating factor (rhGM-CSF) in multiple myeloma patients undergoing ASCT in a I/II phase clinical trial, resulting in a strong B- and T cell based immune response and resulted in stabilization of the disease (Carmon et al., 2015; Avigan and Rosenblatt, 2018; Clinical Trials, 2022).
Moreover, peptide vaccines include those based on testicular cancer antigens, like NY- ESO, MAGE1, and MAGE3 (Avigan and Rosenblatt, 2018). These types of antigens are expressed in male germ cells and are overexpressed in certain pathologies, including multiple myeloma. In addition, they are poorly expressed in healthy tissues, whereas their expression is associated with advanced and more aggressive disease, making these antigens a good material for the creation of vaccines (Hoyos and Borrello, 2016). A phase II trial used a peptide vaccine MAGE-A3 combined with a TLP-3 agonist, the granulocyte and monocyte colony stimulating factor (GM-CSF) and autologous T-cells co-stimulated ex vivo by anti-CD3/CD28, and was administered in patients with multiple myeloma after an ASCT, resulting in a CD8 T cell immune response in the 88% of HLA- A2 positive patients. However, to enter this study, patients could not express MAGE-A3 in myeloid cells, which is a limitation for understanding vaccine-specific T cell and B-cell responses (Rapoport et al., 2014; Clinical Trials, 2022).
Cell-based vaccines are being developed for multiple myeloma, among which can be distinguished two types: Vaccines that use whole tumor cells and vaccines that use antigen-presenting cells generated ex vivo (Avigan and Rosenblatt, 2018). In the case of whole cell vaccines, one strategy is the GVAX platform, a vaccine where primary tumor cells are either genetically modified to express GM-CSF or mixed with cell lines already expressing GM-CSF. For multiple myeloma, this approach is being studied in a phase II clinical trial, using two myeloma tumor cell lines (H929 and U266) and a cell line capable of secreting GM-CSF (K562/GMCSF). The use of this multiple myeloma GVAX vaccine in combination with lenalidomide in patients in near complete remission results in a robust immunity with a durable disease control (Biavati et al., 2021; Clinical Trials, 2022). In the case of antigen-presenting cell-based vaccines, two main approaches can be found in multiple myeloma. First, the idiotype-loaded antigen-presenting cell based-vaccine (APC8020, Mylovenge) that was administered in patients with multiple myeloma after an ASCT in a phase II clinical trial, resulting in an improved overall survival of 5.3 years compared to 3.4 years in patients who underwent only the transplant (Lacy et al., 2009). Second, there are vaccines based on the fusion of whole autologous dendritic cells and patient-derived multiple myeloma cells. Thus, two clinical trials can be found based on this approach. In the first, a phase I clinical trial showed an increase in the percentage of tumor-reactive CD4+ or CD8+T-cells in the 73% of the evaluable patients with a significant stabilization of the disease (Rosenblatt et al., 2011). In the second study, a phase II clinical trial showed the efficacy of this type of vaccine after an autologous stem cell transplantation. As in the previous trial, all evaluable patients were found to have an expansion of the number of myeloma-specific CD4+ and CD8+T-cells. Moreover, complete response and very good partial response was shown in the 68% of the patients and a complete and near complete response was shown in the 47% of the patients (Rosenblatt et al., 2013).
Antibodies
General observations
Antibodies can be used to destroy cancer cells through various mechanisms. They provide a targeting modality to specific antigens in the body that can reduce off-target side effects. Antibodies can be targeted to cell surface receptors to activate or inhibit signaling, activate antibody-dependent cell mediated toxicity, or activate complement-dependent cytotoxicity; alternatively, antibodies can be used to deliver toxins into cancer cells when internalized, or to downregulate cell surface receptors. Their mechanisms of action include direct killing through receptor blockade or agonist activity, inducing apoptosis, delivering drugs, immune-mediated cell killing mechanisms, regulating T cell function, and effecting the tumor vasculature and stroma (Scott et al., 2012).
Monoclonal antibodies (mAbs), shown in Figure 6, are a commonly used immune-therapeutic for cancer treatment. In the body, mAbs are produced by and are specific to unique B cells of the adaptive immune system. These antibodies work by binding to and inhibiting activators of division and angiogenesis signal pathways in cancer cells and induce cytotoxicity to cancer cells. Each mAb is unique to a specific antigen. Scientists can leverage this by designing mAbs that are specific to antigens characteristic to or overexpressed by various forms of cancer (Kimiz-Gebologlu et al., 2018). Tumors contain cancer cells with a wide range of phenotypes, including therapeutic resistance, causing treatments like chemotherapy to have a low efficacy. Monoclonal antibodies provide an intelligent solution to the diversity of cancer cell phenotypes because, the more variance that a single cancer cell has compared to the host, the higher the efficacy of the antibodies against the cell.
Figure 6.

Monoclonal and bispecific antibodies for targeted treatment
Created with BioRender.com.
Immune checkpoints with suppressive characteristics, or negative checkpoints, alter the normal function of the immune system and can serve as a mechanism of protection for cancer cells. The addition of monoclonal antibodies to the tumor environment can reverse these effects by either inhibiting a negative immune checkpoint or stimulating positive immune checkpoints to strengthen the body’s immune response to cancerous tissues (Marhelava et al., 2019). It should be noted, however, that monoclonal antibody therapeutics are still subject to resistance because of existing mutations in the tumor cells or immunoediting of the tumor in response to treatment (Cheson, 2006; Kimiz- Gebologlu et al., 2018).
Bispecific antibodies, shown in Figure 6, are characterized by having binding sites directed to different targets and are thus capable of binding to two antigens or two epitopes of the same antigen simultaneously, presenting improved efficacies compared to mAbs. Bispecific T cell engagers (BiTE) are a specific type of bispecific single chain antibodies, in which the antibody is engineered to have both an arm that binds to a molecule of an immune cell (like CD3, present on T-cells, or CD16 on natural killer cells) and an arm that binds to a tumor-specific antigen in cancer cells, in such a way that the binding of immune cells to cancer cells is achieved, resulting in an immune response. BiTEs have short half-lives, requiring constant administration to maintain the response, and similar toxicity profiles than CAR-T cell therapy (Topp et al., 2011; Minnie and Hill, 2020; Shah and Mailankody, 2020; Yang et al., 2020).
Beyond bispecific antibodies, multispecific antibodies, which are engineered antibodies that combine regions of two or more antibodies to improve binding to tumor cell antigens, activate immune cells, or bring different cell types in proximity with each other, have also been employed (Vago and Gojo, 2020).
Antibodies have promise for use as therapeutics for hematological cancers and have been explored in numerous studies described in the next section. Some advantages that led to their widespread use include high reproducibility, predictable patient response, and many potential targets. However, they have short half-lives in vivo and require high doses that often lead to toxicity and harmful side effects. The fragmentation and multi-functionalization of antibodies and their vehiculization by drug delivery systems are being the main strategies used in improving the outcomes of this immunotherapy approach because they can enhance tumor penetration and have the potential to remedy the aforementioned issues. The use of multispecific antibodies in combination therapies with checkpoint inhibitors, vaccines and/or OVs can overcome the immunosuppressive features of the tumor microenvironment.
Monoclonal antibodies for hematological cancers
Monoclonal antibodies have been utilized for treatment of hematological cancers in many cases. For AML, there have been numerous antigens explored for monoclonal antibody targeting including CD25, CD27/CD70, CD33, CD38, CD44, CD45, CD47, CD123, FLT3, CD135, CD157, CXCR4 (CD184), and CLEC12A, which are reviewed in reference (Morsink and Walter, 2019). In particular, gemtuzumab ozogamicin, a humanized IgG4 CD33 antibody conjugated to a toxic calicheamicin moiety, has led to better survival outcomes for some patients with AML (Godwin et al., 2017), and reduced the risk of relapse in children and adolescents with AML (Gamis et al., 2014). For patients with ALL, treatment with inotuzumab ozogamicin, an anti-CD22 antibody conjugated to calicheamicin, led to more complete remissions than standard chemotherapy treatment (Kantarjian et al., 2012).
Moreover, monoclonal antibody treatment has been used for treatment of CLL as well, and CLL lymphocytes have been associated with antigens expressed on B cells (CD19, CD20, CD79a), CD23, and CD5. Rituximab, a chimeric monoclonal CD20 antibody, has been used for treatment of CLL, as CD20 is expressed on normal and malignant B cells. Rituximab is considered a type I mAb because it causes cytotoxic effects through a direct apoptotic effect, complement-dependent cytotoxicity, and antibody-dependent cellular cytotoxicity (Cheson, 2006). In addition, alemtuzumab, a CD52 antibody, has been used to treat CLL. CD52 is a small glycoprotein that is expressed on almost all lymphocytes, monocytes, macrophages, and eosinophils in relatively high levels on cells from patients with CLL, NHL, and some ALL. Furthermore, cirmutzumab, a humanized monoclonal ROR1 antibody, has been used for CLL treatment, as ROR1 is expressed on neoplastic cells in patients with CLL and is associated with early relapse after therapy or metastasis (Choi et al., 2018).
The CD20 antigen is commonly found to be overproduced in NHL patients as well and is located on all mature B-cells. In fact, rituximab was the first mAb approved for B-cell NHL treatment. Obinutuzumab, a type II mAb that targets CD20, showed promising results in a phase II trial consisting of patients with B-cell NHL. When compared with Rituximab, obinutuzumab demonstrated higher efficacy because of improved antibody-dependent cellular cytotoxicity and direct apoptosis. Obinutuzumab also displays a sufficient safety profile (Morschhauser et al., 2013). Type II mAbs causes cell death through direct apoptotic effect and antibody-dependent cellular cytotoxicity and are advantageous over type I mAbs because of the increase in antibody-dependent cellular cytotoxicity from the lack of complement-resistance factors, increased stability of anti- CD20 mAb complexes, increased binding affinity, and decreased exhaustion of complement proteins (Suresh et al., 2014).
In response to patients that exhibit rituximab resistance, a novel fully human anti-CD20 monoclonal antibody has been developed, called ofatumumab, for use in relapsed or refractory follicular lymphoma. The results of a phase I/II trial showed rapid and significant B-cell depletion, with no significant safety concerns or maximum tolerated dosage limit. When compared with current treatments such as rituximab, ofatumumab displays increased potency and prolonged efficacy (Hagenbeek et al., 2008).
The CD22 antigen is expressed on all B-cells including many malignant B-lymphocytes and functions as an inhibitory co-receptor for the B-cell receptor. Thus, many mAbs target CD22 to effectively target B cells in B-cell lymphoma (Schweizer et al., 2012). Epratuzumab is a human anti-CD22 mAb that is being used to treat aggressive NHL. In a phase I/II trial, Epratuzumab demonstrated a decrease in circulating B cells as well as a 43% response rate in follicular NHL patients (Leonard et al., 2003).
Finally, elotuzumab, a humanized recombinant IgG1 (immunoglobulin G1) mAb that targets the signaling lymphocytic activation molecule F7 (SLAMF7), and daratumumab and isatuximab, humanized IgG1 mAbs that are directed to the transmembrane glycoprotein CD38 antigen, have been reported for the treatment of multiple myeloma. It should be noted that both SLAMF7 and CD38 proteins are highly expressed in multiple myeloma cells and normal plasma cells (Gormley et al., 2017; van de Donk and Usmani, 2018; Minnie and Hill, 2020; Shah and Mailankody, 2020).
Although the activity of elotuzumab is limited, their combination with dexamethasone and lenalidomide or pomalidomide for relapsed and refractory multiple myeloma (RRMM) received the FDA approval in 2015 and 2018, respectively. Also, there is a phase III clinical trial in progress that involve the use of elotuzumab in combination with pomalidomide, dexamethasone and anti-PD1 (nivolumab) in patients with RRMM (Gormley et al., 2017; Lonial et al., 2017; Minnie and Hill, 2020; Shah and Mailankody, 2020; Clinical Trials, 2022).
Likewise, daratumumab, after its first FDA approval in 2015 as monotherapy for patients with three previous treatments, has shown significant progressionfree survivals and high overall response rates in combination with dexamethasone and bortezomib or lenalidomide or pomalidomide or carfilzomib in RRMM patients in several phase III clinical trials that have evolved to the approval of FDA. In the event of newly diagnosed cases, there are phase III clinical trials of daratumumab in combination with dexamethasone and lenalidomide or melphalan, bortezomib and prednisone for transplant ineligible patients and in combination with dexamethasone, thalidomide and bortezomib for transplant eligible patients that also have progressed to the FDA approval. Also, for newly diagnosed cases, an ongoing phase II clinical trial is using four cycles of daratumumab with dexamethasone, lenalidomide and bortezomib, an ASCT and two additional cycles of this combination, with promising results. Recently, in 2020, based on a phase III clinical trial, a new formulation of daratumumab with hyaluronidase-fihj has been approved for subcutaneous administration. In addition, because preclinical models from solid tumors suggest that daratumumab and anti-PD-1 therapy could provide a synergistic response, there are some ongoing clinical trials of daratumumab in combination with several anti-PD1 drugs, like nivolumab, atezolizumab or cetrelimab in patients with RRMM (Dima et al., 2020; Minnie and Hill, 2020; Shah and Mailankody, 2020; Usmani et al., 2021; Clinical Trials, 2022).
Based on two phase III clinical trials, the FDA has recently approved isatuximab in combination with dexamethasone and pomalidomide/carfilzomib in 2020 and 2021, respectively, for patients with RRMM that have previously received specific treatments (Attal et al., 2019; Shah and Mailankody, 2020; Frampton, 2021; Moreau et al., 2021; Clinical Trials, 2022).
In addition, it has been shown in preclinical trials that the use of anti-IL-17A antibodies after ASCT prolonged disease control, so a phase I clinical trial using an anti-IL-17A (CJM112) with an anti-PD-1 (PDR001) is in progress in patients with RRMM, with no reported results up to date (Minnie and Hill, 2020; Clinical Trials, 2022). Also, new monoclonal antibodies against CD38 have appeared: TAK-079, administered subcutaneously as a single treatment in small volumes, and MOR202, used in combination with dexamethasone and immunomodulatory drugs, and, for the first time, a monoclonal antibody against BCMA, SEA-BCMA, is being studied in a phase I clinical trial (Abdallah et al., 2019; Shah and Mailankody, 2020; Clinical Trials, 2022).
All the approved regimens and the main clinical trials that use monoclonal antibodies for the treatment of multiple myeloma are summarized in Table 4.
Table 4.
Approved therapies and main clinical trials with monoclonal antibodies (mAb) in multiple myeloma
| mAb | Target | Conventional regimens | Target patients | Approved/Clinical trials |
|---|---|---|---|---|
| Elotuzumab | SLAMF7 | ELO-DEX-LD | RRMM (1–3 prior therapies) | FDA-approved (2015) |
| ELO-DEX-POM | RRMM (≥2 prior therapies) | FDA-approved (2018) | ||
| ELO-POM-DEX-anti-PD1 (nivolumab) | RRMM (≥2 prior therapies) | Phase III clinical trial (NCT02726581) | ||
| Daratumumab | CD38 | Monotherapy | RRMM (≥3 prior treatments, including a PI and an IMiD; refractory to a PI & an IMiD) | FDA-approved (2015) |
| DARA-DEX-BTZ | RRMM (≥1 prior therapy) | FDA-approved (2016) | ||
| DARA-DEX-CFZ | RRMM (1–3 prior therapies) | FDA-approved (2020) | ||
| DARA-DEX-LD | RRMM (≥1 prior therapy) | FDA-approved (2016) | ||
| DARA-DEX-LD | Newly diagnosed cases (ASCT ineligible) | FDA-approved (2019) | ||
| DARA-DEX-POM | RRMM (≥2 prior treatments, including LD and a PI) | FDA-approved (2017) | ||
| DARA-MEL-BTZ-PRD | Newly diagnosed cases (ASCT ineligible) | FDA-approved (2018) | ||
| DARA-DEX-THAL-BTZ | Newly diagnosed cases (ASCT eligible) | FDA-approved (2019) | ||
| DARA-DEX-LD-BTZ | Newly diagnosed cases (ASCT eligible) | Phase II clinical trial (NCT02874742) | ||
| Subcutaneous formulation of DARA with hyaluronidase-fihj | Newly diagnosed or RRMM | FDA-approved (2020–21) | ||
| DARA in combination with anti- PD1 drugs (nivolumab) | RRMM | Phase I/II clinical trial (NCT01592370) | ||
| DARA in combination with anti- PD1 drugs (atezolizumab) and IMiDs | RRMM or post-ASCT | Phase I clinical trial (NCT02431208) | ||
| DARA in combination with anti- PD1 drugs (cetrelimab) | RRMM | Phase II/III clinical trial (NCT03357952) | ||
| Isatuximab | CD38 | IXZ-DEX-POM | RRMM (≥2 prior therapies, including LD and a PI) | FDA-approved (2020) |
| IXZ-DEX-CFZ | RRMM (1–3 prior therapies) | FDA-approved (2021) | ||
| Anti-IL-17A (CJM112) | IL-17A | CJM112 in combination with an anti-PD-1 (PDR001) | RRMM | Phase I clinical trial (NCT03111992) |
| TAK-079 (subcutaneous) | CD38 | Monotherapy | RRMM | Phase I/II clinical trial (NCT03439280) |
| TAK079-LD-DEX or TAK079-BTZ-LD-DEX | Newly diagnosed cases with not-planned ASCT | Phase I clinical trial (NCT03984097) | ||
| MOR202 | CD38 | Monotherapy or MOR202-DEX or MOR202-DEX-LD/POM | RRMM | Phase I/II clinical trial (NCT01421186) |
| SEA-BCMA | BCMA | Monotherapy or SEA-BCMA-DEX or SEA-BCMA-DEX-POM | RRMM | Phase I clinical trial (NCT03582033) |
PI, Proteasome inhibitor; BTZ, Bortezomib; CFZ, Carfilzomib; IXZ, ixazomib; DEX, Dexamethasone; MEL, melphalan; CP, cyclophosphamide; IMiD, immunomodulatory drugs; THAL, thalidomide; LD, lenalidomide; POM, pomalidomide; PRD, prednisone; mAb, monoclonal antibodies; DARA, daratumumab; ELO, elotuzumab; ISA, isatuximab; ASCT, autologous stem cell transplantation; RRMM, relapsed/refractory multiple myeloma.
Bispecific antibodies for hematological cancers
Blinatumomab is being extensively studied as a BiTE (Topp et al., 2011). With regard to leukemia, blinatumomab has been shown to improve survival times in a phase III trial in patients with ALL (Kantarjian et al., 2017) and, in another study, it allowed more patients to reach a minimal residual disease response (Gökbuget et al., 2018). It has also had success for lymphomas. In a phase I trial, blinatumomab was shown to achieve an objective response rate of 82% at the maximum tolerated dose in relapsed/refractory B-Cell NHL patients. In diffuse large B-cell lymphoma patients, 56% demonstrated a response to blinatumomab (Topp et al., 2011; Suresh et al., 2014). Other studies are being done to find other promising bispecific antibodies for leukemia and lymphoma, and the CD19-specific bispecific antibody (BC250) was found more effective than blinatumomab against ALL xenografts in vivo (Hoseini et al., 2020).
BiTEs are the most common type of bispecific antibodies being used in clinical trials for multiple myeloma as well. The novel BiTE AMG-420, which targets BCMA on cancer cells and CD3 on the T cells, was used in heavily pretreated patients in a phase I trial, showing a response rate of 70% and severe side effects in almost half of the patients (Topp et al., 2018; Clinical Trials, 2022). AMG-420 has been modified to have a longer half-life, resulting in the AMG-701, that is currently under a phase I clinical trial as monotherapy and in combination with pomalidomide and dexamethasone (Cho et al., 2019).
Other BiTEs developed so far that are directed against BCMA and CD3 are analyzed here. CC-93269 has shown promising response rates of 88.9% in early-phase clinical trials in heavily pretreated patients with RRMM (Costa et al., 2019; Clinical Trials, 2022). Furthermore, JNJ-64007957 (teclistamab), is being used in an ongoing phase I clinical trial and achieved response rates of 67% at the maximum dose in patients with RRMM (Usmani et al., 2020; Clinical Trials, 2022). REGN5458 is also under phase I/II clinical trials in patients with RRMM with response rates of 60% at the highest dose, showing the 81.3% of the patients an excellent partial response (Madduri et al., 2020; Clinical Trials, 2022). TNB-383B is another one that is well tolerated in patients with RRMM and shows response rates of 79% in an open phase I clinical trial (Kumar et al., 2021; Clinical Trials, 2022). PF-06863135 (elranatamab) is also being studied in a phase I clinical trial as a monotherapy administered intravenously or subcutaneously or accompanied with immunomodulatory agents in patients with RRMM (Raje et al., 2019; Bahlis et al., 2021; Clinical Trials, 2022).
Even though BCMA has been the most used target for bispecific antibodies, scientists are reporting other binding sites such as CD38. Within this group, AMG-424 that targets CD38 and CD3 has been evaluated in a phase I trial for patients with RRMM, although the trial has been terminated recently by decision of the sponsor company (Zuch de Zafra et al., 2019; Clinical Trials, 2022). In this regard, other targets were recently reported for bispecific antibodies, such as FcRH5 (Fc receptor-homolog 5), towardwhich the BiTE BFCR4350A (cevostamab), that is being studied in a phase I trial, is directed (Li et al., 2017; Clinical Trials, 2022), and GPRC5D (G protein-coupled receptor class C group 5 member D) that is the target for the BiTE JNJ-64407564 (talquetamab) that is under ongoing clinical trials in RRMM patients as monotherapy (phase I/II) or in combination with daratumumab (phase 1b) (Chari et al., 2021; Krishnan et al., 2021; Clinical Trials, 2022).
The main clinical trials that include bispecific antibodies in the treatment of multiple myeloma are summarized in Table 5.
Table 5.
Main clinical trials with bispecific antibodies in multiple myeloma
| Name | Targets (cancer cells/T cells) | Conventional regimens | Target patients | Approved/clinical trials |
|---|---|---|---|---|
| AMG-420 | BCMA/CD3 | Monotherapy | RRMM (≥2 prior therapies, including a PI and an IMiD) | Phase I clinical trial (NCT02514239) |
| AMG-701 | BCMA/CD3 | Monotherapy | RRMM (≥3 prior therapies, including a PI, an IMiD & an anti-CD38 mAb; or refractory to PI, IMiD & anti-CD38 mAb) | Phase I clinical trial (NCT03287908) |
| AMG701-POM-DEX | RRMM (≥2 prior therapies, including a PI, LD &a anti CD38-mAb) | |||
| CC-93269 | BCMA/CD3 | Monotherapy | Heavily pretreated RRMM patients | Phase I clinical trial (NCT03486067) |
| JNJ-64007957 (teclistamab) | BCMA/CD3 | Monotherapy | RRMM (with prior therapies that include a PI, an IMiD & an anti-CD38-mAb) | Phase I clinical trial (NCT03145181) |
| REGN5458 | BCMA/CD3 | Monotherapy | RRMM (≥3 prior therapies, including PI, IMiD & anti-CD38 mAb; or refractory to PI & IMiD after anti-CD38 mAb; phase II: refractory to PI, IMiD & anti-CD38 mAb) | Phase I/II clinical trials (NCT03761108) |
| TNB-383B | BCMA/CD3 | Monotherapy | RRMM (≥3 prior therapies, including PI, IMiD & anti-CD38 mAb) | Phase I clinical trial (NCT03933735) |
| PF-06863135 (elranatamab) | BCMA/CD3 | Monotherapy or PF06863135-DEX or PF06863135-LD or PF06863135-POM | RRMM (with prior therapies including PI, IMiD & anti-CD38 mAb) | Phase I clinical trial (NCT03269136) |
| AMG-424 | CD38/CD3 | Monotherapy | RRMM (≥2 prior therapies, including a PI, IMiD & an anti-CD38-mAb; or refractory to PI, IMiD & anti-CD38 mAb) | Phase I clinical trial (terminated by sponsor) (NCT03445663) |
| BFCR4350A (cevostamab) | FcRH5/CD3 | Monotherapy | RRMM (with no therapy available) | Phase I clinical trial (NCT03275103) |
| JNJ-64407564 (talquetamab) | GPRC5D/CD3 | Monotherapy | RRMM or intolerant to standard therapies | Phase I clinical trial (NCT03399799) Phase II clinical trial (NCT04634552) |
| JNJ64407564-DARA | RRMM (≥3 prior therapies, including a PI & IMiD; or refractory to PI & IMiD) | Phase 1b clinical trial (NCT04108195) |
PI, Proteasome inhibitors; IMiD, immunomodulatory drugs; LD, lenalidomide; POM, pomalidomide; mAb, monoclonal antibodies; DARA, daratumumab; RRMM, relapsed/refractory multiple myeloma; DEX, Dexamethasone.
Cell-based therapies
General observations
In cell-based therapies, immune cells are typically removed from the patient and trained to recognize and destroy the tumor genetic material before being reintroduced into the patient. This is a rapidly growing field of research with high potential for more effective methods of treating cancer. Cellular immunological agent sipuleucel T has been FDA approved for treatment of prostate cancer (Kantoff et al., 2010). Moreover, five drugs based on the CAR (chimeric antigen receptor) T cell therapy for the treatment of hematological cancers were approved between 2017 and 2021. Lisocabtagene maraleucel (Breyanzi) and idecabtagene vicleucel (Abecma) were the latest CAR T therapies to be approved, for the treatment of late-stage lymphoma and multiple myeloma, respectively (Berraondo et al., 2019; Hong and Dobrovolskaia, 2019; Raman et al., 2019; Albinger et al., 2021; U.S. Food and Drug Administration (FDA), 2021a, 2021b).
CAR T cell therapy consists of extracting and genetically modifying patient’s T cells to produce CARs on their surface (Miliotou and Papadopoulou, 2018). The CARs are specific for a tumor antigen and allow the T cells to target, recognize, and attach to the antigen. The CAR T-cells then undergo ex vivo expansion and are infused into the patient to allow for their multiplication and subsequent killing of tumor cells with the specified antigen.
Thus, CAR T-cells utilize both the functionalities of monoclonal antibodies as well as T cells, expressing antigen-binding properties, the ability to lyse cells, and self-renewal. Traditional T cells are subjected to restrictions from the MHC and can therefore have limited response to an antigen unless it is bound to a specific MHC molecule; cytotoxic T cells are MHC-restricted to class I antigens. Thus, if MHC-I expression is not present, it allows tumors to evade recognition by T cells (Garrido et al., 2016). However, CAR T-cells function independently of the MHC and so their tumor-recognition capabilities are not MHC-restricted (Dotti et al., 2014).
CAR T cell therapies have shown promising results in the treatment of many malignancies. Some recent ongoing trials include ZUMA (from Kite) which use CD19− (CD28z) CAR T-cells, TRANSCEND studies (from Juno) and JULIET studies (from Novartis) which both use CD19-(4-1BBz) CARs and have produced promising results. Results of trials have varied based on CAR design, dosage, lymphodepleting strategies and patient tumor burden (Strati et al., 2018). However, CAR T cell therapies are associated with disadvantages, including producing severe off-target side effects and toxicity derived from the cytokine release syndrome (CRS) that is reported after the infusion of the modified T cells. Moreover, many patients face relapse events because of the development of tumor resistance to CAR T-cells, which reduces its targeting capacity because it significantly decreases the antigen expression after T cell administration (antigen escape). There are also associated difficulties in targeting selectively solid tumor antigens that are also expressed in normal cells (on-target off-tumor toxicity), and in entering the tumor tissue because of the immunosuppressive tumor microenvironment and the tumor stroma barrier, making the application of these therapies in solid tumors difficult (Hong and Dobrovolskaia, 2019; Siegler and Kenderian, 2020; Wei et al., 2020; Sterner and Sterner, 2021). Given these disadvantages, there are numerous opportunities for engineers to improve these cell-based therapeutics.
Within cell therapies, natural killer (NK) cells are also beginning to be engineered through the CAR technology (CAR NK-cells). NK cells can kill cells without prior need for antigen priming, with a natural ability to kill abnormal cells based on signals from germline-encoded receptors. Therefore, once NK cells are programmed with specific CARs, they can be triggered by their innate germline receptors with antigen recognition redirected toward CAR-specific targets, which allows them to maintain cytotoxicity against malignant cells even if the target antigen is lost or downregulated. This feature imbues them with greater anti-tumor activities compared to CAR T-cells as they can be activated by a variety of mechanisms (Daher et al., 2021). The use of NK CAR-cells as a new treatment offers other important advantages over CAR T-cells, such as better safety profiles, short-term toxicities as they are short-lived cells, and fewer side effects because they diminish the CRS and the graft-versus-host disease. In addition, their ability to both directly kill the affected cells and to secrete cytokines and chemokines that activate other components of the immune system, stands out (Kriegsmann et al., 2019; Basar et al., 2020; Xie et al., 2020).
Stem cell transplantation is another type of cell-based immunotherapy that can be used to treat cancer (Cieri et al., 2021). Stem cell transplants replenish blood forming stem cells that are lost after chemo- or radiotherapy, allowing for higher doses of treatment to be given to patients. The stem cells can be autologous, allogenic, or syngeneic. In some cases, stem cell transplants can also work against cancer directly in a graft-vs-tumor response. However, sometimes allogenic transplants can cause a graft-vs-host response which causes the patient to reject the transplant. Hematopoietic stem cell transplantation is a widely used approach for treatment of hematological cancers.
Overall, cell-based therapies are commonly associated with developed resistance and CRS because of immune responses to the therapies. They also often have difficulty in selective targeting. Engineering cell-based therapies to be more selective could potentially improve outcomes.
Cell-based therapies in hematological cancers
In recent years, cell-based therapies have been further developed as a therapy for hematological cancers because of their potential, and CAR T cells have generated a lot of interest. The first CAR T cell-based therapies that were approved by the FDA were for treatment of ALL affecting children and advanced lymphoma affecting adults in 2017 (Dotti et al., 2014; Hoffmann et al., 2018; Rafei et al., 2019). In the case of ALL, the two FDA-approved therapies, brexucabtagene autoleucel and tisagenlecleucel, are both CD19-directed T cell immunotherapies. Tisagenlecleucel is also approved for use in treating diffuse large B cell NHL, in addition to axicabtagene ciloleucel (axi-cel). Another CD-19 targeting CAR T cell product, liso-cel, is also receiving promising results in clinical trials for NHL (Chavez et al., 2019). Their successes have led to further investigation into cell-based therapies for leukemias that do not express CD19 (Chavez et al., 2019). Currently, researchers are investigating CD7, CD13, and CD70-specific CAR T-cells for AML (Gomes-Silva et al., 2019; He et al., 2020; Sauer et al., 2021).
In myeloma patients, the most studied target antigen to make these modified T cells is the BCMA, which is expressed to a greater extent on malignant plasma cells than on healthy ones. In fact, numerous BCMA-targeted CAR T-cells have entered in clinical trials, some of them showing very good results in patients with multiple myeloma with an average ORR of 80.5%, demonstrating the 44.8% of the patients a complete response (CR) (Teoh and Chng, 2021). Thus, FDA has recently approved (March 2021) idecabtagene vicleucel (ide-cel, bb2121, Abecma), a BCMA-targeted CAR-T cell therapy that have shown an ORR of 73% in RRMM patients. This is the first FDA-approved CAR T cell therapy for multiple myeloma (Munshi et al., 2021; U.S. Food and Drug Administration (FDA), 2021a). In this regard, it is worth highlighting the new CAR T cell approach ciltacabtagene autoleucel (cilta-cel/JNJ-68284528) that contains two BCMA-targeting domains and is being evaluated in a phase 1b/2 clinical trial in RRMM patients, finding a remarkable ORR of 97% with the 67% of the patients getting a stringent complete response (sCR) (Berdeja et al., 2021; Clinical Trials, 2022). In addition, there are some CAR T-cells designs, which target other multiple myeloma antigens, such as CD19, CD38, CD138, SLAMF7 (signaling lymphocytic activation molecule F7) and GPRC5D, that are in the early stages of testing with very good results in preclinical studies (Teoh and Chng, 2021; van de Donk et al., 2021b).
Numerous efforts are being employed to improve the design of the CAR T-cells to enhance the effectiveness of this therapy. It is worth noting the bispecific CAR T-cells constructs that target two multiple myeloma-specific antigens to avoid antigen escape. In fact, Zah et al. reported CAR T-cells that can target BCMA and SLAMF7 (also called CS1), simultaneously, resulting in an enhanced CAR expression and an increased efficacy to control the disease compared to the constructs that target BCMA or CS1 separately, which prolongs the survival of multiple myeloma-bearing mice (Zah et al., 2020). Thus, the safety, tolerability and efficacy of BCMA/CS1 CAR T cells are being evaluated in phase I/early phase I clinical trials with RRMM patients (Clinical Trials, 2022). Similarly, bispecific CAR-T cells that target BCMA and CD38 have been evaluated recently for the first time in RRMM patients in a Chinese phase I clinical trial, resulting in a high proportion of patients with a significant clinical response (87%) and, more importantly, achieving a sCR in the 52% of the patients (Li et al., 2019; Mei et al., 2021). Likewise, another BCMA-CD38 CAR T-cells construct is being evaluated in RRMM in one more Chinese phase I clinical trial, showing an ORR of 87.5% and a sCR in the 81.3% of the patients, although these promising results are only exploratory because of the small sample size of patients and the non-randomized type of the trial (Tang et al., 2022). Also, a phase I/II clinical trial is being conducted to evaluate the safety and feasibility of BCMA-CD38 CAR T-cells for RRMM (Clinical Trials, 2022).
The use of CAR NK-cells is being analyzed in several studies for hematological cancers. CAR NK-cells that use CS1 as a therapeutic target for treatment of multiple myeloma led to an increased IFN-γ secretion, a specific CS1 mediated cancer cell recognition, and an increased multiple myeloma cells cytolysis. In a multiple myeloma xenograft mouse model, it was found that the use of these CS1-targeted CAR NK-cells inhibited tumor growth (Chu et al., 2014). Moreover, one study used CAR technology to direct NK cells against the CD138 multiple myeloma antigen, being able to induce much greater toxicity in CD138-positive multiple myeloma cells than with the unmodified NK cells accompanied by a high IFN-γ and granzyme B secretion (Jiang et al., 2014). More recently, second generation CAR NK-cells have been reported to enhance their outcomes in the treatment of multiple myeloma. Martín et al. reported the modification of NK-92MI cells to target the conventional multiple myeloma BCMA antigen or the NKG2D one, which is expressed in numerous multiple myeloma cell lines, including resistant ones, showing in both cases much higher oncolytic activity than the respective unmodified NK cells. It is noteworthy that NKG2D-CAR NK-cells and BCMA-CAR NK- cells showed similar toxicities against multiple myeloma cells (Martín et al., 2019). NKG2D-CAR NK-cells have also seen promising results that include greater toxicity against multiple myeloma cells than CD45RA− CAR T-cells expressing NKG2D, increased expression of genes involved in immune processes, and the suppression of tumor growth in a mouse model (Leivas et al., 2021). CD-19 targeting CAR-engineered NK-92 cells also have shown potential for treatment of leukemia and lymphoma (Oelsner et al., 2017). There is currently one clinical trial evaluating the potential of CAR NK-cells, a phase I/II trial where patients with RRMM are treated with BCMA-targeted CAR-NK 92 cells (Clinical Trials, 2022).
Stem cell transplants also have a long history of use in hematological cancers. In the case of multiple myeloma and leukemia, stem cell transplants can work against cancer directly through a graft-versus-tumor response, in addition to the replenishment of stem cells. It has been shown to promote T cell action, alter the microenvironment and establish a balance between myeloma and the immune system in patients who accomplish a long-term control of their disease (Minnie and Hill, 2020). E. Donnall Thomas was awarded the Nobel Prize for his work in developing allogeneic bone marrow transplantations, containing allogeneic hematopoietic stem cells, using HLA-matched donors for patients with acute leukemia (Cieri et al., 2021). Donor lymphocyte infusions were also implemented in 1990 to leukemia patients who relapsed after stem cell transplant (Roddie and Peggs, 2011). Hematological cancers have shown different response rates to donor lymphocyte infusions, with CML showing the highest responses (up to 100%) (Roddie and Peggs, 2011) and acute myeloid and lymphoblastic leukemia showing the lowest responses (10–20%) (Porter et al., 2000; Schmid et al., 2007). Lymphomas and myelomas respond at a rate of approximately 30% (Lokhorst et al., 2004; Peggs et al., 2011).
Oncolytic viruses
Characteristics
There are a series of mutations in the signaling pathways of cancer cells that make them more sensitive to viral infections, as well as an overexpression of molecules on the surface that the viruses can use as receptors to gain access to the intracellular space (Meyers et al., 2017). Thus, OVs have more recently gained credibility among immunotherapy strategies for treating cancer. OVs used in cancer immunotherapy have been engineered from naturally occurring OVs to enhance their therapeutic ability and present a powerful strategy for targeting cancers because of their ability to differentiate normal, healthy cells from cancer cells and kill the cancer cells while leaving normal cells unharmed (Rahman and McFadden, 2021).
OVs target cancer cells specifically and cause immune system activation, converting the “cold” tumor environment with little immunological activity to a “hot” one (Hemminki et al., 2020). The mechanisms through which immune activation can occur are shown in Figure 7. Briefly, after entering the cell, OVs replicate which causes cellular rupture, leading to release of tumor antigens in addition to PAMPs and DAMPs. These antigens activate nearby dendritic cells. Mature dendritic cells then present these antigens to distant T cells, and T cells begin to be attracted to the tumor site. B cells are then activated by CD4+T cells, and release neutralizing antibodies that mark tumor cells for attack by NK cells or phagocytosis by M1 macrophages. Finally, CD8+T cells and NK cells kill the tumor cells (Hemminki et al., 2020). However, this activation of the immune system also determines the efficacy of the virus as a therapy because the OV itself could also be recognized by the immune system and be destroyed. OVs can survive successfully after entering the cells given that many of the signaling pathways used in viral clearance are weakened in cancer cells. It is also important to note that OVs are typically engineered to reduce or eliminate their pathogenic effects (Meyers et al., 2017; Shi et al., 2020; Rahman and McFadden, 2021).
Figure 7.

Summary of immune activation following exposure to OV
(A) Danger signal release and DC maturation. Oncolytic adenoviruses infect tumor cells and cause oncolysis, releasing new virus progeny but also DAMPS and PAMPS, which will activate nearby dendritic cells and foster their maturation by upregulating co-stimulatory markers, such as CD80, CD83, and CD86.
(B Mature dendritic cells will process tumor debris and present tumor-associated and virus antigens to local and distant T cells. Concurrently, the ongoing virus infection attracts T cells to the tumor site.
(C) The activation of B cells by CD4+T cells or BCR-virus interaction causes the release of neutralizing antibodies, which mark infected tumor cells for ADCC by NK cells, or phagocytosis by M1 macrophages.
(D) CD8+T cells and NK cells destroy infected and non-infected tumor cells through INFg/GranzB and GranzB/Perforins, respectively. The oncolytic adenovirus infection also upregulates class I HLA in tumor cells, allowing for increased exposure to CD8+T cells. Overall, the components of this modulation allow the tumor microenvironment to become “hot” with increased immunological activity. DAMP danger-associated molecular patterns, PAMP pathogen-associated molecular patterns, HLA human leukocyte antigen, BCR B cell receptor. Reprinted from reference (Hemminki et al., 2020) with license permission. (http://creativecommons.org/licenses/by/4.0/).
OVs have proved their potential throughout many clinical trials because they are frequently found to be safe and there is generally a high amount of tolerability to treatment using OVs (Hemminki et al., 2020). Because of this, current OV studies tend to focus on both improving their efficacy in current applications and exploring their relevancy in a broader range of applications. One method of increasing the efficacy of OVs that is currently being researched is the effects of adding immunomodulatory transgenes to the viruses. This would allow production of immunostimulatory molecules at the tumor site with reduced systemic side effects (Hemminki et al., 2020; Rahman and McFadden, 2021).
Adenovirus, a DNA-based OV, is the most widely studied. This triggers common influenza type symptoms while representing a significantly versatile platform for cancer therapy because of its ability to infect cells without regard for their division status. The most prevalent backbone for oncolytic virus development is serotype 5 (group c). The design of this backbone involves an icosahedral shaped capsid (made of hexon, penton and fiber proteins) which surrounds a bare double-stranded DNA. Serotype 5 binds preferentially to the coxsackie-and adenovirus receptor CXADR on the surface of tumor cells, after which the penton proteins in the virus interact with the tumor cell integrins (virus internalization). Then transcription of viral proteins begins and eventually disrupts the cell membrane (cell lysis) while infecting surrounding cells and triggers the immune system to disrupt the process (Hemminki et al., 2020).
The use of the measles virus (Edmonston B-strain) (MV-Ed), an RNA-base OV, has also been researched for several decades for its anti-tumor associations (Grote et al., 2001). The reasons for the popularity of this virus are as follows. First, a nonpathogenic strain of MV is available, well characterized, and safe (Norrby and Kristensson, 1997). In addition, in patients with an intact and highly functioning immune system, MV infection is restricted to limited cell types in vivo. In particular, lymphoid organs are popular points of MV replication (Anderson et al., 2004). Furthermore, in measuring the mechanism of specificity, researchers found the correlation of upregulated nectin-4 receptor levels with increasing anti-tumor activity of MV-Edm in human adenocarcinoma cells, suggesting the expression of the receptor in those levels is because of MV-mediated oncolysis (Fujiyuki et al., 2015; Awano et al., 2016).
Though OVs are newer to the immunotherapy field, there already exist three that have been approved globally for use in treating cancer patients (Rahman and McFadden, 2021). Rigvir (an ECHO-7 virus), an oncolytic picornavirus, is the first approved product of virotherapy (Latvia in 2004 and later in other countries) and has some innate tumor selectivity (Alberts et al., 2018). The second, H101 (Oncorine), is an adenovirus that was developed with tumor selectivity guidelines and has been in use in China since 2005 for solid tumors (Liang, 2018). The third type of approved oncolytic virus took arming components (immunological transgenes such as GMCSF) into account, which the first two previously lacked. This oncolytic virus, Talimogene laherparepvec (T-vec, Imlygic), was among the first to produce a boost in immunity and went through clinical trials with 80% complete response. This led to approval by the FDA in 2015, and subsequently the European Medicines Agency (EMA) (Andtbacka et al., 2019). Many more OVs are currently undergoing clinical trials to determine their efficacy in treating various cancers. To improve their efficacy, OVs can be used in combination with other chemotherapeutics and immunotherapies. Studies show it is likely that a combination of OVs and immune checkpoint inhibitors or immunomodulatory drugs could provide a powerful strategy for cancer therapy (Hemminki et al., 2020).
Undoubtedly, the unprecedented progress of genetic engineering is helping the scientific community to face the challenges of this approach and achieve better clinical outcomes. Nevertheless, because of the poor bioavailability of OVs when administered intravenously and their rapid clearance by the reticuloendothelial system, the main problem with OV nowadays is their delivery to the tumor site that is done by local and direct administration to be clinically relevant. In this regard, the role of the drug delivery systems in OVs is clear.
Oncolytic viruses in hematological cancers
Despite the advances that have been reported in the treatment of hematological cancers using OVs, which are described below, the singular features of liquid tumors make the application of this strategy in the treatment of this type of cancer a challenge It must be taken into account that hematological malignancies are systemic diseases, so the intravenous administration is the preferred delivery method for OVs, with drug delivery platforms being a really interesting strategy to consider.
Lymphoma is a developing area of focus for OVs. Currently, several studies have been done that support the potential of using viruses in the therapy of lymphoma. In this regard, reovirus has been studied as an antilymphoma agent with high therapeutic potential because they are able to infect and kill cells with an activated Ras-signaling pathway. These Ras-signaling pathways have been found to be present in transformed B lymphocytes, EBV-infected lymphoblasts, CLL lymphoid cells, and DLBCLs (Alain et al., 2002). In a study done by Alain et al., reovirus infection was demonstrated in vitro in human lymphoma cell lines, including Burkitt lymphoma EBV+, Burkitt lymphoma EBV− and diffuse large B-cell NHL cell lines. In susceptible cell lines, viability decreased 40%–70%. In the same study, the in vivo effects of reovirus were confirmed to follow the in vitro effects. In mice with subcutaneous lymphoma tumors, the virus was able to prevent tumor growth derived from susceptible cell lines. When reovirus was introduced to single-cell suspensions, it was found that reovirus proteins were present in lymphomas but not normal lymphocytes and hematopoietic stem cells. It was also determined that clonogenic progenitors were not significantly susceptible to the reovirus.
Besides, regarding Sindbis Virus (SV), which is a blood-borne alphavirus that is capable of leading to the apoptotic death of tumor cells infected with this OV and results in very few and mild symptoms in humans, Yu et al. found that in mouse models, a combination therapy consisting of SV vectors and α4-1BB mAb was able to achieve complete eradication of B-cell lymphoma. This combination therapy also resulted in enduring anti-tumor immunity in the mice (Yu et al., 2019).
Moreover, letogenic strains of Newcastle disease virus (NDV), an avian paramyxovirus which has been studied recently for its inherent oncolytic and immunostimulatory effects, have been used to explore the in vitro sensitivity of canine and human lymphoma cells, as well as the in vivo distribution of NDV in canine studies. It was seen that the virus treatment decreased human lymphoma cell viability by 52% whereas CHOP chemotherapy treatment (cyclophosphamide-hydroxydaunorubicin-oncovin-prednisone) decreased cell viability by 34%. Multi-parametric flow cytometry confirmed the significant oncolytic effect of NVD in both human and canine cell types. When observing the cytolytic effects of NDV on non-malignant B-cells and peripheral blood mononuclear cells, the studies showed that the healthy human cells were less sensitive than the malignant lymphocytes (Sánchez et al., 2015).
The use of OVs as a treatment for hematological cancers has aroused the interest of many researchers because it has shown to be capable of producing remission in different types of cancer, including multiple myeloma (Meyers et al., 2017; Stewart et al., 2021). In this particular case, current preclinical and clinical efforts are focused on RNA viruses (measles virus, vesicular stomatitis virus and reovirus) and DNA viruses (adenovirus, vaccinia virus and myxoma virus) (Cook et al., 2021).
The measles virus enters multiple myeloma cells, thanks mainly to the CD46, SLAM (signaling lymphocyte-activation molecule or CD150) and nectin-4 receptors, which are upregulated in malignant plasma cells, making them susceptible to infection by this virus. It must be considered that most of the population is vaccinated against this infectious agent, so some strategies are being investigated in preclinical studies to avoid the immune attack, such as surface protein engineering and immune suppression, among others (Meyers et al., 2017; Cook et al., 2021). In the early 2000s, some researchers (Dingli et al., 2004) generated a modified measles virus that expresses the human thyroidal sodium-iodide symporter (MV-NIS), allowing the noninvasive imaging of multiple myeloma by radioactive iodine (123I) and achieving a considerable tumor regression when used in conjunction with beta-emitting radioiodine (131I). The intravenous administration of MV-NIS, with or without cyclophosphamide, in RRMM patients has been reported in a phase I/II clinical trial, and although some grade III and IV toxicities were observed, it was achieved promising results, highlighting the fact that one patient achieved complete remission and others showed temporary reductions in the serum free light chains (Dispenzieri et al., 2017; Packiriswamy et al., 2020; Clinical Trials, 2022). This modified Edmonston strain of measles virus that targets CD46 in multiple myeloma was also applied in combination with cyclophosphamide in a completed phase II clinical trial in patients with RRMM (Cook et al., 2021; Clinical Trials, 2022).
The vesicular stomatitis virus replicates in the cell cytoplasm, so it does not integrate into the genome of the host (Meyers et al., 2017). Because of the specificity of this virus with defective interferon pathways, it has been genetically modified to express interferon-β and amplify the targeting capacity of the virus to the malignant cells. This RNA-based virus that was further modified to express NIS, permitting the in vivo imaging and attenuating its virulence, is under an ongoing phase I clinical trial with multiple myeloma, AML, and T cell lymphoma relapsed/refractory patients, with significant responses so far only in the T cell lymphoma patients (Cook et al., 2021; Cook et al., 2021; Clinical Trials, 2022).
Reovirus is capable of infecting and destroying multiple myeloma cells, observing apoptosis and autophagy processes in early in vitro studies. Recently, it has been discovered that the junctional adhesion molecule-A (JAM-A), which is overexpressed in RRMM patients, and mutated Ras/Raf signaling pathways are the “gates” that facilitate the infection by this RNA-based virus (Meyers et al., 2017; Solimando et al., 2018; Cook et al., 2021; Müller et al., 2021). Some authors demonstrated that reovirus was able to reduce the number of cancer cells as well as the associated bone disease in vivo in a multiple myeloma model. At the cellular level, they observed an increase in NK cells, CD8+T cells and effector memory CD8+T cells. In addition, to observe the effect of this virus in the tumor microenvironment, they cultured multiple myeloma cells with bone marrow stromal cells, observing that, whereas there was resistance to oncolysis, NK cells and T lymphocytes activated by the reovirus were able to kill multiple myeloma cells (Müller et al., 2021). A phase I clinical trial was conducted using Reolysin (pelareorep), a reovirus-based agent, with patients with RRMM who received a previous treatment with bortezomib and lenalidomide, resulting in no significant responses (Sborov et al., 2014). Also, the combination of Reolysin with lenalidomide or pomalidomide, bertezomib and dexamethasone, and carfilzomib and dexamethasone are being studied in other ongoing phase I clinical trials with RRMM patients (Clinical Trials, 2022).
Adenovirus 5 serotype (Ad5), the most common of this type of DNA-based virus, has not been used as a therapy in multiple myeloma because it is internalized by the coxsackie and adenovirus receptor that is overexpressed by the myeloma cells but is not present in the hematopoietic cells (Cook et al., 2021). Although Fernandes et al. reported a genetically modified adenovirus to transport a recombinant CD40 ligand (CD40L) with growth inhibitory activity to multiple myeloma cells because the systemic treatment with CD40L involves significant side effects as the CD40 receptor is also expressed on healthy cells. As a result, these authors found the modification of an adenovirus to contain CD40L inhibited cancer cell growth to a greater extent than the parental virus (Fernandes et al., 2009). Moreover, Tong et al. reported the modification of the adenoviral vector ZD55, an Ad5-based oncolytic virus, with TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) with a demonstrated efficacy in inducing cell apoptosis in multiple myeloma. These authors showed that ZD55 was more cytotoxic in myeloma cells when modified with TRAIL, in both drug-sensitive and drug-resistant cells. The sensibility of this virus to multiple myeloma cells or its apoptotic capacity was enhanced when combined with a PI3K (phosphoinisitide 3-kinase) inhibitor or a proteasome inhibitor, respectively (Tong et al., 2014). To increase the infection capacity of adenovirus to the majority of cell types, including hematopoietic cells, Wenthe et al. switched serotype 5 to 35 to target receptors other than the coxsackie and adenovirus receptor, like the CD46 receptor. Moreover, this chimeric adenovirus was modified to express the trimerized CD40L and 4-1BBL immunostimulatory transgenes that add to the oncolytic capacities of the virus the possibility of increasing the antitumoral immune responses. Different multiple myeloma cell lines were infected and destroyed when cultured with this immunomodulatory oncolytic virus, increasing the activation of cytotoxic T cells (Wenthe et al., 2020).
In vitro and in vivo assays have been performed to test the effects of the modification of vaccinia virus on multiple myeloma. In one of them, this DNA-based virus was modified to express miR-34a or Smac and tested for efficacy separately or in combination in multiple myeloma cell lines and tumor xenograft model in mice, resulting in tumor growth inhibition and apoptosis when used in combination (Lei et al., 2016). Similarly, Futami et al. reported a microRNA-regulated vaccinia virus with capacity to suppress the thymidine kinase gene, which permits to control its virulence and achieve a greater specificity against multiple myeloma cells. The combination of thymidine kinase deletion and microRNA regulation increases the overall survival and tumor regression in RPMI8226 tumor-bearing mice (Futami et al., 2017). Recently, Lei et al. evaluated the use of a modified vaccinia virus expressing Beclin-1, an autophagy-related protein, where thymidine kinase gene deletion was used to increase selectivity against cancer cells, resulting in a significant anti-tumor oncolytic efficacy and autophagic cell death in leukemia and multiple myeloma animal models (Lei et al., 2020). Therefore, the results obtained to date with respect to the different modifications introduced in vaccinia viruses are a promising starting point to continue studying the therapeutic possibilities of these OVs.
Myxoma virus has been found to be capable of infecting and producing apoptosis of all multiple myeloma cell lines in vitro, without infecting non-cancerous cells and producing apoptosis in the malignant cells without virus replication (Bartee et al., 2012; Meyers et al., 2017). In fact, Bartee et al. obtained promising results in this regard to be applied in the treatment of patients with a residual multiple myeloma because they achieved the elimination of 70–90% of cancer cells in 24 h in mice with disseminated multiple myeloma and significant CD8+ T cell-based immune responses (Bartee et al., 2016). Similarly, Villa et al. reported the use of the myxoma virus as a therapeutic agent to combat residual multiple myeloma, which could lead to patient relapse. Specifically, they showed the usefulness of autologous transplantation to deliver myxoma virus to the tumor, using leukocytes as carriers of this DNA-based virus. The authors achieved to eradicate the minimal residual disease in a multiple myeloma animal model and demonstrated that the treatment resulted in the secretion of immune-stimulating molecules into the tumor microenvironment, showing the cured mice immunity when re-exposed to fresh myeloma cells (Villa et al., 2020). Recently, Franco et al. equipped myxoma virus with interleukin-2 (IL-12), which activates T and NK cells, and decorin that induces intracellular signaling processes in the tumor cells, inhibition of the TFG-β pathway and tumor matrix regulation. The authors reported significant oncolytic effects and transgene expression in multiple myeloma cell lines, suggesting that this new approach can be applied in patients resistant to other immunotherapy strategies (Franco et al., 2021).
Conclusions and future perspectives
The complex pathogenesis, diagnosis, and current treatment processes for hematological cancers have been described. Immunotherapies, including checkpoint inhibitors, therapeutic vaccines, antibodies, cell-based therapies, and OVs, can be applied to hematological cancers for better outcomes for some patients. However, these therapies are only effective in specific subsets of patients. To improve outcomes, there are different techniques that can be explored and applied. Importantly, biomarkers to predict outcomes in patients with hematological cancers are needed. In this way, precision medicine will become an important avenue for immunotherapies, with the patient’s own markers dictating which therapy they should receive. Precision medicine will also become important as sex, ancestry, and age-based differences in therapeutic responses are studied. In addition, combination therapies have the potential to further increase the success of immunotherapy. For example, oncolytic virus therapy can be combined with immune checkpoint inhibitors and T cell-based therapies for improved responses (Rahman and McFadden, 2021).
In addition, many immunotherapies are associated with toxicity and adverse side effects. Drug delivery systems such as micro or nanoparticles that directly target cancer cells could also help to improve the outcomes of immunotherapies by increasing efficacy and reducing off-target side effects. Drug delivery systems can protect payloads from premature degradation and allow for controlled release over clinically relevant time periods to reduce required doses. Moreover, new theranostics could be developed which can predict outcome and release appropriate immunotherapy treatment simultaneously. Overall, immunotherapies for hematological cancers show great potential for treatment of hematological cancer and have led to remission in patients that otherwise would not have been possible.
Acknowledgments
NAP’s work was supported in part by the National Institutes of Health; the Office of the Dean of the Cockrell School of Engineering at The University of Texas at Austin for the Institute for Biomaterials, Drug Delivery, and Regenerative Medicine; and the UT-Portugal Collaborative Research Program. OL’s work was supported by the University of Texas at Austin Provost Early Career Postdoctoral Fellowship Program.
Author contributions
O.L.L. and E.P.H. contributed equally to this work and led the effort for writing, editing, and overseeing the manuscript. A.P.D., K.B., E.L., D.M.W., N.A.S,. and S.M.R.M. all worked on writing of various sections and creating figures for the manuscript. N.A.P. oversaw this work and revised the final manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Olivia L. Lanier, Email: olivia.lanier@austin.utexas.edu.
Edgar Pérez-Herrero, Email: eperezhe@ull.edu.es.
Nicholas A. Peppas, Email: peppas@che.utexas.edu.
References
- Abdallah A.-O.A., Hoffman J.E., Schroeder M.A., Jacquemont C., Li H., Wang Y., Van Epps H., Campbell M.S. SGNBCMA-001: a phase 1 study of SEA-BCMA, a non-fucosylated monoclonal antibody, in subjects with relapsed or refractory multiple myeloma. J. Clin. Oncol. 2019;37:TPS8054. doi: 10.1200/JCO.2019.37.15_suppl.TPS8054. [DOI] [Google Scholar]
- Advani R., Flinn I., Popplewell L., Forero A., Bartlett N.L., Ghosh N., Kline J., Roschewski M., LaCasce A., Collins G.P. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N. Engl. J. Med. 2018;379:1711–1721. doi: 10.1056/NEJMoa1807315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S.M., Svane I.M., Andersen M.H. The stimulation of PD-L1-specific cytotoxic T lymphocytes can both directly and indirectly enhance antileukemic immunity. Blood Cancer J. 2014;4:e230. doi: 10.1038/BCJ.2014.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alain T., Hirasawa K., Pon K.J., Nishikawa S.G., Urbanski S.J., Auer Y., Luider J., Martin A., Johnston R.N., Janowska-Wieczorek A., et al. Reovirus therapy of lymphoid malignancies. Blood. 2002;100:4146–4153. doi: 10.1182/BLOOD-2002-02-0503. [DOI] [PubMed] [Google Scholar]
- Alberts P., Tilgase A., Rasa A., Bandere K., Venskus D. The advent of oncolytic virotherapy in oncology: the Rigvir® story. Eur. J. Pharmacol. 2018;837:117–126. doi: 10.1016/J.EJPHAR.2018.08.042. [DOI] [PubMed] [Google Scholar]
- Albinger N., Hartmann J., Ullrich E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021;28:513–527. doi: 10.1038/s41434-021-00246-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison J.P. A brief history of immunotherapy. Target. Oncol. 2014;3 [Google Scholar]
- Anderson B.D., Nakamura T., Russell S.J., Peng K.-W. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64:4919–4926. doi: 10.1158/0008-5472.CAN-04-0884. [DOI] [PubMed] [Google Scholar]
- Andtbacka R.H.I., Collichio F., Harrington K.J., Middleton M.R., Downey G., Ӧhrling K., Kaufman H.L. Final analyses of OPTiM: a randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage IIIIV melanoma. J. Immunother. Cancer. 2019;7:145. doi: 10.1186/s40425-019-0623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansell S.M. Hodgkin lymphoma: 2014 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2014;89:771–779. doi: 10.1002/AJH.23750. [DOI] [PubMed] [Google Scholar]
- Ansell S.M. Hodgkin lymphoma: diagnosis and treatment. Mayo Clin. Proc. 2015;90:1574–1583. doi: 10.1016/J.MAYOCP.2015.07.005. [DOI] [PubMed] [Google Scholar]
- Ansell S.M., Lesokhin A.M., Borrello I., Halwani A., Scott E.C., Gutierrez M., Schuster S.J., Millenson M.M., Cattry D., Freeman G.J., et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015;372:311–319. doi: 10.1056/NEJMOA1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armand P., Lesokhin A., Borrello I., Timmerman J., Gutierrez M., Zhu L., Popa McKiver M., Ansell S.M. A phase 1b study of dual PD-1 and CTLA-4 or KIR blockade in patients with relapsed/refractory lymphoid malignancies. Leukemia. 2021;35:777–786. doi: 10.1038/s41375-020-0939-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attal M., Richardson P.G., Rajkumar S.V., San-Miguel J., Beksac M., Spicka I., Leleu X., Schjesvold F., Moreau P., Dimopoulos M.A., et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): a randomised, multicentre, open-label, phase 3 study. Lancet. 2019;394:2096–2107. doi: 10.1016/S0140-6736(19)32556-5. [DOI] [PubMed] [Google Scholar]
- Avanzi M.P., Brentjens R.J. Emerging role of CAR T cells in non-Hodgkin’s lymphoma. J. Natl. Compr. Canc. Netw. 2017;15:1429–1437. doi: 10.6004/jnccn.2017.7045. [DOI] [PubMed] [Google Scholar]
- Avigan D., Rosenblatt J. Vaccine therapy in hematologic malignancies. Blood. 2018;131:2640–2650. doi: 10.1182/blood-2017-11-785873. https://ashpublications.org/blood/article/131/24/2640/37160/Vaccine-therapy-in-hematologic-malignancies [DOI] [PubMed] [Google Scholar]
- Awano M., Fujiyuki T., Shoji K., Amagai Y., Murakami Y., Furukawa Y., Sato H., Yoneda M., Kai C. Measles virus selectively blind to signaling lymphocyte activity molecule has oncolytic efficacy against nectin-4-expressing pancreatic cancer cells. Cancer Sci. 2016;107:1647–1652. doi: 10.1111/cas.13064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahlis N.J., Raje N.S., Costello C., Dholaria B.R., Solh M.M., Levy M.Y., Tomasson M.H., Dube H., Liu F., Liao K.H., et al. Efficacy and safety of elranatamab (PF-06863135), a B-cell maturation antigen (BCMA)-CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MM) J. Clin. Oncol. 2021;39:8006. doi: 10.1200/JCO.2021.39.15_suppl.8006. [DOI] [Google Scholar]
- Bartee E., Chan W.M., Moreb J.S., Cogle C.R., McFadden G. Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol. Blood Marrow Transplant. 2012;18:1540–1551. doi: 10.1016/J.BBMT.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartee E., Bartee M.Y., Bogen B., Yu X.Z. Systemic therapy with oncolytic myxoma virus cures established residual multiple myeloma in mice. Mol. Ther. Oncolytics. 2016;3:16032. doi: 10.1038/MTO.2016.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basar R., Daher M., Rezvani K. Next-generation cell therapies: the emerging role of CAR-NK cells. Blood Adv. 2020;4:5868–5876. doi: 10.1182/BLOODADVANCES.2020002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdeja J.G., Madduri D., Usmani S.Z., Jakubowiak A., Agha M., Cohen A.D., Stewart A.K., Hari P., Htut M., Lesokhin A., et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398:314–324. doi: 10.1016/S0140-6736(21)00933-8. [DOI] [PubMed] [Google Scholar]
- Berraondo P., Sanmamed M.F., Ochoa M.C., Etxeberria I., Aznar M.A., Pérez-Gracia J.L., Rodríguez-Ruiz M.E., Ponz-Sarvise M., Castañón E., Melero I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer. 2019;120:6–15. doi: 10.1038/s41416-018-0328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biavati L., Huff C.A., Ferguson A., Sidorski A., Stevens M.A., Rudraraju L., Zucchinetti C., Ali S.A., Imus P., Gocke C.B., et al. An allogeneic multiple myeloma GM-CSF–secreting vaccine with lenalidomide induces long-term immunity and durable clinical responses in patients in near complete remission. Clin. Cancer Res. 2021;27:6696–6708. doi: 10.1158/1078-0432.CCR-21-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond D.A., Woyach J.A. Targeting BTK in CLL: beyond ibrutinib. Curr. Hematol. Malig. Rep. 2019;14:197–205. doi: 10.1007/S11899-019-00512-0/FIGURES/1. [DOI] [PubMed] [Google Scholar]
- Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018;68:394–424. doi: 10.3322/CAAC.21492. [DOI] [PubMed] [Google Scholar]
- Cappellano G., Abreu H., Casale C., Dianzani U., Chiocchetti A. Nano-microparticle platforms in developing next-generation vaccines. Vaccines. 2021;9:606. doi: 10.3390/VACCINES9060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmon L., Avivi I., Kovjazin R., Zuckerman T., Dray L., Gatt M.E., Or R., Shapira M.Y. Phase I/II study exploring ImMucin, a pan-major histocompatibility complex, anti-MUC1 signal peptide vaccine, in multiple myeloma patients. Br. J. Haematol. 2015;169:44–56. doi: 10.1111/bjh.13245. [DOI] [PubMed] [Google Scholar]
- Chari A., Hari P., Bahlis N.J., Mateos M.-V., van de Donk N.W., Dholaria B., Garfall A.L., Goldschmidt H., Kortuem K.M., Krishnan A.Y., et al. Phase 1b results for subcutaneous talquetamab plus daratumumab in patients with relapsed/refractory multiple myeloma. Blood. 2021;138:161. doi: 10.1182/BLOOD-2021-148813. [DOI] [Google Scholar]
- Chavez J.C., Bachmeier C., Kharfan-Dabaja M.A. CAR T-cell therapy for B-cell lymphomas: clinical trial results of available products. Ther. Adv. Hematol. 2019;10 doi: 10.1177/2040620719841581. 2040620719841581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Flies D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheson B.D. Monoclonal antibody therapy of chronic lymphocytic leukemia. Cancer Immunol. Immunother. 2006;55:188–196. doi: 10.1007/S00262-005-0010-0/TABLES/4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.-F., Lin L., Xing L., Wen K., Yu T., Hsieh P.A., Li Y., Munshi N.C., Wahl J., Matthes K., et al. AMG 701 potently induces anti-multiple myeloma (MM) functions of T cells and IMiDs further enhance its efficacy to prevent MM relapse in vivo. Blood. 2019;134:135. doi: 10.1182/blood-2019-128528. [DOI] [Google Scholar]
- Choi M.Y., Widhopf G.F., Ghia E.M., Kidwell R.L., Hasan M.K., Yu J., Rassenti L.Z., Chen L., Chen Y., Pittman E., et al. Phase I trial: cirmtuzumab inhibits ROR1 signaling and stemness signatures in patients with chronic lymphocytic leukemia in brief. Cell Stem Cell. 2018;22:951–959.e3. doi: 10.1016/j.stem.2018.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D.T., Nguyen T.T., Tien N.L.B., Tran D.K., Jeong J.H., Anh P.G., Thanh V.V., Truong D.T., Dinh T.C. Recent progress of stem cell therapy in cancer treatment: molecular mechanisms and potential applications. Cells. 2020;9:563. doi: 10.3390/CELLS9030563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J., Deng Y., Benson D.M., He S., Hughes T., Zhang J., Peng Y., Mao H., Yi L., Ghoshal K., et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–927. doi: 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieri N., Maurer K., Wu C.J. 60 Years young: the evolving role of allogeneic hematopoietic stem cell transplantation in cancer immunotherapy. Cancer Res. 2021;81:4373–4384. doi: 10.1158/0008-5472.CAN-21-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinical Trials . 2022. Clinical Trials.https://www.clinicaltrials.gov [Google Scholar]
- Cook J., Peng K.W., Geyer S.M., Ginos B.F., Dueck A.C., Packiriswamy N., Zhang L., Brunton B., Balakrishnan B., Witzig T.E., et al. Clinical activity of systemic VSV-IFNβ-NIS oncolytic virotherapy in patients with relapsed refractory T-cell lymphoma. J. Clin. Oncol. 2021;39:2500. doi: 10.1200/JCO.2021.39.15_suppl.2500. [DOI] [Google Scholar]
- Cook J., Acosta-Medina A.A., Peng K.W., Lacy M., Russell S. Oncolytic virotherapy – forging its place in the immunomodulatory paradigm for Multiple Myeloma. Cancer Treat. Res. Commun. 2021;29:100473. doi: 10.1016/J.CTARC.2021.100473. [DOI] [PubMed] [Google Scholar]
- Cooper M.D., Alder M.N. The evolution of adaptive immune systems. Cell. 2006;124:815–822. doi: 10.1016/J.CELL.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Corradini P., Tarella C., Zallio F., Dodero A., Zanni M., Valagussa P., Gianni A.M., Rambaldi A., Barbui T., Cortelazzo S. Long-term follow-up of patients with peripheral T-cell lymphomas treated up-front with high-dose chemotherapy followed by autologous stem cell transplantation. Leukemia. 2006;20:1533–1538. doi: 10.1038/sj.leu.2404306. [DOI] [PubMed] [Google Scholar]
- Costa L.J., Wong S.W., Bermúdez A., de la Rubia J., Mateos M.-V., Ocio E.M., Rodríguez-Otero P., San-Miguel J., Li S., Sarmiento R., et al. First clinical study of the B-cell maturation antigen (BCMA) 2+1 T cell engager (TCE) CC-93269 in patients (pts) with relapsed/refractory multiple myeloma (RRMM): interim results of a phase 1 multicenter trial. Blood. 2019;134:143. doi: 10.1182/blood-2019-122895. [DOI] [Google Scholar]
- Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/SCIENCE.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- Cox M.C., Castiello L., Mattei M., Santodonato L., D’Agostino G., Muraro E., Martorelli D., Lapenta C., Di Napoli A., Di Landro F., et al. Clinical and antitumor immune responses in relapsed/refractory follicular lymphoma patients after intranodal injections of IFNα-Dendritic cells and rituximab: a phase i clinical trial. Clin. Cancer Res. 2019;25:5231–5241. doi: 10.1158/1078-0432.CCR-19-0709. [DOI] [PubMed] [Google Scholar]
- Daher M., Melo Garcia L., Li Y., Rezvani K. CAR-NK cells: the next wave of cellular therapy for cancer. Clin. Transl. Immunol. 2021;10:e1274. doi: 10.1002/CTI2.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvin P., Toor S.M., Sasidharan Nair V., Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp. Mol. Med. 2018;50:1–11. doi: 10.1038/s12276-018-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daver N., Garcia-Manero G., Basu S., Boddu P.C., Alfayez M., Cortes J.E., Konopleva M., Ravandi-Kashani F., Jabbour E., Kadia T., et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, phase II study. Cancer Discov. 2019;9:370–383. doi: 10.1158/2159-8290.CD-18-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desantis C.E., Lin C.C., Mariotto A.B., Siegel R.L., Stein K.D., Kramer J.L., Alteri R., Robbins A.S., Jemal A. Cancer treatment and survivorship statistics, 2014. CA. Cancer J. Clin. 2014;64:252–271. doi: 10.3322/CAAC.21235. [DOI] [PubMed] [Google Scholar]
- Diefenbach C.S., Hong F., David K.A., Cohen J., Robertson M., Advani R., Palmisiano N.D., Ambinder R.F., Kahl B.S., Ansell S. A phase I study with an expansion cohort of the combination of ipilimumab and nivolumab and brentuximab vedotin in patients with relapsed/refractory Hodgkin lymphoma: a trial of the ECOG-ACRIN cancer research group (E4412 arms D and E) Blood. 2016;128:1106. doi: 10.1182/BLOOD.V128.22.1106.1106. [DOI] [Google Scholar]
- Dima D., Dower J., Comenzo R.L., Varga C. Evaluating daratumumab in the treatment of multiple myeloma: safety, efficacy and place in therapy. Cancer Manag. Res. 2020;12:7891–7903. doi: 10.2147/CMAR.S212526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingli D., Peng K.W., Harvey M.E., Greipp P.R., O’Connor M.K., Cattaneo R., Morris J.C., Russell S.J. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103:1641–1646. doi: 10.1182/BLOOD-2003-07-2233. [DOI] [PubMed] [Google Scholar]
- Dispenzieri A., Tong C., Laplant B., Lacy M.Q., Laumann K., Dingli D., Zhou Y., Federspiel M.J., Gertz M.A., Hayman S., et al. Phase I trial of systemic administration of Edmonston strain of measles virus genetically engineered to express the sodium iodide symporter in patients with recurrent or refractory multiple myeloma. Leukemia. 2017;31:2791–2798. doi: 10.1038/leu.2017.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döhner H., Estey E.H., Amadori S., Appelbaum F.R., Büchner T., Burnett A.K., Dombret H., Fenaux P., Grimwade D., Larson R.A., et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. doi: 10.1182/BLOOD-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- Döhner H., Weisdorf D.J., Bloomfield C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015;373:1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- Dong Y., Shi O., Zeng Q., Lu X., Wang W., Li Y., Wang Q. Leukemia incidence trends at the global, regional, and national level between 1990 and 2017. Exp. Hematol. Oncol. 2020;9:14. doi: 10.1186/s40164-020-00170-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Donk N.W.C.J., Usmani S.Z. CD38 antibodies in multiple myeloma: mechanisms of action and modes of resistance. Front. Immunol. 2018;9:2134. doi: 10.3389/fimmu.2018.02134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Donk N.W.C.J., Pawlyn C., Yong K.L. Multiple myeloma. Lancet. 2021;397:410–427. doi: 10.1016/S0140-6736(21)00135-5. [DOI] [PubMed] [Google Scholar]
- van de Donk N.W.C.J., Usmani S.Z., Yong K. CAR T-cell therapy for multiple myeloma: state of the art and prospects. Lancet. Haematol. 2021;8:e446–e461. doi: 10.1016/S2352-3026(21)00057-0. [DOI] [PubMed] [Google Scholar]
- Dotti G., Gottschalk S., Savoldo B., Brenner M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014;257:107–126. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A.K., Fink J.L., Grady J.P., Morgan G.J., Mullighan C.G., To L.B., Hewett D.R., Zannettino A.C.W. Subclonal evolution in disease progression from MGUS/SMM to multiple myeloma is characterised by clonal stability. Leukemia. 2019;33:457–468. doi: 10.1038/S41375-018-0206-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faderl S.J., Keating M.J. Treatment of chronic lymphocytic leukemia. Curr. Hematol. Rep. 2005;4:31–38. [PubMed] [Google Scholar]
- Fanale M.A., Lai C.-M., McLaughlin P., Romaguera J., Fayad L., Hagemeister F., Samaniego F., Rodriguez M.A., Neelapu S.S., Shah J.J., et al. Outcomes of nodular lymphocyte predominant Hodgkin’s lymphoma (NLPHL) patients treated with R-CHOP. Blood. 2010;116:2812. doi: 10.1182/BLOOD.V116.21.2812.2812. [DOI] [Google Scholar]
- Ferdinand R., Mitchell S.A., Batson S., Tumur I. Treatments for chronic myeloid leukemia: a qualitative systematic review. J. Blood Med. 2012;3:51–76. doi: 10.2147/JBM.S33380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes M.S., Gomes E.M., Butcher L.D., Hernandez-Alcoceba R., Chang D., Kansopon J., Newman J., Stone M.J., Tong A.W. Growth inhibition of human multiple myeloma cells by an oncolytic adenovirus carrying the CD40 ligand transgene. Clin. Cancer Res. 2009;15:4847–4856. doi: 10.1158/1078-0432.CCR-09-0451. [DOI] [PubMed] [Google Scholar]
- Flinn I.W., Van Der Jagt R., Kahl B.S., Wood P., Hawkins T.E., MacDonald D., Hertzberg M., Kwan Y.L., Simpson D., Craig M., et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood. 2014;123:2944–2952. doi: 10.1182/BLOOD-2013-11-531327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton J.E. Isatuximab: a review of its use in multiple myeloma. Target. Oncol. 2021;16:675–686. doi: 10.1007/s11523-021-00827-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco L., Fraser C., Torres-Dominguez L., Grigaitis N., Peter J.S., Abrantes M., Tacner Z., Hrach H., Matos A.d., Sharp L. 744 Multi-armed myxoma virus has therapeutic potential for treatment of multiple myeloma. J. Immunother. Cancer. 2021;9:A775. doi: 10.1136/JITC-2021-SITC2021.744. [DOI] [Google Scholar]
- Fujiyuki T., Yoneda M., Amagai Y., Obayashi K., Ikeda F., Shoji K., Murakami Y., Sato H., Kai C. A measles virus selectively blind to signaling lymphocytic activation molecule shows anti-tumor activity against lung cancer cells. Oncotarget. 2015;6:24895–24903. doi: 10.18632/oncotarget.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futami M., Sato K., Miyazaki K., Suzuki K., Nakamura T., Tojo A. Efficacy and safety of doubly-regulated vaccinia virus in a mouse xenograft model of multiple myeloma. Mol. Ther. Oncolytics. 2017;6:57–68. doi: 10.1016/J.OMTO.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamis A.S., Alonzo T.A., Meshinchi S., Sung L., Gerbing R.B., Raimondi S.C., Hirsch B.A., Kahwash S.B., Heerema-McKenney A., Winter L., et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase iII children’s oncology group Trial AAML0531. J. Clin. Oncol. 2014;32:3021–3032. doi: 10.1200/JCO.2014.55.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido F., Aptsiauri N., Doorduijn E.M., Garcia Lora A.M., van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016;39:44–51. doi: 10.1016/J.COI.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gary E.N., Weiner D.B. DNA vaccines: prime time is now. Curr. Opin. Immunol. 2020;65:21–27. doi: 10.1016/J.COI.2020.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney G.T., Weiner L.M., Atkins M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17:e542–e551. doi: 10.1016/S1470-2045(16)30406-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godwin C.D., Gale R.P., Walter R.B. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia. 2017;31:1855–1868. doi: 10.1038/leu.2017.187. [DOI] [PubMed] [Google Scholar]
- Gökbuget N., Dombret H., Bonifacio M., Reichle A., Graux C., Faul C., Diedrich H., Topp M.S., Brüggemann M., Horst H.A., et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131:1522–1531. doi: 10.1182/BLOOD-2017-08-798322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman J.M., Melo J.V. Chronic myeloid leukemia--advances in biology and new approaches to treatment. N. Engl. J. Med. 2003;349:1451–1464. doi: 10.1056/NEJMRA020777. [DOI] [PubMed] [Google Scholar]
- Gomes-Silva D., Atilla E., Atilla P.A., Mo F., Tashiro H., Srinivasan M., Lulla P., Rouce R.H., Cabral J.M.S., Ramos C.A., et al. CD7 CAR T cells for the therapy of acute myeloid leukemia. Mol. Ther. 2019;27:272–280. doi: 10.1016/J.YMTHE.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gormley N.J., Ko C.-W., Deisseroth A., Nie L., Kaminskas E., Kormanik N., Goldberg K.B., Farrell A.T., Pazdur R. FDA drug approval: elotuzumab in combination with lenalidomide and dexamethasone for the treatment of relapsed or refractory multiple myeloma. Clin. Cancer Res. 2017;23:6759–6763. doi: 10.1158/1078-0432.CCR-16-2870. [DOI] [PubMed] [Google Scholar]
- Greiner T.C., Medeiros L.J., Jaffe E.S. Non-Hodgkin’s lymphoma. Cancer. 1995;75:370–380. doi: 10.1002/1097-0142(19950101)75:1+<370::aid-cncr2820751319>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Greipp P.R., San Miguel J., Durie B.G.M., Crowley J.J., Barlogie B., Bladé J., Boccadoro M., Child J.A., Avet-Loiseau H., Harousseau J.L., et al. International staging system for multiple myeloma. J. Clin. Oncol. 2005;23:3412–3420. doi: 10.1200/JCO.2005.04.242. [DOI] [PubMed] [Google Scholar]
- Grote D., Russell S.J., Cornu T.I., Cattaneo R., Vile R., Poland G.A., Fielding A.K. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood. 2001;97:3746–3754. doi: 10.1182/BLOOD.V97.12.3746. [DOI] [PubMed] [Google Scholar]
- Hagenbeek A., Gadeberg O., Johnson P., Pedersen L.M., Walewski J., Hellmann A., Link B.K., Robak T., Wojtukiewicz M., Pfreundschuh M., et al. First clinical use of ofatumumab, a novel fully human anti-CD20 monoclonal antibody in relapsed or refractory follicular lymphoma: results of a phase 1/2 trial. Blood. 2008;111:5486–5495. doi: 10.1182/BLOOD-2007-10-117671. [DOI] [PubMed] [Google Scholar]
- Hallek M., Cheson B.D., Catovsky D., Caligaris-Cappio F., Dighiero G., Döhner H., Hillmen P., Keating M., Montserrat E., Chiorazzi N., et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745–2760. doi: 10.1182/BLOOD-2017-09-806398. [DOI] [PubMed] [Google Scholar]
- Hargadon K.M., Johnson C.E., Williams C.J. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018;62:29–39. doi: 10.1016/J.INTIMP.2018.06.001. [DOI] [PubMed] [Google Scholar]
- He X., Feng Z., Ma J., Ling S., Cao Y., Gurung B., Wu Y., Katona B.W., O’Dwyer K.P., Siegel D.L., et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood. 2020;135:713–723. doi: 10.1182/BLOOD.2019002779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki O., Dos Santos J.M., Hemminki A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020;13:84. doi: 10.1186/s13045-020-00922-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M., Patel A., Goh C.Y., Moscvin M., Zhang L., Bianchi G. Changing paradigms in diagnosis and treatment of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM) Leukemia. 2020;34:3111–3125. doi: 10.1038/s41375-020-01051-x. [DOI] [PubMed] [Google Scholar]
- Hoffmann J.-M., Schubert M.-L., Wang L., Hückelhoven A., Sellner L., Stock S., Schmitt A., Kleist C., Gern U., Loskog A., et al. Differences in expansion potential of naive chimeric antigen receptor T cells from healthy donors and untreated chronic lymphocytic leukemia patients. Front. Immunol. 2018;8:1956. doi: 10.3389/FIMMU.2017.01956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong E., Dobrovolskaia M.A. Addressing barriers to effective cancer immunotherapy with nanotechnology: achievements, challenges, and roadmap to the next generation of nanoimmunotherapeutics. Adv. Drug Deliv. Rev. 2019;141:3–22. doi: 10.1016/J.ADDR.2018.01.005. [DOI] [PubMed] [Google Scholar]
- Hoseini S.S., Espinosa-Cotton M., Guo H.F., Cheung N.K.V. Overcoming leukemia heterogeneity by combining T cell engaging bispecific antibodies. J. Immunother. Cancer. 2020;8:e001626. doi: 10.1136/JITC-2020-001626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyos V., Borrello I. The immunotherapy era of myeloma: monoclonal antibodies, vaccines, and adoptive T-cell therapies. Blood. 2016;128:1679–1687. doi: 10.1182/BLOOD-2016-05-636357. [DOI] [PubMed] [Google Scholar]
- Hristov A.C., Tejasvi T., Wilcox R.A. Mycosis fungoides and Sézary syndrome: 2019 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2019;94:1027–1041. doi: 10.1002/AJH.25577. [DOI] [PubMed] [Google Scholar]
- Hu Z., Ott P.A., Wu C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018;18:168–182. doi: 10.1038/nri.2017.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber A., Dammeijer F., Aerts J.G.J.V., Vroman H. Current state of dendritic cell-based immunotherapy: opportunities for in vitro antigen loading of different DC subsets? Front. Immunol. 2018;9:2804. doi: 10.3389/FIMMU.2018.02804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T.P., Mauro M.J., Cortes J.E., Minami H., Rea D., DeAngelo D.J., Breccia M., Goh Y.-T., Talpaz M., Hochhaus A., et al. Asciminib in chronic myeloid leukemia after ABL kinase inhibitor failure. N. Engl. J. Med. 2019;381:2315–2326. doi: 10.1056/NEJMOA1902328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunger S.P., Mullighan C.G. Acute lymphoblastic leukemia in children. N. Engl. J. Med. 2015;373:1541–1552. doi: 10.1056/NEJMra1400972. [DOI] [PubMed] [Google Scholar]
- Huse M. The T-cell-receptor signaling network. J. Cell Sci. 2009;122:1269–1273. doi: 10.1242/JCS.042762. [DOI] [PubMed] [Google Scholar]
- Iriguchi S., Kaneko S. Toward the development of true “off-the-shelf” synthetic T-cell immunotherapy. Cancer Sci. 2019;110:16–22. doi: 10.1111/CAS.13892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahanafrooz Z., Baradaran B., Mosafer J., Hashemzaei M., Rezaei T., Mokhtarzadeh A., Hamblin M.R. Comparison of DNA and mRNA vaccines against cancer. Drug Discov. Today. 2020;25:552–560. doi: 10.1016/J.DRUDIS.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Zhang W., Shang P., Zhang H., Fu W., Ye F., Zeng T., Huang H., Zhang X., Sun W., et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 2014;8:297–310. doi: 10.1016/J.MOLONC.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen N.G., Ahmad S.M., Abildgaard N., Straten P.T., Svane I.M., Andersen M.H., Knudsen L.M. Peptide vaccination against multiple myeloma using peptides derived from anti-apoptotic proteins: a phase I trial. Stem Cell Investig. 2016;3:95. doi: 10.21037/sci.2016.11.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampen K.R. The discovery and early understanding of leukemia. Leuk. Res. 2012;36:6–13. doi: 10.1016/J.LEUKRES.2011.09.028. [DOI] [PubMed] [Google Scholar]
- Kantarjian H., Sawyers C., Hochhaus A., Guilhot F., Schiffer C., Gambacorti-Passerini C., Niederwieser D., Resta D., Capdeville R., Zoellner U., et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N. Engl. J. Med. 2002;346:645–652. doi: 10.1056/NEJMOA011573. [DOI] [PubMed] [Google Scholar]
- Kantarjian H., Thomas D., Jorgensen J., Jabbour E., Kebriaei P., Rytting M., York S., Ravandi F., Kwari M., Faderl S., et al. Inotuzumab ozogamicin, an anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. Lancet Oncol. 2012;13:403–411. doi: 10.1016/S1470-2045(11)70386-2. [DOI] [PubMed] [Google Scholar]
- Kantarjian H., Stein A., Gökbuget N., Fielding A.K., Schuh A.C., Ribera J.-M., Wei A., Dombret H., Foà R., Bassan R., et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 2017;376:836–847. doi: 10.1056/NEJMoa1609783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian H., Kadia T., DiNardo C., Daver N., Borthakur G., Jabbour E., Garcia-Manero G., Konopleva M., Ravandi F. Acute myeloid leukemia: current progress and future directions. Blood Cancer J. 2021;11:41. doi: 10.1038/s41408-021-00425-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantoff P.W., Higano C.S., Shore N.D., Berger E.R., Small E.J., Penson D.F., Redfern C.H., Ferrari A.C., Dreicer R., Sims R.B., et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010;363:411–422. doi: 10.1056/NEJMOA1001294. [DOI] [PubMed] [Google Scholar]
- Kimiz-Gebologlu I., Gulce-Iz S., Biray-Avci C. Monoclonal antibodies in cancer immunotherapy. Mol. Biol. Rep. 2018;45:2935–2940. doi: 10.1007/S11033-018-4427-X. [DOI] [PubMed] [Google Scholar]
- Kogut M.H., Lee A., Santin E. Microbiome and pathogen interaction with the immune system. Poult. Sci. 2020;99:1906–1913. doi: 10.1016/J.PSJ.2019.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegsmann K., Kriegsmann M., Cremer M., Schmitt M., Dreger P., Goldschmidt H., Müller-Tidow C., Hundemer M. Cell-based immunotherapy approaches for multiple myeloma. Br. J. Cancer. 2019;120:38–44. doi: 10.1038/S41416-018-0346-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A.Y., Minnema M.C., Berdeja J.G., Oriol A., van de Donk N.W., Rodriguez-Otero P., Askari E., Mateos M.-V., Costa L.J., Verona R.I., et al. Updated phase 1 results from MonumenTAL-1: first-in-human study of talquetamab, a G protein-coupled receptor family C group 5 member D x CD3 bispecific antibody, in patients with relapsed/refractory multiple myeloma. Blood. 2021;138:158. doi: 10.1182/BLOOD-2021-146868. [DOI] [Google Scholar]
- Kumar S., D’Souza A., Shah N., Rodriguez C., Voorhees P.M., Bueno O.F., Buelow B., Freise K.J., Yue S., Pothacamury R.K., et al. A phase 1 first-in-human study of Tnb-383B, a BCMA x CD3 bispecific T-cell redirecting antibody, in patients with relapsed/refractory multiple myeloma. Blood. 2021;138:900. doi: 10.1182/blood-2021-150757. [DOI] [Google Scholar]
- Küppers R. The biology of Hodgkin’s lymphoma. Nat. Rev. Cancer. 2008;9:15–27. doi: 10.1038/nrc2542. [DOI] [PubMed] [Google Scholar]
- Küppers R., Engert A., Hansmann M.L. Hodgkin lymphoma. J. Clin. Invest. 2012;122:3439–3447. doi: 10.1172/JCI61245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy M.Q., Mandrekar S., Dispenzieri A., Hayman S., Kumar S., Buadi F., Dingli D., Litzow M., Wettstein P., Padley D., et al. Idiotype-pulsed antigen-presenting cells following autologous transplantation for multiple myeloma may be associated with prolonged survival. Am. J. Hematol. 2009;84:799–802. doi: 10.1002/ajh.21560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledford H., Else H., Warren M. Cancer immunologists scoop medicine Nobel prize. Nature. 2018;562:20–21. doi: 10.1038/D41586-018-06751-0. [DOI] [PubMed] [Google Scholar]
- Lei W., Wang S., Yang C., Huang X., Chen Z., He W., Shen J., Liu X., Qian W. Combined expression of miR-34a and Smac mediated by oncolytic vaccinia virus synergistically promote anti-tumor effects in Multiple Myeloma. Sci. Rep. 2016;6:32174. doi: 10.1038/srep32174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei W., Wang S., Xu N., Chen Y., Wu G., Zhang A., Chen X., Tong Y., Qian W. Enhancing therapeutic efficacy of oncolytic vaccinia virus armed with Beclin-1, an autophagic Gene in leukemia and myeloma. Biomed. Pharmacother. 2020;125:110030. doi: 10.1016/J.BIOPHA.2020.110030. [DOI] [PubMed] [Google Scholar]
- Leivas A., Valeri A., Córdoba L., García-Ortiz A., Ortiz A., Sánchez-Vega L., Graña-Castro O., Fernández L., Carreño-Tarragona G., Pérez M., et al. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. 2021;11:146. doi: 10.1038/s41408-021-00537-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenk L., Alsadeq A., Schewe D.M. Involvement of the central nervous system in acute lymphoblastic leukemia: opinions on molecular mechanisms and clinical implications based on recent data. Cancer Metastasis Rev. 2020;39:173–187. doi: 10.1007/S10555-020-09848-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard J.P., Coleman M., Ketas J.C., Chadburn A., Ely S., Furman R.R., Wegener W.A., Hansen H.J., Ziccardi H., Eschenberg M., et al. Phase I/II trial of epratuzumab (humanized anti-CD22 antibody) in indolent non-Hodgkin’s lymphoma. J. Clin. Oncol. 2003;21:3051–3059. doi: 10.1200/JCO.2003.01.082. [DOI] [PubMed] [Google Scholar]
- Levy R., Ganjoo K.N., Leonard J.P., Vose J.M., Flinn I.W., Ambinder R.F., Connors J.M., Berinstein N.L., Belch A.R., Bartlett N.L., et al. Active idiotypic vaccination versus control immunotherapy for follicular lymphoma. J. Clin. Oncol. 2014;32:1797–1803. doi: 10.1200/JCO.2012.43.9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Mei H., Hu Y., Guo T., Liu L., Jiang H., Tang L., Wu Y., Ai L., Deng J., Jin D. A bispecific CAR-T cell therapy targeting Bcma and CD38 for relapsed/refractory multiple myeloma: updated results from a phase 1 dose-climbing trial. Blood. 2019;134:930. doi: 10.1182/blood-2019-130340. [DOI] [Google Scholar]
- Li J., Stagg N.J., Johnston J., Harris M.J., Menzies S.A., DiCara D., Clark V., Hristopoulos M., Cook R., Slaga D., et al. Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell. 2017;31:383–395. doi: 10.1016/J.CCELL.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zeng H., Xu R.H., Liu B., Li Z. Vaccination with human pluripotent stem cells generates a broad spectrum of immunological and clinical responses against colon cancer. Stem Cell. 2009;27:3103–3111. doi: 10.1002/STEM.234. [DOI] [PubMed] [Google Scholar]
- Liang M. Oncorine, the world first oncolytic virus medicine and its update in China. Curr. Cancer Drug Targets. 2018;18:171–176. doi: 10.2174/1568009618666171129221503. [DOI] [PubMed] [Google Scholar]
- Liu W., Tang H., Li L., Wang X., Yu Z., Li J. Peptide-based therapeutic cancer vaccine: current trends in clinical application. Cell Prolif. 2021;54:e13025. doi: 10.1111/CPR.13025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loghavi S., Kutok J.L., Jorgensen J.L. B-acute lymphoblastic leukemia/lymphoblastic lymphoma. Am. J. Clin. Pathol. 2015;144:393–410. doi: 10.1309/AJCPAN7BH5DNYWZB. [DOI] [PubMed] [Google Scholar]
- Lokhorst H.M., Wu K., Verdonck L.F., Laterveer L.L., Van De Donk N.W.C.J., Van Oers M.H.J., Cornelissen J.J., Schattenberg A.V. The occurrence of graft-versus-host disease is the major predictive factor for response to donor lymphocyte infusions in multiple myeloma. Blood. 2004;103:4362–4364. doi: 10.1182/BLOOD-2003-11-3862. [DOI] [PubMed] [Google Scholar]
- Long M., Beckwith K., Do P., Mundy B.L., Gordon A., Lehman A.M., Maddocks K.J., Cheney C., Jones J.A., Flynn J.M., et al. Ibrutinib treatment improves T cell number and function in CLL patients. J. Clin. Invest. 2017;127:3052–3064. doi: 10.1172/JCI89756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonial S., Richardson P.G., Reece D.E., Mohamed H., Shelat S., San Miguel J. CheckMate 602: an open-label, randomized, phase 3 trial of combinations of nivolumab, elotuzumab, pomalidomide and dexamethasone in relapsed/refractory multiple myeloma. J. Clin. Oncol. 2017;35:TPS8052. doi: 10.1200/JCO.2017.35.15_suppl.TPS8052. [DOI] [Google Scholar]
- de Miguel M., Calvo E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell. 2020;38:326–333. doi: 10.1016/J.CCELL.2020.07.004. [DOI] [PubMed] [Google Scholar]
- Madduri D., Rosko A., Brayer J., Zonder J., Bensinger W.I., Li J., Xu L., Adriaens L., Chokshi D., Zhang W., et al. REGN5458, a BCMA x CD3 bispecific monoclonal antibody, induces deep and durable responses in patients with relapsed/refractory multiple myeloma (RRMM) Blood. 2020;136:41–42. doi: 10.1182/blood-2020-139192. [DOI] [Google Scholar]
- Mak T.W., Saunders M.E. In: The Immune Response. Mak T.W., Saunders M.E., editors. Academic Press; 2006. Hematopoietic cancers; pp. 1025–1063. [DOI] [Google Scholar]
- Marhelava K., Pilch Z., Bajor M., Graczyk-Jarzynka A., Zagozdzon R. Targeting negative and positive immune checkpoints with monoclonal antibodies in therapy of cancer. Cancers. 2019;11:1756. doi: 10.3390/cancers11111756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín E.M., Encinas J., García-Ortiz A., Ugalde L., Fernández R.A., Leivas A., Paciello M.L., Garrido V., Martín-Antonio B., Suñe G., et al. Exploring NKG2D and BCMA-CAR NK-92 for adoptive cellular therapy to multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2019;19:e24–e25. doi: 10.1016/J.CLML.2019.09.036. [DOI] [Google Scholar]
- Maslak P.G., Dao T., Bernal Y., Chanel S.M., Zhang R., Frattini M., Rosenblat T., Jurcic J.G., Brentjens R.J., Arcila M.E., et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv. 2018;2:224–234. doi: 10.1182/BLOODADVANCES.2017014175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann K.J., Godeseth R., Chudley L., Mander A., Di Genova G., Lloyd-Evans P., Kerr J.P., Malykh V.B., Jenner M.W., Orchard K.H., et al. Idiotypic DNA vaccination for the treatment of multiple myeloma: safety and immunogenicity in a phase I clinical study. Cancer Immunol. Immunother. 2015;64:1021–1032. doi: 10.1007/s00262-015-1703-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei H., Li C., Jiang H., Zhao X., Huang Z., Jin D., Guo T., Kou H., Liu L., Tang L., et al. A bispecific CAR-T cell therapy targeting BCMA and CD38 in relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2021;14:161. doi: 10.1186/s13045-021-01170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meleshko A.N., Petrovskaya N.A., Savelyeva N., Vashkevich K.P., Doronina S.N., Sachivko N.V. Phase I clinical trial of idiotypic DNA vaccine administered as a complex with polyethylenimine to patients with B-cell lymphoma. Hum. Vaccin. Immunother. 2017;13:1398–1403. doi: 10.1080/21645515.2017.1285477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger M.L., Mauz-Körholz C. Epidemiology, outcome, targeted agents and immunotherapy in adolescent and young adult non-Hodgkin and Hodgkin lymphoma. Br. J. Haematol. 2019;185:1142–1157. doi: 10.1111/BJH.15789. [DOI] [PubMed] [Google Scholar]
- Meyers D.E., Thakur S., Thirukkumaran C.M., Morris D.G. Oncolytic virotherapy as an immunotherapeutic strategy for multiple myeloma. Blood Cancer J. 2017;7:640. doi: 10.1038/s41408-017-0020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miliotou A.N., Papadopoulou L.C. CAR T-cell therapy: a new era in cancer immunotherapy. Curr. Pharm. Biotechnol. 2018;19:5–18. doi: 10.2174/1389201019666180418095526. [DOI] [PubMed] [Google Scholar]
- Minnie S.A., Hill G.R. Immunotherapy of multiple myeloma. J. Clin. Invest. 2020;130:1565–1575. doi: 10.1172/JCI129205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau P., Dimopoulos M.A., Mikhael J., Yong K., Capra M., Facon T., Hajek R., Špička I., Baker R., Kim K., et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): a multicentre, open-label, randomised phase 3 trial. Lancet. 2021;397:2361–2371. doi: 10.1016/S0140-6736(21)00592-4. [DOI] [PubMed] [Google Scholar]
- Morschhauser F.A., Cartron G., Thieblemont C., Solal-Céligny P., Haioun C., Bouabdallah R., Feugier P., Bouabdallah K., Asikanius E., Lei G., et al. Obinutuzumab (GA101) monotherapy in relapsed/refractory diffuse large b-cell lymphoma or mantle-cell lymphoma: results from the phase II GAUGUIN study. J. Clin. Oncol. 2013;31:2912–2919. doi: 10.1200/JCO.2012.46.9585. [DOI] [PubMed] [Google Scholar]
- Morsink L.M., Walter R.B. Novel monoclonal antibody-based therapies for acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2019;32:116–126. doi: 10.1016/J.BEHA.2019.05.002. [DOI] [PubMed] [Google Scholar]
- Müller L.M.E., Migneco G., Scott G.B., Down J., King S., Askar B., Jennings V., Oyajobi B., Scott K., West E., et al. Reovirus-induced cell-mediated immunity for the treatment of multiple myeloma within the resistant bone marrow niche. J. Immunother. Cancer. 2021;9:e001803. doi: 10.1136/JITC-2020-001803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi N.C., Anderson L.D., Shah N., Madduri D., Berdeja J., Lonial S., Raje N., Lin Y., Siegel D., Oriol A., et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 2021;384:705–716. doi: 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- Nogai H., Dörken B., Lenz G. Pathogenesis of non-Hodgkin’s lymphoma. J. Clin. Oncol. 2011;29:1803–1811. doi: 10.1200/jco.2010.33.3252. [DOI] [PubMed] [Google Scholar]
- Norrby E., Kristensson K. Measles virus in the brain. Brain Res. Bull. 1997;44:213–220. doi: 10.1016/S0361-9230(97)00139-1. [DOI] [PubMed] [Google Scholar]
- Oelsner S., Friede M.E., Zhang C., Wagner J., Badura S., Bader P., Ullrich E., Ottmann O.G., Klingemann H., Tonn T., Wels W.S. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy. 2017;19:235–249. doi: 10.1016/J.JCYT.2016.10.009. [DOI] [PubMed] [Google Scholar]
- Oiseth S.J., Aziz M.S. Cancer immunotherapy: a brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis Treat. 2017;3:250–261. doi: 10.20517/2394-4722.2017.41. [DOI] [Google Scholar]
- Oka T., Miyagaki T. Novel and future therapeutic drugs for advanced mycosis fungoides and Sézary syndrome. Front. Med. 2019;6:116. doi: 10.3389/fmed.2019.00116/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packiriswamy N., Upreti D., Zhou Y., Khan R., Miller A., Diaz R.M., Rooney C.M., Dispenzieri A., Peng K.W., Russell S.J. Oncolytic measles virus therapy enhances tumor antigen-specific T-cell responses in patients with multiple myeloma. Leukemia. 2020;34:3310–3322. doi: 10.1038/S41375-020-0828-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo A., Avet-Loiseau H., Oliva S., Lokhorst H.M., Goldschmidt H., Rosinol L., Richardson P., Caltagirone S., Lahuerta J.J., Facon T., et al. Revised international staging system for multiple myeloma: a report from international myeloma working group. J. Clin. Oncol. 2015;33:2863–2869. doi: 10.1200/JCO.2015.61.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peggs K.S., Kayani I., Edwards N., Kottaridis P., Goldstone A.H., Linch D.C., Hough R., Morris E.C., Fielding A., Chakraverty R., et al. Donor lymphocyte infusions modulate relapse risk in mixed chimeras and induce durable salvage in relapsed patients after T-cell-depleted allogeneic transplantation for Hodgkin’s lymphoma. J. Clin. Oncol. 2011;29:971–978. doi: 10.1200/JCO.2010.32.1711. [DOI] [PubMed] [Google Scholar]
- Pérez-Herrero E., Fernández-Medarde A. The reversed intra- and extracellular pH in tumors as a unified strategy to chemotherapeutic delivery using targeted nanocarriers. Acta Pharm. Sin. B. 2021;11:2243–2264. doi: 10.1016/J.APSB.2021.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pianko M.J., Moskowitz A.J., Lesokhin A.M. Immunotherapy of lymphoma and myeloma: facts and hopes. Clin. Cancer Res. 2018;24:1002–1010. doi: 10.1158/1078-0432.CCR-17-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter D.L., Collins R.H., Hardy C., Kernan N.A., Drobyski W.R., Giralt S., Flowers M.E., Casper J., Leahey A., Parker P., et al. Treatment of relapsed leukemia after unrelated donor marrow transplantation with unrelated donor leukocyte infusions. Blood. 2000;95:1214–1221. [PubMed] [Google Scholar]
- Raab M.S., Podar K., Breitkreutz I., Richardson P.G., Anderson K.C. Multiple myeloma. Lancet. 2009;374:324–339. doi: 10.1016/S0140-6736(09)60221-X. [DOI] [PubMed] [Google Scholar]
- Rafei H., Mehta R.S., Rezvani K. Editorial: cellular therapies in cancer. Front. Immunol. 2019;10:2788. doi: 10.3389/FIMMU.2019.02788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M.M., McFadden G. Oncolytic viruses: newest frontier for cancer immunotherapy. Cancers. 2021;13:5452. doi: 10.3390/CANCERS13215452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raje N.S., Jakubowiak A., Gasparetto C., Cornell R.F., Krupka H.I., Navarro D., Forgie A.J., Udata C., Basu C., Chou J., et al. Safety, clinical activity, pharmacokinetics, and pharmacodynamics from a phase I study of PF-06863135, a B-cell maturation antigen (BCMA)-CD3 bispecific antibody, in patients with relapsed/refractory multiple myeloma (RRMM) Blood. 2019;134:1869. doi: 10.1182/blood-2019-121805. [DOI] [Google Scholar]
- Raman S.S., Hecht J.R., Chan E. Talimogene laherparepvec: review of its mechanism of action and clinical efficacy and safety. Immunotherapy. 2019;11:705–723. doi: 10.2217/imt-2019-0033. [DOI] [PubMed] [Google Scholar]
- Rapoport A.P., Aqui N.A., Stadtmauer E.A., Vogl D.T., Xu Y.Y., Kalos M., Cai L., Fang H.B., Weiss B.M., Badros A., et al. Combination immunotherapy after ASCT for multiple myeloma using MAGE-A3/poly-ICLC immunizations followed by adoptive transfer of vaccine-primed and costimulated autologous T cells. Clin. Cancer Res. 2014;20:1355–1365. doi: 10.1158/1078-0432.CCR-13-2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner L., Waldmann T.A., Janakiram M., Brammer J.E. Rapid progression of adult T-cell leukemia–lymphoma after PD-1 inhibitor therapy. N. Engl. J. Med. 2018;378:1947–1948. doi: 10.1056/NEJMC1803181. [DOI] [PubMed] [Google Scholar]
- Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020;11:3801. doi: 10.1038/s41467-020-17670-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roddie C., Peggs K.S. Donor lymphocyte infusion following allogeneic hematopoietic stem cell transplantation. Expert Opin. Biol. Ther. 2011;11:473–487. doi: 10.1517/14712598.2011.554811. [DOI] [PubMed] [Google Scholar]
- Rodriguez J., Munsell M., Yazji S., Hagemeister F.B., Younes A., Andersson B., Giralt S., Gajewski J., De Lima M., Couriel D., et al. Impact of high-dose chemotherapy on peripheral T-cell lymphomas. J. Clin. Oncol. 2001;19:3766–3770. doi: 10.1200/JCO.2001.19.17.3766. [DOI] [PubMed] [Google Scholar]
- Röllig C., Knop S., Bornhäuser M. Multiple myeloma. Lancet. 2015;385:2197–2208. doi: 10.1016/S0140-6736(14)60493-1. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J., Vasir B., Uhl L., Blotta S., MacNamara C., Somaiya P., Wu Z., Joyce R., Levine J.D., Dombagoda D., et al. Vaccination with dendritic cell/tumor fusion cells results in cellular and humoral antitumor immune responses in patients with multiple myeloma. Blood. 2011;117:393–402. doi: 10.1182/BLOOD-2010-04-277137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt J., Avivi I., Vasir B., Uhl L., Munshi N.C., Katz T., Dey B.R., Somaiya P., Mills H., Campigotto F., et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin. Cancer Res. 2013;19:3640–3648. doi: 10.1158/1078-0432.CCR-13-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez D., Pelayo R., Medina L.A., Vadillo E., Sánchez R., Núñez L., Cesarman-Maus G., Sarmiento-Silva R.E. Newcastle disease virus: potential therapeutic application for human and canine lymphoma. Viruses. 2015;8:3. doi: 10.3390/V8010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz M.A., Fenaux P., Tallman M.S., Estey E.H., Löwenberg B., Naoe T., Lengfelder E., Döhner H., Burnett A.K., Chen S.J., et al. Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood. 2019;133:1630–1643. doi: 10.1182/BLOOD-2019-01-894980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer T., Parikh K., Sharma S., Omer B., Sedloev D., Chen Q., Angenendt L., Schliemann C., Schmitt M., Müller-Tidow C., et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood. 2021;138:318–330. doi: 10.1182/BLOOD.2020008221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sborov D.W., Nuovo G.J., Stiff A., Mace T., Lesinski G.B., Benson D.M., Efebera Y.A., Rosko A.E., Pichiorri F., Grever M.R., Hofmeister C.C. A phase I trial of single-agent Reolysin in patients with relapsed multiple myeloma. Clin. Cancer Res. 2014;20:5946–5955. doi: 10.1158/1078-0432.CCR-14-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid C., Labopin M., Nagler A., Bornhäuser M., Finke J., Fassas A., Volin L., Gürman G., Maertens J., Bordigoni P., et al. Donor lymphocyte infusion in the treatment of first hematological relapse after allogeneic stem-cell transplantation in adults with acute myeloid leukemia: a retrospective risk factors analysis and comparison with other strategies by the EBMT Acute Leukemia Working Party. J. Clin. Oncol. 2007;25:4938–4945. doi: 10.1200/JCO.2007.11.6053. [DOI] [PubMed] [Google Scholar]
- Schuster S.J., Neelapu S.S., Gause B.L., Janik J.E., Muggia F.M., Gockerman J.P., Winter J.N., Flowers C.R., Nikcevich D.A., Sotomayor E.M., et al. Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J. Clin. Oncol. 2011;29:2787–2794. doi: 10.1200/JCO.2010.33.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer A., Wöhner M., Prescher H., Brossmer R., Nitschke L. Targeting of CD22-positive B-cell lymphoma cells by synthetic divalent sialic acid analogues. Eur. J. Immunol. 2012;42:2792–2802. doi: 10.1002/EJI.201242574. [DOI] [PubMed] [Google Scholar]
- Scott A.M., Allison J.P., Wolchok J.D. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012;12:14. [PMC free article] [PubMed] [Google Scholar]
- Shah U.A., Mailankody S. Emerging immunotherapies in multiple myeloma. BMJ. 2020;370:m3176. doi: 10.1136/bmj.m3176. [DOI] [PubMed] [Google Scholar]
- Shahjahani M., Mohammadiasl J., Noroozi F., Seghatoleslami M., Shahrabi S., Saba F., Saki N. Molecular basis of chronic lymphocytic leukemia diagnosis and prognosis. Cell. Oncol. 2015;38:93–109. doi: 10.1007/S13402-014-0215-3. [DOI] [PubMed] [Google Scholar]
- Shang N., Figini M., Shangguan J., Wang B., Sun C., Pan L., Ma Q., Zhang Z. Dendritic cells based immunotherapy. Am. J. Cancer Res. 2017;7:2091–2102. [PMC free article] [PubMed] [Google Scholar]
- Shankland K.R., Armitage J.O., Hancock B.W. Non-Hodgkin lymphoma. Lancet. 2012;380:848–857. doi: 10.1016/S0140-6736(12)60605-9. [DOI] [PubMed] [Google Scholar]
- Shi T., Song X., Wang Y., Liu F., Wei J. Combining oncolytic viruses with cancer immunotherapy: establishing a new generation of cancer treatment. Front. Immunol. 2020;11:683. doi: 10.3389/fimmu.2020.00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegler E.L., Kenderian S.S. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: insights into mechanisms and novel therapies. Front. Immunol. 2020;11:1973. doi: 10.3389/fimmu.2020.01973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Shaik S., Negi B.S., Rajguru J.P., Patil P.B., Parihar A.S., Sharma U. Non-Hodgkin’s lymphoma: a review. J. Family Med. Prim. Care. 2020;9:1834–1840. doi: 10.4103/JFMPC.JFMPC_1037_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirohi B., Powles R. Multiple myeloma. Lancet. 2004;363:875–887. doi: 10.1016/S0140-6736(04)15736-X. [DOI] [PubMed] [Google Scholar]
- Solimando A.G., Brandl A., Mattenheimer K., Graf C., Ritz M., Ruckdeschel A., Stühmer T., Mokhtari Z., Rudelius M., Dotterweich J., et al. JAM-A as a prognostic factor and new therapeutic target in multiple myeloma. Leukemia. 2018;32:736–743. doi: 10.1038/leu.2017.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner R.C., Sterner R.M. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11:69. doi: 10.1038/s41408-021-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart G., Chantry A., Lawson M. The use of oncolytic viruses in the treatment of multiple myeloma. Cancers. 2021;13:5687. doi: 10.3390/CANCERS13225687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strati P., Patel S., Nastoupil L., Fanale M.A., Bollard C.M., Lin A.Y., Gordon L.I. Beyond chemotherapy: checkpoint inhibition and cell-based therapy in non-hodgkin lymphoma. Am. Soc. Clin. Oncol. Educ. Book. 2018;38:592–603. doi: 10.1200/edbk_200549. [DOI] [PubMed] [Google Scholar]
- Stroopinsky D., Liegel J., Bhasin M., Cheloni G., Thomas B., Bhasin S., Panchal R., Ghiasuddin H., Rahimian M., Nahas M., et al. Leukemia vaccine overcomes limitations of checkpoint blockade by evoking clonal T-cell responses in a murine acute myeloid leukemia model. Haematologica. 2021;106:1330–1342. doi: 10.3324/HAEMATOL.2020.259457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suehiro Y., Hasegawa A., Iino T., Sasada A., Watanabe N., Matsuoka M., Takamori A., Tanosaki R., Utsunomiya A., Choi I., et al. Clinical outcomes of a novel therapeutic vaccine with Tax peptide-pulsed dendritic cells for adult T cell leukaemia/lymphoma in a pilot study. Br. J. Haematol. 2015;169:356–367. doi: 10.1111/BJH.13302. [DOI] [PubMed] [Google Scholar]
- Suresh T., Lee L.X., Joshi J., Barta S.K. New antibody approaches to lymphoma therapy. J. Hematol. Oncol. 2014;7:58. doi: 10.1186/S13045-014-0058-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepański T., van der Velden V.H.J., van Dongen J.J.M. Classification systems for acute and chronic leukaemias. Best Pract. Res. Clin. Haematol. 2003;16:561–582. doi: 10.1016/S1521-6926(03)00086-0. [DOI] [PubMed] [Google Scholar]
- Tang Y., Yin H., Zhao X., Jin D., Liang Y., Xiong T., Li L., Tang W., Zhang J., Liu M., et al. High efficacy and safety of CD38 and BCMA bispecific CAR-T in relapsed or refractory multiple myeloma. J. Exp. Clin. Cancer Res. 2022;41:2. doi: 10.1186/s13046-021-02214-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teachey D.T., O’Connor D. How I treat newly diagnosed T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma in children. Blood. 2020;135:159–166. doi: 10.1182/BLOOD.2019001557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teachey D.T., Pui C.H. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019;20:e142–e154. doi: 10.1016/S1470-2045(19)30031-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teoh P.J., Chng W.J. CAR T-cell therapy in multiple myeloma: more room for improvement. Blood Cancer J. 2021;11:84. doi: 10.1038/s41408-021-00469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakkar S., Sharma D., Kalia K., Tekade R.K. Tumor microenvironment targeted nanotherapeutics for cancer therapy and diagnosis: a review. Acta Biomater. 2020;101:43–68. doi: 10.1016/J.ACTBIO.2019.09.009. [DOI] [PubMed] [Google Scholar]
- Tong Y., Zhu W., Huang X., You L., Han X., Yang C., Qian W. PI3K inhibitor LY294002 inhibits activation of the Akt/mTOR pathway induced by an oncolytic adenovirus expressing TRAIL and sensitizes multiple myeloma cells to the oncolytic virus. Oncol. Rep. 2014;31:1581–1588. doi: 10.3892/OR.2014.3020. [DOI] [PubMed] [Google Scholar]
- Topp M.S., Kufer P., Gökbuget N., Goebeler M., Klinger M., Neumann S., Horst H.A., Raff T., Viardot A., Schmid M., et al. Targeted therapy with the T-cell - engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J. Clin. Oncol. 2011;29:2493–2498. doi: 10.1200/JCO.2010.32.7270. [DOI] [PubMed] [Google Scholar]
- Topp M.S., Duell J., Zugmaier G., Attal M., Moreau P., Langer C., Kroenke J., Facon T., Einsele H., Munzert G. Treatment with AMG 420, an anti-B-cell maturation antigen (BCMA) bispecific T-cell engager (BiTE®) antibody construct, induces minimal residual disease (MRD) negative complete responses in relapsed and/or refractory (R/R) multiple myeloma (MM) patients: results of a first-in-human (FIH) phase I dose escalation study. Blood. 2018;132:1010. doi: 10.1182/blood-2018-99-109769. [DOI] [Google Scholar]
- Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M., et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration (FDA) FDA.GOV; 2021. FDA Approves First Cell-Based Gene Therapy for Adult Patients with Multiple Myeloma.https://www.fda.gov/news-events/press-announcements/fda-approves-first-cell-based-gene-therapy-adult-patients-multiple-myeloma [Google Scholar]
- U.S. Food and Drug Administration (FDA) FDA.GOV; 2021. FDA Approves New Treatment for Adults with Relapsed or Refractory Large-B-Cell Lymphoma.https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-adults-relapsed-or-refractory-large-b-cell-lymphoma [Google Scholar]
- Usmani S.Z., Mateos M.-V., Nahi H., Krishnan A.Y., van de Donk N.W., San Miguel J., Oriol A., Rosiñol L., Chari A., Adams H., et al. Phase I study of teclistamab, a humanized B-cell maturation antigen (BCMA) x CD3 bispecific antibody, in relapsed/refractory multiple myeloma (R/R MM) J. Clin. Oncol. 2020;38:100. doi: 10.1200/JCO.2020.38.15_suppl.100. [DOI] [Google Scholar]
- Usmani S.Z., Quach H., Mateos M.-V., Landgren O., Leleu X., Siegel D., Weisel K., Gavriatopoulou M., Oriol A., Rabin N., et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): updated outcomes from a randomised, multicentre, open-label, phase 3 study. Lancet Oncol. 2021;23:65–76. doi: 10.1016/S1470-2045(21)00579-9. [DOI] [PubMed] [Google Scholar]
- Vaddepally R.K., Kharel P., Pandey R., Garje R., Chandra A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers. 2020;12:738. doi: 10.3390/CANCERS12030738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vago L., Gojo I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Invest. 2020;130:1552–1564. doi: 10.1172/JCI129204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa N.Y., Rahman M.M., Mamola J., D’Isabella J., Goras E., Kilbourne J., Lowe K., Daggett-Vondras J., Torres L., Christie J., et al. Autologous transplantation using donor leukocytes loaded ex vivo with oncolytic myxoma virus can eliminate residual multiple myeloma. Mol. Ther. Oncolytics. 2020;18:171–188. doi: 10.1016/J.OMTO.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vlierberghe P., Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Invest. 2012;122:3398–3406. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahed A., Quesada A., Dasgupta A. In: Hematology and Coagulation. Wahed A., Quesada A., Dasgupta A., editors. Academic Press; 2015. Hodgkin lymphoma; pp. 215–223. [DOI] [Google Scholar]
- Wei J., Liu Y., Wang C., Zhang Y., Tong C., Dai G., Wang W., Rasko J.E.J., Melenhorst J.J., Qian W., et al. The model of cytokine release syndrome in CAR T-cell treatment for B-cell non-Hodgkin lymphoma. Signal Transduct. Target. Ther. 2020;5:134. doi: 10.1038/s41392-020-00256-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen T., Wang J., Shi Y., et al. Inhibitors targeting Bruton’s tyrosine kinase in cancers: drug development advances. Leukemia. 2021;35:312–332. doi: 10.1038/s41375-020-01072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthe J., Naseri S., Hellström A.C., Wiklund H.J., Eriksson E., Loskog A. Immunostimulatory oncolytic virotherapy for multiple myeloma targeting 4-1BB and/or CD40. Cancer Gene Ther. 2020;27:948–959. doi: 10.1038/s41417-020-0176-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G., Dong H., Liang Y., Ham J.D., Rizwan R., Chen J. CAR-NK cells: a promising cellular immunotherapy for cancer. EBioMedicine. 2020;59:102975. doi: 10.1016/j.ebiom.2020.102975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Li Y., Gu H., Dong M., Cai Z. Emerging agents and regimens for multiple myeloma. J. Hematol. Oncol. 2020;13:150. doi: 10.1186/S13045-020-00980-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M., Scherwitzl I., Opp S., Tsirigos A., Meruelo D. Molecular and metabolic pathways mediating curative treatment of a non-Hodgkin B cell lymphoma by Sindbis viral vectors and anti-4-1BB monoclonal antibody. J. Immunother. Cancer. 2019;7:185. doi: 10.1186/S40425-019-0664-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung L., Linch D. Hodgkin’s lymphoma. Lancet. 2003;361:943–951. doi: 10.1016/S0140-6736(03)12777-8. [DOI] [PubMed] [Google Scholar]
- Zah E., Nam E., Bhuvan V., Tran U., Ji B.Y., Gosliner S.B., Wang X., Brown C.E., Chen Y.Y. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun. 2020;11:2283. doi: 10.1038/s41467-020-16160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R., Billingsley M.M., Mitchell M.J. Biomaterials for vaccine-based cancer immunotherapy. J. Control. Release. 2018;292:256–276. doi: 10.1016/J.JCONREL.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuch de Zafra C.L., Fajardo F., Zhong W., Bernett M.J., Muchhal U.S., Moore G.L., Stevens J., Case R., Pearson J.T., Liu S., et al. Targeting multiple myeloma with AMG 424, a novel anti-CD38/CD3 bispecific T-cell–recruiting antibody optimized for cytotoxicity and cytokine release. Clin. Cancer Res. 2019;25:3921–3933. doi: 10.1158/1078-0432.CCR-18-2752. [DOI] [PubMed] [Google Scholar]