

Abstract
The clinical characteristics of autosomal dominant tubulointerstitial kidney disease (ADTKD) include bland urinary sediment, slowly progressive chronic kidney disease (CKD) with many patients reaching end stage renal disease (ESRD) between age 20 and 70 years, and autosomal dominant inheritance. Due to advances in genetic diagnosis, ADTKD is becoming increasingly recognized as a cause of CKD. Pathogenic variants in UMOD, MUC1, and REN are the most common causes of ADTKD. ADTKD‐UMOD is also associated with hyperuricemia and gout. ADTKD‐REN often presents in childhood with mild hypotension, CKD, hyperkalemia, acidosis, and anemia. ADTKD‐MUC1 patients present only with CKD. This review describes the pathophysiology, genetics, clinical manifestation, and diagnosis for ADTKD, with an emphasis on genetic testing and genetic counseling suggestions for patients.
Keywords: Autosomal Dominant Tubulointerstitial Kidney Disease, MUC1, UMOD, REN
1. AUTOSOMAL DOMINANT TUBULOINTERSTITIAL KIDNEY DISEASE
Autosomal dominant tubulointerstitial kidney disease (ADTKD) refers to a group of disorders characterized by: (1) autosomal dominant inheritance, (2) bland urinary sediment (absence of hematuria and urine protein <1+ on dipstick or <200 mg/g creatinine on a spot urine protein creatinine ratio), and (3) slowly progressive chronic kidney disease (CKD) that is highly variable within and between families and leads to the need for kidney replacement therapy (dialysis or kidney transplant) variably between ages 20 and 80 years (Bleyer, Kidd, Zivna, & Kmoch, 2017). This review will concentrate on the clinical characterization, genetic aspects, pathophysiology, biochemical, genetic and histochemical analysis, and treatment. The most common causes of ADTKD include pathogenic variants in the UMOD (Hart et al., 2002), MUC1 (Kirby et al., 2013), and REN (Zivna, Hulkova, Matignon, & Hodanova, 2009) genes. There are other less common causes or diseases in which ADTKD is one of the manifestations (Bolar et al., 2016).
In our clinical experience, we have been referred 859 families with ADTKD (see Figure 1). 169 (20%) of these families were self‐referred, meaning that their clinicians did not have an interest in obtaining a genetic diagnosis for them. 422 (50%) referrals were from academic centers and 249 (30%) from clinicians in private practice. 19 were from genetics counselors. Of the 455 families who provided samples for genetic testing, 207 (38%) were found to have UMOD pathogenic variants, 116 (21%) MUC1 pathogenic variants and 15(3%) REN pathogenic variants. Nine families were found to have a pathogenic variant in MUC1 other than the classically described, most prevalent cytosine duplication. 26 families were found to have other genetic causes of kidney disease. 91 families have ADTKD and do not as yet have a diagnosis.
FIGURE 1.
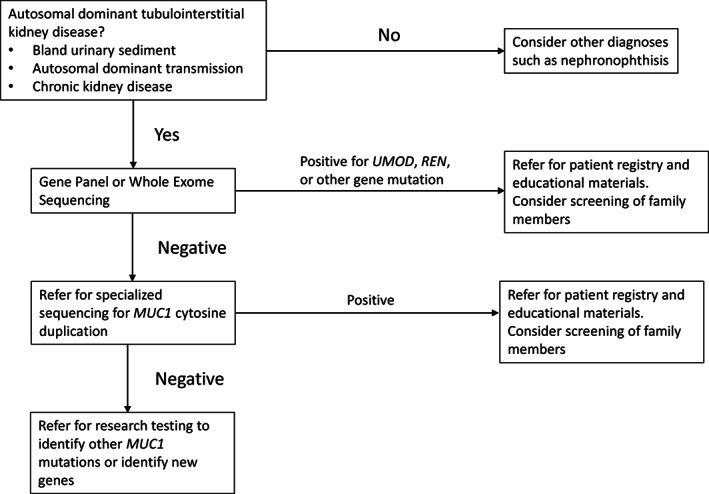
Work flow diagram for genetic testing for Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD)
2. GENERAL CLINICAL FINDINGS IN ADTKD‐UMOD AND ADTKD‐MUC1
2.1. Chronic kidney disease
The glomerular filtration rate (GFR) is the amount of blood that is filtered by the glomeruli to produce the urinary filtrate per unit time. While the kidney has many functions (including excretion of water, maintenance of normal electrolytes, control of blood pressure, and erythropoiesis), glomerular filtration rate has been found to be the best measure of kidney function. Exact measurement of the GFR is possible in a laboratory setting, but in clinical practice the glomerular filtration rate is estimated using equations that take into account age and gender. Estimates of glomerular filtration rate using the current creatinine‐based equations perform better as the eGFR declines and are often not reported if the eGFR is >60 ml/min/1.73m2. In addition, humans have a renal reserve, which results in the initial loss of kidney function not affecting serum creatinine or eGFR (Armenta, Madero, & Rodriguez‐Iturbe, 2022). Thus, detection of loss of kidney function early in the disease course may be difficult. In addition, estimation of GFR is more difficult in children, though recent developments have been most helpful in this area (Pottel et al., 2019).
In ADTKD‐UMOD and ADTKD‐MUC1, the rate of loss of kidney function is variable, with some affected individuals reaching end‐stage kidney disease (need for dialysis or kidney transplant) in their 20's, while individuals from the same family with the same pathogenic variant may not reach end‐stage kidney disease until after age 70 (Olinger et al., 2020) (Bleyer et al., 2014). The reason for this variation in the rate of progression is unknown, though in general patients tend to experience the need for dialysis within a decade of the age of onset of end‐stage kidney disease of their affected parent. The median age of ESRD is approximately 45 years (Kidd et al., 2020; Olinger et al., 2020). In general, decreased eGFR is often first detected about age 20 (Bleyer, Wolf, Kidd, Zivna, & Kmoch, 2021; Stavrou et al., 2002), and there is a very slow rise in the serum creatinine over time, with patients losing approximately 1–3 ml/min/1.73m2/year. Based on data from our registry (Bleyer et al., 2019), the rate of decline is often not linear, and patients may experience several years of decline, followed by stable kidney function for several years. The reasons for this variable rate of change are unclear.
2.2. Bland urinary sediment
Tubulointerstitial kidney diseases such as ADTKD result in pathogenic changes occurring after the glomerulus, where filtration of the blood into the pre‐urine occurs. Thus, tubulointerstitial kidney diseases are usually not associated with the proteinuria or hematuria seen in glomerular diseases (Connaughton et al., 2019). Urinalysis in patients with ADTKD reveals the absence of red or white blood cells in the urine. On urinalysis the dipstick is usually negative for protein, and a spot urine protein: creatinine ratio or urinary albumin: creatinine ratio is in the normal range. Occasionally, as eGFR declines below 60 ml/min/1.73m2, patients may develop mild proteinuria, which is usually less than 500 mg/g creatinine and rarely >1000 mg/g creatinine.
Due to decreased kidney function, patients with ADTKD often undergo kidney ultrasound. Early in the course of disease, patients with ADTKD will usually have a normal kidney ultrasound (Neumann et al., 1997; Stavrou et al., 2002). Some patients may have loss of corticomedullary differentiation, a nonspecific finding seen in all forms of CKD. Later in the course of disease, patients may have small kidneys on ultrasound (Stavrou et al., 2002)—this is also a general finding of CKD. Likewise, cysts may be seen in each kidney as disease progresses, similar to other types of CKD. Thus, kidney ultrasound in this condition shows changes that are commonly found in CKD and is in general not helpful in making this diagnosis (Table 1).
TABLE 1.
Causes of autosomal dominant tubulointerstitial kidney disease (ADTKD)
| Clinical condition | Gene(s) associated | Renal manifestations | Extra‐renal manifestations | Diagnostic considerations in addition to family history of kidney disease |
|---|---|---|---|---|
| ADTKD‐UMOD | UMOD | Hypouricosuric hyperuricemia, CKD | Gout may occur in adolescence | Gout and CKD |
| ADTKD‐MUC1 | MUC1 | CKD | None | Specialized genetic testing. Pathogenic variants not identified by gene panels or whole exome sequencing |
| ADTKD‐REN | REN | Hyperkalemia, acidosis, hypouricosuric hyperuricemia, CKD | Gout, anemia, mild hypotension | Patients are prone to AKI during viral illnesses |
| Alagille syndrome | JAG1 or NOTCH2 | Vesico‐ureteral reflux, CKD | Cardiac abnormalities, butterfly‐shaped thoracic vertebrae, and a prominent forehead | Deep‐set eyes, broad forehead as well as other extra‐renal manifestations. |
| Townes‐brocks syndrome | SALL1 | Solitary kidney, dysplastic kidneys, vesico‐ureteral reflux, CKD | Imperforate anus, dysplastic ears, thumb malformations | Findings of hand and ear abnormalities, imperforate anus |
| HDR syndrome | GATA3 | CKD | H(hypoparathyroidism), D(deafness) R(renal) | Consider this diagnosis in patients with hypocalcemia and deafness |
| ADTKD‐HNF1β | HNF1β | Vesico‐ureteral reflux, solitary kidney, CKD |
MAGIC LUCID Hypomagnesemia, autosomal dominant, gynecologic abnormalities such as a bicornuate uterus, incomplete penetrance, cystic kidneys and other kidney malformations, liver test abnormalities, hyperuricemia and gout, CKD, intellectual disability in patients with a 17q12 deletion, and maturity onset diabetes of youth |
Targeted questioning for family history of associated conditions in patients with kidney cysts |
| Less severe variants of ADPKD | IFT140, DNAJB11 | CKD, may or may not have renal cysts, interstitial kidney fibrosis | May have liver cysts | Consider for families with ADTKD presentation but also history of small renal cysts or polycystic kidneys |
3. ADTKD‐UMOD
ADTKD‐UMOD is caused by pathogenic variants in the UMOD gene encoding the protein uromodulin (Hart et al., 2002). In addition to CKD, patients with ADTKD‐UMOD often develop gout, with 25% of patients who develop gout doing so before age 25 years (Kidd et al., 2020; Warren, Simmonds, Gibson, & Naik, 1981).
Prevalence: About 1% of patients starting on dialysis have causative UMOD pathogenic variants (Cormican et al., 2019; Groopman et al., 2019; Lhotta et al., 2012). Its prevalence in the general population is about 1–3/million population (Devuyst et al., 2019).
3.1. Pathophysiology
Uromodulin is produced exclusively by the thick ascending limb of the loop of Henle in the renal tubules and is the most common protein excreted in normal human urine (Serafini‐Cessi, Bellabarba, Malagolini, & Dall'Olio, 1989; Vyletal, Bleyer, & Kmoch, 2010). Uromodulin is a glycoprotein that provides a protecting coating to the lumen of the thick ascending limb. Uromodulin has a very high cysteine content. The cysteine residues cross‐link in the endoplasmic reticulum (Rampoldi, Scolari, Amoroso, Ghiggeri, & Devuyst, 2011), allowing uromodulin to achieve its molecular structure, which facilitates transport to the cell surface and secretion (Serafini‐Cessi, Malagolini, Hoops, & Rindler, 1993). After folding in the endoplasmic reticulum is complete, the protein is transported to the surface of the thick ascending limb. The protein is then cleaved from the cell membrane and excreted into the urine (Brunati et al., 2015). Uromodulin has a minor role in the prevention of urinary tract infections and kidney stones (Vyletal et al., 2010). Uromodulin facilitates the transport of the sodium potassium NKCC2 transporter to the surface of the thick ascending limb, which increases sodium reabsorption within the kidney (Mutig et al., 2011; Weiss et al., 2020). In a similar way, uromodulin facilitates transport of the renal outer medullary potassium channel 2 (ROMK2) to the tubular cell surface of the thick ascending limb (Renigunta et al., 2011).
In ADTKD‐UMOD, affected individuals have one allele producing normal uromodulin and one mutated allele producing mutated uromodulin. The mutated uromodulin molecule cannot fold properly and is retained within the endoplasmic reticulum (Scolari et al., 2004; Vylet'al et al., 2006; Zaucke et al., 2010), leading to endoplasmic reticulum stress, the unfolded protein response, and accelerated cell death (Bernascone et al., 2006). Gradually over time, tubular cells die, leading to loss of nephron units, decreased glomerular filtration rate, tubulointerstitial fibrosis, and kidney failure. Thus, an essential feature in the pathogenesis of ADTKD‐UMOD is the production of a mutated uromodulin protein that cannot fold properly and deposits within the endoplasmic reticulum. Kidd and colleagues correlated the mean age of onset of end‐stage kidney disease for each pathogenic variant with how much the mutant uromodulin affects uromodulin transport through the endoplasmic reticulum and out of the cell (Kidd et al., 2020).
In addition to the pathophysiologic changes caused by the mutant uromodulin, the decreased synthesis of uromodulin results in decreased transport of the NKCC2 transporter to the surface of the thick ascending limb (Mutig et al., 2011). Affected individuals will experience a mild increase in sodium excretion at the thick ascending limb. However, this is compensated for by increased sodium reabsorption in the proximal tubule, so that patients clinically appear euvolemic (not dehydrated or over‐hydrated), with low normal to normal blood pressure. However, the increased proximal tubular reabsorption of sodium is believed to cause a passive increase in proximal tubule uric acid reabsorption, leading to elevated serum uric acid levels that can lead to gout (Bleyer et al., 2003).
3.2. Genetics
As improper folding of mutated uromodulin is central to the pathogenesis of ADTKD‐UMOD, genetic changes that result in loss of transcription or truncation of the UMOD protein will probably not cause ADTKD‐UMOD. About half of the pathogenic variants causing ADTKD‐UMOD result from a missense mutation leading to either the gain or loss of a cysteine residue (Bollee et al., 2011; Kidd et al., 2020), as the disulfide bridges formed by the 48 cysteine residues in uromodulin are critical to proper folding (Serafini‐Cessi et al., 1993). Approximately 99% of UMOD pathogenic variants occur in exons 3 and 4 (Kidd et al., 2020). The link https://redcap.link/UMOD includes pathogenic variants that have been reported in the literature and identified in our cohort, though they have not all been validated, often due to small family size.
There are a number of common pathogenic variants that have been identified in numerous families, likely representing a common ancestral pathogenic variant. A p.His177‐Arg185del pathogenic variant resulting in a 27‐base pair deletion was first described as a cause of ADTKD‐UMOD and has been identified in over 20 families in the US. While the family is of German descent, no families in Germany with this pathogenic variant have been reported. This pathogenic variant is associated with a median age of ESRD of 46 years and a high prevalence of gout (Bleyer et al., 2003; Kidd et al., 2020). A deletion insertion (p.Val93_Gly97delinsAlaAlaSerCys) has been identified in a number of families from England and families in the US of English descent. This pathogenic variant is associated with a later onset of end‐stage kidney disease (51 years) with a markedly decreased occurrence of gout (Smith et al., 2011). In addition, a p.Arg178Pro pathogenic variant has been identified in 13 US families with a median age of ESRD of 49.7 years. The p.Thr62Pro variant, which has been found in unaffected individuals as well as some individuals with CKD, has an intermediate effect on risk for CKD and is not a sole cause of ADTKD‐UMOD (Olinger et al., 2022). In addition to these pathogenic variants, given the autosomal dominant inheritance, age of end‐stage kidney disease after child‐bearing age in most individuals, and the difficulties in diagnosing this condition, there are a number of distantly related families that share the same pathogenic variant.
3.3. Clinical manifestations
CKD is the most common clinical finding, with patients reaching end‐stage kidney disease at a mean age of between 49 and 54 years (Bollee et al., 2011; Kidd et al., 2020). The rate of progression over time is variable, with some individuals requiring kidney transplantation or dialysis at 20, while other family members may not have reached end‐stage kidney disease at age 75 years. The rate of progression is also variable. For example, patients may have a decline in eGFR to 40 ml/min/1.73m2 and stay stable at this level for 4–5 years. Overall, the rate of decline is ~1–3 ml/min/1.73m2/year.
Approximately 60% of men and 45% of women will develop gout, at a mean age of 29 years for men and 32 years for women (Kidd et al., 2020). Of people who develop gout, 25% will do so before age 20 years. In affected families, parents are vigilant for the development of gout, which they realize is a harbinger of disease. In teenagers, gout is likely to develop in a joint that has been used in physical activity. Thus, patients may develop gout in the big toe after playing soccer, leading to a misdiagnosis of sprain or other trauma.
3.4. Differential diagnosis
For patients presenting with autosomal dominant inheritance, CKD, and a bland urinary sediment, the most common diagnoses will be ADTKD‐UMOD and ADTKD‐MUC1 (Olinger et al., 2020). Gout is much more prevalent in ADTKD‐UMOD and precedes the onset of CKD, whereas in ADTKD‐MUC1, gout occurs only after CKD has been present (Olinger et al., 2020). However, many individuals and some families with ADTKD‐UMOD do not develop gout (Smith et al., 2011), and thus there are many families where it is impossible to differentiate these two disorders based solely on clinical findings. Nephronophthisis is an autosomal recessive disorder that is characterized by a bland urinary sediment and CKD (Wolf, 2015). It can be differentiated from ADTKD due to earlier age of onset (most affected individuals reaching end‐stage kidney disease before age 25) and the absence of evidence of autosomal dominant inheritance. Alagille syndrome (Saleh, Kamath, & Chitayat, 2016) is caused by pathogenic variants in JAG1 or NOTCH2 and also features autosomal dominant transmission, CKD, and a bland urinary sediment. Patients with Alagille syndrome often have cardiac abnormalities, butterfly‐shaped thoracic vertebrae, and a prominent forehead. Townes‐Brocks syndrome is due to SALL1 pathogenic variants (Reardon, Casserly, Birkenhager, & Kohlhase, 2007). This condition is also autosomal dominant and features CKD, but there are associated symptoms such as imperforate anus, dysplastic ears, and thumb malformations. HDR syndrome (Upadhyay, Steenkamp, & Milunsky, 2013) is due to pathogenic variants in the GATA3 gene and is associated with hypoparathyroidism in almost all patients and some degree of deafness. Patients with HNF1B pathogenic variants (Bockenhauer & Jaureguiberry, 2016; Clissold, Hamilton, Hattersley, Ellard, & Bingham, 2015; Faguer et al., 2011) have autosomal dominant inheritance, a bland urinary sediment, and CKD. However, this condition has a spectrum of manifestations that can be remembered by the mnemonic MAGIC LUCID: hypomagnesemia, autosomal dominant, gynecologic abnormalities such as a bicornuate uterus, incomplete penetrance, cystic kidneys and other kidney malformations, liver test abnormalities, hyperuricemia and gout, CKD, intellectual disability if part of the 17q12 recurrent deletion syndrome, and maturity onset diabetes of youth. Thus, the differential of ADTKD‐UMOD includes primary ADTKD‐MUC1 and a number of other rare genetic disorders, most of which have other associated findings. Pathogenic variants in IFT140 (Senum et al., 2022) and DNAJB11 (Cornec‐Le Gall et al., 2018) are classically associated with autosomal dominant polycystic kidney disease, but some patients may not have renal cysts. In addition mitochondrial genetic disorders may present with tubulointerstitial kidney disease (Buglioni et al., 2021; Connor et al., 2017); clinicians must be vigilant for sole maternal transmission of disease.
3.5. Diagnosis
One should consider autosomal dominant kidney disorders whenever both a parent and child present with CKD, even if an alternate diagnosis for kidney disease in either the parent or child has previously been considered. Misdiagnosis of ADTKD is common for the following reasons: (1) Surprisingly, nephrologists often do not pursue a definitive diagnosis for the cause of kidney disease, and a diagnosis that is entered in the chart may be at best an educated guess. For example, hypertensive kidney disease—the diagnosis in 25% of patients starting dialysis in the United States (Table C.11 Patient Counts: by Detailed Primary Diagnosis, 2000)—has no specific diagnostic criteria and could be applied to almost any patient with kidney disease. (2) ADTKD has a nonspecific presentation (CKD, bland urinary sediment). Given the lack of a specific diagnostic characteristic, patients with ADTKD will fit into almost any disease category. Thus, patients with ADTKD can easily be diagnosed with hypertensive kidney disease, diabetic nephropathy (if they also have diabetes), nephrosclerosis, or many other nonspecific conditions. (3) Many nephrologists are not familiar with ADTKD and do not routinely order genetic testing. (4) Even the kidney biopsy in patients with ADTKD is nonspecific, and nephrologists may use a term from the kidney biopsy (for example, nephrosclerosis) that is a pathologic description rather than a clinical diagnosis. Thus, ADTKD has often been misdiagnosed.
3.6. Genetic diagnosis
As genetic techniques evolve, the best methods for diagnosis will change. In addition, geographic variation and local clinical genetic support may result in different approaches. In general, direct sequencing of only the UMOD gene may have a high yield for clinicians with extensive clinical experience with ADTKD, but more often one should consider a broad genetic panel that includes the UMOD gene (all exons), REN gene, nephronophthisis genes (Wolf & Hildebrandt, 2011), and the genes for the rare disorders discussed in the differential (Bleyer et al., 2022). A broader gene panel is optimal because most nephrologists and geneticists may not be familiar with the clinical manifestations of the various rare disorders, and they could be missed without a broader gene panel. Whole exome sequencing (WES) or whole genome sequencing (WGS) also represents a good option if available. Of note, these approaches cannot detect MUC1 pathogenic variants (see below) (Bleyer, Kmoch, & Greka, 2019).
Interpreting variants of uncertain significance: Pathogenic missense variants that result in the gain or loss of a cysteine residue are almost certainly pathogenic. The class p.Thr62Pro variant has an intermediate effect on kidney function and alone does not cause classic ADTKD; one should consider other genetic causes (Olinger et al., 2020). The p.Arg142Gln and p.Val458Leu variants have been identified as less likely pathogenic, and pathogenic variants that occur past position amino acid position 500 are less likely to be pathogenic. As with other variants, sequencing of family members and segregation analysis will be helpful. Pathogenic variants can be tested in vitro as described by Rampoldi (Kidd et al., 2020; Vylet'al et al., 2006). For questionable variants, decreased or absent urinary uromodulin excretion and altered localization of uromodulin in kidney biopsy may aid the diagnosis (Vylet'al et al., 2006) (see Figure 2). Urinary uromodulin excretion in patients after transplantation is not informative, because uromodulin will then be excreted by the transplanted kidney (Vylet'al et al., 2006). Consultation with experts can be helpful. Contact ableyer@wakehealth.edu as needed.
FIGURE 2.
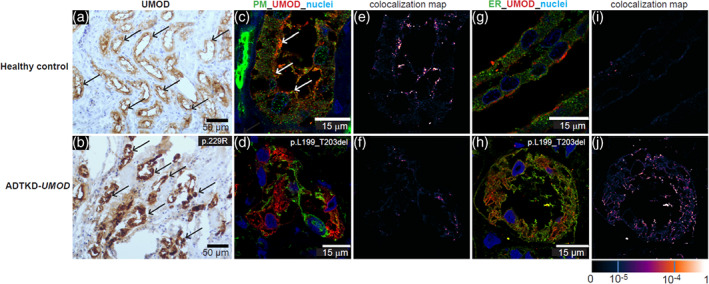
Immunohistochemical and immunoflourescent staining for uromodulin (UMOD) in kidney biopsies, which can be used to help determine if a variant of unknown significance is pathogenic. We do not recommend a kidney biopsy be performed for this purpose, but rather use biopsy material that may have previously been obtained for diagnostic purposes for the patient or another family member. (a) Horseradish peroxidase staining from a healthy control kidney biopsy. Note diffuse staining of uromodulin accentuated at the apical membrane (black arrow) throughout the tubules. In contrast, in (b), a biopsy of ADTKD‐UMOD, one sees clumping of deposits within tubular cells (black arrow). In (c) immunoflourescent staining is performed for uromodulin (in red) and pan cadherin, a marker of the plasma membrane (PM; in green) in control kidney. Uromodulin staining is localized mostly on the apical cell surface (white arrow). In contrast, in (d) uromodulin staining is seen diffusely throughout the cell in ADTKD‐UMOD. (e) Strong colocalization of uromodulin and pan cadherin at the apical surface of control kidney, whereas in (f) almost no colocalization can be seen in the UMOD patient kidney tissue. (g, h) Immunofluorescent staining with protein disulfide‐isomerase (PDI), a marker of the endoplasmic reticulum (ER; green) and uromodulin (red). In the healthy control, uromodulin again stains apically (g), but in ADTKD‐UMOD (h) the red staining is seen diffusely throughout the cell. The colocalization map shows no colocalization of the normal uromodulin with PDI (i), indicating only physiological amounts of uromodulin in the ER. In contrast, in (j) colocalization of mutant uromodulin in the ER can be seen, showing retention of the mutant uromodulin in the ER
3.7. Genetic counseling
Patients benefit from genetic counseling and from learning about this condition from clinicians with expertise in this disorder. Patients should undergo standard pre‐ and post‐genetic counseling. Genetic counseling for individuals who have CKD is straightforward. Pretest genetic counseling for genetic screening in individuals at risk for disease is more complex. First, as with testing for other forms of inherited kidney disease, the patient should be informed that positive test results should not affect health insurance (in the United States) but likely will affect ability to obtain life insurance. In an analysis of our results of genetic testing in ADTKD, we found that ~60% of patients undergoing testing and found to have a positive result thought that they had normal kidney function, when their kidney function was actually decreased (Bleyer et al., 2019). When informed of their results by a clinician with expertise in this area, 99% were happy that they undergone testing (Bleyer, Kidd, Johnson, et al., 2019). Anxiety occurred initially but dissipated over time. Thus, most patients who desire to undergo screening and are found to have a positive result are happy that they underwent testing.
Healthcare professionals should make an effort to inform other family members due to the autosomal dominant inheritance and the frequent presence of inherited kidney disease in other family members. Other family members have often been misdiagnosed or not informed of the cause of kidney disease.
3.8. Testing of minors
American Society of Human Genetics guidelines recommend against testing for children <18 years of age if a specific therapy is not available for the condition. In ADTKD‐UMOD, more than 50% of children will have a reduced eGFR by age 18 years (Bleyer et al., 2021), and ~10% of affected children develop gout before age 18 years (Bleyer et al., 2021). Thus, genetic testing should be individualized and discussed with the patient and then their parents. For example, a 16 year old who is active in sports and has a family history of early gout and kidney disease may wish to be tested and started on allopurinol to prevent gout, which could occur inopportunely before participation in an important sporting event. In contrast, a child whose family has a milder UMOD pathogenic variant, with mean age of ESKD of 55 years and a low prevalence of gout, may be discouraged from testing due to the lack of therapy and the low likelihood the disease with affect him for many years. Our experience with providing results to minors has been positive (Bleyer, Kidd, Johnson, et al., 2019).
3.9. Disease monitoring and treatment
When a positive genetic diagnosis is established, a baseline serum uric acid level and serum creatinine should be obtained. Patients will likely benefit from an initial visit with a nephrologist. The average rate of loss of eGFR is approximately 1 ml/min/year when the eGFR is >60 ml/min/1.73m2. Thus patients with an eGFR >60 ml/min/1.73m2 should have a serum creatinine obtained annually, and with increasing frequency as kidney function declines. Patients with a serum uric acid level > 6 mg/dl should have a discussion about risk factors for gout, likelihood of developing gout, and preventive therapy.
At present, there are no specific therapies for ADTKD‐UMOD. Allopurinol can be used to prevent or treat gout. If patients develop gout, it is important to begin preventive therapy, as they will be at an increasing risk of gout and uric acid burden. Allopurinol in general is well tolerated. For at‐risk groups, one may consider HLA testing to avoid possible severe allergic reactions.
A promising new potential treatment for ADTKD has been identified and hopefully will begin clinical trials in the next several years (Dvela‐Levitt et al., 2019). Treatment with sodium glucose transporter‐2 (SGLT‐2) inhibitors may also be effective and is safe (Heerspink et al., 2020). Patients with ADTKD are in general excellent transplant candidates, as ADTKD will not recur in the transplanted kidney. Patients should be encouraged to remain healthy (prevent obesity, do not smoke) and be placed on the waiting list for a kidney transplant when eGFR falls below 19 ml/min/1.73m2. We highly encourage living donor kidney transplantation from a family member who has tested negative for the family UMOD pathogenic variant or from an unrelated individual.
Patients should be made aware of multiple webinars and educational materials about ADTKD‐UMOD (contact kidney@wakehealth.edu) and a Facebook page for patient support (https://www.facebook.com/groups/1401720046675995).
4. ADTKD‐MUC1
ADTKD‐MUC1 is a pure form of CKD, with all clinical manifestations of the disease being secondary to CKD (Bleyer & Kmoch, 1993; Kiser et al., 2004; Stavrou et al., 2002).
4.1. Clinical characteristics
Similar to patients with ADTKD‐UMOD, patients with ADTKD‐MUC1 have variable progression of CKD, with a median age of end‐stage kidney disease of 45 years (Olinger et al., 2020). CKD rarely begins before age 20 years (Bleyer et al., 2021). The rate of progression is variable and approximates 1–3 ml/min/year, although some patients may remain stable at a given level of kidney function for several years.
There are no other primary clinical manifestations of this disease. Hypertension, anemia, and gout all develop related to decline in eGFR as seen with other conditions.
Mucin‐1 is a membrane‐based mucoprotein that lines the epithelial surfaces of the thick ascending limb and much of the more distal nephron (Knaup et al., 2018). It is expressed on almost all epithelial surfaces, including the nasal mucosa, alveoli, gastrointestinal tract, sebaceous glands, and breasts (Patton, Gendler, & Spicer, 1995).
4.2. Genetics and pathophysiology
MUC1 is located on chromosome 1q21. The gene is highly polymorphic due to the presence of a variable number (20–125) of degenerate 60 ± 3n base pair repeats (VNTR; Gendler, Taylor‐Papadimitriou, Duhig, Rothbard, & Burchell, 1988). Each VNTR repeat has a high (>80%) guanosine/cytosine content and consists of 60 nucleotides encoding 20 amino acids (Kirby et al., 2013). These repetitive units encode for the portion of the protein that is highly O‐glycosylated and give mucin‐1 its characteristic mucinous qualities. The MUC1 gene contains single nucleotide polymorphisms (e.g., rs4072037) that contribute to alternative splicing and production of multiple MUC1 mRNA isoforms (Kumar et al., 2017; Ligtenberg, Gennissen, Vos, & Hilkens, 1991; Zhang, Vlad, Milcarek, & Finn, 2013). The repetitive nature and the high guanosine/cytosine content (>80%) make the analysis of MUC1 sequence extremely difficult. Reference sequences and natural genetic variation of MUC1 are thus poorly characterized.
ADTKD‐MUC1 is always caused by pathogenic variants that result in the creation of the +1 frameshift MUC1 protein (MUC1fs), The most common pathogenic variant causing ADTKD‐MUC1 is the insertion of an extra cytosine within a heptanucleotide cytosine tract that is found in each VNTR unit (Kirby et al., 2013; Wopperer et al., 2022). MUC1fs as a high positive charge (pI) and deposits within the endoplasmic reticulum Golgi intermediate compartment (ERGIC) (Dvela‐Levitt et al., 2019). Human cells do not have a specific method to remove this abnormal protein, and it accumulates over time. MUC1fs accumulates in all cells that normally synthesize mucin‐1. However, only the kidney appears to be affected by the accumulation of MUC1fs, possibly because other epithelial cells turn over more rapidly and are replaced prior to the development of any toxic effects from this protein.
A number of other causative pathogenic variants have been identified. These pathogenic variants include nucleotide deletions, insertions, and duplications within MUC1 that always produce the same MUC1fs (Devuyst et al., 2019), which is toxic to kidney tubular cells (Dvela‐Levitt et al., 2019). The vast majority of these pathogenic variants occur within the variable number of tandem repeats (VNTR) region of MUC1 (Zivna et al., 2018) and very rarely (<1% of cases) before the VNTR (Yamamoto et al., 2017).
Owing to the genomic context, pathogenic variants within VNTR are difficult to genotype using existing technologies such as standard Sanger sequencing or massively parallel Illumina short read sequencing. Five factors make the genetic diagnosis of this disease challenging. First, the MUC1 coding region ranges from 1200 to >8000 base pairs in extreme cases and is difficult to amplify with long range PCR. Second, pathogenic variants are more often located on longer MUC1 alleles and significant difference between VNTR lengths often leads to under‐representation of the longer allele in the PCR amplification step. Third, the high guanosine/cytosine content causes pathogenic variants identical to those causing ADTKD‐MUC1 to be generated randomly in VNTRs during the PCR amplification step. Fourth, the high guanosine/cytosine content affects hybridization protocols that are used for target enrichment in standard panel and exome sequencing protocols. Fifth, each family has a different mutated allele that is composed of a specified number of VNTR units with pathogenic variant located in one of the VNTRs units (for example in one family the mutated MUC1 allele may be composed of 85 VNTR units with the pathogenic variant occurring in the fifth unit, whereas in another family the mutated MUC1 allele may be composed of 35 VNTR units with the pathogenic variant occurring in the fifth unit). These mutated alleles segregate with the disease in families always accompanied by the second wild type MUC1 allele that is inherited from unaffected parents and may have different lengths ranging from 20–125 VNTR units (for example 100 units). Thus the ratio between one mutated VNTR and many wild‐type VNTR copies is very low and differs widely in each affected individual (in our example there is one mutated VNTR and 184 wild type VNTR copies). The low pathogenic variant/wild type allelic ratio limits preparation of the mutated VNTR fragment during polymerase chain reaction (PCR) or hybridization steps and challenges detection of the pathogenic variant by standard sequencing or genotyping protocols where the pathogenic variant will be either not detected or will be interpreted as a sequencing error.
There are only several centers in the world that currently perform genetic analysis of ADTKD‐MUC1. The most prevalent pathogenic variant—the cytosine duplication within the heptanucleotide cytosine tract in one of the VNTRs (Kirby et al., 2013)—is being performed at the Broad Institute of MIT and Harvard (Blumenstiel et al., 2016). This assay is based on the fact that in the affected repeat unit the pathogenic variant destroys a restriction site for the endonuclease MwoI, whereas all other non‐mutated VNTR units are prone to MwoI cleavage. The mutated VNTR fragment that is intact to MwoI cleavage is then specifically PCR amplified with VNTR specific oligonucleotide primers. Next, interrogation for the presence of the extra cytosine is done by oligonucleotide probe extension detected by matrix‐assisted laser desorption/ionization time‐of‐flight (MALDI‐TOF) mass spectrometry. The same assay can also detect the less prevalent duplications or insertions occurring either within or just after the heptanucleotide cytosine tract (Zivna et al., 2018). The oligonucleotide probe extension can be alternatively detected by a mini‐sequencing method with either fluorescent or semiconductor detection (Ekici et al., 2014).
Other pathogenic variants can be identified by Illumina sequencing of the MUC1 with proprietary bioinformatics software at the Charles University in Prague (Zivna et al., 2018). This approach first identifies for each sample all possible reading frame‐changing sequences in the DNA library created by Illumina sequencing of the MUC1 amplicons, and then calculates their percentage from all obtained sequence reads. Because various pathogenic variants (that could also cause a frameshift) can be randomly introduced during the PCR amplification step, real pathogenic variants are considered only where the percentage of reads with a particular putative frameshift pathogenic variant is statistically significantly higher than in control samples.
Other pathogenic variants can also be identified by Single Molecule, Real‐Time (SMRT) Sequencing of MUC1 amplicons with PacBio platform (Wenzel et al., 2018).
While these methods identify many families with MUC1 pathogenic variants, there are still a number of families with characteristic clinical, biochemical and immunohistochemically findings, in whom ADTKD‐MUC1 is highly suspected and a pathogenic variant cannot be identified (Zivna et al., 2018).
4.3. Differential diagnosis
The differential diagnosis is similar to ADTKD‐UMOD (see above). While the presence of gout in younger individuals prior to the onset of CKD is suggestive of ADTKD‐UMOD, the absence of gout does not rule out ADTKD‐UMOD as a potential cause.
4.4. Diagnosis
The diagnosis of ADTKD‐MUC1 is incredibly difficult due to its primary clinical manifestation (CKD) being quite common in the general population and the lack of any specific, unique symptoms. In addition, the age of end‐stage kidney disease is quite variable, and the affected parent of an affected child could have mild CKD while the child has more advanced kidney failure—obscuring the possible diagnosis. The most important clue to the diagnosis is the presence of CKD in the parent and child. As with ADTKD‐UMOD, the cause of kidney disease may have been attributed to conditions such as hypertensive kidney disease or nephrosclerosis. The absence of blood and protein in the urine is also a key diagnostic finding.
Patients in whom ADTKD‐MUC1 is suspected should first undergo genetic testing with a kidney gene panel to rule out other causes. In individuals with a negative gene panel and evidence of autosomal dominant inheritance, free CLIA‐approved genetic testing is available through the Broad Institute of MIT and Harvard in conjunction with Wake Forest University School of Medicine (contact ableyer@wakehealth.edu ). If specific genetic testing for known MUC1 frameshift pathogenic variants is negative, there are several other methods available to diagnose ADTKD‐MUC1. First, as only one allele will produce wild‐type mucin‐1, the plasma levels of mucin‐1 will be approximately half as much in patients with ADTKD‐MUC1 vs. individuals without MUC1 pathogenic variants. While there is overlap, measurement of plasma mucin‐1 levels may be helpful in making a diagnosis (Vylet'al et al., 2021). In addition, even when the pathogenic variant cannot be identified, patients with ADTKD‐MUC1 will still produce the same specific MUC1fs protein, which can be identified with specific antibodies to the protein. Thus, antibody staining of tissues expressing mucin1 (and MUC1fs) can identify the presence of the abnormal MUC1fs protein and be used to diagnose ADTKD‐MUC1 (Zivna et al., 2018). Examples of antibody staining are included in Figure 3. While tissue staining is helpful, staining of urinary cells is often more practical, as urine is readily available. Moreover, as MUC1 is expressed in bladder as well as kidney, urinary cells staining for MUC1fs can be identified in the urine of patients with ADTKD‐MUC1 who have undergone kidney transplantation. Please contact ableyer@wakehealth.edu to arrange testing in individuals suspected of having ADTKD‐MUC1.
FIGURE 3.
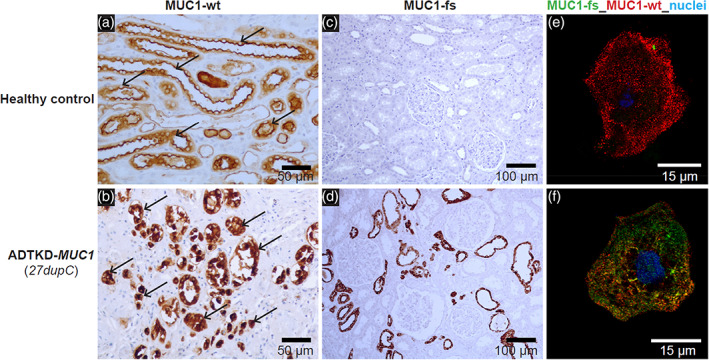
Immunostaining of previously obtained kidney biopsies or urinary cells can be used to identify MUC1 frameshift (MUC1‐fs) protein in cases where the mutation cannot be found genetically. (a) Horseradish peroxidase staining of kidney tissue from a healthy control with antibody to the normal mucin‐1 (MUC1‐wt). One can see apical staining of the MUC1 protein (black arrow), consistent with apical expression of MUC1‐wt. (b) Similar immunostaining for ADTKD‐MUC1: immunostaining shows a clumping of deposits within the cell and absence of normal apical staining (black arrow). (c) Staining with an antibody for the MUC1‐fs protein in the healthy control. No staining is found. In contrast, in (d) immunostaining with the MUC1‐fs is positive in ADTKD‐MUC1. (e, f) Staining of urinary cells for the MUC1‐wt protein (red) and the MUC1‐fs protein (green). In the healthy control, there is only positive staining for the MUC1‐wt (all staining in red). In (f) one sees the MUC1‐fs protein deposited intracellularly. The MUC1‐wt is localized partially at the cytosol and partially on plasma membrane in the urinary cells
4.5. Genetic counseling
Genetic counseling is similar to ADTKD‐UMOD (see above).
Healthcare professionals should make an effort to inform other family members due to the autosomal dominant inheritance and the frequent presence of inherited kidney disease in other family members. Other family members have often been misdiagnosed or not informed of the cause of kidney disease. It is especially important that patients with ADTKD‐MUC1 be aware of the late onset of ESKD in many affected individuals. There can be marked variation between and within families with MUC1 pathogenic variants (Bleyer et al., 2014).
4.6. Testing of minors
American Society of Human Genetics guidelines recommend against testing of children <18 years of age if a specific therapy is unavailable. It is uncommon for children with ADTKD‐MUC1 to have clinical manifestations, and eGFR is usually preserved until after age 20 (Bleyer et al., 2021). Given that an affected child may not have symptoms for up to 30 years after diagnosis, we recommend against the testing of children.
There are no specific treatments for ADTKD‐MUC1, but a promising compound has been identified (Dvela‐Levitt et al., 2019). Clinical trials are anticipated to begin in 2024. Treatment with SGLT‐2 inhibitors may be effective and is safe (Nangaku, 2020). Patients with ADTKD are in general excellent transplant candidates, as ADTKD will not recur in the transplanted kidney. Patients should be encouraged to remain healthy (prevent obesity, do not smoke) and be placed on the waiting list for kidney transplantation when eGFR falls below 19 ml/min/1.73m2. We highly encourage living donor kidney transplantation from a family member who has tested negative for a MUC1 pathogenic variant or from an unrelated individual.
Recently, mucin‐1 has been identified as a critical defense factor for COVID‐19 (Ballester, Milara, & Cortijo, 2021; Biering et al., 2022; Chatterjee, van Putten, & Strijbis, 2020; Kousathanas et al., 2022). In a survey of patients with ADTKD‐MUC1 and ADTKD‐UMOD after the Delta variant, investigators identified a 2.35 increased risk of contracting COVID‐19 for ADTKD‐MUC1 patients vs. ADTKD‐UMOD patients (p = 0.026) (Abstract: American Society of Nephrology Kidney Week,#TH‐PO380). The increased risk occurred primarily in transplant patients. Given these results, we strongly encourage full vaccination, aggressive prevention, and early treatment of COVID‐19 in ADTKD‐MUC1 patients.
Patients should be made aware of multiple webinars and educational materials about ADTKD‐MUC1 (contact kidney@wakehealth.edu) and a Facebook page for patient support (https://www.facebook.com/groups/1401720046675995).
5. ADTKD‐REN
ADTKD‐REN is caused by heterozygous pathogenic variants in the REN gene encoding preprorenin (Schaeffer et al., 2019; Zivna et al., 2009). Pathogenic variants result in decreased production and secretion of wild type prorenin and renin (resulting in the clinical manifestations of hypoaldosteronism) as well as the production of mutant preprorenin, prorenin or renin, leading to slowly progressive CKD. Patients with pathogenic variants in the signal peptide or propeptide of preprorenin present in childhood, whereas patients with pathogenic variants in the prorenin or mature renin peptide present later in life (Zivna et al., 2020).
5.1. Clinical manifestations
Patients with pathogenic variants in the signal peptide or propeptide present as early as infancy with manifestations of hypoaldosteronism and decreased eGFR (Bleyer et al., 2010). Based on an international retrospective cohort study of 111 individuals from 30 families with REN pathogenic variants (Zivna et al., 2020), initial clinical symptoms may include low blood pressure, nausea, and failure to thrive. Patients usually have low normal blood pressure readings and are not overtly hypotensive. Hyperkalemia is often identified on laboratory testing. Serum potassium levels may vary between 4 and 6.5 mEq/L; life‐threatening hyperkalemia is not found. Acidemia is also detected with serum bicarbonate levels between 17 and 24 mEq/L. Anemia also is common in children, with hemoglobin levels between 8 and 11 g/dl. Hyperuricemia may also be present. All patients have low plasma renin levels with mean plasma renin activity 0.5 ± 0.8 ng/mL/h (normal range 2.9 to 24 ng/ml/h) in one study (Zivna et al., 2020).
Patients also have a decreased eGFR, which is often detected in infancy. The decreased eGFR may be due to decreased renin production during embryogenesis leading to decreased kidney mass. In addition, patients behave in many ways as patients receiving angiotensin converting enzyme (ACE) inhibitors. Similar to patients receiving ACE inhibitors, patients have decreased blood pressure, anemia, and decreased glomerular filtration, resulting in decreased eGFR. Similar to patients receiving ACE inhibitors, acute kidney failure can develop in the setting of dehydration or viral illness (Bleyer et al., 2010). Kidney function improves with treatment but does not return to normal. While patients present early in life with a significantly decreased eGFR, kidney function remains relatively stable through childhood, and only rarely do children require renal replacement therapy before age 18, with most patients not reaching end‐stage kidney disease until after age 35 years (Zivna et al., 2020). Fortunately, many of the clinical manifestations can be treated (see below).
Patients with pathogenic variants in the mature renin peptide present in a manner similar to ADTKD‐UMOD (Schaeffer et al., 2019). They often develop hyperuricemia in their twenties and then have slowly progressive CKD, leading to the need for dialysis later in life at >50 years (Zivna et al., 2020).
Genetics and pathophysiology: Patients with pathogenic variants in the signal peptide of renin develop aberrant processing of the mutated renin, with intracellular preprorenin deposition (Zivna et al., 2020). Patients with pathogenic variants in the propeptide also have deposition of the abnormal mutated prorenin in the endoplasmic reticulum Golgi intermediate compartment (ERGIC), and patients with pathogenic variants in mature renin have deposition of mutated renin in the endoplasmic reticulum. Pathogenic variants in all three domains of preprorenin affect the cellular secretion rate of renin and prorenin. Abnormal intracellular deposition leads to intracellular stress and accelerated apoptosis of juxtaglomerular apparatus cells (where renin is predominantly synthesized), together with significantly decreased secretion rate of renin and prorenin, leads to slowly progressive CKD.
5.2. Differential diagnosis
Many of the early clinical findings (anemia, hyperuricemia, hyperkalemia, and acidemia) in ADTKD‐REN can also be found in other forms of CKD. Thus, patients may be considered to have nephronophthisis or other early forms of kidney disease. The presence of CKD, gout, mild hypotension, and anemia in the parent should suggest the diagnosis of ADTKD‐REN.
5.3. Diagnosis
Genetic testing with a gene panel is the best method of making the diagnosis. The position of the REN pathogenic variant will be associated with the clinical characteristics described above. The most common REN pathogenic variant that has been identified is p. Leu16del. Most reported variants and their clinical characteristics are identified in Zivna et al. (Zivna et al., 2020).
5.4. Approach to variants of uncertain significance
Disease‐causing variants result in a change in the amino acid sequence of the protein. Truncating variants or variants that result in loss of allelic production of renin do not result in ADTKD‐REN. Segregation can be determined within families to evaluate possible pathogenic variants. Also, transfection studies of new variants can be performed to help establish pathogenicity. While kidney biopsies should not be performed to diagnose ADTKD‐REN, patients or family members may have already undergone biopsy prior to genetic evaluation. In these individuals, immunohistochemical detection of renin in kidney biopsies can be helpful. Patients with ADTKD‐REN will have abnormal staining for renin (see Figure 4), including antibody staining of endothelial cells. Questions regarding the clinical significance of REN pathogenic variants may be directed to mzivna@lf1.cuni.cz.
FIGURE 4.
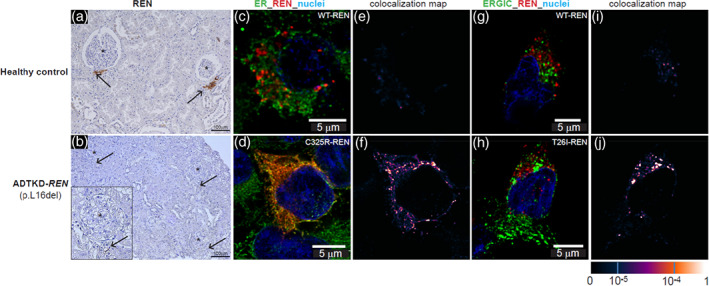
Immunostaining for renin (REN). Immunostaining of previously obtained biopsies can help determine if a variant of uncertain significance is pathogenic. (a) Horseradish peroxidase staining for renin occurring in the juxtaglomerular apparatus (black arrows) in control kidney. (b) Minimal staining of renin is found (black arrows) in the ADTKD‐REN patient kidney. (c) Immunofluorescence staining for wild type renin (WT‐REN) in transiently transfected HEK293 cells together with an ER marker (PDI). There is no colocalization identified (e). In contrast, in (d) immunostaining of the HEK293 cells transiently transfected with the p.Cys325Arg‐REN mutation (mature renin) shows deposition of renin within the ER, as evidenced by colocalization (f). (h) HEK293 cells transiently transfected with the p.Thr26Ile‐REN mutation (prosegment) immunoflourescently stained with an antibody to REN (red) and the endoplasmic reticulum‐Golgi intermediate compartment (ERGIC). In the WT‐REN (g) there is no colocalization (i). In contrast, in the p.Thr26Ile‐REN mutation, there is abundant deposition of REN within the cell (h), colocalizing with the ERGIC (j)
5.5. Treatment
Fortunately, treatments exist for many of the clinical manifestations.
Unlike almost all other forms of CKD, patients with REN pathogenic variants should not be placed on a low sodium diet but rather consume a higher sodium (typical Western) diet. Higher sodium intake will improve blood pressure and help to decrease serum potassium levels (Kmoch, Zivna, & Bleyer, 1993).
Fludrocortisone can correct the clinical manifestations of hypoaldosteronism (Bleyer et al., 2010), including hyperkalemia and acidemia. Patients placed on fludrocortisone will have a small rise in blood pressure, a lowering of serum potassium, and an improvement in acidemia (Bleyer et al., 2010). A benefit of fludrocortisone treatment is the use of one small pill twice a day, which is especially helpful in small children. Alternatively, acidosis can be treated with alkali (sodium citrate) administration (Zivna et al., 2020). However, this requires consumption of a poorly tasting pill or solution, usually in a significant volume. In a retrospective cohort study (Zivna et al., 2020), patients receiving alkali administration did not achieve normalization of serum bicarbonate, which is important for growth (Brown et al., 2020). Hyperkalemia can be treated alternatively with a low potassium diet and potassium‐binding resins—again leading to more discomfort than fludrocortisone treatment.
Anemia in patients with ADTKD‐REN is due to decreased renin production causing decreased erythropoietin production and subsequent anemia (Zivna et al., 2009). Patients may only have mild decreases in hemoglobin levels that may not need to be treated if hemoglobin levels are >10 g/dl. Erythropoietin is an effective therapy for lower hemoglobin levels.
Hyperuricemia can lead to gout, sometimes occurring in later teenage years. Hyperuricemia can be treated easily with daily administration of allopurinol.
5.6. Genetic counseling
Unlike ADTKD‐UMOD and ADTKD‐MUC1, patients with pathogenic variants in the signal peptide or propeptide have a number of treatable manifestations and thus should undergo genetic testing in childhood. Children of affected individuals should have a basic metabolic panel performed within the first several days of life and should undergo genetic testing early in life. Siblings should also be tested to rule out disease.
In families that are affected by pathogenic variants in the mature renin peptide, family members benefit from genetic counseling and from learning about this condition from clinicians with expertise in this disorder. Patients should undergo standard pre‐ and post‐genetic counseling. Genetic counseling for individuals who have CKD is straightforward. Pretest genetic counseling for genetic screening in individuals at risk for disease is more complex. First, as with testing for other forms of inherited kidney disease, the patient should be informed that positive test results should not affect health insurance (in the United States) but likely will affect ability to obtain life insurance. Healthcare professionals should make an effort to inform other family members due to the autosomal dominant inheritance and the frequent presence of inherited kidney disease in other family members. Other family members have often been misdiagnosed or not informed of the cause of kidney disease.
Family members should be made aware of educational information and webinars available (contact kidney@wakehealth.edu).
6. OTHER AUTOSOMAL DOMINANT TUBULOINTERSTITIAL KIDNEY DISEASES
The list of autosomal dominant tubulointerstitial kidney diseases continues to expand over time. Here we provide a brief description of other conditions with helpful references.
ADTKD‐HNF1B. HNF1B is a key regulator of embryonic kidney development, as well as of development in other organs. Heterozygous pathogenic variants in HNF1B are predominantly associated with kidney abnormalities, but also other systemic findings. ADTKD‐HNF1B often affects kidney development and architecture. Common findings include echogenic kidneys found by ultrasound in utero, single kidneys, dysplastic kidneys, or vesocureteral reflux. CKD with bland urinary sediment is also seen (Adalat et al., 2009; Bellanne‐Chantelot et al., 2004; Clissold et al., 2015; Faguer et al., 2011; Horikawa et al., 1997). Hypomagnesemia also occurs, and its treatment may be difficult due to constant urinary losses of magnesium (Adalat et al., 2009; Kolbuc et al., 2020). Asymptomatic elevation in liver function tests and diabetes mellitus occurring in adolescents without obesity also suggest ADTKD‐HNF1B (Edghill et al., 2008; Faguer et al., 2011; Haumaitre et al., 2006; Kolatsi‐Joannou et al., 2001; Montoli et al., 2002). Diagnosis of ADTKD‐HNF1B is difficult, as approximately 50% of the pathogenic variants are de novo (Faguer et al., 2011; Heidet et al., 2010; Izzi et al., 2020; Okorn et al., 2019). In addition, incomplete penetrance is common: for example, one family member may have CKD, diabetes, and gout; another family member may have a solitary kidney; and a third family member may be asymptomatic. Therapy is directed toward clinical manifestations, with no specific treatment available.
SEC61A1 pathogenic variants are a very rare cause of inherited kidney disease and are associated with renal cysts, tubular atrophy, neutropenia, and abscess formation (Bolar et al., 2016). Some affected individuals have intrauterine growth retardation. SEC61A1 encodes the alpha subunit of the endoplasmic reticulum translocon SEC61, and pathogenic variants affect SEC61A1 function, leading to endoplasmic reticulum stress and affecting kidney development. There is no specific treatment for this disorder.
There are a number of pathogenic variants that affect kidney structural development and anatomy, leading to kidney cysts, other structural changes, and CKD. The spectrum varies so that the clinical presentation for some families may for example involve more cystic changes, while others present predominantly with CKD and fewer cysts. Pathogenic variants that may present with CKD and cysts include HNF1B (Bleyer et al., 2021; Bleyer & Kmoch, 2020; Kolbuc et al., 2020; Okorn et al., 2019), DNAJB11(Huynh et al., 2020; Pisani et al., 2022; Senum et al., 2022), ALG5 (Lemoine et al., 2022), and IFT140 (Senum et al., 2022). Similarly, patients with congenital anomalies of the kidney and urinary tract (CAKUT) due to JAG1 (Spinner, Gilbert, Loomes, & Krantz, 1993), NOTCH2 (Kamath et al., 2012), SALL1 (Reardon et al., 2007), and GATA3 (Lemos & Thakker, 2020; Upadhyay et al., 2013) mutations may present in adulthood with CKD.
There are other conditions that are inherited in an autosomal dominant manner and are associated with CKD but also have numerous other manifestations. These conditions have been described in the section on differential diagnosis of ADTKD‐UMOD with appropriate references.
7. SUMMARY
In summary, the clinical manifestations of ADTKD are mundane (CKD with a bland urinary sediment and an unremarkable kidney ultrasound), and the condition is often misdiagnosed by nephrologists. The presence of kidney disease in a parent and child is an important diagnostic clue. A gene panel or whole exome sequencing can identify ADTKD‐UMOD and ADTKD‐REN as well as other conditions that are in the differential. If a gene panel is negative, testing for MUC1 pathogenic variants can be performed at no cost by contacting ableyer@wakehealth.edu. European providers of MUC1 genetic testing can be found at www.orpha.net. Please also contact the Rare Inherited Kidney Disease Team of Wake Forest University School of Medicine and Charles University for aid in interpretation of variants of uncertain significance (ableyer@wakehealth.edu).
CONFLICT OF INTEREST
AJB is a speaker for Natera, a company that provides inherited kidney disease multi‐gene panel testing. MZ, KOK, VB, HH, and SK have no conflicts of interest to report.
ACKNOWLEDGMENTS
SK and colleagues were supported by the grant NU21‐07‐00033 from the Ministry of Health of the Czech Republic, by the National Institute for Treatment of Metabolic and Cardiovascular Diseases (CarDia; LX22NPO5104) and grant LTAUSA19068 from the Ministry of Education, Youth and Sports of the Czech Republic and by institutional program UNCE/MED/007 of Charles University in Prague. AJB was funded by NIH‐NIDDK R21 DK106584, CKD Biomarkers Consortium Pilot and Feasibility Studies Program funded by the NIH‐NIDDK (U01 DK103225), the Slim Health Foundation, the Black‐Brogan Foundation, Soli Deo Gloria.
Živná, M. , Kidd, K. O. , Barešová, V. , Hůlková, H. , Kmoch, S. , & Bleyer, A. J. Sr (2022). Autosomal dominant tubulointerstitial kidney disease: A review. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 190C:309–324. 10.1002/ajmg.c.32008
Martina Zivna and Kendrah O. Kidd contributed equally to the manuscript.
Funding information Black‐Brogan Foundation; CKD Biomarkers Consortium Pilot and Feasibility Studies Program funded by the NIH‐NIDDK; Ministerstvo Zdravotnictví Ceské Republiky; Ministry of Education of the Czech Republic; National Center for Medical Genomics; National Institute for Treatment of Metabolic and Cardiovascular Diseases; NIH‐NIDDK; Slim Health Foundation; Soli Deo Gloria; Univerzita Karlova v Praze
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Adalat, S. , Woolf, A. S. , Johnstone, K. A. , Wirsing, A. , Harries, L. W. , Long, D. A. , … Bockenhauer, D. (2009). HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. Journal of the American Society of Nephrology, 20(5), 1123–1131. 10.1681/ASN.2008060633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenta, A. , Madero, M. , & Rodriguez‐Iturbe, B. (2022). Functional Reserve of the Kidney. Clinical Journal of the American Society of Nephrology, 17(3), 458–466. 10.2215/CJN.11070821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester, B. , Milara, J. , & Cortijo, J. (2021). The role of mucin 1 in respiratory diseases. European Respiratory Review, 30(159), 200149. 10.1183/16000617.0149-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellanne‐Chantelot, C. , Chauveau, D. , Gautier, J. F. , Dubois‐Laforgue, D. , Clauin, S. , Beaufils, S. , … Timsit, J. (2004). Clinical spectrum associated with hepatocyte nuclear factor‐1beta mutations. Annals of Internal Medicine, 140(7), 510–517. [DOI] [PubMed] [Google Scholar]
- Bernascone, I. , Vavassori, S. , Di, P. A. , Santambrogio, S. , Lamorte, G. , Amoroso, A. , … Rampoldi, L. (2006). Defective intracellular trafficking of uromodulin mutant isoforms. Traffic, 7(11), 1567–1579. 10.1111/j.1600-0854.2006.00481.x [DOI] [PubMed] [Google Scholar]
- Biering, S. B. , Sarnik, S. A. , Wang, E. , Zengel, J. R. , Leist, S. R. , Schafer, A. , … Hsu, P. D. (2022). Genome‐wide bidirectional CRISPR screens identify mucins as host factors modulating SARS‐CoV‐2 infection. Nature Genetics, 54(8), 1078–1089. 10.1038/s41588-022-01131-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleyer, A. , Westemeyer, M. , Xie, J. , Bloom, M. , Brossart, K. , Eckel, J. , … McKanna, T. (2022). Genetic etiologies for chronic kidney disease revealed through next‐generation renal gene panel. American Journal of Nephrology, 53(4), 297–306. 10.1159/000522226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleyer, A. J. , Kidd, K. , Johnson, E. , Robins, V. , Martin, L. , Taylor, A. , … Kmoch, S. (2019). Quality of life in patients with autosomal dominant tubulointerstitial kidney disease. Clinical Nephrology, 92(6), 302–311. 10.5414/CN109842 [DOI] [PubMed] [Google Scholar]
- Bleyer, A. J. , Kidd, K. , Robins, V. , Martin, L. , Taylor, A. , Santi, A. , … Kmoch, S. (2019). Outcomes of patient self‐referral for the diagnosis of several rare inherited kidney diseases. Genetics in Medicine, 22, 142–149. 10.1038/s41436-019-0617-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleyer, A. J. , Kidd, K. , Zivna, M. , & Kmoch, S. (2017). Autosomal dominant Tubulointerstitial kidney disease. Advances in Chronic Kidney Disease, 24(2), 86–93. 10.1053/j.ackd.2016.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleyer, A. J. , & Kmoch, S. (1993). Autosomal Dominant Tubulointerstitial Kidney Disease, MUC1‐Related. https://www.ncbi.nlm.nih.gov/books/NBK153723/.
- Bleyer, A. J. , & Kmoch, S. (2020). The varied clinical presentation of autosomal dominant Tubulointerstitial kidney disease due to HNF1beta mutations. Kidney International Reports, 5(12), 2133–2135. 10.1016/j.ekir.2020.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleyer, A. J. , Kmoch, S. , Antignac, C. , Robins, V. , Kidd, K. , Kelsoe, J. R. , … Hart, P. S. (2014). Variable clinical presentation of an MUC1 mutation causing medullary cystic kidney disease type 1. Clinical Journal of the American Society of Nephrology, 9(3), 527–535. 10.2215/CJN.06380613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleyer, A. J. , Kmoch, S. , & Greka, A. (2019). Diagnostic utility of exome sequencing for kidney disease. The New England Journal of Medicine, 380(21), 2080–2081. 10.1056/NEJMc1903250 [DOI] [PubMed] [Google Scholar]
- Bleyer, A. J. , Wolf, M. T. , Kidd, K. O. , Zivna, M. , & Kmoch, S. (2021). Autosomal dominant tubulointerstitial kidney disease: More than just HNF1beta. Pediatric Nephrology, 37, 933–946. 10.1007/s00467-021-05118-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleyer, A. J. , Woodard, A. S. , Shihabi, Z. , Sandhu, J. , Zhu, H. , Satko, S. G. , … Hart, T. C. (2003). Clinical characterization of a family with a mutation in the uromodulin (Tamm‐Horsfall glycoprotein) gene. Kidney International, 64(1), 36–42. [DOI] [PubMed] [Google Scholar]
- Bleyer, A. J. , Zivna, M. , Hulkova, H. , Hodanova, K. , Vyletal, P. , Sikora, J. , … Hart, P. S. (2010). Clinical and molecular characterization of a family with a dominant renin gene mutation and response to treatment with fludrocortisone. Clinical Nephrology, 74(6), 411–422. 10.5414/cnp74411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenstiel, B. , DeFelice, M. , Birsoy, O. , Bleyer, A. J. , Kmoch, S. , Carter, T. A. , … Lennon, N. J. (2016). Development and validation of a mass spectrometry‐based assay for the molecular diagnosis of Mucin‐1 kidney disease. The Journal of Molecular Diagnostics, 18(4), 566–571. 10.1016/j.jmoldx.2016.03.003 [DOI] [PubMed] [Google Scholar]
- Bockenhauer, D. , & Jaureguiberry, G. (2016). HNF1B‐associated clinical phenotypes: The kidney and beyond. Pediatric Nephrology, 31(5), 707–714. 10.1007/s00467-015-3142-2 [DOI] [PubMed] [Google Scholar]
- Bolar, N. A. , Golzio, C. , Zivna, M. , Hayot, G. , Van, H. C. , Schepers, D. , … Loeys, B. L. (2016). Heterozygous loss‐of‐function SEC61A1 mutations cause autosomal‐dominant Tubulo‐interstitial and glomerulocystic kidney disease with anemia. American Journal of Human Genetics, 99(1), 174–187. 10.1016/j.ajhg.2016.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollee, G. , Dahan, K. , Flamant, M. , Moriniere, V. , Pawtowski, A. , Heidet, L. , … Knebelmann, B. (2011). Phenotype and outcome in hereditary tubulointerstitial nephritis secondary to UMOD mutations. Clinical Journal of the American Society of Nephrology, 6(10), 2429–2438. 10.2215/CJN.01220211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, D. D. , Roem, J. , Ng, D. K. , Reidy, K. J. , Kumar, J. , Abramowitz, M. K. , … Melamed, M. L. (2020). Low serum bicarbonate and CKD progression in children. Clinical Journal of the American Society of Nephrology, 15(6), 755–765. 10.2215/CJN.07060619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunati, M. , Perucca, S. , Han, L. , Cattaneo, A. , Consolato, F. , Andolfo, A. , … Rampoldi, L. (2015). The serine protease hepsin mediates urinary secretion and polymerisation of zona Pellucida domain protein uromodulin. eLife, 4, e08887. 10.7554/eLife.08887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buglioni, A. , Hasadsri, L. , Nasr, S. H. , Hogan, M. , Moyer, A. , Siddique, K. , … Alexander, M. P. (2021). Mitochondriopathy manifesting as inherited Tubulointerstitial nephropathy without symptomatic other organ involvement. Kidney International Reports, 6(9), 2514–2518. 10.1016/j.ekir.2021.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee, M. , van Putten, J. P. M. , & Strijbis, K. (2020). Defensive properties of mucin glycoproteins during respiratory infections‐relevance for SARS‐CoV‐2. mBio, 11(6), e02374‐20. 10.1128/mBio.02374-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clissold, R. L. , Hamilton, A. J. , Hattersley, A. T. , Ellard, S. , & Bingham, C. (2015). HNF1B‐associated renal and extra‐renal disease‐an expanding clinical spectrum. Nature Reviews. Nephrology, 11(2), 102–112. 10.1038/nrneph.2014.232 [DOI] [PubMed] [Google Scholar]
- Connaughton, D. M. , Kennedy, C. , Shril, S. , Mann, N. , Murray, S. L. , Williams, P. A. , … Hildebrandt, F. (2019). Monogenic causes of chronic kidney disease in adults. Kidney International, 95(4), 914–928. 10.1016/j.kint.2018.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor, T. M. , Hoer, S. , Mallett, A. , Gale, D. P. , Gomez‐Duran, A. , Posse, V. , … Maxwell, P. H. (2017). Mutations in mitochondrial DNA causing tubulointerstitial kidney disease. PLoS Genetics, 13(3), e1006620. 10.1371/journal.pgen.1006620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormican, S. , Connaughton, D. M. , Kennedy, C. , Murray, S. , Zivna, M. , Kmoch, S. , … Conlon, P. J. (2019). Autosomal dominant tubulointerstitial kidney disease (ADTKD) in Ireland. Renal Failure, 41(1), 832–841. 10.1080/0886022X.2019.1655452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornec‐Le Gall, E. , Olson, R. J. , Besse, W. , Heyer, C. M. , Gainullin, V. G. , Smith, J. M. , … Harris, P. C. (2018). Monoallelic mutations to DNAJB11 cause atypical autosomal‐dominant polycystic kidney disease. American Journal of Human Genetics, 102(5), 832–844. 10.1016/j.ajhg.2018.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devuyst, O. , Olinger, E. , Weber, S. , Eckardt, K. U. , Kmoch, S. , Rampoldi, L. , & Bleyer, A. J. (2019). Autosomal dominant tubulointerstitial kidney disease. Nature Reviews. Disease Primers, 5(1), 60. 10.1038/s41572-019-0109-9 [DOI] [PubMed] [Google Scholar]
- Dvela‐Levitt, M. , Kost‐Alimova, M. , Emani, M. , Kohnert, E. , Thompson, R. , Sidhom, E. H. , … Greka, A. (2019). Small molecule targets TMED9 and promotes lysosomal degradation to reverse Proteinopathy. Cell, 178(3), 521–535.e523. 10.1016/j.cell.2019.07.002 [DOI] [PubMed] [Google Scholar]
- Edghill, E. L. , Oram, R. A. , Owens, M. , Stals, K. L. , Harries, L. W. , Hattersley, A. T. , … Bingham, C. (2008). Hepatocyte nuclear factor‐1beta gene deletions‐‐a common cause of renal disease. Nephrology, Dialysis, Transplantation, 23(2), 627–635. 10.1093/ndt/gfm603 [DOI] [PubMed] [Google Scholar]
- Ekici, A. B. , Hackenbeck, T. , Moriniere, V. , Pannes, A. , Buettner, M. , Uebe, S. , … Wiesener, M. S. (2014). Renal fibrosis is the common feature of autosomal dominant tubulointerstitial kidney diseases caused by mutations in mucin 1 or uromodulin. Kidney International, 86(3), 589–599. 10.1038/ki.2014.72 [DOI] [PubMed] [Google Scholar]
- Faguer, S. , Decramer, S. , Chassaing, N. , Bellanne‐Chantelot, C. , Calvas, P. , Beaufils, S. , … Chauveau, D. (2011). Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney International, 80(7), 768–776. 10.1038/ki.2011.225 [DOI] [PubMed] [Google Scholar]
- Gendler, S. , Taylor‐Papadimitriou, J. , Duhig, T. , Rothbard, J. , & Burchell, J. (1988). A highly immunogenic region of a human polymorphic epithelial mucin expressed by carcinomas is made up of tandem repeats. The Journal of Biological Chemistry, 263(26), 12820–12823. https://www.ncbi.nlm.nih.gov/pubmed/3417635 [PubMed] [Google Scholar]
- Groopman, E. E. , Marasa, M. , Cameron‐Christie, S. , Petrovski, S. , Aggarwal, V. S. , Milo‐Rasouly, H. , … Gharavi, A. G. (2019). Diagnostic utility of exome sequencing for kidney disease. The New England Journal of Medicine, 380(2), 142–151. 10.1056/NEJMoa1806891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart, T. C. , Gorry, M. C. , Hart, P. S. , Woodard, A. S. , Shihabi, Z. , Sandhu, J. , … Bleyer, A. J. (2002). Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. Journal of Medical Genetics, 39(12), 882–892. 10.1136/jmg.39.12.882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haumaitre, C. , Fabre, M. , Cormier, S. , Baumann, C. , Delezoide, A. L. , & Cereghini, S. (2006). Severe pancreas hypoplasia and multicystic renal dysplasia in two human fetuses carrying novel HNF1beta/MODY5 mutations. Human Molecular Genetics, 15(15), 2363–2375. 10.1093/hmg/ddl161 [DOI] [PubMed] [Google Scholar]
- Heerspink, H. J. L. , Stefansson, B. V. , Correa‐Rotter, R. , Chertow, G. M. , Greene, T. , Hou, F. F. , … Investigators . (2020). Dapagliflozin in patients with chronic kidney disease. The New England Journal of Medicine, 383(15), 1436–1446. 10.1056/NEJMoa2024816 [DOI] [PubMed] [Google Scholar]
- Heidet, L. , Decramer, S. , Pawtowski, A. , Moriniere, V. , Bandin, F. , Knebelmann, B. , … Salomon, R. (2010). Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clinical Journal of the American Society of Nephrology, 5(6), 1079–1090. 10.2215/CJN.06810909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikawa, Y. , Iwasaki, N. , Hara, M. , Furuta, H. , Hinokio, Y. , Cockburn, B. N. , … Bell, G. I. (1997). Mutation in hepatocyte nuclear factor‐1 beta gene (TCF2) associated with MODY. Nature Genetics, 17(4), 384–385. 10.1038/ng1297-384 [DOI] [PubMed] [Google Scholar]
- Huynh, V. T. , Audrezet, M. P. , Sayer, J. A. , Ong, A. C. , Lefevre, S. , Le Brun, V. , … Cornec‐Le Gall, E. (2020). Clinical spectrum, prognosis and estimated prevalence of DNAJB11‐kidney disease. Kidney International, 98(2), 476–487. 10.1016/j.kint.2020.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzi, C. , Dordoni, C. , Econimo, L. , Delbarba, E. , Grati, F. R. , Martin, E. , … Scolari, F. (2020). Variable expressivity of HNF1B‐nephropathy: From renal cysts and diabetes to medullary sponge kidney through Tubulo‐interstitial kidney disease. Kidney International Reports, 5(12), 2341–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath, B. M. , Podkameni, G. , Hutchinson, A. L. , Leonard, L. D. , Gerfen, J. , Krantz, I. D. , … Meyers, K. (2012). Renal anomalies in Alagille syndrome: A disease‐defining feature. American Journal of Medical Genetics. Part A, 158A(1), 85–89. 10.1002/ajmg.a.34369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd, K. , Vylet'al, P. , Schaeffer, C. , Olinger, E. , Zivna, M. , Hodanova, K. , … Bleyer, A. J. (2020). Genetic and clinical predictors of age of ESKD in individuals with autosomal dominant Tubulointerstitial kidney disease due to UMOD mutations. Kidney Int Rep, 5(9), 1472–1485. 10.1016/j.ekir.2020.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby, A. , Gnirke, A. , Jaffe, D. B. , Baresova, V. , Pochet, N. , Blumenstiel, B. , … Daly, M. J. (2013). Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nature Genetics, 45(3), 299–303. 10.1038/ng.2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser, R. L. , Wolf, M. T. F. , Martin, J. L. , Zalewski, I. , Attanasio, M. , Hildebrandt, F. , & Klemmer, P. (2004). Medullary cystic kidney disease type 1 in a large native‐American kindred. American Journal of Kidney Diseases, 44, 611–617. [PubMed] [Google Scholar]
- Kmoch, S. , Zivna, M. , & Bleyer, A. J. (1993). Autosomal dominant Tubulointerstitial kidney disease‐REN‐Related. https://www.ncbi.nlm.nih.gov/books/NBK53700/. [DOI] [PMC free article] [PubMed]
- Knaup, K. X. , Hackenbeck, T. , Popp, B. , Stoeckert, J. , Wenzel, A. , Buttner‐Herold, M. , … Wiesener, M. S. (2018). Biallelic expression of Mucin‐1 in autosomal dominant Tubulointerstitial kidney disease: Implications for nongenetic disease recognition. Journal of the American Society of Nephrology, 29(9), 2298–2309. 10.1681/ASN.2018030245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolatsi‐Joannou, M. , Bingham, C. , Ellard, S. , Bulman, M. P. , Allen, L. I. S. , Hattersley, A. T. , & Woolf, A. S. (2001). Hepatocyte nuclear factor ‐1β: A new kindred with renal cysts and diabetes and gene expression in normal human development. Journal of the American Society of Nephrology, 12, 2175–2180. [DOI] [PubMed] [Google Scholar]
- Kolbuc, M. , Lessmeier, L. , Salamon‐Slowinska, D. , Malecka, I. , Pawlaczyk, K. , Walkowiak, J. , … Zaniew, M. (2020). Hypomagnesemia is underestimated in children with HNF1B mutations. Pediatric Nephrology, 35(10), 1877–1886. 10.1007/s00467-020-04576-6 [DOI] [PubMed] [Google Scholar]
- Kousathanas, A. , Pairo‐Castineira, E. , Rawlik, K. , Stuckey, A. , Odhams, C. A. , Walker, S. , … Baillie, J. K. (2022). Whole‐genome sequencing reveals host factors underlying critical COVID‐19. Nature, 607(7917), 97–103. 10.1038/s41586-022-04576-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Cruz, E. , Joshi, S. , Patel, A. , Jahan, R. , Batra, S. K. , & Jain, M. (2017). Genetic variants of mucins: Unexplored conundrum. Carcinogenesis, 38(7), 671–679. 10.1093/carcin/bgw120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine, H. , Raud, L. , Foulquier, F. , Sayer, J. A. , Lambert, B. , Olinger, E. , … Cornec‐Le Gall, E. (2022). Monoallelic pathogenic ALG5 variants cause atypical polycystic kidney disease and interstitial fibrosis. American Journal of Human Genetics, 109(8), 1484–1499. 10.1016/j.ajhg.2022.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos, M. C. , & Thakker, R. V. (2020). Hypoparathyroidism, deafness, and renal dysplasia syndrome: 20 years after the identification of the first GATA3 mutations. Human Mutation, 41(8), 1341–1350. 10.1002/humu.24052 [DOI] [PubMed] [Google Scholar]
- Lhotta, K. , Piret, S. E. , Kramar, R. , Thakker, R. V. , Sunder‐Plassmann, G. , & Kotanko, P. (2012). Epidemiology of uromodulin‐associated kidney disease ‐ results from a nation‐wide survey. Nephron Extra, 2(1), 147–158. 10.1159/000339102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligtenberg, M. J. , Gennissen, A. M. , Vos, H. L. , & Hilkens, J. (1991). A single nucleotide polymorphism in an exon dictates allele dependent differential splicing of episialin mRNA. Nucleic Acids Research, 19(2), 297–301. 10.1093/nar/19.2.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoli, A. , Colussi, G. , Massa, O. , Caccia, R. , Rizzoni, G. , Civati, G. , & Barbetti, F. (2002). Renal cysts and diabetes syndrome linked to mutations of the hepatocyte nuclear factor‐1 beta gene: Description of a new family with associated liver involvement. American Journal of Kidney Diseases, 40(2), 397–402. 10.1053/ajkd.2002.34538 [DOI] [PubMed] [Google Scholar]
- Mutig, K. , Kahl, T. , Saritas, T. , Godes, M. , Persson, P. , Bates, J. , … Bachmann, S. (2011). Activation of the bumetanide‐sensitive Na+, K+, 2Cl‐ cotransporter (NKCC2) is facilitated by Tamm‐Horsfall protein in a chloride‐sensitive manner 1. Journal of Biological Chemistry, 286(34), 30200–30210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangaku, M. (2020). More reasons to use SGLT2 inhibitors: EMPEROR‐reduced and DAPA‐CKD. Kidney International, 98(6), 1387–1389. 10.1016/j.kint.2020.10.002 [DOI] [PubMed] [Google Scholar]
- Neumann, H. P. H. , Zauner, I. , Strahm, B. , Bender, B. U. , Schollmeyer, P. , Blum, U. , … Hildebrandt, F. (1997). Late occurrence of cysts in autosomal dominant medullary cystic kidney disease. Nephrology, Dialysis, Transplantation, 12, 1242–1246. [DOI] [PubMed] [Google Scholar]
- Okorn, C. , Goertz, A. , Vester, U. , Beck, B. B. , Bergmann, C. , Habbig, S. , … Weber, S. (2019). HNF1B nephropathy has a slow‐progressive phenotype in childhood‐with the exception of very early onset cases: Results of the German multicenter HNF1B childhood registry. Pediatric Nephrology, 34(6), 1065–1075. 10.1007/s00467-018-4188-8 [DOI] [PubMed] [Google Scholar]
- Olinger, E. , Hofmann, P. , Kidd, K. , Dufour, I. , Belge, H. , Schaeffer, C. , … Devuyst, O. (2020). Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease due to mutations in UMOD and MUC1. Kidney International, 98(3), 717–731. 10.1016/j.kint.2020.04.038 [DOI] [PubMed] [Google Scholar]
- Olinger, E. , Schaeffer, C. , Kidd, K. , Elhassan, E. A. E. , Cheng, Y. , Dufour, I. , … Devuyst, O. (2022). An intermediate‐effect size variant in UMOD confers risk for chronic kidney disease. Proceedings of the National Academy of Sciences of the United States of America, 119(33), e2114734119. 10.1073/pnas.2114734119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton, S. , Gendler, S. J. , & Spicer, A. P. (1995). The epithelial mucin, MUC1, of milk, mammary gland and other tissues 1. Biochimica et Biophysica Acta, 1241(3), 407–423. [DOI] [PubMed] [Google Scholar]
- Pisani, I. , Allinovi, M. , Palazzo, V. , Zanelli, P. , Gentile, M. , Farina, M. T. , … Manenti, L. (2022). More dissimilarities than affinities between DNAJB11‐PKD and ADPKD. Clinical Kidney Journal, 15(6), 1179–1187. 10.1093/ckj/sfac032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottel, H. , Bjork, J. , Bokenkamp, A. , Berg, U. , Asling‐Monemi, K. , Selistre, L. , … Delanaye, P. (2019). Estimating glomerular filtration rate at the transition from pediatric to adult care. Kidney International, 95(5), 1234–1243. 10.1016/j.kint.2018.12.020 [DOI] [PubMed] [Google Scholar]
- Rampoldi, L. , Scolari, F. , Amoroso, A. , Ghiggeri, G. , & Devuyst, O. (2011). The rediscovery of uromodulin (Tamm‐Horsfall protein): From tubulointerstitial nephropathy to chronic kidney disease. Kidney International, 80(4), 338–347. 10.1038/ki.2011.134 [DOI] [PubMed] [Google Scholar]
- Reardon, W. , Casserly, L. F. , Birkenhager, R. , & Kohlhase, J. (2007). Kidney failure in Townes‐brocks syndrome: An under recognized phenomenon? American Journal of Medical Genetics. Part A, 143A(21), 2588–2591. 10.1002/ajmg.a.31699 [DOI] [PubMed] [Google Scholar]
- Renigunta, A. , Renigunta, V. , Saritas, T. , Decher, N. , Mutig, K. , & Waldegger, S. (2011). Tamm‐Horsfall glycoprotein interacts with renal outer medullary potassium channel ROMK2 and regulates its function 1. Journal of Biological Chemistry, 286(3), 2224–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh, M. , Kamath, B. M. , & Chitayat, D. (2016). Alagille syndrome: Clinical perspectives. The Application of Clinical Genetics, 9, 75–82. 10.2147/TACG.S86420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer, C. , Izzi, C. , Vettori, A. , Pasqualetto, E. , Cittaro, D. , Lazarevic, D. , … Rampoldi, L. (2019). Autosomal dominant Tubulointerstitial kidney disease with adult onset due to a novel renin mutation mapping in the mature protein. Scientific Reports, 9(1), 11601. 10.1038/s41598-019-48014-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scolari, F. , Caridi, G. , Rampoldi, L. , Tardanico, R. , Izzi, C. , Pirulli, D. , … Ghiggeri, G. M. (2004). Uromodulin storage diseases: Clinical aspects and mechanisms. American Journal of Kidney Diseases, 44(6), 987–999. [DOI] [PubMed] [Google Scholar]
- Senum, S. R. , Li, Y. S. M. , Benson, K. A. , Joli, G. , Olinger, E. , Lavu, S. , … Harris, P. C. (2022). Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney‐spectrum phenotype. American Journal of Human Genetics, 109(1), 136–156. 10.1016/j.ajhg.2021.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini‐Cessi, F. , Bellabarba, G. , Malagolini, N. , & Dall'Olio, F. (1989). Rapid isolation of Tamm‐Horsfall glycoprotein (uromodulin) from human urine. Journal of Immunological Methods, 120(2), 185–189. [DOI] [PubMed] [Google Scholar]
- Serafini‐Cessi, F. , Malagolini, N. , Hoops, T. C. , & Rindler, M. J. (1993). Biosynthesis and oligosaccharide processing of human Tamm‐Horsfall glycoprotein permanently expressed in HeLa cells. Biochemical and Biophysical Research Communications, 194(2), 784–790. [DOI] [PubMed] [Google Scholar]
- Smith, G. D. , Robinson, C. , Stewart, A. P. , Edwards, E. L. , Karet, H. I. , Norden, A. G. , … Karet Frankl, F. E. (2011). Characterization of a recurrent in‐frame UMOD indel mutation causing late‐onset autosomal dominant end‐stage renal failure. Clinical Journal of the American Society of Nephrology, 6(12), 2766–2774. 10.2215/CJN.06820711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinner, N. B. , Gilbert, M. A. , Loomes, K. M. , & Krantz, I. D. (1993). Alagille Syndrome. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Mirzaa G., & Amemiya A. (Eds.), GeneReviews([R]). Seattle, WA: University of Washington, Seattle. [PubMed] [Google Scholar]
- Stavrou, C. , Koptides, M. , Tombazos, C. , Psara, E. , Patsias, C. , Zouvani, I. , … Deltas, C. C. (2002). Autosomal‐dominant medullary cystic kidney disease type 1: Clinical and molecular findings in six large Cypriot families. Kidney International, 62(4), 1385–1394. 10.1111/j.1523-1755.2002.kid581.x [DOI] [PubMed] [Google Scholar]
- Table C.11 Patient Counts: by Detailed Primary Diagnosis . (2000). In National Institute of Diabetes and Digestive and Kidney Diseases (Ed.), USRDS 2000 annual data report: Atlas of end‐stage renal disease in the United States (pp. 377–388). (Reprinted from Not in File).
- Upadhyay, J. , Steenkamp, D. W. , & Milunsky, J. M. (2013). The syndrome of hypoparathyroidism, deafness, and renal anomalies. Endocrine Practice, 19(6), 1035–1042. [DOI] [PubMed] [Google Scholar]
- Vylet'al, P. , Kidd, K. , Ainsworth, H. C. , Springer, D. , Vrbacka, A. , Pristoupilova, A. , … Bleyer, A. J. (2021). Plasma Mucin‐1 (CA15‐3) levels in autosomal dominant Tubulointerstitial kidney disease due to MUC1 mutations. American Journal of Nephrology, 52(5), 378–387. 10.1159/000515810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vylet'al, P. , Kublova, M. , Kalbacova, M. , Hodanova, K. , Baresova, V. , Stiburkova, B. , … Kmoch, S. (2006). Alterations of uromodulin biology: A common denominator of the genetically heterogeneous FJHN/MCKD syndrome. Kidney International, 70(6), 1,155–1,169. [DOI] [PubMed] [Google Scholar]
- Vyletal, P. , Bleyer, A. J. , & Kmoch, S. (2010). Uromodulin biology and pathophysiology‐‐an update. Kidney and Blood Pressure Research, 33(6), 456–475. 10.1159/000321013 [DOI] [PubMed] [Google Scholar]
- Warren, D. J. , Simmonds, H. A. , Gibson, T. , & Naik, R. B. (1981). Familial gout and renal failure. Archives of Disease in Childhood, 56, 699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, G. L. , Stanisich, J. J. , Sauer, M. M. , Lin, C.‐W. , Eras, J. , Zyla, D. S. , … Glockshuber, R. (2020). Architecture and function of human uromodulin filaments in urinary tract infections. Science, 369(6506), 1,005–1,010. 10.1126/science.aaz9866 [DOI] [PubMed] [Google Scholar]
- Wenzel, A. , Altmueller, J. , Ekici, A. B. , Popp, B. , Stueber, K. , Thiele, H. , … Beck, B. B. (2018). Single molecule real time sequencing in ADTKD‐MUC1 allows complete assembly of the VNTR and exact positioning of causative mutations. Scientific Reports, 8(1), 4,170. 10.1038/s41598-018-22428-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, M. T. (2015). Nephronophthisis and related syndromes. Current Opinion in Pediatrics, 27(2), 201–211. 10.1097/MOP.0000000000000194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, M. T. , & Hildebrandt, F. (2011). Nephronophthisis. Pediatric Nephrology, 26(2), 181–194. 10.1007/s00467-010-1585-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wopperer, F. J. , Knaup, K. X. , Stanzick, K. J. , Schneider, K. , Jobst‐Schwan, T. , Ekici, A. B. , … Wiesener, M. S. (2022). Diverse molecular causes of unsolved autosomal dominant tubulointerstitial kidney diseases. Kidney International, 102(2), 405–420. 10.1016/j.kint.2022.04.031 [DOI] [PubMed] [Google Scholar]
- Yamamoto, S. , Kaimori, J. Y. , Yoshimura, T. , Namba, T. , Imai, A. , Kobayashi, K. , … Isaka, Y. (2017). Analysis of an ADTKD family with a novel frameshift mutation in MUC1 reveals characteristic features of mutant MUC1 protein. Nephrology Dialysis Transplantation, 32(12), 2010–2017. 10.1093/ndt/gfx083 [DOI] [PubMed] [Google Scholar]
- Zaucke, F. , Boehnlein, J. M. , Steffens, S. , Polishchuk, R. S. , Rampolid, L. , Fischer, A. , … Wolf, M. T. (2010). Uromodulin is expressed in renal primary cilia and UMOD mutations result in decreased ciliary uromodulin expression. Human Molecular Genetics, 19(10), 1985–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Vlad, A. , Milcarek, C. , & Finn, O. J. (2013). Human mucin MUC1 RNA undergoes different types of alternative splicing resulting in multiple isoforms. Cancer Immunology, Immunotherapy, 62(3), 423–435. 10.1007/s00262-012-1325-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivna, M. , Hulkova, H. , Matignon, M. , Hodanova, K. , Vylet'al, P. , Kalbacova, M. , … Kmoch, S. (2009). Dominant renin gene mutations associated with early‐onset hyperuricemia, anemia, and chronic kidney failure. American Journal of Human Genetics, 85(2), 204–213. 10.1016/j.ajhg.2009.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivna, M. , Kidd, K. , Pristoupilova, A. , Baresova, V. , DeFelice, M. , Blumenstiel, B. , … Kmoch, S. (2018). Noninvasive Immunohistochemical diagnosis and novel MUC1 mutations causing autosomal dominant Tubulointerstitial kidney disease. Journal of the American Society of Nephrology, 29(9), 2,418–2,431. 10.1681/Asn.2018020180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivna, M. , Kidd, K. , Zaidan, M. , Vyletal, P. , Baresova, V. , Hodanova, K. , … Bleyer, A. J. (2020). An international cohort study of autosomal dominant Tubulointerstitial kidney disease due to REN mutations identifies distinct clinical subtypes. Kidney International, 98(6), 1589–1604. 10.1016/j.kint.2020.06.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.