

SUMMARY
Direct reprogramming of glia into neurons is a potentially promising approach for the replacement of neurons lost to injury or neurodegenerative disorders. Knockdown of the polypyrimidine tract-binding protein Ptbp1 has been recently reported to induce efficient conversion of retinal Müller glia into functional neurons. Here, we use a combination of genetic lineage tracing, single-cell RNA sequencing (scRNA-seq), and electroretinogram analysis to show that selective induction of either heterozygous or homozygous loss-of-function mutants of Ptbp1 in adult retinal Müller glia does not lead to any detectable level of neuronal conversion. Only a few changes in gene expression are observed in Müller glia following Ptbp1 deletion, and glial identity is maintained. These findings highlight the importance of using genetic manipulation and lineage-tracing methods in studying cell-type conversion.
Graphical Abstract
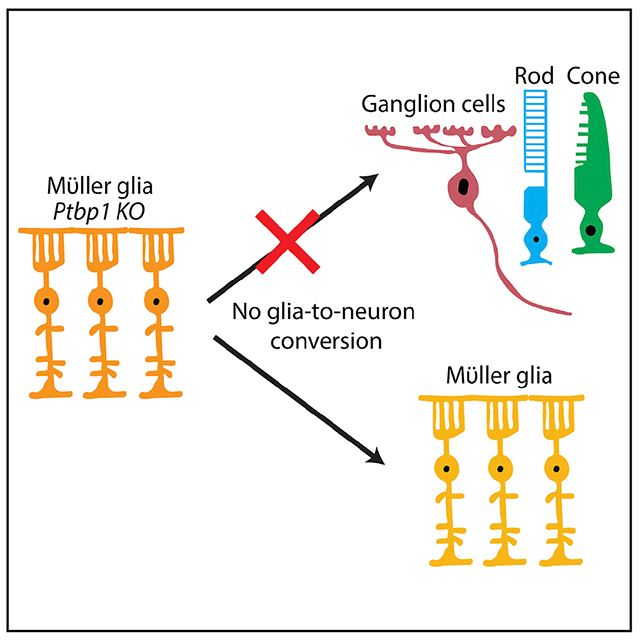
In brief
Using genetic loss of function of Ptbp1 in adult retinal Müller glia, Hoang et al. show that Ptbp1 deletion does not convert adult retinal Müller glia into neurons or significantly change Müller glial transcriptional identity.
INTRODUCTION
Neurodegenerative disorders are clinically diverse and represent a huge public-health burden. To address this, considerable effort has been focused on directed transdifferentiation of endogenous glial cells into functional neurons that were lost to disease. Most strategies to achieve this have included glial-specific overexpression of genes that promote neuronal identity, including transcription factors and microRNAs (miRNAs) (Berninger et al., 2007; Caiazzo et al., 2011; Cervo et al., 2017; Jorstad et al., 2017; Liu et al., 2015; Niu et al., 2013), or genetic or small-molecule-based manipulation of extracellular signaling pathways (Zamboni et al., 2020; Zhang et al., 2015). These approaches have achieved variable rates of success (Vignoles et al., 2019). Many treatments often work only in vitro or in immature glia or else induce only partial reprogramming. Furthermore, studies that reported highly efficient reprogramming of glia into photoreceptors or retinal ganglion cells have been criticized for lacking data that unambiguously demonstrate a lineage relationship between reprogrammed glia and neurons (Blackshaw and Sanes, 2021; Qian et al., 2021).
The ability to reliably and efficiently induce glial reprogramming in vivo by manipulation of a single factor would be a major advance towards effective cell-based therapy for neurodegenerative disorders. Several recent reports have claimed to achieve exactly this outcome through knockdown of Ptbp1 expression in retinal Müller glia and brain astrocytes (Fu et al., 2020; Maimon et al., 2021; Qian et al., 2020; Zhou et al., 2020). Ptbp1 is an RNA-binding protein and splicing regulator that is broadly expressed in non-neuronal and neuronal progenitor cells and represses neuronal-specific alternative splicing (Boutz et al., 2007; Ling et al., 2016; Makeyev et al., 2007). Neural progenitor-specific deletion of Ptbp1 leads to precocious neurogenesis (Shibasaki et al., 2013), and knockdown of Ptbp1 has been reported to be sufficient to convert both fibroblast and N2a cells into neurons in vitro (Xue et al., 2013). Furthermore, several recent papers reported that knockdown of Ptbp1 is sufficient to induce glial conversion into neurons. One study reported that Ptbp1 knockdown in retinal Müller glia using adeno-associated virus (AAV)-mediated CasRx led to rapid and efficient transdifferentiation of Müller glia into retinal ganglion cells, which were then able to efficiently innervate targets in the brain following excitotoxic inner retinal injury (Zhou et al., 2020). This same study also reported that Ptbp1 knockdown was sufficient for the efficient conversion of brain astrocytes into dopaminergic neurons in the striatum and rescued function in a 6- hydroxydopamine (6-OHDA)-induced mouse model of Parkinson’s disease (PD). A second study showed that lentiviral-mediated short hairpin RNA (shRNA) and antisense oligonucleotide (ASO)-mediated knockdown of Ptbp1 in astrocytes in the cortex, striatum, and substantia nigra all induced efficient reprogramming of astrocytes into functional neurons, which in turn also rescued neurological defects in this same PD model (Qian et al., 2020). Other studies have reported that Ptbp1 knockdown can convert retinal Müller glia to photoreceptors (Fu et al., 2020) and restore neurogenic competence in neural progenitor cells in the dentate gyrus of aged mice (Maimon et al., 2021).
This simple and elegant loss-of-function approach potentially addresses many of the difficulties associated with previous efforts toward directed glial reprogramming, particularly in a clinical setting, such as low reprogramming efficiencies and the use of complex overexpression constructs. However, several major concerns remain to be addressed. First, none of these approaches convincingly demonstrated a reduction of Ptbp1 expression in glial cells in situ. Second, lineage relationships between glia and neurons were inferred through the use of GFAP promoter-based AAV constructs or transgenes, which are known to show neuronal expression in some contexts (Fujita et al., 2014; Su et al., 2004). Third, convincing evidence for direct glia-to-neuron conversion using reliable genetic lineage analysis and/or single-cell RNA sequencing (scRNA-seq)-based trajectory analysis is missing. These concerns need to be addressed before the Ptbp1 knockdown approach can further advance toward clinical applications.
Here, we address the question of knockdown specificity and efficiency of glia-to-neuron conversion upon Ptbp1 reduction through the use of glial-specific conditional mutants of Ptbp1. We combined both genetic lineage and scRNA-seq analysis of adult wild-type Müller glia, as well as Müller glia carrying heterozygous or homozygous mutants of Ptbp1. Although we observe efficient and cell-specific disruption of Ptbp1, we observe no evidence for conversion of Müller glia into neurons in either heterozygous or homozygous Ptbp1 mutants. We do not find significant changes in either dark- or light-adapted electroretinogram (ERG) responses in mice with Müller-glial-specific Ptbp1 deletion. scRNA-seq analysis reveals only subtle changes in gene expression in mutant Müller glia. Our data indicate that the Müller-glia-to-neuron conversion reported in previous studies following Ptbp1 knockdown does not reflect the effects of Ptbp1 loss of function.
RESULTS
Genetic loss of function of Ptbp1 in retinal Müller glia did not lead to glia-to-neuron conversion
To simultaneously disrupt Ptbp1 in adult retinal Müller glia and irreversibly label these cells with a visible marker, we used three transgenic lines: GlastCreERT2, which efficiently and selectively induces Cre-dependent recombination in Müller glia following tamoxifen treatment in the adult retina (Hoang et al., 2020; de Melo et al., 2012), Sun1-GFPlox/lox, which expresses GFP targeted to the nuclear envelope under the ubiquitous CAG promoter following Cre activation (Mo et al., 2015), and Ptbp1lox/lox, in which loxP sites flank the promoter and the first coding exon of Ptbp1 and Cre activation disrupts transcription (Shibasaki et al., 2013). We then generated wild-type (GlastCreERT2;Sun1-GFPlox/lox; Ptbp1+/+), heterozygous (GlastCreERT2;Sun1-GFPlox/lox; Ptbp1lox/+), and homozygous (GlastCreERT2;Sun1-GFPlox/lox; Ptbp1lox/lox) mutant mice. Starting at ~5 weeks old, we induced Cre activation by using 4 doses of daily injections of 4-hydroxytamoxifen (4-OHT) and conducted immunohistochemical analysis 2 and 4 weeks later (Figure 1A).
Figure 1. Ptbp1 deletion does not result in the conversion of Müller glia into retinal neurons in uninjured adult mice.
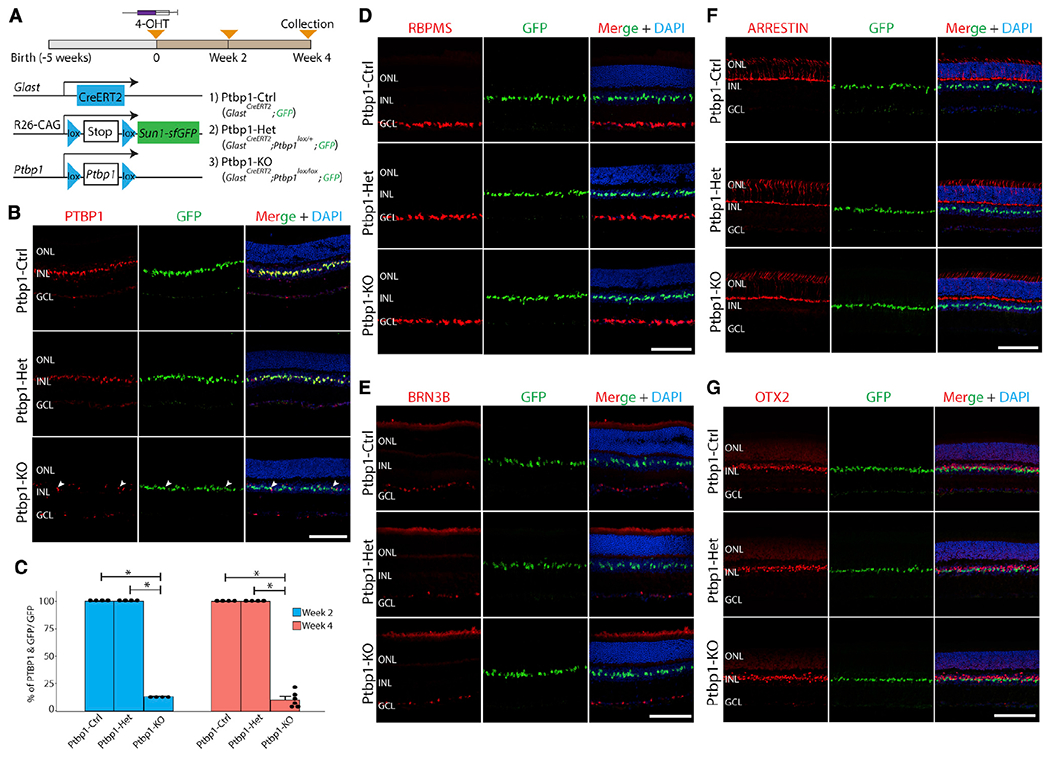
(A) A schematic diagram of the generation of specific deletion of Ptbp1 and lineage tracing of Müller glia.
(B) Representative immunohistochemistry of PTBP1 expression in wild-type (Ptbp1-Ctrl), heterozygous (Ptbp1-Het), and homozygous (Ptbp1-knockout [KO]) Ptbp1 retinas after 4 weeks of 4-OHT intraperitoneal (i.p.) injection. White arrowheads indicate residual Ptbp1-expressing Müller glial cells.
(C) Quantification of the percentage of PTBP1/GFP-double positive Müller glial cells after 2 and 4 weeks of 4-OHT i.p. injection (n ≥ 4 retinas/genotype).
(D) Representative immunohistochemistry for RBPMS, a pan-retinal ganglion cell marker, in retinal sections from three genotypes (n ≥ 6 retinas/genotype) after 4 weeks of 4-OHT i.p. injection.
(E-G) Representative images of immunohistochemical analysis of BRN3B (E), cone arrestin (F), and OTX2 (G) in retinal sections from three genotypes after 4 weeks of 4-OHT i.p. injection (n ≥ 3 retinas/genotype). ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. Scale bars, 100 μm.
Immunohistochemistry data showed that PTBP1 protein expression is enriched in Müller glia and some other non-glial cells in adult wild-type retinas (Figure 1B). We observed that Cre activation led to a ~90% reduction in the number of PTBP1-positive Müller glia cells at both 2 and 4 weeks following 4-OHT induction in the homozygous retinas (Figures 1B and 1C). GFP-positive nuclei of Müller glia remained confined to the inner nuclear layer, implying that they did not generate either retinal ganglion cells or photoreceptors (Figure 1B). To confirm this finding, we then conducted immunostaining for the retinal-ganglion-cell-specific markers RBPMS (Figures 1D, S1A, and S1B) and BRN3B (Figures 1E and S1C). In contrast to a recent report (Zhou et al., 2020), we did not observe any colocalization of these markers with GFP following Ptbp1 deletion. We likewise observed no colocalization of the cone-specific marker arrestin (Figure 1F) or the photoreceptor and bipolar marker OTX2 (Figure 1G) with GFP, in contrast to another recent study (Fu et al., 2020).
To determine whether loss of function of Ptbp1 disrupts retinal function, we performed ERG analysis to measure both rod- and cone-induced light responses in uninjured mice. We did not reveal any obvious defects or significant differences between control and Ptbp1-deficient retinas in photoreceptor-derived a-or bipolar-cell-derived b-wave amplitude dark-adapted animals or in b-wave amplitude in light-adapted animals (Figures 2A–2D).
Figure 2. Ptbp1 deletion does not affect ERG response in the uninjured retina and does not lead to the conversion of Müller glia into retinal neurons following NMDA injury.
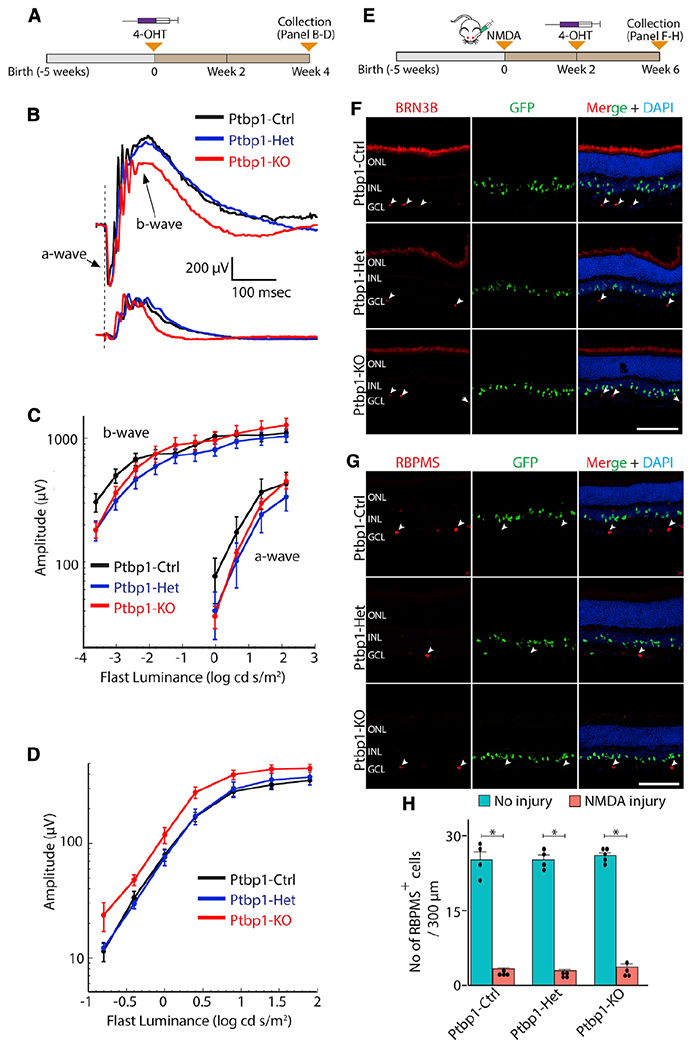
(A–D) A schematic diagram of the generation of specific Ptbp1 deletion (A) and experimental timeline for ERG response (B–D).
(B) Representative waveforms for dark- and light-adapted ERGs. No obvious defect is seen in Ptbp1-deficient retinas.
(C and D) Dark- (C) and light- (D) adapted ERGs as a function of stimulus intensity (n = 5 mice/genotype). No differences are seen between Ptbp1-deficient retinas and controls (p > 0.06).
(E–H) A schematic diagram of the generation of specific Ptbp1 deletion (E) and experimental timeline of lineage tracing of Müller glia following NMDA injury (F–H).
(F) Efficient depletion of BRN3B-positive retinal ganglion cells in all three genotypes after 6 weeks of intravitreal NMDA injection. White arrowheads indicate remaining BRN3B-positive retinal ganglion cells that survived after intravitreal NMDA injection.
(G) Representative immunohistostaining of RBPMS in retinal sections from the three genotype groups at 6 weeks after NMDA injury (n ≥ 3 retinas/genotype). White arrowheads indicate residual RBPMS-expressing ganglion cells.
(H) Quantification of RBPMS-positive cells in wild-type, heterozygous, and homozygous Ptbp1 mutant retinas (n ≥ 4 retinas/genotype) with and without NMDA injury.
ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. Scale bars, 100 μm.
It had also been previously reported that the robust conversion of Müller glia to retinal ganglion cells induced by Ptbp1 depletion resulted in the restoration of visual function following excitotoxic NMDA damage (Zhou et al., 2020). Although NMDA treatment led to extensive loss of RBPMS-positive retinal ganglion cells, we likewise did not observe any GFP/BRN3B-positive or GFP/RBPMS-positive cells nor the recovery of retinal ganglion cells after 4 weeks following NMDA injury (Figures 2E–2H). Together, our data demonstrate that loss of Ptbp1 does not convert Müller glia into retinal neurons either in healthy or damaged retinas.
Genetic loss of function of Ptbp1 leads to only subtle changes in the gene-expression profile of Müller glia
Previous studies did not characterize the gene-expression profile of glial cells after Ptbp1 knockdown (Qian et al., 2020; Zhou et al., 2020). To comprehensively profile the cellular phenotype induced by Ptbp1 deletion in Müller glia, we performed scRNA-seq analysis of retina 2 weeks following tamoxifen treatment of wild-type, heterozygous, and homozygous mice (Figures 3A–3C). The relative fraction of either Müller glia or any major subtype of retinal neurons did not change among any of the genotypes (Figure 3D). Müller glia were then subsetted for further analysis (Figure 3E). We saw a consistent reduction in Ptbp1 expression levels in Müller glia in heterozygous and homozygous mice (Figure 3F).
Figure 3. scRNA-seq analysis of mouse retinas after Müller-glia-specific Ptbp1 deletion reveals that glial identity is maintained.
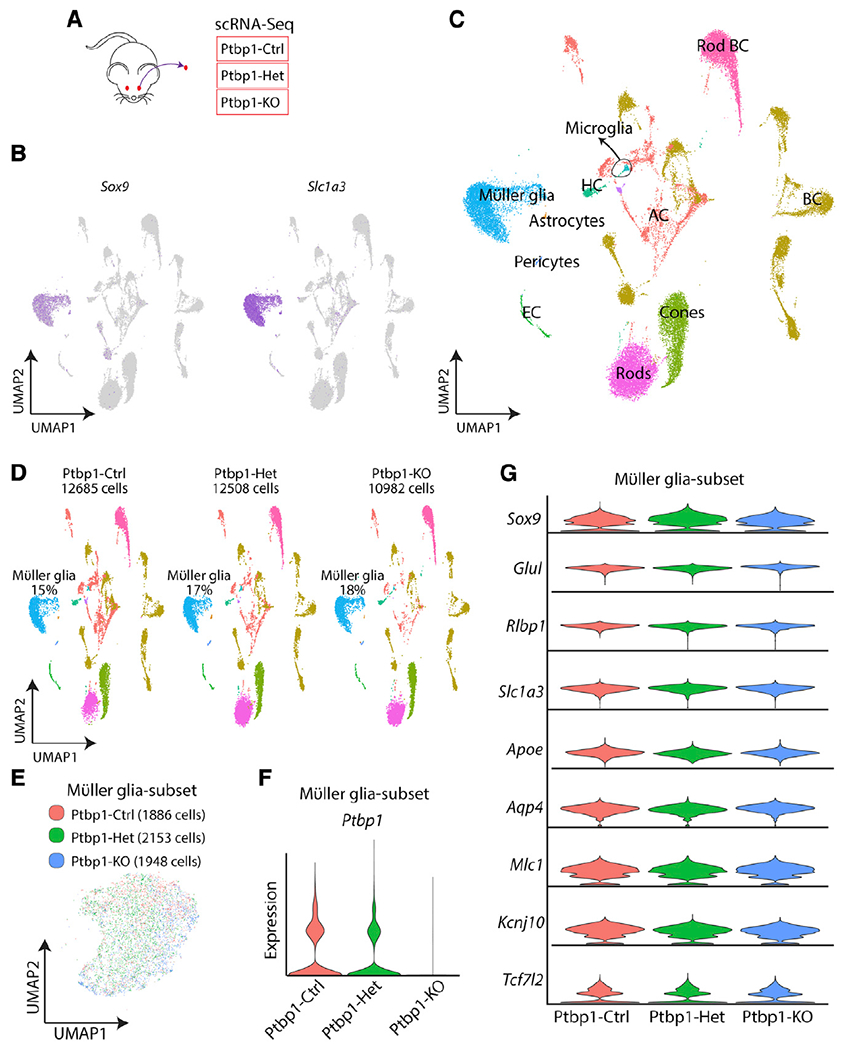
(A) A schematic diagram of scRNA-seq experiments in GlastCreERT2;Sun1-GFPlox/lox; Ptbp1+/+ (Ptbp1-Ctrl), GlastCreERT2;Sun1-GFPlox/lox; Ptbp1lox/+ (Ptbp1-Het), and GlastCreERT2;Sun1-GFPlox/lox; Ptbp1lox/lox (Ptbp1-KO) retina.
(B) Gene plots of representative Müller glia markers, Sox9 and Slc1a3.
(C) Uniform manifold approximation and projection (UMAP) plots showing scRNA-seq clusters of all three combined genotypes.
(D) UMAP plots showing scRNA-seq clusters of all three genotypes separately and the percentage of Müller glia in each scRNA-seq dataset.
(E) UMAP plot showing subsetted retinal Müller glia from all 3 genotypes. Note that there is no separated Ptbp1 knockout Müller glia cluster(s).
(F) Violin plots showing Ptbp1 RNA expression in Müller glia across the 3 genotypes.
(G) Violin plots showing no significant change in the expression of Müller glia markers across three genotypes.
We did not observe either significant reductions of expression of Müller-glia-specific markers, including Sox9, Glul, Rlbp1, Slc1a3, Apoe, Aqp4, Mlc1, Kcnj10, and Tcf7l2, or induction of genes specific to retinal progenitors or mature neurons in heterozygous and homozygous mutants (Figure 3G; Table S1). Immunostaining confirmed that Ptbp1-deficient Müller glia retained expression of the glial marker SOX9 (Figure 4A). No induction of GFAP expression was observed in Ptbp1-deficient Müller glia (Figure 4B), indicating that Ptbp1 depletion did not initiate reactive gliosis. In all cases, the changes in gene expression were quite modest (Table S1). We observed an increased expression of the Ptbp1 paralogue Ptbp2 expression level following Ptbp1 disruption (Figure 4C). Ptbp2 upregulation has previously been reported in neural progenitors following Ptbp1 loss of function (Boutz et al., 2007), and since Ptbp1 and Ptbp2 show partially redundant functions (Vuong et al., 2016), this may provide some functional compensation for Ptbp1 loss of function. We also observed increased expression of Id3, Trf, Eno1, Mt3, and Lgals3, with reduced expression of Msi2, Dio2, Rnf121, and Atp1a2 (Figure 4C). Together, our data demonstrate that loss of function of Ptbp1 does not dramatically alter gene expression in Müller glia or cause these cells to lose their identity.
Figure 4. Only subtle changes in gene expression in Müller glia are observed after Ptbp1 deletion.
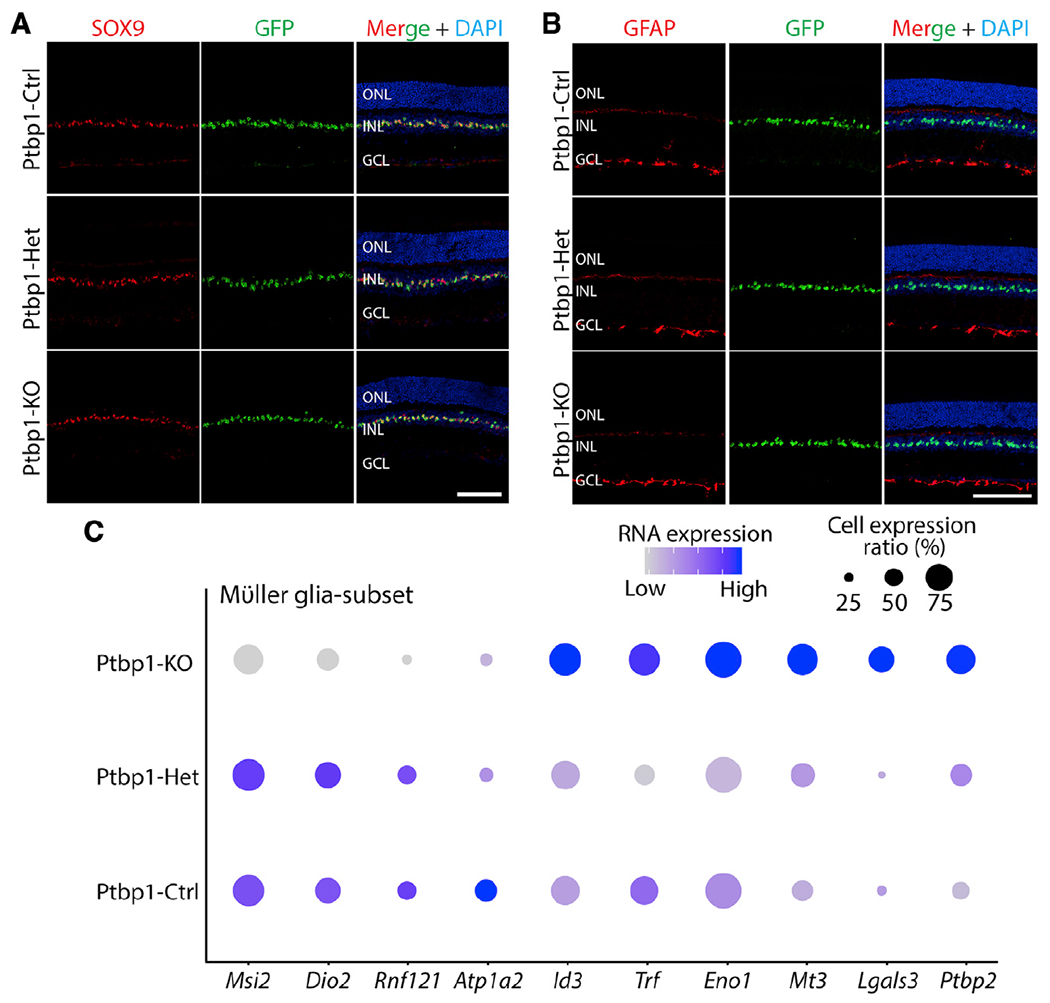
(A) Representative SOX9 and GFP immunostaining images of the retina from 3 genotypes at 4 weeks after 4-OHT i.p. injection (n ≥ 4 mice/genotype).
(B) Representative images of immunohistochemical analysis for GFAP expression in the retinas of the three genotypes after 4 weeks of 4-OHT i.p. injection (n ≥ 3 mice/genotype).
(C) Dot plots showing some mildly changed gene expression of Müller glia across the three genotypes in scRNA-seq dataset.
ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. Scale bars, 100 μm.
DISCUSSION
Using the genetic loss-of-function and cell-lineage analyses, in combination with scRNA-seq analysis, we observed no evidence that either partial or complete loss of function of Ptbp1 induces glia-to-neuron conversion in the retina. Our data contrast sharply with several recent studies that analyzed the effects of Ptbp1 knockdown using ASO, shRNA, and/or CasRx (Fu et al., 2020; Maimon et al., 2021; Qian et al., 2020; Zhou et al., 2020) but is in agreement with recent studies that reexamined previous studies reporting astrocyte-to-neuron conversion following either Neurod1 overexpression or shRNA- or CasRx-mediated Ptbp1 knockdown in brain (Chen et al.; Wang et al., 2021). These latter studies concluded that reports of glia-to-neuron conversion in these two models represented a leaky neuronal expression of GFAP-based AAV reagents used to label astrocytes. Leaky neuronal expression of GFAP-based reagents has been previously reported in other contexts (Fujita et al., 2014; Su et al., 2004), and caution should be used when interpreting results obtained using these methods without corroborating data obtained using more strongly glial-specific minipromoters and Cre lines, such as the extensively validated GlastCreERT2 line used here.
Our data show that the previous reports of Müller-glia-to-neuron conversion are unlikely to have resulted from Ptbp1 loss of function in Müller glia cells. Then, what could have accounted for the recovery of visual function following NMDA excitotoxicity? Ectopic neuronal expression of GFAP-based reagents in native neurons represents one possible explanation. The inclusion of the Ptbp1-dependent splicing of proapoptotic gene Bak1 is essential for neuronal and animal survival (Lin et al., 2020). Knockdown of residual Ptbp1 expression in neurons may therefore further promote neuronal survival. Another possibility is unexpected beneficial off-target effects of reagents targeting Ptbp1 in the previous reports. In either case, genetic methods should be used to validate future reports of glia-to-neuron conversion.
Limitations of the study
Our genetic loss-of-function approach does not precisely replicate the knockdown of Ptbp1 reported in previous studies, which used either ASOs, shRNA, or CasRx (Fu et al., 2020; Maimon et al., 2021; Qian et al., 2020; Zhou et al., 2020). It is formally possible that glia-to-neuron conversion is only efficiently induced by a reduction in Ptbp1 rather than complete loss of function. However, in Ptbp1 heterozygous conditional mutants, which show a ~50% reduction in Ptbp1 mRNA expression, we did not observe any conversion of Müller glia to retinal neurons. It is also possible that the upregulation of Ptbp2 that is seen in Ptbp1-deleted Müller glia in our study may have a potential compensatory function in maintaining glial identity, if CasRx-mediated Ptbp1 knockdown in Zhou et al. (2020) does not result in Ptbp2 upregulation. However, other studies have reported that shRNA-mediated Ptbp1 knockdown induces both Ptbp2 upregulation and neurogenesis (Qian et al., 2020; Xue et al., 2013), making this also unlikely. Finally, AAV infection might in some way promote reprogramming following Ptbp1 knockdown. However, others have recently failed to observe astrocyte-to-neuron reprogramming following AAV-mediated delivery of either Ptbp1 shRNA or CasRx constructs (Chen et al.; Wang et al., 2021), implying that AAV infection does not promote neurogenesis in combination with Ptbp1 knockdown.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Seth Blackshaw (sblack@jhmi.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
All single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon request.
No unique code is described in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PTBP1 | Proteintech | #125821-AP |
| RBPMS | Proteintech | #151871-AP |
| BRN3B | Santa Cruz | #sc6026 |
| Cone arrestin | Millipore Sigma | #AB15282 |
| OTX2 | R&D systems | #AF1979 |
| GFAP | Dako | #z0334 |
| SOX9 | Millipore Sigma | #AB5535 |
| GFP | Life technologies | #A6455 |
| GFP | Thermo Fisher Scientific | #A10262 |
| Chemicals, peptides, and recombinant proteins | ||
| NMDA | Millipore Sigma | M3262-25MG |
| Tamoxifen | Millipore Sigma | H6278-50MG |
| Papain Dissociation System | Worthington | LK003150 |
| Hibernate™-A media | Thermo Fisher Scientific | A1247501 |
| GlutaMAX™ | Thermo Fisher Scientific | 35050061 |
| B-27™ Supplement (50X), serum-free | Thermo Fisher Scientific | 17504044 |
| RNasin® Ribonuclease Inhibitor | Promega | N2615 |
| Critical commercial assays | ||
| 10× scRNA-Seq 3’ v3.1 | 10× Genomics | 1000268 |
| Deposited data | ||
| All scRNA-Seq data | GEO | GSE184933 |
| Experimental models: Organisms/strains | ||
| GlastCreERT2 | Dr. Jeremy Nathans | (de Melo et al., 2012) |
| Sun1-GFPlox/lox | Dr. Jeremy Nathans | (Mo et al., 2015) |
| Ptbp1lox/lox | Dr. Manabu Ozawa | (Shibayama et al., 2009) |
| Software and algorithms | ||
| Cell Ranger | 10× Genomics | Version 6.1.1 |
| Seurat | https://github.com/satijalab/seurat | Version 3.4 |
| ImageJ/Fiji | https://imagej.net/software/fiji/ | N/A |
| Adobe Illustrator | http://www.adobe.com | V25.2.1 |
| R | https://www.r-project.org | v3.6.1 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All experimental procedures were pre-approved by the Institutional Animal Care and Use Committee (IACUC) of the Johns Hopkins University School of Medicine and/or the Cleveland Clinic. GlastCreERT2 and Sun1-GFPlox/lox transgenic mice were provided by Dr. Jeremy Nathans (de Melo et al., 2012; Mo et al., 2015). Ptbp1lox/lox mice carrying loxP sites that flank the promoter and 1st exon of Ptbp1 were generated as described previously (Shibayama et al., 2009). To induce specific Cre activation in adult Müller, 4 consecutive doses of 4-Hydroxytamoxifen (4-OHT) intraperitoneal (i.p) injection (30 mg/kg) were performed in adult wildtype, heterozygous and homozygous mice at ~5 weeks old. Mice were sacrificed at the indicated time for analysis. All procedures were approved by the Johns Hopkins University Animal Care and Use Committee.
METHOD DETAILS
Intravitreal NMDA injection
We followed a previously described protocol (Zhou et al., 2020). Briefly, adult mice were anesthetized with 4% isoflurane inhalation. 1.5 μL of 200 mM NMDA in PBS was injected intravenously into the retinas. 4 consecutive doses of 4-OHT i.p injection (30 mg/kg) were performed at 2 weeks after NMDA injection. Mice were sacrificed, and retinas were collected at 4 weeks after 4-OHT injection.
Immunohistochemical and imaging analysis
Retinal collection and immunohistochemical analysis were performed as described previously (Hoang et al., 2020). Briefly, mice were anesthetized by CO2, and eye globes were fixed in 4% paraformaldehyde in PBS for 4 h at room temperature. Retinas were dissected and placed into 30% sucrose in PBS at 4°C overnight. Retinas were embedded in OCT, sectioned at 16 μm thickness, and stored at −20°C. Retinal sections were air-dried at 37°C for 20 min, washed 3 times × 5 min with PBS, and incubated in a blocking buffer (0.4% Triton X-100, 10% horse serum in PBS) for 1 h at room temperature. Primary antibodies were incubated in the blocking buffer at the indicated concentration overnight at 4°C. Primary antibodies used in this study were: rabbit anti-Ptbp1(1:200, Proteintech, #125821-AP), rabbit anti-RBPMS (1:200, Proteintech, #151871-AP), goat anti-BRN3B (1:200, Santa Cruz, #sc6026), rabbit anti-cone arrestin (1:200, Millipore Sigma, #AB15282), goat anti-OTX2 (1:200, R&D systems, #AF1979), rabbit anti-GFAP (1; 300, Dako, #z0334), rabbit anti-SOX9 (1:200, Millipore Sigma, #AB5535), rabbit anti-GFP (1:400, Life technologies, #A6455) and chicken anti-GFP (1:400, Thermo Fisher Scientific, #A10262). Secondary antibodies were incubated at 1:400 dilution in the blocking buffer. Retinal sections were then incubated with DAPI, washed 4 times × 5 min in PBS, mounted with ProLong Gold mounting media (ThermoFisher Scientific, #P36935), air-dried, and stored at 4°C. Images were acquired using Zeiss LSM700 confocal microscope at > 6 random regions for each retina. Images were processed using ImageJ.
Electroretinogram analysis
Mouse ERGs were recorded using a published procedure (Kinoshita and Peachey, 2018). After overnight dark adaptation, mice were anesthetized using intraperitoneal injection of ketamine (80 mg/kg) and xylazine (16 mg/kg) and placed on a temperature-controlled heating pad. The pupils were dilated with eye drops (1% tropicamide, 1% cyclopentolate, 2.5% phenylephrine HCl). Strobe flash stimuli were presented within UTAS Bigshot system (LKC Technologies) first in darkness (−3.6 to 2.1 log cd s/m2) and then superimposed upon a steady 20 cd/m2 achromatic background (−0.8 to 1.9 log cd s/m2). ERGs were recorded (0.3–1,500 Hz) using a stainless-steel wire electrode contacting the anesthetized (proparacaine HCl) corneal surface through a layer of 1% methylcellulose. The a-wave amplitude was measured at 8 msec after flash onset relative to the pre-stimulus baseline. The b-wave amplitude was measured from the a-wave trough to the peak of the b-wave, or from the pre-stimulus baseline if the a-wave was not detectable.
Cell dissociation and scRNA-Seq
Retinal cell dissociation was performed as described previously (Hoang et al., 2020). Briefly, one female mouse per genotype was euthanized by CO2, and eye globes were removed and placed in ice-cold PBS. Retinas were dissected, cells were dissociated using Papain Dissociation System (LK003150, Worthington). Dissociated cells were resuspended in a buffer containing 9.8 mL Hibernate A, 200 μL B27, 20 μL GlutaMAX and 0.5 U/μL RNAse inhibitor. Cells were filtered through a 50 μm filter. Cell count and viability were determined by using 0.4% Trypan blue.
Cells were then loaded into the 10× Genomics Chromium Single Cell System (10× Genomics) and libraries were generated using v3.1 chemistry following the manufacturer’s instructions. Libraries were sequenced on the Illumina NovaSeq platform (500 million reads per library).
QUANTIFICATION AND STATISTICAL ANALYSIS
ScRNA-Seq data analysis
Sequencing data were first processed through the Cell Ranger (v6.0.1, 10× Genomics) with default parameters, aligned to the mm10 genome (refdata-gex-mm10-2020-A), and matrix files were used for subsequent bioinformatic analysis.
Matrix data were further processed using Seurat 3.4 version (Stuart et al., 2019). Low quality cells with <500 genes, < 2000 UMI and >30% mitochondrial genes were removed. Datasets were normalized using Seurat ‘scTransform’ function. Cells were clustered and visualized using UMAP. Retinal Müller glia (Sox9+ & Slc1a3+) clusters were subsetted for further analysis to compare differential gene expression across genotypes. Differential gene expression between different genotypes was calculated using the Seurat ‘FindMarker’ function.
Image analysis
All image data were analyzed statistically by one-way ANOVA with a post hoc t test for multiple comparisons using Excel. In all tests, values of p < 0.05 were considered to indicate significance.
Supplementary Material
Highlights.
Ptbp1 is genetically disrupted selectively in adult mouse Müller glia
The fate of cells lacking Ptbp1 is analyzed with lineage tracing and molecular markers
Ptbp1 deletion does not lead to glia-to-neuron conversion in retina
scRNA-seq shows that glial identity is maintained after Ptbp1 deletion
ACKNOWLEDGMENTS
We thank F. Zhou, A. Fischer, J. Ling, Cheng Qian, and W. Yap for their comments on the manuscript. We thank the Single Cell & Transcriptomics Core (Johns Hopkins) for sequencing of scRNA-seq libraries. This work was supported by NIH National Eye Institute grants R01EY020560 and U01EY027267 to S.B. and R24EY027283 to S.B. and N.S.P., CORE grant P30EY025585 to N.S.P., and the Maryland Stem Cell Research Fund (2019-MSCRFF-5124) to D.W.K. S.B. is supported by a Stein Innovation Award from Research to Prevent Blindness.
DECLARATION OF INTERESTS
S.B. is a co-founder of, and shareholder in, CDI Labs, LLC, and receives financial support from Genentech.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110849.
REFERENCES
- Berninger B, Costa MR, Koch U, Schroeder T, Sutor B, Grothe B, and Götz M (2007). Functional properties of neurons derived from in vitro reprogrammed postnatal astroglia. J. Neurosci 27, 8654–8664. 10.1523/jneurosci.1615-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackshaw S, and Sanes JR (2021). Turning lead into gold: reprogramming retinal cells to cure blindness. J. Clin. Invest 131, e146134. 10.1172/jci146134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Stoilov P, Li Q, Lin C-H, Chawla G, Ostrow K, Shiue L, Ares M Jr., and Black DL (2007). A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 21, 1636–1652. 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caiazzo M, Dell’Anno MT, Dvoretskova E, Lazarevic D,Taverna S, Leo D, Sotnikova TD, Menegon A, Roncaglia P, Colciago G, et al. (2011). Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476, 224–227. 10.1038/nature10284. [DOI] [PubMed] [Google Scholar]
- Cervo PR, di V, di Val Cervo PR, Romanov RA, Spigolon G, Masini D, Martín-Montañez E, Toledo EM, La Manno G, Ng YH, et al. (2017). Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol 35, 444–452. 10.1038/nbt.3835. [DOI] [PubMed] [Google Scholar]
- Chen W, Zheng Q, Huang Q, Ma S, and Li M Repressing PTBP1 is incapable to convert reactive astrocytes to dopaminergic neurons in a mouse model of Parkinson’s disease. Preprint at bioRxiv. 10.1101/2021.11.12.468309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Zhu J, Duan Y, Li G, Cai H, Zheng L, Qian H, Zhang C, Jin Z, Fu X-D, et al. (2020). Visual function restoration in genetically blind mice via endogenous cellular reprogramming. Preprint at bioRxiv. 10.1101/2020.04.08.030981. [DOI] [Google Scholar]
- Fujita T, Chen MJ, Li B, Smith NA, Peng W, Sun W, Toner MJ, Kress BT, Wang L, Benraiss A, et al. (2014). Neuronal transgene expression in dominant-negative SNARE mice. J. Neurosci 34, 16594–16604. 10.1523/jneurosci.2585-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang T, Wang J, Boyd P, Wang F, Santiago C, Jiang L, Yoo S, Lahne M, Todd LJ, Jia M, et al. (2020). Gene regulatory networks controlling vertebrate retinal regeneration. Science 370, eabb8598. 10.1126/science.abb8598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorstad NL, Wilken MS, Grimes WN, Wohl SG, VandenBosch LS, Yoshimatsu T, Wong RO, Rieke F, and Reh TA (2017). Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature 548, 103–107. 10.1038/nature23283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita J, and Peachey NS (2018). Noninvasive electroretinographic procedures for the study of the mouse retina. Curr. Protoc. Mouse Biol 8, 1–16. 10.1002/cpmo.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Zhang M, Stoilov P, Chen L, and Zheng S (2020). Developmental attenuation of neuronal apoptosis by neural-specific splicing of Bak1 microexon. Neuron 107, 1180–1196.e8. 10.1016/j.neuron.2020.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JP, Chhabra R, Merran JD, Schaughency PM, Wheelan SJ, Corden JL, and Wong PC (2016). PTBP1 and PTBP2 repress nonconserved cryptic exons. Cell Rep. 17, 104–113. 10.1016/j.celrep.2016.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Miao Q, Yuan J, Han S’E, Zhang P, Li S, Rao Z, Zhao W, Ye Q, Geng J, et al. (2015). Ascl1 converts dorsal midbrain astrocytes into functional neurons in vivo. J. Neurosci 35, 9336–9355. 10.1523/jneurosci.3975-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maimon R, Chillon-Marinas C, Snethlage CE, Singhal SM, McAlonis-Downes M, Ling K, Rigo F, Bennett CF, Da Cruz S, Hnasko TS, et al. (2021). Therapeutically viable generation of neurons with antisense oligonucleotide suppression of PTB. Nat. Neurosci 24,1089–1099. 10.1038/s41593-021-00864-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makeyev EV, Zhang J, Carrasco MA, and Maniatis T (2007). The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27, 435–448. 10.1016/j.molcel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Melo J, Miki K, Rattner A, Smallwood P, Zibetti C, Hirokawa K, Monuki ES, Campochiaro PA, and Blackshaw S (2012). Injury-independent induction of reactive gliosis in retina by loss of function of the LIM homeodomain transcription factor Lhx2. Proc. Natl. Acad. Sci. U S A 109, 4657–4662. 10.1073/pnas.1107488109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, et al. (2015). Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86, 1369–1384. 10.1016/j.neuron.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W, Zang T, Zou Y, Fang S, Smith DK, Bachoo R, and Zhang C-L (2013). In vivo reprogramming of astrocytes to neuroblasts in the adult brain. Nat. Cell Biol 15, 1164–1175. 10.1038/ncb2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian C, Dong B, Wang X-Y, and Zhou F-Q (2021). In vivo glial trans-differentiation for neuronal replacement and functional recovery in central nervous system. FEBS J. 288, 4773–4785. 10.1111/febs.15681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H, Kang X, Hu J, Zhang D, Liang Z, Meng F, Zhang X, Xue Y, Maimon R, Dowdy SF, et al. (2020). Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 582, 550–556. 10.1038/s41586-020-2388-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki T, Tokunaga A, Sakamoto R, Sagara H, Noguchi S, Sasaoka T, and Yoshida N (2013). PTB deficiency causes the loss of adherens junctions in the dorsal telencephalon and leads to lethal hydrocephalus. Cereb. Cortex 23, 1824–1835. 10.1093/cercor/bhs161. [DOI] [PubMed] [Google Scholar]
- Shibayama M, Ohno S, Osaka T, Sakamoto R, Tokunaga A, Nakatake Y, Sato M, and Yoshida N (2009). Polypyrimidine tract-binding protein is essential for early mouse development and embryonic stem cell proliferation. FEBS J. 276, 6658–6668. 10.1111/j.1742-4658.2009.07380.x. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M, Hu H, Lee Y, d’Azzo A, Messing A, and Brenner M (2004). Expression specificity of GFAP transgenes. Neurochem. Res 29, 2075–2093. 10.1007/s11064-004-6881-1. [DOI] [PubMed] [Google Scholar]
- Vignoles R, Lentini C, d’Orange M, and Heinrich C (2019). Direct lineage reprogramming for brain repair: breakthroughs and challenges. Trends Mol. Med 25, 897–914. 10.1016/j.molmed.2019.06.006. [DOI] [PubMed] [Google Scholar]
- Vuong JK, Lin C-H, Zhang M, Chen L, Black DL, and Zheng S (2016). PTBP1 and PTBP2 serve both specific and redundant functions in neuronal pre-mRNA splicing. Cell Rep. 17, 2766–2775. 10.1016/j.cel-rep.2016.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L-L, Serrano C, Zhong X, Ma S, Zuo Y, and Zhang C-L (2021). Revisiting astrocyte to neuron conversion with lineage tracing in vivo. Cell 184, 5465–5481.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ouyang K, Huang J, Zhou Y, Ouyang H, Li H, Wang G, Wu Q, Wei C, Bi Y, et al. (2013). Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 152, 82–96. 10.1016/j.cell.2012.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamboni M, Llorens-Bobadilla E, Magnusson JP, and Frisén J (2020). A widespread neurogenic potential of neocortical astrocytes is induced by injury. Cell Stem Cell 27, 605–617.e5. 10.1016/j.stem.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yin J-C, Yeh H, Ma N-X, Lee G, Chen XA, Wang Y, Lin L, Chen L, Jin P, and Wu GY (2015). Small molecules efficiently reprogram human astroglial cells into functional neurons. Cell Stem Cell 17, 735–747. 10.1016/j.stem.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Su J, Hu X, Zhou C, Li H, Chen Z, Xiao Q, Wang B, Wu W, Sun Y, et al. (2020). Glia-to-Neuron conversion by CRISPR-CasRx alleviates symptoms of neurological disease in mice. Cell 181, 590–603. 10.1016/j.cell.2020.03.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon request.
No unique code is described in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PTBP1 | Proteintech | #125821-AP |
| RBPMS | Proteintech | #151871-AP |
| BRN3B | Santa Cruz | #sc6026 |
| Cone arrestin | Millipore Sigma | #AB15282 |
| OTX2 | R&D systems | #AF1979 |
| GFAP | Dako | #z0334 |
| SOX9 | Millipore Sigma | #AB5535 |
| GFP | Life technologies | #A6455 |
| GFP | Thermo Fisher Scientific | #A10262 |
| Chemicals, peptides, and recombinant proteins | ||
| NMDA | Millipore Sigma | M3262-25MG |
| Tamoxifen | Millipore Sigma | H6278-50MG |
| Papain Dissociation System | Worthington | LK003150 |
| Hibernate™-A media | Thermo Fisher Scientific | A1247501 |
| GlutaMAX™ | Thermo Fisher Scientific | 35050061 |
| B-27™ Supplement (50X), serum-free | Thermo Fisher Scientific | 17504044 |
| RNasin® Ribonuclease Inhibitor | Promega | N2615 |
| Critical commercial assays | ||
| 10× scRNA-Seq 3’ v3.1 | 10× Genomics | 1000268 |
| Deposited data | ||
| All scRNA-Seq data | GEO | GSE184933 |
| Experimental models: Organisms/strains | ||
| GlastCreERT2 | Dr. Jeremy Nathans | (de Melo et al., 2012) |
| Sun1-GFPlox/lox | Dr. Jeremy Nathans | (Mo et al., 2015) |
| Ptbp1lox/lox | Dr. Manabu Ozawa | (Shibayama et al., 2009) |
| Software and algorithms | ||
| Cell Ranger | 10× Genomics | Version 6.1.1 |
| Seurat | https://github.com/satijalab/seurat | Version 3.4 |
| ImageJ/Fiji | https://imagej.net/software/fiji/ | N/A |
| Adobe Illustrator | http://www.adobe.com | V25.2.1 |
| R | https://www.r-project.org | v3.6.1 |