

Nonalcoholic fatty liver disease (NAFLD) is an umbrella term for hepatic abnormalities, including steatosis (fat accumulation, NAFL) and nonalcoholic steatohepatitis (NASH), which is NAFL plus hepatic injury, inflammation, and fibrosis (1). NAFLD has a prevalence of ~25% worldwide and results from the inability of the liver to maintain lipid homeostasis, leading to accumulation of triglyceride (TG), the major energy-storage molecule in mammals. Obesity, insulin resistance, and diabetes mellitus are drivers of NAFLD, so it is not surprising that mechanistic target of rapamycin (mTOR), which sits at the crossroads of nutrient signaling (2), plays a critical role in its etiology. It was also expected that the role of mTOR would be complex, but the extent of this complexity seems endless. On page 364 of this issue, Gosis et al. (3) present evidence that selective inhibition of a noncanonical arm of mTOR complex 1 (mTORC1) signaling inhibits hepatic de novo lipogenesis (DNL) and protects mice from NAFLD.
The normal liver contains between 15 and 75 g of hepatic TG (1 to 5% of an ~1500-g total liver weight). In NAFL, liver fat may increase to 20 to 30% of a 2000-g liver, or ~500 g of hepatic TG. Steatosis can lead to substantial hepatic pathology, including cirrhosis and hepatocellular carcinoma. Four major metabolic processes regulate hepatic TG amounts. The major driver of TG accumulation, which accounts for 65 to 70% of hepatic TG, is delivery of plasma albumin-bound fatty acids (FAs), which are derived mainly from adipose tissue (4). The second pathway for accumulation is DNL, which is the synthesis of TG from acetyl–coenzyme A derived mainly from metabolism of glucose in the mitochondria. DNL can account for 5 to 30% of hepatic TG (4, 5). The two pathways responsible for “disposing” of hepatic TG and maintaining normal hepatic TG content are oxidation of FAs and secretion of TG in very-low-density lipoprotein (VLDL). All of these pathways, which are altered in individuals with insulin resistance, obesity, and diabetes mellitus, are regulated, at least in part, by mTOR.
mTOR was identified in the mid-1990s as a protein kinase that was the target of the immunosuppressive drug rapamycin, when in complex with 12-kDa FK506-binding protein (FKBP12) (2, 6, 7). Subsequently, the involvement of mTOR in many central cellular functions beyond immunosuppression was identified, as were two key regulatory components. mTOR exists in two distinct complexes: regulatory-associated protein of mTOR (RAPTOR) “defines” mTORC1, and rapamycin-insensitive companion of mTOR (RICTOR) defines mTORC2. There is a detailed understanding of the regulation of each mTORC by numerous proteins as well as the many downstream processes regulated by each, depending on signals from hormones, nutrients, and energy-producing pathways (2). The number of molecules involved, as well as the many autoregulatory feedback loops, suggests that there are more molecules and pathways left to be discovered, as in the case of NAFLD and DNL.
A link between insulin signaling and the sterol-regulatory element binding proteins (SREBPs), particularly SREBP-1c, was demonstrated in the late 1990s (8, 9). Subsequent studies showed that insulin signaling, through its hepatic receptor, is required for the proteolytic processing and transport to the nucleus of SREBP-1c, where it transcriptionally activates several genes required for DNL. Further studies generated inconsistent and sometimes conflicting data regarding the regulation of DNL by mTORC1. For example, studies indicated that deletion of Raptor, which reduced mTORC1 activity, or deletion of tuberous sclerosis complex 1 (Tsc1) or Tsc2, which activated mTORC1, both resulted in reduced DNL and protection from hepatic steatosis in mice (10, 11). Gosis et al. attempted to clarify these conflicting data. They identified a noncanonical pathway involving the protein folliculin (FLCN) that, when depleted in livers of mice, results in suppressed SREBP-1c activity and DNL, with protection against NAFLD.
The mechanism they uncovered involves activation of the canonical mTORC1-S6 kinase (S6K) pathway, leading to “feedback” down-regulation of FLCN and suppression of mTORC1-mediated phosphorylation of the transcription factor TFE3 by a noncanonical pathway. This results in translocation of unphosphorylated TFE3 into the nucleus, where it increases expression of insulin-induced gene 2 (Insig2), ultimately leading to inhibition of the proteolytic processing of SREBP-1c in the endoplasmic reticulum and Golgi apparatus that is necessary for its transcriptional activity (see the figure). This finding is consistent with the demonstration that activation of mTORC1 by disruption of TSC1 and TSC2 is insufficient for stimulation of DNL and requires suppression of Insig2 (11).
Figure 1. Complex regulation of de novo lipogenesis.
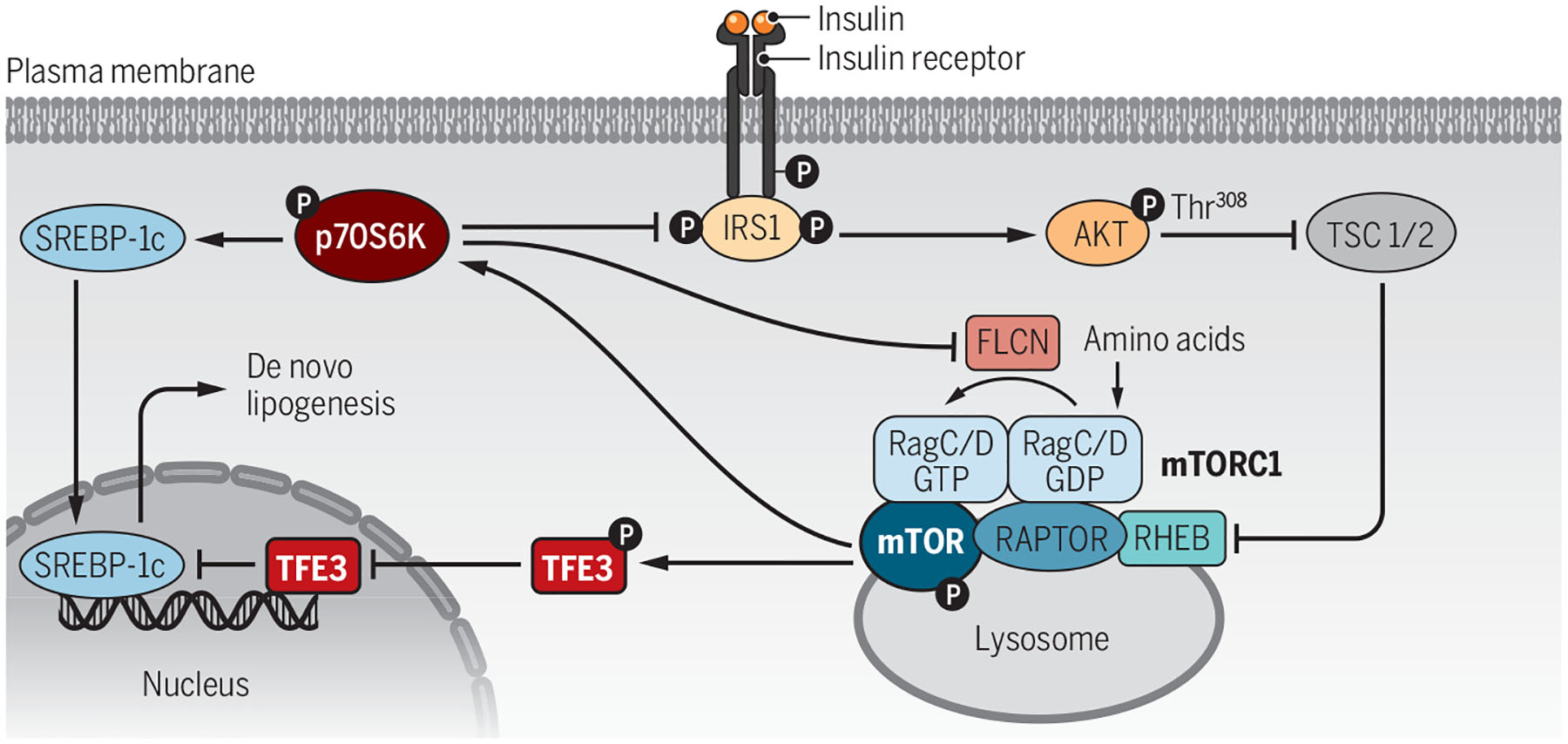
Insulin activates the insulin receptor on hepatocytes, which activates the mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) and folliculin (FLCN). FLCN inhibits transcription factor E3 (TFE3) nuclear localization and its suppression of sterol-regulatory element binding protein 1c (SREBP-1c) and de novo lipogenesis through a noncanonical pathway. Disruption of FLCN allows TFE3 to enter the nucleus and inhibit SREBP-1c without affecting p70 S6 kinase (p70S6K) activity, including its negative feedback of insulin signaling.
GDP, guanosine diphosphate; GTP, guanosine triphosphate; IRS1, insulin receptor substrate 1; P, phosphorylation; RAPTOR, regulatory-associated protein of mTOR; RHEB, Ras homolog enriched in brain; TSC, tuberous sclerosis complex.
Although Gosis et al. used mice in which Flcn was ablated for most of their work, they raised the possibility that targeting FLCN could be an effective therapy to prevent or diminish NAFLD. Targeting FLCN could avoid inhibiting targets of the canonical mTORC1 pathway, including p70S6K, which can both stimulate and inhibit DNL. It is unlikely, based on the complexity of the metabolic pathways regulated by mTORC1, that inhibiting FLCN, and thereby activating TFE3, alone would completely inhibit DNL.
There are additional insights into the complexity of hepatic mTOR signaling. The activity of mTORC1 was found to be important for the maintenance of VLDL secretion by increasing the expression of CTP:phosphocholine cytidyltransferase a (CCTa), a key enzyme in the generation of phosphatidylcholine, which is required for VLDL assembly and secretion (12). Another study focused on mTORC2 and the dual-specificity tyrosine phosphorylation-regulated kinase (DYRK1B), which has been linked to metabolic syndrome in several kindreds (13). Increasing DYRK1B expression in the livers of mice that were fed high-fat, high-sucrose diets increased hepatic DNL, FA uptake, and TG secretion, concomitant with development of hyperlipidemia and NASH (14). Disruption of mTORC2 reversed these abnormalities.
Knowledge about the role of each mTORC and their components in the regulation of DNL and the development of NAFLD remains far from complete. Additional studies, hopefully not never-ending, are needed for the development of therapies that can target components of mTOR and prevent the development of NAFLD without inhibiting the many critical roles of this master regulator of cell metabolism.
REFERENCES AND NOTES
- 1.Loomba R, Friedman SL, Shulman GI, Cell 184, 2537 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Liu GY, Sabatini DM, Nat. Rev. Mol. Cell Biol 21, 183 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gosis BS et al. , Science 376, eabf8271 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donnelly KL et al. , J. Clin. Invest 115, 1343 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith GI et al. , J. Clin. Invest 130, 1453 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown EJ et al. , Nature 369, 756 (1994). [DOI] [PubMed] [Google Scholar]
- 7.Sabers CJ et al. , J. Biol. Chem 270, 815 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Horton JD, Bashmakov Y, Shimomura I, Shimano H, Proc. Natl. Acad. Sci. U.S.A 95, 5987 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foretz M et al. , Mol. Cell. Biol 19, 3760 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peterson TR et al. , Cell 146, 408 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yecies JL et al. , Cell Metab. 14, 21 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quinn WJ 3rd et al. , J. Clin. Invest 127, 4207 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keramati AR et al. , N. Engl. J. Med 370, 1909 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhat N et al. , J. Clin. Invest 132, e153724 (2022).34855620 [Google Scholar]