

Abstract

When the nucleoside analogue acyclovir was introduced in the early 1980s, it presented a game-changing treatment modality for herpes simplex virus infections. Since then, work has been ongoing to improve the weaknesses that have now been identified: a narrow time window for therapeutic success, resistance in immunocompromised patients, little influence on frequency of recurrences, relatively fast elimination, and poor bioavailability. The present Drug Annotation focuses on the helicase–primase inhibitor pritelivir currently in development for the treatment of acyclovir-resistant HSV infections and describes how a change of the molecular target (from viral DNA polymerase to the HSV helicase–primase complex) afforded improvement of the shortcomings of nucleoside analogs. Details are presented for the discovery process leading to the final drug candidate, the pivotal preclinical studies on mechanism of action and efficacy, and on how ongoing clinical research has been able to translate preclinical promises into clinical use.
1. Introduction
Herpesviruses have been constant companions of many animal species for millions of years. Of the two subtypes of herpes simplex viruses (HSV), which are specialized to humans, herpes simplex virus type 1 (HSV-1) has coevolved longest with our human ancestors, while herpes simplex virus type 2 (HSV-2) is a later acquisition and genetically closer to chimpanzee HSV.1 HSV-1 and HSV-2 share the ability to cause local lesions and spread to peripheral neurons, where they mainly remain quiescent, awaiting reactivation following yet unknown molecular triggers. It seems that HSV-1, as the primary invader, has occupied the niche of the oral cavity, with trigeminal ganglia as the site of latency, while HSV-2, being the later intruder, has adapted to the genitalia and adjacent skin, becoming latent in the sacral ganglia.2 These localizations are not exclusive, though, and the historical assignment of HSV-1 and HSV-2 to labial and genital herpes, respectively, has been eroded to reveal substantial overlap (summarized by Whitley and Baines, 2018).3
There is no doubt that the anti-HSV drug acyclovir represents one of the few game-changing medicines introduced in the past 50 years. In terms of prescribed packages, acyclovir (together with its prodrug, valacyclovir) accounted for more than 9 million of prescriptions in the U.S. in 2019, making it no. 84 in that year’s ranking (ClinCalc DrugStats Database). Almost 40 years of clinical use have formed a clear picture of the drug’s merits but also of its weaknesses, e.g., limited oral bioavailability, the narrow time window for effective administration, suboptimal efficacy, and the potential for resistance development.
Despite a continuous drive to address these shortcomings,4 no major breakthroughs have been achieved in HSV treatment since introduction of (val)acyclovir.5,6 This may in part be attributed to the continuing focus on acyclovir’s target, the viral DNA polymerase. The only approved HSV drug to deviate from this approach is the long-chain alcohol, n-docosanol, which, while being an effective inhibitor of viral cell entry, is limited to topical administration on account of its very poor oral bioavailability.7
After years of sluggish and stepwise innovations, a new and promising avenue for development has been discovered, and it may not come as a surprise that this approach is based on a new target, i.e., the viral helicase–primase. Since the first publications of inhibitors of this enzyme complex in the mid-90s,8,9 the helicase–primase has been developed as a validated target for anti-HSV therapy.10−14 It is the scope of the present drug annotation to describe the journey of one such drug, pritelivir (synonyms BAY 57-1293, AIC316), from its discovery through its relevant chemistry and preclinical hurdles and up to its current status as a clinical phase 3 drug.
1.1. The Medical Need
While often transient and more a nuisance than a threat in immunocompetent persons, HSV infections and reactivations can take a dramatic, even life-threatening course in the setting of immune suppression, be it of iatrogenic, pathogenic, or constitutional etiology. This is the setting of most urgent medical need. However, otherwise healthy persons may also suffer painful lesions or indeed develop severe complications such as herpes simplex encephalitis, not to speak of the burden of social stigmatization that can be associated with overt manifestations.
Existing treatment modalities need to be used at the early stage (i.e., as early as possible after infection or reactivation) to alleviate the condition. It may be due to the particular characteristics of HSV infection that shortening of acute episodes and the extension of symptom-free intervals have been difficult or elusive with the current therapeutic armamentarium. Treatment success is further jeopardized by drug resistant HSV strains, a condition that poses a special threat to the immunocompromised.15,16 Thus, a wider time window for successful treatment, shortening of acute disease, more efficient suppression of viral shedding and outbreaks of latent infections, as well as therapeutic options to tackle resistant HSV strains are clear desiderata for new treatment modalities for HSV infections.
1.2. Guidepost for a New Drug
As acyclovir and its congeners are notably safe drugs, any new medicine in the field of HSV infections should reach or exceed this safety bar, at least where uncomplicated disorders in immunocompetent patients are concerned.
To justify development of a new drug for indications related to HSV infections, candidates should
be active against both HSV-1 and HSV-2,
show higher potency, especially in settings of a compromised immune system (section 2.2.1),
be efficacious without being restricted by the time elapsed since symptom onset (section 2.2.2),
prevent relapses or extend the intervals between outbreaks (section 2.2.3),
offer a resistance-breaking alternative to existing medicines, especially in immunocompromised patients (section 2.2.4),
display suitable pharmacokinetics (good oral bioavailability and sufficiently slow elimination to permit convenient dosing schedules) (section 2.2.6),
have a virus-specific mechanism of action, i.e., sparing host cells (sections 2.1.4 and 2.2.7).
These aspects are taken up in sections indicated in parentheses, demonstrating how preclinical development of pritelivir has addressed these needs.
2. Results
2.1. Discovery
The discovery of pritelivir was the result of systematic research rather than an accidental discovery. Unfortunately, systematic approaches do not guarantee a straightforward path to the ultimate drug candidate that meets all requirements. Therefore, the drug discovery process is often characterized by iterative cycles of optimizing various properties and characteristics of small molecules. The medicinal chemistry leading to pritelivir showed specific iterations along the path with some of the compounds having evolved in parallel and successful/beneficial modifications being implemented together in a later iteration. Unavoidably, several paths of development turned out to be dead ends.
2.1.1. Basic Screening
As reported in a seminal paper11 and later complemented,17 compounds of a large chemical library containing unsorted small molecules were screened for anti-HSV properties using a high-throughput screening technique in cultured cells infected with HSV-1. Basically, the scan of approximately 420 000 compounds relied on a quenched dye (fluorescein diacetate) that, upon activation by intracellular esterases, delivers a fluorescent signal. Loss of cell viability, which occurs after 5–6 days of incubation with HSV-1 or HSV-2, will suppress this signal, while the presence of an effective antiviral will preserve it, thus revealing antiviral activities regardless of the mechanism of action. The same system was used to define a selectivity index via quantification of any inherent cytotoxicity of effective test items, thus permitting the comparison of antiviral potency (EC50 of inhibition of viral damage) with the inherent cytotoxicity (CC50) of the compound alone. On this first sweep, the approximate hit rate was 1 per 1000 compounds screened; compound BAY 38-9489 with an initial activity of 0.5 μM against HSV-1 and 0.7 μM against HSV-2 was identified as the primary lead (Figure 1).
Figure 1.

Schematic overview of the discovery steps leading to first in vivo active analogues.11
2.1.2. Medicinal Chemistry: Optimization of the Primary Lead
Kleymann and colleagues11 subsequently optimized the primary lead compound in a multicycle process involving synthesis and testing of approximately 3500 analogues and derivatives, aiming not only for high in vitro potency and tolerability but also for in vivo efficacy and druggability (e.g., with a view to solubility; Figure 2).
Figure 2.

Various analogues of pritelivir (16) developed during the discovery process. Circles mark the sites of modifications, with pink ones leading to reduced activity (after data from Kleymann et al.11 and data on file).
As summarized in Figure 2, N-methylated urea in morpholino thiazole 1 (BAY 44-5138) displayed better in vitro potency than the demethylated compound 2. In addition, the methyl substituent greatly increased the free fraction when compared to the original hit BAY 38-9498 carrying a chlorine in the same position (Figure 1). Also an in vivo demethylation of N,N-dimethyl sulfonamide unit in 1 was observed that consequently led the further optimization to 3, wherein the morpholino group was replaced with a lipophilic phenyl ring and the compound exhibited a superior in vivo efficacy than valacyclovir in a murine lethal challenge model.11 Introduction of N-cyclopropyl in the sulfonamide moiety in 4 reduced the antiviral activity. A urea-to-amide transformation afforded 5 with improved in vitro potency. Replacement of N-methyl sulfonamide in 5 with other lipophilic as well as polar groups (6–8) did not yield more potent candidates. Resorting to a primary sulfonamide afforded 9, a compound with high in vitro and in vivo activity. However, solubility remained a challenging issue with several potent molecules including 9, and therefore the latter was not a suitable candidate for further development.
Introduction of substitutions on the phenyl ring (10–12) did influence the in vitro potency of the molecules but the solubility remained critical. Introduction of five-membered heterocycles in place of phenyl, for instance, a pyrazole or isoxazole in 13 and 14, respectively, afforded less potent molecules, although with improved solubility. While 3-pyridyl substituent in para-position of the phenyl ring in 15 displayed good potency, the compound was both less soluble and less active than the 2-pyridyl counterpart (16) which exhibited much better solubility and activity. Compound 16 (pritelivir) eventually combined good druggability with excellent in vivo activity (ED50 in mg/kg): 0.5 (HSV-1 and -2, see section 2.2.1 for more detail). A comparison of the respective ED50 values from murine lethal challenge assays with acyclovir/valacyclovir/pritelivir (HSV-1, 22/17/0.5; HSV-2, 16/14/0.5)11 highlights the progress achieved, and therefore 16 was selected as the drug candidate for further development.
Development of a hit to lead and lead to candidate is obviously not a predictable and straightforward way to success but an iterative process guided by trial, error, and educated guesses. On the route to pritelivir, a number of highly interesting molecules marked their presence although they were not followed up for various reasons. To set this in perspective, some efforts are exemplified in Figure 3 that in hindsight proved to be aberrant. In particular, the inverse amide idea unraveled intriguing antiviral compounds. For instance, compound 17 with an inversed-amide bond was found to be less potent than compound 9. Replacement of the thiazole core with indoline brought the urea moiety back and could retain the in vitro potency with differently substituted sulfonamides (18, 19) but did not improve the solubility of the compounds. Further replacement of phenyl group with pyrrolidine in the indoline sulfonamide (20) improved the solubility but yielded a less potent molecule both in vitro and in vivo experiments and was therefore not followed up (data from Kleymann et al.11 and on file).
Figure 3.

Some representative inverse amide/urea analogues of pritelivir (16) during the discovery process (data from Kleymann et al.11 and on file).
2.1.3. Chemistry of Pritelivir
Synthesis of pritelivir and its analogues is based on the reported methods in the literature18−20 and presented in Figure 4. A simple retrosynthetic disconnection of the target compound suggests a coupling of the thiazolyl sulfonamide and diaryl acetic acid (Figure 4a). During the course of development an optimized route applying the principles of green chemistry was developed and will be used for the commercial phase.
Figure 4.

Synthesis of pritelivir (16) and analogues: (a) disconnection approach to target molecules; (b) synthesis of thiazolyl sulfonamide reagents; (c) synthesis of diaryl acetic acids; (d) synthesis of some representative examples of pritelivir and analogues.18−20
The synthesis of the thiazolyl sulfonamide reagents begins with a reaction of chloroacetone (17) and potassium thiocyanide to give an intermediate ketone which was cyclized to the thiazole 18 by treatment with gaseous hydrochloride (Figure 4b). Chlorosulfonylation with chlorosulfonic acid and thionyl chloride resulted in the sulfonyl chloride 19 that was converted to the corresponding sulfonamides 20 and 21 after treatment with ammonia or methylamine, respectively. The 2-chloro substituent in 20 and 21 was converted to the methyl amine in an SNAr reaction to deliver the building blocks 22–23. Diaryl acetic acid reagents were synthesized using palladium catalyzed coupling reactions with organometallic intermediates formed from the corresponding halo-aryl esters (Figure 4c). Ester saponification then delivered the corresponding carboxylic acids (e.g., 26 and 28, Figure 4c). Finally, the target molecules, for instance, 5, 11, 16, and 9, were obtained using amide coupling reaction conditions with corresponding diaryl acetic acids and the thiazolyl sulfonamides (Figure 4d).18−20
2.1.4. Target and Mechanism of Action
As mentioned above, the screening method used can identify compounds acting by any mechanism able to counter or prevent the deleterious effects of HSV on cell viability. Thus, any stage of the cycle of infection and replication could have been targeted by pritelivir.
The intricate temporal organization of the replication cycle of HSV has been reviewed by Ibanez and colleagues.21 Dedicated studies revealed that pritelivir, while allowing cell infection and initiation of expression of viral immediate early genes, was associated with reduction, suppression, or loss of initiation of early and late gene expression. This was in agreement with the absence of DNA-containing C-capsids in electron microscopy.11 These findings pointed to a target (UL5 and UL52, see Figure 5) functionally localized early in the replication sequence, consistent with a report on 2-amino thiazoles, which were found to reversibly suppress HSV DNA replication by inhibition of the helicase–primase complex.9 The function of this complex is illustrated in Figure 5, showing that operation of the helicase–primase complex precedes that of the DNA polymerase, the target of nucleoside analogue drugs like acyclovir.
Figure 5.

Herpes simplex virus DNA replication as targeted by nucleoside triphosphates, for example, acyclovir, and helicase–primase inhibitors like pritelivir. The heterotrimeric helicase–primase complex is formed by UL5 (the helicase subunit, which unwinds and separates the duplex DNA to form the replication fork, exposing the two individual DNA strands), UL52 (the primase, which synthesizes short RNA primers as substrates for DNA polymerase), and UL8 (noncatalytic subunit, essential for coordination between UL5 and UL52). Single strand DNA is protected from spontaneous realignment by ICP8, also known as single-stranded DNA-binding protein. The heterodimeric DNA polymerase is composed of UL30 (catalytic subunit) and UL42 (processivity subunit). Arrows denote the direction in which the DNA polymerase complements the single strand templates. This enzyme strictly operates in the 5′–3′ direction so that one strand is processed continuously (toward the fork) and therefore faster (“leading strand”) and the other strand discontinuously (“lagging strand”). The latter process starts at multiple primed sites to produce short 5′–3′ segments, the so-called Okazaki fragments, which are linked subsequently. Detailed descriptions of these processes and proteins have been provided previously.22,23
More direct clues to the site of action of pritelivir were gained using cells transfected with one or more PCR fragments coding for the components of the helicase–primase complex (UL5, UL8, and UL52, cf. Figure 5). These were derived from either wild-type virus or HSV-1 viruses resistant to one of the discussed identified inhibitors (1).11 Infection of such pretreated cells with wild-type virus in the presence of inhibitor 5 had outcomes dependent on the type of transfection: mutant UL5 and UL52 genes conditioned the cells for appearance of viral plaques, indicating that the inhibitor had been rendered ineffective, whereas wild-type gene transfection alone, i.e., in the absence of mutant UL5 and UL52 variants, did not enable plaque formation. The results of these complementation experiments were in agreement with further direct genomic analyses of virus resistant to 5 and led to the conclusion that the helicase–primase complex is the target of these thiazole-type inhibitors.11
Resistance mutation studies (see section 2.2.4 for more detail) suggest that pritelivir interacts with the helicase–primase complex at its DNA-binding site, binding simultaneously to the helicase and primase components, thereby stabilizing the interaction between the HSV helicase–primase complex and nucleic acid,11,24 essentially freezing it in this conformation (see also section 2.2.4 for more details on resistance). A graphical representation of the molecular interactions between pritelivir and the UL5 helicase of the helicase–primase complex is presented in Figure 6.
Figure 6.

Interactions between pritelivir and amino acid residues of HSV-1 UL5 (helicase) visualized as pharmacophore features derived by LigandScout (version 4.4.7, Inte:Ligand GmbH, Vienna, Austria): hydrophobic interactions (yellow spheres) with Phe351 and Phe375, H-bond donor (green arrow) with His368, H-bond acceptor with Gln370 (red arrow), and π–cation interaction with Arg874 and Lys356 (blue ring with arrow pointing toward positively charged amino acids). Amino acids highlighted with a blue box represent those pritelivir resistant mutations in the UL5 gene protein in relation to the binding of pritelivir. The interaction features were obtained from the homology model built by Biswas and colleagues.25
2.2. Fields of Improvement
2.2.1. Potency
Increased potency was one of the goals of the discussed screening and selection process of thiazole-type helicase–primase inhibitors (cf. Figure 1). Translation of a high in vitro activity to the in vivo situation was investigated in mice, rats (this section), and guinea pigs (section 2.2.2).
Mice and rats were exposed to severe, ultimately lethal HSV infections with survival as the primary end point.11,26 ED50 values determined in mice are shown in Figure 7. The left panel presents potency of analogue compounds, which increased in the course of development. The right panel contrasts the ED50 of pritelivir with ED50 values of existing drugs. Pritelivir showed a vastly superior potency except for the comparison with ganciclovir, which was surpassed by factors of 5 (HSV-1) and 2 (HSV-2).11,26 However, these comparisons are based on mg/kg doses. Using molar doses, which seem a more appropriate basis of comparison, the respective factors were 8- and 3-fold vs ganciclovir and 42- and 37-fold higher potencies of pritelivir relative to valacyclovir.
Figure 7.

Murine lethal challenge assays comparing developmental compounds of the thiazole type (left panel) and existing HSV infection treatment modalities (right panel). Six hours after intranasal infection with HSV-1 or HSV-2, mice (n = 10 per group) were orally administered test items for 5 consecutive days, three times a day. Three-week survival rates were recorded. ED50 is the dose at which 50% of the infected animals survived (after data from refs (11) and (26)).
A similar study on pritelivir in rats comparing three daily oral doses of pritelivir (0.5 and 2 mg/kg) and valacyclovir (45 and 135 mg/kg) confirmed the superior efficacy of pritelivir.26 Consistent effects were observed in studies on the importance of treatment timing (see section 2.2.2).
Encouraging results were obtained in a mouse model using BALB/c mice,27 in which both immunocompetent and immunodeficient (athymic-nude) animals were investigated. Immunocompetent mice, infected with HSV-1 via scarification of a neck skin site, developed progressive ear thickness, marked body weight loss, and high virus titers in skin, ear pinna, and brainstem samples; 4 out of 5 mice had to be sacrificed in extremis on day 7 postinfection. As anticipated, all these effects were prevented by pritelivir in immunocompetent animals (15 mg/kg, administered orally or intraperitoneally once daily on days 1–4 postinfection; 5 mice per group). Viral titers were reduced below the level of detection after 2 days of treatment (skin) or replication was prevented entirely (ear pinna and brainstem).
Immunodeficient mice were studied in group sizes of 2 animals. Pritelivir was administered in single doses by intraperitoneal injection at 1.5 or 15 mg/kg on postinfection day 3 or on days 3 and 4. The more extensive treatment (higher dose, 2 days) caused impressive reductions of the virus yield at the inoculation and secondary sites (ear and brainstem). Also, in contrast to the lower dose groups, it prevented the formation of visible lesions and allowed body weight gain. Lower drug exposures appeared less effective but were still associated with favorable trends in virus burden.27
Although this latter set of results must be considered preliminary, as only 2 mice per group could be investigated, the data on HSV-1 shedding, in conjunction with similar results in other studies (cf. section 2.2.2, HSV-2, and section 2.2.4, HSV-1) foreshadowed corresponding outcomes in human patients (HSV-2; see section 3.6).28,29
2.2.2. Therapeutic Time Window
Particular attention was paid to the therapeutic time window. In a first dedicated study, the murine lethal challenge assay reported in section 2.2.1 was essentially repeated with a treatment delay of 72 h postinfection,30 a setting that is considered to mimic the typical clinical situation in which patients first develop symptoms and then seek treatment. As illustrated in Figure 8, pritelivir decreased the 3-week mortality already at b.i.d. doses of 0.3 mg/kg (HSV-1) or 1 mg/kg (HSV-2). These reductions were comparable to the effects of 50 mg/kg acyclovir and, in the HSV-2 arm, were enhanced at higher doses (no mortality at 10 and 30 mg/kg, twice a day). Findings of this study in experiments with HSV strains resistant to acyclovir are addressed in section 2.2.4.
Figure 8.

Potency of pritelivir therapy in murine lethal challenge studies after delayed treatment onset (n = 15 per group). Acyclovir (ACV) was used as a reference compound in ACV-sensitive and -resistant HSV strains. Drugs were orally administered (twice daily), starting 72 h after intranasal infection with HSV-1 (left panel) or HSV-2 (right panel). Three-week survival rates were recorded (after data from ref (30)) ■ = ACV sensitive HSV, treatment ACV; □ = ACV resistant HSV, treatment ACV; ● =ACV sensitive HSV, treatment pritelivir; ○ = ACV resistant HSV, treatment pritelivir.
Delayed treatment was also investigated in a model of genital herpes infection.31 Guinea pigs (10 animals per group) were infected intravaginally with HSV-2 and received oral valacyclovir (150 mg/kg) or pritelivir (20 mg/kg) twice a day, either on day 0 (one dose, 6 h postinfection), continued until day 4, or on day 4 postinfection for 10 days.
Pritelivir caused the expected drop of the lesion score not only when given as early treatment but also when therapy was commenced with a delay (Figure 9).
Figure 9.
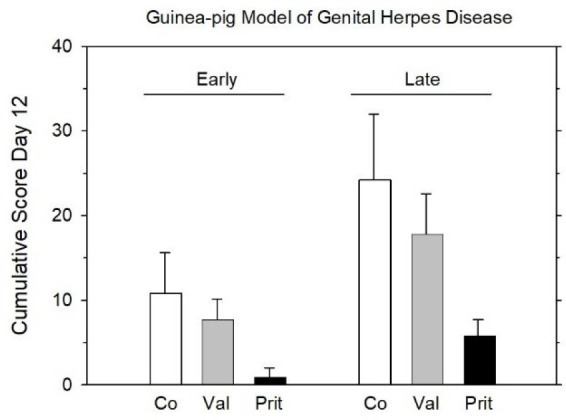
Effect of early (days 0–4 postinfection) and late treatment (starting on day 4 postinfection for 10 days) in a guinea pig model of genital herpes disease. Mean values ± SD (n = 10 per group) of cumulative lesion scores on day 12 are shown, i.e., on day 8 post-treatment with early and on day 7 of treatment with late treatment. Co, Control; Val, valacyclovir (150 mg/kg, b.i.d.); Prit, pritelivir (20 mg/kg, b.i.d.) (after data from ref (31)).
These favorable effects were accompanied by a marked decrease in viral shedding, monitored by means of daily vaginal swabs. On day 2 postinfection, shedding decreased by 3 orders of magnitude in the pritelivir group compared to controls (20 mg/kg b.i.d.). In contrast, there was no discernible effect from valacyclovir. Likewise, viral load in sacral dorsal root ganglia, determined in different sets of animals on days 7, 25, and 85 postinfection, was considerably lower in the pritelivir group than in the valacyclovir group or in the control group.31
Thus, not surprisingly, pritelivir proved to be more efficacious than valacyclovir with both early and delayed dosing schedules (Figure 9).
Finally, the retention of efficacy despite a delayed onset of therapy was also demonstrated in a murine model of cutaneous infection with HSV-2.26 These experiments are further addressed in section 2.2.3.
2.2.3. Duration, Frequency, and Suppression of Recurrences
Intuitively, it may be assumed that strong reductions in viral load (cf. sections 2.2.1 and 2.2.2) could not only lower the risk of transmission but also may shorten symptomatic episodes and delay or even prevent recurrences. Indeed, a clear reduction of the cumulative disease score, representing the composite of less episodes and reduced severity of symptoms, has been confirmed during medium- and long-term surveillance after treatment of the primary infection.31
The guinea pig model of genital herpes described in section 2.2.2, which resembles its human counterpart in terms of self-limitation and spontaneous recurrence, was used in a long-term observational study. After resolution of a primary HSV-2 infection following early drug or placebo treatment (days 0–4), guinea pigs (10 per group) were followed up for up to 85 days and recurrence rates were determined for 10-day periods. Rates for pritelivir (20 and 30 mg/kg, b.i.d.) were 0.4 and 0.3, respectively, marking statistically significant difference vs valacyclovir (0.9; 100 mg/kg t.i.d. or 150 mg/kg, b.i.d.) which was very similar to the control (1.0). Similar effects and differences were also obvious from comparison of the mean cumulative disease scores.31 This outcome is in line with the lower copy number of viral genomes detected in ganglia after pritelivir treatment compared to treatment with valacyclovir or placebo (section 2.2.2).
The above long-term observation indicates a clear reduction of recurrence rates and cumulative disease scores. A lower disease burden is also evident from delayed treatment of an acute episode, i.e., when lesions were already present, as also investigated in the guinea pig model.31 Oral treatment with pritelivir (20 mg/kg b.i.d., administered from day 4 to day 14 postinfection) halved the time to return to a disease score of zero from 14 to 7 days (n = 10 per group). Valacyclovir (150 mg/kg b.i.d.) decreased the time by only 2 days (12 vs 14 days).
Furthermore, suppression of recurrences was demonstrated in a murine model of recurrent cutaneous disease.26 HSV-2 was applied to a scarified area of the body flank, and the severity of the unfolding disease was quantified using a scoring system. Oral drug treatment (n = 10 per group) was initiated on day 3 postinfection and continued to day 7.
Valacyclovir (60 or 240 mg/kg t.i.d.) treatment led to a temporary decrease in lesion score, followed by an increase. Ultimately, animals suffered substantial mortality by encephalitis (100% at the lower dose level), sharing the fate of controls. By contrast, no such recurrence was observed with pritelivir (15 or 60 mg/kg t.i.d.). Instead, the drug produced a continuous improvement of the disease score, which approached zero at the end of the observation period. Mortality was at 10% in both dose groups. Cumulative disease scores revealed a statistically significant difference between the low dose of pritelivir and the high dose of valacyclovir.26
As another example, Kaufmann and colleagues studied the ability of pritelivir to suppress reactivation of ocular herpes in latently infected mice.32 Mouse corneas were infected with HSV-1, and infection was confirmed by determination of virus load in tear fluid. Thirty-five days later, latent mice were heat-stressed for virus reactivation. Active treatment consisted of two oral doses of pritelivir (50 mg/kg) administered immediately after stress and 12 hours later. Twenty-four hours after reactivation, eyes were swabbed for virus detection. Of the vehicle-treated mice, 63% (19/39) tested positive for HSV-1; this rate was halved to 32% (16/50) in the pritelivir group.32
This outcome was in agreement with an assay of viral DNA content of trigeminal ganglia. Mice were infected, stressed, and treated as in the first set, and ganglia of pritelivir-treated animals as well as vehicle-treated and untreated animals were analyzed 24 h after heat stress. The mean genome copy number in the two control groups was ∼5800-fold higher than after pritelivir treatment (1.4 × 106 vs 2.4 × 102). The authors concluded that pritelivir might prevent recurrencies in a more effective way than do available treatment modalities.32
2.2.4. Resistance
The vast majority of resistance to anti-HSV nucleoside analogues results from mutations in the viral thymidine kinase gene or, to a lesser extent, the viral DNA polymerase, resulting in impaired activity or loss of function of the affected protein.15 These changes, however, are not relevant to the action of helicase–primase inhibitors. Not surprisingly, mutations in HSV strains that confer resistance to acyclovir do not affect the efficacy of pritelivir, both in vitro(11,33) and in vivo(30) (Figure 8). It is also important to note that resistance to foscarnet, which plays a role as emergency medication in particular in immunocompromised patients with acyclovir -resistant HSV strains, can also be overcome by treatment with pritelivir (ref (33); polymerase mutant strain KD3 in their Table 1).
Experiments in a small number of immunodeficient mice (cf. section 2.2.1) suggest that there is no fast development of resistance to pritelivir in this setting. This is based on viral samples collected on day 6 postinfection in mice that had received either lower doses of pritelivir (still permitting considerable virus replication) or a single high dose.27 Furthermore, no emergence of resistant virus has been observed so far in vivo, including specifically under suboptimal treatment.34
Even so, therapy in humans with pritelivir might encounter pre-existing resistance or induce resistance by prolonged exposure. Such resistances are mostly based on single amino acid exchanges. To date, all mutations in the HSV genome conferring resistance to pritelivir (and other helicase–primase inhibitors) are located in the UL5 gene that codes for the helicase and/or at a single locus in the UL52 gene coding for the primase gene (A899 for HSV-1, A905 for HSV-2). A synopsis of helicase point mutations from different strains reveals clusters of affected sites that are obviously essential to helicase activity and hence gives clues to the localization of the target site of pritelivir (cf. Figure 6).25,35,36 No virus isolate or strain has been identified so far, for which resistance to pritelivir was mediated by an amino acid change in the noncatalytic subunit, UL8.37 For the major part, this information originates from HSV-1, but HSV-2 resistances, though less extensively researched, seem to align with the pattern reported for HSV-1.
Resistance to pritelivir might be overcome by modifying dose level and treatment duration. This possibility was tested in the murine neck infection model34 described in section 2.2.1.27 With wild-type HSV-1 infection, oral doses of pritelivir administered from postinfection day 1 to day 4 were largely (5 mg/kg) or fully (10 mg/kg) effective in terms of a favorable impact on lesion score and virus shedding but had only marginal (10 mg/kg) or no influence in a pritelivir-resistant HSV-1 strain characterized by a 27-fold higher in vitro ED50. However, pushing the dose to 60 mg/kg and the treatment period to 8 days prevented mortality and weight loss, more than halved lesion score and ear thickness compared to control, and enhanced the suppression of infectious virus titers in skin (inoculation site), ear pinna, and brainstem to below the detection limit.
Infection with an entirely resistant HSV population represents a worst-case scenario rather than a real-world situation. In vitro, the frequency of pritelivir-resistant HSV mutants among plaque-purified virus was determined to be 0.5 to 4.5 × 10–6.10 This is at least 1 order of magnitude lower than that for acyclovir, for which a resistance frequency of 10–3–10–4 was reported.38
According to Biswas and colleagues,39,40 HSV-1 resistant to helicase primase inhibitors has been detected at a frequency of up to 10–4 plaque forming units in clinical isolates from immunocompromised subjects and may have been pre-existing or favored by tissue culture conditions.41 On these grounds, studies of mixtures of wild-type HSV-1 and a pritelivir-resistant mutant, conducted in the same murine neck skin model as above, carry special interest.42 The employed mutant commanded an in vitro EC50 of >248 μmol/L compared to a wild-type EC50 of 0.03 μmol/L.
Wild-type, mutant/wild-type mixtures of 1:50 and 1:500, and mutant alone were studied in groups of 5–6 mice. The respective mortalities at 8 days without active treatment were 100, 100, 80, and 60%, respectively. Oral treatment with 5 mg/kg pritelivir from day 1 to day 4 postinfection caused the expected favorable responses in the wild type and the two mixture groups (except for some small lesions in 1/5 mice of each group), whereas infection with the HSV-1 mutant without wild-type mixture rendered the drug practically ineffective, even at the high dose of 60 mg/kg.42
Pritelivir evoked dramatic decreases of infectious virus titers in wild-type and mutant/wild-type mixtures. Values fell below the detection limit in skin samples and ear pinna derived from the 1:500 mutant/wild-type mixture group. In the 1:50 group, measurable though reduced titers, detectable resistant virus were only observed on day 5 but not days 3 and 7.42
The authors reasoned that a low fraction of pre-existing pritelivir resistant mutants should still pose no therapeutical problem, as the 1:500 mutant/wild-type mixture, which did not interfere with any metric of pritelivir efficacy, still carried a 20-fold mutant excess over the highest clinical isolate (10–4).
2.2.5. Combination Therapy
Under certain disease conditions, e.g., severe and potentially fatal infections like herpes simplex encephalitis, a combination treatment with more than one active drug could be beneficial for the treatment success. Combinations of pritelivir with acyclovir were tested in a murine model of herpes simplex encephalitis.30 It was shown that suboptimal doses of both drugs in combination have a positive effect on survival of infected animals, indicative for additive or even synergistic activity. Additional studies are needed to further investigate the interaction between both drugs.
2.2.6. Pharmacokinetics
An overview of pritelivir formulations used in pharmacokinetic studies in different animal species as well as in humans with corresponding pharmacokinetic parameters is listed in Table 1. Oral bioavailability of pritelivir was 65, 83, and 63% in rat, dog, and monkey, respectively. The drug was eliminated with half-lives of 5–10 h in rodents, 22–39 h in dogs, and 30 h in monkeys. When compared, human data in healthy volunteers (bioavailability, 73%; t1/2z, 60–70 h; see Discussion, section 3.4) appear similar or even more favorable.43
Table 1. Dose, Formulation, and Pharmacokinetic Parameters of Pritelivir after Single Oral Administration to Various Species.
| species | dose | formulation | AUC [mg·h/L] | T1/2 [h] | F [%] |
|---|---|---|---|---|---|
| Rat | 1 mg/kg | Solution of pritelivir free base in 2.5% ethanol, 40% solutol, 57.5% aqua dest. | 12.9 | 5–10 | 65 |
| Dog | 1 mg/kg | Solution of pritelivir free base in PEG400 | 54.2 | 22–39 | 83 |
| Monkey | 1 mg/kg | Suspension of pritelivir free base in 0.5% aqueous methylcellulose solution (w/v) | 39.5 | 30 | 63 |
| Human | 100 mg | Tablet containing pritelivir mesylate, microcrystalline cellulose, croscarmellose sodium, mannitol, colloidal anhydrous silica, magnesium stearate, hydroxymethylpropyl cellulose, polyethylene glycol, and titanium dioxide | 90.8 | 60–70 | 73 |
The range of percentages of plasma protein binding in different species (species, % unbound: mice, 0.97; rats, 1.2; guinea pigs, 5.9; rabbits, 3.4; dogs, 1.0, minipigs, 2.5, monkeys, 0.75) encompassed the value of 2.7% for humans.43
For more than 50%, elimination of pritelivir in humans involves mainly nonenzymatic hydrolysis yielding pyridinyl phenyl acetic acid (26) and amino thiazole sulfonamide (23, Figure 10). Quantitatively less important are hydroxylation by CYP3A4 and glucuronidation. In vitro experiments in microsomes and hepatocytes confirmed that cytochrome P450-dependent metabolism is not the major route of metabolic clearance of pritelivir.43
Figure 10.
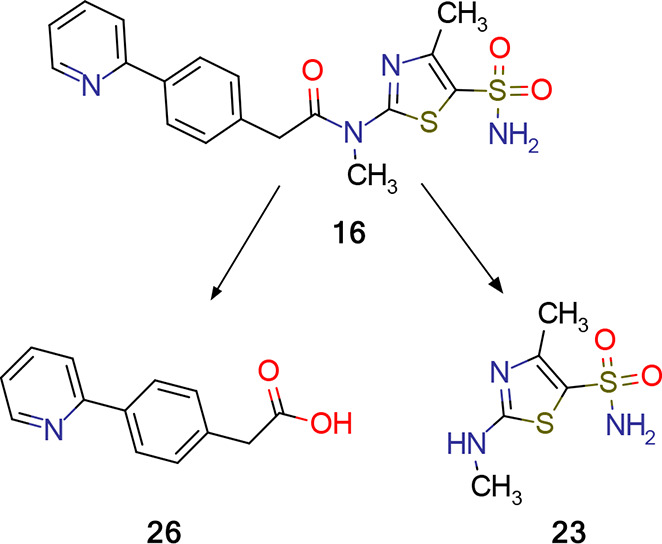
Nonenzymatic hydrolysis of pritelivir in humans.
2.2.7. Selectivity and Specificity of Target
There is a multitude of different helicases, and these are classified in various enzyme superfamilies. They are distinguished by sequential and structural differences that open the possibility of species-specific effects. Possible interactions between pritelivir and human helicases were studied with two human helicases, one nuclear and one located in the mitochondria.43 Neither of these enzymes was inhibited by pritelivir, confirming selectivity between human and HSV helicases which is also in line with the lack of cytotoxicity on a panel of different cell types (liver, heart muscle, kidney, macrophages, monocytes, T lymphocytes, fibroblasts, and neuronal cells)43 and its selective activity against HSV-1 and HSV-2 and only weak (if any) activity against other herpesviruses.11
A potential effect of pritelivir on human ether-à-go-go-related gene (hERG) encoded potassium channels was investigated by whole-cell patch-clamp technique in stably transfected HEK293 cells (human embryonic kidney cells).43 A 50% inhibitory concentration of approximately 160 μM was determined which is more than 100-fold higher than the maximum observed plasma concentration at therapeutic doses after correction for protein binding. Therefore, the interaction between pritelivir and hERG potassium channels is of negligible clinical relevance in humans.
Furthermore, the genotoxic potential of pritelivir free base and pritelivir mesylate monohydrate was examined in vitro and pritelivir was found to be nongenotoxic in a standard ICH battery comprising bacterial reversion and mammalian cell chromosome aberration assays.43
Primary sulfonamides are well-known for their affinity to carbonic anhydrases (CAs). Pritelivir is an inhibitor of human CAs in vitro with Ki values ranging from 12.8 to 474.0 nM against the various CA isoforms, determined by assaying the CA-catalyzed CO2 hydration activity using phenol red as an indicator.44 Of note, in an esterase assay investigating the activity against the human CA isoform II, IC50 values of 2–5 μmol/L were reported.11 However, the hydration assay is considered more physiologic since carbonic anhydrases catalyze CO2 hydration and not the hydrolysis of p-nitrophenyl acetate as used in the esterase assay.
2.3. Pharmaceutical Development and Optimization
Physicochemical properties like crystallinity, solid phase stability (including thermal stability and photostability), and thermodynamic solubility served to guide the preparation and investigation of several acceptable salt forms including maleate, sulfate, and mesylate salts.45 During preliminary polymorphism studies of the latter species, the mesylate monohydrate form was selected for further development due to its improved profile in terms of excipient-compatibility and stability (accelerated and long-term) and its optimized in vitro dissolution profile (cf. Figure 11).
Figure 11.
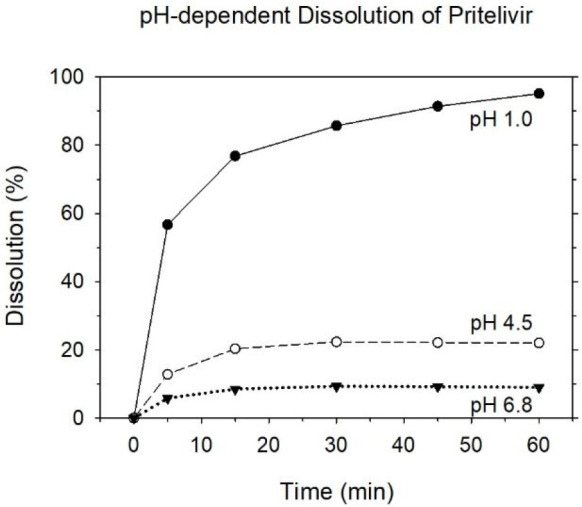
pH dependent dissolution of pritelivir mesylate monohydrate 100 mg film-coated tablets (1000 mL, 37 ± 0.5 °C, 50 rpm, USP apparatus 2 (paddle)).51
The pritelivir mesylate monohydrate polymorph was selected for further development from many other pharmaceutical salts and salt/hydrates for several reasons.45 The mesylate monohydrate showed the highest stability under forced degradation conditions and was the most suitable crystalline form for the tableting process. In addition, the pH-dependent solubility profile was more suitable for achieving the desired bioavailability. The solid form was therefore selected by pharmaceutical properties with respect to drug product manufacturing, thermodynamic solubility, and chemical stability under ICH storage conditions.
Pritelivir mesylate monohydrate is formulated as a 100 mg immediate release round biconvex film-coated tablet. The initial tablet formulation, back in 2004, used a fluid bed granulation process, which was later improved by using a direct compaction process. For manufacturing, pritelivir mesylate monohydrate is blended with microcrystalline cellulose and colloidal silicon and sieved. The remaining excipients are blended and sieved as well, and the final blend is then directly compacted into tablets. The resulting core tablets are then film coated. During upscaling, further optimization was performed following the quality by design approach,46−48 although the initial composition remained largely unchanged.
Pritelivir mesylate monohydrate 100 mg film-coated tablets are fast disintegrating, i.e., within 1–2 min. Drug dissolution properties are therefore mainly driven by the drug substance characteristics. Pritelivir mesylate monohydrate is a BCS class 2 drug substance.49,50 As a weak acid with a pKa value of 4.6 and an octanol–water partition coefficient of log P = 1.745 ± 0.564 (at 25 °C), it is well soluble below pH 2–3. With increasing pH value, the solubility decreases substantially (Figure 11). Nonetheless, oral bioavailability was determined to be 73% (cf. Discussion, section 3.4), indicating that the goal of a satisfactory oral bioavailability had been achieved.
3. Discussion
3.1. Potency and Therapeutic Time Window
It has been learned early on that nucleoside analogue treatment must be initiated as early as possible in the course of an episode52 if they are to deliver on their full therapeutic potential, a goal hard to achieve in real life. Indeed, their dependence on expression of a viral protein (the thymidine kinase engaged in the first activation step) leads to a delayed onset of action until the respective protein has been expressed, which occurs in the early phase of gene expression.21 In contrast, there is no delay in the onset of action in the case of the helicase–primase inhibitors, which attack viral replication earlier than acyclovir-type inhibitors and do not need an activation step. The same conclusion derives from the finding that polymerase is recruited to the replication fork only in the presence of an active primase subunit and probably only once an RNA primer has been synthesized.53 Preclinical studies have shown consistently that pritelivir surpasses valacyclovir and related compounds in anti-HSV potency, especially when initiation of treatment is delayed. This may relate to an inherently higher potency, one of the goals of this drug’s development process, but may also derive from its target being located further upstream in the viral replication cycle than that of the nucleoside analogues.
Irrespective of the specific contributions of these mechanisms, the superior potency of pritelivir in both early and delayed treatment studies adds to the range of achieved development aims.
3.2. Duration and Frequency of Recurrences
There are numerous examples of pritelivir’s ability to prevent early recurrence of HSV lesions and to shorten such episodes. Longer-term experiments would be helpful to understand whether suppression of early relapse just reflects a delay of reactivation, that would occur at a later time nonetheless or an ability to cause a long-lasting suppression, i.e., to confirm that pritelivir treatment indeed antagonizes the establishment of latency when administered early during primary infection in the clinic. To date, there is no convincing clinical evidence yet that helicase–primase inhibitors have an impact on viral latency.
3.3. Resistance
As it is well-known, the immune system makes an important contribution to the task of an anti-infective agent, which is to rid the organism of microorganisms. HSV infections are a good example of this interaction, where recurrences are mostly self-limiting disorders when the immune system is competent but can be complicated, painful, and even life-threatening in the context of a dysfunctional immune response. In the absence of synergies from the host’s immune defense, the impact of antiherpetic drugs has been limited, giving the virus time to develop resistance and achieve survival. Frequent exposure to nucleoside antivirals puts greater selective pressure on HSV to develop resistance, mainly affecting expression or function of the viral thymidine kinase, which is needed for initiation of activation but is not essential for HSV for replicating in dividing cells.
Thus, nucleoside analogue resistance is foremost a problem in immunocompromised patients, where prevalence between 2.5 and 10% or even as high as 25% has been reported, depending on the underlying disorder and in stark contrast to immunocompetent patients, where values of 0.5% seem to mark the upper end.54
Preclinical studies have identified HSV strains resistant to pritelivir. Quite early it was found that such resistance is typically secondary to mutations of the genes coding for the helicase and/or the primase.11 Since pritelivir-resistance and acyclovir-resistance mediating mutations are located in different genes in different genomic loci (cf. section 2.1.4), the anti-HSV potency of pritelivir is independent of the resistance status to acyclovir (Figure 8).33 Exactly this was one of the aims of the discussed drug development effort: identifying a drug that retains full activity against strains resistant to nucleoside analogues.
It is too early to form an opinion on the development of resistance to pritelivir in the clinical setting. There seem to be good arguments suggesting that pritelivir-resistant mutants detected so far have been pre-existing or fostered by the environment of cell cultures.39 Potential emergence of resistance was analyzed in a clinical trial in immunocompetent patients with genital herpes, who were treated with different daily doses of pritelivir (5, 25, or 75 mg) for 4 weeks.28 Daily swabs from the genital area were used to sequence resistance regions from 87 participants. There was no evidence of treatment-emergent resistance, let alone time-dependent genetic variations at any site. The authors emphasized that this also included patients who have received an apparently insufficient dose, as evidenced by persistent viral shedding despite continued treatment.37
Thus, while the issue of resistance to pritelivir induced over the course of long-term exposure, particularly in the immunocompromised, awaits the test of time, it is clear that the present drug development yielded an effective compound for the treatment of patients with HSV strains, which are resistant to nucleoside analogues.
3.4. Pharmacokinetics
Excessively fast elimination has been identified as a major obstacle to treatment success with current (nucleoside-type) anti-HSV drugs.55 Indeed, after oral dosing with the prodrug valacyclovir, elimination half-lives of acyclovir determined in rats and monkeys (1.0 and 1.3–1.5 h, respectively) are remarkably short56,57 and the corresponding values in humans (2.7–3.6 h)58,59 are not much longer. Half-lives of 2.0–2.5 h determined for penciclovir after dosing with the prodrug famciclovir fall into the same range.60−62 Against this background, results for pritelivir (at least 5 h in rodents and 22 h in non-rodents; section 2.2.6) suggested a favorably prolonged exposure of humans. Indeed, a terminal elimination half-life of 60–70 h determined in human volunteers at the therapeutic dose level (100 mg, single dose)34,63 and predictable in otherwise healthy subjects with genital herpes also confirms achievement of the first of the pharmacokinetic goals of the present development.
A good oral bioavailability was the other developmental goal in pharmacokinetics. Animal data (rat, 65%; dog, 83%; monkey, 63%) fell into the range of acyclovir bioavailability reported after administration of its prodrug valacyclovir in experimental animals (58–84%)64 and heralded acceptable human values. An oral bioavailability of 73% in human volunteers43 meets this expectation, comparing well with bioavailability achieved using the prodrug approach: acyclovir (via valacyclovir), 54%59 and penciclovir (via famciclovir), 60–77%.61,62
Thus, the development process described led to a compound well-suited for oral and once daily (or even weekly) dosing.
3.5. Specificity of Target as a Safety Factor
The hallmark of the safety profile of acyclovir and its congeners is integration of a specifically viral enzyme in their activation cascade. An equivalent distinction is evident for helicase–primase inhibitors. Kwong and colleagues65 dwelled on substantial differences in primary sequence and tertiary structure between viral and cellular helicases to make the point that such differences can be exploited to confer specificity to an antiviral inhibitor. Thus, it was a predictable outcome that pritelivir showed high specificity and failed to inhibit helicases of human origin in vitro.43
As of today, no findings (either pharmacological or safety-related) in clinical trials could be attributed to inhibition of CA isoforms following administration of pritelivir, though the trials were not designed to detect potential pharmacological benefits of CA inhibitors such as an impact on obesity or certain cancers. In line with this, clinical trials revealed no adverse effects attributable to sequelae of CA inhibition such as electrolyte disturbances.
In summary, it seems safe to conclude that human cells are safe against off-target effects of pritelivir on the host’s genetic machinery, another core objective of the present development process.
3.6. Translation to the Clinics
The process of translation from laboratory and preclinical studies to the clinical setting has likewise shown promising results. First trials confirmed the favorable pharmacokinetics (cf. section 2.2.6) and the powerful reduction of virus burden and lesion recrudescence (cf. section 2.2.3). The latter capability was investigated in a randomized, parallel group, double-blind, placebo-controlled trial in patients with a history of recurrent genital herpes.28 Over a period of 28 days, patients took daily swabs of genital skin and mucosa, carried a diary of genital signs and symptoms, and returned to the clinic within 24 h of a recurrence for examination. Pritelivir (100 mg, oral) caused dose-dependent and apparently inter-related decreases in the number of days with genital lesions and virus shedding when administered in suppressive fashion over 28 days (Figure 12).
Figure 12.
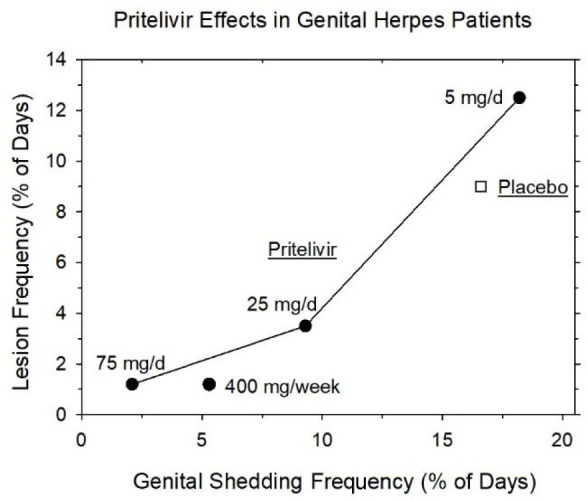
Dose-dependent decrease of virus shedding and lesion frequency in patients with recurrent genital herpes under treatment with pritelivir over 28 days (after data from ref (28)).
Similar results were obtained in a further trial with viral shedding as primary end point, which directly compared the efficacy of pritelivir versus valacyclovir.29 Designed with a high time resolution secured by four times daily swabbing, this trial also pointed to a superior efficacy vs valacyclovir in terms of both viral shedding rate and days with lesions. This trial was prematurely terminated due to a clinical hold imposed because of hematological and skin related findings in a concurrent chronic toxicity study in monkeys.29 However, based on further in-depth investigations and the beneficial outcome of an additional chronic toxicity study in monkeys, where no comparable findings were observed, and the overall favorable clinical data, the hold was subsequently lifted.
3.7. Prospects
As summarized in this Drug Annotation, in vitro, in vivo, and clinical data suggest that pritelivir is more potent than acyclovir or its prodrug valacyclovir for the treatment of HSV infections. Due to its different mode of action, the drug does not need to become activated by viral or cellular enzymes and is active against nucleoside analogue-resistant virus. Currently, pritelivir is under development for the treatment of acyclovir-resistant mucocutaneous HSV infection in immunocompromised patients, a patient population where the highest medical need exists (ClinicalTrials.gov identifier: NCT03073967). Indeed, the drug was granted fast track designation for this indication by the FDA. More recently, pritelivir has been admitted to the FDA process of a Breakthrough Therapy designation that is “designed to expedite the development of drugs intended to treat a serious condition, where preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant end point”.66 This step was taken in consideration of interim data from a phase 2 open-label clinical trial comparing pritelivir and foscarnet in immunocompromised patients with acyclovir resistant, mucocutaneous HSV infection and, in a separate arm, studying pritelivir in such patients, who were also either intolerant to foscarnet or resistant to it (i.e., were infected with a double-resistant strain). Preliminary data from this trial showed that pritelivir was well tolerated without significant adverse effects. Of the 23 subjects treated with pritelivir, 19 demonstrated full resolution of their HSV-related lesions within the treatment period of 28 days and in 4 subjects lesions improved but did not heal completely during the observation period.67
Experience from compassionate use of pritelivir is in line with the results from clinical trials. A recent case report concerns an immunosuppressed (HIV negative) allogenic peripheral blood stem cell transplant recipient with acyclovir-resistant HSV-2 infection, which was refractory to second-line therapy (infusions with foscarnet and cidofovir with oral probenecid). Treatment with pritelivir provided symptomatic control for the duration of its use, turning PCR assays of genital swabs negative.68 Another recent article reported multiple successful treatment courses with pritelivir in two patients who developed acyclovir-resistant genital HSV-2 infection after hematopoietic stem cell transplantation.69
Growing experience with pritelivir might lead to expansion of its indications and modes of use in the future. A potential option could be, for example, treatment of herpes simplex encephalitis, either as monotherapy or in combination with polymerase inhibitors, as demonstrated in a respective disease model.30
4. Conclusion
It seems more than likely that the ongoing clinical development of pritelivir will result in a long-awaited addition to the anti-HSV armamentarium by providing a novel and effective treatment option for immunocompromised patients infected with acyclovir and/or foscarnet resistant strains. In addition, there is an exciting prospect that the favorable properties of pritelivir demonstrated during preclinical development could lead to use in indications beyond the present scope.
Acknowledgments
The authors thank T. Langer for the pharmacophore model, and K. U. Petersen, R. Saunders, K. Kumar, B. Lesch, and H. Trübel for their support with generating and improving the manuscript.
Glossary
Abbreviations Used
- CA
carbonic anhydrase
- CC50
50% cytotoxic concentration
- ICH
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
- UL5
HSV helicase
- UL52
HSV primase
- UL8
HSV noncatalytic subunit of the helicase–primase complex
- USP
United States Pharmacopeia
The authors declare the following competing financial interest(s): The authors are or have been employees at AiCuris, the company that develops pritelivir.
References
- Wertheim J. O.; Smith M. D.; Smith D. M.; Scheffler K.; Kosakovsky Pond S. L. Evolutionary origins of human herpes simplex viruses 1 and 2. Mol. Biol. Evol. 2014, 31, 2356–2364. 10.1093/molbev/msu185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underdown S. J.; Kumar K.; Houldcroft C. Network analysis of the hominin origin of herpes simplex virus 2 from fossil data. Virus Evol. 2017, 3 (2), vex026. 10.1093/ve/vex026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley R.; Baines J. Clinical management of herpes simplex virus infections: past, present, and future. F1000Research 2018, 7, 1726. 10.12688/f1000research.16157.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski L. A.; Upadhyay R.; Greeley Z. W.; Margulies B. J. Current drugs to treat infections with herpes simplex viruses-1 and −2. Viruses. 2021, 13, 1228. 10.3390/v13071228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkmann A.; Zimmermann H. HSV antivirals - current and future treatment options. Curr. Opin Virol. 2016, 18, 9–13. 10.1016/j.coviro.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Birkmann A.Herpes Simplex Viruses. In New Drug Development for Known and Emerging Viruses; Ruebsamen-Schaeff H., Buschmann H., Eds.; Wiley VCH: Weinheim, Germany, 2021. [Google Scholar]
- FDA. n-Docosanol. Pharmacology Review (1999). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2000/020941s000_PharmR.pdf (accessed Sep 11, 2021).
- Crute J. J.; Lehman I. R.; Gambino J.; Yang T. F.; Medveczky P.; Medveczky M.; Khan N. N.; Mulder C.; Monroe J.; Wright G. E. Inhibition of herpes simplex virus type 1 helicase-primase by (dichloroanilino)purines and -pyrimidines. J. Med. Chem. 1995, 38, 1820–1825. 10.1021/jm00010a027. [DOI] [PubMed] [Google Scholar]
- Spector F. C.; Liang L.; Giordano H.; Sivaraja M.; Peterson M. G. Inhibition of herpes simplex virus replication by a 2-amino thiazole via interactions with the helicase component of the UL5-UL8-UL52 complex. J. Virol. 1998, 72, 6979–6987. 10.1128/JVI.72.9.6979-6987.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crute J. J.; Grygon C. A.; Hargrave K. D.; Simoneau B.; Faucher A. M.; Bolger G.; Kibler P.; Liuzzi M.; Cordingley M. G. Herpes simplex virus helicase-primase inhibitors are active in animal models of human disease. Nat. Med. 2002, 8, 386–391. 10.1038/nm0402-386. [DOI] [PubMed] [Google Scholar]
- Kleymann G.; Fischer R.; Betz U. A.; Hendrix M.; Bender W.; Schneider U.; Handke G.; Eckenberg P.; Hewlett G.; Pevzner V.; Baumeister J.; Weber O.; Henninger K.; Keldenich J.; Jensen A.; Kolb J.; Bach U.; Popp A.; Mäben J.; Frappa I.; Haebich D.; Lockhoff O.; Rübsamen-Waigmann H. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nat. Med. 2002, 8, 392–398. 10.1038/nm0402-392. [DOI] [PubMed] [Google Scholar]
- Chono K.; Katsumata K.; Kontani T.; Kobayashi M.; Sudo K.; Yokota T.; Konno K.; Shimizu Y.; Suzuki H. ASP2151, a novel helicase-primase inhibitor, possesses antiviral activity against varicella-zoster virus and herpes simplex virus types 1 and 2. J. Antimicrob. Chemother. 2010, 65, 1733–1741. 10.1093/jac/dkq198. [DOI] [PubMed] [Google Scholar]
- Gege C.; Bravo F. J.; Uhlig N.; Hagmaier T.; Schmachtenberg R.; Elis J.; Burger-Kentischer A.; Finkelmeier D.; Hamprecht K.; Grunwald T.; Bernstein D. I.; Kleymann G. A helicase-primase drug candidate with sufficient target tissue exposure affects latent neural herpes simplex virus infections. Sci. Transl. Med. 2021, 13, eabf8668. 10.1126/scitranslmed.abf8668. [DOI] [PubMed] [Google Scholar]
- Hou J.; Gao H.; Fan Y.; Wang Y.; Qin M.; Di Y.; Wang L.; Zhou X.; Zhou Y.; Qin D.; Hill G. Pharmacokinetics and safety study of HN0037, a novel anti-human herpes simplex virus inhibitor, in healthy volunteers. Clin. Pharmacol. Drug Dev. 2022, 10.1002/cpdd.1138. [DOI] [PubMed] [Google Scholar]
- Schmidt S.; Bohn-Wippert K.; Schlattmann P.; Zell R.; Sauerbrei A. Sequence analysis of herpes simplex virus 1 thymidine kinase and DNA polymerase genes from over 300 clinical isolates from 1973 to 2014 finds novel mutations that may be relevant for development of antiviral resistance. Antimicrob. Agents Chemother. 2015, 59, 4938–4945. 10.1128/AAC.00977-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajfr M.; Pliskova L.; Bolehovská R.; Uhlířová Z.; Vrbacký F. Herpes simplex virus resistant to acyclovir: A single-centre experience from the Czech Republic. J. Glob Antimicrob Resist. 2019, 19, 269–273. 10.1016/j.jgar.2019.05.004. [DOI] [PubMed] [Google Scholar]
- Kleymann G. Discovery, SAR and medicinal chemistry of Herpesvirus helicase primase inhibitors. Curr. Med. Chem. - Anti-Infective Agents. 2004, 3, 69–83. 10.2174/1568012043354242. [DOI] [Google Scholar]
- Fischer R.; Bender W.; Henninger K.; Eckenberg P.; Handke G.; Keldenich J.; Weber O.; Kleymann G.; Betz U.; Hendrix M.; Schneider U.; Jensen A.; Baumeister J.. WO 0147904, 2001. (Bayer AG).
- Betz U.; Laich T.; Bender W.; Fischer R.; Hendrix M.; Kleymann G.. WO03000259, 2003. (Bayer AG).
- Schohe-Loop R.; Betz U.; Fischer R.; Hendrix M.; Kleymann G.; Baumeister J.; Bender W.; Eckenberg P.; Handke-Ergüden G.; Henninger K.; Jensen A.; Keldenich J.; Schneider U.; Weber O.. WO03000260, 2003. (Bayer AG).
- Ibáñez F. J.; Farías M. A.; Gonzalez-Troncoso M. P.; Corrales N.; Duarte L. F.; Retamal-Díaz A.; González P. A. Experimental dissection of the lytic replication cycles of herpes simplex viruses in vitro. Front Microbiol. 2018, 9, 2406. 10.3389/fmicb.2018.02406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic S.; Cohen S.; Singh M.; Tam B.; Dayan A.; Akabayov B. DnaG primase - A target for the development of novel antibacterial agents. Antibiotics (Basel). 2018, 7, 72. 10.3390/antibiotics7030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermek O.; Williams R. S. The three-component helicase/primase complex of herpes simplex virus-1. Open Biol. 2021, 11, 210011. 10.1098/rsob.210011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S.; Kleymann G.; Swift M.; Tiley L. S.; Lyall J.; Aguirre-Hernández J.; Field H. A single drug-resistance mutation in HSV-1 UL52 primase points to a difference between two helicase-primase inhibitors in their mode of interaction with the antiviral target. J. Antimicrob. Chemother. 2008, 61, 1044–1047. 10.1093/jac/dkn057. [DOI] [PubMed] [Google Scholar]
- Biswas S.; Miguel R. N.; Sukla S.; Field H. J. A mutation in helicase motif IV of herpes simplex virus type 1 UL5 that results in reduced growth in vitro and lower virulence in a murine infection model is related to the predicted helicase structure. J. Gen Virol. 2009, 90, 1937–1942. 10.1099/vir.0.011221-0. [DOI] [PubMed] [Google Scholar]
- Betz U. A.; Fischer R.; Kleymann G.; Hendrix M.; Rübsamen-Waigmann H. Potent in vivo antiviral activity of the herpes simplex virus primase-helicase inhibitor BAY 57-1293. Antimicrob. Agents Chemother. 2002, 46, 1766–1772. 10.1128/AAC.46.6.1766-1772.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S.; Jennens L.; Field H. J. The helicase primase inhibitor, BAY 57-1293 shows potent therapeutic antiviral activity superior to famciclovir in BALB/c mice infected with herpes simplex virus type 1. Antiviral Res. 2007, 75, 30–35. 10.1016/j.antiviral.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Wald A.; Corey L.; Timmler B.; Magaret A.; Warren T.; Tyring S.; Johnston C.; Kriesel J.; Fife K.; Galitz L.; Stoelben S.; Huang M. L.; Selke S.; Stobernack H. P.; Ruebsamen-Schaeff H.; Birkmann A. Helicase-primase inhibitor pritelivir for HSV-2 infection. N Engl J. Med. 2014, 370, 201–210. 10.1056/NEJMoa1301150. [DOI] [PubMed] [Google Scholar]
- Wald A.; Timmler B.; Magaret A.; Warren T.; Tyring S.; Johnston C.; Fife K.; Selke S.; Huang M. L.; Stobernack H. P.; Zimmermann H.; Corey L.; Birkmann A.; Ruebsamen-Schaeff H. Effect of pritelivir compared with valacyclovir on genital hsv-2 shedding in patients with frequent recurrences: A randomized clinical trial. JAMA 2016, 316, 2495–2503. 10.1001/jama.2016.18189. [DOI] [PubMed] [Google Scholar]
- Quenelle D. C.; Birkmann A.; Goldner T.; Pfaff T.; Zimmermann H.; Bonsmann S.; Collins D. J.; Rice T. L.; Prichard M. N. Efficacy of pritelivir and acyclovir in the treatment of herpes simplex virus infections in a mouse model of herpes simplex encephalitis. Antiviral Res. 2018, 149, 1–6. 10.1016/j.antiviral.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister J.; Fischer R.; Eckenberg P.; Henninger K.; Ruebsamen-Waigmann H.; Kleymann G. Superior efficacy of helicase-primase inhibitor BAY 57-1293 for herpes infection and latency in the guinea pig model of human genital herpes disease. Antiviral Chem. Chemother. 2007, 18, 35–48. 10.1177/095632020701800104. [DOI] [PubMed] [Google Scholar]
- Kaufman H. E.; Varnell E. D.; Gebhardt B. M.; Thompson H. W.; Atwal E.; Rübsamen-Waigmann H.; Kleymann G. Efficacy of a helicase-primase inhibitor in animal models of ocular herpes simplex virus type 1 infection. J. Ocul Pharmacol Ther. 2008, 24, 34–42. 10.1089/jop.2007.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field H. J.; Huang M. L.; Lay E. M.; Mickleburgh I.; Zimmermann H.; Birkmann A. Baseline sensitivity of HSV-1 and HSV-2 clinical isolates and defined acyclovir-resistant strains to the helicase-primase inhibitor pritelivir. Antiviral Res. 2013, 100, 297–299. 10.1016/j.antiviral.2013.08.024. [DOI] [PubMed] [Google Scholar]
- Biswas S.; Sukla S.; Goldner T.; Field H. J.; Kropeit D.; Paulsen D.; Welbers A.; Ruebsamen-Schaeff H.; Zimmermann H.; Birkmann A. Pharmacokinetics-pharmacodynamics of the helicase-primase inhibitor pritelivir following treatment of wild-type or pritelivir-resistant virus infection in a murine herpes simplex virus 1 infection model. Antimicrob. Agents Chemother. 2014, 58, 3843–3852. 10.1128/AAC.02641-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field H. J.; Biswas S. Antiviral drug resistance and helicase-primase inhibitors of herpes simplex virus. Drug Resist Updat. 2011, 14, 45–51. 10.1016/j.drup.2010.11.002. [DOI] [PubMed] [Google Scholar]
- Ligat G.; Da Re S.; Alain S.; Hantz S. Identification of amino acids essential for viral replication in the hcmv helicase-primase complex. Front Microbiol. 2018, 9, 2483. 10.3389/fmicb.2018.02483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlefsen P. T.; Birkmann A.; Huang M. L.; Magaret C. A.; Kee J. J.; Diem K.; Goldner T.; Timmler B.; Stoelben S.; Ruebsamen-Schaeff H.; Zimmermann H.; Warren T.; Wald A.; Corey L. No evidence of pritelivir resistance among herpes simplex virus type 2 isolates after 4 weeks of daily therapy. J. Infect Dis. 2016, 214, 258–264. 10.1093/infdis/jiw129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon T. H.; Levin M. J.; Leary J. J.; Sarisky R. T.; Sutton D. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin Microbiol Rev. 2003, 16, 114–128. 10.1128/CMR.16.1.114-128.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S.; Swift M.; Field H. J. High frequency of spontaneous helicase-primase inhibitor (BAY 57–1293) drug-resistant variants in certain laboratory isolates of HSV-1. Antivir Chem. Chemother. 2007, 18, 13–23. 10.1177/095632020701800102. [DOI] [PubMed] [Google Scholar]
- Biswas S.; Smith C.; Field H. J. Detection of HSV-1 variants highly resistant to the helicase-primase inhibitor BAY 57-1293 at high frequency in 2 of 10 recent clinical isolates of HSV-1. J. Antimicrob. Chemother. 2007, 60, 274–279. 10.1093/jac/dkm182. [DOI] [PubMed] [Google Scholar]
- Biswas S.; Jennens L.; Field H. J. Single amino acid substitutions in the HSV-1 helicase protein that confer resistance to the helicase-primase inhibitor BAY 57–1293 are associated with increased or decreased virus growth characteristics in tissue culture. Arch. Virol. 2007, 152, 1489–1500. 10.1007/s00705-007-0964-7. [DOI] [PubMed] [Google Scholar]
- Sukla S.; Biswas S.; Birkmann A.; Lischka P.; Ruebsamen-Schaeff H.; Zimmermann H.; Field H. J. Effects of therapy using a helicase-primase inhibitor (HPI) in mice infected with deliberate mixtures of wild-type HSV-1 and an HPI-resistant UL5 mutant. Antiviral Res. 2010, 87, 67–73. 10.1016/j.antiviral.2010.04.008. [DOI] [PubMed] [Google Scholar]
- AiCuris Anti-Infective Cures AG, unpublished results.
- Carta F.; Birkmann n A.; Pfaff T.; Buschmann H.; Schwab W.; Zimmermann H.; Maresca A.; Supuran C. T. Lead development of thiazolylsulfonamides with carbonic anhydrase inhibitory action. J. Med. Chem. 2017, 60, 3154–3164. 10.1021/acs.jmedchem.7b00183. [DOI] [PubMed] [Google Scholar]
- Ruebsamen-Schaeff H.; Buschmann H. Different solid forms for optimizing route of administration of the herpes drug Pritelivir. Medchem Comm. 2019, 10, 1867–1870. 10.1039/C9MD00233B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ICH guideline Q8 (R2) on pharmaceutical development (2014).
- ICH Q9 on quality risk management, step 5 (2014).
- ICH Q10 on pharmaceutical quality system, step 5 (2014).
- Amidon G. L.; Lennernas H.; Shah V. P.; Crison J. R. A theoretical basis for a biopharmaceutics drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- CDER/FDA . The Biopharmaceutics Classification System Guidance; Office of Pharmaceutical Science, U.S. Food and Drug Administration, 2006. [Google Scholar]
- USP 42, General Chapter ⟨711⟩ Dissolution. USP, 2019. [Google Scholar]
- Reichman R. C.; Badger G. J.; Mertz G. J.; Corey L.; Richman D. D.; Connor J. D.; Redfield D.; Savoia M. C.; Oxman M. N.; Bryson Y.; Tyrrell D. L.; Portnoy J.; Creigh-Kirk T.; Keeney R. E.; Ashikaga T.; Dolin R. Treatment of recurrent genital herpes simplex infections with oral acyclovir. A controlled trial. JAMA 1984, 251, 2103–2107. 10.1001/jama.1984.03340400031020. [DOI] [PubMed] [Google Scholar]
- Carrington-Lawrence S. D.; Weller S. K. Recruitment of polymerase to herpes simplex virus type 1 replication foci in cells expressing mutant primase (UL52) proteins. J. Virol. 2003, 77, 4237–4247. 10.1128/JVI.77.7.4237-4247.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frobert E.; Burrel S.; Ducastelle-Lepretre S.; Billaud G.; Ader F.; Casalegno J. S.; Nave V.; Boutolleau D.; Michallet M.; Lina B.; Morfin F. Resistance of herpes simplex viruses to acyclovir: an update from a ten-year survey in France. Antiviral Res. 2014, 111, 36–41. 10.1016/j.antiviral.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Schiffer J. T.; Swan D. A.; Corey L.; Wald A. Rapid viral expansion and short drug half-life explain the incomplete effectiveness of current herpes simplex virus 2-directed antiviral agents. Antimicrob. Agents Chemother. 2013, 57, 5820–5829. 10.1128/AAC.01114-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnette T. C.; de Miranda P. Metabolic disposition of the acyclovir prodrug valaciclovir in the rat. Drug Metab. Dispos. 1994, 22, 60–64. [PubMed] [Google Scholar]
- de Miranda P.; Burnette T. C. Metabolic fate and pharmacokinetics of the acyclovir prodrug valaciclovir in cynomolgus monkeys. Drug Metab. Dispos. 1994, 22, 55–59. [PubMed] [Google Scholar]
- Wang L. H.; Schultz M.; Weller S.; Smiley M. L.; Blum M. R. Pharmacokinetics and safety of multiple-dose valaciclovir in geriatric volunteers with and without concomitant diuretic therapy. Antimicrob. Agents Chemother. 1996, 40, 80–85. 10.1128/AAC.40.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soul-Lawton J.; Seaber E.; On N.; Wootton R.; Rolan P.; Posner J. Absolute bioavailability and metabolic disposition of valaciclovir, the L-valyl ester of acyclovir, following oral administration to humans. Antimicrob. Agents Chemother. 1995, 39, 2759–2764. 10.1128/AAC.39.12.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filer C. W.; Allen G. D.; Brown T. A.; Fowles S. E.; Hollis F. J.; Mort E. E.; Prince W. T.; Ramji J. V. Metabolic and pharmacokinetic studies following oral administration of 14C-famciclovir to healthy subjects. Xenobiotica. 1994, 24, 357–368. 10.3109/00498259409045899. [DOI] [PubMed] [Google Scholar]
- Gill K. S.; Wood M. J. The clinical pharmacokinetics of famciclovir. Clin Pharmacokinet. 1996, 31, 1–8. 10.2165/00003088-199631010-00001. [DOI] [PubMed] [Google Scholar]
- Luber A. D.; Flaherty J. F. Jr. Famciclovir for treatment of herpesvirus infections. Ann. Pharmacother. 1996, 30, 978–985. 10.1177/106002809603000913. [DOI] [PubMed] [Google Scholar]
- Birkmann A.; Kropeit D.; McCormick D.; Zimmermann H.; Ruebsamen-Schaeff H. Abstract 21, Safety and human pharmacokinetics of AIC316, a potent helicase-primase inhibitor of herpes simplex virus (HSV). Antiviral Res. 2011, 90, A25. 10.1016/j.antiviral.2011.03.014. [DOI] [Google Scholar]
- Szczech G. M. Preclinical development of antiviral drugs. Clin Infect Dis. 1996, 22, 355–360. 10.1093/clinids/22.2.355. [DOI] [PubMed] [Google Scholar]
- Kwong A. D.; Rao B. G.; Jeang K. T. Viral and cellular RNA helicases as antiviral targets. Nat. Rev. Drug Discovery 2005, 4, 845–853. 10.1038/nrd1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA. Breakthrough Therapy (2018)/ https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/breakthrough-therapy (accessed Nov 24, 2021).
- Workowski K.; Albrecht J.; Avery R.; Chandrasekar P.; Chemaly R.; Issa N.; Kotton C.; Kumar P.; Ramesh M.; Ramgopal M.; Schiffer J.; Schreibman T.; Wald A.; Ison M.. Pritelivir in immunocompromised patients with mucocutaneous acyclovir resistant herpes simplex virus infections—first case series. Presented at ID Week, Sept 29–Oct 03, 2021; virtual event.
- Cannon L.; Tholouli E.; Ward C.; Farooq H.; Kingston M. Use of pritelivir in refractory aciclovir-resistant herpes simplex virus type 2. Int. J. STD AIDS. 2021, 32, 978–980. 10.1177/09564624211006568. [DOI] [PubMed] [Google Scholar]
- Serris A.; Pouvaret A.; Loiseau C.; Abid H.; Burrel S.; Fourgeaud J.; Rouzaud C.; Lanternier F.; Boutolleau D.; Frange P. Pritelivir for recurrent aciclovir-resistant herpes simplex virus 2 infections in immunocompromised patients. J. Antimicrob. Chemother. 2022, 77, 2303–2305. 10.1093/jac/dkac165. [DOI] [PubMed] [Google Scholar]