

Abstract
Signal transduction pathways post-translationally regulating nucleotide metabolism remain largely unknown. Guanosine monophosphate reductase (GMPR) is a nucleotide metabolism enzyme that decreases GTP pools by converting GMP to IMP. We observed that phosphorylation of GMPR at Tyr267 is critical for its activity, and found that this phosphorylation by ephrin receptor tyrosine kinase EPHA4 decreases GTP pools in cell protrusions and levels of GTP-bound RAC1. EPHs possess oncogenic and tumor-suppressor activities, although the mechanisms underlying switches between these two modes are poorly understood. We demonstrated that GMPR plays a key role in EPHA4-mediated RAC1 suppression. This supersedes GMPR-independent activation of RAC1 by EPHA4 resulting in a negative overall effect on melanoma cell invasion and tumorigenicity. Accordingly, EPHA4 levels increase during melanoma progression and inversely correlate with GMPR levels in individual melanoma tumors. Therefore, phosphorylation of GMPR at Tyr267 is a metabolic signal transduction switch controlling GTP biosynthesis and transformed phenotypes.
Keywords: EPHA4, RAC1, GMPR, GTP, GEVAL GTP sensors
Graphical Abstract
eTOC blurb
Wolff et al. demonstrate that phosphorylation of guanosine monophosphate reductase (GMPR) by Ephrin receptor A4 (EPHA4) regulates GMPR enzymatic activity, intracellular GTP levels, RAC1 activation, and melanoma cell invasion. The phosphorylation status of GMPR acts as a switch influencing the net pro- versus anti-oncogenic effects of EPHA4.
INTRODUCTION
Acquisition of invasive capability is a critical event driving progression from primary to metastatic disease in multiple cancers including melanoma (Gaggioli and Sahai, 2007; Leong et al., 2012; Arozarena and Wellbrock, 2017). Cell invasion is to a large degree controlled by the activity of several members of the RHO GTPase family of small G proteins (Arozarena and Wellbrock, 2017; Lawson and Ridley, 2018; Clayton and Ridley, 2020). Of these, RAC1 has the most prominent role in melanoma progression, as the gain-of-function P29S mutation in RAC1 is one of the most common recurring mutations in sun-exposed melanomas, present in approximately 5-9% of cases (Krauthammer et al., 2012; Davis et al., 2013; Halaban, 2015).
EPH receptors comprise the largest known family of receptor tyrosine kinases (Tuzi and Gullick, 1994; Liang et al., 2019). EPH receptor signaling primarily occurs following activation by membrane-bound ephrin ligands (ephrins, EFN) which trigger EPH-dependent phosphorylation of intracellular effectors (Poliakov et al., 2004; Niethamer and Bush, 2019). These include members of the SRC kinase family, PI3 kinase, and guanine nucleotide exchange factors such as VAV proteins (Cowan et al., 2005; Hunter et al., 2006; Hjorthaug and Aasheim, 2007; Holen et al., 2008), which in turn control the activity of RHO GTPases including RAC1 (Liu and Burridge, 2000; Rossman et al., 2005). Intriguingly, EFN-EPH signaling has been reported to either activate or suppress RHO GTPases depending on the cellular context and the specific EFN/EPH pathway activated (Wahl et al., 2000; Cowan et al., 2005; Shi et al., 2007; Pasquale, 2008). This dualism contributes in part to the ability of EPH receptors to display tumor-suppressor or oncogenic functions, depending on context (Pasquale, 2010; Kaenel et al., 2012; Buckens et al., 2020). However, the mechanisms underlying the switch from pro- to anti- oncogenic EFN/EPH activity are not well understood.
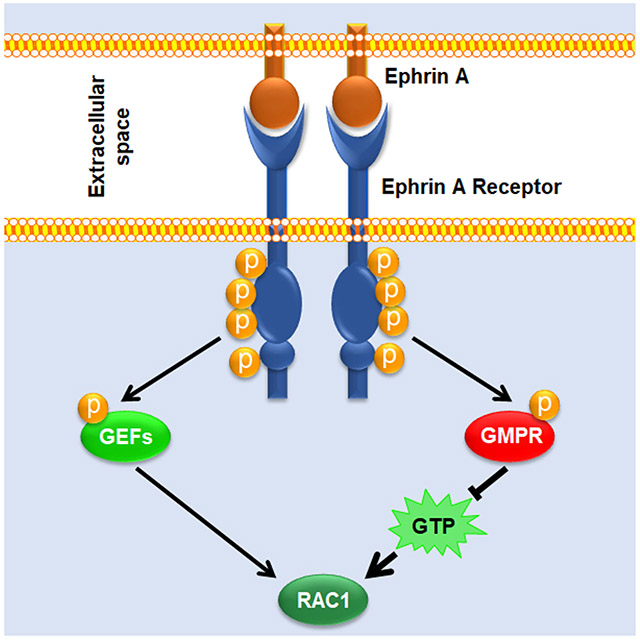
Studies by us an others have reported that the activity of small G proteins and cell invasion can be regulated by enzymes involved in the biosynthesis of guanosine triphosphate (GTP) (Wawrzyniak et al., 2013; Bianchi-Smiraglia et al., 2015; Kollareddy et al., 2015; Bianchi-Smiraglia et al., 2017a; Emmanuel et al., 2017; Bianchi-Smiraglia et al., 2021; Huang et al., 2021). Importantly, these studies demonstrated that manipulating GTP levels in cells through pharmacological inhibition of GTP synthesis or addition of guanosine to culture media is sufficient to alter activity levels of GTPases such as RAC1, RHOA, and RHEB (Wawrzyniak et al., 2013; Bianchi-Smiraglia et al., 2017a; Emmanuel et al., 2017). We found that guanosine monophosphate reductase (GMPR), an enzyme that converts GMP to IMP (Figure 1A), moderately decreases intracellular GTP pools and suppresses the activity of several RHO GTPases, most prominently RAC1 (Wawrzyniak et al., 2013). While the levels of enzymes promoting de novo biosynthesis of guanylates were shown to increase in the course of tumor progression (Bianchi-Smiraglia et al., 2015; Huang et al., 2018; Kofuji et al., 2019; Huang et al., 2021), GMPR protein levels are downregulated during melanoma progression from non-invasive to invasive primary melanomas and further in melanoma metastases (Wawrzyniak et al., 2013; Bianchi-Smiraglia et al., 2017a). However, the post-translational regulation of GMPR activity is completely unknown. In fact, signaling pathways that influence GTP pools via modification of guanylate metabolism enzymes also remain, with few exceptions, largely unknown (Ingley and Hemmings, 2000; Plana-Bonamaiso et al., 2020).
Figure 1. De novo guanine biosynthesis enzymes.

A. Schematic representation of de novo GTP biosynthesis. B. GMPR functional domains and phosphorylation sites. Shown are GMPR phosphorylation sites identified via mass spectrometry analysis of GMPR-fl protein immunoprecipitated with anti-FLAG antibodies from SK-Mel-103 cells. Relevant regions of GMPR are denoted.
In this study, we report a clinically relevant mechanism whereby EFN/EPHA4 signaling directly regulates the activity of GMPR via a post-translational modification that leads to suppression of GTP levels in cell protrusions, thus lowering RAC1 activity and invasive capacity of melanoma cells. This occurs independently from the previously reported EPHA4-mediated RAC1 activation, demonstrating a role for GMPR in determining the net effect of EPHA4 signaling on melanoma cells.
RESULTS
Phosphorylation of Tyr267 is critical for GMPR-mediated regulation of GTP levels and invasion in melanoma cells
To identify signal transduction pathways that control GMPR activity, we studied GMPR post-translational modifications. To this end, GMPR-negative SK-Mel-103 melanoma cells were transduced with a lentivirus encoding Flag-tagged GMPR (GMPR-fl) followed by isolation of GMPR-fl by immunoprecipitation with anti-Flag antibodies and mass spectrometry analysis. The post-translational modifications revealed with high confidence by this analysis were phosphorylation of tyrosine 267 (Y267), serine 271 (S271), and threonine 273 (T273) (Figure 1B). These residues are located within the ‘flexible binding region’ of GMPR, which interacts with the active site located at amino acids 179-188 (Figure 1B, Figure S1) (Li et al., 2006; Rosenberg et al., 2018).
To interrogate the functional role of each phosphorylation site, we generated GMPR mutants that could not undergo phosphorylation at the mutagenized residues including GMPRY267F, GMPRS271A and GMPRT273A. SK-Mel-103 cells expressing mutant or wild-type GMPR (GMPRWT) proteins were probed in a Boyden’s chamber invasion assay, since we have previously demonstrated that GMPRWT suppresses melanoma cell invasion (Wawrzyniak et al., 2013). The assay revealed that GMPRY267F lacks the ability to suppress melanoma cell invasion, whereas the GMPRS271A and GMPRT273A mutants partially retain this ability (Figure 2A). Therefore, we focused on Tyr267 phosphorylation as a major regulator of GMPR function in melanoma cell invasion. Importantly, overexpression of GMPR bearing a phospho-mimetic Y267E substitution suppressed invasion to a similar extent as GMPRWT in melanoma cells (Figure 2B). These results were recapitulated in SK-Mel-147, another GMPR-negative melanoma cell line (Figure S2).
Figure 2. Phosphorylation of tyrosine 267 is critical for GMPR activity.
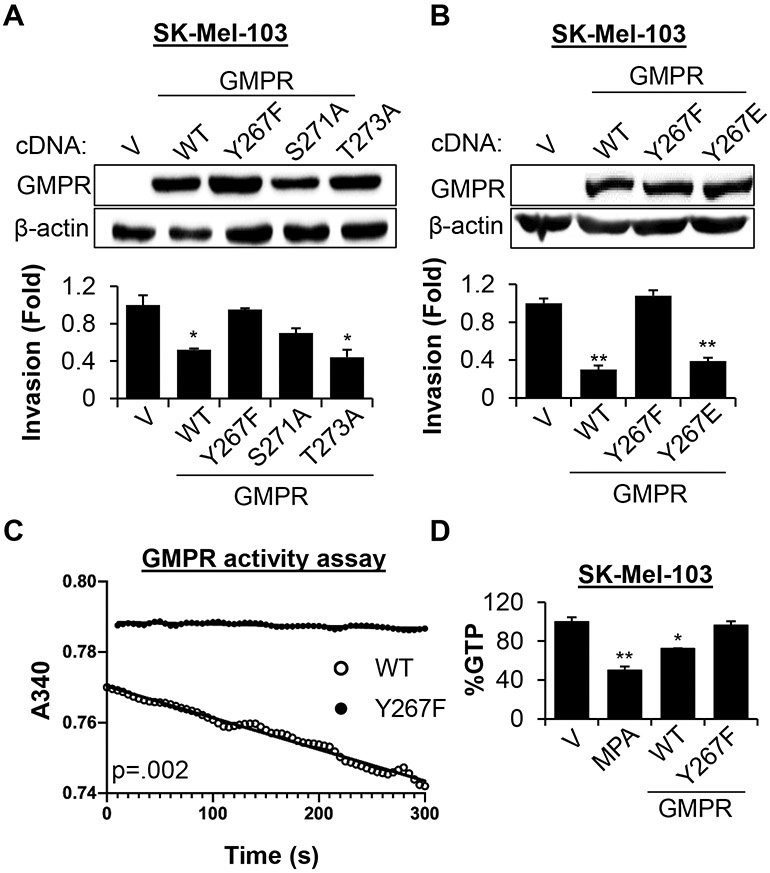
A-B. SK-Mel-103 cells transduced with empty lentivirus vector (V) or vectors expressing cDNA encoding the indicated GMPR mutants were probed in immunoblotting with the indicated antibodies (top) or in Boyden’s chamber invasion assay (bottom, average +/− SEM of at least two biological replicas, Student’s t-test). C. SK-Mel-103 cells stably expressing the indicated flag-tagged proteins were used for immunoprecipitation with anti-FLAG antibodies followed by an in vitro GMPR activity assay measuring the rate of NADPH oxidation during GMPR-mediated conversion of GMP to IMP (see STAR Methods). Representative reaction progress curves are shown. The average specific activity of at least three determinations were 0.82 x 10−3 s−1 and 5.1 x 10−3 s−1 for GMPRY267F and GMPRWT, respectively (Student’s t-test; p = 0.002). D. SK-Mel-103 cells transduced with the indicated constructs or treated with mycophenolic acid (MPA, 1 μM for 24 hours) were probed in mass spectrometry to determine GTP content (average +/− SEM of ≥ 2 biological replicas, Student’s t-test). All immunoblots shown are representative images from ≥2 independent experiments.
Enzymatically, GMPR reduces guanosine monophosphate (GMP) to inosine monophosphate (IMP), acting as a negative regulator of de novo GTP synthesis (Figure 1A) (Spector et al., 1979). Thus, we overexpressed GMPRWT or GMPRY267F in SK-Mel-103 cells followed by immunoprecipitation and probing of the isolated proteins in an in vitro GMPR enzymatic activity assay (Patton et al., 2011). This analysis revealed that the enzymatic activity of GMPRY267F was less than that of GMPRWT by a factor of ~6 (Figure 2C). In parallel, we assessed the ability of GMPRY267F to decrease intracellular GTP levels in melanoma cells, since we previously found that overexpression of GMPR in SK-Mel-103 cells reduced intracellular GTP pools (Wawrzyniak et al., 2013). Consistent with the in vitro data, GMPRWT overexpression in melanoma cells decreased total GTP by approximately 28% compared to empty vector, whereas GMPRY267F failed to suppress GTP levels (Figure 2D). Mycophenolic acid, an inhibitor of IMPDH enzymes (Verham et al., 1987), was used as a positive control for suppression of intracellular GTP (Figure 2D). Taken together, these results demonstrate that phosphorylation of Tyr267 is critical for the ability of GMPR to regulate intracellular GTP pools and cell invasion.
The ephrinA5-EPHA4 axis regulates GMPR phosphorylation on Tyr267 in melanoma cells
The above data suggest that GMPR activity in melanoma cells is controlled by one or more signaling pathways culminating in GMPR phosphorylation on Tyr267. To identify these pathways, we utilized an in vitro screening of tyrosine kinases capable of phosphorylating a 23-mer peptide consisting of GMPR amino acids 256-277, encompassing a single tyrosine corresponding to GMPR Tyr267. A total of 94 tyrosine kinases were screened in a radiometric assay by ProQinase, GmbH (Freiburg im Breisgau, Germany). Ten primary hits were identified with an activity ratio of ≥2 comparing the reaction mixtures with or without the GMPR-derived peptide (Table S1). Four out of ten hits belonged to the EPH receptor family: EPHA4, EPHA5, EPHA6 and EPHB1.
To test whether EPHs phosphorylate GMPR on Tyr267, we first generated GMPR Tyr267 phospho-specific antibodies using an 11-mer peptide containing GMPR amino acids 263-273 with a phosphorylated tyrosine corresponding to Tyr267. The specificity of the antibodies was confirmed in an ELISA utilizing this peptide and the corresponding non-phosphorylated peptide (Table S2), and via immunoblotting where the antibodies readily detected GMPRWT but not GMPRY267F immunoprecipitated from SK-Mel-103 cells (Figure 3A). In addition, treatment of the immunoprecipitated GMPRWT with alkaline phosphatase prior to immunoblotting decreased the antibody signal, confirming the specificity of these antibodies for GMPR phosphorylated on Tyr267 (pGMPRY267).
Figure 3. The EFNA5-EPHA4 axis drives GMPRY267 phosphorylation in melanoma cells.
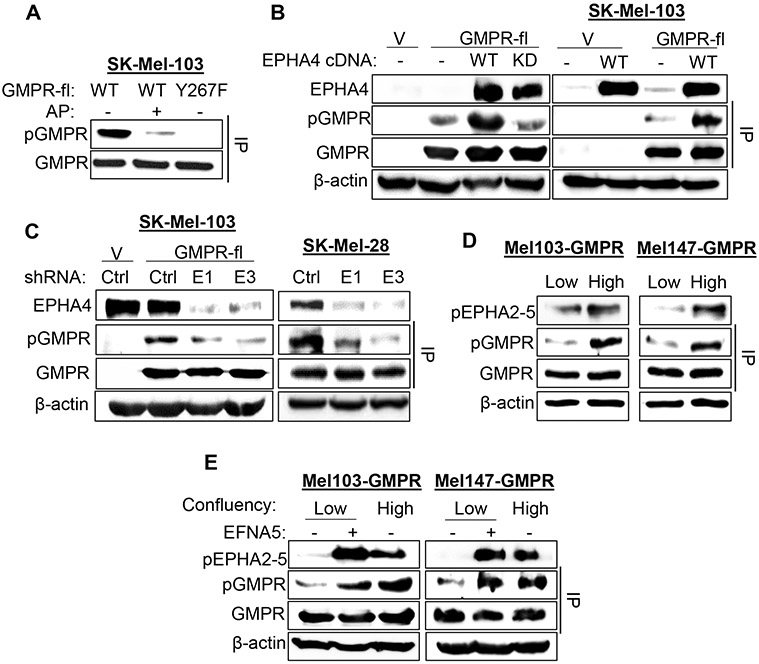
A. SK-Mel-103 cells stably expressing the indicated flag-tagged proteins were used in immunoprecipitation with anti-FLAG antibodies followed by immunoblotting with anti-GMPR (GMPR) or anti-phosphoGMPRY267 (pGMPR) antibodies. Prior to SDS-PAGE, immunoprecipitates were subjected or not to treatment with alkaline phosphatase (1 unit for 1 hour at 37°C) as indicated. B. Left: 293FT cells were transfected with the following plasmids (empty lentiviral vector - V, wildtype EPHA4 - WT, catalytically inactive EPHA4 mutant – Mut, FLAG-tagged GMPR (GMPR-fl)). Cell extracts were subjected or not to immunoprecipition with anti-FLAG antibodies (IP) and probed in immunoblotting with the indicated antibodies. Right: Cell extracts from SK-Mel-103 cells stably expressing the indicated constructs were subjected or not to anti-FLAG IP and probed in immunoblotting with the indicated antibodies. C. Left: SK-Mel-103 cells stably expressing or co-expressing the indicated constructs (Control vector- V, GMPR-fl, Ctrl - control shRNA, E1 - EPHA4 shRNA1, E3 – EPHA4 shRNA3) were probed in immunoblotting with the indicated antibodies. Right: Lysates from SK-Mel-28 cells expressing endogenous GMPR and the indicated shRNA were subjected or not to anti-GMPR IP and probed in immunoblotting as indicated. D. GMPR-expressing SK-Mel-103 and SK-Mel-147 cells were harvested at low or high confluency and subjected or not to anti-FLAG IP and immunoblotting with the indicated antibodies. E. Cells cultured as in (D) and treated or not with 2 μg/mL EFNA5 for 30 minutes prior to harvesting. PBS was used as a vehicle control. Cell extracts were subjected or not to anti-FLAG IP and probed in immunoblotting with the antibodies indicated above and with antibodies against phosphorylated EPHA2-5 (pEPHA2-5). All immunoblots shown are representative images from ≥ 2 independent experiments.
Next, expression vectors encoding Flag-tagged EPHA4, EPHA5, or EPHB1 were co-transfected with GMPRWT-fl in 293FT cells. EPHA6 was excluded since it is not expressed in melanoma (data not shown), and thus could not be responsible for GMPR phosphorylation in these cells. Immunoblot analysis of immunoprecipitated GMPRWT protein demonstrated elevated pGMPRY267 signal in cells expressing any of the three EPH receptors relative to the negative control, with EPHA4 showing the strongest ability to phosphorylate GMPR on Tyr267 (Figure S3A). Thus, EPHA4 was chosen for further studies.
To determine whether phosphorylation of GMPR depends on EPHA4 kinase activity, 293FT cells were co-transfected with vectors encoding GMPRWT and either EPHA4 wild-type or the K653M kinase-inactive mutant (Kullander et al., 2001). Elevated pGMPRY267 levels were detected only in cells expressing wild-type EPHA4 (Figure 3B, left panel). In addition, we found that EPHA4 overexpression also increases phosphorylation of GMPRWT stably expressed in SK-Mel-103 cells (Figure 3B, right panel). Reciprocally, shRNA-mediated depletion of EPHA4 in SK-Mel-103 cells stably expressing GMPRWT decreased GMPR Tyr267 phosphorylation relative to cells transduced with control shRNA (Figure 3C, left panel). Similar results were obtained for endogenous GMPR protein in SK-Mel-28 cells, the only melanoma cell line in our collection that expresses moderate levels of GMPR (Figure 3C, right panel) (Wawrzyniak et al., 2013).
EPH receptors including EPHA4 are activated via interactions with their ligands, the ephrins (EFNs) (Niethamer and Bush, 2019). Like EPHs, EFNs are membrane-bound proteins, and activation of EPHs by EFNs typically involves cell-cell contact (Pasquale, 2008; Singh et al., 2012). Therefore, we tested the hypothesis that GMPR phosphorylation is regulated by EFN-mediated activation of EPHs. We compared the levels of pGMPRY267 and activated EPHA receptors in melanoma cells cultured at low or high confluency. The levels of activated (tyrosine phosphorylated) EPHA receptors were determined using a phosphospecific antibody that recognizes the phosphorylated Tyr779 motif in the activation loop of EPHA4 and the corresponding conserved motif in EPHA2, EPHA3, and EPHA5 (Binns et al., 2000). The levels of activated EPHAs and pGMPRY267 in SK-Mel-103 and SK-Mel-147 cells stably expressing GMPR were higher when the cells were cultured at high confluency compared low confluency (Figure 3D). Furthermore, short-term treatment of low confluency SK-Mel-103 and SK-Mel-147 cells stably expressing GMPR and with soluble EFNA5 (2 μg/mL) activated EPHAs and increased pGMPRY267 levels (Figure 3E, S3B). GMPR expression did not affect EPHA activation by EFNA5 treatment (Figure S3C). Treatment with soluble EFNA5 also increased phosphorylation of endogenous GMPR in SK-Mel-28 cells (Figure S3D). Collectively, these data argue that the EFNA5-EPHA4 signaling axis controls GMPR Tyr267 phosphorylation in melanoma cells.
EPHA4 interacts with and phosphorylates GMPR
To investigate the mechanisms underlying GMPR phosphorylation by EPHA4, we assessed GMPR-EPHA4 association. Immunoprecipitation of endogenous EPHA4 from SK-Mel-103 melanoma cells stably expressing GMPR demonstrated that GMPR co-immunoprecipitates with EPHA4-specific but not control (IgG) antibodies (Figure 4A). A reciprocal experiment demonstrated that EPHA4 co-immunoprecipitates with GMPR (Figure 4A). Additionally, we analyzed the same cells via co-immunofluorescence utilizing GMPR antibodies and antibodies recognizing the EPHA4 intracellular region (EPHA4-ICD). The results demonstrated co-localization of GMPR and EPHA4 in lamellipodia and at the leading edges of cells. (Figure 4B). We next performed proximity ligation assays (PLA) using the same antibodies in SK-Mel-103 with or without stable GMPR expression. A strong PLA signal was detected only in GMPR-expressing cells that were probed with both EPHA4-ICD and GMPR antibodies (Figure 4C), indicating a close association between these two proteins in live cells.
Figure 4. EPHA4 interacts with and phosphorylates GMPR and regulates intracellular GTP levels.

A. SK-Mel-103 cells stably expressing FLAG-tagged GMPR were subjected to immunoprecipitation with the indicated antibodies followed by immunoblotting with the indicated antibodies. B. Cells described in (A) were grown on chambered coverslips and subjected to co-immunofluorescence analysis with GMPR antibodies and antibodies to the intracellular portion of EPHA4 (EPHA4-ICD), and counterstained with DAPI. Images were acquired at 60x magnification. Scale bars 10 μm. C. SK-Mel-103 cells expressing either GMPR or V control were grown on chambered coverslips and subjected to proximity ligation assay using the indicated antibody combinations, and counterstained with DAPI. Images were acquired at 20x magnification. Scale bars 20 μm. D. Bacterially purified GST-tagged GMPR protein was subjected to an in vitro kinase reaction with or without purified EPHA4 kinase. The resulting reaction mixtures were probed in immunoblot analysis using the indicated antibodies. E. SK-Mel-103 cells transduced with the indicated constructs were analyzed by mass spectrometry to determine GTP content (average +/− SEM of ≥ 2 biological replicas, Student’s t-test). F. Bacterially purified GST-GMPR was phosphorylated or not as in (D), and subjected to GMPR activity assays. The lines are fits to the Michaelis-Menton equation. See also Table S3. All immunoblots shown are representative images from ≥2 independent experiments.
To determine whether EPHA4 directly phosphorylates GMPR, we probed bacterially-expressed recombinant GMPR protein in an in vitro phosphorylation assay with recombinant EPHA4-ICD protein. Immunoblot analysis demonstrated a dose-dependent increase in pGMPRY267 in the reactions containing EPHA4-ICD (Figure 4D). An in vitro binding assay further showed that purified EPHA4 co-precipitated in equivalent amounts with bacterially purified GMPR and GMPRY267F (Figure S4A), indicating that the interaction is not dependent on Tyr267. To visualize the interaction between EPHA4 and GMPR, a molecular docking approach was employed based on previously established methods (Galeazzi et al., 2018). Computational protein-protein docking, with constraints to target the interaction to the substrate binding site of EPHA4 (Davis et al., 2009) and the proposed GMPR phosphorylation site (Tyr267), was performed between the crystal structure of the EPHA4 kinase domain (PDB file 2Y6M) and a single monomer of GMPR (PDB file 2BLE) to generate an initial complex. This initial complex was energy minimized and subjected to molecular dynamics simulation for further refinement. The refined model was used as the basis for additional modeling of the interaction of EPHA4 with the GMPR homotetramer (Figure S4B). This computational model suggests that an interaction between EPHA4 and GMPR is plausible and that EPHA4 binds to two monomers of the GMPR homotetramer on the outer upper edge, away from the cavity of the GMPR α/β-barrel core (Li et al., 2006). In the model, the GMPR monomer with the putative target phosphorylation site (Tyr267) contributes 11 residues to stabilize the EPHA4-GMPR interface (including 9 hydrogen bonds), whereas the neighboring monomer provides nearly twice as many contacts (21 residues), including 8 hydrogen bonds. Cumulatively, these results indicate that EPHA4 directly interacts with and phosphorylates GMPR.
To test the hypothesis that EPHA4 modulates GTP pools in melanoma cells, SK-Mel-103 cells stably expressing GMPR or vector control were transduced with empty vector or EPHA4 cDNA followed by assessment of intracellular GTP levels by mass spectroscopy. GMPR overexpression decreased total GTP by ~20% compared to empty vector. Ectopic expression of EPHA4 in control cells did not affect GTP levels, but EPHA4 expression further decreased GTP levels in GMPR-overexpressing cells (~25% total) (Figure 4E). Thus, EPHA4 controls intracellular GTP levels in a GMPR-dependent manner.
Additionally, we performed in vitro GMPR activity assays utilizing bacterially isolated GMPR protein phosphorylated or not in vitro with purified EPHA4 kinase as in Figure 4D. This analysis revealed that GMPR phosphorylation increased the activity of GMPR by a factor of ~2 due to increasing the value of Vmax with no apparent effect on the value of Km for NADPH, while the value of Km for GMP was too low to measure (Figure 4F, Table S3). Taken together, these results demonstrate that phosphorylation by EPHA4 positively regulates GMPR enzymatic activity.
GMPR levels define EPHA4 anti- and pro-oncogenic functions
Several members of the EPH family, including EPHA4, can have tumor suppressor or oncogenic functions, including the ability to regulate cell migration and invasion in opposite ways, although the exact mechanisms are not fully understood (Pasquale, 2008, 2010). On the other hand, we have demonstrated that GMPR suppresses invasion in several cancer cell types (Wawrzyniak et al., 2013). Thus, we were intrigued by the possibility that GMPR expression influences regulation of cell invasion by EPHA4.
To test this hypothesis, we first depleted EPHA4 via shRNA-mediated knockdown in the GMPR-positive SK-Mel-28 cells and the GMPR-negative SK-Mel-103 cells, and examined them in a Boyden’s chamber invasion assay. EPHA4 depletion increased invasion in SK-Mel-28 cells (expressing GMPR) but decreased invasion in SK-Mel-103 cells (not expressing GMPR) (Figures 5A, B). Reciprocally, overexpression of EPHA4 suppressed invasion in SK-Mel-28 and upregulated invasion in SK-Mel-103 (Figures 5C, D). To determine whether GMPR expression accounts for the observed difference in EPHA4-dependent invasion, we transduced SK-Mel-103 cells with control or GMPR-encoding vectors, followed by infection with control or EPHA4 shRNAs (as in Figure 3C). Knockdown of EPHA4 increased invasion in SK-Mel-103 cells expressing GMPR compared to control shRNA (Figure 5E), despite suppressing invasion in parental cells (Figure 5B). Importantly, overexpression of EPHA4 increased cell invasion in melanoma cells expressing the GMPRY267F mutant but decreased it in cells expressing GMPRWT (Figures 5F, G).
Figure 5. GMPR status controls EPHA4-dependent cell invasion.

A. SK-Mel-103 and SK-Mel-28 cells were transduced with the indicated constructs (Ctrl - control shRNA, E1 - EPHA4 shRNA1, E3 – EPHA4 shRNA3) followed by immunoblotting with the indicated antibodies. B. Cells described in (A) were probed in the Boyden’s chamber invasion assay. C. SK-Mel-103 and SK-Mel-28 cells were transduced with control vector (V) or EPHA4-expressing vector (A4), followed by immunoblotting with the indicated antibodies. D. Cells described in (C) were probed in the Boyden’s chamber invasion assay. E. SK-Mel-103 cells generated as in Figure 3C were probed in the Boyden’s chamber invasion assay. F. SK-Mel-103 and SK-Mel-147 cells were transduced with the indicated constructs followed by immunoblotting with the indicated antibodies. G. Cells described in (F) were probed in the Boyden’s chamber invasion assay. For invasion assays, data represent average +/− SEM from ≥ 2 biological replicas, Student’s t-test. All immunoblots shown are representative images of ≥ 2 independent experiments.
Enhanced cell invasion can overcome spatial constraints and increase tumor xenograft growth in immunocompromised mice in vivo (Hotary et al., 2003; Wawrzyniak et al., 2013). Therefore, we examined the ability of GMPR to modulate EPHA4-dependent melanoma cell tumorigenicity in SCID mice. Ectopic expression of GMPR suppressed SK-Mel-103 and SK-Mel-147 cell xenograft growth (Figures 6A, B), in agreement with previously published data (Wawrzyniak et al., 2013). Importantly, EPHA4 overexpression increased the tumorigenicity of empty vector cells, but suppressed it in cells expressing GMPR (Figures 6A, B). Taken together, these data strongly suggest that the pro- and anti-oncogenic activities of EPHA4 are determined in part by the expression level and phosphorylation status of GMPR.
Figure 6. GMPR influences the pro- and anti-oncogenic functions of EPHA4.

A. SK-Mel-103 and SK-Mel-147 cells stably expressing or co-expressing control vector (V), EPHA4 (A4) and/or GMPR, were implanted into both flanks of SCID mice (n=5) and measured by digital caliper for indicated time periods once palpable tumors formed. Statistical analysis was performed by two-way ANOVA. B. Pictures of representative tumors harvested after experimental end-points were reached. C. Expression of GMPR and EPHA4 in primary melanomas (n=54) and melanoma metastases (n=63). The Tukey’s boxplots represent the distribution of the IHC index. D. Samples were separated into “high” and “low” GMPR expression using the average GMPR IHC index in metastasis (~4.8) as a cut-off. EPHA4 IHC index values from each group are plotted. Statistics for IHC index comparisons were performed using Mann-Whitney U test. E. Chi-Squared analysis of individual melanoma samples with EPHA4 IHC scores ≤1 (low) and 3 (high) compared to GMPR ≤4 (low) and ≥6 (high). F. Analysis of 363 Skin Cutaneous Melanoma (SKCM) samples from the TCGA PanCancer Atlas. Shown are log2-transformed normalized counts for EFNA5 and GMPR mRNA.
EPHA4 and GMPR expression levels are inversely correlated in human melanoma specimens
Based on the in vivo tumorigenicity assay data, we hypothesized that expression levels of EPHA4 and GMPR are inversely correlated in human primary melanoma specimens. To test this hypothesis, we analyzed EPHA4 expression by immunohistochemistry (IHC) in 54 primary and 63 metastatic human melanoma specimens. Tumors were scored for the intensity of staining and the percentage of labelled cells (see Materials and Methods). An IHC index corresponding to the product of these parameters was calculated and used to semicategorically assess EPHA4 expression levels. The expression of the membrane-bound EPHA4 was significantly higher in the metastatic melanoma cohort compared to the primary melanoma cutaneous cohort (Figure 6C, left panel). In contrast, GMPR expression was significantly lower in the metastatic melanoma cohort compared to primary melanoma cohort (Figure 6C, right panel), as we previously reported (Wawrzyniak et al., 2013; Bianchi-Smiraglia et al., 2017a). Furthermore, comparing GMPR and EPHA4 expression levels in individual specimens using different statistical methods revealed an inverse correlation (Figures 6D, E). We also uncovered an inverse correlation between GMPR and EFNA5 mRNA expression in melanoma specimens by analyzing publicly-available data from The Cancer Genome Atlas (TCGA) (Figure 6F) (Cancer Genome Atlas, 2015; Goldman et al., 2020). Together, these results demonstrate that EPHA4 activity increases during progression to metastatic melanoma and that this increase is accompanied by a downregulation of GMPR expression.
EPHA4 suppresses RAC1 activity via phosphorylation of GMPR
We have previously reported that GMPR suppresses invasion in melanoma cells by inhibiting several RHO GTPases, most prominently RAC1 (Wawrzyniak et al., 2013). To evaluate the functional role of GMPR phosphorylation in the regulation of RAC1 by EPHA4, we compared RAC1 activity in cells expressing GMPRWT or GMPRY267F using RAC1 pull-down assays. This demonstrated that GMPRY267F fails to suppress RAC1 activity (Figures 7A, B). These data suggest that phosphorylation of GMPR on Tyr267 is important for RAC1 inhibition.
Figure 7. Phosphorylation of GMPR by EPHA4 suppresses RAC1 activation.

A. SK-Mel-103 cells transduced with the indicated constructs were probed in immunoblotting with the indicated antibodies. B. Cells described in (A) were probed in RAC1 activity pulldown assay followed by immunoblotting. Quantification represents average +/− SEM of 3 independent RAC1 pull-down experiments with a student’s t-test. C. SK-Mel-103 cells transduced with the indicated constructs (V, GMPR, Ctrl - control shRNA, E1 - EPHA4 shRNA1, E3 – EPHA4 shRNA3) were probed in immunoblotting with the indicated antibodies. D. The cells described in (C) were probed in RAC1 activity pulldown assay followed by immunoblotting. Quantification represents average +/− SEM of 3 independent RAC1 pull-down experiments with a student’s t-test. E. Cells stably expressing the indicated constructs were probed in immunoblotting with the indicated antibodies. F. Cells described in (E) were probed in RAC1 activity pulldown assays followed by immunoblotting Quantification represents average +/− SEM of 3 independent RAC1 pull-down experiments with a student’s t-test. G. Cell bodies (CB) and cell protrusions (CP) were harvested from SK-Mel-103 and SK-Mel-147 cells stably expressing GMPR as described in STAR Methods. Equal amounts of protein were probed in immunoblotting with the indicated antibodies (top) or stained with Ponceau S (bottom) as a loading control. H. SK-Mel-103 cells stably expressing the indicated constructs (control vector - V, GMPR, GMPRY267F) were probed in GEVAL-activity assays as described in STAR Methods. Shown is dot-plot quantification of GEVAL activity in individual CBs and CPs of the indicated cells. Bars represent mean value as a percentage of CB signal for each condition. Statistical analysis was performed by using the Mann-Whitney U test. See also Figs. S6 and S7. All immunoblots shown are representative of ≥2 independent experiments.
Next, we examined if GMPR expression determines whether EPHA4 activates or suppresses RAC1. To this end, we treated GMPR-positive SK-Mel-28 and GMPR-negative SK-Mel-103 with soluble EFNA5, and measured RAC1 activity. EFNA5 treatment resulted in decreased RAC1 activity in SK-Mel-28, but increased RAC1 activity in SK-Mel-103 (Figures S5 A, B). Additionally, SK-Mel-103-Vector or SK-Mel-103-GMPR cells were infected with control or EPHA4 shRNAs lentiviruses and probed in RAC1-GTP pulldown assays. Depletion of EPHA4 decreased RAC1-GTP levels in SK-Mel-103-Vector cells, but increased them in SK-Mel-103-GMPR cells (Figures 7C, D). Conversely, overexpression of EPHA4 increased RAC1-GTP levels in SK-Mel-103-Vector or SK-Mel-103-GMPRY267F cells but decreased RAC1-GTP levels in SK-Mel-103-GMPRWT cells (Figures 7E, F). Similar results were observed for other members of RHO-GTPase family, namely RHO-A and RHO-C, in SK-Mel-103 cells expressing ectopic EPHA4 with or without GMPR (Figures S5C, D).
EPHA4 positively regulates the RAC1 guanine nucleotide exchange factor VAV2 via direct phosphorylation on tyrosine 172 (pVAV2Y172) (Ogita et al., 2003; Cowan et al., 2005). To test whether GMPR expression disrupts this regulation, we assayed the status of pVAV2Y172 in melanoma cells depleted of EPHA4 in the presence and absence of GMPR. GMPR overexpression did not affect the levels of VAV2 phosphorylation (Figure S5E). Moreover, a similar reduction of pVAV2Y172 in SK-Mel-103 and SK-Mel-147 following depletion of EPHA4 with shRNA was detected in both control and GMPR-expressing cells (Figure S5E). These data suggest that EPHA4 suppresses RAC1 activity via GMPR phosphorylation on Tyr267 independently from its ability to activate RAC1 via phosphorylation of VAV2 on Tyr172.
GMPR phosphorylation results in depletion of GTP levels in cell protrusions of invading cells
To gain mechanistic insights into how EPHA4 suppresses RAC1 activity and melanoma cell invasion, we first tested whether EPHA4 colocalizes with RAC1 and GMPR in the protrusions of melanoma cells (Bianchi-Smiraglia et al., 2021). To isolate cell bodies (CBs) and cell protrusions (CPs), we applied a previously reported methodology with some modifications (Figure S6A) (Wang et al., 2007; Mardakheh et al., 2015). Equal amounts of material from CB and CP lysates were analyzed by immunoblotting. We found that EPHA4, RAC1, and GMPR were enriched in the CP fraction of SK-Mel-103 and SK-Mel-147 cells (Figure 7G). The nuclear aryl hydrocarbon receptor nuclear translocator (ARNT) and Integrin β1 (ITGB1) were used as positive controls for CB and CP fractions, respectively (Cox et al., 2001; Bersten et al., 2013).
We recently generated genetically-encoded ratiometric fluorescent biosensors capable of detecting changes in free intracellular GTP (termed GEVAL for GTP evaluator) (Bianchi-Smiraglia et al., 2017b). Interaction with free GTP results in a ratiometric change in GEVAL fluorescence (Figure S6B). One of the sensors, GEVAL30 with Keff=32.3μM (GTP concentration required to obtain 50% of the maximal ratiometric signal) detects changes in GTP levels starting from 4μM with saturation starting at ~100μM, thus enabling us to detect GTP fluctuations in live cells at concentrations in this range(Bianchi-Smiraglia et al., 2017b). The GEVALNull sensor, incapable of binding GTP, serves as a critical control which rules out GTP-unrelated changes in GEVAL activity. Utilizing these tools, we assessed relative GTP content in CPs versus CBs in SK-Mel-103 cells expressing empty vector, GMPRWT or GMPRY267F. Local GTP levels were higher in CPs than CBs in cells expressing vector or GMPRY267F, but not in cells expressing GMPRWT (Figure 7H, Figure S7A), thus demonstrating that GMPR phosphorylation on Tyr267 is an important mechanism regulating GTP levels in cell protrusions. These data also correlate with the previously reported increase in RAC1 activity in cell protrusions (Machacek et al., 2009; MacNevin et al., 2016; Bianchi-Smiraglia et al., 2021). No significant differences in GEVAL activity were observed between CPs and CBs when the same experiment was performed in cells expressing the GTP-insensitive GEVALNull sensor (Figure S7A, B).
DISCUSSION
Aberrant signaling of RHO GTPases, particularly RAC1, is a hallmark of cancer that drives several malignant phenotypes including invasion and metastasis (Bid et al., 2013; Lawson and Ridley, 2018; Clayton and Ridley, 2020). RAC1 activity can be elevated in solid tumors through numerous mechanisms, including gene amplification, gain-of-function mutations, dysregulated upstream signaling, and others (Davis et al., 2013; Marei and Malliri, 2017; De et al., 2019). This is particularly relevant for sun-exposed melanoma, where RAC1P29S is reported to be the third most common driver mutation, promoting drug resistance and an invasive mesenchymal phenotype (Krauthammer et al., 2012; Watson et al., 2014; Halaban, 2015; Lionarons et al., 2019).
Physiological changes in intracellular GTP pools were historically considered insufficient to affect RHO GTPase association with GTP. This is because GTP concentrations measured by HPLC or mass spectrometry suggested cellular concentrations in the 0.5-1.5mM range (Woodland and Pestell, 1972; Traut, 1994) and, when assayed in vitro using non-hydrolyzable guanylate analogs, the dissociation constants (Kd) for GTP and GDP binding to RHO GTPases are very similar and typically reported to be in the sub-micromolar range (Zhang et al., 2000). The activity of GEFs increases the KdGTP of GTPases such as RAC1, but not beyond the low micromolar range in vitro (Goody and Hofmann-Goody, 2002; Haeusler et al., 2003). However, studies by us and others have established that GTP production by guanylate metabolism influences cell motility and invasion via modulation of small GTPase activity (Wawrzyniak et al., 2013; Bianchi-Smiraglia et al., 2015; Kollareddy et al., 2015; Bianchi-Smiraglia et al., 2017a; Bianchi-Smiraglia et al., 2021). Although we cannot pinpoint the exact mechanism at present, several lines of evidence suggest that GTP availability is a relevant factor. Previous methodologies used to measure intracellular GTP fail to account for heterogeneous distribution or to distinguish free versus sequestered GTP. Our recently developed intracellular fluorescent GTP sensors (GEVALs) revealed that free GTP gradients exist in live cells in the range of 30μM (Bianchi-Smiraglia et al., 2017b; Bianchi-Smiraglia et al., 2021). We further demonstrated that RAC1 activity is sensitive to local changes in GTP biosynthesis (Bianchi-Smiraglia et al., 2021). Additionally, the KdGTP of RAC1 measured in vitro with non-hydrolyzable guanylate analogues does not account for all variables within the dynamic environment of the cell, such as the effects of rapid GTP consumption driven by GTPase activating proteins (GAPs) (Zhang et al., 1998; Zhang et al., 2000), and thus may not represent the GTP demands of RAC1 to sustain high activity in vivo. Likewise, the effect of GEFs on GTPases measured in vitro may not account for areas of the cell with highly concentrated GEF and RAC1 activities, often co-localized near the membrane (Azoitei et al., 2019; Marston et al., 2020). The data presented herein demonstrate that RAC1 activation by EPHA4 is prevented by GMPR-mediated suppression of local GTP pools, primarily in cell protrusions where RAC1 is most active. Although it is possible that reduced GTP levels indirectly suppress RAC1 through other signaling events, we previously showed that GMPR has no detectable effects on activity of relevant signaling molecules such as AKT or SRC (Wawrzyniak et al., 2013). Thus, we propose that the most probable interpretation of this data is that suppression of RAC1 by GMPR occurs through limited GTP availability for RAC1 binding.
Cancer cells possess high levels of the enzymes involved in the de novo biosynthesis of nucleotides, including guanylates, which are important for a multitude of processes activated in cancer cells (Wawrzyniak et al., 2013; Bianchi-Smiraglia et al., 2015; Valvezan et al., 2017; Kofuji et al., 2019; Villa et al., 2019; Zhou et al., 2020; Huang et al., 2021). However, despite a long-standing interest in the regulation of de novo guanylate biosynthesis, the vast majority of the available literature addresses transcriptional mechanisms of regulation (Liu et al., 2008; Mannava et al., 2008; Kollareddy et al., 2015; Bianchi-Smiraglia et al., 2017a; Huang et al., 2021). In contrast, post-translational modifications controlling the activity of these enzymes are unknown, with few exceptions (Ingley and Hemmings, 2000; Plana-Bonamaiso et al., 2020). Our finding that phosphorylation of GMPR on Tyr267 regulates its activity and therefore intracellular GTP pools provides insight into these understudied mechanisms. Intriguingly, the key enzymes involved in guanine nucleotide biosynthesis and recycling, GMPR and IMPDH, are derived from the same protein ancestor, yet catalyze opposing reactions: where the action of GMPR reduces the guanine nucleotide pool, IMPDH catalyzes the first committed step in guanine nucleotide biosynthesis. Our experiments demonstrate that phosphorylation of Tyr267 activates GMPR. This residue is near a mobile loop close to the cofactor binding site (Figure S1), and phosphorylation may induce a more productive binding mode for NADPH. The analogous loop undergoes a critical conformational change during the IMPDH catalytic cycle (Hedstrom, 2012). Additionally, Tyr267 is immediately adjacent to a conserved phosphorylation "hotspot" previously identified in the IMPDH/GMPR family (Strumillo et al., 2019). However, whereas Tyr267 phosphorylation activates GMPR, the phosphorylation of the hotspot serine in IMPDH1 decreases activity (Plana-Bonamaiso et al., 2020). Potentially coordinated regulation of GMPR and IMPDH by phosphorylation, perhaps by the same upstream signals, is an attractive model. Regarding the GMPR-EPHA4 interaction, the modeled complex (Figure S5) provides a starting point for assessing which residues are critical for the interaction between the two proteins. Targeted mutations in critical secondary structural elements of either GMPR or EPHA4 can be used to evaluate key interactions, particularly focusing on the newly proposed interaction between the non-phosphorylated GMPR monomer (residues in helix α10) and EPHA4 (loop between β4 and β5).
EPHs primarily affect cell behavior at sites of cell-cell contact by interacting with membrane-bound EFN ligands (Pasquale, 2008; Singh et al., 2012), as is exemplified by the regulation of axon guidance and growth cone collapse during the development of the olfactory, auditory, and visual sensory systems (Suetterlin et al., 2012; Triplett and Feldheim, 2012; Cramer and Miko, 2016), as well as motor control (Jiang et al., 2020). Multiple studies reported that these processes occur through alterations of RHO GTPase activity at the distal tips of growing neurites (Wahl et al., 2000; Sahin et al., 2005; Iwasato et al., 2007; Shi et al., 2007; Kao et al., 2015). Here, we demonstrate that EPHA4 activity leads to suppression of RAC1 activity in a GMPR-dependent manner. As a key enzyme in guanylate metabolism, GMPR is expressed in most cell types including neurons (Liu et al., 2018). Therefore, it will be interesting in the future to explore the role of GMPR and GTP metabolism in the regulation of EPH-dependent neuronal development.
Consistent with the multi-faceted roles of ephrin signaling during embryonic development, the function of EFNs and EPHs in tumorigenesis appears to be complex, involving both oncogenic and tumor suppressive activities. In melanoma, EPH receptor signaling has primarily been reported to promote tumor growth and aggressiveness (Yang et al., 2006; Udayakumar et al., 2011; Miao et al., 2015; Zhang et al., 2020), although tumor suppressor effects have also been described (Guo et al., 2021). Similarly, EPHA4 has been shown to increase proliferation, motility and/or invasion in colorectal (de Marcondes and Morgado-Diaz, 2017), breast (Dong et al., 2018), and pancreatic cancer cells (Iiizumi et al., 2006; Furuhashi et al., 2021), but suppresses migration and invasion of lung adenocarcinoma cells (Saintigny et al., 2012). In melanoma cells, the role of EPHA4 is understudied with one publication reporting that suppression of EPHA4 by oncogenic miR-519d promotes transformed phenotypes in cultured cells and metastasis in vivo (Hua et al., 2018), suggesting that EPHA4 acts as a tumor suppressor in this context. Interestingly, this study utilized the A2058 human melanoma cell line, which retains moderate GMPR expression according to publicly available expression profiling (data not shown). On the other hand, high EPHA4 expression has been detected in a highly metastatic melanoma cell subpopulation (Snyder et al., 2020). Also, EPHA4 mutations reportedly occur preferentially in the absence of the main melanoma driver mutations in BRAF and NRAS (Xia et al., 2014), with at least one recurrent mutation resulting in elevated kinase activity (Light et al., 2021), suggesting a potential influence of EPHA4 mutations on melanoma progression.
Here we identify a mechanism in melanoma cells through which GMPR phosphorylation by EPHA4 converts EPHA4-mediated RAC1 activation into RAC1 inhibition. We found that EPHA4 promotes RAC1 activity and invasion in GMPR-negative melanoma cells through mechanisms described in other contexts, such as the phosphorylation of VAV2 (Cowan et al., 2005; Hunter et al., 2006). EPHA4-mediated phosphorylation of VAV2 is not altered by GMPR, suggesting that the suppression of RAC1 by GMPR counteracts the activity of GEFs such as VAV2. This scenario might explain the observed increase in EPH levels in melanoma specimens with decreased GMPR levels, which is associated with metastatic progression. It should be noted that EPHA4 can also inhibit RAC1 through a GMPR-independent mechanism involving the RAC1 GAP α2-chimaerin (Beg et al., 2007; Shi et al., 2007; Wegmeyer et al., 2007). While α2-chimaerin can suppress RAC1 activity, its function in melanoma is undescribed.
While GMPR appears to be a critical co-factor for EPHA4-dependent regulation of RAC1 in melanoma, the role for guanylate metabolism in the control of RHO GTPases by EPHs in general remains to be explored. It is conceivable that GMPR phosphorylation could mediate signaling by other EPHs, including EPHA5 and EPHB1, since these kinases are capable of phosphorylating GMPR on Tyr267. GMPR phosphorylation on Ser271 also appears to be relevant for the ability of GMPR to suppress melanoma cell invasion, further supporting the notion that GMPR may mediate the effects of multiple kinases on transformed phenotypes.
In summary, our study describes a previously unknown role of guanylate metabolism in determining the phenotypic consequence of upstream signaling pathways. GMPR expression and phosphorylation affect the regulation of RAC1, melanoma cell invasion, and tumorigenicity induced by EPHA4-dependent signaling. This occurs through a phosphorylation event that promotes the enzymatic activity of GMPR, thus decreasing GTP levels and suppressing RAC1 activation. Since sustained RAC1 activity plays a major role in promoting an invasive phenotype and progression to metastasis in melanoma, targeting guanylate metabolism may represent an important approach for the treatment of melanoma.
STAR METHODS
Resource availability
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Mikhail A. Nikiforov (mikhail.nikiforov@duke.edu).
Materials availability
Aliquots of the phospho-GMPRY267 antibody will be available in limited quantity upon request.
Data and code availability
This paper analyzes existing, publicly available data. The accession numbers are listed in the key resource table.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rac1 | ProteinTech | Cat# 66122; RRID:AB_2881521 |
| GMPR | ProteinTech | Cat# 15683; RRID:AB_2111072 |
| anti-FLAG | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| EPHA4 | BD Biosciences | Cat# 610471; RRID:AB_397843 |
| EPHA4 | Thermo Fisher | Cat# 37-1600; RRID:AB_2533301 |
| β-ACTIN | ProteinTech | Cat# HRP-66009; RRID:AB_2883836 |
| phospho-EPHA3 (Tyr779) | Cell Signaling | Cat# 8862; RRID:AB_2797673 |
| phospho-VAV2 (Tyr172) | ECM Biosciences | Cat# VP2641; RRID:AB_2213715 |
| Integrin Beta 1 | ProteinTech | Cat# 26918; RRID:AB_2880685 |
| ARNT1 | Santa Cruz | Cat# sc-17811; RRID:AB_626698 |
| RhoA | Cell Signaling | Cat# 2117; RRID:AB_10693922 |
| RhoC | Cell Signaling | Cat# 3430; RRID:AB_2179246 |
| phospho-GMPR (Tyr267) | GenScript | This paper |
| Bacterial strains | ||
| DH5α Competent E. coli | New England BioLabs | Cat# C2987 |
| BL21 Competent E. coli | New England BioLabs | Cat# C2527 |
| Stable Competent E. coli | New England BioLabs | Cat# C3040 |
| Recombinant proteins | ||
| GST-GMPR | This paper | N/A |
| GST-GMPRY267F | This paper | N/A |
| GST/His-EPHA4 (aa 570-986) | SinoBiological | Cat# 11314-H20B1 |
| Critical commercial assays | ||
| Rac1 activity assay | Cell Biolabs | Cat# STA-401-1 |
| RhoA/C activity assay | Cell Biolabs | Cat# STA-403-A |
| Proximity ligation assay | Millipore Sigma | Cat# DUO92101 |
| 0.4 μm transwells (fractionation) | Corning | Cat# 3412 |
| 8.0 μm transwells (invasion) | Corning | Cat# 354480 |
| Tyrosine kinase screen | ProQinase | N/A |
| Deposited data | ||
| EPHA4 crystal structure | Protein Data Bank | PDB: 2Y6M |
| GMPR crystal structure | Protein Data Bank | PDB: 2BLE |
| Melanoma expression dataset | TCGA and Xena | GDC TCGA Melanoma (SKCM) |
| Experimental models: Cell lines | ||
| HEK293FT | Thermo Fisher | Cat# R70007 |
| SK-Mel-103 | MSKCC | RRID:CVCL_6069 |
| SK-Mel-147 | MSKCC | RRID:CVCL_3876 |
| SK-Mel-28 | ATCC | Cat# HTB-72; RRID:CVCL_0526 |
| Experimental models: Organisms/strains | ||
| ICR scid mice | Taconic | Cat# ICRSC |
| Recombinant DNA | ||
| pCMV-deltaR8.2 | Didier Trono | RRID:Addgene_12263 |
| pCMV-VSVG | Bob Weinberg | RRID:Addgene_8454 |
| pLV-SV40-puro | Peter Chumakov | PMID: 11854510 |
| pLV-GMPR-flag | In-house | PMID: 24139804 |
| pLV-GMPRY267F-flag | This paper | N/A |
| pLKO/TRC1-shEPHA4 (E1) (GATTGGCTCCAGGCCATTAAA) | Millipore Sigma | TRC0000220021 |
| pLKO/TRC2-shEPHA4 (E3) (GCAATTGCCTATCGTAAATTC) | Millipore Sigma | TRC00003828495 |
| pLenti-UbC-EPHA4 | Eric Haura | PMID: 20190765 |
| pLenti-UbC-EPHA4K653M | Eric Haura | PMID: 20190765 |
| pLVX-EPHA4-flag | In-house | PMID: 34139238 |
| pcDNA3-EPHA5-flag | This paper | N/A |
| pCMV3-EPHB1-flag | SinoBiological | Cat# MG50479-NF |
| pLenti6.2-EmGFP | Thermo Fisher | Cat# V36920 |
| pGEX-4T1-GMPR | This paper | N/A |
| pGEX-4T1-GMPRY267F | This paper | N/A |
| Software and algorithms | ||
| FIJI | Public domain | https://imagej.net/software/fiji/ |
| GraphPad Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| ChemiDoc MP Imaging System | Bio-Rad | Cat# 17001402 |
| Confocal microscope | Leica | TCS SP5 |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Cell lines
Human embryonic kidney 293FT cells expressing SV40 large T antigen were purchased from Thermo Fisher. Human melanoma SK-Mel-103 and SK-Mel-147 cells, which both harbor mutations encoding for NRASQ61R expression, were obtained from the Memorial Sloan Kettering Cancer Center. Human melanoma SK-Mel-28 cells, which express BRAFV600E, were purchased from the ATCC. All cells were cultured in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and penicillin-streptomycin antibiotics at 37°C and 5% CO2 in a humidified incubator. All cell lines have been recently authenticated and verified as mycoplasma-free using MycoAlert mycoplasma detection Kit purchased from Lonza (Allendale, NJ, USA, Cat # LT07-318).
Mice
Animals were maintained and all experiments were conducted according to a protocol approved by the Institute Animal Care and Use Committee at Wake Forrest Baptist Health Science. Genetically modified SK-Mel-103 or SK-Mel-147 cells expressing lentiviral constructs were inoculated subcutaneously in both flanks (1.0x106 cells/flank) of female ICR scid mice (Taconic, Model #ICRSC). For all cohorts, the time of the appearance of tumor ≥ 2 mm in at least one dimension was recorded. SK-Mel-103 or SK-Mel-147 tumors were measured thereafter every other day. Mice were euthanized when total tumor burden reached ~2000mm3.
Method details
Immunoblotting
Membranes were developed with horseradish peroxidase-conjugated secondary antibodies and signals were visualized using the ChemiDoc Imaging System (Bio-Rad). The following commercial antibodies were used in the study: RAC1 (ProteinTech, Cat. #66122), GMPR (ProteinTech Cat. #15683), anti-Flag (Millipore Sigma, Cat. #F1804), EPHA4 (BD Biosciences, Cat. #610471), EPHA4 (Thermo Fisher, Cat. #37-1600), Beta Actin-HRP (Proteintech, Cat. #66009), phospho-EPHA3 (Tyr779) (Cell Signaling, Cat. #8862), phospho-VAV2 (Y172) (ECM Biosciences, Cat. #VP2641), Integrin β1 (ProteinTech, Cat. #26918), ARNT (Santa Cruz, Cat. #sc-17811), RHO-A (Cell Signaling, Cat. #2117), RHO-C (Cell Signaling, Cat. #3430).
Immunohistochemical Analysis
Formalin fixed paraffin sections were cut at 4 μm, placed on charged slides, and dried at 60°C for one hour. Slides were cooled to room temperature and added to the Leica Bond Rx, where they were deparaffinized with Bond Dewax Solution (Leica AR9222) and rinsed in water. Bond Epitope Retrieval 2 (Leica AR9640) was used for target retrieval for 30 minutes. Slides were blocked using peroxide block from Bond Polymer Refine Detection kit (Leica DS9800) for 5 minutes. The following reagents are from the Bond Polymer Refine Detection kit (Leica DS9800). Post Primary was applied for 8 minutes followed by HRP polymer for 8 minutes. DAB (Diaminobenzidine) was applied for 10 minutes for visualization. Slides were counterstained with Hematoxylin for 8 minutes then placed into water. After removing slides from the bond they were dehydrated, cleared, and coverslipped. Positive and negative control slides were supplied by the Pathology Core Facility and were included with every immunochemistry run. For EPHA4 antibodies (Invitrogen PA5-14578) at 1/850 for 45 min) and GMPR antibodies (Sigma-Aldrich, HPA021476), the Novocastra PowerVision kit was used for visualization, followed by Fast Red (Thermo Scientific). The slides were manually counterstained with hematoxylin. Analysis of the GMPR-specific staining was described previously (Wawrzyniak et al., 2013). For EPHA4 analysis, samples were digitally scanned using Aperio Scanscope (Aperio Technologies, Inc., Vista, CA) with 20x bright-field microscopy. These images are then accessible using Spectrum (Aperio Technologies, Inc., Vista, CA), a web-based digital pathology information management system. Once slides are scanned, Aperio ImageScope version 12 (Aperio Technologies, Inc., Vista, CA) was to view images for image analysis. An annotation layer was created for each core of interest in the TMA targeting the melanoma cells for image analysis. Similarly, multiple regions of melanoma where circled on the whole sections for image analysis. Regions were identified and annotated to appropriately represent the heterogeneity of staining of each slide and TMA core and to reduce irrelevant regions from image analysis calculations. Special attention was made to avoid areas of strong melanin pigmentation or areas with heavy infiltration of melanin-laden macrophages. The Aperio image analysis platform was used to develop quantitative image analysis algorithm macros for the quantification of immunohistochemistry (IHC) slides. A Membrane Algorithm was developed to detect membrane staining for individual tumor cells and quantifies the intensity and completeness of the membrane staining. Tumor cells were individually classified as 0, 1+, 2+ and 3+ based on their membrane staining intensity and completeness, using the HER2 scoring scheme. Image analysis data were exported from Spectrum as a .csv file and converted to an Excel file and formatted using Microsoft Excel 2010.
Plasmids and Lentiviral Transduction
The pCMV-deltaR8.2 plasmid was a gift from Didier Trono (Addgene plasmid #12263). The pCMV-VSV-G vector was a gift from Bob Weinberg (Addgene plasmid #8454) (Stewart et al., 2003). The pLV-SV40-puro lentiviral vector was obtained from Dr. Peter Chumakov, Cleveland Clinic (Cleveland, OH, USA). Lentiviral shRNAs targeting EPHA4 were purchased from Sigma Aldrich (shRNA “E1”: TRC0000220021, “E3”: TRC00003828495). Lentiviral vector expressing GMPR with N-terminal Flag tag was generated by inserting human GMPR cDNA into pLV-SV40-puro via a PCR-based cloning strategy as described previously (Wawrzyniak et al., 2013), and GMPR mutants were generated via site-directed mutagenesis (New England Biolands, cat. #E0554S). Lentiviral constructs expressing wild-type or catalytically-inactive K653M EPHA4 mutant were kindly provided by Dr. Eric Haura (Moffitt Cancer Center). Human EPHA4 cDNA was cloned into pLVX-IRES-Neo lentiviral vector with an N-terminal Flag tag as described previously (Light et al., 2021). A pcDNA3 plasmid expressing N-terminal Flag-tagged EPHA5 was generated via a PCR-based cloning strategy. The pCMV3 plasmid expressing Flag-tagged EPHB1 was purchased from SinoBiological (Cat. # MG50479-NF). GFP-expressing lentiviral control vector was purchased from Thermo Fisher (Cat. #V36920). Lentivirus was generated following transfection of 293FT cells using polyethylenimine (PEI). Melanoma cells were transduced overnight in the presence of 8 μg/mL polybrene, and selected with appropriate antibiotics.
LC-MS analysis of GMPR modifications
GMPR-fl protein immunoprecipitated from SK-Mel-103 cells with and without GMPR-fl expression was processed for LC-MS analysis using a surfactant-aided precipitation/on-pellet digestion protocol. Samples were first spiked with SDS to a final concentration of 0.5%. Protein was reduced by 10 mM dithiothreitol (DTT) under 56°C and alkylated by 25 mM iodoacetamide (IAM) under 37°C to dissociate protein disulfide bonds. Both steps were performed in a covered thermomixer (Eppendorf, Hauppauge, NY) for 30 min with constant shaking. Protein was precipitated by two-step addition of 1 and 5 volumes of chilled acetone with vortexing, and was incubated under - 20°C for 3 hr. Precipitated protein was pelleted by centrifugation at 18,000 g under 4°C for 30 min, and pelleted protein was washed by 400 μL methanol. After wetting the protein with 46 μL pH 8.4 Tris-formic acid (FA), 4 μL 0.25 μg/μL trypsin (Sigma-Aldrich, St. Louis, MO) was added to the samples, and tryptic digestion was performed under 37°C overnight (~16 hr) in a thermomixer with constant shaking. Digestion was terminated by addition of 0.5 μL FA, and samples were centrifuged at 18,000 g under 4°C for 30 min. Supernatant was carefully transferred to LC vials for analysis.The LC-MS system consists of a SCIEX ekspert nanoLC 425 system (SCIEX, Redwood City, CA) and a Thermo Fisher Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA). For each sample, 4 μL of digested protein was injected for analysis. Samples were first loaded onto a large-inner diameter (i.d.) trapping column (300 μm i.d. × 5 mm), and then back-flushed onto a meter-long nano LC column (75 μm i.d × 100 cm) for separation. Mobile phase A and B were 0.1% FA in 2% acetonitrile (ACN) and 0.1% FA in 88% ACN. The 132-min LC gradient profile was: 3 – 5% B in 5 min, 5 – 24% B in 117 min, 24 – 50% in 10 min. MS was operated under data-dependent acquisition (DDA) mode with a maximal duty cycle time of 3 sec. MS1 spectra were acquired in profile mode by Orbitrap in an m/z range of 400 – 1,500 Th under 120k resolution with the following settings: Automatic gain control (AGC) target = 5E5, Maximum injection time = 50 ms, dynamic exclusion duration & m/z width = 20 s & 10 ppm. Precursor ions were filtered by quadrupole using a 1.6-Th window and fragmented in parallel by 1) collision-induced dissociation (CID) with a 30% collision energy; 2) electron transfer dissociation (ETD) with an ETD reagent target of 2E5, and 200 ms for both ETD reagent injection and reaction time. CID-MS2 spectra were acquired in centroid mode by Ion Trap in an m/z range of 350 – 1,400 with an AGC target of 1E4 and a maximal injection time of 50ms; ETD-MS2 spectra were acquired in centroid mode by Ion Trap with an AGC target of 1E4 and a maximal injection time of 200 ms. LC-MS files were searched against Homo Sapiens GMPR1 protein sequence using Sequest HT embedded in Proteome Discoverer 1.4 (Thermo Fisher Scientific). The searching parameters are lists as follows: 1) Precursor mass tolerance: 20 ppm; 2) Fragment mass tolerance: 0.8 Da; 3) Maximum missed cleavage sites: 2; 4) Dynamic modifications: methionine oxidation, serine/threonine/tyrosine phosphorylation; 5) Static modification: cysteine carbamidomethylation. Search results were imported Scaffold 4 (Proteome Software Inc.) for result filtering, FDR control and data export.
LC-MS/MS analysis of GTP pools
Cell pellets were thawed and suspended in 200 μL of PBS (Lonza, Basel, Switzerland) before being transferred to homogenization tubes containing 0.5 mm glass beads (Thermo Fisher Scientific). An 800 μL aliquot of chilled methanol (Optima, Thermo Fisher Scientific, Waltham, MA, USA) and 5 μL of MES (Thermo Fisher Scientific, Waltham, MA, USA) internal standard solution (10 ng/μL) was then introduced to the cell suspension before pulverization using a bead mill homogenizer (Bead Ruptor 24, OMNI International, Kennesaw, Georgia, USA). The homogenates were cooled for at least thirty minutes on ice before being centrifuged at 16,000 x g for 15 minutes. The supernatant was removed and dried under vacuum and frozen at −80°C until LC-MS/MS analysis.
Samples were reconstituted for analysis in 50 μL of mobile phase A. The analysis was performed on a Shimadzu Nexera UHPLC system coupled with a Shimadzu LCMS-8050 triple-quadrupole mass spectrometer (Kyoto, Japan). All standards were purchased from Sigma (St. Louis, MO, USA) with the exception of UTP purchased from Thermo Fisher Scientific (Alfa Aesar, Thermo Fisher Scientific, Haverhill, MA, USA). An ion-pairing LC-MS/MS method was used to measure ATP, UTP, CTP, and GTP. A mobile phase gradient of ultrapure water (Optima, Thermo Fisher Scientific, Waltham, MA, USA) with 10 mM ammonium acetate (J.T. Baker, Thermo Fisher Scientific, Waltham, MA, USA) and 50 mM tributylamine (Acros Organics, Thermo Fisher Scientific, Fair Lawn NJ, USA) (mobile phase A) and methanol (Optima, Thermo Fisher Scientific, Waltham, MA, USA) with 50 mM tributylamine (mobile phase B) was used to separate the analytes. Separation was performed at 0.3 ml/min on a Zorbax Eclipse Plus C18 column (1.8 μm, 2.1 x 100 mm; Agilent, Santa Clara, CA USA) using the following gradient: 2 minutes at 0% B, a ramp to 25% B at 8 minutes, another ramp to 98%B at 12 minutes, a 3 minute hold until 15 minutes, and then a drop back to 0% B at 15.1 minutes and allowed to equilibrate there until 25 minutes. All analytes were monitored in negative mode. The following MRM transitions were used: CTP, 481.90>159.00, 481.90>384.10; GTP, 521.90>159.00, 521.90>423.95; UTP, 482.90>159.00, 482.90>384.90; ATP 505.90>159.05, 505.90>407.90; and the internal standard MES, 194.10>80.15, 194.10>107.10 m/z.
GTP-bound RHO-GTPase pull-down assay
The assay was performed with 500 μg of protein from lysed cells using RAC1 activation assay kit (Cell Biolabs, Catalog #STA-401-1) in accordance with the manufacturer’s instructions. Cells were washed with ice-cold PBS and lysed in ice-cold lysis buffer containing protease inhibitors in a cold room. After one hour rotation with PAK1-PBD Agarose beads at 4°C, the lysate was discarded and beads were washed 3 times for 5 minutes each with lysis buffer. GTP-bound RAC1 levels were assessed via immunoblotting, and signal quantified relative to input RAC1 using FIJI (Schindelin et al., 2012). RHO-A and RHO-C activities were determined in the same manner except Rho activation assay kit (Cell Biolabs, Catalog #STA-403-A) was used.
Generation of Y267-GMPR phospho-specific antibodies
Generation of GMPRY267 phospho-specific antibodies was done by GenScript. A peptide consisting of amino acids 263-273 of GMPR containing a phosphorylated tyrosine at position 267 (LKLF[pY]GMSSDC) was used to immunize New Zealand Rabbits and polyclonal antibodies were isolated following three immunizations and verified by ELISA assay relative to unphosphorylated peptide.
Immunofluorescence and proximity ligation assay
For IF and PLA, cells were fixed by incubation with 4% paraformaldehyde for 15 minutes at room temperature. Cells were washed three times with PBS, blocked with solution containing 0.1% Triton™-X for one hours at room temperature, and incubated with primary antibodies overnight at 4°C. For IF, cells were washed three times for 5 minutes with PBS prior to addition of secondary antibodies for one hour at room temperature. Samples were washed and mounted on slides with mounting medium containing DAPI. For PLA, samples after primary antibody incubation were processed using a Duolink® PLA Kit (Millipore Sigma) according to manufacturer’s instructions.
Cell body (CB) and cell protrusion (CP) fractionation
Confluent cells were cultured in DMEM containing 0.5% FBS overnight. The following day, Corning Costar® 0.4 μm pore polycarbonate membrane transwells (catalog #3412) were coated with 10μg/mL Poly-L-lysine (R&D Systems, catalog #3438) for 30 minutes at room temperature. Trypsinized cells were pelleted and resuspended in serum-free DMEM at 1.5x106 cells per mL. Serum-free DMEM was added to the bottom chamber, and 1 mL of suspended cells added to the top. Cells attached for 2 hours at 37°C. Transwells were washed twice with PBS and transferred to a new 6-well plate with 10% FBS DMEM added to the bottom chamber. Pseudopods were allowed to form for 2 hours at 37°C. Transwells were washed with PBS and fixed with −20°C methanol (top and bottom of chamber) for 30 minutes at 4°C. To collect CPs, material from the top of the membrane was scraped off prior to adding RIPA buffer containing protease inhibitors to harvest material from the bottom of the membrane. CBs were collected in the opposite manner.
GEVAL activity assay
Cell protrusions were formed similarly as above except 3 μM pore transwells were used which were coated with a 10 μg/mL solution of rat tail collagen type 1 (Thermo Fisher). The collagen was stained with far-red dye for visualization during microscopy. GEVAL sensors were designed to detect changes in the amounts of free (unbound) GTP (Bianchi-Smiraglia et al., 2017b). GEVAL30 detects as low as 4μM GTP and its saturation starts at around 100μM GTP. The Keff of GEVAL30 (GTP concentrations required to obtain 50% of the maximal ratiometric signal) is 32.3μM GTP, and GEVAL30 is most responsive to changes in GTP concentrations around its Keff. The GEVALNull sensor serves as a negative control to rule out GTP-independent fluctuations in GEVAL activity. GEVAL activity was measured as previously described (Bianchi-Smiraglia et al., 2017b). In brief, cells were washed with PBS and cultured in FluoroBrite media (Thermo Fisher, cat #A1896701) 1 hour prior to imaging. Cells were imaged with a Leica AOBS SP5 confocal microscope equipped with a multi-line Argon laser, a 405 diode laser, and a 594 helium-neon laser, with a 20x dry lens. The GEVALs were excited sequentially at 405 nm and 488 nm wavelengths and emissions from each excitation were acquired from 502 to 544 nm. Pictures were taken in a Z-stack spanning the top (CB) and bottom (CP) portions of the cells. Ratiometric image analysis and false coloring was performed with FIJI as previously described (Bianchi-Smiraglia et al., 2017b). In brief, the ratio (Ex405/Ex488) of fluorescence intensity was determined by tracing individual CBs and CPs and measuring the mean fluorescence for each channel. The mean ratio for CBs from each experimental condition was set to 100% and individual ratios were plotted and analyzed as shown (see Figures 7H and S7). False coloring was added to the displayed images (Figure S7) using the FIRE LUT tool in FIJI.
Tyrosine kinase screening
A panel of 94 tyrosine kinases was screened by ProQinase (Freigburg, Germany). In brief, 1 μM biotinylated peptide corresponding to amino acids 256-277 of GMPR, which contains tyrosine 267 ([Bio]FERNGRKLKLFYGMSSDTAMNKH, purchased from Thermo Fisher), was incubated with individual kinases in a radiometric assay based on streptavidin-coated FlashPlate® PLUS plates (Perkin Elmer) in a 96 well format. Incorporation of 33Pi was determined by scintillation counter.
GMPR activity assay
Experiments were performed in assay buffer (75 mM Tris-HCl, pH 7.8, 100 mM KCl, 1 mM DTT, and 0.5 mM EDTA) with 3.3-7.8 μg/mL enzyme at 25°C. The consumption of NADPH was monitored by changes in absorbance at 340 nm (ϵ340 = 6.22 mM−1cm−1) on a Shimadzu UV-2600 UV-Vis spectrophotometer with a temperature controlled cell holder or a BioTek uv/vis/fluorescence plate reader with a temperature controlled chamber. Activity was determined by collecting initial velocity data at 100 μM of GMP and 100 μM NADPH, or with varying concentrations of NADPH. Experiments were repeated at least three times.
Invasion assay
Invasion assays were performed as previously described (Wawrzyniak et al., 2013; Bianchi-Smiraglia et al., 2017a). In brief, pelleted cells were washed with PBS and counted before resuspension in serum-free DMEM at 5-10x104 cells per mL (varying by cell line). Cells (500μL of suspension) were loaded into the upper chamber of 8.0 μm BioCoat Matrigel® invasion chambers (Corning, catalog #354480) in technical duplicates. DMEM containing 10% FBS was used as a chemoattractant in the lower chamber. Chambers were then incubated at 37°C overnight, scraped off the top of the membrane, fixed, and stained with a the Three-Step Stain Set (Thermo Scientific, catalog #3300) according to manufacturer’s protocol. After drying, images were acquired from 5 view fields per transwell, and cells were counted using FIJI.
GMPR-EPHA4 Docking Model
Structures for EPHA4 (2Y6M) and GMPR (2BLE) were obtained from the Protein Data Bank (rcsb.org) (Berman et al., 2000). The 2BLE monomer was used for generating docking solutions. Structures were prepared for docking in UCSF Chimera (Pettersen et al., 2004) using the Model/Refine Loops and Dock Prep tools. The PatchDock Webserver (Schneidman-Duhovny et al., 2005) was used to generate initial docking solutions, designating 2Y6M the receptor (receptor binding site residues 745, 782, 785, 823, 827, and 830 (Davis et al., 2009)) and 2BLE the ligand (ligand binding site residues 267, 271, and 273; this work). PatchDock generated 16 docking solutions, which were all submitted to the FiberDock Webserver (Mashiach et al., 2010) for refinement. Two FiberDock solutions were selected based on negative global energy values for further refinement via minimization and molecular dynamics simulations using GROMACS 2020 (Abraham et al., 2015), following the procedure and parameters as described (Galeazzi et al., 2018). The final docked structure displayed is derived from the FiberDock solution with the lowest global energy. To generate an optimized candidate structure for the binding of EPHA4 to the homotetramer of GMPR, the refined docked complex was structurally aligned in Chimera to the 2BLE biological assembly, which was then subjected to another round of minimization and molecular dynamics simulations. Cluster analysis of the resulting trajectory was performed with a 0.2nm cutoff and the average structure from the most populated cluster was used for visual inspection. Chimera Clashes/Contacts and HBond tools were used to analyze interactions.
GST-tagged purification and in vitro binding assay
For GST-tagged bacterial expression, GMPR and GMPRY267F were cloned into pGEX-4T1 from pLV-SV40-puro by PCR-based cloning. The IPTG-induced GST-recombinant proteins were captured by the Pierce™ glutathione-agarose beads (Thermo Scientific). GST-tagged protein was purified from bacterial lysate following IPTG induction with the Pierce™ glutathione-agarose beads (Thermo Scientific) according to the manufacturer’s instruction. For in vitro binding, EPHA4-Flag protein was purified from the lysates of transfected 293FT cells using M2 Flag beads. Binding was assessed with GST-pull down assay as described previously (Deng et al., 2012). Briefly, 0.5 μg EPHA4-Flag protein was incubated with the beads containing GST-GMPR, GST-GMPRY267F, or GST alone and rotated overnight at 4°C. The beads were stringently washed with wash buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 20% Tween-20, 0.1 M DTT) 6 times (10 min/wash) on a rotator. The beads were boiled with 1 x SDS sample loading dye for western blot detection.
In vitro kinase assay
The tyrosine kinase reaction cocktail was prepared with 1x kinase buffer (50 mM Tris, pH 7.5, 20 mM MgCl2, 0.1 mM MnCl2, 0.2 mM Na3VO4), 300 mM ATP and appropriate amount (0.05-0.1 μg) of purified EPHA4 recombinant protein (Sino Biological, #11314-H20B1). 1 μg purified GST-GMPR protein was then added into the kinase cocktail for 1 hour incubation at room temperature. The reaction samples terminated by adding 2x SDS loading dye were directly used for western blotting.
Quantification and statistical analysis
Experiments were each performed at least two independent times, and the results are expressed as the mean +/− SEM unless otherwise indicated. GraphPad Prism software (GraphPad Software, Inc.) was used to employ a student’s t-test, a 2-way ANOVA, or a nonparametric Mann-Whitney U test where appropriate. A p value of <0.05 was considered statistically significant. In each case, * denotes p<0.05; ** denotes p<0.01; *** denotes p<0.001; **** denotes p<0.0001.
Supplementary Material
HIGHLIGHTS.
GMPR is activated by phosphorylation at Tyr267 causing depletion of GTP pools
EPHA4, GMPR, and RAC1 colocalize in the cell
EPHA4 phosphorylates GMPR leading to decreased RAC1 activity and cell invasion
EPHA4 pro- versus anti-oncogenic function depends on GMPR phosphorylation status
SIGNIFICANCE.
Dysregulated nucleotide biosynthesis plays major functional and regulatory roles in cancer progression. Unlike other metabolic processes, posttranslational regulation of nucleotide biosynthesis in cancer cells remain virtually unknown. The data described in this study uncover a signal transduction pathway that connects ephrin (EFN) - ephrin receptor (EPH) signaling to the depletion of intracellular guanosine triphosphate (GTP) pools. This occurs via an EPH-mediated activating phosphorylation of guanosine monophosphate reductase (GMPR), an enzyme that downregulates GTP pools by converting guanosine monophosphate to inosine monophosphate. Previous data implicated GTP metabolism in regulation of several members of small RHO-GTPase family, including RAC1, a protein with a critical role in cancer progression in multiple malignancies. Despite multiple attempts, chemotherapeutic agents directly inhibiting RAC1 have not yet entered the clinical trials. Thus, identification of novel regulatory pathways of GMPR activity could lead to the development of long-sought after RAC1-suppressing anticancer drugs. Furthermore, the EFN-EPH-GMPR axis provides mechanistic explanation to the dualistic function of ephrin signaling in suppression and activation of transformed phenotypes in cancer cells.
ACKNOWLEDGEMENTS
This work has been supported by NIH grants to M.A.N. (CA224434, CA264984), A.B.S. (CA248018), L.H. (GM054403, AI125362), and E.B.P (AG062617), a T32 training grant to D.W.W. (CA247819), the Roswell Park Alliance Foundation to A.B-S., and in part by NCI Cancer Center Support Grant P30CA16056 to the Roswell Park Cancer Institute. The authors wish to acknowledge the Wake Forest Baptist Comprehensive Cancer Center Proteomics and Metabolomics Shared Resource, supported by the National Cancer Institute’s Cancer Center Support Grant award number P30CA012197. Some results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Abraham MJ, Murtola T, Schulz R, Pall S, Smith JC, Hess B, and Lindahl E (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25. [Google Scholar]
- Arozarena I, and Wellbrock C (2017). Targeting invasive properties of melanoma cells. FEBS J 284, 2148–2162. [DOI] [PubMed] [Google Scholar]
- Azoitei ML, Noh J, Marston DJ, Roudot P, Marshall CB, Daugird TA, Lisanza SL, Sandi MJ, Ikura M, Sondek J, et al. (2019). Spatiotemporal dynamics of GEF-H1 activation controlled by microtubule- and Src-mediated pathways. J Cell Biol 218, 3077–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Sommer JE, Martin JH, and Scheiffele P (2007). alpha2-Chimaerin is an essential EphA4 effector in the assembly of neuronal locomotor circuits. Neuron 55, 768–778. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000). The Protein Data Bank. Nucleic Acids Res 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersten DC, Sullivan AE, Peet DJ, and Whitelaw ML (2013). bHLH-PAS proteins in cancer. Nat Rev Cancer 13, 827–841. [DOI] [PubMed] [Google Scholar]
- Bianchi-Smiraglia A, Bagati A, Fink EE, Moparthy S, Wawrzyniak JA, Marvin EK, Battaglia S, Jowdy P, Kolesnikova M, Foley CE, et al. (2017a). Microphthalmia-associated transcription factor suppresses invasion by reducing intracellular GTP pools. Oncogene 36, 84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi-Smiraglia A, Rana MS, Foley CE, Paul LM, Lipchick BC, Moparthy S, Moparthy K, Fink EE, Bagati A, Hurley E, et al. (2017b). Internally ratiometric fluorescent sensors for evaluation of intracellular GTP levels and distribution. Nat Methods 14, 1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi-Smiraglia A, Wawrzyniak JA, Bagati A, Marvin EK, Ackroyd J, Moparthy S, Bshara W, Fink EE, Foley CE, Morozevich GE, et al. (2015). Pharmacological targeting of guanosine monophosphate synthase suppresses melanoma cell invasion and tumorigenicity. Cell Death Differ 22, 1858–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi-Smiraglia A, Wolff DW, Marston DJ, Deng Z, Han Z, Moparthy S, Wombacher RM, Mussell AL, Shen S, Chen J, et al. (2021). Regulation of local GTP availability controls RAC1 activity and cell invasion. Nat Commun 12, 6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bid HK, Roberts RD, Manchanda PK, and Houghton PJ (2013). RAC1: an emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol Cancer Ther 12, 1925–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns KL, Taylor PP, Sicheri F, Pawson T, and Holland SJ (2000). Phosphorylation of tyrosine residues in the kinase domain and juxtamembrane region regulates the biological and catalytic activities of Eph receptors. Mol Cell Biol 20, 4791–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckens OJ, El Hassouni B, Giovannetti E, and Peters GJ (2020). The role of Eph receptors in cancer and how to target them: novel approaches in cancer treatment. Expert Opin Investig Drugs 29, 567–582. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas, N. (2015). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton NS, and Ridley AJ (2020). Targeting Rho GTPase Signaling Networks in Cancer. Front Cell Dev Biol 8, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan CW, Shao YR, Sahin M, Shamah SM, Lin MZ, Greer PL, Gao S, Griffith EC, Brugge JS, and Greenberg ME (2005). Vav family GEFs link activated Ephs to endocytosis and axon guidance. Neuron 46, 205–217. [DOI] [PubMed] [Google Scholar]
- Cox EA, Sastry SK, and Huttenlocher A (2001). Integrin-mediated adhesion regulates cell polarity and membrane protrusion through the Rho family of GTPases. Mol Biol Cell 12, 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer KS, and Miko IJ (2016). Eph-ephrin signaling in nervous system development. F1000Res 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Ha BH, Holman EC, Halaban R, Schlessinger J, and Boggon TJ (2013). RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc Natl Acad Sci U S A 110, 912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TL, Walker JR, Allali-Hassani A, Parker SA, Turk BE, and Dhe-Paganon S (2009). Structural recognition of an optimized substrate for the ephrin family of receptor tyrosine kinases. FEBS J 276, 4395–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Marcondes PG, and Morgado-Diaz JA (2017). The Role of EphA4 Signaling in Radiation-Induced EMT-Like Phenotype in Colorectal Cancer Cells. J Cell Biochem 118, 442–445. [DOI] [PubMed] [Google Scholar]
- De P, Aske JC, and Dey N (2019). RAC1 Takes the Lead in Solid Tumors. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Sui G, Rosa PM, and Zhao W (2012). Radiation-induced c-Jun activation depends on MEK1-ERK1/2 signaling pathway in microglial cells. PLoS One 7, e36739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Liu Y, Jiang A, Li R, Yin M, and Wang Y (2018). MicroRNA-335 suppresses the proliferation, migration, and invasion of breast cancer cells by targeting EphA4. Mol Cell Biochem 439, 95–104. [DOI] [PubMed] [Google Scholar]
- Emmanuel N, Ragunathan S, Shan Q, Wang F, Giannakou A, Huser N, Jin G, Myers J, Abraham RT, and Unsal-Kacmaz K (2017). Purine Nucleotide Availability Regulates mTORC1 Activity through the Rheb GTPase. Cell Rep 19, 2665–2680. [DOI] [PubMed] [Google Scholar]
- Furuhashi S, Morita Y, Ida S, Muraki R, Kitajima R, Takeda M, Kikuchi H, Hiramatsu Y, Setou M, and Takeuchi H (2021). Ephrin Receptor A4 Expression Enhances Migration, Invasion and Neurotropism in Pancreatic Ductal Adenocarcinoma Cells. Anticancer Res 41, 1733–1744. [DOI] [PubMed] [Google Scholar]
- Gaggioli C, and Sahai E (2007). Melanoma invasion - current knowledge and future directions. Pigment Cell Res 20, 161–172. [DOI] [PubMed] [Google Scholar]
- Galeazzi R, Laudadio E, Falconi E, Massaccesi L, Ercolani L, Mobbili G, Minnelli C, Scire A, Cianfruglia L, and Armeni T (2018). Protein-protein interactions of human glyoxalase II: findings of a reliable docking protocol. Org Biomol Chem 16, 5167–5177. [DOI] [PubMed] [Google Scholar]
- Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al. (2020). Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 38, 675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goody RS, and Hofmann-Goody W (2002). Exchange factors, effectors, GAPs and motor proteins: common thermodynamic and kinetic principles for different functions. Eur Biophys J 31, 268–274. [DOI] [PubMed] [Google Scholar]
- Guo Y, Shi W, and Fang R (2021). miR18a5p promotes melanoma cell proliferation and inhibits apoptosis and autophagy by targeting EPHA7 signaling. Mol Med Rep 23, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler LC, Blumenstein L, Stege P, Dvorsky R, and Ahmadian MR (2003). Comparative functional analysis of the Rac GTPases. FEBS Lett 555, 556–560. [DOI] [PubMed] [Google Scholar]
- Halaban R (2015). RAC1 and melanoma. Clin Ther 37, 682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom L (2012). The dynamic determinants of reaction specificity in the IMPDH/GMPR family of (beta/alpha)(8) barrel enzymes. Crit Rev Biochem Mol Biol 47, 250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjorthaug HS, and Aasheim HC (2007). Ephrin-A1 stimulates migration of CD8+CCR7+ T lymphocytes. Eur J Immunol 37, 2326–2336. [DOI] [PubMed] [Google Scholar]
- Holen HL, Shadidi M, Narvhus K, Kjosnes O, Tierens A, and Aasheim HC (2008). Signaling through ephrin-A ligand leads to activation of Src-family kinases, Akt phosphorylation, and inhibition of antigen receptor-induced apoptosis. J Leukoc Biol 84, 1183–1191. [DOI] [PubMed] [Google Scholar]
- Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, and Weiss SJ (2003). Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell 114, 33–45. [DOI] [PubMed] [Google Scholar]
- Hua KT, Hong JB, Sheen YS, Huang HY, Huang YL, Chen JS, and Liao YH (2018). miR-519d Promotes Melanoma Progression by Downregulating EphA4. Cancer Res 78, 216–229. [DOI] [PubMed] [Google Scholar]
- Huang F, Huffman KE, Wang Z, Wang X, Li K, Cai F, Yang C, Cai L, Shih TS, Zacharias LG, et al. (2021). Guanosine triphosphate links MYC-dependent metabolic and ribosome programs in small-cell lung cancer. J Clin Invest 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Ni M, Chalishazar MD, Huffman KE, Kim J, Cai L, Shi X, Cai F, Zacharias LG, Ireland AS, et al. (2018). Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers. Cell Metab 28, 369–382 e365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter SG, Zhuang G, Brantley-Sieders D, Swat W, Cowan CW, and Chen J (2006). Essential role of Vav family guanine nucleotide exchange factors in EphA receptor-mediated angiogenesis. Mol Cell Biol 26, 4830–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iiizumi M, Hosokawa M, Takehara A, Chung S, Nakamura T, Katagiri T, Eguchi H, Ohigashi H, Ishikawa O, Nakamura Y, et al. (2006). EphA4 receptor, overexpressed in pancreatic ductal adenocarcinoma, promotes cancer cell growth. Cancer Sci 97, 1211–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingley E, and Hemmings BA (2000). PKB/Akt interacts with inosine-5' monophosphate dehydrogenase through its pleckstrin homology domain. FEBS Lett 478, 253–259. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Katoh H, Nishimaru H, Ishikawa Y, Inoue H, Saito YM, Ando R, Iwama M, Takahashi R, Negishi M, et al. (2007). Rac-GAP alpha-chimerin regulates motor-circuit formation as a key mediator of EphrinB3/EphA4 forward signaling. Cell 130, 742–753. [DOI] [PubMed] [Google Scholar]
- Jiang J, Kullander K, and Alstermark B (2020). EphA4 Is Required for Neural Circuits Controlling Skilled Reaching. J Neurosci 40, 7091–7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaenel P, Mosimann M, and Andres AC (2012). The multifaceted roles of Eph/ephrin signaling in breast cancer. Cell Adh Migr 6, 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao TJ, Nicholl GC, Johansen JA, Kania A, and Beg AA (2015). alpha2-chimaerin is required for Eph receptor-class-specific spinal motor axon guidance and coordinate activation of antagonistic muscles. J Neurosci 35, 2344–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji S, Hirayama A, Eberhardt AO, Kawaguchi R, Sugiura Y, Sampetrean O, Ikeda Y, Warren M, Sakamoto N, Kitahara S, et al. (2019). IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma. Nat Cell Biol 21, 1003–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, Carrero ZI, Ramakrishnan G, Watabe K, Haupt Y, et al. (2015). Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun 6, 7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. (2012). Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 44, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullander K, Mather NK, Diella F, Dottori M, Boyd AW, and Klein R (2001). Kinase-dependent and kinase-independent functions of EphA4 receptors in major axon tract formation in vivo. Neuron 29, 73–84. [DOI] [PubMed] [Google Scholar]
- Lawson CD, and Ridley AJ (2018). Rho GTPase signaling complexes in cell migration and invasion. J Cell Biol 217, 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong SP, Mihm MC Jr., Murphy GF, Hoon DS, Kashani-Sabet M, Agarwala SS, Zager JS, Hauschild A, Sondak VK, Guild V, et al. (2012). Progression of cutaneous melanoma: implications for treatment. Clin Exp Metastasis 29, 775–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wei Z, Zheng M, Gu X, Deng Y, Qiu R, Chen F, Ji C, Gong W, Xie Y, et al. (2006). Crystal structure of human guanosine monophosphate reductase 2 (GMPR2) in complex with GMP. J Mol Biol 355, 980–988. [DOI] [PubMed] [Google Scholar]
- Liang LY, Patel O, Janes PW, Murphy JM, and Lucet IS (2019). Eph receptor signalling: from catalytic to non-catalytic functions. Oncogene 38, 6567–6584. [DOI] [PubMed] [Google Scholar]
- Light TP, Gomez-Soler M, Wang Z, Karl K, Zapata-Mercado E, Gehring MP, Lechtenberg BC, Pogorelov TV, Hristova K, and Pasquale EB (2021). A cancer mutation promotes EphA4 oligomerization and signaling by altering the conformation of the SAM domain. J Biol Chem 297, 100876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionarons DA, Hancock DC, Rana S, East P, Moore C, Murillo MM, Carvalho J, Spencer-Dene B, Herbert E, Stamp G, et al. (2019). RAC1(P29S) Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance. Cancer Cell 36, 68–83 e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu BP, and Burridge K (2000). Vav2 activates Rac1, Cdc42, and RhoA downstream from growth factor receptors but not beta1 integrins. Mol Cell Biol 20, 7160–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Luo K, and Luo D (2018). Guanosine monophosphate reductase 1 is a potential therapeutic target for Alzheimer's disease. Sci Rep 8, 2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, Sedivy JM, Zeller KI, and Dang CV (2008). Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One 3, e2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, and Danuser G (2009). Coordination of Rho GTPase activities during cell protrusion. Nature 461, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNevin CJ, Toutchkine A, Marston DJ, Hsu CW, Tsygankov D, Li L, Liu B, Qi T, Nguyen DV, and Hahn KM (2016). Ratiometric Imaging Using a Single Dye Enables Simultaneous Visualization of Rac1 and Cdc42 Activation. J Am Chem Soc 138, 2571–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannava S, Grachtchouk V, Wheeler LJ, Im M, Zhuang D, Slavina EG, Mathews CK, Shewach DS, and Nikiforov MA (2008). Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 7, 2392–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardakheh FK, Paul A, Kumper S, Sadok A, Paterson H, McCarthy A, Yuan Y, and Marshall CJ (2015). Global Analysis of mRNA, Translation, and Protein Localization: Local Translation Is a Key Regulator of Cell Protrusions. Dev Cell 35, 344–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marei H, and Malliri A (2017). Rac1 in human diseases: The therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases 8, 139–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston DJ, Vilela M, Huh J, Ren J, Azoitei ML, Glekas G, Danuser G, Sondek J, and Hahn KM (2020). Multiplexed GTPase and GEF biosensor imaging enables network connectivity analysis. Nat Chem Biol 16, 826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiach E, Nussinov R, and Wolfson HJ (2010). FiberDock: a web server for flexible induced-fit backbone refinement in molecular docking. Nucleic Acids Res 38, W457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao B, Ji Z, Tan L, Taylor M, Zhang J, Choi HG, Frederick DT, Kumar R, Wargo JA, Flaherty KT, et al. (2015). EPHA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov 5, 274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethamer TK, and Bush JO (2019). Getting direction(s): The Eph/ephrin signaling system in cell positioning. Dev Biol 447, 42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogita H, Kunimoto S, Kamioka Y, Sawa H, Masuda M, and Mochizuki N (2003). EphA4-mediated Rho activation via Vsm-RhoGEF expressed specifically in vascular smooth muscle cells. Circ Res 93, 23–31. [DOI] [PubMed] [Google Scholar]
- Pasquale EB (2008). Eph-ephrin bidirectional signaling in physiology and disease. Cell 133, 38–52. [DOI] [PubMed] [Google Scholar]
- Pasquale EB (2010). Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer 10, 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton GC, Stenmark P, Gollapalli DR, Sevastik R, Kursula P, Flodin S, Schuler H, Swales CT, Eklund H, Himo F, et al. (2011). Cofactor mobility determines reaction outcome in the IMPDH and GMPR (beta-alpha)8 barrel enzymes. Nat Chem Biol 7, 950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Plana-Bonamaiso A, Lopez-Begines S, Fernandez-Justel D, Junza A, Soler-Tapia A, Andilla J, Loza-Alvarez P, Rosa JL, Miralles E, Casals I, et al. (2020). Post-translational regulation of retinal IMPDH1 in vivo to adjust GTP synthesis to illumination conditions. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliakov A, Cotrina M, and Wilkinson DG (2004). Diverse roles of eph receptors and ephrins in the regulation of cell migration and tissue assembly. Dev Cell 7, 465–480. [DOI] [PubMed] [Google Scholar]
- Rosenberg MM, Redfield AG, Roberts MF, and Hedstrom L (2018). Dynamic Characteristics of Guanosine-5'-monophosphate Reductase Complexes Revealed by High-Resolution (31)P Field-Cycling NMR Relaxometry. Biochemistry 57, 3146–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, and Sondek J (2005). GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 6, 167–180. [DOI] [PubMed] [Google Scholar]
- Sahin M, Greer PL, Lin MZ, Poucher H, Eberhart J, Schmidt S, Wright TM, Shamah SM, O'Connell S, Cowan CW, et al. (2005). Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse. Neuron 46, 191–204. [DOI] [PubMed] [Google Scholar]
- Saintigny P, Peng S, Zhang L, Sen B, Wistuba II, Lippman SM, Girard L, Minna JD, Heymach JV, and Johnson FM (2012). Global evaluation of Eph receptors and ephrins in lung adenocarcinomas identifies EphA4 as an inhibitor of cell migration and invasion. Mol Cancer Ther 11, 2021–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, and Wolfson HJ (2005). PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res 33, W363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Fu WY, Hung KW, Porchetta C, Hall C, Fu AK, and Ip NY (2007). Alpha2-chimaerin interacts with EphA4 and regulates EphA4-dependent growth cone collapse. Proc Natl Acad Sci U S A 104, 16347–16352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Winterbottom E, and Daar IO (2012). Eph/ephrin signaling in cell-cell and cell-substrate adhesion. Front Biosci (Landmark Ed) 17, 473–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder D, Wang Y, and Kaetzel DM (2020). A rare subpopulation of melanoma cells with low expression of metastasis suppressor NME1 is highly metastatic in vivo. Sci Rep 10, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector T, Jones TE, and Miller RL (1979). Reaction mechanism and specificity of human GMP reductase. Substrates, inhibitors, activators, and inactivators. J Biol Chem 254, 2308–2315. [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumillo MJ, Oplova M, Vieitez C, Ochoa D, Shahraz M, Busby BP, Sopko R, Studer RA, Perrimon N, Panse VG, et al. (2019). Conserved phosphorylation hotspots in eukaryotic protein domain families. Nat Commun 10, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suetterlin P, Marler KM, and Drescher U (2012). Axonal ephrinA/EphA interactions, and the emergence of order in topographic projections. Semin Cell Dev Biol 23, 1–6. [DOI] [PubMed] [Google Scholar]
- Traut TW (1994). Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 140, 1–22. [DOI] [PubMed] [Google Scholar]
- Triplett JW, and Feldheim DA (2012). Eph and ephrin signaling in the formation of topographic maps. Semin Cell Dev Biol 23, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzi NL, and Gullick WJ (1994). eph, the largest known family of putative growth factor receptors. Br J Cancer 69, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udayakumar D, Zhang G, Ji Z, Njauw CN, Mroz P, and Tsao H (2011). EphA2 is a critical oncogene in melanoma. Oncogene 30, 4921–4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valvezan AJ, Turner M, Belaid A, Lam HC, Miller SK, McNamara MC, Baglini C, Housden BE, Perrimon N, Kwiatkowski DJ, et al. (2017). mTORC1 Couples Nucleotide Synthesis to Nucleotide Demand Resulting in a Targetable Metabolic Vulnerability. Cancer Cell 32, 624–638 e625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verham R, Meek TD, Hedstrom L, and Wang CC (1987). Purification, characterization, and kinetic analysis of inosine 5'-monophosphate dehydrogenase of Tritrichomonas foetus. Mol Biochem Parasitol 24, 1–12. [DOI] [PubMed] [Google Scholar]
- Villa E, Ali ES, Sahu U, and Ben-Sahra I (2019). Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl S, Barth H, Ciossek T, Aktories K, and Mueller BK (2000). Ephrin-A5 induces collapse of growth cones by activating Rho and Rho kinase. J Cell Biol 149, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ding SJ, Wang W, Jacobs JM, Qian WJ, Moore RJ, Yang F, Camp DG 2nd, Smith RD, and Klemke RL (2007). Profiling signaling polarity in chemotactic cells. Proc Natl Acad Sci U S A 104, 8328–8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson IR, Li LR, Cabeceiras PK, Mahdavi M, Fang ZN, Stemke-Hale K, Mills GB, and Chin L (2014). The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawrzyniak JA, Bianchi-Smiraglia A, Bshara W, Mannava S, Ackroyd J, Bagati A, Omilian AR, Im M, Fedtsova N, Miecznikowski JC, et al. (2013). A purine nucleotide biosynthesis enzyme guanosine monophosphate reductase is a suppressor of melanoma invasion. Cell Rep 5, 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmeyer H, Egea J, Rabe N, Gezelius H, Filosa A, Enjin A, Varoqueaux F, Deininger K, Schnutgen F, Brose N, et al. (2007). EphA4-dependent axon guidance is mediated by the RacGAP alpha2-chimaerin. Neuron 55, 756–767. [DOI] [PubMed] [Google Scholar]
- Woodland HR, and Pestell RQ (1972). Determination of the nucleoside triphosphate contents of eggs and oocytes of Xenopus laevis. Biochem J 127, 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Jia P, Hutchinson KE, Dahlman KB, Johnson D, Sosman J, Pao W, and Zhao Z (2014). A meta-analysis of somatic mutations from next generation sequencing of 241 melanomas: a road map for the study of genes with potential clinical relevance. Mol Cancer Ther 13, 1918–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang NY, Pasquale EB, Owen LB, and Ethell IM (2006). The EphB4 receptor-tyrosine kinase promotes the migration of melanoma cells through Rho-mediated actin cytoskeleton reorganization. J Biol Chem 281, 32574–32586. [DOI] [PubMed] [Google Scholar]
- Zhang B, Chernoff J, and Zheng Y (1998). Interaction of Rac1 with GTPase-activating proteins and putative effectors. A comparison with Cdc42 and RhoA. J Biol Chem 273, 8776–8782. [DOI] [PubMed] [Google Scholar]
- Zhang B, Zhang Y, Wang Z, and Zheng Y (2000). The role of Mg2+ cofactor in the guanine nucleotide exchange and GTP hydrolysis reactions of Rho family GTP-binding proteins. J Biol Chem 275, 25299–25307. [DOI] [PubMed] [Google Scholar]
- Zhang C, Smalley I, Emmons MF, Sharma R, Izumi V, Messina J, Koomen JM, Pasquale EB, Forsyth PA, and Smalley KSM (2020). Noncanonical EphA2 Signaling Is a Driver of Tumor-Endothelial Cell Interactions and Metastatic Dissemination in BRAF InhibitorResistant Melanoma. J Invest Dermatol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Yao Y, Scott AJ, Wilder-Romans K, Dresser JJ, Werner CK, Sun H, Pratt D, Sajjakulnukit P, Zhao SG, et al. (2020). Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat Commun 11, 3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. The accession numbers are listed in the key resource table.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rac1 | ProteinTech | Cat# 66122; RRID:AB_2881521 |
| GMPR | ProteinTech | Cat# 15683; RRID:AB_2111072 |
| anti-FLAG | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| EPHA4 | BD Biosciences | Cat# 610471; RRID:AB_397843 |
| EPHA4 | Thermo Fisher | Cat# 37-1600; RRID:AB_2533301 |
| β-ACTIN | ProteinTech | Cat# HRP-66009; RRID:AB_2883836 |
| phospho-EPHA3 (Tyr779) | Cell Signaling | Cat# 8862; RRID:AB_2797673 |
| phospho-VAV2 (Tyr172) | ECM Biosciences | Cat# VP2641; RRID:AB_2213715 |
| Integrin Beta 1 | ProteinTech | Cat# 26918; RRID:AB_2880685 |
| ARNT1 | Santa Cruz | Cat# sc-17811; RRID:AB_626698 |
| RhoA | Cell Signaling | Cat# 2117; RRID:AB_10693922 |
| RhoC | Cell Signaling | Cat# 3430; RRID:AB_2179246 |
| phospho-GMPR (Tyr267) | GenScript | This paper |
| Bacterial strains | ||
| DH5α Competent E. coli | New England BioLabs | Cat# C2987 |
| BL21 Competent E. coli | New England BioLabs | Cat# C2527 |
| Stable Competent E. coli | New England BioLabs | Cat# C3040 |
| Recombinant proteins | ||
| GST-GMPR | This paper | N/A |
| GST-GMPRY267F | This paper | N/A |
| GST/His-EPHA4 (aa 570-986) | SinoBiological | Cat# 11314-H20B1 |
| Critical commercial assays | ||
| Rac1 activity assay | Cell Biolabs | Cat# STA-401-1 |
| RhoA/C activity assay | Cell Biolabs | Cat# STA-403-A |
| Proximity ligation assay | Millipore Sigma | Cat# DUO92101 |
| 0.4 μm transwells (fractionation) | Corning | Cat# 3412 |
| 8.0 μm transwells (invasion) | Corning | Cat# 354480 |
| Tyrosine kinase screen | ProQinase | N/A |
| Deposited data | ||
| EPHA4 crystal structure | Protein Data Bank | PDB: 2Y6M |
| GMPR crystal structure | Protein Data Bank | PDB: 2BLE |
| Melanoma expression dataset | TCGA and Xena | GDC TCGA Melanoma (SKCM) |
| Experimental models: Cell lines | ||
| HEK293FT | Thermo Fisher | Cat# R70007 |
| SK-Mel-103 | MSKCC | RRID:CVCL_6069 |
| SK-Mel-147 | MSKCC | RRID:CVCL_3876 |
| SK-Mel-28 | ATCC | Cat# HTB-72; RRID:CVCL_0526 |
| Experimental models: Organisms/strains | ||
| ICR scid mice | Taconic | Cat# ICRSC |
| Recombinant DNA | ||
| pCMV-deltaR8.2 | Didier Trono | RRID:Addgene_12263 |
| pCMV-VSVG | Bob Weinberg | RRID:Addgene_8454 |
| pLV-SV40-puro | Peter Chumakov | PMID: 11854510 |
| pLV-GMPR-flag | In-house | PMID: 24139804 |
| pLV-GMPRY267F-flag | This paper | N/A |
| pLKO/TRC1-shEPHA4 (E1) (GATTGGCTCCAGGCCATTAAA) | Millipore Sigma | TRC0000220021 |
| pLKO/TRC2-shEPHA4 (E3) (GCAATTGCCTATCGTAAATTC) | Millipore Sigma | TRC00003828495 |
| pLenti-UbC-EPHA4 | Eric Haura | PMID: 20190765 |
| pLenti-UbC-EPHA4K653M | Eric Haura | PMID: 20190765 |
| pLVX-EPHA4-flag | In-house | PMID: 34139238 |
| pcDNA3-EPHA5-flag | This paper | N/A |
| pCMV3-EPHB1-flag | SinoBiological | Cat# MG50479-NF |
| pLenti6.2-EmGFP | Thermo Fisher | Cat# V36920 |
| pGEX-4T1-GMPR | This paper | N/A |
| pGEX-4T1-GMPRY267F | This paper | N/A |
| Software and algorithms | ||
| FIJI | Public domain | https://imagej.net/software/fiji/ |
| GraphPad Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| ChemiDoc MP Imaging System | Bio-Rad | Cat# 17001402 |
| Confocal microscope | Leica | TCS SP5 |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.