

Abstract
The current study examined the roles of Alpha4, a non-canonical subunit of protein phosphatase 2A, in the regulation of acute (insulin secretion) and chronic (cell dysfunction) effects of glucose in pancreatic beta cells. Alpha4 is expressed in human islets, rat islets and INS-1832/13 cells. Incubation of INS-1832/13 cells and rat islets with high glucose (HG) significantly increased the expression of Alpha4. C2-Ceramide, a biologically active sphingolipid, also increased the expression of Alpha4 in INS-1832/13 cells and rat islets. Subcellular distribution studies of Alpha4 in low glucose (LG) and HG exposed INS-1832/13 cells revealed that it is predominantly cytosolic, and its expression is significantly increased in the non-nuclear/cytosolic fractions in cells exposed to HG. siRNA-mediated knockdown of Alpha4 exerted minimal effects on glucose- or KCl-induced insulin secretion. siRNA-mediated deletion of Alpha4 significantly increased p38MAPK and JNK1/2 phosphorylation under LG conditions, comparable to the degree seen under HG conditions. Paradoxically, a significant potentiation of HG-induced p38MAPK and JNK2 phosphorylation was noted following Alpha4 deletion. HG-induced CHOP expression (ER stress marker) and caspase-3 activation were markedly attenuated in cells following Alpha4 knockdown. Deletion of Alpha4 in INS-1832/13 cells prevented HG-induced loss in the expression of Connexin36, a gap junction channel protein, which has been implicated in normal beta cell function. Lastly, depletion of endogenous Alpha4 significantly reduced HG-induced cell death in INS-1832/13 cells. Based on these findings we conclude that Alpha4 contributes to HG-induced metabolic dysfunction of the islet beta cell.
Keywords: Pancreatic islet, Alpha4, PP2A or Protein-serine/threonine phosphatase, Connexin36, Metabolic stress, Diabetes
1. Introduction
Physiological (i.e., Glucose-stimulated) insulin secretion (GSIS) from islet beta cells involves generation and mobilization of a variety of hydrophobic and hydrophilic second messengers, including ions, cyclic nucleotides, and lipid hydrolytic products of phospholipases (Prentki et al., 2013; Macdonald, 1990; Newgard and Mcgarry, 1995; Thompson and Satin, 2021; Kowluru, 2010, 2020a; Islam, 2020). It is well recognized that some of the known actions of these second messengers include functional regulation of a variety of protein kinases indigenous to the islet beta cells. Earlier studies in this field have demonstrated localization of functionally-regulable protein kinases in human and rodent islets as well as clonal β-cells; these include Ca2+-, Ca2+/calmodulin-, cAMP-, and phospholipid-dependent protein kinases (Tengholm and Gylfe, 2017; Jones and Persaud, 1998; Santo-Domingo et al., 2019; Nesher et al., 2002). The overall phosphorylation status of proteins is tightly regulated by net activities of protein kinases and phosphatases.
Despite a growing body of evidence suggesting key roles for protein kinases in islet beta-cell function, protein phosphatases remain an under-studied class of signaling proteins in islet biology (Kowluru, 2005; Ortsäter et al., 2014). Protein serine/threonine phosphatase 2A (PP2A), which accounts for 80% of total serine/threonine phosphatases, has been implicated in the regulation of cell proliferation, survival and apoptosis. PP2A is a heterotrimeric holoenzyme consisting of the structural (A), regulatory (B) and catalytic (C) subunits. Post-translational methylation and phosphorylation of PP2Ac have been shown to increase and decrease the catalytic function of PP2A, respectively (Kowluru, 2005). Earlier investigations from our laboratory have revealed that PP2A-like enzymes play critical regulatory roles in GSIS (Kowluru et al., 2001; Kowluru, 2005). In addition, our previous observations have shown sustained activation of PP2A in beta-cell models of metabolic stress (Kowluru, 2020b; Arora et al., 2014; Kowluru and Matti, 2012); based on these findings we proposed that hyperactivation of PP2A could lead to dephosphorylation of key proteins requisite for physiological insulin secretion and beta cell proliferation (Arora et al., 2014).
In the context of functional regulation of protein serine/threonine phosphatases (e.g., PP2A), studies in other cell types have identified novel regulatory roles for non-canonical subunits of PP2A, including Alpha4 and TOR signaling pathway regulator (TIPRL1 (Sents et al., 2013; Zolnierowicz, 2000; Janssens and Goris, 2001);). Alpha4, the focus of the current set of investigations, which is also referred to as immunoglobulin binding protein 1 (IGBP1) and PP2A-associated protein of 42 kDa (Tap42; in yeast), has been shown to form complexes with PP2Ac independent of the A and B subunits of PP2A (Sents et al., 2013; Janssens and Goris, 2001; Zolnierowicz, 2000). Published evidence also suggests that, in addition to PP2Ac, Alpha4 interacts with and regulates functions of other phosphatases, including PP4 and PP6; such regulatory events have been shown to involve interactions between Alpha4 and the catalytic subunits of PP4 (PP4c) and PP6 (PP6c) (Mcconnell et al., 2010; Sents et al., 2013, Lenoue-Newton et al., 2016; Chen et al., 1998, Kloeker et al., 2003). Despite the evidence in other cell types, very little is known about the putative regulatory roles, if any, of Alpha4 in the functional regulation of pancreatic beta cell exposed to acute (i.e., GSIS) and chronic hyperglycemic (i.e., cell demise) conditions. Therefore, we undertook the current studies to decipher regulatory roles of Alpha4 in islet beta cell function under conditions favorable to physiological insulin secretion and cellular dysfunction induced by exposure to metabolic stress conditions.
2. Materials and methods
2.1. Chemicals and reagents
An antibody directed against Alpha4 (A300–471A) was from Bethyl Laboratories Inc. (Montgomery, TX, USA). Total- and phospho-specific antibodies for p38MAPK [p38 MAPK; 9212S, and P-p38 MAPK (Thr180/Tyr182); 9211S], for JNK1/2 [SAPK/JNK; 9258S, and P-SAPK/JNK (Thr183/Tyr185); 9251S], and antibodies against cleaved Caspase-3 (Asp175; 9661S), CHOP (2895S), GAPDH (5174S) and HRP-conjugated secondary antibodies were from Cell Signaling Technology, Inc. (Danvers, MA, USA). Antibodies against lamin B (sc-374015), E-Cadherin (sc-8426), gp91-phox [(54.1); sc-130543], PP2A-Cα/β antibody [(1D6); sc-80665], Cx36 (sc-398063), Cx43 (sc-13558), Histone H3 (sc-517576) and β-PIX [(H-3); sc-393184] were from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-Rac1 antibody (610650) was from BD Transductions Laboratories (San Jose, CA, USA). β-actin antibody (A1978) and Cell Death Detection ELISAPLUS Roche kit (11774425001) were from Sigma Aldrich (St. Louis, MO, USA). ON-TARGETplus Non-targeting siRNA (Control siRNA: Con-si; D001810-01-20), ON-TARGETplus siRNA, Igbp1 (immunoglobulin binding protein 1), Rat, SMARTpool (Alpha4-si; L-100452-02-0020) and DharmaFect1 transfection reagent (T-2001–03) were from Horizon Discovery (Lafayette, CO, USA). The cell permeable C2-ceramide was from Cayman Chemicals (Ann Arbor, MI, USA). NE-PER Nuclear and Cytoplasmic Extraction Kit (78835) and Mem-PER Plus Kit (89842Y) were from Thermo Fisher Scientific (Waltham, MA, USA). Rat High Range Insulin ELISA kit (80-INSRTH-E01) was from ALPCO (Salem, NH, USA). Normal mouse IgG polyclonal antibody (12–371) was from EMD Milli-pore (Temecula, CA, USA).
2.2. Culture of INS-1832/13 cells and rat islets
INS-1832/13 cells (passage #s 50–60) were cultured in RPMI-1640 medium containing 10% FBS supplemented with 100 IU/ml penicillin and 100 IU/ml streptomycin, 1 mM sodium pyruvate, 50 μM 2-mercapto-ethanol (not added in medium for rat and human islets), and 10 mM HEPES (pH 7.4). Cells were treated overnight with low serum/low glucose media prior to each experiment. Cells were incubated in the presence of LG (2.5 mM) or HG (20 mM) for 24 h, unless specified otherwise. Islets were isolated from Sprague-Dawley rats using the collagenase digestion method (Kowluru et al., 1996; Syed et al., 2011; Veluthakal et al., 2015). All protocols received approvals from IACUC (Wayne State University and the JDD VA Medical Center, Detroit, MI, USA). Human islets were obtained from Prodo Labs (Aliso Viejo, CA, USA).
2.3. siRNA-mediated deletion of Alpha4 in INS-1832/13 cells
INS-1832/13 cells were transfected with Alpha4-siRNA (Alpha4-si; 100 nM) using the DharmaFect1 reagent. Non-targeting control siRNA (Con-si) was used as a control. Cells transfected with Con-si or Alpha4-si were maintained for an additional 48 h in complete growth medium. The efficiency of Alpha4 knockdown was determined by western blotting and densitometry.
2.4. Insulin secretion assays
The transfected cells were cultured overnight in the presence of 2.5 mM glucose and 2.5% fetal bovine serum. After pre-incubation in Krebs Ringer Bicarbonate buffer for 1 h cells were employed for quantification of glucose- or KCl-stimulated insulin secretion. For GSIS, cells were incubated in the presence of LG (2.5 mM) or HG (20 mM) glucose for 45 min at 37 °C. For KCl-induced insulin secretion, cells were incubated in the presence of LG (2.5 mM) or KCl (60 mM) for 60 min. The supernatants were then removed, and the amount of insulin released was quantified by ELISA (Veluthakal et al., 2015; Syed et al., 2011; Kowluru and Veluthakal, 2005).
2.5. Isolation of non-nuclear and nuclear or cytosolic and membrane fractions from INS-1832/13 cells
INS-1832/13 cells were incubated under LG (2.5 mM) or HG (20 mM) exposure conditions for 24 h. To obtain the nuclear and non-nuclear fractions, cell fractionation was carried out using NE-PER Nuclear and Cytoplasmic Extraction kit as previously described (Baidwan et al., 2017). To isolate the cytosolic and membrane fractions, cell fractionation was carried out using Mem-PER kit (Thamilselvan et al., 2020; Thamilselvan and Kowluru, 2019). The purity of these fractions was assessed using specific protein markers (GAPDH, E-Cadherin and lamin B (Khadija et al., 2014; Baidwan et al., 2017; Thamilselvan and Kowluru, 2019; Thamilselvan et al., 2020);).
2.6. Co-immunoprecipitation assay
Immunoprecipitation was performed according to our previously published method (Gamage et al., 2022). Briefly, lysates (500 μg protein) derived from LG- or HG-treated INS-1832/13 cells were incubated overnight with Alpha4 antibody. Pre-cleared lysates were incubated (continuous agitation) with agarose beads for 3 h at 4°C. After multiple washes with lysis buffer, the resulting immunoprecipitates were subjected to SDS-PAGE and western blotting (below) to detect and quantify the abundance of proteins of interest in the immunoblots.
2.7. Western blotting
Cell lysates (20–50 μg of protein), prepared in RIPA buffer containing protease and phosphatase inhibitors, were resolved by SDS-PAGE and electro-transferred onto the nitrocellulose membrane. The membranes were blocked with 3% BSA in PBS-T at room temperature for 1 h and were then incubated with the corresponding primary antibodies overnight at 4 °C. Following this incubation, the membranes were washed (3 × 5 min each) with PBS-T and probed with the appropriate HRP-conjugated secondary antibody in 1.5% BSA in PBS-T for 1 h at room temperature. After washing (3 × 10 min each), the target protein signal band was detected by ECL chemiluminescence using a kit from Thermo Fisher Scientific (Waltham, MA, USA). Quantification of the band intensities was done by densitometry using Image Studio Lite (Li-COR Biosciences, Lincoln, NE, USA).
2.8. Cell death detection assay
INS-1832/13 cells were incubated with LG (2.5 mM) or HG (20 mM) for 24 h and analyzed for degree of cell death using a Cell Death Detection ELISAplus kit (Chundru et al., 2021). Briefly, cells were lysed using the lysis buffer, the cell lysates were centrifuged at 200g for 10 min. The supernatants were collected and incubated in a streptavidin-coated microplate along with the immuno-reagent containing anti-histone-biotin and anti-DNA-peroxidase for 2 h. The complexes were further detected photometrically using ABTS substrate. Absorbance was measured at 405 nm (Chundru et al., 2021).
2.9. Statistical analysis
Data are presented as mean ± SEM. Statistical analysis was done using the student t-test. A p-value of <0.05 was considered statistically significant.
3. Results
3.1. Alpha4 is expressed in a variety of insulin-secreting cells, and plays minimal regulatory roles in glucose- or KCl-induced insulin secretion from INS-1832/13 cells
At the outset, we determined the expression of Alpha4 in a variety of insulin secreting cells. The antibody used in the current studies reacted with a protein band at 45 kDa in lysates from INS-1832/13 cells, which corresponds to the apparent molecular weight of Alpha4. Further, this antibody reacted with Alpha4 in lysates derived from THP-1 cells (used as a positive control; Fig. 1; Panel A). Data depicted in Fig. 1 (Panel B) affirmed that Alpha4 is expressed in INS-1832/13 cells, normal rat islets and human islets.
Fig. 1. Alpha4 is expressed in a variety of insulin-secreting cells, and siRNA-mediated knockdown Alpha4 exerted no significant effects on GSIS and KCl-induced insulin secretion in INS-1832/13 cells.
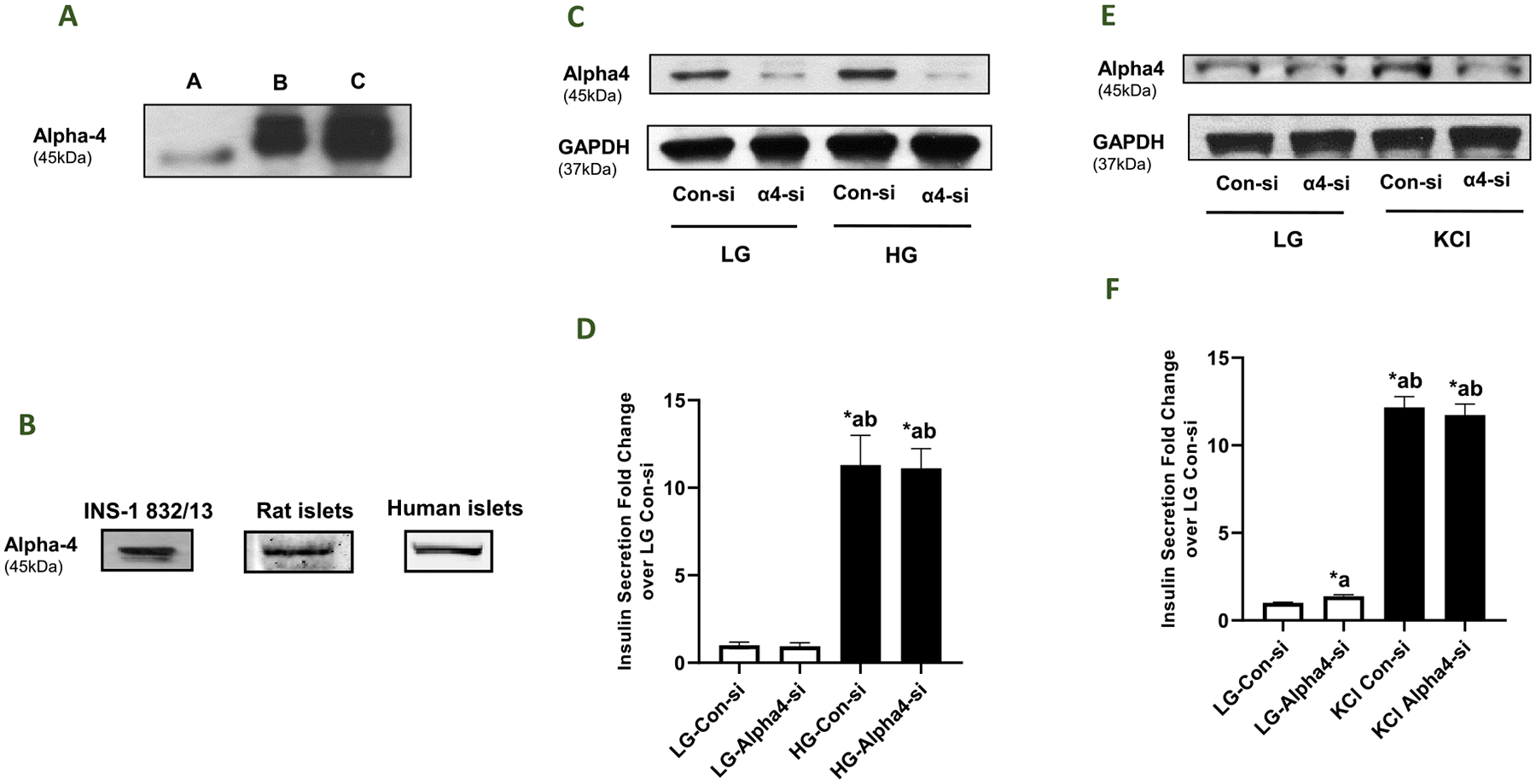
Panel A: A representative Western blot depicting the specificity of the Alpha4 antibody employed in the current studies. (A) THP-1 cell lysate (positive control; 50 μg/lane), (B) 15 μg INS-1832/13 cell lysate protein, (C) 30 μg INS-1832/13 cell lysate protein. Panel B: Western blot data demonstrating expression of Alpha4 in lysates derived from INS-1832/13 cells, rat islets and human islets. Panel C: INS-1832/13 cells were transfected with control-siRNA or Alpha4-siRNA. Knockdown of Alpha4 expression was assessed by Western blotting. GAPDH was used as the control for protein loading. Panel D: Control-siRNA or Alpha4-siRNA transfected cells were treated with LG (2.5 mM) or HG (20 mM) for 45min and insulin released into the media was quantified by ELISA. The data are expressed as fold change relative to LG-Con-si. Data are mean ± SEM from two experiments; 12 replicates per condition. (Comparisons: a - significant compared with LG-Con-si; b - significant compared with LG-Alpha4-si; *p < 0.05). Panel E: INS-1832/13 cells were transfected with control-siRNA or Alpha4-siRNA. Knockdown of Alpha4 expression was assessed by Western blotting. GAPDH was used as the control for protein loading. Panel F: Control-siRNA or Alpha4-siRNA transfected cells were treated with LG (2.5 mM) or KCl (60 mM) for 60 min and insulin released into the media was quantified by ELISA. The data are expressed as fold change relative to LG-Con-si. Data are mean ± SEM from two experiments; 12 replicates per condition. (Comparisons: a - significant compared with LG-Con-si; b - significant compared with LG-Alpha4-si; *p < 0.05).
We next asked if Alpha4 contributes to insulin secretion elicited by glucose and/or a membrane depolarizing concentration of KCl. We addressed this question by deleting the expression of Alpha4 using Alpha4-si. A non-targeting siRNA is used as control in these studies (Con-si). Data from multiple experiments indicated ~80% knockdown of Alpha4 in INS-1832/13 cells under our experimental conditions (n = 15 experiments; additional data not shown). Data in Fig. 1 (Panel D) demonstrated a robust stimulation of insulin secretion in INS-1832/13 cells transfected with Con-si in the presence of glucose (20 mM; 45 min exposure). In addition, a similar degree of GSIS was seen in these cells following transfection with Alpha4-si. No significant differences in basal insulin secretion were noted in Con-si or Alpha4-si transfected cells. Extent of knockdown of Alpha4 in these studies is shown in Fig. 1 (Panel C). Furthermore, in a manner akin to data obtained in GSIS studies, transfection of INS-1832/13 cells with Alpha4-si had minimal effects on insulin secretion promoted by a membrane depolarizing concentration of KCl (Fig. 1; Panel F). A representative Western blot demonstrating knockdown of Alpha4 under these experimental conditions in shown in Fig. 1 (Panel E). Together, these data indicated no contributory roles for Alpha4 in insulin secretion elicited by either a physiological concentration of glucose or a membrane-depolarizing concentration of KCl.
3.2. Chronic exposure of INS-1832/13 cells to high glucose or a cell-permeable C2-ceramide increases the expression of Alpha4 in INS-1832/13 cells and rat islets
Earlier studies have reported an increase in the expression of Alpha4/IGBP1 in pathological states, including human lung and breast cancers (Chen et al., 2011; Jiang et al., 2020). Therefore, we asked if the expression of Alpha4 is altered in pancreatic beta cells following exposure to metabolic stress conditions. Data in Fig. 2 (Panel A) demonstrate a greater than two-fold increase in the expression of Alpha4 following exposure to HG conditions (20 mM; 24 h). As shown in Fig. 2 (Panel A), maximal effects of HG on the expression of Alpha4 were seen at 24 h. In addition to HG, exposure of these cells to a cell permeable analogue of ceramide (Cer), a biologically-active sphingolipid (50 μM; 24 h), also resulted in increase in the expression of Alpha4 (Fig. 2; Panel B). Pooled data from multiple experiments are graphed in Fig. 2 (Panel C). Lastly, cycloheximide, a protein biosynthesis inhibitor, prevented HG-induced expression of Alpha4 suggesting that the increase in the expression of Alpha4 by HG is due to increased de novo biosynthesis of Alpha4 (Fig. 2; Panel D). In addition, HG and Cer significantly increased expression of Alpha4 in rat islets (Fig. 2; Panel E). Taken together, these data demonstrate a significant increase in the expression of Alpha4 under conditions of exposure to HG or Cer. Please note that, in order to gain better mechanistic insights, we have limited our current studies toward assessing the regulatory roles of Alpha4 in the metabolic dysregulation of pancreatic beta cells under the duress of HG conditions (see Discussion).
Fig. 2. Exposure of INS-1832/13 cells to high glucose and a cell-permeable analogue of ceramide results in increased expression of Alpha4.
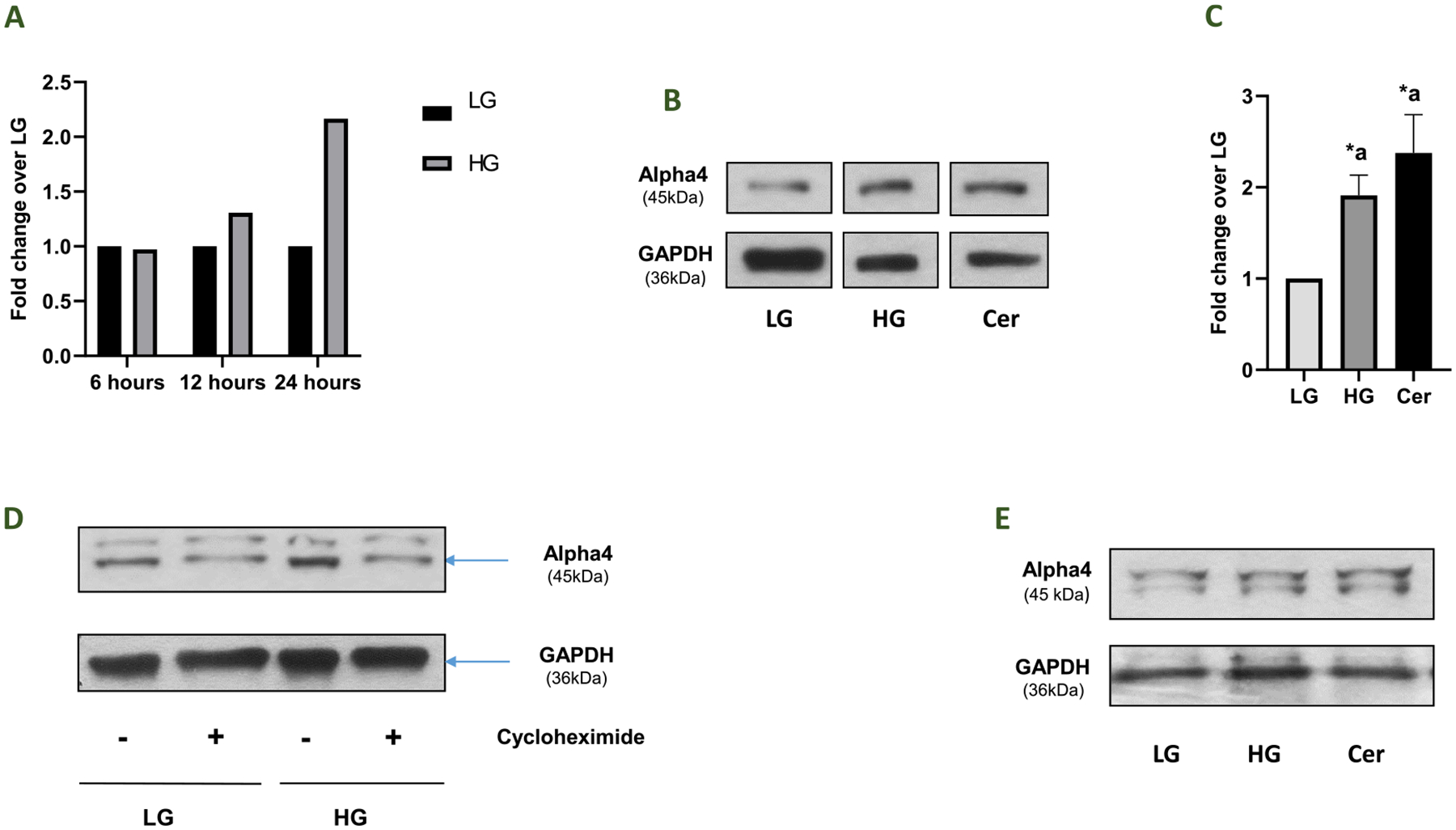
Panel A: INS-1832/13 cells were incubated with basal glucose (LG, 2.5 mM) or high glucose (HG, 20 mM) for various time intervals as indicated in the figure. Densitometric quantification of Alpha4 bands under these conditions is provided herein. Panel B: INS-1832/13 cells were incubated with basal glucose (LG, 2.5 mM), high glucose (HG, 20 mM), or ceramide (Cer, 50 μM) for 24 h and lysates were analyzed by Western blotting for the expression of Alpha4. Corresponding GAPDH levels are provided in this blot. Panel C: Densitometric quantification of Alpha4 band intensities is shown here. The results (Alpha4/GAPDH ratios) are presented as means ± SEM. The data are expressed as fold change relative to LG (n = 5; Comparisons: a – significant compared with LG-treated lysates; *p < 0.05). Panel D: INS-1832/13 cells were incubated with basal glucose (LG, 2.5 mM) and high glucose (HG, 20 mM) in the absence or presence of cycloheximide (7 μM) for 12 h. A representative Western blot is shown here. GAPDH was used as the corresponding loading control. Panel E: Islets isolated from normal rats were incubated with basal glucose (LG, 2.5 mM), high glucose (HG, 20 mM), or ceramide (Cer, 50 μM) for 24 h and lysates were analyzed by Western blotting for the expression of Alpha4. Corresponding GAPDH levels are provided in this blot.
3.3. Subcellular distribution of Alpha4 in INS-1832/13 cells exposed to HG conditions
The next set of experiments were undertaken to determine potential subcellular distribution of Alpha4, if any, in pancreatic beta cells exposed to HG conditions. We addressed this question by determining the relative degree of abundance of Alpha4 in the membrane vs. cytosolic fractions (Fig. 3; Panel A) or nuclear vs. non-nuclear fractions (Fig. 3; Panel B). These fractions were isolated from INS-1832/13 cells exposed to LG and HG (see Methods for additional details). Purity of the fractions was assessed by association of these fractions with respective marker proteins (i.e., GAPDH for the cytosolic fraction; E-cadherin for the membrane fraction; and lamin B for the nuclear fraction). Data accrued in these studies indicated that Alpha4 is predominantly soluble in nature, and that its localization is not significantly altered under HG exposure conditions. However, HG exposure resulted in increased expression of Alpha4 in the cytosolic (~1.45 fold) and non-nuclear (~2.3 fold) fractions. Together, these studies demonstrate no major alterations in the subcellular localization of Alpha4 under the duress of HG conditions. They also confirm the data highlighted in Fig. 3 that HG promotes expression of Alpha4 following exposure to HG.
Fig. 3. HG exposure conditions do not promote alterations in subcellular distribution of Alpha4 in INS-1832/13 cells.
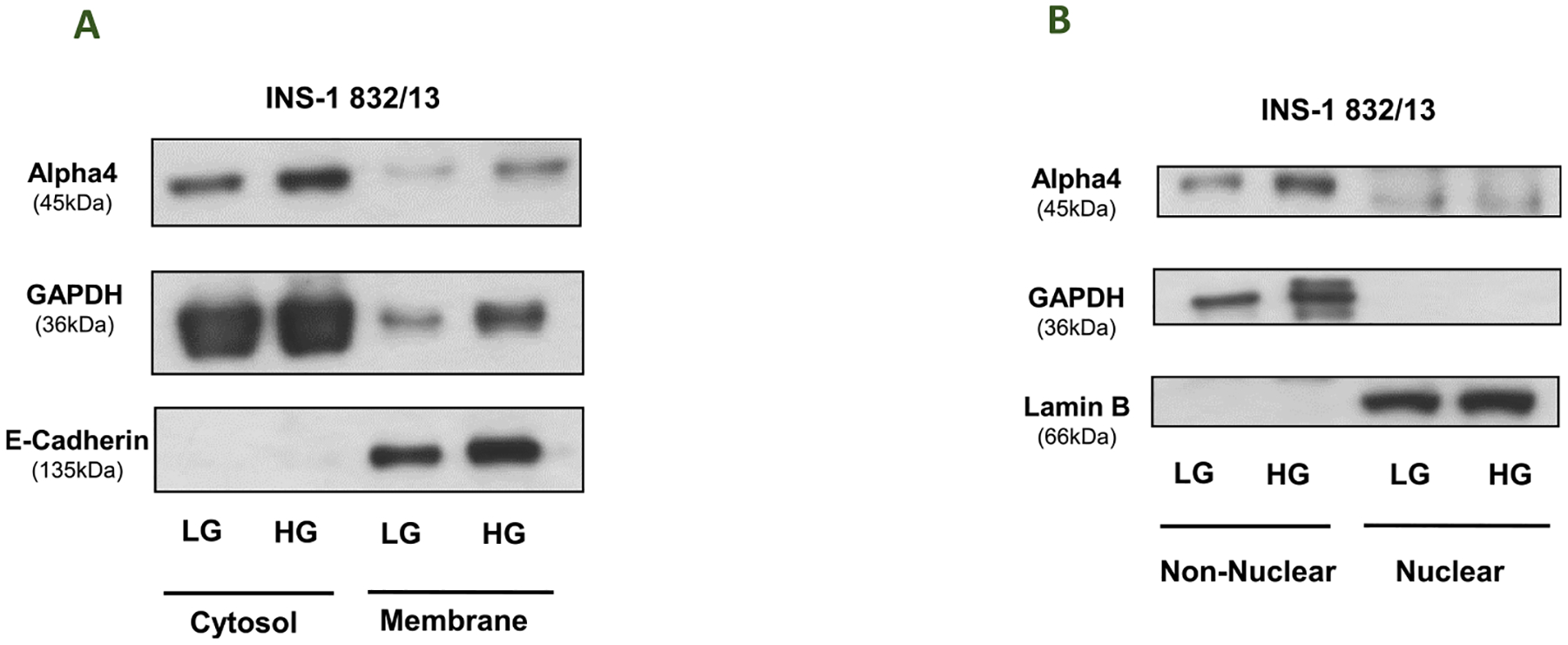
Panel A: Distribution of Alpha4 in membrane and cytosolic fractions derived from INS-1832/13 cells exposed to LG and HG: INS-1832/13 cells treated with low glucose (LG, 2.5 mM) or high glucose (HG, 20 mM) for 24hrs. and subjected to subcellular fractionation yielding cytosolic and membrane fractions. Western blot analysis was done on the cytosolic and membrane fractions. E-Cadherin was done to see the purity of the membrane fractions and GAPDH was done as loading control. Panel B: Distribution of Alpha4 in non-nuclear and nuclear fractions in INS-1832/13 cells exposed to LG and HG: INS-1832/13 cells treated with low glucose (LG, 2.5 mM) or high glucose (HG, 20 mM) for 24 h and subjected to subcellular fractionation yielding non-nuclear and nuclear fractions. Western blot analysis was done on the non-nuclear and nuclear fractions. Lamin B was done to see the purity of the nuclear fractions and GAPDH was done as loading control.
3.4. siRNA-mediated knockdown of Alpha4 increases p38 activation under LG conditions and potentiates its activation induced by HG in INS-1832/13 cells
We have reported increased stress kinase (p38 MAPK and JNK1/2) activation in pancreatic beta cells under the duress of metabolic stress, including chronic hyperglycemia (Sidarala and Kowluru, 2017; Gamage et al., 2022; Syed et al., 2011). Herein, we investigated relative contributory roles of Alpha4 in HG-induced p38 MAPK activation by quantifying the later in INS-1832/13 cells following deletion of Alpha4. Data in Fig. 4 (Panel A) show a significant increase in p38 phosphorylation in these cells following Alpha4 knockdown under LG conditions. Interestingly, the extent of phosphorylation was comparable to that seen in the presence of HG in cells transfected with Con-si. It is noteworthy that phosphorylation of p38 MAPK in HG treated cells was potentiated following knockdown of Alpha4 (Fig. 4; Panel B). These studies demonstrate potential dual regulatory roles of Alpha4 in pancreatic beta cells in that deletion of this protein promoted p38MAPK activation under LG conditions, and potentiated HG-induced activation of p38 MAPK (see Discussion).
Fig. 4. Contributory roles of Alpha4 to HG-induced activation of p38 MAPK in INS-1832/13 cells.
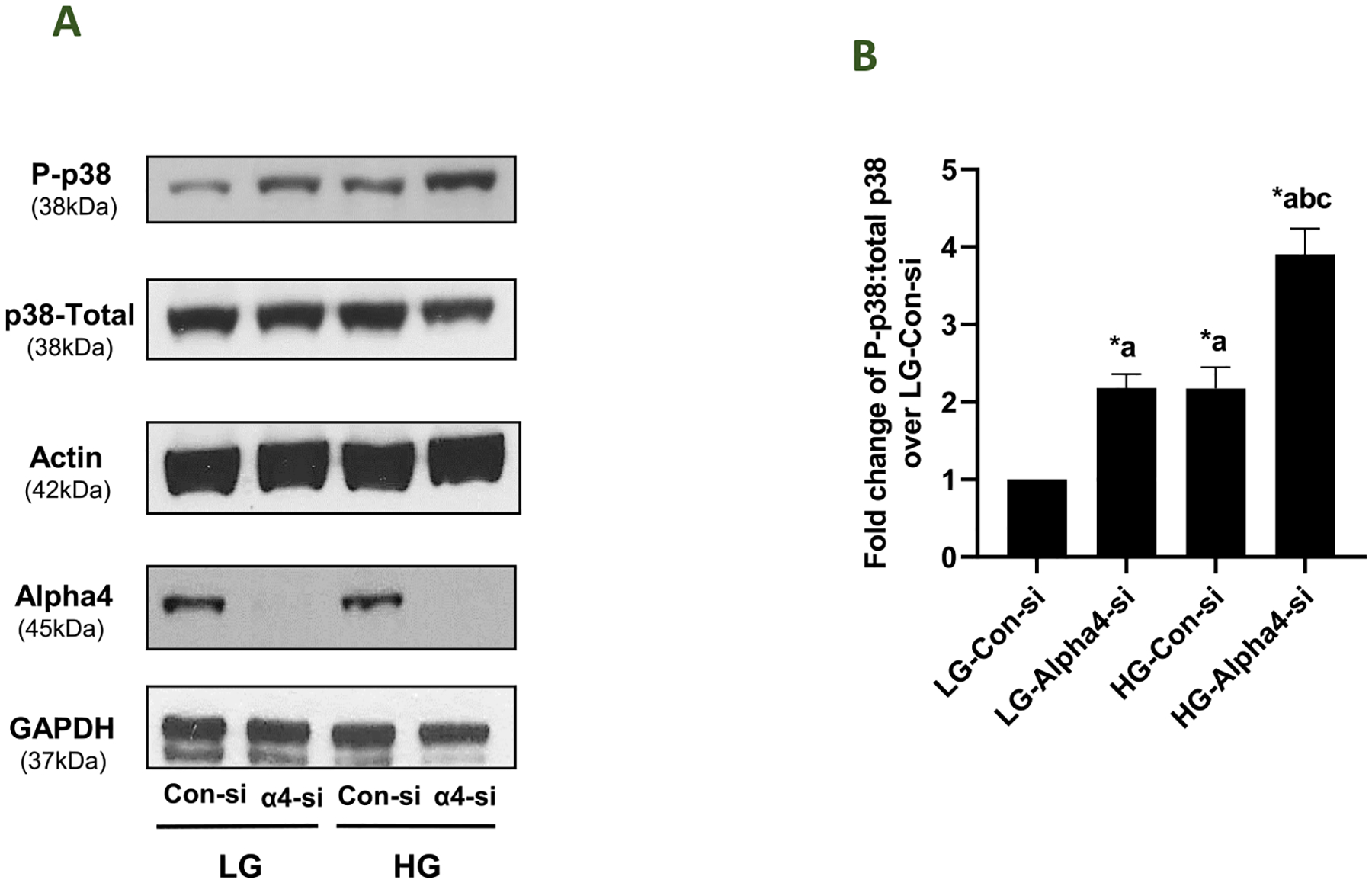
Panel A: INS-1832/13 cells were transfected with Con-si or Alpha4-si. Forty eight hrs. post transfection the cells were treated with LG or HG for 24 h. Relative abundance of phospho-p38 and total-p38 in lysates from these cells was assessed by western blotting. Western blot data for Alpha4 expression in these cell lysates is shown to demonstrate knockdown of Alpha4 under these experimental conditions. β- Actin and GAPDH were used as loading control for these conditions. A representative blot from four independent studies is shown here. Panel B: Band intensities (from data in Panel A) were quantified by densitometry for phospho-p38MAPK and Total-p38MAPK and the ratios (phospho-p38MAPK: Total-p38MAPK) are provided herein. Data are represented as fold change from LG- Con-si and are given as mean ± SEM (n = 4). (Comparisons: a: significant compared with LG-Con-si; b: significant compared with LG-Alpha4-si; c: significant compared with HG-Con-si; *p < 0.05).
3.5. siRNA-mediated knockdown of Alpha4 increases basal JNK1/2 activation and potentiates HG-induced JNK-2 phosphorylation in INS-1832/13 cells
In the next series of studies, we examined the roles of Alpha4 in HG-mediated regulation of JNK1/2 in INS-1832/13 cells. To address this, we utilized the same experimental approach that we employed in our p38 MAPK studies (see Fig. 4). Data obtained in these studies suggested a marked increase in the phosphorylation of both JNK1 and JNK2 in INS-1832/13 cells under basal conditions, following depletion of endogenous Alpha4 (Fig. 5; Panels B and C). As in the case of p38 MAPK, the degree of stimulation was comparable to that observed in these cells transfected with Con-si exposed to HG. Interestingly, the phosphorylation of JNK2, but not JNK1, was potentiated in HG-exposed cells following knockdown of Alpha4 (Fig. 5; Panel C). Taken together, data in Figs. 4 and 5 suggest that Alpha4 might contribute to the survival of beta cell under normal (basal) conditions, and appears to suppress putative regulatory factors that might potentiate HG-induced phosphorylation of stress kinase since knockdown of Alpha4 resulted in stress kinase activation to a large degree (see Discussion).
Fig. 5. Regulatory roles of Alpha4 in HG-induced activation of JNK1/2 in INS-1832/13 cells.
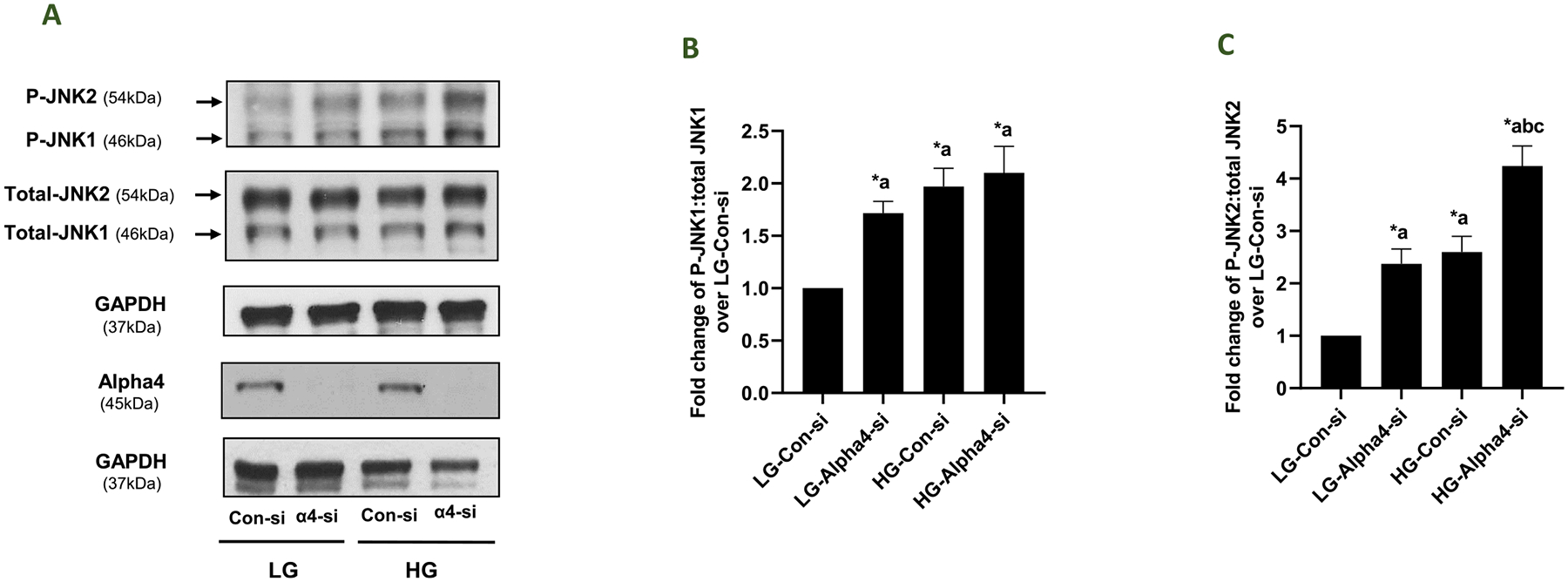
Panel A: INS-1832/13 cells were transfected with Con-si or Alpha4-si. Forty eight hrs. post transfection the cells were treated with LG or HG for 24 h. Relative abundance of phospho- and Total-JNK1 and JNK2 in lysates from these cells was assessed by western blotting. Western blot data for Alpha4 expression in these cell lysates is shown to demonstrate knockdown of Alpha4 under these experimental conditions. GAPDH was used as loading control for these conditions. A representative blot from four independent studies is shown here. Panels B and C: Densitometric analysis of pooled data from four independent experiments (in Panel A) is provided in Panel B (JNK1) and Panel C (JNK2). The data are shown as fold change from LG-Con-si (mean ± SEM; n = 4). (Comparisons: a: significant compared with LG-Con-si; b: significant compared with LG-Alpha4-si; c: significant compared with HG-Con-si; *p < 0.05).
3.6. siRNA-mediated depletion of Alpha4 inhibits HG-induced CHOP (ER stress marker) expression and caspase 3 activation in INS-1832/13 cells
As a logical extension to the above studies, we investigated putative regulatory roles of Alpha4 in HG-induced CHOP expression (a marker for endoplasmic reticulum stress) and caspase-3 activation (a marker of mitochondrial defects) in INS-1832/13 cells following transfection with Con-si and Alpha4-si. Data highlighted in Fig. 6 (Panels A and B) demonstrate a significant augmentation of CHOP expression in HG treated cells transfected with Con-si. Transfection of these cells with Alpha4-si markedly attenuated HG-induced CHOP expression. Likewise, HG-induced Caspase-3 activation was also suppressed in these cells following deletion of Alpha4 (Fig. 6; Panels C and D). Together, these data provide support that Alpha4 promotes metabolic dysregulation (e. g., ER stress and Caspase 3 activation) in INS-1832/13 cells under the duress of HG conditions.
Fig. 6. Alpha4 mediates HG-induced Caspase- 3 activation and CHOP expression in INS-1832/13 cells.
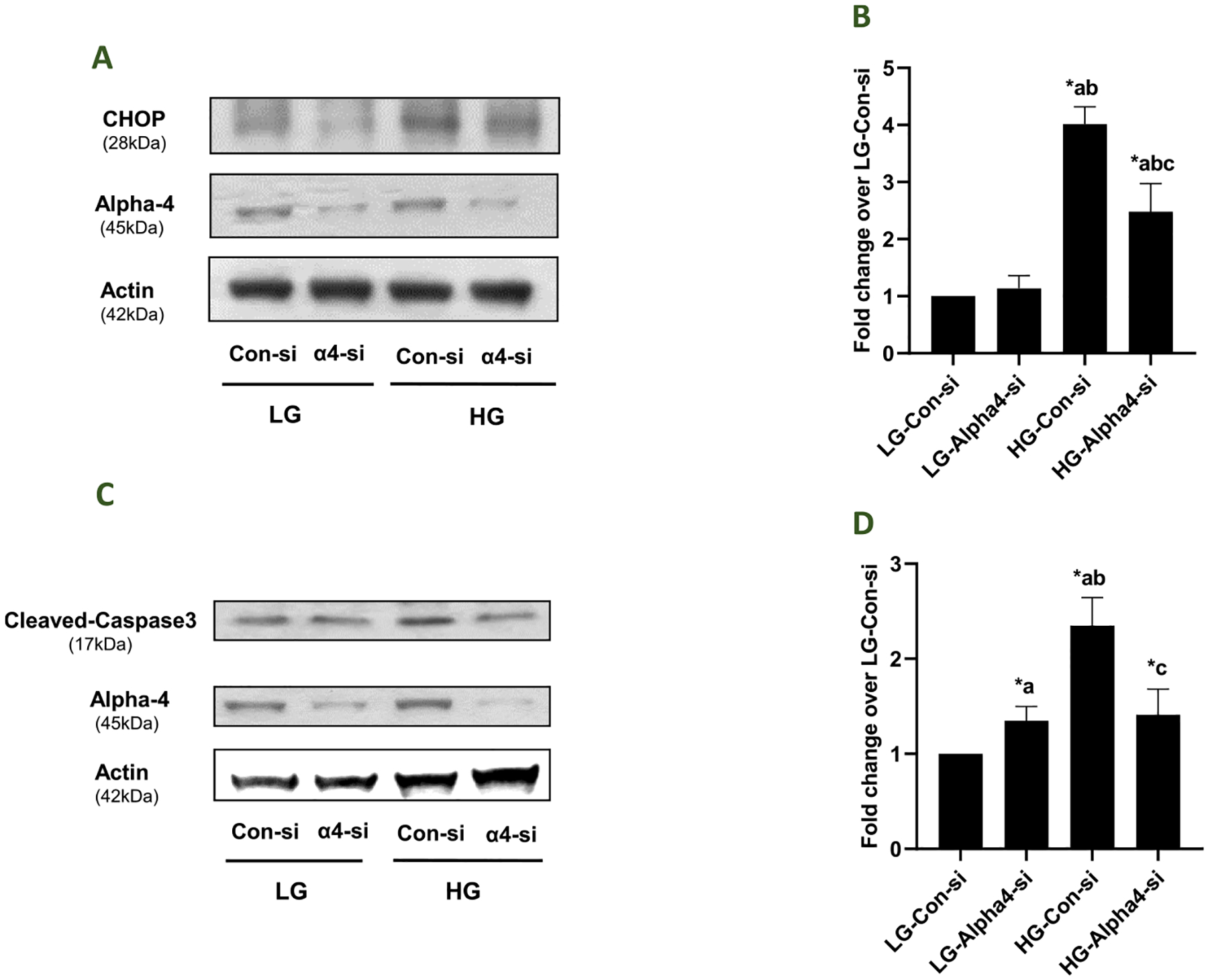
Panel A: Following transfection with Con-si or Alpha4-si, INS-1832/13 cells were treated with LG or HG for 24 h. Relative abundance of CHOP and Alpha4 in the cell lysates was determined by western blotting. β- Actin was used as the control for protein loading. A representative blot from 4 independent experiments is shown here. Panel B: Densitometric quantification of CHOP expression in 4 independent experiments is shown here. Data are expressed as fold change relative to LG-Con-si (mean ± SEM; n = 4). (Comparisons: a: significant compared with LG-Con-si; b: significant compared with LG-Alpha4-si; c: significant compared with HG-Con-si; *p < 0.05). Panel C: Following transfection with Con-si or Alpha4-si, INS-1832/13 cells were treated with LG or HG for 24 h. Relative abundance of cleaved Caspase-3 (active form) and Alpha4 in the cell lysates was determined by western blotting. β- Actin was used as the control for protein loading. A representative blot from 5 independent experiments is shown here. Panel D: Densitometric quantitation of cleaved Caspase-3 in Panel A is depicted herein. The pooled data are expressed as fold change relative to LG-Con-si (mean ± SEM; n = 5). (Comparisons: a: significant compared with LG-Con-si; b: significant compared with LG-Alpha4-si; c: significant compared with HG-Con-si; *p < 0.05).
3.7. Putative roles of Alpha4 in HG-mediated effects on the expression of specific signaling proteins involved in islet beta cell function
Earlier studies in other cell types have identified Rac1, a small G protein, and β-PIX, a known guanine nucleotide exchange factor for small G proteins (e.g., Rac1 and Cdc42), as interacting/binding partners and downstream signaling proteins for Alpha4 (Kong et al., 2007; Chung et al., 2018). Recent studies in pancreatic beta cells have demonstrated gp91phox (Nox2) as one of the proteins that is regulated by Rac1 (Newsholme et al., 2009; Kowluru, 2011; Kowluru and Kowluru, 2014). Therefore, we undertook a study to determine the impact of Alpha4 deletion on the expression of Rac1, gp91phox and β-PIX in INS-1832/13 cells following exposure to LG and HG. Data in Fig. 7 (Panel B) indicated no significant changes in the expression of Rac1 in INS-1832/13 cells exposed to LG and HG following depletion of Alpha4. Data shown in Fig. 7 (Panel D) showed a significant increase in the expression of gp91phox in HG-treated cells transfected with Con-si. However, Alpha4-si transfection failed to exert any significant effects in these cells under HG exposure conditions. Lastly, we observed a significant increase in the expression of β-PIX in LG-treated cells transfected with Alpha4-si, and HG-treated cells transfected with either Con-si or Alpha4-si (Panel F). Taken together, despite its varying effects on the expression of Rac1, gp91phox and β-PIX expression in INS-1832/13 cells under LG conditions, Alpha4 exerted no significant effects on the expression of these proteins under HG conditions.
Fig. 7. Depletion of Alpha4 exerted no significant effects on the expression of specific signaling proteins involved in its signaling modules.
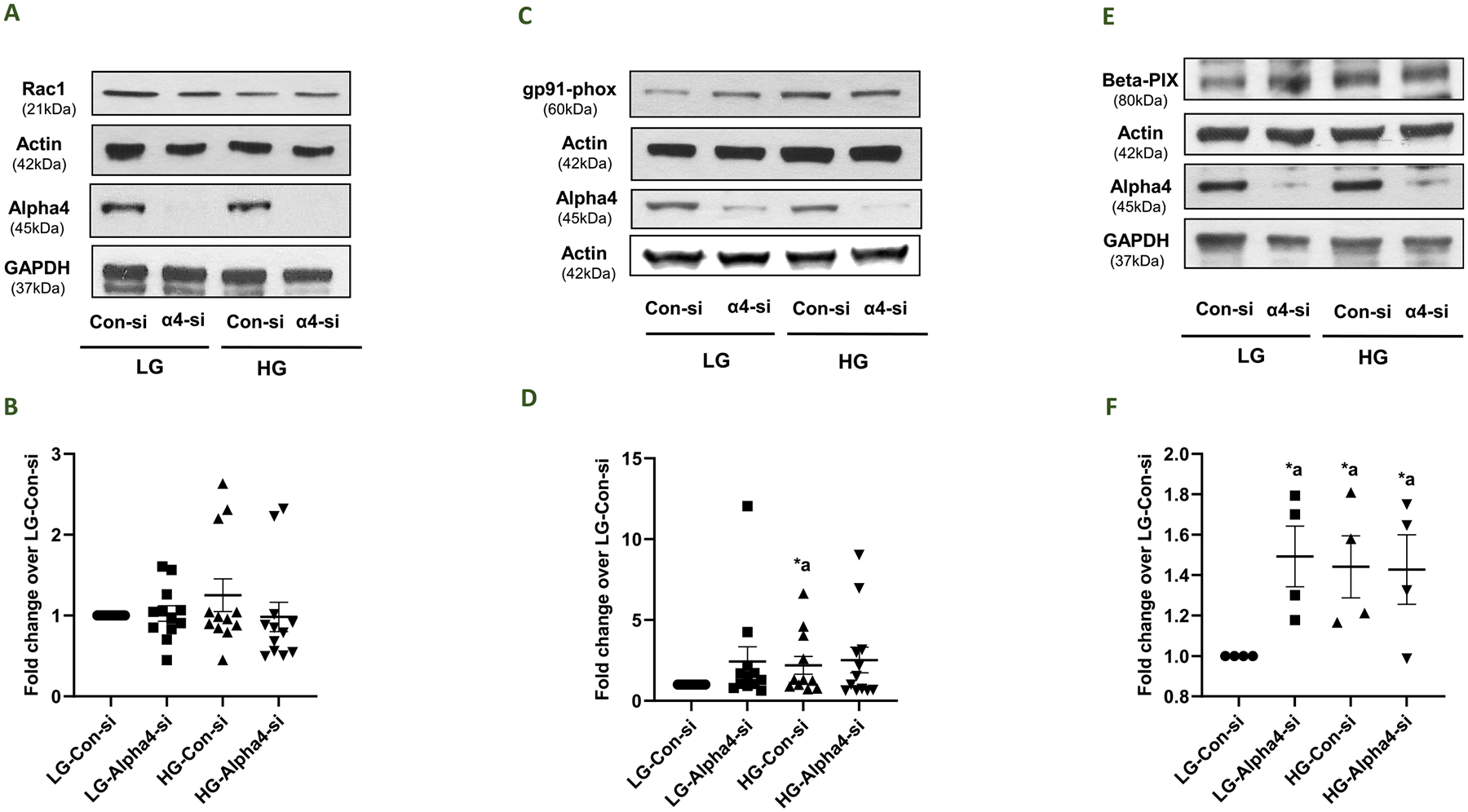
Panel A: Impact of Alpha4 knockdown on the expression of Rac1 in INS-1832/13 cells. Following transfection with Con-si or Alpha4-si, INS-1832/13 cells were treated with LG or HG for 24 h. Western blotting was done to determine the relative abundance of Rac1 and Alpha4 in cell lysates. β- Actin and GAPDH were used as controls for protein loading. A representative blot from 12 independent experiments is shown here. Panel B: Densitometric quantification of Rac1 expression in 12 independent experiments is shown here. Data are expressed as fold change relative to LG-Con-si (mean ± SEM; n = 12). Panel C: Impact of Alpha4 knockdown on the expression of gp91-phox in INS-1832/13 cells. Following transfection with Con-si or Alpha4-si, INS-1832/13 cells were treated with LG or HG for 24 h. Western blotting was done to determine the relative abundance of gp91phox and Alpha4 in cell lysates. β- Actin and GAPDH were used as controls for protein loading. A representative blot from 12 independent experiments is shown here. Panel D: Densitometric quantification of gp91-phox expression in 12 independent experiments is shown here. Data are expressed as fold change relative to LG-Con-si (mean ± SEM; n = 12). (Comparisons: a: significant compared with LG-Con-si; *p < 0.05). Panel E: Impact of Alpha4 knockdown on the expression of β-PIX in INS-1832/13 cells. Following transfection with Con-si or Alpha4-si, INS-1832/13 cells were treated with LG or HG for 24 h. Western blotting was done to determine the relative abundance of β-PIX and Alpha4 in cell lysates. β- Actin and GAPDH were used as controls for protein loading, respectively. A representative blot from 4 independent experiments is shown here. Panel F: Densitometric quantification of β-PIX expression in 4 independent experiments is shown here. Data are expressed as fold change relative to LG-Con-si (mean ± SEM; n = 4). (Comparisons: a: significant compared with LG-Con-si; *p < 0.05).
3.8. Regulatory roles of Alpha4 in HG-mediated effects on the expression of specific connexins (Cxs) involved in islet beta cell function
Earlier observations from multiple laboratories suggested key regulatory roles for gap junctions in the cascade of events leading to GSIS in pancreatic beta cells (Umrani et al., 2017; Farnsworth and Benninger, 2014; Benninger et al., 2011; Meda, 2018). Several gap junction channel proteins have been identified in pancreatic beta cells, among which Connexin36 (Cx36) is the most studied for its role in physiological insulin secretion (Umrani et al., 2017; Farnsworth and Benninger, 2014; Benninger et al., 2011; Pérez-Armendariz, 2013; Cigliola et al., 2013; Bosco et al., 2011; Serre-Beinier et al., 2000). Other Cxs, such as Cx43 have also been proposed to contribute to beta cell function, but published evidence suggests that Cx43 is expressed only in INS-1 beta cells, but not in primary beta cells and mouse-derived MIN6 cells (Jain and Lammert, 2009; Vozzi et al., 1995). In the context of current studies, previous studies in cultured beta cells and rodent islets reported significant downregulation of Connexin 36 under hyperglycemic conditions (Umrani et al., 2017; Farnsworth and Benninger, 2014; Haefliger et al., 2013). Therefore, we undertook a study to assess potential impact of Alpha4 knockdown on HG-mediated effects on the expression of Cx36 and Cx43 in INS-1832/13 cells exposed to LG and HG conditions. Data in Fig. 8 (Panel A) indicated nearly 60% inhibition in the expression of Cx36 in HG-treated cells transfected with control-si. Interestingly, deletion of Alpha4 in these cells significantly protected the loss of expression (~30% inhibition) of Cx36 under HG-treatment conditions. Pooled data from four experiments are provided in Panel B. It is noteworthy that, unlike Cx36, the expression of Cx43 remained unchanged under these experimental conditions (Panels C and D). Together, our findings suggest specific regulatory roles of Alpha4 in HG-induced loss in the expression of Cx36 (see Discussion).
Fig. 8. Impact of Alpha4 on HG-mediated effects on the expression of Cx36 and Cx43 in INS-1832/13 cells.
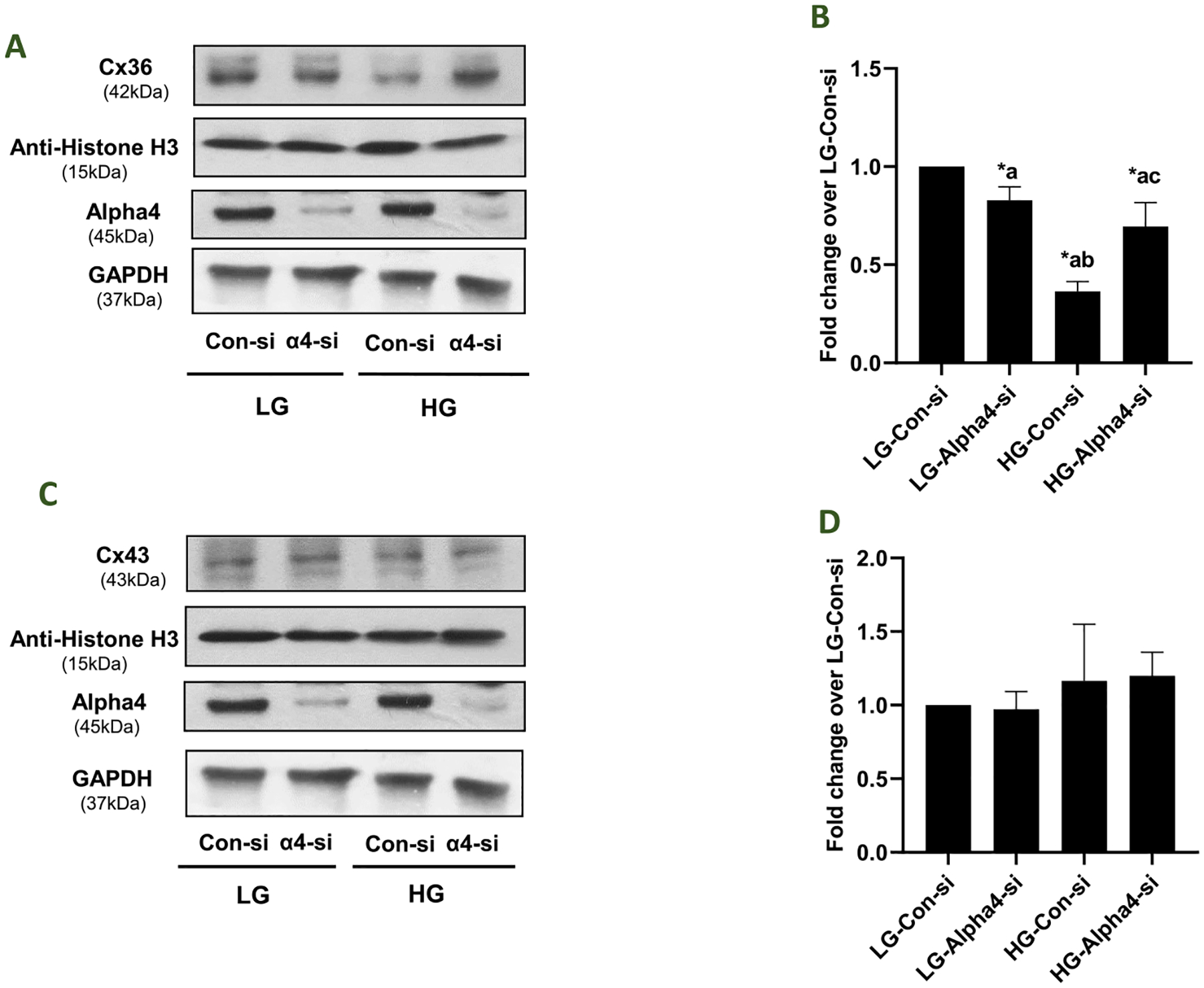
Panel A: Following transfection with Con-si or Alpha4-si, INS-1832/13 cells were treated with LG or HG for 24 h. Relative abundance of Cx36 and Alpha4 in the cell lysates was determined by western blotting. Histone H3 and GAPDH were used to verify equal protein loading. A representative blot from 4 independent experiments is shown here. Panel B: Densitometric quantitation of Cx36 in Panel A is depicted herein. The pooled data are expressed as fold change relative to LG-Con-si (mean ± SEM; n = 4). (Comparisons: a: significant compared with LG-Con-si; b: significant compared with LG-Alpha4-si; c: significant compared with HG-Con-si; *p < 0.05). Panel C: Following transfection with Con-si or Alpha4-si, INS-1832/13 cells were treated with LG or HG for 24 h. Relative abundance of Cx43 and Alpha4 in the cell lysates was determined by western blotting. Histone H3 and GAPDH were used as loading controls. A representative blot from 4 independent experiments is shown here. Panel D: Densitometric quantitation of Cx43 in Panel C is depicted herein. The pooled data are expressed as fold change relative to LG-Con-si (mean ± SEM; n = 4).
3.9. Co-immunoprecipitation (Co-ip) studies reveal increased interaction of PP2Ac with Alpha4 under HG conditions in INS-1832/13 cells
Several lines of published evidence in multiple cell types suggest that Alpha4 is a key regulator of serine/threonine phosphatase (e.g., PP2A, PP4 and PP6) activities (Chen et al., 1998; Lenoue-Newton et al., 2016; Kong et al., 2009; Eleftheriadou et al., 2017). Previous proteomic studies from our laboratory identified Alpha4 as one of the interacting partners of PP2Ac in pancreatic beta cells. Therefore, in the next set of studies, we determined the degree of association between Alpha4 and PP2Ac in INS-1832/13 cells exposed to LG and HG conditions. To address this, we quantified the relative abundance of PP2Ac in the immunoprecipitates obtained from lysates from LG and HG treated cells incubated with Alpha4 antibody (see Methods for additional details). Data shown in Fig. 9 (Panel A) suggest increased abundance of PP2Ac in Alpha4 immunoprecipitates from HG treated cells. Pooled data from multiple studies indicated ~ 2-fold increase in interaction between Alpha4 and PP2Ac under current experimental conditions. These data provide evidence in support of the formulation that Alpha4 might contribute to the sustained activation of PP2A in pancreatic beta cells under metabolic stress conditions ((Arora et al., 2014); see Discussion).
Fig. 9. Increased interaction between PP2A-Cα/β and Alpha4 under HG conditions.
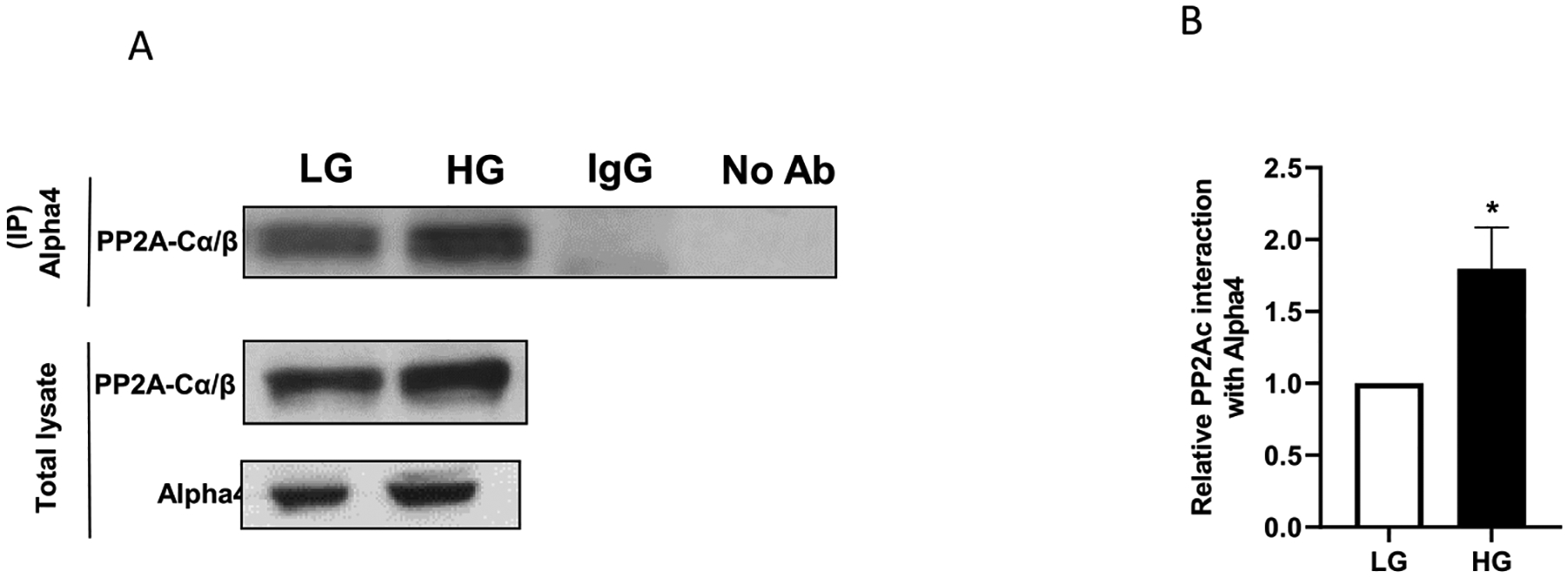
Panel A: INS-1832/13 cells were treated with LG or HG for 24 h and lysed in IP lysis buffer. Alpha4 or normal mouse IgG polyclonal antibody was immunoprecipitated from equal volumes of pre-cleared cell lysates. The immunoprecipitates were resolved by SDS-PAGE and immune-blotted for PP2A-Cα/β. A representative blot from 3 independent experiments is shown here. Panel B: Densitometric quantification of PP2A-Cα/β abundance in the immunoprecipitates of Alpha4 from LG and HG treated cells in 3 independent experiments is shown here. Data are expressed as fold change relative to LG (mean ± SEM; n = 3). (Comparisons: *significant compared with LG; *p < 0.05).
3.10. Alpha4 contributes to HG-induced cell death in INS-1832/13 cells
In the last set of studies, we asked if Alpha4 plays critical regulatory roles in HG-induced cell death. To address this, cell death was quantified in LG and HG treated INS-1832/13 cells following transfection with Con-si and Alpha4-si by quantifying the histone-complexed DNA fragments (mono- and oligonucleosomes) in the cytoplasm of cells following the induction of apoptosis of INS-1832/13 cells by HG. Data depicted in Fig. 10 demonstrated a significant increase in the cell death in HG-treated cells transfected with Con-si. Transfection of these cells with Alpha4-si markedly reduced HG-induced cell death, suggesting that Alpha4 might mediate HG-induced cell demise. It should be noted that Alpha4 depletion modestly, but significantly, reduced the basal values seen under LG conditions.
Fig. 10. Alpha4 contributes to HG-induced cell death in INS-1832/13 cells.
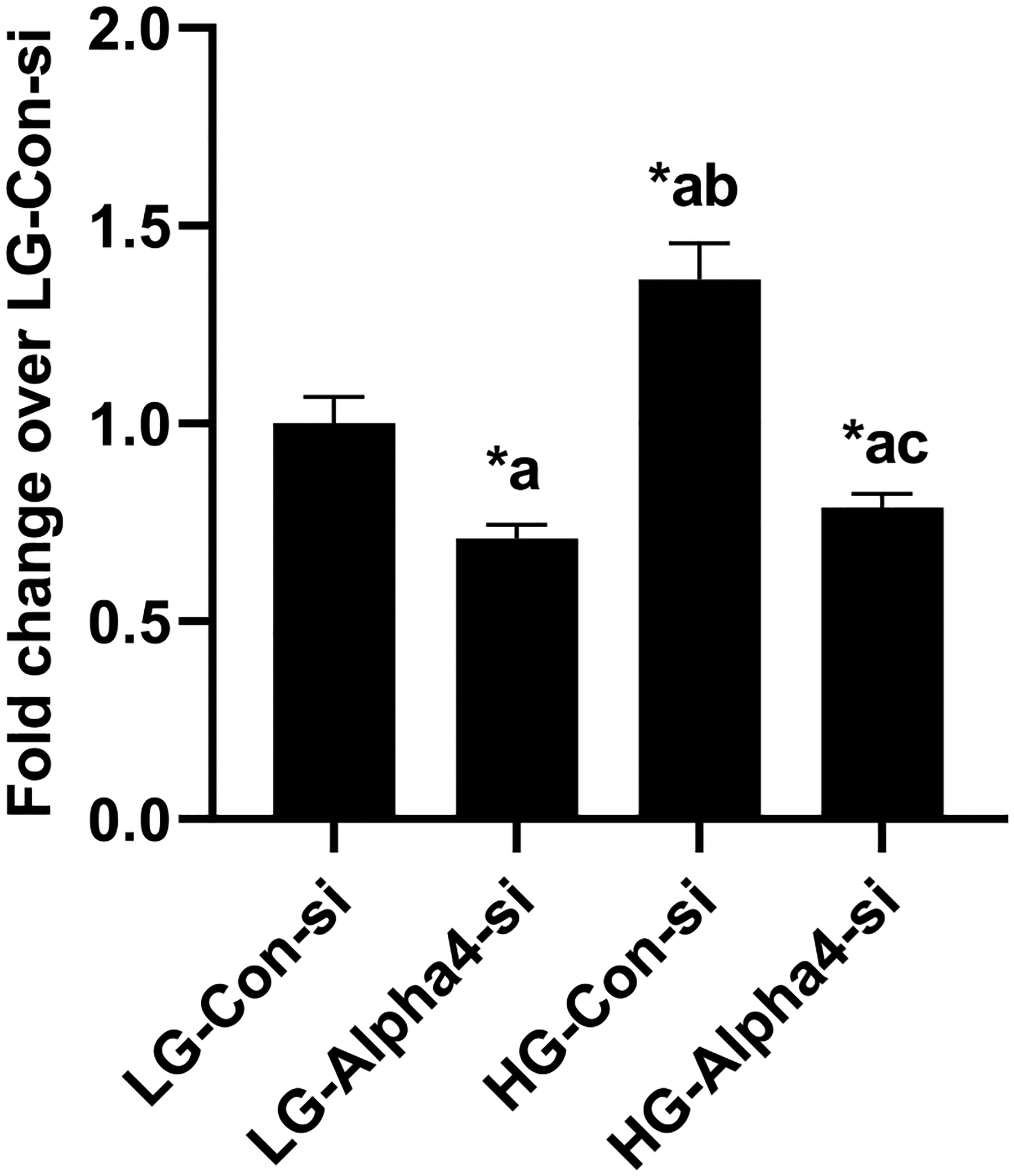
Following transfection with Con-si or Alpha4-si, INS-1832/13 cells were treated with LG or HG for 24 h. Degree of cell death was quantified using Cell Death detection ELISAplus kit. Absorbance was measured at 405 nm and data are expressed as fold change relative to LG-Con-si. Data are expressed as mean ± SEM from two experiments with 14 replicates per condition. (Comparisons: a: significant compared with LG-Con-si; b: significant compared with LG-Alpha4-si; c: significant compared with HG-Con-si; *p < 0.05).
4. Discussion
Published evidence in several cell types implicates critical roles for Alpha4 in the biogenesis, stability and activation of protein phosphatases (e.g., PP2A, PP4, PP6 etc.) (Sents et al., 2013; Zolnierowicz, 2000; Kloeker et al., 2003, Mcconnell et al., 2010). Despite the known regulatory roles of protein phosphatases in islet beta cell function and dysfunction (see refs. for reviews (Ortsäter et al., 2014; Kowluru and Matti, 2012; Kowluru, 2005)), limited information is available on roles of Alpha4 in islet function, including GSIS (i.e., acute effects of glucose) or beta cell dysfunction under metabolic stress conditions (chronic effects of glucose). Using co-ip and tandem mass spectrometry approaches, our previous proteomic studies identified Alpha4 as one of the interaction partners of PP2Ac in insulin-secreting pancreatic β-cells (Zhang et al., 2016). As a logical extension to these investigations, we have undertaken the current study to further assess the contributory roles of Alpha4 in acute and chronic effects of glucose in pancreatic beta cells. Salient findings from the current studies are: [i] Alpha4 is expressed in a variety of insulin-secreting cells, including human islets, rat islets and INS-1832/13 cells; [ii] expression of Alpha4 is significantly increased in INS-1832/13 cells following exposure to HG; [iii] siRNA-mediated depletion of Alpha4 exerted minimal effects on glucose- or KCl-induced insulin secretion in INS-1832/13 cells; [iv] deletion of Alpha4 significantly increased stress kinase (p38MAPK and JNK1/2) phosphorylation; and [v] siRNA-mediated knockdown of Alpha4 markedly attenuated HG-induced CHOP expression, caspase 3 activation and cell death. Based on these observations, we propose that Alpha4 contributes to HG-induced metabolic dysfunction of the islet beta cell. Potential implications and significance of these findings are discussed below.
Several earlier studies have affirmed roles for Alpha4 in the regulation of various protein phosphatase activities. For example, using the yeast two-hybrid system, Chen and coworkers reported association of Alpha4 with PP2A, PP4 and PP6 (Chen et al., 1998). Kong et al. showed that deletion of Alpha4 results in progressive loss of all of the above phosphatase complexes, including a significant reduction in the abundance of post-translationally methylated (Leu-309) PP2Ac. Based on additional evidence accrued in other cells, under normal health and cell stress conditions, these authors surmised that Alpha4 plays key regulatory roles in the assembly and maintenance of PP2A complexes (Kong et al., 2009). Using immunodepletion approaches in HEK293T cells, LeNoue-Newton et al. have reported that Alpha4 preferentially affects the expression of PP4c and PP6c compared to the levels of expression of PP2Ac (Lenoue-Newton et al., 2016). Moreover, Eleftheriadou and coworkers proposed major regulatory roles of PP2A-Alpha4 complex in healthy and hypertrophied myocadia (Eleftheriadou et al., 2017). Based on data accrued from immunological, biochemical, cell biological and pharmacological investigations, we previously reported regulatory roles for PP2A-like phosphatases in islet function (e.g., GSIS) and dysfunction under metabolic stress conditions (Kowluru and Matti, 2012; Kowluru, 2005). We also demonstrated a significant increase in the methylation of PP2Ac and catalytic activation of PP2A in human islets, rat islets and INS-1832/13 cells exposed to hyperglycemic conditions. In follow-up studies, we demonstrated that siRNA-mediated knockdown of PP2Ac markedly attenuated glucose-mediated sustained activation of PP2A in INS-1832/13 cells. Based on these findings, we concluded that exposure of the islet beta-cell to chronic hyperglycemic conditions leads to accelerated PP2A signaling pathway, culminating in loss in GSIS and the onset of metabolic dysfunction (Arora et al., 2014). In an attempt to further demonstrate functional relationships between PP2A and Alpha4, using co-ip and tandem mass spectrometry approaches, we identified Alpha4 as one of the interacting partners of PP2Ac in insulin-secreting cells (Zhang et al., 2016). Data from our co-ip experiments reported in the current study further affirm increased association between Alpha4 and PP2Ac in pancreatic beta cells under HG conditions. Lastly, our recent findings suggesting that siRNA-mediated knockdown of endogenous expression of Alpha4 resulted in significant inhibition (~60%) of high glucose-induced PP2A activity in INS-1832/13 cells further substantiate key modulatory roles for Alpha4 in the sustained activation of PP2A under glucotoxic conditions (Kowluru, 2018). Further studies are needed to define putative signaling mechanisms underlying Alpha4-mediated regulation of PP2A activation including its roles in facilitating post-translational methylation of PP2Ac and the holoenzyme assembly of PP2A. Lastly, potential regulation, by Alpha4, of other phosphatases in the islet beta cell (e.g., PP4c (Veluthakal et al., 2006);), remain to be investigated.
Data accrued in our current studies have shown a significant increase in the expression of Alpha4 in rodent islets and INS-1832/13 cells exposed to HG and Cer. It appears that such an increase may, in part be, due to de novo synthesis of Alpha4 since cycloheximide significantly inhibited high glucose-induced expression of Alpha4. Interestingly, albeit in the context of other pathologies, Chen and coworkers have reported high degree of expression of α4 in a variety of hepatic and lung cancer cell lines (Chen et al., 2011). Furthermore, they observed overexpression of α4 in primary hepatocellular carcinomas, primary lung cancers and primary breast cancers; these data indicated that α4 is ubiquitously highly expressed in human cancers. Functional studies revealed that elevated α4 expression results in an increase in cell proliferation, promotion of cell survival and decreased PP2A-attributable activity. Studies by Sakashita and associates have investigated the expressional characteristics of IGBP-1 and PP2Ac during pulmonary adenocarcinogenesis (Sakashita et al., 2011). Their data provided compelling evidence to indicate that the positivity rate of IGBP-1 significantly increased during the course of sequential progression from non-invasive to invasive adenocarcinoma, and that IGBP-1 positivity was significantly correlated with a poor prognosis. Thus, it appears that Alpha4 expression is significantly increased in various pathological states. Future studies, including determination of Alpha4 expression levels in islets derived from animal models of impaired insulin secretion and T2DM, and islets from humans with T2DM, are needed to further assess the roles of Alpha4 in the pathogenesis of metabolic defects in the islet beta cell in diabetes.
Our current findings also indicated significant association of Alpha4 in the nuclear fraction derived from pancreatic beta cells exposed to HG conditions. Putative roles of nuclear Alpha4 in in cell (dys) function remain unknown at this time. Some potential possibilities might include regulation of nuclear PP4 activity that we reported in islet beta cells earlier (Veluthakal et al., 2006), and regulation of transcription of various apoptotic genes involved in cell dysfunction and demise (Krauss et al., 2008; Fielhaber et al., 2009). Lastly, it may be likely that nuclear Alpha4 might contribute to the accelerated PP2A-Rac1 signaling pathway in the induction of cell dysfunction under metabolic dysfunction that we proposed recently (Kowluru, 2020b).
Several earlier studies have implicated potential regulatory roles of Alpha4 (and PP2A) in the regulation of several downstream signaling pathways, including activation of small G proteins (e.g., Rac1 (Kong et al., 2007);), NADPH oxidase (e.g., Nox2 (Kowluru, 2020b; Cheng and Li, 2020);), and other signaling molecules involved in G protein activation (β-PIX (Chung et al., 2018);). Data from our current investigations revealed no clear impact of Alpha4 deletion on the expression of Rac1, gp91phox, β-PIX in pancreatic beta cells. Additional studies are needed to further understand potential roles of these signaling pathways in Alpha4-mediated beta cell dysfunction under conditions of HG exposure.
We also provided evidence to suggest that depletion of Alpha4 partially protects/restores HG-induced loss of Cx36 in INS-1832/13 cells, thus suggesting novel roles for Alpha4 in HG-induced loss in expression of this gap junction channel protein in beta cells. Several lines of evidence suggest defects in Cx36 signaling module in clonal beta cells and rodent models of impaired insulin secretion and diabetes (Farnsworth and Benninger, 2014; Umrani et al., 2017; Cigliola et al., 2013; Meda, 2018). However, putative mechanisms and/or the identity of target proteins that mediate such defects remain only partially understood. Along these lines, studies by Haefliger and coworkers implicated upregulation of ICER-1/ICER-1γ in hyperglycemia-mediated downregulation of expression of Cx36 (Haefliger et al., 2013). From a mechanistic standpoint, investigations by Allagnat et al. demonstrated siRNA- or adenoviral-mediated depletion of Cx36 in INS-1E cells further increased apoptosis induced by pro-inflammatory cytokines. They also reported that overexpression of Cx36 prevented cytokine-induced ER and oxidative stress and associated cell dysfunction (Allagnat et al., 2013). Emerging evidence also implicates novel regulatory roles for specific microRNAs in the expression of Cxs in pancreatic beta cells (Umrani et al., 2017). Potential defects in these signaling modules leading to decreased expression of Cx36 in beta cells under metabolic stress and diabetes remain to be explored. In summary, data from our studies identify Alpha4 as a potential regulator of HG-induced loss in expression of Cx36. Furthermore, it should be noted that our studies have yielded no significant effects of HG conditions on the expression of Cx43 in INS-1832/13 cells (control or Alpha4 depleted). Additional studies are needed to further affirm roles of Alpha4 in the onset of functional defects in gap junctions culminating in metabolic dysfunction of the islet beta cell in metabolic stress and diabetes.
It is important to note that, in addition to HG conditions, we observed a significant increase in the expression of Alpha4 in INS-1832/13 cells and normal rat islets exposed to a cell permeable analogue of Cer. Even though, the current investigations were focused primarily on mechanisms involved in Alpha4-mediated pancreatic beta cell dysfunction under conditions of HG conditions, we propose that Alpha4 might also contribute to Cer-induced islet beta cell dysregulation. For example, it might be involved in the regulation of the Cer-activated protein phosphatase, a PP2A-like phosphatase that we have identified and characterized in the beta cell previously (Kowluru and Metz, 1997). Ceramide/PP2A mediated impairment in gap junction functions has been reported under specific experimental conditions (Squecco et al., 2021); these data raise an interesting possibility for additional regulatory mechanisms underlying abnormalities in Cx36 signaling modules in pancreatic beta cells under metabolic stress conditions. In addition, our published evidence also implicates critical roles for Cer in promoting other signaling steps (e.g., sustained activation of Rac1, and generation of Nox2-derived ROS, and mitochondrial defects) leading to the metabolic dysfunction and demise of the pancreatic beta-cell (Syed et al., 2010, 2012; Veluthakal et al., 2005, 2016; Kowluru and Kowluru, 2018). It is noteworthy that a growing body of evidence implicates sphingolipids in the onset of insulin resistance and type 2 diabetes (Chaurasia and Summers, 2021; Roszczyc-Owsiejczuk and Zabielski, 2021). Methodical investigations on putative roles of Alpha4 in Cer-mediated beta cell dysregulation and demise are being undertaken in our laboratory.
5. Conclusions
We provide the first evidence to implicate Alpha4, a non-canonical subunit of PP2A, in the onset of metabolic dysfunction (stress kinase activation, increased ER stress and onset of mitochondrial defects) and cell demise of the pancreatic beta cell under chronic metabolic stress conditions. We believe that these findings will form the basis for future investigations toward the identification of novel interacting partners and signaling pathways that are precisely regulated by the Alpha4 signaling module in the islet beta cell.
Acknowledgements
These studies are supported (to AK) by Merit Review (I01 BX004663) and Senior Research Career Scientist (IK6 BX005383) awards from the US Department of VA, and an R01 grant from the NIH/NEI (EY022230). AK would like to thank Wayne State University for the Distinguished Professorship award. Early work of BW on Alpha4 was supported by grants from NIH (GM015366 and DK070787). The authors would like to thank Dr. Vijayalakshmi Thamilselvan and Mr. Noah Gleason for advice and help during the course of these investigations.
Abbreviations used
- Alpha4
A non-canonical subunit of protein phosphatases (also referred to as Igbp-1, immunoglobulin-binding protein 1)
- β-PIX
β-PAK-interacting exchange factor
- Cer
ceramide
- CHOP
C/EBP homologous protein
- Co-IP
co-immunoprecipitation
- Cx36
Connexin36
- Cx43
Connexin43
- ER stress
endoplasmic reticulum stress
- GSIS
glucose-induced insulin secretion
- HG
high glucose
- JNK1/2
c-Jun N-terminal kinases 1 and 2
- LG
low glucose
- Nox2
Phagocyte-like NADPH oxidase 2
- p38MAPK
p38 mitogen-activated protein kinase
- PP2A
protein serine/threonine phosphatase 2A
- PP2Ac
PP2A catalytic subunit
- PP4
protein serine/threonine phosphatase 4
- PP6
protein serine/threonine phosphatase 6
- Rac1
Ras-related C3 botulinum toxin substrate 1
- siRNA
small interfering RNA
- T2DM
type 2 diabetes mellitus
- Tap42
type 2A-associated protein of 42 kDa
- TIPRL1
target of rapamycin signaling pathway regulator-like 1
- TOR
target of rapamycin
Footnotes
portion of this work is accepted for presentation in the 58th Annual Meetings of the European Association for the Study of Diabetes to be held in Stockholm, September 2022.
Statement of ethics
All protocols involving animal care and use were reviewed and approved by Wayne State University and John D. Dingell VA Medical Center Institutional Animal Care and Use Committees. Studies involving human islets were approved by the Biosafety Committee at the John D. Dingell VA Medical Center.
CRediT authorship contribution statement
Mirabela Hali: participated in data curation, Formal analysis, Investigation, Methodology, Validation, Visualization. Brian E. Wadzinski: participated in methodology, Writing – review & editing. Anjaneyulu Kowluru: participated in conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
Data will be made available on request.
References
- Allagnat F, Klee P, Cardozo AK, Meda P, Haefliger JA, 2013. Connexin36 contributes to INS-1E cells survival through modulation of cytokine-induced oxidative stress, ER stress and AMPK activity. Cell Death Differ. 20, 1742–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora DK, Machhadieh B, Matti A, Wadzinski BE, Ramanadham S, Kowluru A, 2014. High glucose exposure promotes activation of protein phosphatase 2A in rodent islets and INS-1 832/13 beta-cells by increasing the posttranslational carboxylmethylation of its catalytic subunit. Endocrinology 155, 380–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baidwan S, Chekuri A, Hynds DL, Kowluru A, 2017. Glucotoxicity promotes aberrant activation and mislocalization of Ras-related C3 botulinum toxin substrate 1 [Rac1] and metabolic dysfunction in pancreatic islet β-cells: reversal of such metabolic defects by metformin. Apoptosis 22, 1380–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninger RK, Head WS, Zhang M, Satin LS, Piston DW, 2011. Gap junctions and other mechanisms of cell-cell communication regulate basal insulin secretion in the pancreatic islet. J. Physiol 589, 5453–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco D, Haefliger JA, Meda P, 2011. Connexins: key mediators of endocrine function. Physiol. Rev 91, 1393–1445. [DOI] [PubMed] [Google Scholar]
- Chaurasia B, Summers SA, 2021. Ceramides in metabolism: key lipotoxic players. Annu. Rev. Physiol 83, 303–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Peterson RT, Schreiber SL, 1998. Alpha 4 associates with protein phosphatases 2A, 4, and 6. Biochem. Biophys. Res. Commun 247, 827–832. [DOI] [PubMed] [Google Scholar]
- Chen LP, Lai YD, Li DC, Zhu XN, Yang P, Li WX, Zhu W, Zhao J, Li XD, Xiao YM, Zhang Y, Xing XM, Wang Q, Zhang B, Lin YC, Zeng JL, Zhang SX, Liu CX, Li ZF, Zeng XW, Lin ZN, Zhuang ZX, Chen W, 2011. α4 is highly expressed in carcinogen-transformed human cells and primary human cancers. Oncogene 30, 2943–2953. [DOI] [PubMed] [Google Scholar]
- Cheng G, Li L, 2020. High-glucose-induced apoptosis, ROS production and pro-inflammatory response in cardiomyocytes is attenuated by metformin treatment via PP2A activation. J. Biosci 45. [PubMed] [Google Scholar]
- Chundru SA, Harajli A, Hali M, Gleason N, Gamage S, Kowluru A, 2021. RhoG-Rac1 signaling pathway mediates metabolic dysfunction of the pancreatic beta-cells under chronic hyperglycemic conditions. Cell. Physiol. Biochem 55, 180–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HK, Wang SR, Xiao L, Rathor N, Turner DJ, Yang P, Gorospe M, Rao JN, Wang JY, 2018. α4 coordinates small intestinal epithelium homeostasis by regulating stability of HuR. Mol. Cell Biol 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigliola V, Chellakudam V, Arabieter W, Meda P, 2013. Connexins and β-cell functions. Diabetes Res. Clin. Pract 99, 250–259. [DOI] [PubMed] [Google Scholar]
- Eleftheriadou O, Boguslavskyi A, Longman MR, Cowan J, Francois A, Heads RJ, Wadzinski BE, Ryan A, Shattock MJ, Snabaitis AK, 2017. Expression and regulation of type 2A protein phosphatases and alpha4 signalling in cardiac health and hypertrophy. Basic Res. Cardiol 112, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth NL, Benninger RK, 2014. New insights into the role of connexins in pancreatic islet function and diabetes. FEBS Lett. 588, 1278–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielhaber JA, Han YS, Tan J, Xing S, Biggs CM, Joung KB, Kristof AS, 2009. Inactivation of mammalian target of rapamycin increases STAT1 nuclear content and transcriptional activity in alpha4- and protein phosphatase 2A-dependent fashion. J. Biol. Chem 284, 24341–24353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamage S, Hali M, Chen F, Kowluru A, 2022. CARD9 mediates pancreatic islet beta-cell dysfunction under the duress of hyperglycemic stress. Cell. Physiol. Biochem 56, 120–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haefliger JA, Rohner-Jeanrenaud F, Caille D, Charollais A, Meda P, Allagnat F, 2013. Hyperglycemia downregulates Connexin36 in pancreatic islets via the upregulation of ICER-1/ICER-1γ. J. Mol. Endocrinol 51, 49–58. [DOI] [PubMed] [Google Scholar]
- Islam MS, 2020. Stimulus-secretion coupling in beta-cells: from basic to bedside. Adv. Exp. Med. Biol 1131, 943–963. [DOI] [PubMed] [Google Scholar]
- Jain R, Lammert E, 2009. Cell-cell interactions in the endocrine pancreas. Diabetes Obes. Metabol 11 (Suppl. 4), 159–167. [DOI] [PubMed] [Google Scholar]
- Janssens V, Goris J, 2001. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J 353, 417–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Li D, Liang Z, Wang Y, Pei X, Tang J, 2020. High expression of IGBP1 correlates with poor prognosis in esophageal squamous cell carcinoma. Int. J. Biol. Markers 35, 33–40. [DOI] [PubMed] [Google Scholar]
- Jones PM, Persaud SJ, 1998. Protein kinases, protein phosphorylation, and the regulation of insulin secretion from pancreatic beta-cells. Endocr. Rev 19, 429–461. [DOI] [PubMed] [Google Scholar]
- Khadija S, Veluthakal R, Sidarala V, Kowluru A, 2014. Glucotoxic and diabetic conditions induce caspase 6-mediated degradation of nuclear lamin A in human islets, rodent islets and INS-1 832/13 cells. Apoptosis 19, 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloeker S, Reed R, Mcconnell JL, Chang D, Tran K, Westphal RS, Law BK, Colbran RJ, Kamoun M, Campbell KS, Wadzinski BE, 2003. Parallel purification of three catalytic subunits of the protein serine/threonine phosphatase 2A family (PP2A(C), PP4(C), and PP6(C)) and analysis of the interaction of PP2A(C) with alpha4 protein. Protein Expr. Purif 31, 19–33. [DOI] [PubMed] [Google Scholar]
- Kong M, Bui TV, Ditsworth D, Gruber JJ, Goncharov D, Krymskaya VP, Lindsten T, Thompson CB, 2007. The PP2A-associated protein alpha4 plays a critical role in the regulation of cell spreading and migration. J. Biol. Chem 282, 29712–29720. [DOI] [PubMed] [Google Scholar]
- Kong M, Ditsworth D, Lindsten T, Thompson CB, 2009. Alpha4 is an essential regulator of PP2A phosphatase activity. Mol. Cell 36, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru A, 2005. Novel regulatory roles for protein phosphatase-2A in the islet beta cell. Biochem. Pharmacol 69, 1681–1691. [DOI] [PubMed] [Google Scholar]
- Kowluru A, 2010. Small G proteins in islet beta-cell function. Endocr. Rev 31, 52–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru A, 2011. Friendly, and not so friendly, roles of Rac1 in islet β-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem. Pharmacol 81, 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru A, 2018. Novel roles of alpha-4, non-canonical scaffolding subunit of protein phosphatase 2A, in the onset of beta cell dysfunction under glucotoxic conditions. Diabetologia 61, S1–S620. [Google Scholar]
- Kowluru A, 2020a. GPCRs, G proteins, and their impact on β-cell function. Compr. Physiol 10, 453–490. [DOI] [PubMed] [Google Scholar]
- Kowluru A, 2020b. Potential roles of PP2A-Rac1 signaling axis in pancreatic β-cell dysfunction under metabolic stress: progress and promise. Biochem. Pharmacol 180, 114138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru A, Chen HQ, Modrick LM, Stefanelli C, 2001. Activation of acetyl-CoA carboxylase by a glutamate- and magnesium-sensitive protein phosphatase in the islet beta-cell. Diabetes 50, 1580–1587. [DOI] [PubMed] [Google Scholar]
- Kowluru A, Kowluru RA, 2014. Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem. Pharmacol 88, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru A, Kowluru RA, 2018. RACking up ceramide-induced islet beta-cell dysfunction. Biochem. Pharmacol 154, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru A, Matti A, 2012. Hyperactivation of protein phosphatase 2A in models of glucolipotoxicity and diabetes: potential mechanisms and functional consequences. Biochem. Pharmacol 84, 591–597. [DOI] [PubMed] [Google Scholar]
- Kowluru A, Metz SA, 1997. Ceramide-activated protein phosphatase-2A activity in insulin-secreting cells. FEBS Lett. 418, 179–182. [DOI] [PubMed] [Google Scholar]
- Kowluru A, Seavey SE, Li G, Sorenson RL, Weinhaus AJ, Nesher R, Rabaglia ME, Vadakekalam J, Metz SA, 1996. Glucose- and GTP-dependent stimulation of the carboxyl methylation of CDC42 in rodent and human pancreatic islets and pure beta cells. Evidence for an essential role of GTP-binding proteins in nutrient-induced insulin secretion. J. Clin. Invest 98, 540–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru A, Veluthakal R, 2005. Rho guanosine diphosphate-dissociation inhibitor plays a negative modulatory role in glucose-stimulated insulin secretion. Diabetes 54, 3523–3529. [DOI] [PubMed] [Google Scholar]
- Krauss S, Foerster J, Schneider R, Schweiger S, 2008. Protein phosphatase 2A and rapamycin regulate the nuclear localization and activity of the transcription factor GLI3. Cancer Res. 68, 4658–4665. [DOI] [PubMed] [Google Scholar]
- Lenoue-Newton ML, Wadzinski BE, Spiller BW, 2016. The three Type 2A protein phosphatases, PP2Ac, PP4c and PP6c, are differentially regulated by Alpha4. Biochem. Biophys. Res. Commun 475, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald MJ, 1990. Elusive proximal signals of beta-cells for insulin secretion. Diabetes 39, 1461–1466. [DOI] [PubMed] [Google Scholar]
- Mcconnell JL, Watkins GR, Soss SE, Franz HS, Mccorvey LR, Spiller BW, Chazin WJ, Wadzinski BE, 2010. Alpha4 is a ubiquitin-binding protein that regulates protein serine/threonine phosphatase 2A ubiquitination. Biochemistry 49, 1713–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meda P, 2018. Gap junction proteins are key drivers of endocrine function. Biochim. Biophys. Acta Biomembr 1860, 124–140. [DOI] [PubMed] [Google Scholar]
- Nesher R, Anteby E, Yedovizky M, Warwar N, Kaiser N, Cerasi E, 2002. Beta-cell protein kinases and the dynamics of the insulin response to glucose. Diabetes 51 (Suppl. 1), S68–S73. [DOI] [PubMed] [Google Scholar]
- Newgard CB, Mcgarry JD, 1995. Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu. Rev. Biochem 64, 689–719. [DOI] [PubMed] [Google Scholar]
- Newsholme P, Morgan D, Rebelato E, Oliveira-Emilio HC, Procopio J, Curi R, Carpinelli A, 2009. Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia 52, 2489–2498. [DOI] [PubMed] [Google Scholar]
- Ortsäter H, Grankvist N, Honkanen RE, Sjöholm Å, 2014. Protein phosphatases in pancreatic islets. J. Endocrinol 221, R121–R144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Armendariz EM, 2013. Connexin 36, a key element in pancreatic beta cell function. Neuropharmacology 75, 557–566. [DOI] [PubMed] [Google Scholar]
- Prentki M, Matschinsky FM, Madiraju SR, 2013. Metabolic signaling in fuel-induced insulin secretion. Cell Metabol. 18, 162–185. [DOI] [PubMed] [Google Scholar]
- Roszczyc-Owsiejczuk K, Zabielski P, 2021. Sphingolipids as a culprit of mitochondrial dysfunction in insulin resistance and type 2 diabetes. Front. Endocrinol 12, 635175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakashita S, Li D, Nashima N, Minami Y, Furuya S, Morishita Y, Tachibana K, Sato Y, Noguchi M, 2011. Overexpression of immunoglobulin (CD79a) binding protein1 (IGBP-1) in small lung adenocarcinomas and its clinicopathological significance. Pathol. Int 61, 130–137. [DOI] [PubMed] [Google Scholar]
- Santo-Domingo J, Galindo AN, Cominetti O, De Marchi U, Cutillas P, Dayon L, Wiederkehr A, 2019. Glucose-dependent phosphorylation signaling pathways and crosstalk to mitochondrial respiration in insulin secreting cells. Cell Commun. Signal 17, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sents W, Ivanova E, Lambrecht C, Haesen D, Janssens V, 2013. The biogenesis of active protein phosphatase 2A holoenzymes: a tightly regulated process creating phosphatase specificity. FEBS J. 280, 644–661. [DOI] [PubMed] [Google Scholar]
- Serre-Beinier V, Le Gurun S, Belluardo N, Trovato-Salinaro A, Charollais A, Haefliger JA, Condorelli DF, Meda P, 2000. Cx36 preferentially connects beta-cells within pancreatic islets. Diabetes 49, 727–734. [DOI] [PubMed] [Google Scholar]
- Sidarala V, Kowluru A, 2017. The regulatory roles of mitogen-activated protein kinase (MAPK) pathways in health and diabetes: lessons learned from the pancreatic beta-cell. Recent Pat. Endocr. Metab. Immune Drug Discov 10, 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squecco R, Pierucci F, Idrizaj E, Frati A, Lenci E, Vicenti C, Iachini MC, Martinesi M, Garella R, Baccari MC, Francini F, Meacci E, 2021. Ceramide/protein phosphatase 2A axis is engaged in gap junction impairment elicited by PCB153 in liver stem-like progenitor cells. Mol. Cell. Biochem 476, 3111–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed I, Jayaram B, Subasinghe W, Kowluru A, 2010. Tiam1/Rac1 signaling pathway mediates palmitate-induced, ceramide-sensitive generation of superoxides and lipid peroxides and the loss of mitochondrial membrane potential in pancreatic beta-cells. Biochem. Pharmacol 80, 874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed I, Kyathanahalli CN, Jayaram B, Govind S, Rhodes CJ, Kowluru RA, Kowluru A, 2011. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes 60, 2843–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed I, Szulc ZM, Ogretmen B, Kowluru A, 2012. L- threo -C6-pyridinium-ceramide bromide, a novel cationic ceramide, induces NADPH oxidase activation, mitochondrial dysfunction and loss in cell viability in INS 832/13 beta-cells. Cell. Physiol. Biochem 30, 1051–1058. [DOI] [PubMed] [Google Scholar]
- Tengholm A, Gylfe E, 2017. cAMP signalling in insulin and glucagon secretion. Diabetes Obes. Metabol 19 (Suppl. 1), 42–53. [DOI] [PubMed] [Google Scholar]
- Thamilselvan V, Gamage S, Harajli A, Chundru SA, Kowluru A, 2020. P-Rex1 mediates glucose-stimulated Rac1 activation and insulin secretion in pancreatic β-cells. Cell. Physiol. Biochem 54, 1218–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thamilselvan V, Kowluru A, 2019. Paradoxical regulation of glucose-induced Rac1 activation and insulin secretion by RhoGDIbeta in pancreatic beta-cells. Small GTPases 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson B, Satin LS, 2021. Beta-cell ion channels and their role in regulating insulin secretion. Compr. Physiol 11, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umrani MR, Joglekar MV, Somerville Glover E, Wong W, Hardikar AA, 2017. Connexins and microRNAs: interlinked players in regulating islet function? Islets 9, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veluthakal R, Arora DK, Goalstone ML, Kowluru RA, Kowluru A, 2016. Metabolic stress induces caspase-3 mediated degradation and inactivation of farnesyl and geranylgeranyl transferase activities in pancreatic β-cells. Cell. Physiol. Biochem 39, 2110–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veluthakal R, Palanivel R, Zhao Y, Mcdonald P, Gruber S, Kowluru A, 2005. Ceramide induces mitochondrial abnormalities in insulin-secreting INS-1 cells: potential mechanisms underlying ceramide-mediated metabolic dysfunction of the beta cell. Apoptosis 10, 841–850. [DOI] [PubMed] [Google Scholar]
- Veluthakal R, Tunduguru R, Arora DK, Sidarala V, Syeda K, Vlaar CP, Thurmond DC, Kowluru A, 2015. VAV2, a guanine nucleotide exchange factor for Rac1, regulates glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia 58, 2573–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veluthakal R, Wadzinski BE, Kowluru A, 2006. Localization of a nuclear serine/threonine protein phosphatase in insulin-secreting INS-1 cells: potential regulation by IL-1beta. Apoptosis 11, 1401–1411. [DOI] [PubMed] [Google Scholar]
- Vozzi C, Ullrich S, Charollais A, Philippe J, Orci L, Meda P, 1995. Adequate connexin-mediated coupling is required for proper insulin production. J. Cell Biol 131, 1561–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Damacharla D, Ma D, Qi Y, Tagett R, Draghici S, Kowluru A, Yi Z, 2016. Quantitative proteomics reveals novel protein interaction partners of PP2A catalytic subunit in pancreatic beta-cells. Mol. Cell. Endocrinol 424, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolnierowicz S, 2000. Type 2A protein phosphatase, the complex regulator of numerous signaling pathways. Biochem. Pharmacol 60, 1225–1235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.