

Abstract
Drug discovery and development for stroke is challenging as evidenced by few drugs that have advanced beyond a Phase III clinical trial. Memantine is a N-methyl-D-aspartate (NMDA) receptor antagonist that has been shown to be neuroprotective in various preclinical studies. We have identified an endogenous BBB uptake transport system for memantine: organic cation transporters 1 and 2 (Oct1/Oct2). Our goal was to evaluate Oct1/Oct2 as a required BBB mechanism for memantine neuroprotective effects. Male Sprague-Dawley rats (200-250 g) were subjected to middle cerebral artery occlusion (MCAO) for 90 min followed by reperfusion. Memantine (5 mg/kg, i.v.) was administered 2 h following intraluminal suture removal. Specificity of Oct-mediated transport was evaluated using cimetidine (15 mg/kg, i.v.), a competitive Oct1/Oct2 inhibitor. At 2 h post-MCAO, [3H]memantine uptake was increased in ischemic brain tissue. Cimetidine inhibited blood-to-brain uptake of [3H]memantine, which confirmed involvement of an Oct-mediated transport mechanism. Memantine reduced post-MCAO infarction and brain edema progression as well as improved neurological outcomes during post-stroke recovery. All positive effects of memantine were attenuated by co-administration of cimetidine, which demonstrates that Oct1/Oct2 transport is required for memantine to exert neuroprotective effects in ischemic stroke. Furthermore, Oct1/Oct2-mediated transport was shown to be the dominant mechanism for memantine brain uptake in the MCAO model despite a concurrent increase in paracellular “leak.” These novel and translational findings provide mechanistic evidence on the critical role of BBB transporters in CNS delivery of stroke therapeutics, information that can help such drugs advance in clinical trials.
Keywords: Blood-Brain Barrier, Drug Delivery, Endothelial Cell, Ischemic Stroke, Memantine, Organic Cation Transporters
Introduction
One of the primary objectives of preclinical stroke research is to discover and develop novel therapeutic agents. Such endeavors have proven to be extremely challenging as evidenced by the fact that no drugs have advanced beyond a Phase III clinical trial despite encouraging results from in vivo studies on neuroprotection and/or neural repair (1, 2). Several reasons have been proposed for the poor translation from preclinical studies to clinical trials including i) lack of detailed behavioral assessment of functional neurocognitive outcomes; ii) efficacy in preclinical models only achieved using doses higher than those that can be safely administered to patients; and iii) initial in vivo evaluations are routinely conducted in young male animals but subsequent follow-up studies do not often consider comorbid conditions (i.e., diabetes mellitus, obesity, tobacco smoking, hypertension, atrial fibrillation) or biological variables such as age and sex (1–4). Additionally, it is particularly striking that studies examining neuroprotection and/or neural repair in experimental models of ischemic stroke have not evaluated specific mechanisms that can be targeted for CNS drug delivery (i.e., endogenous blood-brain barrier (BBB) transporters). Our laboratory is currently addressing this knowledge gap through our work on endogenous solute carrier (SLC) transporters at the BBB. Much of our recent work has focused on organic anion transporting polypeptides (OATPs in humans; Oatps in rodents). Included amongst OATP/Oatp transport substrates are the 3-hydroxyl-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (i.e., statins), drugs that are effective in stroke patients (5–8). Results from our preclinical in vivo studies have shown that blood-to-brain transport of currently marketed statins (i.e., atorvastatin, pravastatin) requires functional expression of Oatp1a4 (9–11). More recently, we performed detailed transport studies in human endothelial cells and reported that statin transport properties of OATP1A2 (i.e., the human orthologue of Oatp1a4) are consistent with those of Oatp1a4 (12). Taken together, these data imply that targeting endogenous BBB transporters represents an approach that can be developed for CNS drug delivery in treating ischemic stroke.
While our observations on BBB transport of statins are highly compelling, it is important to consider that not all therapeutics can access the brain via OATP/Oatp-mediated transport. For example, memantine is a small molecule antagonist of N-methyl-D-aspartate (NMDA) receptors, which enables it to protect against excitotoxicity resulting from cerebral ischemia (13–16). Physiochemical properties of memantine such as molecular weight (179 Da), lipophilicity (cLogP =2.07) and a single hydrogen bond acceptor on its structure suggests that this compound can successfully permeate the BBB (17); however, at physiological pH, the majority of memantine molecules are protonated and, therefore, possess a positive charge as demarcated by a highly basic pKa of 10.7 (17). This fact suggests that memantine requires a specific transport mechanism for cationic solutes to cross biological membranes, including the brain microvascular endothelium. At present, transport properties for memantine at the BBB have not been fully elucidated; however, it has been reported to be a substrate for cationic solute transporters in cerebral endothelial cells (18, 19). Such cation transporters include the organic cation transporters (OCTs in humans; Octs in rodents). OCTs/Octs, as well as novel organic cation transporters (OCTNs in humans; Octns in rodents), are two subfamilies of the solute carrier 22 (SLC22) family of proteins that have considerable potential to mediate brain uptake of positively charged, zwitterionic, and uncharged drugs (20). Studies in Swiss outbred mice showed that blood-to-brain transport of memantine could involve a selective proton-cation antiporter (18). Similarly, Higuchi and colleagues demonstrated that cellular memantine uptake in cultured human brain endothelial cells involved a proton-coupled organic cation transport system (19). Additionally, studies in Xenopus laevis oocytes showed that memantine is a transport substrate for the polyspecific and electrogenic transporter OCT2 (21). Using confocal microscopy, our laboratory has previously observed localization of Oct1 in rat brain microvessels (22). Western blot analyses on isolated luminal and abluminal membrane fractions of human and rat brain microvessel endothelial cells reported expression of OCT1/Oct1 and OCT2/Oct2 at the BBB (23, 24). Additionally, global proteomic analysis of cerebral microvasculature collected postmortem from healthy human subjects revealed measurable abundance of OCT1 protein (25). These data on localization and expression of OCTs/Octs in cerebral microvasculature piqued our interest and provided impetus for an evaluation of Oct1/Oct2-mediated transport of memantine in the setting of ischemic stroke.
In this study, we hypothesize that neuroprotective efficacy of memantine for ischemic stroke treatment requires functional expression of Oct1/Oct2 at the BBB. Here, we show for the first time that Oct-mediated transport enabled memantine to improve functional neurological outcomes during the acute and subacute phase of post-stroke recovery. We also report that Oct-mediated transport of memantine was absolutely required for this therapeutic compound to reduce cerebral infarction volume and decrease the brain edema ratio. These unique and translational observations were obtained via pharmacological inhibition of Oct-mediated transport with cimetidine, a strategy that attenuated all beneficial effects of memantine on neurological outcomes and infarction progression in our MCAO model. The transport effect of Oct1/Oct2 persisted despite MCAO-induced increases in BBB paracellular permeability (i.e., non-selective “leak”). It is well known that an ischemic insult results in BBB dysfunction and subsequent increases in paracellular permeability (26–29); however, our novel study demonstrated that specific BBB transport conferred by Oct1/Oct2 can dominate over non-selective brain uptake via opening of a paracellular route for passive diffusion. Overall, this study shows, for the first time, that membrane transport mediated by Oct transporters is the critical mechanism for memantine uptake into the brain and, subsequently, therapeutic efficacy in ischemic stroke.
Methods
Animals and Drug Treatments
Animal use was approved by the University of Arizona Institutional Animal Care and Use Committee (IACUC; Protocol #11-252) and experiments were designed in accordance with the Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines. Male Sprague-Dawley rats (200-250 g; 3-months-old; Envigo, Denver, CO) were used in all experiments that comprised this study. We exclusively used male rats to reduce variability of our results due to sex as a biological variable. All animals were housed under standard 12 h light/12 h dark conditions and received food and water ad libitum. Animals were randomly assigned into treatment groups and subjected to transient middle cerebral artery occlusion (MCAO), or Sham surgery as described below. For memantine treatment experiments, animals were injected intravenously (i.v.) with either memantine (5 mg/kg; 100 μl injection volume) in 65% ethanol/35% saline; Millipore-Sigma, St. Louis, MO) or vehicle (65% ethanol/35% saline; 100 μl injection volume) via the tail vein. The dose of memantine chosen for our studies is clinically relevant and based on CNS effects observed in previous in vivo studies in rats (30, 31) The vehicle dose of ethanol selected for our experiments has been demonstrated to not induce clinical impairment via reduced motor activity or to affect respiratory drive in vivo (https://echa.europa.eu/registration-dossier/-/registered-dossier/16105/7/3/1). Additionally, we did not observe any overt injection site injury in any of the experimental animals included in our study. The selection of i.v. dosing for our experiments is based on the pharmacokinetic principal that therapeutically effective plasma concentrations can be rapidly achieved via i.v. dosing, which will allow administration of memantine at early time points following onset of reperfusion. This is a critical translational consideration because it is well documented that many stroke patients suffer from dysphagia and are unable to swallow in the hours immediately following onset of stroke symptoms (32–35). Therefore, both memantine and vehicle were administered to experimental animals at 2 h following onset of reperfusion. Specificity of Oct-mediated transport was determined using cimetidine (15 mg/kg; 100 μl injection volume, i.v.; Millipore-Sigma), a compound that is well-known to block Oct1/Oct2-mediated transport of drugs (36–38). For these experiments, cimetidine was administered to experimental animals at the same time as memantine (i.e., 2 h following initiation of reperfusion). The concentration of cimetidine was selected to be sufficiently greater than published Ki values for OCT transporter inhibition (39), thereby enabling maximal blockade of Oct-mediated memantine uptake at the BBB.
Transient Middle Cerebral Artery Occlusion (MCAO) Model
MCAO surgery was performed in male Sprague-Dawley rats according to a previously published method (40, 41) with a few modifications. Surgeries were conducted using a Leica M80 surgical microscope (Leica Biosystems, Wetzlar, Germany). Rats were induced to a surgical plane of anesthesia using 4% isoflurane and maintained at 2.5-3% isoflurane anesthesia in O2 using a VetFlo™ vaporizer (Kent Scientific Corporation, Torrington, CT). Continuous blood flow was measured using a laser-doppler probe (Moor Instruments, Wilmington, DE). The probe was placed on the skull directly above the left middle cerebral artery (MCA) region (MCA: 2 mm posterior, 6 mm lateral to Bregma). Body temperature was maintained at 37°C using a thermoregulated surgical pad (Scintica Instrumentation, London, ON, Canada) and continuously monitored using a rectal probe thermometer. Temperature maintenance is a critical consideration in MCAO studies as reduced body temperature can confound experimental results obtained from this model (42). After shaving and aseptic preparation of the surgical site with betadine and 70% ethanol, bupivacaine (0.5% (w/v), 200 μl injection volume, s.c.; Millipore-Sigma) was administered for pain management. At this time, a ventral midline incision was made on the midline over the trachea. Careful blunt and sharp dissection was performed to expose the left common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) from the surrounding superficial fascia, digastric, sternohyoid, and sternomastoid muscles. After permanent ligation of the distal ECA and temporary ligation of the CCA, a small incision is made in the ECA and a silicone-coated intraluminal monofilament (0.33 mm tip diameter; Cat #503323PK10; Doccol Corporation, Sharon, MA) was inserted and advanced approximately 2 cm into the ICA to occlude blood flow to the middle cerebral artery (MCA). A decrease in blood flow of at least 75% from baseline was considered a successful occlusion via the laser-doppler probe.
After 90 min of occlusion, the intraluminal filament was carefully removed to allow for restoration of blood flow (i.e., reperfusion). This MCAO time point was selected to enable generation of a severe and reproducible ischemic injury in rat brain. A successful reperfusion was characterized by an increase of 70% or more of occluded blood flow. If this benchmark was not achieved, animals were excluded from further analysis. Sham control animals underwent the same surgical procedure as MCAO animals except for insertion of the intraluminal filament. Upon removal of the intraluminal filament, the incision site was closed with absorbable sutures and all animals received local injection of bupivacaine (0.5% (w/v), 200 μl injection volume, s.c.) for post-operative pain management. All animals were placed in a socially housed recovery cage (≤ 4 animals per cage) with clean bedding, medicated wet food (Nutra-Gel Diet™ (F5769-KIT); formulated with 2.15 mg carprofen; Bio-serv, Flemington, NJ) available ad libitum, and thermal support via a heating pad.
In situ brain perfusion
In situ brain perfusion was performed as described previously by our laboratory (10, 11). Following 90min MCAO/2 h reperfusion or time-matched Sham surgery, animals were anesthetized and heparinized (10,000 U/kg i.p.). The common carotid arteries were visualized using a Leica A60 microscope and cannulated with silicone tubing connected to a perfusion circuit. Since the in situ perfusion approach was performed on animals previously subjected to transient MCAO, both cannulas were inserted into the artery approximately 1.5 cm below the carotid bifurcation. This strategy ensured that the tip of the cannula would not contact the cauterized ECA stump resulting from the MCAO surgery. Visual inspection of the ECA stump ensured that no leak of perfusion buffer was present during the entire perfusion experiment. Perfusion pressure and flow rate were maintained at 95 to 105 mmHg and 3.1 ml/min, respectively. Both jugular veins were severed to allow for perfusate drainage. The perfusate was an erythrocyte-free modified mammalian Ringer’s solution consisting of 117 mM NaCl, 4.7 mM KCl, 0.8 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 10 mM d-glucose, 3.9% (w/v) dextran (molecular weight 60,000), and 1.0 g/liter bovine serum albumin (type IV), pH 7.4, warmed to 37°C and continuously oxygenated with 95% O2/5% CO2. Evan’s blue dye (55 mg/liter) was added to the perfusate to serve as a visual marker of BBB integrity. Using a slow-drive syringe pump (Harvard Apparatus Inc., Holliston, MA), [3H]memantine (0.5 μCi/ml; Moravek Biochemicals Inc, Brea, Ca) was added to the inflowing perfusion solution at a rate of 0.5 ml/min per cerebral hemisphere, which resulted in a total flow rate of 3.6 ml/min. For inhibition studies, animals were perfused with Ringer’s solution containing transport inhibitor (i.e., 25 μM cimetidine) for 10 min prior to perfusion with [3H]memantine. We have previously confirmed the stability of transport substrates and sucrose in both perfusion medium and in jugular vein venous outflow and have shown that they remain intact in our in situ perfusion experiments (9, 43, 44). Since the BBB is known to be compromised in the setting of experimental stroke, all perfusions conducted in tMCAO or Sham-operated animals involved co-perfusion with [14C]sucrose (0.5 μCi/ml; PerkinElmer Life Sciences, Boston, MA). Sucrose is a small molecular weight vascular marker that does not cross the intact BBB (45). Therefore, this approach enables us to account for changes in paracellular “leak”, which can affect drug uptake into the brain in the tMCAO model.
Immediately following perfusion for the desired time (i.e., 10 min), rats were decapitated, and brains were removed. The meninges and choroid plexus were excised, and cerebral hemispheres were sectioned and labeled as ipsilateral or contralateral. TS2 tissue solubilizer (1.0 ml; Research Products International, Mt. Prospect, IL) was added to the tissue samples, which were allowed to solubilize for 2 days at room temperature. To eliminate chemiluminescence, 100 μl of 30% glacial acetic acid was added, along with 2.0 ml of Optiphase SuperMix liquid scintillation cocktail (PerkinElmer Life and Analytical Sciences, Boston, MA). Radioactivity was measured using a model 1450 Liquid Scintillation and Luminescence Counter (PerkinElmer Life and Analytical Sciences). Results were reported as picomoles of radiolabeled transport substrate (i.e., memantine or sucrose) per milligram of brain tissue (RBr; pmol/mg brain tissue), which is equal to the total amount of radioisotope in the brain [RBrain; dpm/mg tissue] divided by the amount of radioisotope in the perfusate [RPerfusate; dpm/pmol]:
| (Equation 1) |
The brain vascular volume in rats has been previously shown to range between 6 and 9 μl/g brain tissue in perfusion studies utilizing a saline-based bicarbonate buffer (46). Since brain tissue was processed immediately after perfusion with radiolabeled substrate, all uptake values require correction for brain vascular volume (i.e., 8.0 μl/g brain tissue as calculated from data reported by Takasato and colleagues) from whole-brain uptake data obtained using radiolabeled memantine or sucrose.
Brain Microvessel Isolation
Microvessels were isolated from male Sprague-Dawley rat brain tissue using a protocol developed in our laboratory (47). All steps were performed on ice at 4°C to minimize degradation of proteins during tissue processing. Following euthanasia by decapitation, brains were isolated from the skull and meninges and choroid plexus were removed. Cerebral hemispheres were separated into ipsilateral and contralateral cortices and then homogenized at 3,700 x g in 5 ml brain microvessel buffer (300 mM mannitol, 5 mM EGTA, 12 mM Tris HCl, pH 7.4) containing 0.1% protease inhibitor cocktail (Sigma-Aldrich). At this time, 8 ml 26% (w/v) dextran (MW 75,000; Spectrum Chemical Manufacturing Corporation, Gardena, CA) solution was added to each homogenate sample. Samples were then thoroughly vortexed and centrifuged at 5,000 x g for 15 min at 4°C. The supernatant was then aspirated, and capillary pellets were resuspended in 5 ml of brain microvessel buffer. Dextran homogenization and centrifugation steps were repeated an additional three times to ensure appropriate quality of microvessels. Following completion of dextran homogenization and centrifugation, the supernatant was aspirated and the microvessel pellet was resuspended in 5 ml of brain microvessel buffer. At this time, samples consisting of enriched whole microvessels were subjected to total membrane isolation. For total membrane isolation, samples were homogenized at 3,700 x g, placed into ultracentrifuge tubes, and centrifuged at 150,000 x g for 60 min at 4°C. Pellets containing total cellular membranes were resuspended in 500 μl of storage buffer (50% brain microvessel isolation buffer; 50% diH2O, v/v) containing 0.1% protease inhibitor cocktail. Samples were stored at −80°C until further use. We have previously confirmed purity of our microvessel preparations by demonstrating enrichment in platelet endothelial cell adhesion molecule-1 (PECAM-1; also known as CD31) as compared to expression of astrocyte marker proteins (i.e., glial fibrillary acidic protein) or neuronal marker proteins (i.e., synaptophysin) (11, 47, 48).
Western Blotting
Western blotting was performed as previously described(10) with a few modifications. Total cellular membrane samples from isolated microvessels were quantified for total protein using Bradford reagent (Sigma-Aldrich) and heated at 95°C for 5 min under reducing conditions (i.e., 2.5% (v/v) 2-mercaptoethanol (Sigma-Aldrich) in 1X Laemmli sample buffer (Bio-Rad, Hercules, CA). Following SDS-PAGE and transfer, PVDF membranes were incubated overnight at 4°C with primary antibodies against Oct1 (anti-Oct1; 0.27 mg/ml at 1:750 dilution; Cat #24617-1-AP; ThermoFisher Scientific, Waltham, MA), or Oct2 (anti-Oct2; 4.0 mg/ml at 1:1000 dilution; Cat #PA5-112715; ThermoFisher Scientific). For western blot experiments on microvessel samples from control (i.e., untreated) rats, Oct1 and Oct2 bands were normalized to α-tubulin, which was detected using a primary antibody previously validated in our laboratory (anti-α tubulin; 1 mg/mL at 1:20,000 dilution; Cat #ab7291; Abcam, Cambridge, MA). Since ischemic conditions can cause fluctuations in loading control proteins commonly used for brain microvascular samples (49), all western blot data derived from microvessel samples from Sham and tMCAO animals were normalized to total protein using Ponceau S staining as recommended by Fosang and Colbran (50). Membranes were washed and incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:40,000 dilution; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) or anti-mouse IgG (1:50,000 dilution; Jackson ImmunoResearch) for 60 min at room temperature. Control western blot experiments to confirm antibody specificity were performed in the presence of secondary antibody only (i.e., absence of primary antibody). Protein bands were visualized using enhanced chemiluminescence (Super Signal West Pico, ThermoFisher Scientific, Waltham, MA). Bands were quantitated using ImageJ software (Wayne Rasband, Research Services Branch, National Institute of Mental Health, Bethesda, MD), normalized to tubulin or total protein, and reported as relative values.
TTC staining
To identify viable and nonviable brain tissue, TTC (2,3,5-triphenyltetrazolium chloride) staining was used in accordance with a previously published method (41, 51). TTC staining provides a reliable colorimetric redox indicator of healthy versus damaged (infarct core and penumbra) brain tissue that results from an ischemic insult. Specifically, tissue containing viable mitochondria is stained dark red while infarcted brain tissue remains unstained (i.e., white).
After 90 min MCAO followed by 70.5 h reperfusion, animals were anesthetized with ketamine/xylazine (K: 50 mg/kg; X: 10 mg/kg, i.p.) and decapitated using a guillotine. The brain was quickly harvested and sectioned using an ice-cold (4°C) brain matrix that enables preparation of 1 mm brain slices in the rostral-to-caudal direction. The slices were then incubated in TTC (2% (w/v) in phosphate buffered saline (PBS) at 37°C) for approximately 5 minutes. Slices were then removed from TTC stain, rinsed with PBS (pH 7.4), and imaged. Brain slice images were analyzed for infarction volume and cerebral edema using Image J software. As previously described(41), we measured three areas of each brain slice: infarct area (X) (mm2), area of the infarcted cerebral (i.e., ipsilateral) hemisphere (Y) (mm2), and the area of the non-infarcted (i.e., contralateral) hemisphere (Z) (mm2). Brain infarction areas were corrected for brain edema and percent (%) brain edema was calculated using:
| (Equation 2) |
Neurological Deficit Scoring
Stroke severity was assessed after 90 min MCAO/22.5 h reperfusion (24 h total time point) and 90 min MCAO/70.5 h reperfusion (72 h total time point) using neurological deficit scores (i.e., functional neuroscore analysis) modeled upon the modified Rankin Scale (mRS). This four-point scale awarded a single point for each one of three identifiable motor impairments: i) unilateral circling; ii) retraction of the right front paw; and iii) resistance to horizontal push on the laboratory bench. An animal with a score of “0” was deemed to be not impaired while an animal with a score of “3” was reported as severely impaired.
Rotarod Motor Performance Test
Functional motor performance was assessed using the Rotarod motor performance instrument (760772 Harvard Apparatus, Holliston, MA). Rats were placed on a constantly rotating drums at a speed of 8 rpm. Animals were trained on the Rotarod instrument at least 24 h prior to MCAO or Sham surgery. The time each rat maintains balance on the rod was recorded and defined as a “latency to fall (LTF)” score. An experimental cut off at 180 s was used for all rotarod experiments.
Randomization, Blinding, and Statistical Analysis
A pre-determined randomization schedule was generated based on treatment groups and the randomization schedule was followed with an intent to treat. Animals that did not complete the protocol because of mortality, early euthanasia, or technical problems were replaced before continuing the randomization sequence. The individual performing the experimental protocol was blinded to drug treatment and to treatment with vehicle. All data are reported as mean ± SD of six individual animals per treatment group (n = 6). Statistical significance was determined using one-way ANOVA followed by post hoc Dunnett’s Multiple Comparison test. A value of p < 0.05 was accepted as statistically significant.
Results
Organic Cation Transporters 1 and 2 are Functionally Expressed at the Blood-Brain Barrier in Male Sprague-Dawley Rats
Previous data has shown that Oct isoforms (i.e., Oct1 and Oct2) are expressed at the BBB (23, 24); however, expression of these cation transporters has not been confirmed in brain microvessels isolated from male Sprague-Dawley rats. We report protein expression of Oct1 and Oct2 in our microvessel samples by western blot analysis as indicated by single bands at approximately 67 kDa and 66 kDa, respectively (Figure 1A). To study Oct1/Oct2-mediated transport activity at the BBB, we measured brain uptake of [3H]memantine (Figure 1B), an established transport substrate for these cation transporters (52–54). Additionally, we purposely chose to analyze Oct1/Oct2 transport function using memantine due to clinical evidence that it may be effective as a neuroprotective agent in patients with mild-to-moderate ischemic stroke (55). In control (i.e., untreated) male Sprague-Dawley rats, [3H]memantine uptake was 84.59 ± 9.73 pmol/mg brain tissue (95% CI: 74.38, 94.80) (Figure 1C). In the presence of the established Oct1/Oct2 transport inhibitor cimetidine (25 μM), brain accumulation of [3H]memantine was decreased by 36% to 54.14 ± 8.35 pmol/mg brain tissue (95% CI: 45.38, 62.90). Taken together, these observations provide clear evidence for functional expression of Oct1/Oct2 at the BBB in male Sprague-Dawley rats.
Figure 1: Functional Expression of Oct1 and Oct2 at the BBB in Male Sprague-Dawley Rats.
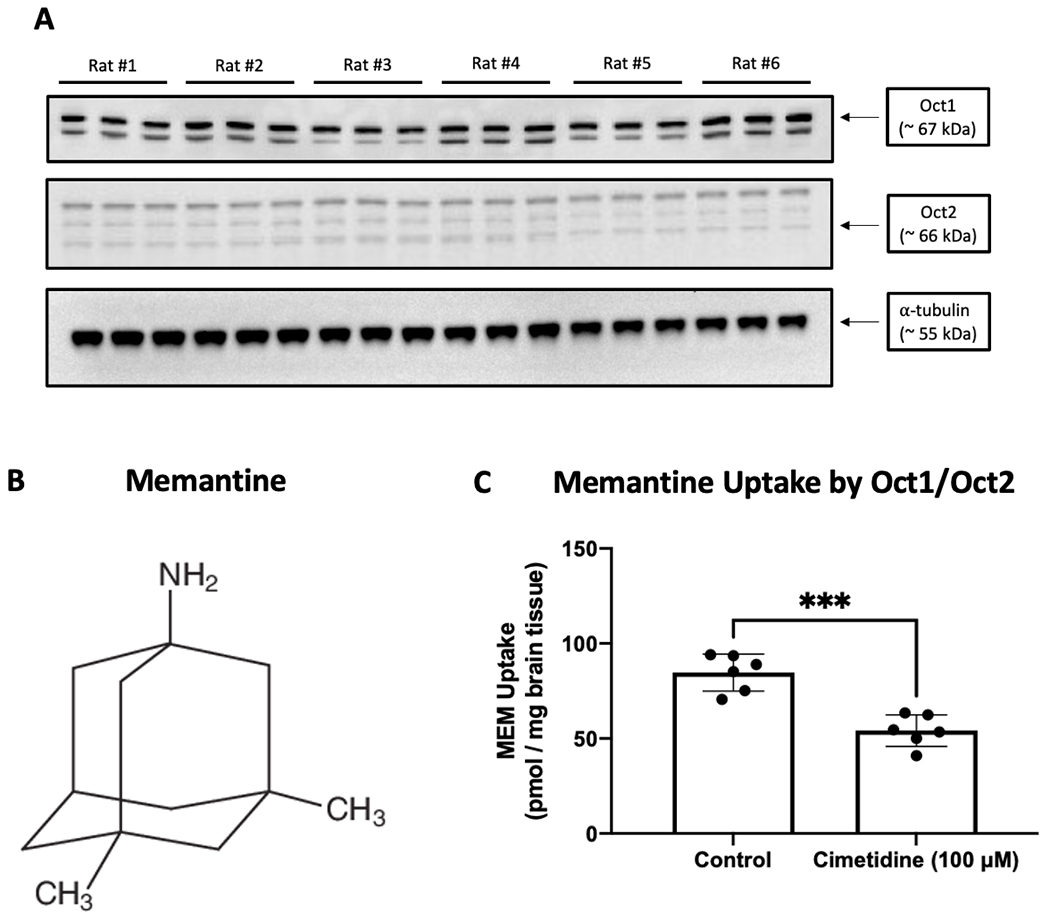
A: Oct1 and Oct2 protein expression was measured by western blot analysis of brain microvessels isolated from control (i.e., untreated) male Sprague-Dawley rats. Isolated microvessels (10 μg) were resolved on a 4-12% SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and analyzed for expression of Oct1, Oct2, or α-tubulin (i.e., the loading control). Each lane triplet on the depicted western blot corresponds to a microvessel sample obtained from a single experimental animal. The image depicts a representative blot showing transporter expression data from six experimental animals (n = 6). B: Chemical structure of memantine. C: Uptake of [3H]memantine (0.5 μCi/ml) in brain tissue isolated from control (i.e., untreated) male Sprague-Dawley rats. Inhibition experiments were conducted in the presence and absence of cimetidine (25 μM), a competitive Oct transport inhibitor, that was perfused for 10 min prior to administration of [3H]memantine. Results are expressed as mean ± SD of six animals per time point (n = 6). Asterisks represent statistical significance between animals perfused with [3H]memantine and animals perfused with [3H]memantine in the presence of cimetidine (*** p < 0.001).
Oct1 and Oct2 Protein Expression are Not Altered in Cortical Microvessels Following MCAO (90 min) and Reperfusion (2 h).
The primary objective of this study was to show that BBB transport mediated by Oct isoforms is a critical mechanism that enables memantine to confer neuroprotective effects in the setting of ischemic stroke. To accomplish this goal, we designed our experiments to inject memantine (5 mg/kg, i.v.) after 90 min MCAO followed by 2 h reperfusion. This time point was selected because it is within the therapeutic window for stroke fibrinolytic therapy where neuroprotection is necessary. Therefore, it is essential to evaluate protein expression of Oct1 and Oct2 in ischemic and non-ischemic brain tissue. To assess whether differences in transporter expression exist between microvessels isolated from ipsilateral and contralateral cerebral cortices, we used western blot analysis to measure Oct1 protein expression at the time of memantine administration in rats subjected to tMCAO and in Sham-operated rats (Figure 2A). Densitometric analysis of our western blot data showed no statistically significant change in Oct1 protein expression (normalized to total protein) in either ipsilateral or contralateral cortical microvessels from male Sprague-Dawley rats subjected to MCAO (90 min) followed by reperfusion (2 h) (Figure 2B). Similar to our Oct1 experiments, we used western blot analysis to measure Oct2 protein expression in rats subjected to tMCAO and in Sham-operated controls (Figure 3A). Densitometric analysis of our western blot data showed no statistically significant change in Oct2 protein expression (normalized to total protein) in either ipsilateral or contralateral cortical microvessels from male Sprague-Dawley rats subjected to MCAO (90 min) followed by reperfusion (2 h) (Figure 3B).
Figure 2: Protein Expression of Oct1 in Ipsilateral and Contralateral Cerebral Cortical Microvessels after MCAO.

A: Oct1 protein expression was measured by western blot analysis of brain microvessels isolated from ipsilateral and contralateral cerebral cortex following transient MCAO (90 min) with reperfusion (2 h). Isolated microvessels (10 μg) were resolved on a 4-12% SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and analyzed for expression of Oct1. Each lane doublet on the depicted western blot corresponds to a microvessel sample obtained from a single experimental animal. The image depicts a representative blot from six individual animals (n = 6). B: Relative levels of Oct1 protein expression in ipsilateral and contralateral microvessels were determined by densitometric analysis and normalized to total protein. Quantitative results are expressed as mean ± SD from six individual animals (n = 6).
Figure 3: Protein Expression of Oct2 in Ipsilateral and Contralateral Cerebral Cortical Microvessels after MCAO.
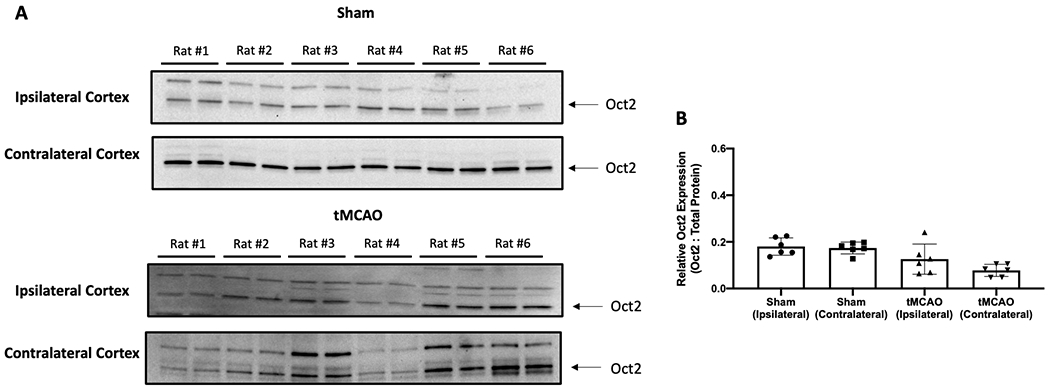
A: Oct2 protein expression was measured by western blot analysis of brain microvessels isolated from ipsilateral and contralateral cerebral cortex following transient MCAO (90 min) with reperfusion (2 h). Isolated microvessels (10 μg) were resolved on a 4-12% SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and analyzed for expression of Oct2. Each lane doublet on the depicted western blot corresponds to a microvessel sample obtained from a single experimental animal. The image depicts a representative blot from six individual animals (n = 6). B: Relative levels of Oct2 protein expression in ipsilateral and contralateral microvessels were determined by densitometric analysis and normalized to total protein. Quantitative results are expressed as mean ± SD from six individual animals (n = 6).
Blood-to-Brain Delivery of Memantine to Ischemic Brain Tissue Requires Transport Mediated by Oct1 and Oct2 at the BBB
To study Oct-mediated transport at the BBB after 90 min MCAO followed by 2 h reperfusion, brain uptake of [3H]memantine was studied in Sham-operated and MCAO rats in the presence and absence of cimetidine, a competitive Oct1/Oct2 transport inhibitor. Our study represents the first application of the in situ brain perfusion approach for the study of endogenous BBB uptake transporters in the MCAO model. Accumulation of [3H]memantine following 90 min MCAO/2 h reperfusion was 116.57 ± 11.54 pmol/mg brain tissue (95% CI: 104.47, 128.68), a value that was significantly increased (p < 0.0001) as compared to Sham-operated controls (85.34 ± 10.90 pmol/mg brain tissue; 95% CI: 73.90, 96.77; Figure 4A). In the presence of cimetidine (25 μM), brain uptake of [3H]memantine was decreased to 68.78 ± 5.72 pmol/mg brain tissue (95% CI: 62.77, 74.79) in tMCAO animals and to 49.29 ± 6.69 pmol/mg brain tissue (95% CI: 42.26, 56.31) in Sham-operated controls, thereby confirming involvement of Oct1/Oct2 in brain uptake of memantine. Since these data reflected total uptake of [3H]memantine into whole brain tissue, we performed additional analyses to determine drug uptake into ipsilateral and contralateral cerebral cortices, respectively. As shown in Figure 4B, [3H]memantine accumulation was increased (p < 0.0001) into ipsilateral cortical tissue, but not into contralateral cerebral cortices, after 90 min MCAO/2 h reperfusion as compared to Sham-operated controls. Within the cohort of animals subjected to tMCAO, [3H]memantine accumulation was also observed to be enhanced (p = 0.0268) in ipsilateral cortex (125.78 ± 12.88 pmol/mg brain tissue; 95% CI: 112.27, 139.30) as compared to contralateral cortex (107.36 ± 10.54 pmol/mg brain tissue; 95% CI: 96.31, 118.42). As expected, there was no difference in ipsilateral (85.15 ± 11.39 pmol/mg brain tissue; 95% CI: 73.19, 97.10) versus contralateral (85.53 ± 10.67 pmol/mg brain tissue; 95% CI: 74,33, 96.72) cortical [3H]memantine uptake in Sham-operated control rats. Under both experimental conditions, blood-to-brain transport of [3H]memantine was significantly reduced (p < 0.0001) in the presence of cimetidine (25 μM), which implies that memantine requires a selective transport process (i.e., Oct-mediated transport) to traverse the BBB and accumulate in ischemic brain tissue (Figure 4B).
Figure 4: CNS Delivery of Memantine by Oct-Mediated Transport and Paracellular Sucrose “Leak” in Male Sprague-Dawley Rats Subjected to MCAO.

A: Uptake of [3H]memantine (0.5 μCi/ml) in whole brain tissue from male Sprague-Dawley rats subjected to transient MCAO (90 min) with reperfusion (2 h) and Sham-operated controls. Inhibition experiments were conducted in the presence and absence of cimetidine (25 μM), a competitive Oct transport inhibitor, that was perfused for 10 min prior to administration of [3H]memantine. B: Uptake of [3H]memantine (0.5 μCi/ml) in ipsilateral and contralateral cortical brain tissue from experimental animals subjected to transient MCAO (90 min) with reperfusion (2 h) and Sham-operated controls. Inhibition experiments were conducted in the presence and absence of cimetidine (25 μM). C: Uptake of [3H]sucrose (0.5 μCi/ml) in whole brain tissue from male Sprague-Dawley rats subjected to transient MCAO (90 min) with reperfusion (2 h) and Sham-operated controls. D: Uptake of [3H]sucrose (0.3 μCi/ml) in ipsilateral and contralateral cortical brain tissue from experimental animals subjected to transient MCAO (90 min) with reperfusion (2 h) and Sham-operated controls. Results are expressed as mean ± SD of six animals per time point (n = 6). Asterisks represent data points that were significantly different from control animals (* p < 0.05; ** p < 0.01; **** p < 0.0001; ns = not significant).
Paracellular Permeability to Sucrose is Increased in Male Sprague-Dawley Rats Subjected to MCAO
MCAO/reperfusion conditions are well known to cause BBB injury (56–58). Specifically, modulation of tight junction protein complexes in the setting of focal cerebral ischemia can cause increased paracellular diffusion (i.e., “leak”) of small molecules. Relevant to our study, this process must be considered as a contributor to non-selective permeation of memantine across the BBB via the paracellular route. To address this issue, we co-perfused [14C]sucrose with [3H]memantine to enable a quantitative assessment of paracellular BBB permeability (i.e., “leak”) in animals subjected to tMCAO and in Sham-operated controls. In whole brain cortical tissue, [14C]sucrose uptake was significantly increased (p < 0.0001) in animals subjected to 90 min MCAO/2 h reperfusion as compared to Sham-operated controls (Figure 4C). When we compared permeability between cerebral cortices, we showed that brain [14C]sucrose accumulation was significantly increased (p < 0.0001) in ipsilateral cortices versus contralateral cortical tissue collected from animals subjected to MCAO (Figure 4D). There was no statistically significant change in [14C]sucrose brain uptake between ipsilateral and contralateral cortices isolated from Sham-operated controls or between contralateral cortical tissue from animals subjected to MCAO and Sham-operated controls. Despite our observation of enhanced paracellular “leak” in male Sprague-Dawley rats after 90 min MCAO/2 h reperfusion, the decrease in [3H]memantine uptake in the presence of cimetidine reveals, for the first time, that Oct-mediated transport is required for this neuroprotective drug to permeate the BBB in the setting of ischemic stroke. Hence, selective transport mechanisms must be considered as critical determinants of drug delivery to the brain in ischemic stroke.
Memantine Requires Oct-Mediated Transport to Reduce Neurological Dysfunction and Motor Impairment in Male Sprague-Dawley Rats Subjected to MCAO.
To evaluate neurological outcomes, we measured neurological deficit scores and motor performance via the rotarod test in male MCAO rats administered memantine or memantine/cimetidine. For these experiments, memantine or memantine/cimetidine were injected 2 h following onset of reperfusion. Behavioral readouts were measured both at an acute time point (i.e., 24 h post-stroke) and at a subacute time point (i.e., 72 h post-stroke). Under our MCAO conditions, neurological scores at 24 h post-MCAO (Figure 5A) and 72 h post-MCAO (Figure 5B) were significantly increased (p < 0.0001) as compared to Sham-operated controls. When memantine was injected (5 mg/kg, i.v.), an improvement in neurological scoring was only detected at 72 h post-stroke. While memantine/cimetidine had no effect on neurological scoring at 24 h post-stroke, a clear significant effect was apparent at 72 h post-stroke. Beneficial effects of memantine were more apparent when motor performance was measured via the rotarod test (Figure 6A-B). Following i.v. injection of memantine (5 mg/kg), rotarod performance was significantly enhanced at 24 h (p = 0.0002) and at 72 h (p = 0.0088); however, cimetidine (15 mg/kg, i.v.) attenuated this improvement in motor function at both time points. Taken together, these data provide the first evidence that memantine requires selective BBB transport by Oct1/Oct2 to improve neurological scoring and motor performance during recovery from ischemic stroke.
Figure 5: Memantine Improves Neurological Scoring via Oct1/Oct2 in Male Sprague-Dawley Rats Subjected to MCAO.

Neurological deficit score assessment in male Sprague-Dawley rats at 24 h post-MCAO (A) or 72 h post-MCAO (B). MCAO animals were administered vehicle, memantine (MEM, 5 mg/kg, i.v.) or memantine/cimetidine (MEM, 5 mg/kg, i.v.; cimetidine, 15 mg/kg, i.v.). Sham-operated animals were used as experimental controls. Results are expressed as mean ± SD of six animals per treatment group (n = 6). Asterisks represent data points that were significantly different from control animals (* p < 0.05; **** p < 0.0001; ns = not significant).
Figure 6: Memantine Improves Motor Performance via Oct1/Oct2 in Male Sprague-Dawley Rats Subjected to MCAO.

Motor performance was assessed by rotarod performance testing in animals at 24 h post-MCAO (A) and 72 h post-MCAO (B). MCAO animals were administered vehicle, memantine (MEM, 5 mg/kg, i.v.) or memantine/cimetidine (MEM, 5 mg/kg, i.v.; cimetidine, 15 mg/kg, i.v.). Sham-operated animals were used as experimental controls. Results are expressed as mean ± SD of six animals per treatment group (n = 6). Asterisks represent data points that were significantly different from control animals (** p < 0.01; *** p < 0.001; **** p < 0.0001; ns = not significant).
Positive Effects of Memantine on Cerebral Infarction Volume and Edema Ratio in Male Sprague-Dawley Rats Subjected to MCAO is Dependent Upon Oct-Mediated Transport at the BBB
To further evaluate whether Oct-mediated transport of memantine is required for this compound to exert beneficial effects in ischemic stroke, we performed TTC-staining of brain tissue and measured brain infarction volumes and edema ratios following MCAO. At 24 h post-MCAO, memantine had no effect on infarction volumes or edema ratios (data not shown). In contrast, brain infarction volumes and cerebral edema ratios were both significantly reduced (p < 0.0001) at 72 h post-MCAO (Figure 7). Of translational importance, the beneficial effects of memantine in MCAO/reperfusion animals were attenuated in the presence of cimetidine. Taken together, these results are highly novel and translational and support our hypothesis that CNS delivery of memantine by Oct1/Oct2 (i.e., a selective BBB transport mechanism) is required for this neuroprotective drug to reduce expansion of infarction volume and decrease cerebral edema following an ischemic stroke.
Figure 7: Memantine Attenuates Cerebral Infarction Volume and Edema Ratio via Oct1/Oct2 in Male Sprague-Dawley Rats Subjected to MCAO.

A: Representative TTC staining of brain tissue collected from experimental animals subjected to transient MCAO (90 min) with reperfusion (70.5 h) (i.e., 72 h post-MCAO). Animals were administered vehicle, memantine (MEM, 5 mg/kg, i.v.) or memantine/cimetidine (MEM, 5 mg/kg, i.v.; cimetidine, 15 mg/kg, i.v.). Sham-operated animals were used as experimental controls. Arrows indicate regions of infarction in individual brain slides as detected by TTC staining. B: Measurement of cerebral infarction volume in TTC-stained brain tissue. C: Measurement of brain edema ratio in TTC-stained brain tissue. Results are expressed as mean ± SD of six animals per treatment group (n = 6). Asterisks represent data points that were significantly different from control animals (** p < 0.01; *** p < 0.001; **** p < 0.0001; ns = not significant).
Discussion
The pathophysiology of ischemic stroke is characterized by impaired blood supply that decreases delivery of oxygen and glucose to an affected brain region. Both oxygen and glucose are essential in production of ATP, the biological fuel that, in part, ensures maintenance of monovalent and divalent ion gradients (i.e., Na+, K+, Ca2+) (59, 60). Collapse of ion gradients in response to stroke pathophysiology can have profound consequences for brain function such as an increased intracellular Ca2+ (61, 62). Rapid elevation of intracellular Ca2+ in excitatory neurons triggers release of neurotransmitters such as glutamate and dopamine. High extracellular concentrations of glutamate and dopamine are neurotoxic and cause increased neuronal cell death and infarction progression (60, 63). Excitotoxicity induced by glutamate is particularly injurious to the brain due to overstimulation of metabotropic glutamate receptors as well as extensive activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptors (59, 64, 65). It is at this interface where memantine has been proposed as a therapeutic agent for ischemic stroke. While excessive activation of NMDA receptors is pathological, physiological activity of this receptor system is critical for neuronal function. As such, complete blockade of NMDA receptor can result in serious adverse effects including drowsiness, hallucinations, and potentially coma (66). For example, clinical development of the NMDA receptor antagonist aptiganel hydrochloride was halted due to safety concerns including dose-dependent CNS side effects such as confusion, somnolence and stupor as well as cerebral edema (67). Additionally, selfotel was shown in clinical trials to be associated with increased patient mortality during post-stroke recovery, which implied that NMDA receptor blockade could be associated with neurotoxicity (68). Memantine has several pharmacological advantages over other NMDA receptor antagonists such as fast on-off kinetics, low-to-moderate NMDA receptor affinity, and the ability to attenuate glutamate release without interfering with basal receptor activation (69). Indeed, several preclinical studies have reported positive effects of memantine on cerebral infarction expansion and functional neurocognitive outcomes. For example, Hau and colleagues showed that memantine (20 mg/kg) improved corner test performance in female CD-1 mice subjected to tMCAO as compared to untreated control animals (70). At the biochemical level, treatment with memantine (0.2 mg/kg/day; 3 day treatment course via osmotic minipump) reduced striatal and striato-cortical lesions following MCAO in male C57BL6 mice purchased from Charles River Laboratories (Oxford, UK) (71). Additionally, chronic high-dose memantine treatment (i.e., 20 mg/kg/day; treatment started at 72 h post-MCAO and given once daily to 28 days post-MCAO) improved post-MCAO motor performance and spatial memory in male C57BL6/J mice (72). Of additional importance, in vivo studies in a thrombotic model of ischemic stroke demonstrated that memantine reduced the negative effects of delayed thrombolysis on infarction volume and post-stroke neurocognitive performance (73). Clinically, memantine has been demonstrated to improve neurocognitive performance and be well-tolerated in a small cohort of stroke patients as measured by the National Institutes of Health Stroke Scale (NIHSS) and the Barthel Index (55). Taken together, these observations support the therapeutic potential of memantine in treatment of ischemic stroke.
Drugs for stroke treatment can only be effective if they are able to attain efficacious concentrations in the brain. This objective requires a rigorous understanding of transport mechanisms at the BBB that control movement of such therapeutics from blood into brain parenchyma. Currently, preclinical stroke research is afflicted by a lack of studies designed to evaluate mechanisms for CNS delivery of stroke therapeutics such as BBB transporters. Our laboratory’s research is focused on addressing this knowledge gap via detailed study of endogenous BBB transport mechanisms that can be exploited for selective blood-to-brain delivery of neuroprotective drugs. In this study, we report that memantine, a transport substrate for BBB cation transport systems (18), can improve functional neurological outcomes, reduce cerebral infarction progression, and decrease the brain edema ratio when administered as a single acute dose following 90 min MCAO and 2 h reperfusion. Our observations are consistent with previous preclinical studies that have shown improvement in infarction volumes and neurological deficit scores following transient MCAO in Sprague-Dawley rats (74, 75). Additionally, memantine is known to prevent cerebral edema following experimental ischemic stroke (76, 77) and intracerebral hemorrhage (78), an effect that may reflect the capability of this drug to protect against stroke-induced BBB dysfunction. Interestingly, the Oct transport inhibitor cimetidine attenuated all positive effects in our MCAO model, suggesting that Oct1/Oct2 transport is a primary mechanism for neuroprotective effects of memantine. This is the first time that an endogenous BBB transport system has been shown to be required for a stroke drug to be effective. At the time of memantine administration (i.e., following 90 min MCAO and 2 h reperfusion), protein expression of Oct1 and Oct2 at the BBB were not statistically different from Sham-operated controls; however, we observed considerable variation in transporter protein levels in ipsilateral cerebral cortical microvessels, particularly for Oct1. A strength of our approach is that the lanes in each of our western blots corresponds to a protein sample prepared from a single experimental animal. This strategy enables us to properly detect variability in transporter expression at the BBB, especially that induced by disease states such as focal cerebral ischemia. It is apparent that protein expression changes in Oct transporters following MCAO are evolving and may show statistically significant differences at later time points post-MCAO. As such, our laboratory is currently evaluating the complex time course of BBB transporter changes that result from an ischemic insult and/or reperfusion because such transporter expression variability can dramatically affect the time of drug dosing during post-stroke recovery. Our in situ brain perfusion data shows that memantine brain uptake into whole brain tissue as well as into ipsilateral and contralateral cerebral cortices was blocked by cimetidine, results that support the critical role for Oct1/Oct2 in CNS delivery of this cationic neuroprotective drug. Taken together, our findings have profound implications for improvement of ischemic stroke treatment. Targeting BBB transporters such as Oct1/Oct2 provides a unique opportunity to determine what types of drugs can be successfully delivered to the brain, thereby providing safer and more effective pharmacotherapy. This can include many small molecule therapeutics that are Oct1/Oct2 transport substrates across multiple diverse therapeutic classes such as NMDA receptor antagonists (i.e., amantadine, ketamine), natural product compounds (i.e., berberine), smoking cessation aids (i.e., varenicline), monoamine oxidase inhibitors (i.e., selegiline), and the biguanide anti-diabetic drug metformin (79–84).
Our in situ perfusion data shows that cimetidine can block brain uptake of memantine in ipsilateral and contralateral cerebral cortices from both MCAO animals and Sham-operated controls; however, the magnitude of memantine uptake in ipsilateral cortex was greater than that measured in contralateral cortex under MCAO conditions. This finding suggests that a component of blood-to-brain memantine uptake results from non-selective paracellular diffusion (i.e., “leak”). Indeed, we demonstrated increased brain uptake of sucrose, a small molecule vascular tracer that does not permeate the intact BBB (45), following MCAO in whole brain tissue and in ipsilateral cerebral cortex. It must be emphasized that cimetidine treatment blocked the ability of memantine to improve neurological performance and to limit progression of cerebral infarction and brain edema. These data demonstrate that Oct-mediated transport is required for neuroprotective effects of memantine, an effect that persists despite concurrent paracellular “leak” at the BBB. The concept that endogenous brain microvascular endothelial transporters can overcome passive paracellular diffusion has been previously reported for other disease states. For example, our laboratory has shown increased paracellular permeability and simultaneous induction of the critical efflux transporter P-glycoprotein (P-gp) at the BBB in female Sprague-Dawley rats subjected to pain/inflammation (44, 85–87). One fascinating observation derived from this work was that morphine (i.e., a P-gp transport substrate) brain exposure in animals subjected to pain/inflammation could only be enhanced in the presence of the competitive P-gp inhibitor cyclosporine A (85). This finding implies that transcellular transport remains the principal determinant of blood-to-brain morphine delivery despite knowledge that pain/inflammation also opens a non-selective passive paracellular diffusion route between adjacent cerebrovascular endothelial cells. Taken together, our work indicates the essential need to consider BBB uptake transport mechanisms during preclinical drug development for ischemic stroke. The consideration of endothelial cell transport kinetics will enable discovery of novel therapeutic agents that are both effective at discrete molecular targets and capable of reaching those targets at efficacious concentrations.
An important consideration in the understanding of drug transport dynamics at the BBB in the setting of ischemic stroke is knowledge that brain microvascular endothelial cells express selective transporters that both move solutes into the brain (i.e., influx transporters such as Octs) and prevent substances from accessing brain parenchyma (i.e., efflux transporters). Ischemic stroke is associated with altered functional expression of the critical efflux transporter P-glycoprotein (P-gp). Of particular significance, Spudich and colleagues demonstrated that P-gp protein expression is elevated at the BBB as early as 3 h following transient middle cerebral artery occlusion (MCAO) (88). Additionally, western blot analyses showed increased P-gp protein expression in cerebral microvasculature following MCAO (90 min) as well as in cultured brain endothelial cells subjected to hypoxia/aglycemia (89). Altered functional expression of influx and efflux BBB transporters can have profound implications for brain disposition and therapeutic effectiveness of drugs designed to treat ischemic stroke. Indeed, several drugs with neuroprotective properties are established P-gp transport substrates. For example, P-gp is involved in brain uptake and distribution of tanshinone IIA, a natural product compound that has shown effectiveness against ischemia/reperfusion injury in preclinical studies (90). More recently, in vitro studies in human umbilical vein endothelial cells (HUVECs) have identified atorvastatin and rosuvastatin as P-gp transport substrates (12). In contrast to P-gp, little is known as to how ischemic stroke affects other efflux transporters such as breast cancer resistance protein (Bcrp). Using bovine brain endothelial cells that were co-cultured with rat astrocytes, Tornabene and colleagues showed decreased Abcg2 mRNA in response to oxygen/glucose deprivation (OGD) (91). Interestingly, this reduction in transporter gene expression occurred as early as 4 h post-OGD treatment but ABCG2 transcript expression returned to control levels after 24 h reoxygenation (91). In contrast, Abcg2 mRNA was shown to be upregulated in blood vessels within the peri-infarction region at 3 days and 14 days post-MCAO in young male Sprague-Dawley rats (92). Since Bcrp functions in synergy with P-gp to restrict BBB permeability of many currently marketed drugs (2, 93), it is critical that the role of Bcrp in CNS drug delivery be rigorously studied in animal models of experimental ischemic stroke.
Generally, transport of cationic drugs across a polarized epithelial layer involves a two-step process in which Octs mediate initial cellular uptake. This principal has been demonstrated in human renal proximal tubule epithelial cells where uptake of solutes from blood is mediated by OCT2 and extrusion into urinary filtrate involves the multidrug and toxin extrusion transporter 1 (MATE1 in humans; Mate1 in rodents) (94). At the BBB, evaluation of OCT/Oct localization has indicated that these transporters are preferentially expressed at the luminal plasma membrane of brain microvascular endothelial cells (20). Functional expression of Octs has been demonstrated at the BBB as evidenced by Oct1 and Oct2-mediated uptake of N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a compound that can easily accumulate in brain parenchyma and does not remain “trapped” within the endothelial cell (23). This study suggests that additional transporters (i.e., MATEs) may be required to ensure effective delivery of cationic substances to brain tissue. Recently, protein expression of MATE1 and MATE2 has been reported in the human brain endothelial cell line hCMEC/d3 (95). Additionally, Mate1 and Mate2 protein expression was detected in cerebral microvessels isolated from C57BL6/129 mice (95) and Mate1 mRNA and protein expression was observed in brain capillaries from male ddY mice (96). More recently, Mate1 mRNA was shown to be expressed in murine microvessels isolated from various mouse strains including Swiss, FVB, and C57BL6/JRj (97). Of particular significance, MATE1 and MATE2 protein was detected in capillaries collected from human frontal cortex, caudate nucleus, and putamen (95). These results are consistent with immunofluorescence microscopy studies on human brain microvessels that demonstrated protein expression of MATE1 (98). Using quantitative proteomic analysis, the relative abundance of MATE1 was below the limit of detection in microvessels isolated from human temporal lobe glioma specimens (97). Since these data were derived from human tumor tissue, it is possible that transporters for cationic substrates such as MATEs were downregulated in response to cancer pathophysiology or to chemotherapy. The variability in MATE/Mate expression data represented by these studies indicates a critical need to further clarify involvement of Mate isoforms as transporters that function in concert with Octs to deliver cationic drugs to the brain. This information is essential for development of cation transport systems as targets for CNS drug delivery. Our laboratory is currently undertaking rigorous studies on Mate localization, expression, and transport activity at the BBB. These studies will enable our laboratory to achieve the paramount goal of exploiting Octs/Mates for effective drug delivery to the ischemic brain.
Conclusion
The present study has identified BBB transport mediated by Oct1/Oct2 as a critical mechanism that facilitates CNS drug delivery in the setting of ischemic stroke. Specifically, we have shown for the first time that blood-to-brain transport via Oct1/Oct2 is absolutely required for memantine to exert neuroprotective effects in the ischemic brain and to promote post-stroke recovery. This effect occurred despite concurrent paracellular “leak”, which demonstrates that selective BBB transporters dominate over non-selective drug uptake pathways. Additionally, we showed that reperfusion is an essential process for transporters to efficiently deliver therapeutic agents into ischemic brain tissue. This is consistent with previous reports in the literature, which have demonstrated that circulating solutes (i.e., ions, insulin) as well as many small molecules that are transporter substrates cross the BBB in a manner dependent upon cerebral blood flow (99–101). Our data also emphasize the need to assess brain penetration of therapeutic agents during preclinical development, a consideration that will likely enable new drugs for ischemic stroke to advance further in clinical trials. Overall, results presented in this paper strengthen the novel and translational evidence generated by our laboratory that endogenous BBB transport systems are primary determinants of CNS drug uptake and can be targeted for CNS drug delivery, thereby providing a platform for development of novel treatment strategies for ischemic stroke.
Highlights.
Memantine requires organic cation transporters (Octs) at the blood-brain barrier (BBB) for neuroprotection in stroke.
Oct-mediated transport is the primary mechanism for memantine effects despite stroke-induced paracellular BBB “leak”.
Memantine improved neurological scoring and rotarod performance up to 72 h post-MCAO.
Memantine reduced infarction volume and brain edema ratio at 72 h post-MCAO.
Pharmacological inhibition of Octs prevented memantine effects in the middle cerebral artery occlusion (MCAO) rat model.
Funding
This work is funded by grants from the National Institutes of Neurological Diseases and Stroke (NINDS; R01 NS084941) and the American Heart Association (19TPA34910113) to PTR.
Footnotes
Disclosures
No author has an actual or perceived conflict of interest with the contents of this article.
References
- 1.Shi L, Rocha M, Leak RK, Zhao J, Bhatia TN, Mu H, et al. A new era for stroke therapy: Integrating neurovascular protection with optimal reperfusion. J Cereb Blood Flow Metab. 2018;38(12):2073–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nilles KL, Williams EI, Betterton RD, Davis TP, Ronaldson PT. Blood-Brain Barrier Transporters: Opportunities for Therapeutic Development in Ischemic Stroke. Int J Mol Sci. 2022;23(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turner RC, Lucke-Wold B, Lucke-Wold N, Elliott AS, Logsdon AF, Rosen CL, et al. Neuroprotection for ischemic stroke: moving past shortcomings and identifying promising directions. Int J Mol Sci. 2013;14(1):1890–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lucke-Wold BP, Logsdon AF, Turner RC, Rosen CL, Huber JD. Aging, the metabolic syndrome, and ischemic stroke: redefining the approach for studying the blood-brain barrier in a complex neurological disease. Adv Pharmacol. 2014;71:411–49. [DOI] [PubMed] [Google Scholar]
- 5.Ishikawa H, Wakisaka Y, Matsuo R, Makihara N, Hata J, Kuroda J, et al. Influence of Statin Pretreatment on Initial Neurological Severity and Short-Term Functional Outcome in Acute Ischemic Stroke Patients: The Fukuoka Stroke Registry. Cerebrovasc Dis. 2016;42(5-6):395–403. [DOI] [PubMed] [Google Scholar]
- 6.Malhotra K, Safouris A, Goyal N, Arthur A, Liebeskind DS, Katsanos AH, et al. Association of statin pretreatment with collateral circulation and final infarct volume in acute ischemic stroke patients: A meta-analysis. Atherosclerosis. 2019;282:75–9. [DOI] [PubMed] [Google Scholar]
- 7.Lee M, Saver JL, Wu YL, Tang SC, Lee JD, Rao NM, et al. Utilization of Statins Beyond the Initial Period After Stroke and 1-Year Risk of Recurrent Stroke. J Am Heart Assoc. 2017;6(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Montaner J, Bustamante A, Garcia-Matas S, Martinez-Zabaleta M, Jimenez C, de la Torre J, et al. Combination of Thrombolysis and Statins in Acute Stroke Is Safe: Results of the STARS Randomized Trial (Stroke Treatment With Acute Reperfusion and Simvastatin). Stroke. 2016;47(11):2870–3. [DOI] [PubMed] [Google Scholar]
- 9.Thompson BJ, Sanchez-Covarrubias L, Slosky LM, Zhang Y, Laracuente ML, Ronaldson PT. Hypoxia/reoxygenation stress signals an increase in organic anion transporting polypeptide 1a4 (Oatp1a4) at the blood-brain barrier: relevance to CNS drug delivery. J Cereb Blood Flow Metab. 2014;34(4):699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdullahi W, Brzica H, Hirsch NA, Reilly BG, Ronaldson PT. Functional Expression of Organic Anion Transporting Polypeptide 1a4 Is Regulated by Transforming Growth Factor-beta/Activin Receptor-like Kinase 1 Signaling at the Blood-Brain Barrier. Mol Pharmacol. 2018;94(6):1321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brzica H, Abdullahi W, Reilly BG, Ronaldson PT. Sex-specific differences in organic anion transporting polypeptide 1a4 (Oatp1a4) functional expression at the blood-brain barrier in Sprague-Dawley rats. Fluids Barriers CNS. 2018;15(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ronaldson PT, Brzica H, Abdullahi W, Reilly BG, Davis TP. Transport Properties of Statins by Organic Anion Transporting Polypeptide 1A2 and Regulation by Transforming Growth Factor-beta Signaling in Human Endothelial Cells. J Pharmacol Exp Ther. 2021;376(2):148–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Culmsee C, Junker V, Kremers W, Thal S, Plesnila N, Krieglstein J. Combination therapy in ischemic stroke: synergistic neuroprotective effects of memantine and clenbuterol. Stroke. 2004;35(5):1197–202. [DOI] [PubMed] [Google Scholar]
- 14.Volbracht C, van Beek J, Zhu C, Blomgren K, Leist M. Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur J Neurosci. 2006;23(10):2611–22. [DOI] [PubMed] [Google Scholar]
- 15.Shih AY, Blinder P, Tsai PS, Friedman B, Stanley G, Lyden PD, et al. The smallest stroke: occlusion of one penetrating vessel leads to infarction and a cognitive deficit. Nat Neurosci. 2013;16(1):55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu SX, Sonkar VK, Katare PB, Kumar R, Kruger WD, Arning E, et al. Memantine Protects From Exacerbation of Ischemic Stroke and Blood Brain Barrier Disruption in Mild But Not Severe Hyperhomocysteinemia. J Am Heart Assoc. 2020;9(4):e013368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Summerfield SG, Zhang Y, Liu H. Examining the Uptake of Central Nervous System Drugs and Candidates across the Blood-Brain Barrier. J Pharmacol Exp Ther. 2016;358(2):294–305. [DOI] [PubMed] [Google Scholar]
- 18.Mehta DC, Short JL, Nicolazzo JA. Memantine transport across the mouse blood-brain barrier is mediated by a cationic influx H+ antiporter. Mol Pharm. 2013;10(12):4491–8. [DOI] [PubMed] [Google Scholar]
- 19.Higuchi K, Kitamura A, Okura T, Deguchi Y. Memantine transport by a proton-coupled organic cation antiporter in hCMEC/D3 cells, an in vitro human blood-brain barrier model. Drug Metab Pharmacokinet. 2015;30(2):182–7. [DOI] [PubMed] [Google Scholar]
- 20.Betterton RD, Davis TP, Ronaldson PT. Organic Cation Transporter (OCT/OCTN) Expression at Brain Barrier Sites: Focus on CNS Drug Delivery. Handb Exp Pharmacol. 2021;266:301–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Busch AE, Karbach U, Miska D, Gorboulev V, Akhoundova A, Volk C, et al. Human neurons express the polyspecific cation transporter hOCT2, which translocates monoamine neurotransmitters, amantadine, and memantine. Mol Pharmacol. 1998;54(2):342–52. [DOI] [PubMed] [Google Scholar]
- 22.Brzica H, Abdullahi W, Ibbotson K, Ronaldson PT. Role of Transporters in Central Nervous System Drug Delivery and Blood-Brain Barrier Protection: Relevance to Treatment of Stroke. J Cent Nerv Syst Dis. 2017;9:1179573517693802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin CJ, Tai Y, Huang MT, Tsai YF, Hsu HJ, Tzen KY, et al. Cellular localization of the organic cation transporters, OCT1 and OCT2, in brain microvessel endothelial cells and its implication for MPTP transport across the blood-brain barrier and MPTP-induced dopaminergic toxicity in rodents. J Neurochem. 2010;114(3):717–27. [DOI] [PubMed] [Google Scholar]
- 24.Sekhar GN, Georgian AR, Sanderson L, Vizcay-Barrena G, Brown RC, Muresan P, et al. Organic cation transporter 1 (OCT1) is involved in pentamidine transport at the human and mouse blood-brain barrier (BBB). PLoS One. 2017;12(3):e0173474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Majdoub ZM, Al Feteisi H, Achour B, Warwood S, Neuhoff S, Rostami-Hodjegan A, et al. Proteomic Quantification of Human Blood-Brain Barrier SLC and ABC Transporters in Healthy Individuals and Dementia Patients. Mol Pharm. 2019;16(3):1220–33. [DOI] [PubMed] [Google Scholar]
- 26.Underly RG, Levy M, Hartmann DA, Grant RI, Watson AN, Shih AY. Pericytes as Inducers of Rapid, Matrix Metalloproteinase-9-Dependent Capillary Damage during Ischemia. J Neurosci. 2017;37(1):129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang X, Andjelkovic AV, Zhu L, Yang T, Bennett MVL, Chen J, et al. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol. 2018;163-164:144–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sayeed I, Turan N, Stein DG, Wali B. Vitamin D deficiency increases blood-brain barrier dysfunction after ischemic stroke in male rats. Exp Neurol. 2019;312:63–71. [DOI] [PubMed] [Google Scholar]
- 29.Edwards DN, Salmeron K, Lukins DE, Trout AL, Fraser JF, Bix GJ. Integrin alpha5beta1 inhibition by ATN-161 reduces neuroinflammation and is neuroprotective in ischemic stroke. J Cereb Blood Flow Metab. 2020;40(8):1695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marvanova M, Lakso M, Pirhonen J, Nawa H, Wong G, Castren E. The neuroprotective agent memantine induces brain-derived neurotrophic factor and trkB receptor expression in rat brain. Mol Cell Neurosci. 2001;18(3):247–58. [DOI] [PubMed] [Google Scholar]
- 31.Marvanova M, Lakso M, Wong G. Identification of genes regulated by memantine and MK-801 in adult rat brain by cDNA microarray analysis. Neuropsychopharmacology. 2004;29(6):1070–9. [DOI] [PubMed] [Google Scholar]
- 32.Lawrence ES, Coshall C, Dundas R, Stewart J, Rudd AG, Howard R, et al. Estimates of the prevalence of acute stroke impairments and disability in a multiethnic population. Stroke. 2001. [DOI] [PubMed] [Google Scholar]
- 33.Martino R, Foley N, Bhogal S, Diamant N, Speechley M, Teasell R. Dysphagia after stroke: Incidence, diagnosis, and pulmonary complications. Stroke. 2005. [DOI] [PubMed] [Google Scholar]
- 34.Toscano M, Cecconi E, Capiluppi E, Vigano A, Bertora P, Campiglio L, et al. Neuroanatomical, Clinical and Cognitive Correlates of Post-Stroke Dysphagia. Eur Neurol. 2015;74(3-4):171–7. [DOI] [PubMed] [Google Scholar]
- 35.Cohen DL, Roffe C, Beavan J, Blackett B, Fairfield CA, Hamdy S, et al. Post-stroke dysphagia: A review and design considerations for future trials. Int J Stroke. 2016;11(4):399–411. [DOI] [PubMed] [Google Scholar]
- 36.Burt HJ, Neuhoff S, Almond L, Gaohua L, Harwood MD, Jamei M, et al. Metformin and cimetidine: Physiologically based pharmacokinetic modelling to investigate transporter mediated drug-drug interactions. Eur J Pharm Sci. 2016;88:70–82. [DOI] [PubMed] [Google Scholar]
- 37.Nishiyama K, Toshimoto K, Lee W, Ishiguro N, Bister B, Sugiyama Y. Physiologically-Based Pharmacokinetic Modeling Analysis for Quantitative Prediction of Renal Transporter-Mediated Interactions Between Metformin and Cimetidine. CPT Pharmacometrics Syst Pharmacol. 2019;8(6):396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koepsell H Update on drug-drug interaction at organic cation transporters: mechanisms, clinical impact, and proposal for advanced in vitro testing. Expert Opin Drug Metab Toxicol. 2021;17(6):635–53. [DOI] [PubMed] [Google Scholar]
- 39.Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 2007;24(7):1227–51. [DOI] [PubMed] [Google Scholar]
- 40.Hom S, Fleegal MA, Egleton RD, Campos CR, Hawkins BT, Davis TP. Comparative changes in the blood-brain barrier and cerebral infarction of SHR and WKY rats. Am J Physiol Regul Integr Comp Physiol. 2007;292(5):R1881–92. [DOI] [PubMed] [Google Scholar]
- 41.Villalba H, Shah K, Albekairi TH, Sifat AE, Vaidya B, Abbruscato TJ. Potential role of myo-inositol to improve ischemic stroke outcome in diabetic mouse. Brain Res. 2018;1699:166–76. [DOI] [PubMed] [Google Scholar]
- 42.Barber PA, Hoyte L, Colbourne F, Buchan AM. Temperature-regulated model of focal ischemia in the mouse: a study with histopathological and behavioral outcomes. Stroke. 2004;35(7):1720–5. [DOI] [PubMed] [Google Scholar]
- 43.Ronaldson PT, Finch JD, Demarco KM, Quigley CE, Davis TP. Inflammatory pain signals an increase in functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier. J Pharmacol Exp Ther. 2011;336(3):827–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ronaldson PT, Demarco KM, Sanchez-Covarrubias L, Solinsky CM, Davis TP. Transforming growth factor-beta signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain. J Cereb Blood Flow Metab. 2009;29(6):1084–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lochhead JJ, Yang J, Ronaldson PT, Davis TP. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front Physiol. 2020;11:914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takasato Y, Rapoport SI, Smith QR. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol. 1984;247(3 Pt 2):H484–93. [DOI] [PubMed] [Google Scholar]
- 47.Brzica H, Abdullahi W, Reilly BG, Ronaldson PT. A Simple and Reproducible Method to Prepare Membrane Samples from Freshly Isolated Rat Brain Microvessels. J Vis Exp. 2018(135). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abdullahi W, Brzica H, Ibbotson K, Davis TP, Ronaldson PT. Bone morphogenetic protein-9 increases the functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier via the activin receptor-like kinase-1 receptor. J Cereb Blood Flow Metab. 2017;37(7):2340–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Comajoan P, Gubern C, Huguet G, Serena J, Kadar E, Castellanos M. Evaluation of common housekeeping proteins under ischemic conditions and/or rt-PA treatment in bEnd.3 cells. J Proteomics. 2018;184:10–5. [DOI] [PubMed] [Google Scholar]
- 50.Fosang AJ, Colbran RJ. Transparency Is the Key to Quality. J Biol Chem. 2015;290(50):29692–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang L, Islam MR, Karamyan VT, Abbruscato TJ. In vitro and in vivo efficacy of a potent opioid receptor agonist, biphalin, compared to subtype-selective opioid receptor agonists for stroke treatment. Brain Res. 2015;1609:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fourie J, Escobar MR, Sitar DS. NMDA receptor antagonists to characterize rat renal organic cation transporter function. Eur J Pharmacol. 2002;452(1):1–10. [DOI] [PubMed] [Google Scholar]
- 53.Kitamura A, Okura T, Higuchi K, Deguchi Y. Cocktail-Dosing Microdialysis Study to Simultaneously Assess Delivery of Multiple Organic-Cationic Drugs to the Brain. J Pharm Sci. 2016;105(2):935–40. [DOI] [PubMed] [Google Scholar]
- 54.Muller F, Weitz D, Derdau V, Sandvoss M, Mertsch K, Konig J, et al. Contribution of MATE1 to Renal Secretion of the NMDA Receptor Antagonist Memantine. Mol Pharm. 2017;14(9):2991–8. [DOI] [PubMed] [Google Scholar]
- 55.Beladi Moghadam N, Pourheidar E, Ahmadpour F, Kafi H, Salamzadeh J, Nasiri S, et al. The effects of memantine on the serum concentrations of matrix metalloproteinases and neurologic function of patients with ischemic stroke. J Clin Neurosci. 2021;90:268–72. [DOI] [PubMed] [Google Scholar]
- 56.Kim KA, Kim D, Kim JH, Shin YJ, Kim ES, Akram M, et al. Autophagy-mediated occludin degradation contributes to blood-brain barrier disruption during ischemia in bEnd.3 brain endothelial cells and rat ischemic stroke models. Fluids Barriers CNS. 2020;17(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Winkler L, Blasig R, Breitkreuz-Korff O, Berndt P, Dithmer S, Helms HC, et al. Tight junctions in the blood-brain barrier promote edema formation and infarct size in stroke - Ambivalent effects of sealing proteins. J Cereb Blood Flow Metab. 2021;41(1):132–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao Q, Yu S, Ling Y, Hao S, Liu J. The Protective Effects of Dexmedetomidine against Hypoxia/Reoxygenation-Induced Inflammatory Injury and Permeability in Brain Endothelial Cells Mediated by Sigma-1 Receptor. ACS Chem Neurosci. 2021;12(11):1940–7. [DOI] [PubMed] [Google Scholar]
- 59.Thompson BJ, Ronaldson PT. Drug delivery to the ischemic brain. Adv Pharmacol. 2014;71:165–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abdullahi W, Tripathi D, Ronaldson PT. Blood-brain barrier dysfunction in ischemic stroke: targeting tight junctions and transporters for vascular protection. Am J Physiol Cell Physiol. 2018;315(3):C343–C56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kiedrowski L NCX and NCKX operation in ischemic neurons. Ann N Y Acad Sci. 2007;1099:383–95. [DOI] [PubMed] [Google Scholar]
- 62.Luo J, Wang Y, Chen H, Kintner DB, Cramer SW, Gerdts JK, et al. A concerted role of Na+ -K+ -Cl− cotransporter and Na+/Ca2+ exchanger in ischemic damage. J Cereb Blood Flow Metab. 2008;28(4):737–46. [DOI] [PubMed] [Google Scholar]
- 63.Adibhatla RM, Hatcher JF. Tissue plasminogen activator (tPA) and matrix metalloproteinases in the pathogenesis of stroke: therapeutic strategies. CNS Neurol Disord Drug Targets. 2008;7(3):243–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arai K, Lok J, Guo S, Hayakawa K, Xing C, Lo EH. Cellular mechanisms of neurovascular damage and repair after stroke. J Child Neurol. 2011;26(9):1193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu QJ, Tymianski M. Targeting NMDA receptors in stroke: new hope in neuroprotection. Mol Brain. 2018;11(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lipton SA. Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx. 2004;1(1):101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Albers GW, Goldstein LB, Hall D, Lesko LM, Aptiganel Acute Stroke I. Aptiganel hydrochloride in acute ischemic stroke: a randomized controlled trial. JAMA. 2001;286(21):2673–82. [DOI] [PubMed] [Google Scholar]
- 68.Davis SM, Lees KR, Albers GW, Diener HC, Markabi S, Karlsson G, et al. Selfotel in acute ischemic stroke : possible neurotoxic effects of an NMDA antagonist. Stroke. 2000;31(2):347–54. [DOI] [PubMed] [Google Scholar]
- 69.Lopez-Valdes HE, Clarkson AN, Ao Y, Charles AC, Carmichael ST, Sofroniew MV, et al. Memantine enhances recovery from stroke. Stroke. 2014;45(7):2093–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hao J, Mdzinarishvili A, Abbruscato TJ, Klein J, Geldenhuys WJ, Van der Schyf CJ, et al. Neuroprotection in mice by NGP1-01 after transient focal brain ischemia. Brain Res. 2008;1196:113–20. [DOI] [PubMed] [Google Scholar]
- 71.Trotman M, Vermehren P, Gibson CL, Fern R. The dichotomy of memantine treatment for ischemic stroke: dose-dependent protective and detrimental effects. J Cereb Blood Flow Metab. 2015;35(2):230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang YC, Sanchez-Mendoza EH, Doeppner TR, Hermann DM. Post-acute delivery of memantine promotes post-ischemic neurological recovery, peri-infarct tissue remodeling, and contralesional brain plasticity. J Cereb Blood Flow Metab. 2017;37(3):980–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Montagne A, Hebert M, Jullienne A, Lesept F, Le Behot A, Louessard M, et al. Memantine improves safety of thrombolysis for stroke. Stroke. 2012;43(10):2774–81. [DOI] [PubMed] [Google Scholar]
- 74.Chen B, Wang G, Li W, Liu W, Lin R, Tao J, et al. Memantine attenuates cell apoptosis by suppressing the calpain-caspase-3 pathway in an experimental model of ischemic stroke. Exp Cell Res. 2017;351(2):163–72. [DOI] [PubMed] [Google Scholar]
- 75.Wang F, Liang W, Lei C, Kinden R, Sang H, Xie Y, et al. Combination of HBO and Memantine in Focal Cerebral Ischemia: Is There a Synergistic Effect? Mol Neurobiol. 2015;52(3):1458–66. [DOI] [PubMed] [Google Scholar]
- 76.Dogan A, Eras MA, Rao VL, Dempsey RJ. Protective effects of memantine against ischemia-reperfusion injury in spontaneously hypertensive rats. Acta Neurochir (Wien). 1999;141(10):1107–13. [DOI] [PubMed] [Google Scholar]
- 77.Gorgulu A, Kins T, Cobanoglu S, Unal F, Izgi NI, Yanik B, et al. Reduction of edema and infarction by Memantine and MK-801 after focal cerebral ischaemia and reperfusion in rat. Acta Neurochir (Wien). 2000;142(11):1287–92. [DOI] [PubMed] [Google Scholar]
- 78.Chen X, Xiang X, Xie T, Chen Z, Mou Y, Gao Z, et al. Memantine protects blood-brain barrier integrity and attenuates neurological deficits through inhibiting nitric oxide synthase ser1412 phosphorylation in intracerebral hemorrhage rats: involvement of peroxynitrite-related matrix metalloproteinase-9/NLRP3 inflammasome activation. Neuroreport. 2021;32(3):228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ruhl L, Kuramatsu JB, Sembill JA, Kallmunzer B, Madzar D, Gerner ST, et al. Amantadine treatment is associated with improved consciousness in patients with non-traumatic brain injury. J Neurol Neurosurg Psychiatry. 2022;93(6):582–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xiong Z, Chang L, Qu Y, Pu Y, Wang S, Fujita Y, et al. Neuronal brain injury after cerebral ischemic stroke is ameliorated after subsequent administration of (R)-ketamine, but not (S)-ketamine. Pharmacol Biochem Behav. 2020;191:172904. [DOI] [PubMed] [Google Scholar]
- 81.Zhao Y, Li Z, Lu E, Sheng Q, Zhao Y. Berberine exerts neuroprotective activities against cerebral ischemia/reperfusion injury through up-regulating PPAR-gamma to suppress NF-kappaB-mediated pyroptosis. Brain Res Bull. 2021;177:22–30. [DOI] [PubMed] [Google Scholar]
- 82.Chen S, Bennet L, McGregor AL. Delayed Varenicline Administration Reduces Inflammation and Improves Forelimb Use Following Experimental Stroke. J Stroke Cerebrovasc Dis. 2017;26(12):2778–87. [DOI] [PubMed] [Google Scholar]
- 83.Bartolo M, Zucchella C, Capone A, Sandrini G, Pierelli F. An explorative study regarding the effect of l-deprenyl on cognitive and functional recovery in patients after stroke. J Neurol Sci. 2015;349(1-2):117–23. [DOI] [PubMed] [Google Scholar]
- 84.Sharma S, Nozohouri S, Vaidya B, Abbruscato T. Repurposing metformin to treat age-related neurodegenerative disorders and ischemic stroke. Life Sci. 2021;274:119343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Seelbach MJ, Brooks TA, Egleton RD, Davis TP. Peripheral inflammatory hyperalgesia modulates morphine delivery to the brain: a role for P-glycoprotein. J Neurochem. 2007;102(5):1677–90. [DOI] [PubMed] [Google Scholar]
- 86.Lochhead JJ, McCaffrey G, Quigley CE, Finch J, DeMarco KM, Nametz N, et al. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010;30(9):1625–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McCaffrey G, Staatz WD, Sanchez-Covarrubias L, Finch JD, Demarco K, Laracuente ML, et al. P-glycoprotein trafficking at the blood-brain barrier altered by peripheral inflammatory hyperalgesia. J Neurochem. 2012;122(5):962–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Spudich A, Kilic E, Xing H, Kilic U, Rentsch KM, Wunderli-Allenspach H, et al. Inhibition of multidrug resistance transporter-1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat Neurosci. 2006;9(4):487–8. [DOI] [PubMed] [Google Scholar]
- 89.DeMars KM, Yang C, Hawkins KE, McCrea AO, Siwarski DM, Candelario-Jalil E. Spatiotemporal Changes in P-glycoprotein Levels in Brain and Peripheral Tissues Following Ischemic Stroke in Rats. J Exp Neurosci. 2017;11:1179069517701741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen X, Zhou ZW, Xue CC, Li XX, Zhou SF. Role of P-glycoprotein in restricting the brain penetration of tanshinone IIA, a major active constituent from the root of Salvia miltiorrhiza Bunge, across the blood-brain barrier. Xenobiotica. 2007;37(6):635–78. [DOI] [PubMed] [Google Scholar]
- 91.Tornabene E, Helms HCC, Pedersen SF, Brodin B. Effects of oxygen-glucose deprivation (OGD) on barrier properties and mRNA transcript levels of selected marker proteins in brain endothelial cells/astrocyte co-cultures. PLoS One. 2019;14(8):e0221103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dazert P, Suofu Y, Grube M, Popa-Wagner A, Kroemer HK, Jedlitschky G, et al. Differential regulation of transport proteins in the periinfarct region following reversible middle cerebral artery occlusion in rats. Neuroscience. 2006;142(4):1071–9. [DOI] [PubMed] [Google Scholar]
- 93.Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, et al. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino }methyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab Dispos. 2009;37(2):439–42. [DOI] [PubMed] [Google Scholar]
- 94.Sandoval PJ, Zorn KM, Clark AM, Ekins S, Wright SH. Assessment of Substrate-Dependent Ligand Interactions at the Organic Cation Transporter OCT2 Using Six Model Substrates. Mol Pharmacol. 2018;94(3):1057–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sekhar GN, Fleckney AL, Boyanova ST, Rupawala H, Lo R, Wang H, et al. Region-specific blood-brain barrier transporter changes leads to increased sensitivity to amisulpride in Alzheimer’s disease. Fluids Barriers CNS. 2019;16(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hiasa M, Matsumoto T, Komatsu T, Moriyama Y. Wide variety of locations for rodent MATE1, a transporter protein that mediates the final excretion step for toxic organic cations. Am J Physiol Cell Physiol. 2006;291(4):C678–86. [DOI] [PubMed] [Google Scholar]
- 97.Chaves C, Campanelli F, Chapy H, Gomez-Zepeda D, Glacial F, Smirnova M, et al. An Interspecies Molecular and Functional Study of Organic Cation Transporters at the Blood-Brain Barrier: From Rodents to Humans. Pharmaceutics. 2020;12(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Geier EG, Chen EC, Webb A, Papp AC, Yee SW, Sadee W, et al. Profiling solute carrier transporters in the human blood-brain barrier. Clin Pharmacol Ther. 2013;94(6):636–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hom S, Egleton RD, Huber JD, Davis TP. Effect of reduced flow on blood-brain barrier transport systems. Brain Res. 2001;890(1):38–48. [DOI] [PubMed] [Google Scholar]
- 100.Banks WA. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009;9 Suppl 1:S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chowdhury EA, Noorani B, Alqahtani F, Bhalerao A, Raut S, Sivandzade F, et al. Understanding the brain uptake and permeability of small molecules through the BBB: A technical overview. J Cereb Blood Flow Metab. 2021;41(8):1797–820. [DOI] [PMC free article] [PubMed] [Google Scholar]