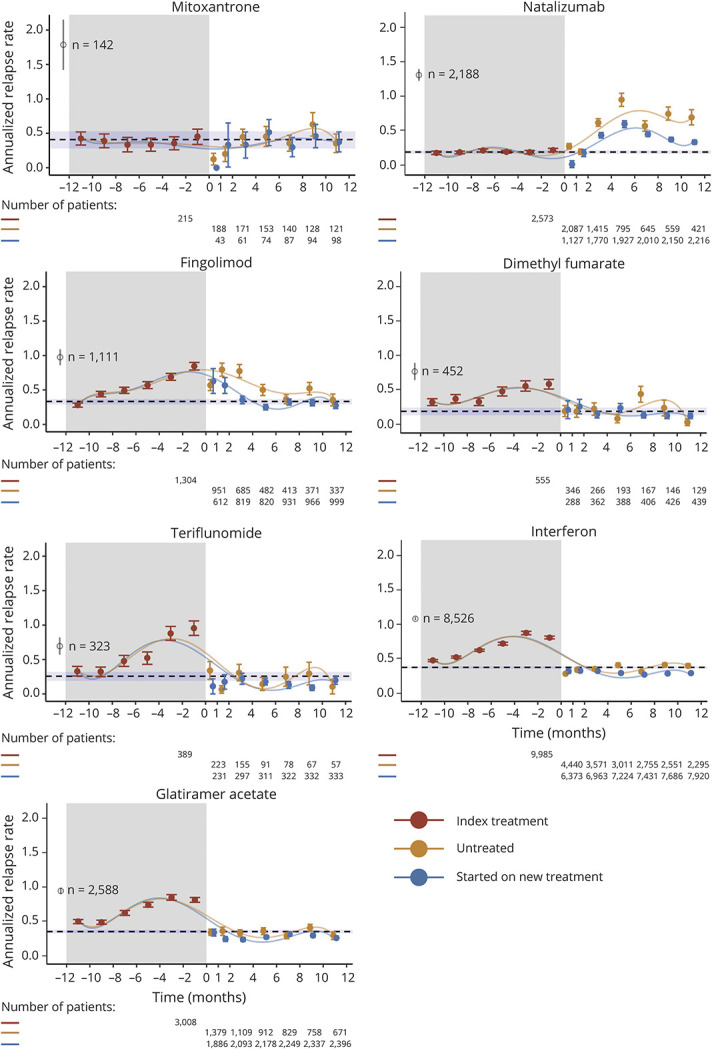
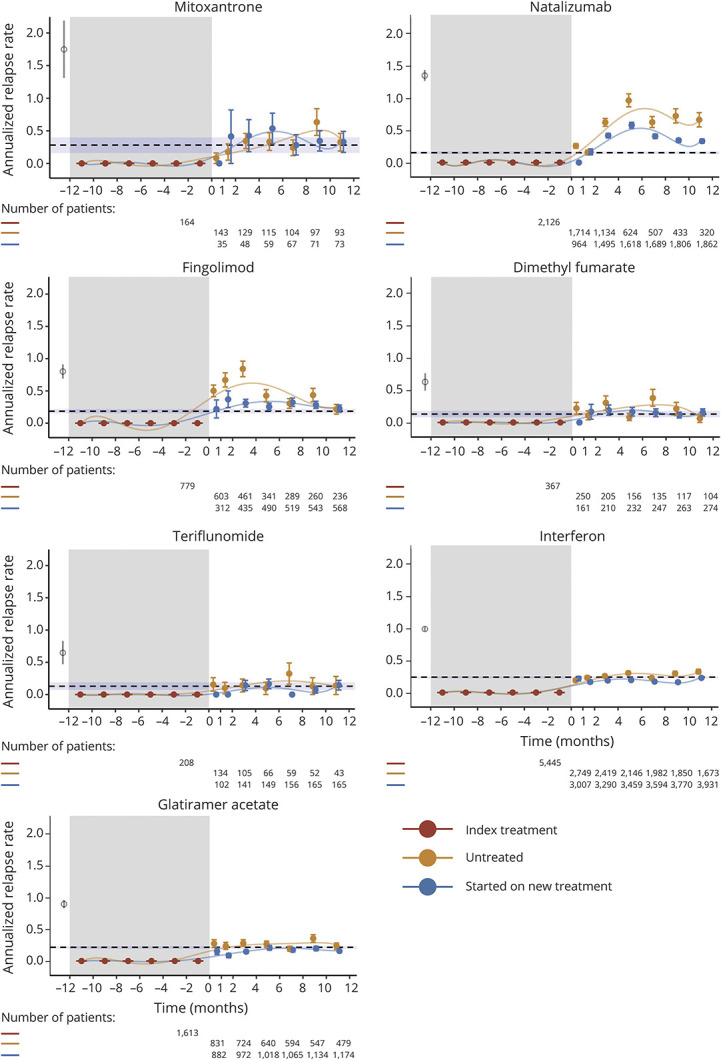
Izanne Roos
Izanne Roos, MBChB, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Charles Malpas
Charles Malpas, MPsych PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Emmanuelle Leray
Emmanuelle Leray, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Romain Casey
Romain Casey, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Dana Horakova
Dana Horakova, MD, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Eva Kubala Havrdova
Eva Kubala Havrdova, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Marc Debouverie
Marc Debouverie, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Francesco Patti
Francesco Patti, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Jerome De Seze
Jerome De Seze, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Guillermo Izquierdo
Guillermo Izquierdo, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Sara Eichau
Sara Eichau, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Gilles Edan
Gilles Edan, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Alexandre Prat
Alexandre Prat, MD, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Marc Girard
Marc Girard, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Serkan Ozakbas
Serkan Ozakbas, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Pierre Grammond
Pierre Grammond, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Helene Zephir
Helene Zephir, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Jonathan Ciron
Jonathan Ciron, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Elisabeth Maillart
Elisabeth Maillart, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Thibault Moreau
Thibault Moreau, PHD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Maria Pia Amato
Maria Pia Amato, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Pierre Labauge
Pierre Labauge, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Raed Alroughani
Raed Alroughani, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Katherine Buzzard
Katherine Buzzard, MBBS, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Olga Skibina
Olga Skibina, MBBS, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Murat Terzi
Murat Terzi, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
David Axel Laplaud
David Axel Laplaud, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Eric Berger
Eric Berger, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Francois Grand'Maison
Francois Grand'Maison, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Christine Lebrun-Frenay
Christine Lebrun-Frenay, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Elisabetta Cartechini
Elisabetta Cartechini, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Cavit Boz
Cavit Boz, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Jeannette Lechner-Scott
Jeannette Lechner-Scott, MD, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Pierre Clavelou
Pierre Clavelou, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Bruno Stankoff
Bruno Stankoff, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Julie Prevost
Julie Prevost, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Ludwig Kappos
Ludwig Kappos, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Jean Pelletier
Jean Pelletier, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Vahid Shaygannejad
Vahid Shaygannejad, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Bassem I Yamout
Bassem I Yamout, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Samia J Khoury
Samia J Khoury, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Oliver Gerlach
Oliver Gerlach, MD, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Daniele LA Spitaleri
Daniele LA Spitaleri, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Vincent Van Pesch
Vincent Van Pesch, MD, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Olivier Gout
Olivier Gout, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Recai Turkoglu
Recai Turkoglu, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Olivier Heinzlef
Olivier Heinzlef, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Eric Thouvenot
Eric Thouvenot, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Pamela Ann McCombe
Pamela Ann McCombe, MBBS
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Aysun Soysal
Aysun Soysal, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Bertrand Bourre
Bertrand Bourre, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Mark Slee
Mark Slee, BMBS, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Tamara Castillo-Trivino
Tamara Castillo-Trivino, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Serge Bakchine
Serge Bakchine, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Radek Ampapa
Radek Ampapa, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Ernest Gerard Butler
Ernest Gerard Butler, MBBS
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Abir Wahab
Abir Wahab, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Richard A Macdonell
Richard A Macdonell, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Eduardo Aguera-Morales
Eduardo Aguera-Morales, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Philippe Cabre
Philippe Cabre, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Nasr Haifa Ben
Nasr Haifa Ben, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Anneke Van der Walt
Anneke Van der Walt, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Guy Laureys
Guy Laureys, MD, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Liesbeth Van Hijfte
Liesbeth Van Hijfte
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Cristina M Ramo-Tello
Cristina M Ramo-Tello, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Nicolas Maubeuge
Nicolas Maubeuge, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Suzanne Hodgkinson
Suzanne Hodgkinson, MBBS, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
José Luis Sánchez-Menoyo
José Luis Sánchez-Menoyo, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Michael H Barnett
Michael H Barnett, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Celine Labeyrie
Celine Labeyrie, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Steve Vucic
Steve Vucic, MBBS, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Youssef Sidhom
Youssef Sidhom, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Riadh Gouider
Riadh Gouider, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Tunde Csepany
Tunde Csepany, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Javier Sotoca
Javier Sotoca, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Koen de Gans
Koen de Gans, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Abdullah Al-Asmi
Abdullah Al-Asmi, MD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Yara Dadalti Fragoso
Yara Dadalti Fragoso, MSc, MD, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Sandra Vukusic
Sandra Vukusic, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Helmut Butzkueven
Helmut Butzkueven, MBBS, PhD
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,
Tomas Kalincik
Tomas Kalincik, MD, PhD, PGCertBiostat
1From the CORe (I.R., C.M., T.K.), Department of Medicine, University of Melbourne, Australia; Melbourne MS Centre (I.R., C.M., T.K.), Department of Neurology, Royal Melbourne Hospital, Australia; Rennes, University (E.L.), EHESP, REPERES EA 7449, France; Univ Rennes (E.L.), CHU Rennes, Inserm, CIC 1414 ([Centre d'Investigation Clinique de Rennes]), France; Université de Lyon (R.C.), Université Claude Bernard Lyon 1, France; Hospices Civils de Lyon (R.C.), Service de Neurologie, Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Bron, France; Observatoire Français de La Sclérose en Plaques (R.C.), Centre de Recherche en Neurosciences de Lyon, INSERM 1028 et CNRS UMR 5292, France; Eugène Devic EDMUS Foundation Against Multiple Sclerosis (R.C.), State-approved Foundation, Bron, France; Department of Neurology and Center of Clinical Neuroscience (D.H., E.K.H.), First Faculty of Medicine, Charles University in Prague and General University Hospital, Czech Republic; Nancy University Hospital (M.D.), Department of Neurology, Nancy, France; Université de Lorraine (M.D.), APEMAC, Nancy, France; Department of Medical and Surgical Sciences and Advanced Technologies (F.P.), GF Ingrassia, Catania, Italy; Multiple Sclerosis Center (F.P.), University of Catania, Italy; CHU de Strasbourg (J.D.S.), Department of Neurology and Clinical Investigation Center, CIC 1434, INSERM 1434, Strasbourg, France; Hospital Universitario Virgen Macarena (G.I., S.E.), Sevilla, Spain; CHU Pontchaillou (G.E.), CIC1414 INSERM, Rennes, France; CHUM MS Center and Universite de Montreal (A.P., M.G.), Canada; Dokuz Eylul University (S.O.), Konak/Izmir, Turkey; CISSS Chaudière-Appalache (P.G.), Levis, Canada; CHU Lille (H.Z.), CRCSEP Lille, Univ Lille, U1172, France; CHU de Toulouse (J.C.), Hôpital Pierre-Paul Riquet, Department of Neurology, CRC-SEP, France; Département de Neurologie (E.M.), Hôpital Pitié-Salpêtrière, APHP, Paris; CHU de Dijon (T.M.), Department of Neurology, EA4184, France; Department NEUROFARBA (M.P.A.), University of Florence, Italy; CHU de Montpellier (P.L.), MS Unit, France; University of Montpellier (MUSE) (P.L.), France; Division of Neurology (Raed Alroughani), Department of Medicine, Amiri Hospital, Sharq, Kuwait; Department of Neurology (K.B., O.S.), Box Hill Hospital, Melbourne, Australia; Monash University (K.B., O.S.), Melbourne, Australia; Melbourne MS Centre (K.B.), Royal Melbourne Hospital, Australia; The Alfred Hospital (O.S.), Melbourne, Australia; Medical Faculty (M.T.), 19 Mayis University, Samsun, Turkey; CHU de Nantes (D.A.L.), Service de Neurologie & CIC015 INSERM, France; CRTI-Inserm U1064 (D.A.L.), Nantes, France; CHU de Besançon (E.B.), Service de Neurologie 25 030 Besançon, France; Neuro Rive-Sud (F.G.M.), Quebec, Canada; Neurology (C.L.-F.), UR2CA, Centre Hospitalier Universitaire Pasteur2, Université Nice Côte d'Azur, Nice, France; UOC Neurologia (E.C.), Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy; KTU Medical Faculty Farabi Hospital (C.B.), Trabzon, Turkey; School of Medicine and Public Health (J.L.-S.), University Newcastle, Australia; Department of Neurology (J.L.-S.), John Hunter Hospital, Hunter New England Health, Newcastle, Australia; CHU Clermont-Ferrand (Pierre Clavelou), Department of Neurology; Université Clermont Auvergne, Inserm, Neuro-Dol, Clermont-Ferrand, France; Sorbonne Universités (B.S.), UPMC Paris 06, Brain and Spine Institute, ICM, Hôpital de La Pitié Salpêtrière, Inserm UMR S 1127, CNRS UMR 7225, and Department of Neurology, AP-HP, Saint-Antoine Hospital, Paris, France; CSSS Saint-Jérôme (Julie Prevost), Saint-Jerome, Canada; Neurologic Clinic and Policlinic (L.K.), Departments of Medicine and Clinical Research, University Hospital and University of Basel, Switzerland; Aix Marseille Univ (Jean Pelletier), APHM, Hôpital de La Timone, Pôle de Neurosciences Cliniques, Service de Neurologie, France; Isfahan University of Medical Sciences (V.S.), Iran; Nehme and Therese Tohme Multiple Sclerosis Center (B.I.Y., S.J.K.), American University of Beirut Medical Center, Beirut, Lebanon; Department of Neurology (Oliver Gerlach), Zuyderland Medical Center, Sittard-Geleen, Netherlands; Azienda Ospedaliera di Rilievo Nazionale San Giuseppe Moscati Avellino (D.L.A.S.), Italy; Cliniques Universitaires Saint-Luc (V.V.P.), Université Catholique de Louvain, Brussels, Belgium; Fondation Rotschild (Olivier Gout), Department of Neurology, Paris, France; Haydarpasa Numune Training and Research Hospital (R.T.), Istanbul, Turkey; Hôpital de Poissy (O.H.), Department of Neurology, France; Department of Neurology (E.T.), Nimes University Hospital, France; Institut de Génomique Fonctionnelle (E.T.), UMR5203, INSERM 1191, Univ. Montpellier, France; University of Queensland (P.A.M.), Brisbane, Australia; Royal Brisbane and Women's Hospital (P.A.M.), Australia; Bakirkoy Education and Research Hospital for Psychiatric and Neurological Diseases (A.S.), Istanbul, Turkey; CHU de Rouen (B.B.), Department of Neurology, France; Flinders University (M.S.), Adelaide, Australia; Instituto de Investigación Sanitaria Biodonostia (T.C.-T.), Hospital Universitario Donostia, San Sebastián, Spain; CHU de Reims (S.B.), Department of Neurology, France; Nemocnice Jihlava (Radek Ampapa), Czech Republic; Monash Medical Centre (E.G.B.), Melbourne, Australia; APHP (A.W.), Hôpital Henri Mondor, Department of Neurology, Créteil, France; Austin Health (R.A.M.), Melbourne, Australia; University Hospital Reina Sofia (E.A.-M.), Cordoba, Spain; CHU de La Martinique (Philippe Cabre), Department of Neurology, Fort-de-France, France; Hôpital Sud Francilien (N.H.B.), Department of Neurology, Corbeil Essonnes, France; Department of Neurology (A.V.W., H.B.), The Alfred Hospital, Melbourne, Australia; Central Clinical School (A.V.W., H.B.), Monash University, Melbourne, Australia; Department of Neurology (G.L., L.V.H.), University Hospital Ghent, Belgium; Hospital Germans Trias I Pujol (C.M.R.-T.), Badalona, Spain; CHU La Milétrie (N.M.), Hôpital Jean Bernard, Department of Neurology, Poitiers, France; Liverpool Hospital (S.H.), Sydney, Australia; Hospital de Galdakao-Usansolo (J.L.S.-M.), Spain; Brain and Mind Centre (M.H.B.), Sydney, Australia; CHU Bicêtre (C.L.), Department of Neurology, F-94275 Le Kremlin Bicêtre, France; Westmead Hospital (Steve Vucic), Sydney, Australia; Department of Neurology (Y.S., R.G.), Razi Hospital, Manouba, Tunisia; Department of Neurology (T.C.), Faculty of Medicine, University of Debrecen, Hungary; Hospital Universitari MútuaTerrassa (J.S.), Barcelona, Spain; Groene Hart Ziekenhuis (K.G.), Gouda, Netherlands; Sultan Qaboos University Hospital (A.A.-A.), Al-Khodh, Oman; Universidade Metropolitana de Santos (Y.D.F.), Santos, Brazil; Service de Neurologie (Sandra Vukusic), Sclérose en Plaques, Pathologies de La Myéline et Neuro-inflammation, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, Bron, France; Centre des Neurosciences de Lyon (Sandra Vukusic), Observatoire Français de La Sclérose en Plaques, INSERM 1028 et CNRS UMR5292, France; and Université Claude Bernard Lyon 1 (Sandra Vukusic), Faculté de Médecine Lyon Est, France.
1,✉;
on behalf of MSBase and OFSEP1