

DIFFERENT WAYS TO DIE
So far, we have mostly focused on apoptosis, also known as type I cell death.1 Along the way, we touched on other forms of cell death, in particular those most related to apoptosis—pyroptosis, caused by the action of caspase-1, -4, -5, or -11, and caspase-independent cell death, caused by mitochondrial outer membrane permeabilization (MOMP) when subsequent caspase activation is blocked. Two other major classes of cell death mainly concern us here: autophagic cell death (also called type II cell death) and necrosis (also called type III cell death). There is another type of cell death that may or may not fall into any of these categories—termed “mitotic catastrophe”—that we also consider in this review. Note that, although one pathway or type of cell death can appear to dominate in a particular setting, this might only be because it happens to be faster.
ACCIDENTS WILL HAPPEN
Cells are highly complex, and any of a wide variety of accidental events can compromise them, leading to cell death. There have been attempts to classify cell death on the basis of whether it is “accidental” or “active.” But where do we draw the line? If a cell is ruptured, it dies immediately, and this death appears distinct from apoptosis. But a milder insult can result in the cell engaging apoptosis or another cell death pathway. Is this “intentional”? In the end, the distinction can be too artificial to be useful. Furthermore, below we consider necrosis, a form of death that can be “accidental” but, as we will see, is not always so.
NECROSIS (TYPE III CELL DEATH)
During necrosis, an influx of water causes organelles to enlarge and the cell swells, ultimately lysing. Unlike cells undergoing apoptosis, chromatin does not condense and the plasma membrane does not form blebs (Fig. 1).
Figure 1.

A fibroblast cell dying by necrosis. (Reprinted from Edinger and Thompson 2004, ©2004 with permission from Elsevier.)
Much of the energy in a cell is devoted to powering ion pumps that sustain gradients across the plasma membrane. If ATP levels become depleted because of a lack of nutrients or the actions of toxins, the cell can swell and rupture, undergoing necrotic cell death. But this and more direct forms of damage are not the only way necrosis comes about.
One way that necrosis can occur is as a consequence of apoptosis. Normally, if a cell dies by apoptosis, it is rapidly removed and degraded by phagocytic cells (discussed in detail in Green 2022b). If for any reason this clearance does not occur (e.g., in tissue culture or because of defects in the clearance mechanisms), the integrity of the plasma membrane of the apoptotic cell is ultimately lost and necrosis ensues. This is referred to as secondary necrosis and is a confounding variable in many studies of cell death. Cells that undergo secondary necrosis have features of both apoptotic cells (e.g., condensed chromatin) and necrotic cells (e.g., a ruptured plasma membrane) (Fig. 2). In many cases, necrosis proceeds independently of apoptosis, or the two forms of cell death can be mixed and difficult to tease apart. As we will see, the key to understanding what is happening is the molecular mechanism underlying how the cell died.
Figure 2.

Apoptosis and secondary necrosis. Living cell (left), apoptotic cell (center), and secondary necrosis (right). Note that, in secondary necrosis, the nucleus is condensed, as in apoptosis. (Reprinted with permission from Springer Science+Business Media: Silva et al. 2008, ©2008.)
As it turns out, however, secondary necrosis of apoptotic cells can also be an active process. One of the gasdermins (see Green 2022c), DNFA5 (also called gasdermin E), is cleaved and activated by active caspase-3 or caspase-7, much as gasdermin D is activated by inflammatory caspases. Like active gasdermin D, active DNFA5 targets the plasma membrane, promoting secondary necrosis. Apoptotic cells that lack DNFA5 remain intact for some time, often breaking into small, membrane-bound pieces (although again if these are not cleared, the plasma membrane integrity will eventually be lost). In contrast, apoptotic cells that contain DNFA5 rapidly undergo secondary necrosis. Nevertheless, as in the situation in which clearance does not occur, these dying cells show features of both apoptosis and necrosis.
NECROSIS CAN BE AN ACTIVE PROCESS
In the above example of secondary necrosis, the death of a cell undergoing necrosis can be an active process. That is, a cell can participate in its death by virtue of molecularly controlled processes. Several forms of regulated (or “programmed”) necrosis exist, and we discuss those that are best understood in this review. In considering these, it might be helpful to think about the concepts of suicide and sabotage discussed in Green (2022d); it could well be that some of these processes did not evolve for the “purpose” of killing the cell but rather represent a normal cellular activity gone awry. In at least one case, however, regulated necrosis appears to have arisen as a bona fide cell suicide. This is the process of necroptosis.
DEATH RECEPTORS CAN CAUSE NECROPTOSIS
Death receptors of the tumor necrosis factor receptor (TNFR) family can trigger an apoptotic pathway (Green 2022a). However, in some cases, they can also induce necrosis, although this form of cell death has some features of type II (autophagic) cell death (discussed later in this review). The necrotic form of cell death induced by death receptors is referred to as necroptosis.2
When TNF engages its death receptor (TNFR1), this activates a complex of TRADD–TRAF–RIPK1 that dissociates from the receptor and recruits the adapter FADD and caspase-8 (see Green 2022a), and apoptosis proceeds. However, in many cells, if caspase-8 is inhibited, death ensues nevertheless. This death is necrotic and requires RIPK1. Unlike the activation of NF-κB by this receptor, wherein RIPK1 performs a scaffolding function (see Green 2022a), necroptosis induced through TNFR1 requires the serine/threonine (Ser/Thr) kinase activity of RIPK1 (Fig. 3).
Figure 3.

Tumor necrosis factor receptor (TNFR) signaling engages necroptosis if FADD–caspase-8–FLIP activity is blocked or overwhelmed.
Inhibitors called necrostatins block the kinase activity of RIPK1 and prevent necrosis induced by TNF. They can also reduce cell death caused by ischemia–reperfusion injury (discussed below) and pathological cell death in other settings, although, in these cases, whether this is caused by TNF, another death ligand, or other signals that engage RIPK1 is not known.
RIPK1 recruits and activates another Ser/Thr-protein kinase, RIPK3, that is also required for TNF-induced necrosis. RIPK1 does not phosphorylate RIPK3, but instead the active RIPK1 binds to RIPK3, and this binding activates it. Cells lacking RIPK1 or RIPK3 do not die by TNF-induced necroptosis, and, if caspase-8-induced apoptosis is also inhibited, the cells survive TNF treatment.
OTHER STIMULI CAN INDUCE NECROPTOSIS
As it turns out, it is the activation of RIPK3 that is important for necroptosis, and there are ways in which RIPK3 can be activated independently of a requirement for RIPK1 (although, see below). Both RIPK1 and RIPK3 contain a small region called the RIP-homology interaction motif (RHIM) that mediates the interaction of RIPK3 with RIPK1 and other proteins. One of these is TIR-domain-containing adapter-inducing interferon (TRIF), an adapter molecule engaged by some Toll-like receptors (TLRs). Another is an intracellular sensor of viral nucleic acids called Z-DNA-binding protein 1 (ZBP1, also called DAI) (Fig. 4). By the binding of these proteins to RIPK3 through RHIM–RHIM interactions, RIPK3 oligomerizes and activates its kinase activity to promote necroptosis. Interferons can also induce the activation of RIPK3, although the mechanism is less clear.
Figure 4.

Other pathways can activate RIPK3 and necroptosis. Some Toll-like receptors (e.g., TLR3, TLR4) engage the adapter protein TRIF when they are activated by their ligands. TRIF contains a RIP-homology interaction motif (RHIM) that interacts with the RHIM of RIPK3 directly, activating the kinase. ZBP1 (DAI) is an intracellular sensor of viral nucleic acids, and it too can directly activate RIPK3 via RHIM–RHIM interactions. The figure shows activation of necroptosis in the absence of RIPK1. When RIPK1 is present, the interactions are more complex. The function of MLKL is discussed below.
MLKL IS THE “WEAPON” OF NECROPTOSIS
The crucial substrate for RIPK3 in necroptosis is a pseudokinase called MLKL (for “mixed-lineage kinase domain-like protein”). A pseudokinase structurally resembles kinases but lacks kinase activity. When MLKL is phosphorylated by RIPK3, it undergoes a conformational change (essentially identical to that of an activated kinase) that exposes a unique region of the protein, located near its amino terminus (Fig. 5). This region comprises a “bundle” and a “brace.” The brace region can self-associate, and this functions to oligomerize the activated protein.
Figure 5.

Structure of the mixed-lineage kinase domain-like protein MLKL. (Left) The amino-terminal bundle, brace, and pseudokinase domain are shown. (Right) MLKL is the weapon of necroptosis. (Model made by Dr. Scott Brown, St. Jude Children's Research Institute.)
One way that the function of the amino terminus has been studied is by the use of artificial dimerization methods (further discussed in Green 2022e). If only the amino-terminal bundle of MLKL is fused to a so-called dimerization domain, a dimeric drug that binds this domain will force the bundle into dimers, and this is sufficient to kill the cell (Fig. 6).
Figure 6.

Dimerizing the amino-terminal bundle of the mixed-lineage kinase domain-like protein MLKL. (A) Scheme of a chimeric protein that can be expressed in cells. The amino-terminal bundle of MLKL is fused to a domain (FKBP) that can be bound by a cell-permeable dimeric drug that can connect two chimeric monomers. (B) A three-dimensional electron micrograph of cells expressing the construct in A. Without the dimerizer agent, the plasma membrane remains intact (left), but, upon addition of the dimerizer (right), the plasma membrane is rapidly destroyed, although intracellular membranes appear unaffected. (3D scanning electron micrographs by Dr. Giovanni Quarato and Dr. Sharon Frase, St. Jude Children's Research Institute.)
When activated, MLKL oligomerizes, and the amino terminus of the protein interacts with the plasma membrane, disrupting it. As a result, the cell dies by necroptosis. But, along the way, something interesting happens. As in apoptosis, this cell death process induces the exposure of phosphatidylserine on the outer leaflet of the plasma membrane, before the loss of plasma membrane integrity. Neither the lipid scramblase involved in apoptosis (Xkr8; see Green 2022f) nor the calcium-ion-responsive scramblase (TMEM16F) are involved in this effect, and it is possible that it is active MLKL, itself, that causes the externalization of phosphatidylserine.
It is likely, although not definitively known, that the activation of MLKL is the final step in necroptosis—that is, that MLKL is the “weapon” that destroys the plasma membrane (Fig. 5). However, MLKL might have one or more additional targets that actually do the killing. Some studies have suggested that MLKL interacts with and opens ion channels in the cell, and these are what ultimately kill the cell. It remains possible, however, that this is a side effect of the action of MLKL as attempts to prevent death by reducing ions in the extracellular milieu (thus limiting their influx) appear to be cell-type-specific, and at best only partially work. Furthermore, although there is an influx of calcium ions upon MLKL activation, this is dispensable for the externalization of phosphatidylserine that precedes the loss of plasma membrane integrity.
RIPK3 is the only kinase known to activate MLKL (the pathways are shown in Figs. 3 and 4). But RIPK3 (and RIPK1) have other effects in the cell, including the induction of inflammatory mediators. It is therefore difficult to define a role for necroptosis in a biological process (or disease) based on roles for RIPK1 or RIPK3. MLKL involvement is perhaps a more definitive measure, and the field has moved to define necroptosis as “cell death that is dependent on MLKL.” Should we find conditions in which MLKL is activated independently of RIPK3, we will most likely consider the resulting cell death necroptosis.
Necroptosis appears to be a fairly recent evolutionary “invention,” found only in some vertebrates. The key components, RIPK1, RIPK3, and MLKL, are found in bony-jawed fishes, amphibians, and reptiles, but RIPK3 is absent in birds, and MLKL is present in some fish but not others. Although most mammals have these components, marsupials lack RIPK3 and MLKL, and carnivores lack MLKL. Why some vertebrates have the necroptosis machinery and others lack it is a mystery.
Why do cells have this backup mechanism for cell death? One likely answer relates to viruses. As we mentioned, viruses deploy strategies to inhibit caspase-8 as a way to block apoptosis that is induced in host cells by cells of the immune system. When this occurs, the activation of MLKL and the destruction of the cell by necroptosis is a good alternative to combat the infection.
CASPASE-8 AND FLIP INHIBIT NECROPTOSIS
Inhibition of caspase-8 allows TNF to induce necroptosis rather than apoptosis, and therefore caspase-8 would appear to inhibit this pathway. Indeed, caspase-8 cleaves RIPK1, preventing its activity, and this appears to be how caspase-8 blocks necrosis. But, if activation of caspase-8 commits the cell to die by apoptosis anyway, can inhibition of necroptosis preserve the cell? When TNF binds to TNFR1, it induces the activation of nuclear factor-κB (NF-κB), and this transcription factor induces the expression of FLIP (FLICE-like inhibitory protein), which, as we have seen, is a protein that is related to the caspases but lacks a catalytic site. Like caspase-8, FLIP can be recruited to the death-inducing signaling complex (DISC) in the death receptor pathway. At the DISC, FLIP forms a complex with caspase-8 that is proteolytically active but unstable because caspase-8 is not cleaved; consequently, FLIP prevents apoptosis. (As discussed in Green [2022a], FLIP can prevent apoptosis by not allowing the formation of oligomers of caspase-8.) However, this complex also prevents necroptosis. The caspase-8–FLIP dimer cleaves RIPK1 and RIPK3 in the complex and thereby ensures that the cell undergoes neither apoptosis nor necrosis and therefore survives (Fig. 7). Caspase-8–FLIP also cleaves CYLD, the deubiquitinase (DUB) that removes ubiquitin from RIPK1 (discussed in Green 2022a), and this cleavage might also function to restrict necroptosis.
Figure 7.

The FADD–caspase-8–FLIP complex inhibits necroptosis. RIPK1 binds to FADD, which in turn is bound to caspase-8–FLIP. The catalytically active caspase-8–FLIP cleaves RIPK1 to prevent necroptosis.
Mice lacking caspase-8, FADD, or FLIP die during embryonic development because of extensive cell death in the endothelium (discussed in more detail in Green 2022g). Strikingly, if mice lacking FADD or caspase-8 also lack RIPK3 or MLKL, they survive. Furthermore, mice lacking FLIP can also survive if both FADD (promoting apoptosis) and either RIPK3 or MLKL are removed. Formally, these data allow the conclusion that the embryonic lethality caused by loss of caspase-8 or FADD is dependent on RIPK3 and MLKL. This makes sense in the context of the pathways we discussed. In the case of the embryonic lethality caused by loss of FLIP, we can say that it is dependent on RIPK3 and MLKL, and on FADD (and probably caspase-8).3
Again, this makes sense.
In each case, the mice develop and are born normally, but, as they age, they display the symptoms of acute lymphoproliferative syndrome (ALPS; see Green 2022a), with massively enlarged lymphoid organs. This disease is seen in mice and humans with defects in CD95 or its ligand CD95-L. We can conclude from these observations that the function of CD95 and CD95-L in controlling ALPS is likely to be dependent on cell death signaling by the receptor.
TWO FACES OF RIPK1
Mice that lack RIPK1 do not die during development but instead die shortly after birth. Deletion of RIPK3 or MLKL does not prevent this perinatal lethality, but animals lacking RIPK1, either FADD or caspase-8, and either RIPK3 or MLKL survive well into adulthood, until they succumb to ALPS.
These findings tell us several important things, all of which have been confirmed in experiments with cells: RIPK3 can be activated (and can engage MLKL) without a requirement for RIPK1 (see Fig. 8); RIPK1 can inhibit such RIPK3 activation; and RIPK1 also functions to inhibit apoptosis mediated by FADD and caspase-8. When RIPK1 is absent, cells become sensitive to both caspase-8-dependent apoptosis and some triggers of necroptosis.
Figure 8.

The two faces of RIPK1. (A) The complex inhibitory “balancing act” between FADD–caspase-8–FLIP and RIPK3–MLKL interactions depends on RIPK1 to prevent both apoptosis and necroptosis, resulting in normal development. (B) When RIPK1 is removed, both apoptosis and necroptosis occur, and signals promote lethality. (C) Removal of either RIPK3 or MLKL in RIPK1-deficient animals (or cells) does not restore survival in RIPK1-deficient animals (or cells), as apoptosis still occurs. (D) Removal of either FADD or caspase-8 in RIPK1-deficient animals does not restore survival, as necroptosis still occurs. (E) Removal of either RIPK3 or MLKL plus either caspase-8 or FADD allows RIPK1-deficient animals to survive.
It is likely that RIPK1 inhibits necroptosis in at least two ways. First, it recruits FADD through DD–DD interactions, which recruits caspase-8 and FLIP (as we have seen). When RIPK1 is absent, necroptosis induced by signals that directly engage RIPK3 (such as TLR, ZBP1, or interferon signaling) proceeds even if caspase activity is not prevented. Second, when RIPK1 is in its inactive conformation (e.g., upon addition of necrostatins), the RIPK1 appears to block RIPK3 activation. Again, when RIPK1 is absent, necrostatins have no effect on necroptosis induced by direct activation of RIPK3.
RIPK1 also prevents the activation of apoptosis, especially by the ligation of death receptors. Most likely this is because RIPK1 functions in the activation of NF-κB (as we have seen), which promotes expression of anti-apoptotic proteins, such as FLIP.
But RIPK1 can also promote both necroptosis and apoptosis in some settings. As described above, necroptosis induced by the ligation of death receptors requires RIPK1 to bind and activate RIPK3. When the kinase activity of RIPK1 is inhibited (e.g., by necrostatin), it prevents not only death-receptor-induced necroptosis, but also necroptosis induced by signals that directly engage RIPK3 (such as TLR, ZBP1, or interferon signaling). This inhibitory activity of RIPK1 has confused the interpretation of effects of RIPK1 inhibitors, leading to conclusions that RIPK1 is always required for necroptosis.4 The studies with RIPK1-deficient animals and cells show that this is often not the case.
Because RIPK1 also binds to FADD (to recruit and activate caspase-8–FLIP heterodimers), RIPK1 can also promote apoptosis when FLIP is limiting. Interestingly, RIPK3 that is kinase inactive (owing to use of inhibitors or mutations) can activate this pro-apoptotic effect of caspase-8. Mice with a kinase-inactivating mutation in RIPK3 die during embryogenesis at the same time as do FADD- or caspase-8-deficient mice. However, removing caspase-8 protects these RIPK3 mutant animals, which are born and develop to adulthood normally (but, as with animals deficient in caspase-8 and RIPK3, these mice succumb to ALPS).
NECROPTOSIS AND DISEASE
There is compelling evidence that necroptosis plays roles in many diseases, but this is complicated. All of the proteins involved in necroptosis (RIPK1, RIPK3, and MLKL) have additional signaling features that can induce inflammation by the expression of cytokines and other mediators. Therefore, any interpretation that “necroptosis causes disease” must be treated with care.
Conditional deletion of caspase-8 in the intestinal epithelium or in the skin causes massive inflammation that is completely prevented by removing RIPK3, and therefore might be due to necroptosis in these tissues (with the caveat above). Similarly, conditional deletion of caspase-8 in endothelium causes embryonic lethality, which is again dependent on RIPK3 and MLKL. In contrast, several tissues and cell types are resistant to the effects of deletion of caspase-8, including heart, skeletal muscle, liver, and neurons.
Although the liver (in mice) appears to be resistant to necroptosis, this is not the case when animals are fed a high-fat diet. Animals with conditional deletion of caspase-8 in the liver become sensitive to liver damage and inflammation when fed on such a diet, and this is prevented by genetic removal of RIPK3. One effect of the diet is to induce expression of MLKL in the liver, thus sensitizing it for necroptosis. Another consequence of a high-fat diet is atherosclerosis, and animals that are genetically predisposed to this disease are protected by deletion of RIPK3.
Other liver diseases also appear to depend on RIPK3, including ethanol- and acetaminophen-induced liver damage (however, given the resistance of liver to necroptosis in conventionally fed animals, it is possible that this is not due to necroptosis).
Other models of disease also appear to be dependent (at least in part) on RIPK3. These include necrotizing pancreatitis (Fig. 9), retinal detachment, and acute peritonitis. Intriguingly, in two mouse models of amyotrophic lateral sclerosis (ALS), animals with a kinase-inactive RIPK1 (which inhibits necroptosis) were strikingly protected.
Figure 9.

RIPK3 and pancreatic injury. Wild-type (left) or RIPK3-deficient (right) mice were treated with an agent that induces necrotic injury in the pancreas. Animals without RIPK3 were protected. This protection has also been described in animals lacking the mixed-lineage kinase-like protein MLKL. (Reprinted from He et al. 2009, ©2009 with permission from Elsevier.)
Necroptosis can also be protective. As we noted, most evidence suggests that necroptosis evolved as a mechanism of defense against viruses, and the death of an infected cell can limit disease. Animals lacking RIPK3 are sensitive to several types of viruses, including vaccinia, influenza, and West Nile virus. Interestingly, mice lacking MLKL do not show this sensitivity to influenza, but mice lacking both FADD and MLKL are as sensitive (or more so) as animals lacking RIPK3. At least in this case, the role for RIPK3 in influenza infection might be both in apoptosis and in necroptosis. In the case of West Nile virus, however, the protective role of RIPK3 ablation does not arise through the inhibition of cell death but, rather, because RIPK3 promotes harmful inflammation. Again, it is important that care is used when concluding that necroptosis is involved in a pathology based on effects of removing RIPK3.
Antibodies that detect the active, phosphorylated form of human MLKL are likely to be useful as indicators of where necroptosis occurs in diseased human tissues. These studies are under way.
PARP AND NECROSIS
The repair of damaged DNA is energetically costly, and, if the costs of repair exceed the resources available to the cell (ATP and NADH), necrosis can result. In particular, the DNA repair enzyme poly(ADP-ribose) polymerase 1 (PARP1) engages processes that consume a large amount of NAD+, which the enzyme uses to generate the ADP-ribose needed for the repair process. But NAD+ is also crucial for energy metabolism, and many metabolic reactions are dependent on a form of this molecule (NAD+, NADH, NADP+, and NADPH). PARP can therefore promote necrotic cell death in some circumstances by using up NAD+, especially if nutrients are limited.
DNA damage can also trigger apoptosis through signaling mechanisms, including the action of p53 (see Green 2022h). However, if DNA damage is very extensive, this can result in a form of necrosis5 that can be blocked by pharmacological inhibitors of PARP or additional sources of NADH. For example, the addition of nicotinamide can reduce necrosis induced by high-dose radiation.6
Reactive oxygen species (ROS) can damage DNA, and high levels of these can also cause cell death that depends on PARP. Mice lacking PARP develop normally but are resistant to neuronal damage induced by hydrogen peroxide, one source of reactive oxygen. We will see below other ways in which ROS that lead to necrosis can be generated.
EXCITOTOXICITY
Neurons can undergo active necrosis in response to high levels of glutamate, which functions as a neurotransmitter in the brain. This can occur as a consequence of ischemic injury (i.e., stroke) in the brain (discussed in detail below). Glutamate-induced neuronal necrosis is often referred to as excitotoxicity or excitotoxic death.
When neurons are exposed to high levels of glutamate, this causes an influx of calcium ions into the cells. This, in turn, activates a complex enzyme, NADPH oxidase, that produces ROS. Originally, NADPH oxidase was said to occur only in neutrophils, which use it to destroy bacteria and other pathogens. But we now realize that many cells in the body express this or a related form of this enzyme, which is more simply referred to as Nox.
Nox produces ROS by moving an electron from NADPH to oxygen to make superoxide. But, unlike the electron-transport chain of mitochondria, this does not generate energy but instead produces protons. These must be transported out of the cell, which generates a high charge difference across the plasma membrane. This charge is seen as excitation in glutamate-exposed neurons. As a result of the superoxide, PARP is activated, and NAD+ is depleted (Fig. 10). The loss of energy in the cells promotes their death by necrosis. Inhibitors of Nox prevent both PARP activation and death by excitotoxicity.
Figure 10.

One view of excitotoxicity. Calcium influx triggers NADPH oxidase, which produces reactive oxygen species (damaging DNA and inducing the poly(ADP-ribose) polymerase PARP) and hydrogen ions (creating plasma membrane charge). Necrosis occurs as a consequence of energy depletion.
ISCHEMIA–REPERFUSION INJURY
Ischemic injury occurs when the blood supply to a tissue, such as the heart or brain, is disrupted. Not surprisingly, deprivation of oxygen and nutrients can cause necrosis. However, when the blood supply is restored (“reperfusion”), this often causes a wave of cell death that can have catastrophic consequences. In the heart, for example, the extent of cell death is directly linked to remodeling events that, over time, cause heart enlargement, accompanied by thinning of the muscle walls and ultimately heart failure. This is why heart attacks so often predispose individuals to subsequent heart attacks. Controlling the extent of damage from ischemia–reperfusion injury is a major therapeutic goal.
Why does ischemia–reperfusion cause so much damage? Part of the answer relates to changes in the levels of potassium ions in the cells. Following reperfusion, potassium channels open in the plasma membrane, causing a drop in intracellular potassium that triggers a range of changes, including an increase in intracellular calcium ions. As we have seen, this can activate NADPH oxidase.7 In addition, the increase in calcium can also activate the protease calpain, which at high levels can cause necrosis (Fig. 11). Animals that are treated with inhibitors of calpain, or that lack this enzyme, show reduced damage as a result of ischemia–reperfusion injury.
Figure 11.

One view of ischemia–reperfusion injury. Reperfusion induces an opening in potassium channels, and, in turn, the change in potassium opens calcium channels. The influx of calcium triggers NADPH oxidase, and its consequences, and activates calpain, a protease. Death can be a consequence of energy depletion, calpain action, or both. Other forms of cell death contribute to this form of injury, including apoptosis and probably necroptosis and ferroptosis (see below).
Mitochondria are important in apoptosis (see Green 2022i,j), but they can also have an important role in necrosis. As discussed in Green (2022i), mitochondria can respond to high levels of calcium, ROS, and other signals by opening a channel in the inner membrane, the permeability transition pore (PTP). This causes the mitochondrial matrix of the organelle to swell, eventually rupturing its membranes, resulting in the so-called mitochondrial permeability transition (MPT) (Fig. 12). One protein that we saw to be clearly involved is the peptidylproline isomerase cyclophilin D. Mice lacking cyclophilin D are developmentally normal, but mitochondria from these mice show defective PTPs.8 These mice are strikingly resistant to ischemia–reperfusion injury of the heart and brain. The simplest explanation for this is that the MPT has a role in the damage. The MPT can contribute to necrosis by both destroying mitochondrial energy generation and ablating the ability of mitochondria to scavenge reactive oxygen (Fig. 12).
Figure 12.

The mitochondrial permeability transition (MPT) in ischemia–reperfusion injury. Elevated calcium induces the MPT, which disrupts energy generation and curtails scavenging of reactive oxygen species (ROS). Mice lacking the MPT are resistant to ischemia–reperfusion injury.
Cyclosporin A, a drug that inhibits cyclophilins, including cyclophilin D, limits ischemia–reperfusion injury in rodents and can reduce heart damage in humans. It is tempting to believe that this is because the drug blocks the MPT. However, it might not be so simple. Cyclosporin A undergoes a conformational change when it binds to cyclophilins, and the complex of the drug and cyclophilins in the cytoplasm is a potent inhibitor of the enzyme calcineurin, and this effect might be more important.9
Ischemia–reperfusion injury might also involve necroptosis. Animals lacking RIPK3 are somewhat protected from such injury in the kidney and heart. Such injury involves inflammation, and it is possible that this is an effect of TNF and perhaps other death ligands on the endothelium and the kidney epithelium.
The damage in ischemia–reperfusion injury is not limited to necrosis; indeed, the area around the initial damage undergoes extensive apoptosis. This might be because of signals such as ROS released from the necrotic cells or the effects of inflammatory cells that are activated by the necrosis (discussed in more detail in Green 2022g). Inflammatory cells also produce ligands for death receptors (see Green 2022a). Mice lacking one of the death receptors, CD95, show reduced injury following ischemia–reperfusion. As we have seen, however, signaling from death receptors can also induce necroptosis. As mentioned above, necroptosis can also contribute to the damage observed in ischemia–reperfusion injury.
Another type of cell death that can be important in ischemia–reperfusion injury is ferroptosis. This form of regulated necrosis is considered next.
FERROPTOSIS
All eukaryotes and some bacteria synthesize glutathione, which in its reduced form (GSH) is an important antioxidant for scavenging ROS. The oxidized form (GSSG) can be enzymatically converted to the active, reduced form if NADPH is available. Therefore, glutathione and NADPH are essential to control ROS in our cells.
Glutathione is produced by an enzymatic pathway from three amino acids: glutamate, cysteine, and glycine. Cysteine is taken up in cells as cystine, in large part by the function of a cell-surface transporter, known as the cystine/glutamate antiporter or simply system Xc−. If cells are deprived of cystine, or system Xc− is impaired, glutathione levels decline and cells die.
It turns out that this cell death is not apoptotic, but is instead a regulated form of necrosis, called ferroptosis. Iron that is taken up in cells by transferrin and its receptor is required for ferroptosis (hence the name), and this metal reacts with hydrogen peroxide to generate ROS. These act on lipids, in particular polyunsaturated fatty acids in the cell, to generate lipid peroxides that are toxic (Fig. 13). This is because, if unchecked, lipid peroxides can act on other lipids to create more lipid peroxides, in a chain reaction that can damage the membranes of the cell, as well as producing other toxic species.
Figure 13.

Ferroptosis. Iron (Fe2+) is transported into cells by transferrin, where it can catalyze the oxidation of lipids by hydrogen peroxide (H2O2) in the cell to form toxic lipid peroxides. System Xc− imports cystine (exporting glutamate), which is converted to cysteine and used in the generation of glutathione. The enzyme GPX4 reduces lipid peroxides but requires glutathione to regenerate its reducing potential. Disruption of System Xc−, glutathione synthesis, or GPX4 function can therefore result in ferroptosis.
A glutathione peroxidase enzyme, GPX4, uses glutathione to reduce lipid peroxides and is the only known way that these can be reduced in cells. Inhibition of GPX4 also induces ferroptosis, without a concomitant loss of glutathione.
Ferroptosis appears to be important in many forms of ischemia–reperfusion injury. It also suggests an alternative mechanism for glutamate-induced excitotoxicity (discussed above) as excess extracellular glutamate can inhibit the function of system Xc− to import cystine.
AUTOPHAGIC (TYPE II) CELL DEATH
Autophagic cell death is characterized by the presence of large vacuoles in the cytoplasm, as well as molecular markers of autophagy (see below). In general, the characteristic features of apoptosis, including plasma membrane blebbing, nuclear condensation, and chromatin fragmentation, are not seen, and caspases do not have a role.
Autophagic cell death might be a “victim” of its name; for the most part, neither autophagy (primarily a survival mechanism) nor molecular components of the autophagy pathway are responsible for autophagic cell death. There are exceptions and some tantalizing observations, as we will see. But most instances of autophagic cell death appear to be accompanied by autophagy, rather than being caused by it. In fact, autophagy appears to antagonize cell death, and inhibition of autophagy can dramatically increase it. This is a controversial area, and there are sure to be important revelations in the coming years.
AUTOPHAGY AS A SURVIVAL MECHANISM
In general, autophagy is considered a survival mechanism for cells. Formally, there are three types, but here we are only concerned with macroautophagy.10 This is usually referred to simply as “autophagy,” and we use this nomenclature here.
Autophagy is found throughout the eukaryotes, and there is remarkable conservation of the components of the central pathway. It provides energy when external nutrients are depleted or the pathways for taking them up are disrupted, removes excess or damaged organelles and other cellular components, and is involved in the degradation of long-lived proteins and protein aggregates in cells. Autophagy also functions in cellular defense to isolate and destroy invading organisms.
Because autophagy is an important survival mechanism, it is useful to go into some detail about how it works. This will also help us to understand how it might be engaged to kill cells as well.
THE AUTOPHAGY PATHWAY
Autophagy involves the generation of membrane vesicles in the cell, called autophagosomes, that enclose cytoplasm, organelles, protein aggregates, or invading organisms and carry them to lysosomes, with which the autophagosomes fuse (forming autophagolysosomes). The degradative enzymes in the lysosomes break down the contents that are then reused as sources of energy and raw materials by the cell (Fig. 14).
Figure 14.
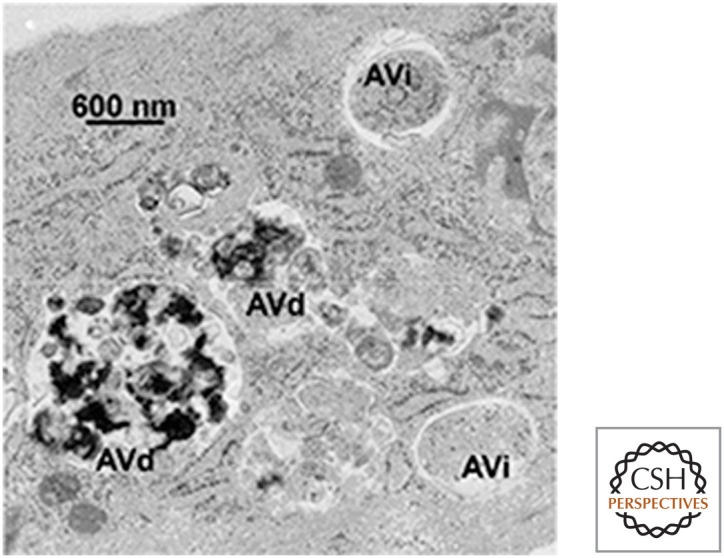
Autophagosomes. Shown are early autophagosomes (AVi) and autophagolysosomes (AVd). (Reprinted from Eskelinen 2008, ©2008 with permission from Elsevier.)
Autophagy in mammals occurs by the hierarchical action of three complexes that form in a region of the endoplasmic reticulum (facing the cytosol). The first is a complex that contains a Ser/Thr-protein kinase (ULK1) together with proteins required for its kinase function. This activates a second complex that contains a lipid kinase, the phosphoinositide 3-kinase (PI3K) VPS34, together with additional proteins, including beclin-1. The action of this complex recruits a third complex, which functions as a ligase (akin to the E1–E2–E3 ligases of the ubiquitylation pathway). Its function is to place a small protein (LC3 or a related protein) onto a lipid (phosphatidylethanolamine) present in the endoplasmic reticulum membrane. It is likely that cytoskeletal components now “pull” the membrane as a sheet. Each complex recruits the next,11 ultimately extruding a double-membrane structure called a “phagophore.” The phagophore seals to engulf cytoplasm, the complexes dissociate, and the result is an autophagosome (Fig. 15).
Figure 15.

Simplified, hierarchical autophagy pathway. (Left) At the cytosolic face of the endoplasmic reticulum (ER), the serine kinase complex, activated, for example, by conditions of nutrient restriction, recruits and activates the phosphoinositide 3-kinase complex, which, in turn, promotes the assembly of the ligase complex. The latter directly conjugates the small molecules of the LC3 family to lipid in the ER membrane. (Center) The membrane is then extruded to form the phagophore. (Right) The phagophore seals to form a double-membrane structure, the autophagosome, trapping cytosolic material.
MAMMALIAN AUTOPHAGY IN DETAIL.
Autophagy in mammals is engaged by the pre-initiation complex.12 This contains the kinase mentioned above, ULK1, plus ATG13 and a protein called FIP200. When autophagy is activated by starvation or cellular damage, ATG13 dissociates and recruits the next complex in the pathway. This complex contains beclin-1 (see Green 2022j) and a class III PI3K called VPS34, together with other components. This generates the lipid phosphatidylinositol 3-phosphate (PtdIns3P, or PI3P) that recruits the next components of the autophagy pathway by a bridge molecule (WIPI2) that binds to PI3P.
ATG7, an enzyme similar to a ubiquitin E1-ligase, binds to a small protein called ATG12 and passes it to an E2-like enzyme, ATG10, which then places it on another protein, ATG5. The ATG5–ATG12 complex recruits a further protein, ATG16, and this forms the E3-ligase, necessary for the next step in the process (Fig. 16). ATG16, carrying ATG5–12, is bound by WIPI2, bringing the E3-ligase to the membrane.
Figure 16.

The initial steps in autophagy.
ATG7 again acts as a ligase, passing LC3 or related molecules13 to ATG3, which then places LC3 onto a lipid, phosphatidylethanolamine (PE).14 This LC3–PE complex is the building block of the phagophore, which begins to grow (Fig. 17). When it is complete, the other proteins peel off from the membrane, and it is now an autophagosome, ready to fuse with a lysosome.
Figure 17.

LC3 and the phagophore. The interaction of motor proteins with lipidated LC3 in the membrane to promote membrane extrusion is speculative. LC3 proteins also function at a later step in the fusion of the phagophore to form the autophagosome.
ENGAGING THE AUTOPHAGY PATHWAY
Autophagy is inhibited in several ways, and how this inhibition is itself regulated provides some clues as to how autophagy is engaged in the cell. Although we consider two mechanisms for induction of autophagy, it is worth noting that there are almost certainly other ways in which this pathway can be triggered.
One primary regulator of autophagy is also a key regulator of cell growth, metabolism, and protein synthesis, a kinase called mTOR. It is found in two different protein complexes, TORC1 and TORC2, but it appears to be TORC1 that controls autophagy by phosphorylating and inhibiting ATG13, a component of the ULK1 kinase complex. When nutrients are plentiful, mTOR is active—cells grow and autophagy is inhibited. However, when energy levels are low, adenosine monophosphate (AMP) accumulates, and this activates a Ser/Thr-protein kinase, AMPK, that inhibits mTOR, and autophagy is engaged. AMPK also phosphorylates ATG13, but this is at a site different from that used by TORC1, and this actually activates ATG13. Similarly, growth-factor receptor signaling activates AKT (see Green 2022j), and this inhibits a complex, TSC1/2, that is an inhibitor of mTOR. When AKT is inactive, TORC1 is inhibited, and autophagy proceeds(Fig. 18). The drug rapamycin is a potent inhibitor of TORC1 (in fact, mTOR stands for “mammalian target of rapamycin”), and the addition of this drug to cells induces autophagy.
Figure 18.

mTOR and autophagy.
Another way in which autophagy is regulated involves BCL-2 (and possibly other anti-apoptotic BCL-2 family proteins), which can bind and sequester beclin-1 using the same BH groove used to sequester BH3-only proteins in apoptosis. Indeed, beclin-1 has a BH3 domain, although this binds weakly to BCL-2 compared with the binding strength of others discussed in Green (2022j). It seems that if BH3-only proteins are expressed, they can neutralize the interaction between BCL-2 and beclin-1, and, if MOMP does not occur (i.e., BAX and BAK are not activated), autophagy can result.15
Autophagy can also be caused by DNA damage. This induces UVRAG, a protein that promotes the activity of VPS34 in the beclin–PI3K complex (see above). Autophagy in this setting might provide additional energy needed for DNA repair.
In each of these scenarios, autophagy protects cells. If it is blocked, apoptosis ensues. However, when apoptosis is blocked and autophagy is active (e.g., by removal of BAX and BAK), autophagy will keep the cells alive for extended periods, wearing them down to mere “skeletons” of their former selves (Fig. 19). If autophagy in stressed cells is also blocked, the cells die by necrosis.
Figure 19.
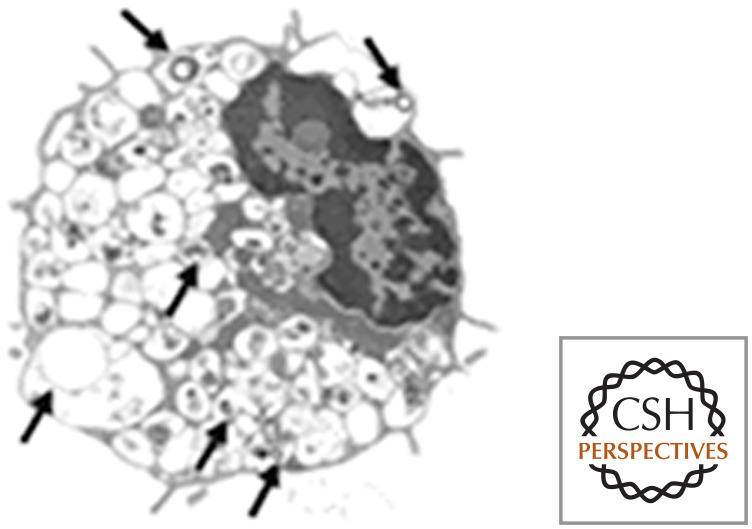
An autophagic survivor. Arrows indicate autophagosomes. (Reprinted from Lum et al. 2005, ©2005 with permission from Elsevier.)
MITOPHAGY
Mitophagy is a selective form of autophagy that specifically captures damaged mitochondria in autophagosomes and destroys them in the lysosomes. It is important for the quality control of mitochondria; defects in mitophagy lead to accumulation of defective mitochondria that produce ROS and can inflict damage on the cell. As noted in Green (2121j), mitophagy is also engaged under conditions in which the numbers of mitochondria must be reduced, such as in hypoxia and in the development of some cell types such as red cells.
There are several mechanisms by which mitophagy is engaged, one of which is through the functions of BNIP3 and NIX (Green 2022j). The targeting of defective mitochondria, however, operates through a different process.
Healthy mitochondria generate a charge across the inner mitochondrial membrane (ΔΨm) that is produced as a consequence of the electron-transport chain or by reversal of the ATPase complex (if electron transport is not active). ΔΨm is essential for the import of proteins into the mitochondria and for other mitochondrial functions. One protein that is imported into mitochondria is a Ser/Thr-protein kinase, PINK1. When PINK1 is imported into the mitochondria, it is degraded by proteases; however, if the import machinery is inactive (owing to loss of ΔΨm), PINK1 accumulates on the surface of the damaged mitochondrion. PINK1 then recruits and activates an E3-ligase, parkin, which ubiquitylates proteins on the mitochondria, including a protein involved in mitochondrial fusion, MFN2, which is also involved in this form of mitophagy (in this case, the fusion activity of MFN2 is not involved in the process). The autophagy machinery is then recruited to the damaged mitochondrion, which is then removed (Figs. 20 and 21).
Figure 20.

Mitophagy mediated by PINK1–PARKIN.
Figure 21.

Mitophagy in action. Immunoelectron microscopy showing double-membrane autophagosome with associated LC3 (black dots), enclosing a damaged mitochondrion. (Courtesy of Ynjie Wei and Beth Levine.)
Because mitochondria are important in several forms of cell death, the process of mitophagy and its role in sustaining mitochondrial quality is thereby linked to cell death pathways.
WHAT IS AUTOPHAGIC CELL DEATH?
The above considerations help to clarify why a stressed cell that can undergo apoptosis engages the autophagic pathway as a result of the stress. Therefore, autophagy and apoptosis can occur in the same cell, but in such cases this is apoptosis and not autophagic cell death. However, cells often die without displaying features of apoptosis, instead displaying features of autophagy. In such cases, this is called autophagic cell death (type II cell death), and it is seen in the response of tumor cells to several therapeutic agents. Usually, this is cell death that is associated with, but not caused by, autophagy, and if autophagy is blocked, the cells die by necrosis. Most autophagic cell death has this feature.16
WHEN AUTOPHAGY KILLS
Nevertheless, in some cases, it appears that autophagy can indeed promote (type II) cell death. The best-studied case is in the metamorphosis of the fruit-fly Drosophila (discussed in Green 2022h). Larvae have very large salivary glands that die during metamorphosis. Some of this death is by apoptosis, but most of it has the form of type II cell death. If autophagy genes are defective in the developing fly, much of the cell death does not occur and the development is prevented.
In mammals, there are several examples wherein type II cell death appears to be promoted by autophagy.17 One way that this can occur is if the autophagic process is activated but for some reason is not completed before the contents of the autophagosome are degraded in lysosomes. As a result, the cytoplasm is disrupted, but the cell does not gain nutrients from the process, leading to death.18 Alternatively, it has been proposed that another way in which autophagy promotes cell death is by the removal of catalase, a long-lived protein that neutralizes some ROS. We can also imagine that, if autophagy removes other key survival proteins as well, cell death could result.
Although mitophagy, as discussed above, is important in sustaining healthy mitochondria in a cell, excessive mitophagy can kill a cell, either by apoptosis or necrosis. The antibiotic valinomycin is an ionophore that induces apoptosis that is dependent on PINK1 and parkin (see above), and causes widespread mitophagy. It is not known how this results in apoptosis, but it is likely that cytochrome c is released in the process, as the apoptosis depends on APAF1. In other settings, extensive mitophagy can compromise the metabolic fitness of a cell, resulting in a loss of ATP and necrosis.
There is another interesting possibility, albeit untested. As we have discussed, autophagy involves fusion of the autophagosome with lysosomes in the cell. It is conceivable that, if the process accelerates, so that incompletely formed autophagosomes engage lysosomes, the contents of the latter might be released into the cytosol. This might result in autophagic cell death as the destructive contents of the lysosome set to work. As mentioned above, large vacuoles are often associated with autophagic cell death, and it is possible that they are produced by such an interaction. Support for this notion comes from studies on a protein called DRAM. DRAM is associated with lysosomal membranes, and its expression is induced in some pathways leading to cell death. In some cases, inhibition of the expression of DRAM can protect cells from what appears to be type II cell death.
Perhaps the most interesting possibility relates to necroptosis. As we discussed, cells lacking caspase-8 can die following exposure to TNF in a manner that depends on RIPK1 kinase activity. In some studies, inhibition of autophagy prevents this cell death. Similarly, T lymphocytes that lack caspase-8 or FADD fail to proliferate and die following activation, and this cell death is blocked (and proliferation restored) by inhibition of RIPK1 kinase. Intriguingly, there is evidence that disruption of autophagy can also rescue these T cells. It appears that the ATG5–ATG12 complex in the autophagic pathway can recruit the adapter FADD. FADD, caspase-8, and FLIP associated with ATG5–ATG12, might cleave and inhibit RIPK1 kinase to prevent cell death when autophagy is engaged. If caspase-8 is not active, RIPK3-mediated necroptosis ensues. This intriguing, but untested, scenario links at least some forms of autophagic cell death to necroptosis (Fig. 22).
Figure 22.

Hypothetical connection between autophagy and necrosis. Abundant ATG5–ATG12 on the phagophore can bind FADD. This might recruit RIPK1 and RIPK3 to signal for necroptosis.
Normally, during autophagy, ATG5–ATG12 dissociates from the membrane when the autophagosome forms. If the model shown in Figure 22 is correct, it might be that a determining factor in whether autophagy contributes to cell death is the continued presence of ATG5–ATG12 on the autophagosome membrane. This is only speculation, however.
MITOTIC CATASTROPHE
During mitosis, the nuclear membrane dissolves and chromosomes are segregated to the poles of the dividing cell. If this process is disrupted, cells die, and this is often called “mitotic catastrophe.” This can occur if, for example, the DNA is extensively damaged, the cell cycle machinery is stalled, or microtubules are dysfunctional.
Often, mitotic catastrophe results in apoptosis. How the mitotic machinery is linked to the control of apoptosis is not clear, but one possible mechanism is intriguing. Cyclin-dependent kinase 1 (CDK1), the kinase that orchestrates mitosis, phosphorylates caspase-2 (see Green 2022c), preventing its activation. When mitosis is not completed on schedule, CDK1 activity declines and caspase-2 becomes active. Cells might therefore use caspase-2 to activate apoptosis when mitosis fails. There are likely other mechanisms as well.
If cells are faced with defective mitosis and apoptosis is blocked, the cells die nevertheless, showing features of autophagic cell death or necrosis. An example of this is seen in the intestines of mice lacking BAX and BAK following irradiation (Fig. 23). How this cell death occurs remains obscure.
Figure 23.

γ-Irradiation causes nonapoptotic cell death in intestines lacking BAX and BAK. Mice with (left) or without (right) intestinal BAX and BAK were irradiated. Although apoptosis (green stars) was decreased, cell death nevertheless occurred in the deficient cells. This was associated with abnormal mitosis. (Reprinted from Kirsch et al. 2010, ©2010 with permission from AAAS.)
In this review, we considered forms of cell death that are not apoptosis. Different paths to cell death are interesting (and can have fundamental repercussions for therapeutic intervention), but does it matter to the body how a cell dies? The consequences of cell death go beyond the cell, and, indeed, the mode of cell death can influence these sequelae. Such consequences are explored in Green (2022b).
Footnotes
From the recent volume Cell Death: Apoptosis and Other Means to an End by Douglas R. Green
Additional Perspectives on Cell Death available at www.cshperspectives.org
It is unfortunate that “type I” can refer to a form of cell death as well as a way in which cells respond to death receptor signaling, leading to apoptosis (Green 2022a). In this review, all reference to “type I, II, or III” is to a form of cell death, not to the death receptor signaling response, per se. It is easy to get confused!
The term necroptosis is admittedly unwieldy, but useful for searching the literature for this particular form of cell death.
This was not tested for the technical reason that FLIP and caspase-8 are very close to each other in the genome, and therefore the double-null allele cannot be generated by simple crosses.
There is another problem with a commonly used RIPK1 inhibitor, necrostatin-1. It turns out that this inhibitor can have effects that are independent of RIPK1 (e.g., it can inhibit ferroptosis). This has led to gross misinterpretations in the literature. A more specific inhibitor of RIPK1 is necrostatin-1s. Thus, care should be taken in considering conclusions that are based only on the use of necrostatin-1.
It can be confusing to suggest that apoptosis is superseded by necrosis under some circumstances, and, in real life, the effects can be mixed. However, as we noted, the mechanisms are the key, and blocking one mechanism (e.g., apoptosis) will not necessarily block the other (e.g., necrosis).
Nicotinamide has multiple roles in cells, and it might be simplistic to conclude that its effect on cell death is only as a precursor for NADH and NADPH.
This is Nox1, a relative of the Nox that we have been discussing.
Remember that cells from these animals display normal apoptosis, and we therefore suspect that this is not an important mechanism for MOMP in apoptosis (see Green 2022i).
Calcineurin is important for the activation of a set of transcription factors called NFAT (it is the inhibition of NFAT activation that accounts for the ability of cyclosporin A to block T-lymphocyte activation and tissue rejection, for which it is used). NFAT also has important roles in the development of blood vessels and other functions, and, at this point, we simply do not know how much these contribute to the beneficial effects of the drug in ischemia–reperfusion injury.
The other two types are called microautophagy and chaperone-mediated autophagy.
This is only a model. However, the model is useful for understanding how autophagy might, in principle, work, and there is evidence to support it.
Also called the ULK1 complex because ULK1 is the catalytic subunit.
There are several LC3-like molecules that function in this regard. LC3 is actually MAP1LC3 and has several family members: LC3A, LC3B, and LC3C. Other LC3-like molecules that are similarly lapidated are GABARAP, GABARAPL1 and Gate16 (also called GABARAPL2), and GABARAPL3. It is possible that the LC3 and GABARAP subfamilies have different functions in the process, and both subfamilies are required for the biogenesis of autophagosomes
Another view holds that the ATG5–12 pathway and the LC3–PE pathways are parallel, both contributing to the construction of the phagophore without the hierarchical organization proposed here.
In Green (2022cj), one case in which this effect could be important was outlined. A BH3-only protein called NIX, which is not a potent potentiator of apoptosis, is important for the removal of mitochondria during the development of mammalian red blood cells. It is possible (although not formally proven) that NIX does this by disrupting the interaction between beclin-1 and BCL-2. Another BH3-only protein, BNIP3 (which is also a poor inducer of apoptosis), is related to NIX, and this protein might also be involved in the autophagic removal of mitochondria in some settings.
This might change as more systems are explored in vivo and new situations in which autophagy actually kills cells are identified.
Often, this has been shown by knocking down levels of expression of key proteins with siRNA. This is not without problems because siRNA can have off-target effects, including interferon responses and other unexpected consequences that could affect cell death. Here, however, we will assume that there is something to the idea that autophagy can promote cell death in mammals.
This can be performed pharmacologically, by the addition of an autophagy inducer such as rapamycin and a lysosome inhibitor such as chloroquine. This strategy is being explored as a therapeutic regimen for cancer. Some viruses also inhibit lysosomes, and the metabolic stress caused by infection might result in defective autophagy that kills the cell in this manner.
ADDITIONAL READING
Tonnus W, Linkermann A. 2017. The in vivo evidence for regulated necrosis. Immunol Rev 277: 128–149.
Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. 2016. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat Rev Drug Discov 15: 348–366.
Secondary Necrosis
Silva MT, do Vale A, dos Santos NM. 2008. Secondary necrosis in multicellular animals: an outcome of apoptosis with pathogenic implications. Apoptosis 13: 463–482.
Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. 2017. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 8: 14128.
One of two papers showing that caspase-3 induces secondary necrosis via cleavage of DFNA5/gasdermin E.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao, F. 2017. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547: 99–103.
See above.
Necroptosis
Weinlich R, Oberst A, Beere HM, Green DR. 2017. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 18: 127–136.
Vanden Berghe T, Hassannia B, Vandenabeele P. 2016. An outline of necrosome triggers. Cell Mol Life Sci 73: 2137–2152.
Brault M, Oberst, A. 2017. Controlled detonation: evolution of necroptosis in pathogen defense. Immunol Cell Biol 95: 131–136.
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. 2000. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1: 489–495.
The first description of RIPK-dependent cell death, in this case induced by CD95 (called “Fas”).
Xie T, Peng W, Liu Y, Yan C, Maki J, Degterev A, Yuan J, Shi Y. 2013. Structural basis of RIP1 inhibition by necrostatins. Structure 21: 493–499.
He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. 2009. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell 137: 1100–1111.
One of three papers that initially defined the role of RIPK3 in necroptosis.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. 2009. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112–1123.
See above.
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. 2009. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325: 332–336.
See above. The proposed mechanism of action of RIPK3 to promote necroptosis was ultimately shown to be incorrect, but the role of RIPK3 was demonstrated.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. 2012. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213–227.
The first paper to define MLKL as the target of RIPK3 in necroptosis.
Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. 2013. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39: 443–453.
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. 2011. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471: 363–367.
One of two papers that initially showed that ablation of RIPK3 prevents embryonic lethality in caspase-8-deficient mice.
Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. 2011. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471: 368–372.
See above.
Dillon CP, Tummers B, Baran K, Green DR. 2016. Developmental checkpoints guarded by regulated necrosis. Cell Mol Life Sci 73: 2125–2136.
Weinlich R, Green DR. 2014. The two faces of receptor interacting protein kinase-1. Mol Cell 56: 469–480.
Ferroptosis
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et al. 2017. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171: 273–285.
Other Forms of Regulated Necrosis
Bouchard VJ, Rouleau M, Poirier GG. 2003. PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol 31: 446–454.
Szydlowska K, Tymianski M. 2010. Calcium, ischemia and excitotoxicity. Cell Calcium 47: 122–129.
Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, et al. 1997. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 3: 1089–1095.
A connection between ischemia/reperfusion injury and PARP.
Autophagy
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, et al. 2017. Molecular definitions of autophagy and related processes. EMBO J 36: 1811–1836.
Green DR, Levine B. 2014. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157: 65–75.
Mitophagy
Zimmermann M, Reichert AS. 2017. How to get rid of mitochondria: crosstalk and regulation of multiple mitophagy pathways. Biol Chem 399: 29–45.
Nguyen TN, Padman BS, Lazarou M. 2016. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol 26: 733–744.
Autophagic Cell Death
Fulda S, Kogel D. 2015. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene 34: 5105–5113.
Zhang H, Baehrecke EH. 2015. Eaten alive: novel insights into autophagy from multicellular model systems. Trends Cell Biol 25: 376–387.
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. 2004. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304: 1500–1502.
The first paper suggesting that caspase-8 inhibits autophagy-dependent cell death, which ultimately turned out to be RIPK-dependent necroptosis. The relationships between these modes of cell death remain unresolved.
Mitotic Catastrophe
Vakifahmetoglu H, Olsson M, Zhivotovsky B. 2008. Death through a tragedy: mitotic catastrophe. Cell Death Differ 15: 1153–1162.
Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. 2004. Cell death by mitotic catastrophe: a molecular definition. Oncogene 23: 2825–2837.
Kirsch DG, Santiago PM, di Tomaso E, Sullivan JM, Hou WS, Dayton T, Jeffords LB, Sodha P, Mercer KL, Cohen R, et al. p53 controls radiation-induced gastrointestinal syndrome in mice independent of apoptosis. Science 327: 593–596.
Cell death in the intestines of BAX–BAK double-deficient mice may be a form of mitotic catastrophe. p53 is discussed in Green (2022h).
FIGURE CREDITS
Edinger AL, Thompson CB. 2004. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 16: 663–669. doi:10.1016/j.ceb.2004.09.011
Eskelinen E-L. 2008. New insights ito the mechanisms of macroautophagy in mammalian cells. Intl Rev Cell Mol Biol 266: 207–247. doi:10.1016/S1937-6448(07)66005-5
He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. 2009. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell 137: 1100–1111. doi:10.1016/j.cell.2009.05.021
Kirsch D, Santiago PM, di Tomaso E, Sullivan JM, Hou WS, Dayton T, Jeffords LB, Sodha P, Mercer KL, Cohen R, et al. 2010. p53 controls radiation-induced gastrointestinal syndrome in mice independent of apoptosis. Science 327: 593–596. doi:10.1126/science.1166202
Lum J, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. 2005. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120: 237–248. doi: 10.1016/j.cell.2004.11.046
Silva MT, do Vale A, dos Santos NMN. 2008. Secondary necrosis in multicellular animals: an outcome of apoptosis with pathogenic implications. Apoptosis 13: 463–482. doi:10.1007/s10495-008-0187-8
REFERENCES
*Reference is also in this collection.
- *.Green DR. 2022a. The death receptor pathway of apoptosis. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022b. The burial: clearance and consequences. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022c. Inflammasomes and other caspase-activation platforms. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022d. A matter of life and death. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022e. The future of death. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022f. Caspases and their substrates. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022g. Cell death in development. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022h. Cell death and cancer. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041103 [DOI] [Google Scholar]
- *.Green DR. 2022i. The mitochondrial pathway of apoptosis, Part 1: MOMP and beyond. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022j. The mitochondrial pathway of apoptosis, Part II: the BCL-2 protein family. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041046 [DOI] [PMC free article] [PubMed] [Google Scholar]