

Summary
Nearly one-third of children with medulloblastoma, a malignant embryonal tumor of the cerebellum, succumb to their disease. Conventional response monitoring by imaging and cerebrospinal fluid (CSF) cytology remains challenging and a marker for measurable residual disease (MRD) is lacking. Here, we show the clinical utility of CSF-derived cell-free DNA (cfDNA) as a biomarker of MRD in serial samples collected from children with medulloblastoma (123 patients, 476 samples) enrolled on a prospective trial. Using low-coverage whole-genome sequencing, tumor-associated copy-number variations (CNVs) in CSF-derived cfDNA are investigated as an MRD surrogate. MRD is detected at baseline in 85% and 54% of patients with metastatic and localized disease, respectively. The number of MRD-positive patients decline with therapy, yet those with persistent MRD have significantly higher risk of progression. Importantly, MRD detection precedes radiographic progression in half who relapse. Our findings advocate for the prospective assessment of CSF-derived liquid biopsies in future trials for medulloblastoma.
Keywords: childhood cancer, medulloblastoma, liquid biopsy, cell-free DNA, measurable residual disease, cerebrospinal fluid, biomarkers, relapse disease
Graphical Abstract
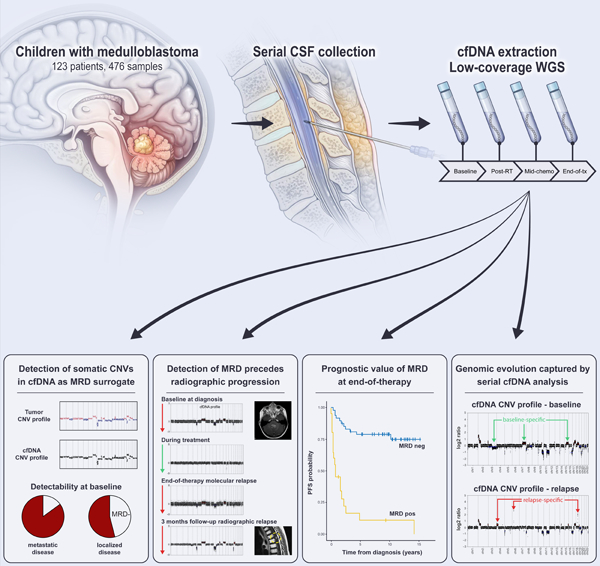
eTOC Blurb
Liu et al. describe the utility of profiling CSF-derived cfDNA from 123 children with medulloblastoma. Use of low-coverage whole-genome sequencing captures tumor-associated CNVs as MRD surrogate, allowing correlation with tumor burden and prediction of disease progression. Serial analysis of cfDNA reflects evolution of the medulloblastoma genome in response to therapy.
Introduction
Medulloblastoma is a malignant (WHO Grade IV) embryonal central nervous system (CNS) tumor of childhood with propensity to metastasize along the leptomeninges. Frontline multimodal therapy with surgery, radiotherapy (RT), and chemotherapy cures ~70% of patients (Gajjar et al., 2006; Gajjar et al., 2021; Kortmann et al., 2000; Lannering et al., 2012; Packer et al., 2006). Patients experiencing progression almost universally succumb to their disease, while survivors often endure long-term treatment-related toxicities, highlighting the importance of effective and well-stratified frontline therapy (Kumar et al., 2021; Mulhern et al., 2005; Ramaswamy et al., 2013; Sabel et al., 2016). While genomic analyses have informed risk-stratification by defining prognostically-relevant molecular groups (WNT, SHH, Group 3 and Group 4) (Taylor et al., 2012), there are currently no molecular biomarkers to monitor response beyond magnetic resonance imaging (MRI) and cerebrospinal fluid (CSF) cytology (Fouladi et al., 1999). Reliance on these modalities precludes the identification of patients with persistent microscopic disease, for whom treatment intensity could be modified, and delays the diagnosis of relapse until the disease is macroscopic and often widely disseminated. Therapy intensification might enable salvage of patients with persistent disease, whereas early detection of relapse would allow timely initiation of second-line or experimental therapy. Furthermore, assessment of tumor-derived molecular profiles at recurrence currently requires invasive neurosurgical procedures with possible morbidities (Kumar et al., 2021; Ramaswamy et al., 2013). Thus, robust, minimally invasive, and clinically actionable biomarkers are necessary to improve outcomes for medulloblastoma patients who fail frontline therapy.
Cell-free DNA (cfDNA) represents DNA fragments released during cellular turnover into biofluids (Jahr et al., 2001). Recent studies in cancer patients have demonstrated feasibility of capturing tumor-associated genomic alterations in cfDNA isolated from bodily fluids (i.e., plasma, CSF, urine) (Merker et al., 2018; Wang and Bettegowda, 2017). In patients with CNS malignancies, CSF-derived cfDNA has been shown to be of greater utility than cfDNA isolated from plasma (De Mattos-Arruda et al., 2015; Escudero et al., 2020; Wang et al., 2015). To date, cfDNA analysis for CNS tumors has been largely restricted to studies using targeted sequencing approaches in patients with high-grade gliomas, where a few recurrent driver mutations dominate the disease. However, data on children with CNS embryonal tumors is limited (Table S1) (Bonner et al., 2018; Escudero et al., 2020; Huang et al., 2017; Miller et al., 2019; Mouliere et al., 2018; Nevel et al., 2018; Panditharatna et al., 2018). Medulloblastoma genomes harbor few hotspot driver mutations and are instead characterized by pervasive chromosomal copy-number variations (CNVs), limiting the generalizability of cfDNA mutational profiling in this particular entity (Escudero et al., 2020; Northcott et al., 2017). Here, we leverage the preponderance of chromosomal instability in medulloblastoma to evaluate the clinical utility of assessing tumor-associated CNVs in CSF-derived cfDNA as a marker of measurable residual disease (MRD). Using serially banked CSF samples from a large cohort of children enrolled on a prospective clinical trial (Gajjar et al., 2021), the predictive and prognostic values of cfDNA analysis are investigated (Figure 1).
Figure 1.
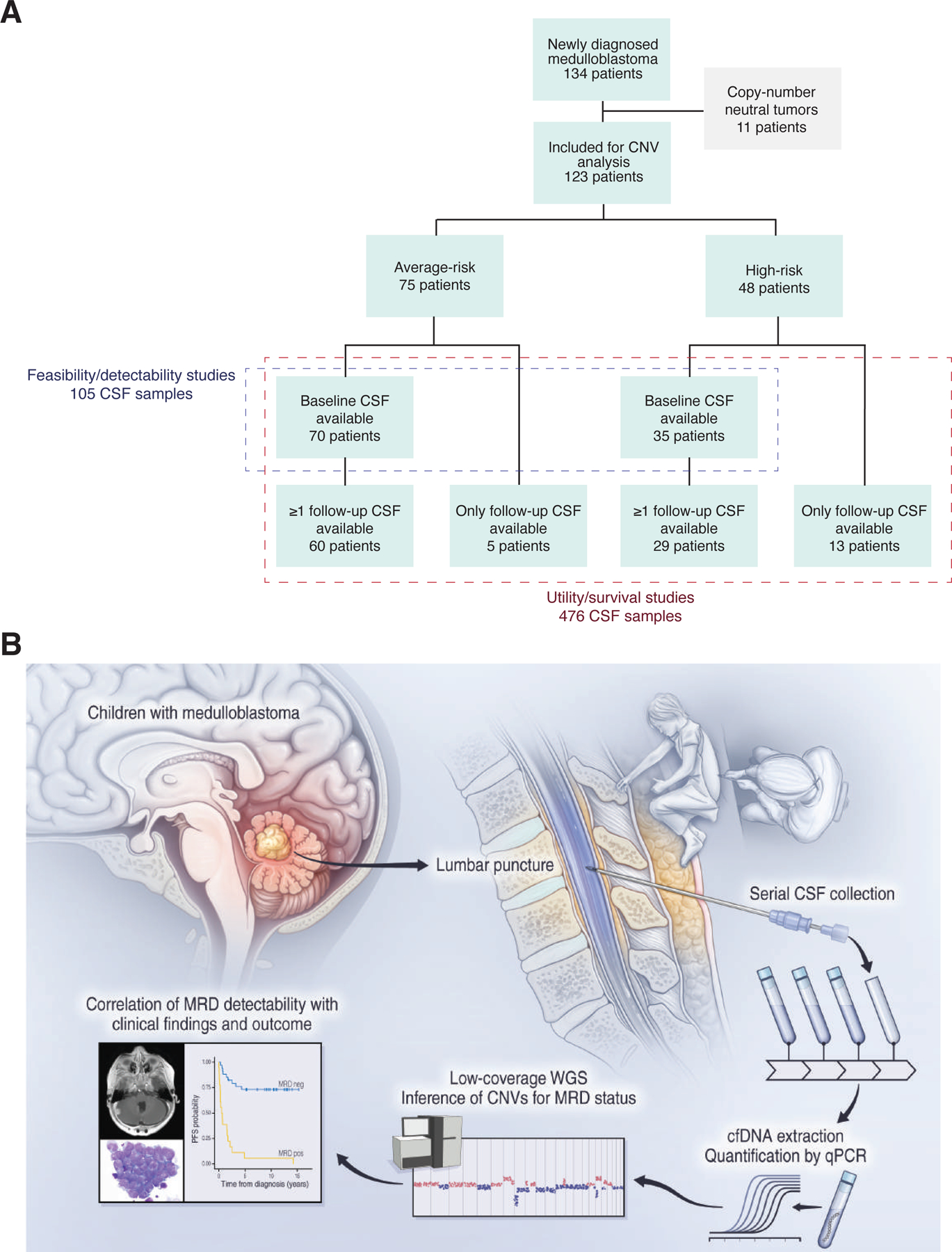
Detection of MRD from serial CSF-derived cfDNA samples in children with medulloblastoma. (A) Study cohort composition with patients stratified by disease risk and sample availability, see also Table S2. (B) Outline of experimental workflow, comprising prospective, serial CSF banking, cfDNA extraction and quantification, low-coverage whole-genome sequencing, CNV calling and clinical correlation. See also Figure S1. cfDNA; cell-free DNA; CSF, cerebrospinal fluid; CNV, copy-number variation; MRD, minimal residual disease; WGS, whole-genome sequencing.
Results
Medulloblastoma cohort composition
The study cohort consisted of 134 children with newly diagnosed medulloblastoma based on availability of banked CSF samples. After excluding 11 patients with copy-number neutral primary tumors, 123 patients remained for analysis, including 105 patients with baseline CSF samples (Figure 1A). Demographics, clinical, and molecular characteristics of the cohort are summarized in Table S2 in comparison with all medulloblastoma patients treated on SJMB03. In total, we generated low-coverage whole-genome sequencing (lcWGS) data on cfDNA isolated from 476 unique CSF samples for downstream analyses (Figure S1).
Detectability of CNVs from CSF-derived cfDNA
To determine the utility of cfDNA-derived CNVs as a biomarker of MRD, available baseline CSF samples (n=105) were studied (Figure 2, Table 1). MRD was detected in 67/105 (64%) baseline samples. In contrast, none of seven control CSF samples obtained from non-oncologic patients displayed cfDNA-derived CNVs (Figure S2). Pairwise correlation of CNV profiles between MRD-positive CSF samples collected at baseline and corresponding primary tumors indicated a high degree of concordance (Figure 3; median Pearson coefficient 0.855). Baseline MRD detectability was significantly associated with metastatic status, molecular subgroup, and tumor location (Table 1). In patients with metastatic disease, 29/34 (85%) baseline samples were positive as compared to 38/71 (54%) of those with non-metastatic disease (Fisher’s exact p=0.003). Detection of baseline MRD was lowest in patients with SHH subgroup tumors (SHH, 32%; WNT 63%; Group 3, 77%; Group 4, 75%; Fisher’s exact p=0.004). Similarly, baseline MRD detectability was lower in patients with tumors centered within the cerebellar hemisphere or vermis, when compared to those within the fourth ventricle (44% vs. 76%; Fisher’s exact p=0.004). Using multivariate logistic regression, metastatic status and subgroup, or metastatic status and tumor location, in two separate models, were significantly associated with baseline MRD detectability (Table S3). In contrast, age, sex, extent of resection, cytologic findings, and duration between resection and CSF sampling were not associated with MRD detection. Of note, MRD was detected at similar rates in patients with high-risk disease regardless of the corresponding CSF cytologic findings (MRD detectability in cytology negative vs. positive: 18/20 [90%] vs. 11/14 [79%]; Fisher’s exact p=0.35). Irrespective of disease risk, 56/91 (62%) cytology negative CSF samples collected at baseline were positive for MRD.
Figure 2.
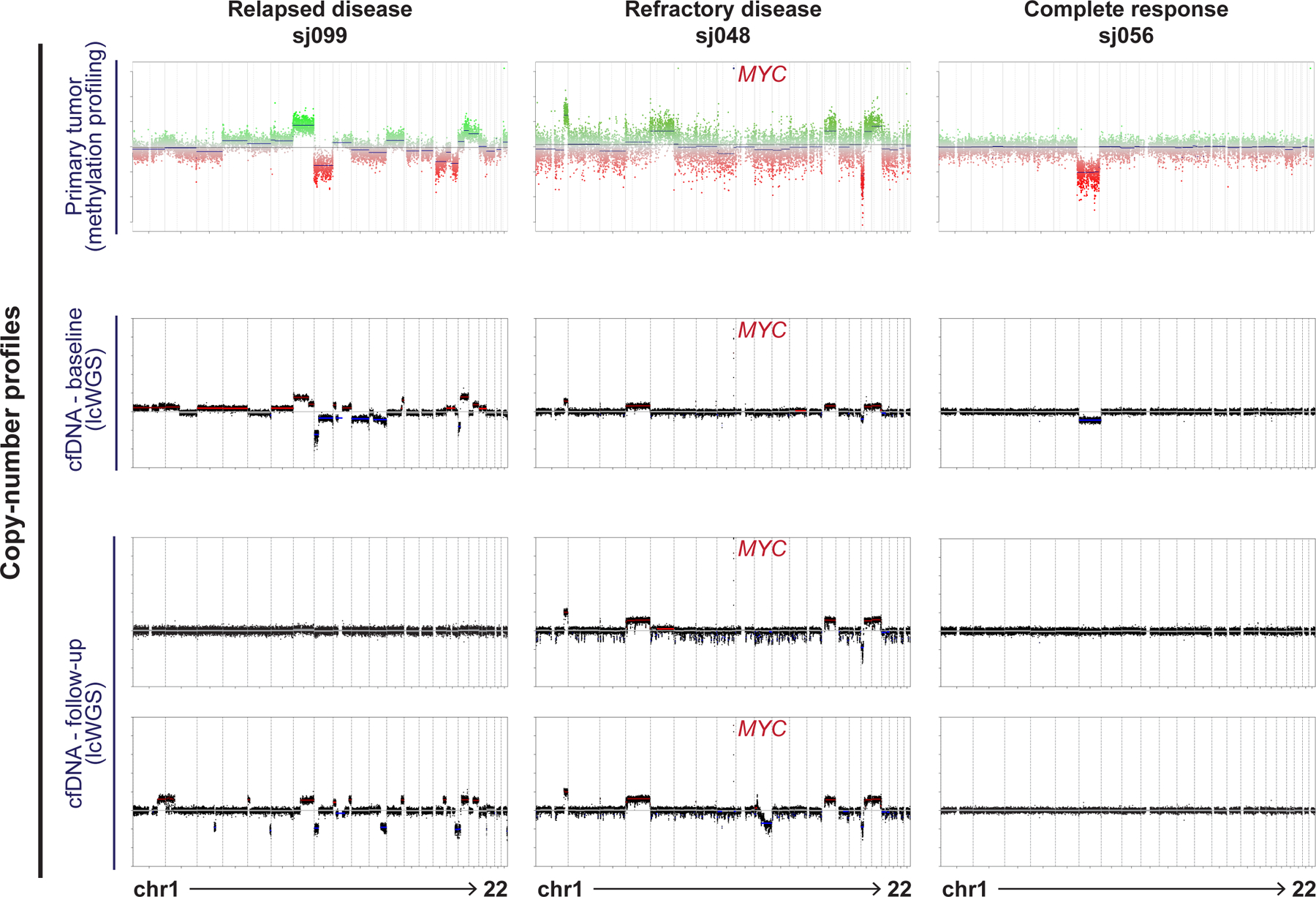
Tumor-associated CNVs are detectable in CSF-derived cfDNA. Exemplary data comparing CNV profiles of primary tumor and longitudinal cfDNA samples in three patients: sj099 (disease relapse after initial response), sj048 (persistent refractory disease) and sj056 (complete response).
Table 1.
Association between MRD detectability at baseline and clinical-molecular features IQR, interquartile range; M0, non-metastatic; M+, metastatic; no. number; RBC, red blood cell; WBC, white blood cell
| Characteristics | MRD neg N=38 |
MRD pos N=67 |
P Value |
|---|---|---|---|
|
Median age at diagnosis (IQR) - years |
8.74 (6.67–11.7) |
8.78 (6.74–11.2) |
0.86 |
| Sex – no. (%) | 0.60 | ||
| Female | 12 (31.6%) | 26 (68.4%) | |
| Male | 26 (38.8%) | 41 (61.2%) | |
| Risk group – no. (%) | 0.002 | ||
| Average | 33 (47.1%) | 37 (52.9%) | |
| High | 5 (14.3%) | 30 (85.7%) | |
| Metastasis – no. (%) | 0.003 | ||
| M0 | 33 (46.5%) | 38 (53.5%) | |
| M+ | 5 (14.7%) | 29 (85.3%) | |
| Chang stage – no. (%) | <0.001 | ||
| M0 | 33 (46.5%) | 38 (53.5%) | |
| M1 | 2 (66.7%) | 1 (33.3%) | |
| M2 | 3 (20.0%) | 12 (80.0%) | |
| M3 | 0 | 16 (100%) | |
| CSF cytology at diagnosis – no. (%) | 0.35 | ||
| Negative | 35 (38.5%) | 56 (61.5%) | |
| Positive | 3 (21.4%) | 11 (78.6%) | |
| CSF cytology at diagnosis in M+ disease – no. (%) | 0.63 | ||
| Negative | 2 (10.0%) | 18 (90.0%) | |
| Positive | 3 (21.4%) | 11(78.6%) | |
| Methylation subgroup – no. (%) | 0.004 | ||
| WNT | 6 (37.5%) | 10 (62.5%) | |
| SHH | 15 (68.2%) | 7 (31.8%) | |
| G3 | 4 (23.5%) | 13 (76.5%) | |
| G4 | 13 (26.0%) | 37 (74.0%) | |
| Primary location$ – no. (%) | 0.008* | ||
| Fourth ventricle | 22 (28.6%) | 55 (71.4%) | |
| Cerebellar hemispheric/vermis | 15 (60.0%) | 10 (40.0%) | |
| Cerebellopontine angle | 1 (50.0%) | 1 (50.0%) | |
| Extent of resection before CSF collection – no. (%) | 1.00 | ||
| Gross/near-total resection | 35 (36.5%) | 61 (63.5%) | |
| Subtotal resection | 3 (33.3%) | 6 (66.7%) | |
| Median time from resection to CSF collection (IQR) – days | 17.0 (13.2–22.0) |
18.0 (14.0–22.5) |
0.52 |
| Median CSF WBC (IQR) – /mm3 | 10.0 (5.50–35.0) |
7.00 (2.00–16.0) |
0.05 |
| Median CSF RBC (IQR) – /mm3 | 12.0 (2.50–65.5) |
21.0 (5.00–110) |
0.33 |
Comparison between primaries in fourth ventricle and cerebellar hemisphere/vermis
One patient without specific primary location annotated
Figure 3.
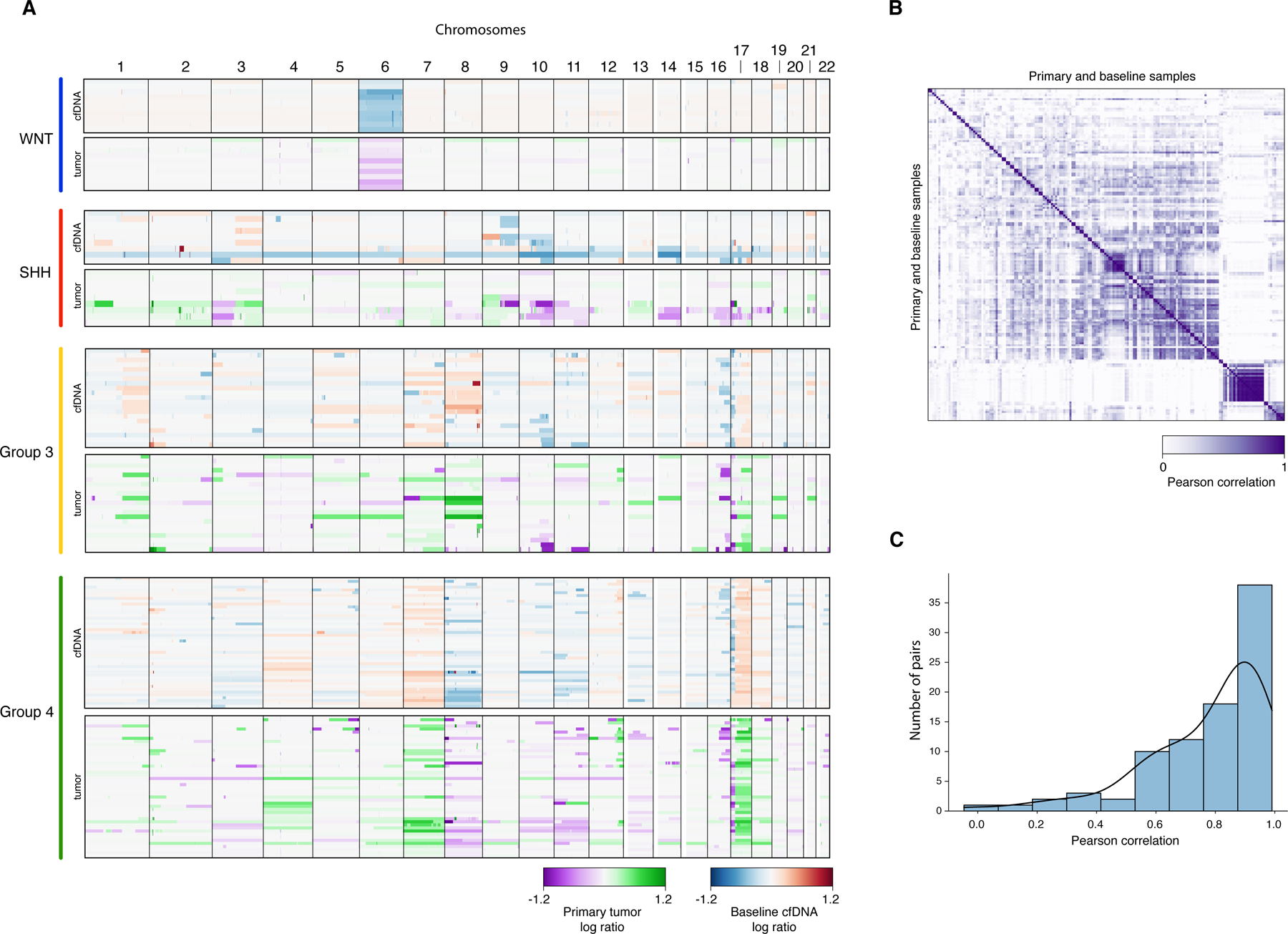
Patient-matched comparison of CNVs from baseline cfDNA and primary tumors. (A) Genome-wide CNV profiles arranged by molecular subgroup and sample origin (low-coverage whole-genome sequencing of cfDNA vs. methylation profiling of primary tumors). (B) Heatmap and (C) histogram summarization of pairwise Pearson correlation between CNVs from baseline cfDNA and primary tumors.
Utility of serial MRD assessment and association with relapse
Longitudinally, MRD was detected in 30/77 (39%) samples available post-RT, 21/75 (28%) mid-chemotherapy, and 20/68 (34%) at end-of-therapy. Patients who eventually progressed had significantly higher rates of MRD persistence during therapy than those without (Kendall’s Tau p<0.0001 at post-RT, mid-chemotherapy, and end-of-therapy; Figure 4A-D). In 16/32 patients (50%) who achieved complete radiographic response before disease relapse, detection of MRD in the absence of radiographic and/or cytologic abnormalities preceded relapse by ≥ 3 months (median 8.3 months, range, 3.0–29.4) (Figure 4A-B). All studied CSF samples (n=27) in the 12 patients with persistent radiographic and/or cytologic disease were positive for MRD. CSF collected within 3 months of disease progression were MRD positive in 24/25 patients (Figure 4B). The pattern of failure in these 25 patients were distant in 22 (including extra-CNS in 2), mixed distant and local in 1, and local only in 2, with the latter including the only patient where MRD was not detected at progression (sj034). In patients without progression, 193/209 CSF samples (92%) collected after baseline were MRD negative (Figure 4C).
Figure 4.
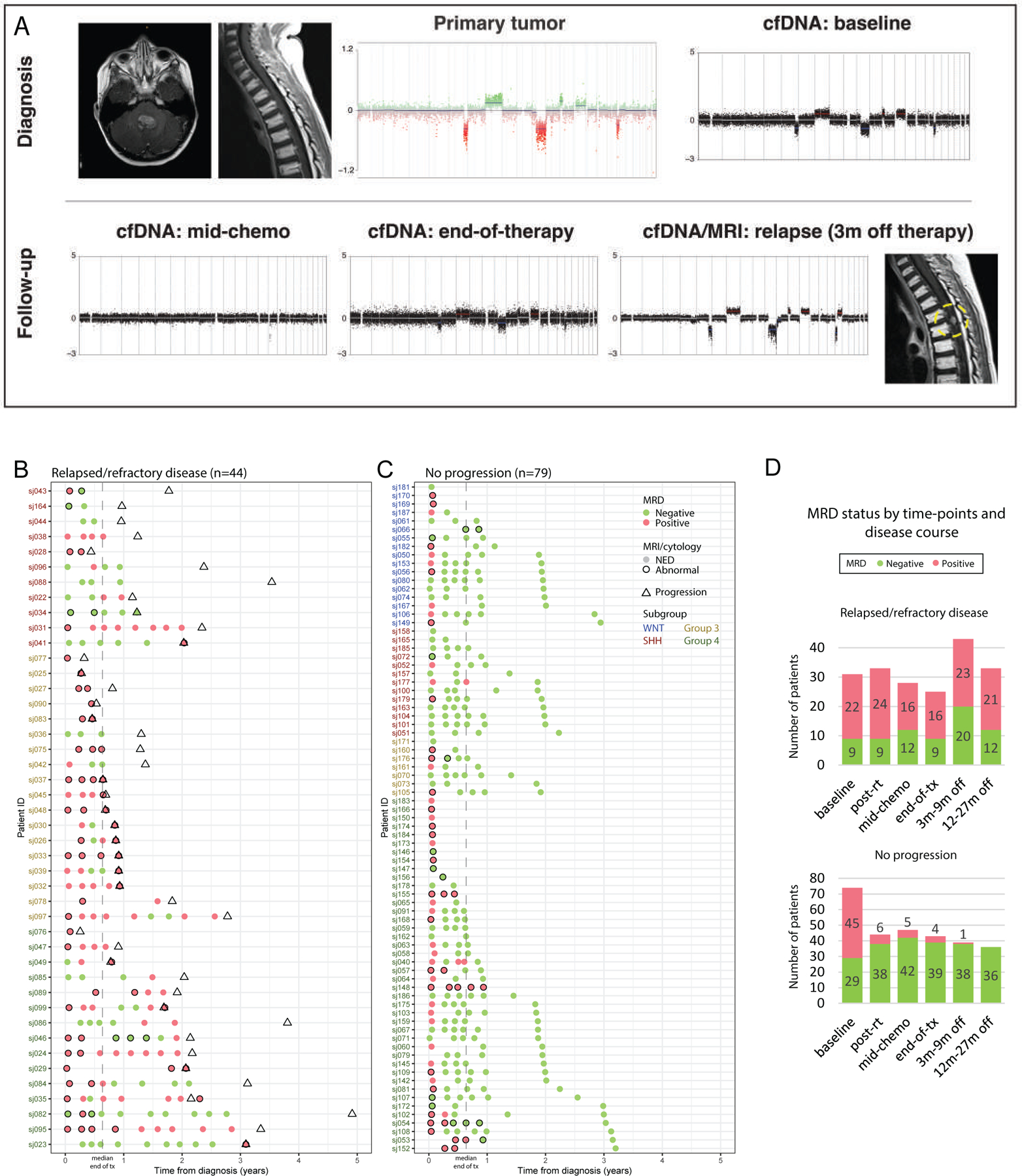
MRD detectability in serial CSF mirrors disease course. (A) MRD re-emergence 3-months before radiographic diagnosis of a solitary metastatic relapse in a patient with Group 3-MB (sj026). (B-C) MRD detectability and corresponding MRI and CSF cytology findings in patients with (B) relapsed or refractory disease, or (C) without progression. Evaluation time-points are only shown where data from both MRD and conventional assessments are available. (D) Number of patients with MRD detectability summarized according to protocol time-points and disease status. m, month; NED, no evidence of disease; RT, radiotherapy; tx, treatment.
Prognostic value of serial MRD assessments
MRD detectability at baseline was not prognostic (hazard ratio 1.36, 95%CI 0.67–2.76; log-rank p=0.4); however, patients with MRD detected post-RT (hazard ratio 6.53, 95%CI 3.22–13.26; log-rank p<0.0001), mid-chemotherapy (hazard ratio 6.35, 95%CI 3.06–13.19; log-rank p<0.0001), or at end-of-therapy (hazard ratio 8.94, 95%CI 4.10–19.49; log-rank p<0.0001) had significantly worse progression-free survival (PFS) than patients without detectable MRD (Figure 5A-D). Compared to standard evaluation with MRI and CSF cytology at end-of-therapy, MRD detectability at the same time-point better stratified patients according to PFS and had higher sensitivity (64% vs 24%) for residual disease (Figure S3A-B, Table S4 and S5). At end-of-therapy, 12/20 (60%) MRD positive patients had normal MRI/cytology among which 10/12 subsequently progressed; at the same time-point, two patients who had abnormal MRI findings but were MRD negative did not progress (Table S4).
Figure 5.
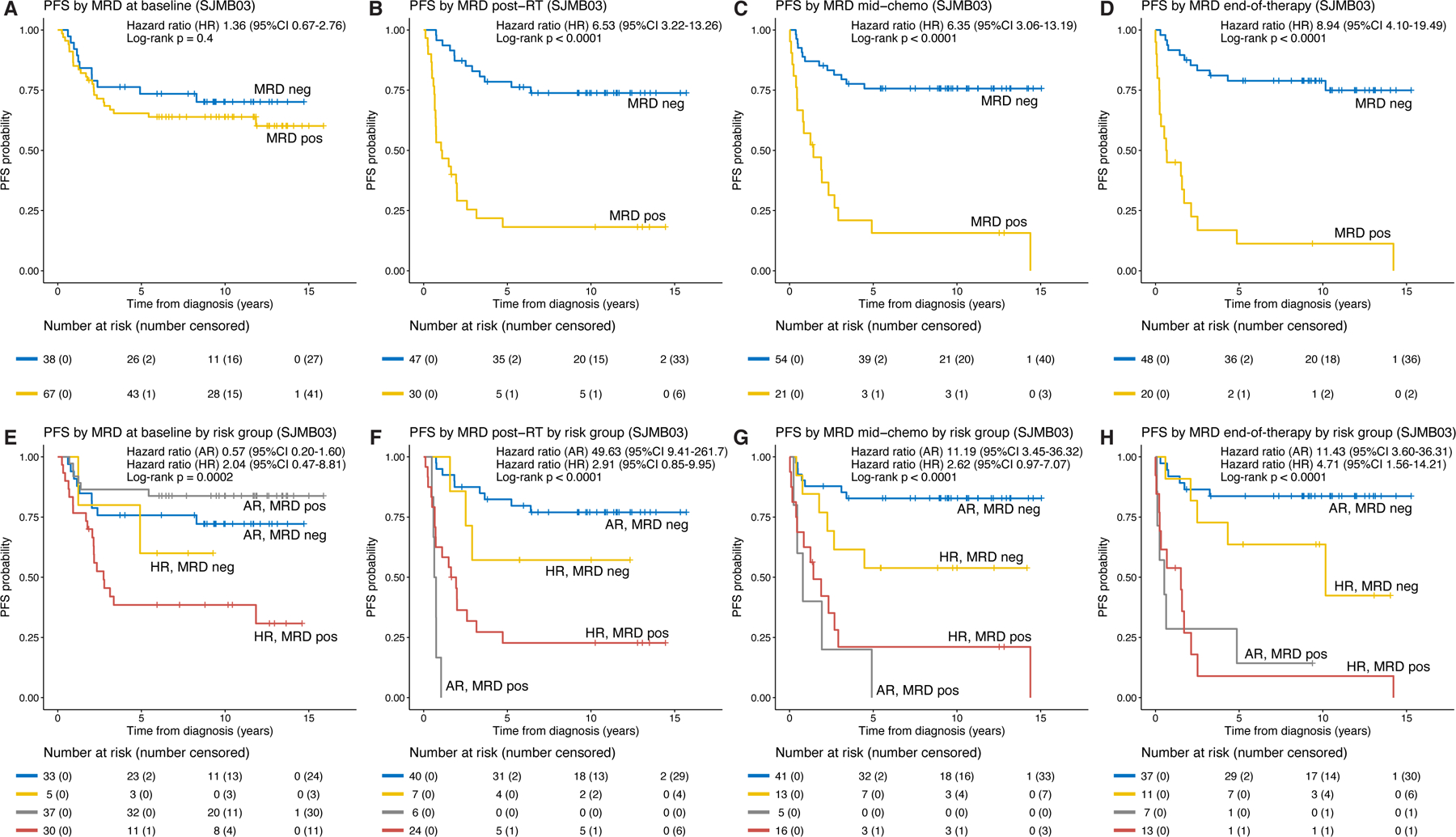
MRD detectability stratifies patients by risk of progression. (A-D) Progression-free survival (PFS) for patients from the longitudinal cohort stratified by MRD at baseline, post-radiotherapy (RT), mid-chemotherapy, and end-of-therapy. (E-H) PFS for patients from the longitudinal cohort stratified by MRD and clinical risk group at the respective time-points. See also Figures S3 and S4. AR, average-risk; CI, confidence interval; HR, high-risk; neg, negative; pos, positive.
Accounting for risk-stratification, MRD detectability post-RT, mid-chemotherapy, and end-of-therapy were significantly associated with PFS in patients with average-risk disease (post-RT: hazard ratio 49.63, 95%CI 9.41–261.7; mid-chemotherapy: hazard ratio 11.19, 95%CI 3.45–36.32; end-of-therapy: hazard ratio 11.43, 95%CI 3.60–36.31).
Among high-risk patients, MRD at end-of-therapy was significantly associated with PFS (hazard ratio 4.71, 95%CI 1.56–14.21); MRD positive high-risk patients post-RT (hazard ratio 2.91, 95%CI 0.85– 9.95) and mid-chemotherapy (hazard ratio 2.62, 95%CI 0.97–7.07) trended towards significance (Figure 5E-H). The cumulative incidence of progression was significantly higher in patients with MRD detected during and at end-of-therapy, with or without accounting for the effect of risk groups (Figure S4A-H). MRD detectability at end-of-therapy was significantly associated with PFS in patients with Group 3 (hazard ratio 4.07, 95%CI 1.04–16.01; log-rank p=0.03) or Group 4 medulloblastoma (hazard ratio 8.58, 95%CI 2.10–35.14; log-rank p<0.001, Figure S5A-C). As a time-dependent variable, MRD detectability throughout follow-up was significantly associated with PFS (Cox regression p<0.0001 in univariate analysis and multivariable analysis with risk group or molecular subgroup included as part of the models) (Table S6).
Acquisition of tumor-associated molecular profiles at relapse
For study patients with progressive disease (PD) and MRD-positive CSF at both an early time-point (baseline or post-RT) as well as when disease progressed, we compared CNV profiles derived from patient-matched CSF samples. Comparative analysis of this limited cohort revealed divergent chromosomal aneuploidy, suggestive of clonal selection or evolution in 12/15 patients (80%, Figure 6). Loss of chromosome 10q was a recurrent event at relapse, while relapse-specific focal genetic alterations (i.e., SMAD2, PIM1 amplifications) were mostly anecdotal. In select cases, cfDNA analysis allowed early detection of tumor clones that predominated at relapse (Figure 7A).
Figure 6.
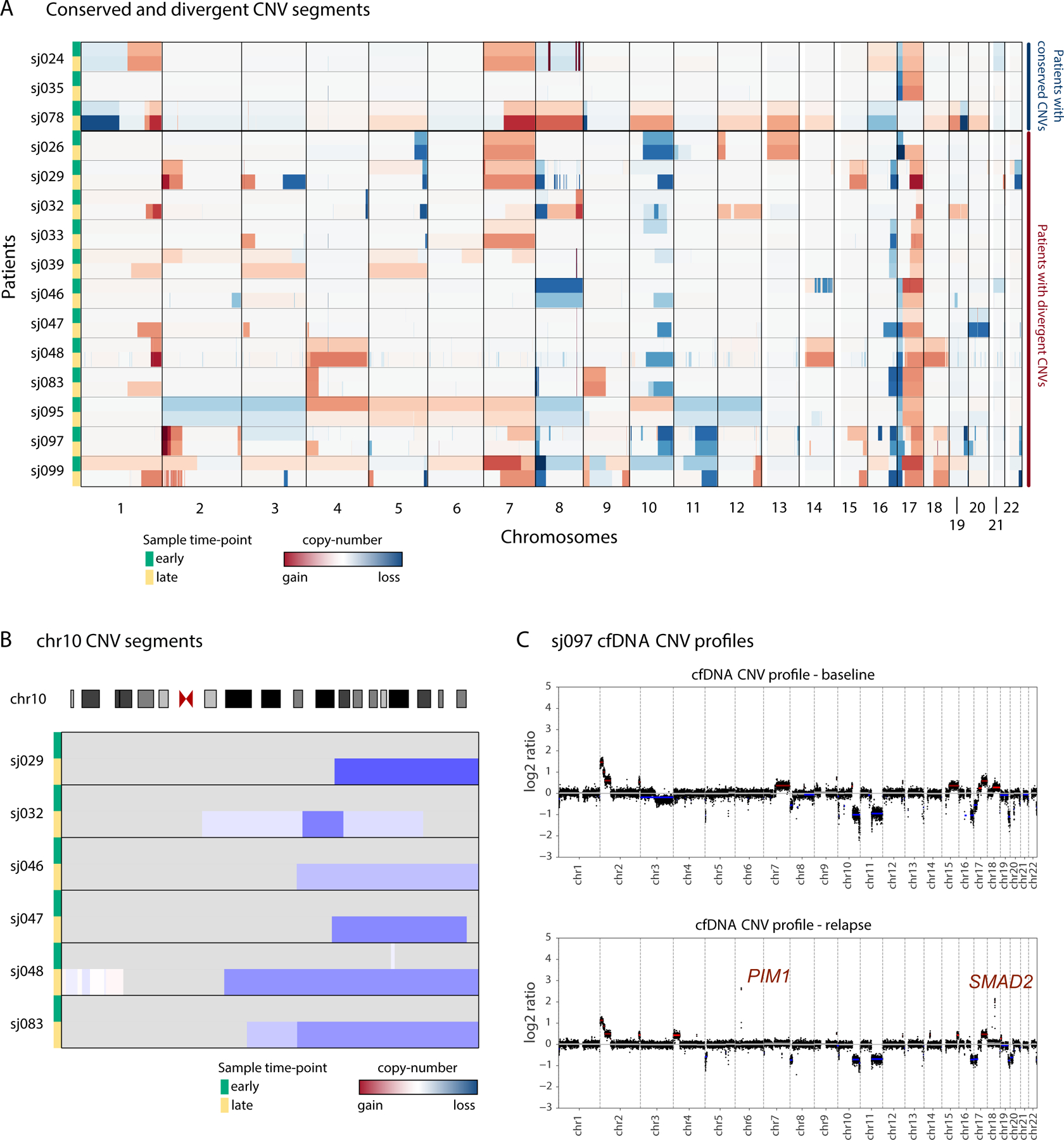
Serial cfDNA profiles reveal tumor evolution at progression. (A) Heatmap depicting evolution of genome-wide CNV profiles in paired early and late cfDNA samples in patients with progression. Patients are grouped by conserved (sj024-sj078) or divergent (sj026-sj099) CNV profiles. (B) Loss of chromosome 10q acquired in cfDNA samples from 6 patients at relapse. (C) Comparison of cfDNA profiles at diagnosis and progression in patient sj097 illustrating relapse-specific focal amplicons on chromosomes 6 and 18 that target PIM1 and SMAD2, respectively.
Figure 7.
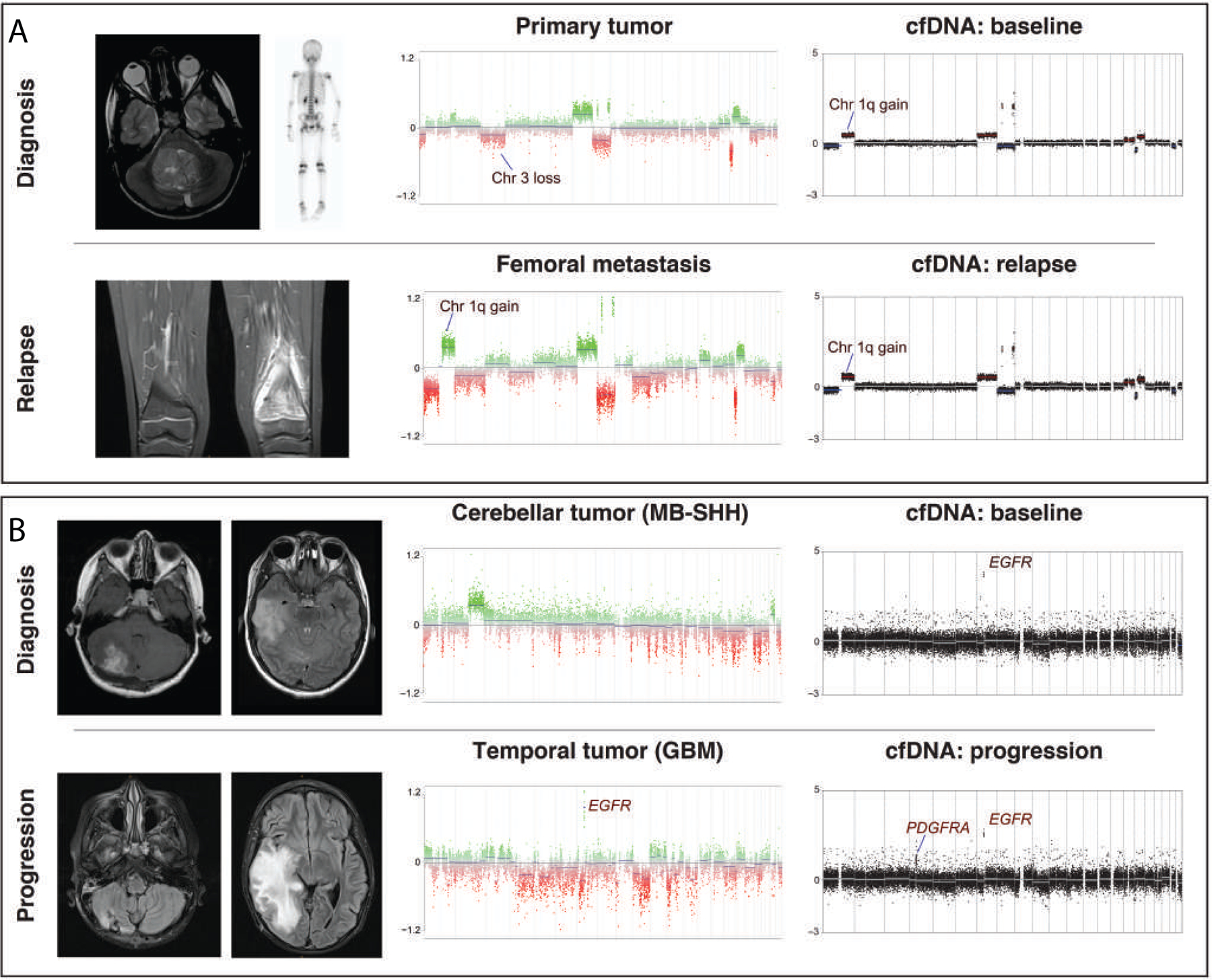
Illustrative vignettes highlighting the utility of serial cfDNA profiling in children with medulloblastoma. (A) CNV profiles of bulk tumor and CSF-derived cfDNA at diagnosis/baseline and metastatic relapse in a patient with Group 4-MB (sj024). (B) Patient with SHH-MB and rapidly progressive right temporal lesion histologically diagnosed as high-grade neuroepithelial tumor with EGFR and PDGFRA amplification (sj028). cfDNA analysis at baseline retrospectively indicated presence of PDGFRA and EGFR amplification, suggesting concurrent medulloblastoma and high-grade glioma (HGG).
Discordant CNVs detected in a subset of primary tumor vs. baseline cfDNA comparisons informed challenging diagnostic vignettes. For example, patient sj024 was diagnosed with Group 4 medulloblastoma and subsequently a metastatic, skeletal relapse (Figure 7A). The patient’s baseline cfDNA-derived CNV profile was more congruent with that of the relapsed tumor than the corresponding primary (i.e., gain of chromosome 1q, balanced chromosome 3), suggesting that the patient’s CSF sample at baseline included the aggressive subclone that drove disease progression. As a second example, patient sj028 was diagnosed with SHH subgroup medulloblastoma with temporal lobe metastasis. Rapid progression of the temporal lesion necessitated surgical resection that was histologically confirmed as high-grade glioma with EGFR and PDGFRA amplifications (Figure 7B; data not shown). cfDNA analysis of baseline CSF revealed presence of the EGFR amplicon, retrospectively confirming the co-occurrence of SHH subgroup medulloblastoma and high-grade glioma. Germline sequencing revealed a pathogenic MSH2 mutation (NM_000251.2:c.998G>A), diagnostic of constitutional mismatch repair deficiency in this patient.
Integrating mutational studies for MRD analysis
As orthogonal validation of our study workflow, we performed panel sequencing on 13 baseline CSF samples using targeted enrichment of NGS libraries prepared for lcWGS. Considering putative driver gene alterations among these samples (tumor variant allele frequency [VAF] 20–100%), we detected the corresponding mutations from 7 of 8 CSF samples that were MRD positive by CNVs, and only 1 of 5 samples where MRD was negative by CNVs (Table S7). Significant correlation was observed between the corresponding cfDNA mutant VAF and tumor fraction estimated by CNV analysis (Pearson’s r=0.7611, p=0.0025).
Applicability of MRD studies in other pediatric CNS embryonal tumors
To further evaluate the applicability of lcWGS-based cfDNA profiling in other childhood brain tumor entities, we analyzed a cohort of 17 patients with non-medulloblastoma CNS embryonal tumors treated on SJMB03 (Table S8). All patients included in this cohort harbored chromosomal and/or focal CNVs in their corresponding primary tumors. Based on lcWGS of CSF-derived cfDNA in this non-medulloblastoma cohort, 13 (76%) post-operative baseline samples were MRD positive, including all three with metastatic disease. Furthermore, we observed molecular responses from serial cfDNA samples that mirrored clinical course, analogous to our findings in the medulloblastoma study cohort (Figure S6). As an example, one patient (sj214) with posterior fossa atypical teratoid rhabdoid tumor (ATRT) demonstrated resurgence of MRD seven months before eventual radiographic progression in the spinal cord (Figure S7).
Discussion
MRD-guided therapy has revolutionized the management of pediatric leukemia (Coustan-Smith et al., 2000; Rubnitz et al., 2010; Wood et al., 2018). Protocols for acute leukemia incorporating MRD assessment are now the standard-of-care, sparing good-responders from unnecessarily toxic therapy and identifying poor-responders for escalated, intensified consolidation treatment. Such an approach is currently unavailable for children with CNS tumors, including medulloblastoma, due to the lack of an effective biomarker for MRD detection. As a result, the extent of tumor eradication cannot be assessed beyond the resolution of MRI (Gajjar et al., 2004; Ramaswamy et al., 2016). Too often patients are either being overtreated, and incur unnecessary toxicity from excess therapy, or undertreated, and fail to have their disease sufficiently eliminated by the end-of-therapy. The collection of CSF for cytology is routinely performed for staging and disease assessment based on the presence of circulating tumor cells by morphology and represents an optimal medium for assessment of cfDNA as a potential biomarker for MRD in this population (Fouladi et al., 1999). Leveraging a large, longitudinal cohort of CSF samples collected from medulloblastoma and other CNS embryonal tumor patients enrolled on a prospective clinical trial (Gajjar et al., 2021), the current report is the first to effectively and systematically demonstrate the clinical utility of serial CSF-derived cfDNA profiling for detection of MRD in childhood CNS cancer.
In the current study, 62% of cytologic-negative CSF samples harbored tumor-derived cfDNA, demonstrating the superior sensitivity of CSF liquid biopsy analysis over conventional CSF cytology, in agreement with recent findings based on a limited sample size (Escudero et al., 2020). Moreover, we showed that persistent MRD positivity during or at the end of adjuvant therapy was highly predictive of progression, regardless of initial risk-grouping, offering treating physicians an unprecedented opportunity to identify poor-responders for whom additional, intensified, and/or experimental therapy can be implemented to mitigate risk of disease progression. On the contrary, early MRD negativity, such as at the post-RT timepoint, could be evaluated as an additional criterion for treatment de-escalation in patients who are otherwise considered to have low-risk disease. The lack of an association between baseline MRD detectability and outcome is expected, as the baseline signal merely represents post-operative residual disease detectable by our molecular assay, which in a significant proportion of patients, could be effectively eradicated by subsequent adjuvant treatment. Molecular disease positivity was detected before frank radiographic progression in half of patients, enabling the potential for second-line therapy to be initiated at a lower tumor burden. These findings establish serial MRD assessment as a promising complementary tool to molecular and clinical risk-stratification defined at diagnosis that warrants integration into future medulloblastoma clinical trials.
Although different liquid biopsy techniques are available for evaluating tumor-derived material, the use of lcWGS for the detection of tumor-associated CNVs in CSF-derived cfDNA is well-suited for medulloblastoma (Bonner et al., 2018; Escudero et al., 2020; Nevel et al., 2018; Siravegna et al., 2017). First, the yield of cfDNA in CSF of patients with resected medulloblastoma is significantly lower than that reported for patients with other brain tumors, and in plasma of patients with non-CNS solid tumors (Diaz Jr and Bardelli, 2014; Wang et al., 2015). These sub-nanogram yields represent a challenge for other platforms, such as epigenomic profiling and mutational studies, but are uniquely tolerated by lcWGS. Second, chromosomal CNVs are nearly ubiquitous in childhood medulloblastoma, affecting 95% of patient tumors studied in the SJMB03 trial (Gajjar et al., 2021; Northcott et al., 2017). Capturing genome-wide CNVs obviates the need for targeting a spectrum of different mutational drivers using custom probe designs, as in the case of droplet digital PCR, and accommodates the significant proportion of medulloblastoma samples that lack known driver gene mutations. For the minority of patients with copy-number neutral tumors, recent studies have established feasibility of tracking mutations in medulloblastoma CSF liquid biopsies (Escudero et al., 2020; Miller et al., 2020). Similar detection rates of known medulloblastoma-associated driver mutations were verified in our limited gene panel sequencing analysis of baseline CSF samples. On the other hand, mutational profiling allows the detection of alterations that are known to be targetable, such as PTCH1 in SHH subgroup medulloblastomas. Thus, combination CNV and mutational-based approaches will likely be complementary and compatible with the streamlined experimental workflow described herein.
The exact mechanism of cfDNA release remains elusive. It has been proposed that cfDNA results from the controlled release of fragmented DNA during cellular turnover (Jahr et al., 2001). The detectability of cfDNA in CSF of medulloblastoma patients was associated with metastatic status, tumor location and molecular subgroup, with an obvious interaction between the latter two as cerebellar hemispheric tumors are exclusively SHH-activated (Perreault et al., 2014). The tumor-CSF interface of ventricular tumors and (microscopic) metastatic lesions might have promoted the release of cfDNA into the CSF compartment leading to higher detectability in Group 3 and 4 subgroups. It is also possible that cfDNA profiles are dominated by signatures from more aggressive subclones present in heterogeneous bulk tumors, as supported by an illustrative vignette in the current study. cfDNA profiling might thus more genuinely represent the genomic fingerprint of malignant cells that eventually drives disease progression. Furthermore, future studies may evaluate genomic evolution with serial liquid biopsies without the need for repeated surgical biopsies. This enables an avenue for less-invasive analysis of the relapsed medulloblastoma genome to facilitate the rational prioritization of experimental salvage therapies based on molecular composition of the tumor at progression. The use of cfDNA profiling would thus inform design of novel therapeutics for the management of relapsed/refractory medulloblastoma which has thus far been largely based on the genomics of primary untreated tumors.
This study did have some notable limitations. First, for a number of reasons, the availability of CSF for this study only encompassed 40% of the trial population: collection and banking were recommended on the trial but not mandated; additional CSF for research was not always collected; lumbar punctures were omitted due to physician discretion; and logistical challenges arose in coordinating, shipping, maintaining, and storing hundreds of CSF samples from multiple sites over a 10-year period. Secondly, because the value of continued surveillance was unknown, CSF was not collected in average-risk patients beyond two years from diagnosis and in high-risk patients beyond three years. The result was a smaller study cohort than was possible and missed opportunities to study CSF samples before or around the time of progression in some patients. Another important consideration is that our CNV-based approach excludes a small proportion of patients with copy-number neutral tumors, especially from the infant age-group. Since mutations in known medulloblastoma-associated genes are often identified in these tumors, targeted sequencing approaches may complement lcWGS to further enhance generalizability (Escudero et al., 2020; Robinson et al., 2018). Furthermore, we acknowledge that our findings warrant replication in an independent trial cohort as validation with an optimized assay, which might be achieved through concomitant capturing of CNVs and mutational events. To maximize clinical utility, the cfDNA assay should allow greater sensitivity for MRD detection which is especially crucial for selection of patients for treatment de-escalation. Such developments will be essential before longitudinal CSF-derived cfDNA profiling can be recommended as standard-of-care.
In conclusion, we demonstrate that lcWGS of serial CSF-derived cfDNA allows MRD detection and predicts treatment response in children with medulloblastoma. This study supports incorporating prospective cfDNA evaluation into medulloblastoma clinical trials for further investigation and technical refinement such that future medulloblastoma management may personalize therapy according to MRD response. Broadly, our findings provide a solid foundation for the future deployment of CSF-derived liquid biopsies as an actionable biomarker for malignant CNS tumors in children and adults.
STAR Methods
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Paul Northcott, PhD (paul.northcott@stjude.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The code generated during this study is available through Github at https://github.com/kylessmith/cfdna. Sequencing data used in the manuscript have been deposited to European Genome-Phenome Archive (EGAS00001005592) and are publicly available as of the date of publication. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
Experimental Model and Subject Details
Human subjects
This study was conducted according to the principles expressed in the Declaration of Helsinki. Ethical approval was obtained from the Institutional Review Board of St. Jude Children’s Research Hospital under protocol #Pro00008912. All patients provided written informed consent for sample and clinical data collection and subsequent analyses on the SJMB03 study, being enrolled between 2003 and 2016 (NCT00085202) (Gajjar et al., 2021). CSF samples were collected by lumbar punctures performed only with clinical indications at diagnosis and follow-up. Samples from 134 medulloblastoma patients were studied and those from 123 included for analysis. For the non-medulloblastoma CNS embryonal tumor cohort, samples from 17 patients were studied and included for analysis. Detailed demographic characteristics for the medulloblastoma cohort and CNS embryonal tumor cohort are listed in Tables S2 and S7 respectively. CSF from individuals without malignancies (n=7) were collected from German Cancer Research Center with ethnical approval and informed consent.
Medulloblastoma Cell lines
For tumor-normal mixing experiments (Figure S8), CHLA-01R-MED human medulloblastoma cells were purchased from ATCC and were cultured according to the manufacturer’s instructions. Cells were cultured in DMEM:F12 Medium with 20 ng/mL human recombinant EGF, 20 ng/mL human recombinant basic FGF, and B-27 Supplement to a final concentration of 2% under controlled temperature (37C) and CO2 (5%).
Method Details
Study design
This study was approved by the Institutional Review Board of St. Jude Children’s Research Hospital (Review Board number Pro00008912). The main study cohort included children ≥3 years of age with medulloblastoma enrolled on a concluded, multi-institutional, prospective, risk-adapted trial for newly diagnosed CNS embryonal tumors (SJMB03, NCT00085202, Figure 1A) (Gajjar et al., 2021). Serially banked CSF samples available from post-operative baseline (before start of adjuvant therapy), during therapy and follow-up after completion of therapy were used. A non-medulloblastoma CNS embryonal tumor cohort, inclusive of patients with pineoblastoma (n=9), embryonal tumors with multilayered rosettes (ETMR, n=3), atypical teratoid/rhabdoid tumor (ATRT, n=5), from the same trial was included as validation. CSF samples from seven patients with non-oncologic indications collected at German Cancer Research Center were studied as controls.
Treatment protocol for children enrolled on SJMB03
Maximal safe resection was performed for all patients prior to treatment initiation, with extent of resection based on post-operative imaging and classified as gross-total resection (GTR), if no tumor remained; near-total resection (NTR), if residual tumor measured ≤1.5cm2; or subtotal resection (STR), if residual tumor measured >1.5cm2. All patients underwent magnetic resonance imaging (MRI) of the entire spinal axis and lumbar puncture for CSF cytology (collected at a median of 18 days from the prior surgical resection, range 5 to 30 days) for staging. Disease stage was assigned according to the Chang’s criteria.(Chang et al., 1969) Patients were stratified as having “average-risk” disease if they had no evidence of metastasis (M0) and GTR/NTR, or as “high-risk” if they had metastatic disease (M1–3) and/or STR. All patients received risk-adapted craniospinal irradiation (CSI, average-risk: 23.4Gy, high-risk: 36–39.6Gy) with tumor boost (total dose: 55.8–59.4Gy), followed by four, 28-day cycles of chemotherapy consisting of cisplatin (75mg/m2 on day −4), cyclophosphamide (2g/m2 daily on days −3 and −2) and vincristine (1mg/m2 on days −4 and +6) with autologous hematopoietic stem cell rescue (day 0).
Response evaluation on SJMB03
Serial disease evaluation was pre-defined by the protocol and varied according to risk status. Complete disease evaluations with MRI of the brain and spine and cytologic assessment of CSF collected by lumbar puncture was recommended on all patients at post-operative baseline before the start of adjuvant therapy (baseline), after radiotherapy (post-RT), after two courses of high-dose chemotherapy (mid-chemotherapy), at end of treatment (end-of-therapy), and at 12 months from diagnosis (3 months off therapy). Thereafter average-risk patients were followed with complete disease evaluations every 6 months until 24 months from diagnosis (9 and 15 months off therapy) and high-risk patients were followed with complete disease evaluations every 3 months until 24 months from diagnosis (6, 9, 12 and 15 months off therapy) and then every 6 months until 36 months from diagnosis (21 and 27 months off therapy). All patients continued to be followed with serial MRI evaluations until 72 months from diagnosis or progression, however no further CSF collection was recommended unless suspicion of relapse was identified on surveillance. According to MRI and cytologic findings, patients were classified as having complete response (CR) or having no evidence of disease (NED) with disappearance of all lesions on MRI and negative CSF cytology; partial response (PR) with ≥50% reduction in sum of the products of the maximum perpendicular diameters of all measurable lesions, or two consecutively negative CSF cytologies (if initially cytology positive) plus a <50% reduction in tumor dimensions; stable disease (SD) with <50% reduction in tumor dimensions and persistently negative or positive CSF cytology; and progressive disease (PD) with >25% increase in the size of any lesion, new lesion, or newly positive CSF cytology.
With informed consent, 1–2 ml of CSF was obtained in addition to cytologic specimens during these assessments and banked for research. CSF samples were collected in plain sterile tubes and centrifuged at 10,000 rpm for 10 minutes at 4°C. The supernatant was removed and aliquoted for storage at −80°C.
cfDNA extraction and low-coverage whole-genome sequencing (lcWGS) for MRD detection
Supernatants from pre-processed CSF samples were thawed and processed using the NucleoSnap cfDNA kit (Macherey-Nagel) according to the manufacturer’s instruction (Figure 1B). Elution was performed with 50 µl of 5 mM Tris-HCL. Extracted cfDNA was quantified by qPCR using primers for the ALU sequences referenced against serially diluted human genomic DNA (1 pg/µl – 10 ng/µl, 10-fold dilutions, Promega) (Iqbal et al., 2015). Size distribution of cfDNA was assessed using the TapeStation System and High Sensitivity D1000 Assay (Agilent). For MRD detection using lcWGS, up to 2 ng or 40 µl of cfDNA was used without fragmentation as input for library preparation with the Accel-NGS 2S Hyb DNA Library Kit (Swift Biosciences). Dephosphorylation, end repair, and ligation of single-indexing adapters were performed according to protocol while library amplification was guided by a pilot quantitative PCR run. No template controls were included in each experiment. Libraries were quantified by the PicoGreen dsDNA Assay (Invitrogen), pooled, and subjected to 100bp paired-end sequencing on the NovaSeq 6000 instrument (Illumina) targeting for 3x coverage.
Panel sequencing of cfDNA
Target-enrichment of the prepared DNA libraries was performed using a customized gene panel (cfDNA300, 2.3Mb) covering 300 tumor-associated genes (Twist Bioscience) and Twist Target Enrichment reagents (Twist Bioscience). Amplified, Molecular Identifier (MID)-containing libraries prepared by the Accel-NGS 2S Hyb DNA Library Kit were pooled in an equimolar fashion to achieve a target of 1500 ng per reaction. The combined libraries were then hybridized in solution with the capture probes in the presence of universal blockers at 70°C for 16 hour, and were then bound to streptavidin beads for washing. The captured libraries were then PCR-amplified, cleaned, and sequenced 100bp paired-end on the NovaSeq 6000 instrument (Illumina) targeting for 250M reads per sample.
CNV analysis of whole-genome sequencing data
To infer disease status from CSF-derived whole-genome sequencing data, we developed a computational framework to detect large-scale CNVs (cfdna v0.1.0). Reads were mapped to the human reference genome (hg19) via BWA, de-duplicated (Picard), and down-sampled to 50 million paired reads if necessary. For each sample, mean fragment coverage was calculated for 100kb non-overlapping bins throughout the genome and filtered to remove common blacklisted regions. Bin sums were then corrected for GC content and mapping using LOWESS regression and normalized by taking the log2 ratio between each bin and the median bin coverage (ngsfragments v0.1.0). Copy-number segments were determined from resulting bins using a Bayesian change point approach with a heuristic probability threshold of 0.3 (bcpseg v0.1.0). The ichorCNA probabilistic model was re-implemented in Python in order to run faster on our large cohort and integrate into our downstream analysis pipeline. The resulting Python package was used to predict tumor purity and ploidy based on larger 1Mb bins (hmmcnv v0.1.0). CNV plots were created and manually curated by reviewers blinded to the sample time-point, patient identifier, and disease status. Copy-number segments determined from lcWGS of cfDNA taken at baseline were correlated with matched CNVs predicted from methylation array profiling of patient-matched primary tumors. Each chromosome was binned into 1Mb segments and assigned the mean log2 ratio within each window. Pearson correlation coefficients were calculated for all matched autosomal segments between primary tumor and baseline cfDNA.
Sensitivity benchmarking utilizing tumor-normal mixing
The sensitivity of our pipeline was benchmarked with mixing of tumor-normal genomic DNA, indicating the limit of detection for broad CNVs and focal amplifications to be at 10% and 1% tumor purity, respectively. Genomic DNA from CHLA-01R-MED (CRL-3034, ATCC) and normal human (G3041, Promega) were sheared to a fragment size of 175 bp utilizing M220 focused ultrasonicator (Covaris) according to manufacturer’s instructions. Fragment size distributions were confirmed using TapeStation D1000 high-sensitivity reagents (Agilent) prior to quantification by Qubit dsDNA HS assay (Thermo Fisher). Sheared tumor DNA was admixed with sheared normal human DNA at constant total masses (100 pg, 500 pg, 1 ng) to achieve the following tumor fractions: 0%, 1%, 5%, 10%, 25%, 50%, 100%. Tumor-normal DNA mixtures were again quantified using qPCR prior to library preparation, sequencing, and downstream analyses, as described in Methods for processing and analysis of CSF-derived cfDNA samples.
Mutational analysis of panel sequencing data
All NGS reads were trimmed of adaptors and mapped to the human reference genome (hg38) via BWA-mem. UMI collapsing was conducted using fgbio into higher quality, de-duplicated consensus reads and re-mapped to the human reference genome (hg38). Locations of previously annotated mutations from patient matched primary tumors were inspected in the UMI collapsed bams for each sample using pysam. Variant allele frequencies (VAFs) were calculated as the proportion of reads containing the variant allele to all other observed alleles.
Genome-wide DNA methylation profiling and exome-sequencing of primary tumors
Genomic DNA was extracted from frozen or formalin-fixed, paraffin-embedded (FFPE) tumor tissue for Illumina 450k methylation array. DNA methylation-based classification, exome sequencing and calling was performed using methods described previously (Gajjar et al., 2021; Northcott et al., 2017; Robinson et al., 2018). Somatic genome-wide CNVs of tumor samples were inferred from Illumina normalized probe intensities using the Conumee R package (v1.24.0) with default parameters.
Quantification and Statistical Analysis
Continuous variables were summarized via medians with interquartile range, and categorical variables were summarized as frequencies and percentages. Kruskal–Wallis test, Fisher’s exact test, and Kendall’s Tau were used for comparison of continuous, categorical and ordinal variables respectively. Multivariate logistic regression was used to investigate the association of clinical and molecular factors with MRD detectability. Survival probabilities were estimated using the Kaplan-Meier method. Progression-free survival (PFS) was defined as the duration from diagnosis to disease relapse, progression, death due to any cause, or last follow-up (censored), while overall survival (OS) was defined as the duration from diagnosis to death due to any cause or last follow-up (censored). To evaluate the association between MRD detectability at on-therapy time-points and outcome, PFS and OS duration was calculated from end of radiotherapy (post-RT MRD), end of cycle 2 chemotherapy (mid-chemotherapy MRD), and end of last chemotherapy cycle (end-of-therapy MRD). MRD detectability was also analyzed as a time-dependent covariate in univariable and multivariable Cox models. The cumulative incidence of progression was assessed with the Fine and Gray model in the presence of the competing risk of death from other causes. For studying the relationship between MRD detectability and results based on conventional disease evaluation, a patient was considered as having no evidence of disease (NED) if MRI and CSF cytology were reported as CR or NED, but having an abnormal evaluation if MRI showed active or residual disease (PR, SD, PD) and/or CSF cytology was positive for malignant cells. Two-tailed p-values <0.05 were considered significant. Analyses were performed using R version 3.6.3 (Bioconductor).
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Cerebrospinal fluid samples | Participating study centers | N/A |
| Tumor samples | Participating study centers | N/A |
| Critical commercial assays | ||
| NucleoSnap cfDNA kit | Macherey-Nagel | 740300.50 |
| Tapestation High Sensitivity Kit | Agilent | 5067–5584 / 5067–5585 |
| Accel-NGS 2S Hyb DNA Library Kit | Swift Biosciences | 23096 |
| Twist Hybridization and Wash Kit | Twist Bioscience | 101025 |
| Deposited data | ||
| Raw sequencing data | This paper | EGA: EGAS00001005592 |
| Code for data analysis | This paper | Github: https://github.com/kylessmith/cfdna |
| Experimental models: Cell lines | ||
| Human: CHLA-01R-MED | ATCC | CRL-3034 |
| Software and algorithms | ||
| R software (version 3.6.3) | R Project for Statistical Computing | https://www.r-project.org/ |
Highlights.
Biomarkers for response monitoring are lacking in medulloblastoma
CSF-derived cfDNA profiling captures chromosomal landscapes of primary tumors
Detectability of CNVs in cfDNA carries clinical utility as an MRD biomarker
Liquid biopsy analyses should be incorporated into future medulloblastoma trials
Acknowledgements
The authors acknowledge patients and families, physicians, nursing, research, and administrative staff from all participating institutions. In particular, we thank Aksana Vasilyeva, PhD and Jana Freeman, CCRP from the St. Jude Division of Neuro-Oncology for coordination of study material, the St. Jude Biorepository for archiving and providing patient samples, Scott Olsen and Sanchit Trivedi from the St. Jude Hartwell Center, for their help with next-generation sequencing. We thank Brandon Stelter for assistance with figure preparation and artwork. This study was principally supported by the National Cancer Institute (P.A.N.; R21CA256386). Additional funding was provided by the American Lebanese Syrian Associated Charities, National Cancer Institute Cancer Center Grant (P30CA021765), St. Jude Comprehensive Cancer Center Developmental Funds (P.A.N. & G.W.R.) and Conquer Cancer – #cureMEdullo powered by Carson Leslie Foundation Young Investigator Award (A.P.Y.L.). A.P.Y.L. is supported by the Li Shu Pui Medical Foundation Training Grant and the Lin Kin Pang-HKU Foundation Scholarship. P.A.N. is a Pew-Stewart Scholar for Cancer Research (Margaret and Alexander Stewart Trust), Sontag Foundation Distinguish Scientist, and recipient of the Robert J. Arceci Innovation Award (St. Baldrick’s Foundation).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
Title for Supplementary Table 1. Summary of literature on cfDNA studies for CNS tumors. Related to Figure 1.
References
- Bonner ER, Bornhorst M, Packer RJ, and Nazarian J (2018). Liquid biopsy for pediatric central nervous system tumors. NPJ precision oncology 2, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Housepian EM, and Herbert C (1969). An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology 93, 1351–1359. [DOI] [PubMed] [Google Scholar]
- Coustan-Smith E, Sancho J, Hancock ML, Boyett JM, Behm FG, Raimondi SC, Sandlund JT, Rivera GK, Rubnitz JE, and Ribeiro RC (2000). Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood 96, 2691–2696. [PubMed] [Google Scholar]
- De Mattos-Arruda L, Mayor R, Ng CK, Weigelt B, Martínez-Ricarte F, Torrejon D, Oliveira M, Arias A, Raventos C, and Tang J (2015). Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nature Communications 6, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA Jr, and Bardelli A (2014). Liquid biopsies: genotyping circulating tumor DNA. Journal of Clinical Oncology 32, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudero L, Llort A, Arias A, Diaz-Navarro A, Martínez-Ricarte F, Rubio-Perez C, Mayor R, Caratù G, Martínez-Sáez E, and Vázquez-Méndez É (2020). Circulating tumour DNA from the cerebrospinal fluid allows the characterisation and monitoring of medulloblastoma. Nature Communications 11, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouladi M, Gajjar A, Boyett JM, Walter AW, Thompson SJ, Merchant TE, Jenkins JJ, Langston JW, Liu A, Kun LE, and Heideman RL (1999). Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumor. Journal of Clinical Oncology 17, 3234–3237. [DOI] [PubMed] [Google Scholar]
- Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, Woo S, Wheeler G, Ahern V, Krasin MJ, et al. (2006). Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. The Lancet Oncology 7, 813–820. [DOI] [PubMed] [Google Scholar]
- Gajjar A, Hernan R, Kocak M, Fuller C, Lee Y, McKinnon PJ, Wallace D, Lau C, Chintagumpala M, and Ashley DM (2004). Clinical, histopathologic, and molecular markers of prognosis: toward a new disease risk stratification system for medulloblastoma. Journal of Clinical Oncology 22, 984–993. [DOI] [PubMed] [Google Scholar]
- Gajjar A, Robinson GW, Smith KS, Lin T, Merchant TE, Chintagumpala M, Mahajan A, Su J, Bouffet E, Bartels U, et al. (2021). Outcomes by Clinical and Molecular Features in Children With Medulloblastoma Treated With Risk-Adapted Therapy: Results of an International Phase III Trial (SJMB03). J Clin Oncol, JCO2001372. [DOI] [PMC free article] [PubMed]
- Huang TY, Piunti A, Lulla RR, Qi J, Horbinski CM, Tomita T, James CD, Shilatifard A, and Saratsis AM (2017). Detection of Histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathologica Communications 5, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal S, Vishnubhatla S, Raina V, Sharma S, Gogia A, Deo SS, Mathur S, and Shukla NK (2015). Circulating cell-free DNA and its integrity as a prognostic marker for breast cancer. Springerplus 4, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch R-D, and Knippers R (2001). DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Research 61, 1659–1665. [PubMed] [Google Scholar]
- Kortmann R-D, Kühl J, Timmermann B, Mittler U, Urban C, Budach V, Richter E, Willich N, Flentje M, Berthold F, et al. (2000). Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: results of the german prospective randomized trial hit ‘91. International Journal of Radiation Oncology*Biology*Physics 46, 269–279. [DOI] [PubMed] [Google Scholar]
- Kumar R, Smith KS, Deng M, Terhune C, Robinson GW, Orr BA, Liu APY, Lin T, Billups CA, Chintagumpala M, et al. (2021). Clinical Outcomes and Patient-Matched Molecular Composition of Relapsed Medulloblastoma. J Clin Oncol, JCO2001359. [DOI] [PMC free article] [PubMed]
- Lannering B, Rutkowski S, Doz F, Pizer B, Gustafsson G, Navajas A, Massimino M, Reddingius R, Benesch M, and Carrie C (2012). Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. Journal of clinical oncology 30, 3187–3193. [DOI] [PubMed] [Google Scholar]
- Merker JD, Oxnard GR, Compton C, Diehn M, Hurley P, Lazar AJ, Lindeman N, Lockwood CM, Rai AJ, and Schilsky RL (2018). Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists joint review. Archives of Pathology & Laboratory Medicine 142, 1242–1253. [DOI] [PubMed] [Google Scholar]
- Miller A, Szalontay L, Ahmad H, Bouvier N, Rodriguez-Sanchez I, Bale T, Benayed R, Arcila M, Donzelli M, Dunkel I, et al. (2020). BIOM-56. The integration of a liquid biopsy program into the care of pediatric brain tumor patients. Neuro-Oncology 22, ii13–ii14. [Google Scholar]
- Miller AM, Shah RH, Pentsova EI, Pourmaleki M, Briggs S, Distefano N, Zheng Y, Skakodub A, Mehta SA, Campos C, et al. (2019). Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 565, 654–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouliere F, Mair R, Chandrananda D, Marass F, Smith CG, Su J, Morris J, Watts C, Brindle KM, and Rosenfeld N (2018). Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Molecular Medicine 10, e9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulhern RK, Palmer SL, Merchant TE, Wallace D, Kocak M, Brouwers P, Krull K, Chintagumpala M, Stargatt R, and Ashley DM (2005). Neurocognitive consequences of risk-adapted therapy for childhood medulloblastoma. Journal of Clinical Oncology 23, 5511–5519. [DOI] [PubMed] [Google Scholar]
- Nevel KS, Wilcox JA, Robell LJ, and Umemura Y (2018). The utility of liquid biopsy in central nervous system malignancies. Current Oncology Reports 20, 60. [DOI] [PubMed] [Google Scholar]
- Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, Gröbner S, Segura-Wang M, Zichner T, Rudneva VA, et al. (2017). The whole-genome landscape of medulloblastoma subtypes. Nature 547, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer RJ, Gajjar A, Vezina G, Rorke-Adams L, Burger PC, Robertson PL, Bayer L, LaFond D, Donahue BR, and Marymont MH (2006). Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. Journal of Clinical Oncology 24, 4202–4208. [DOI] [PubMed] [Google Scholar]
- Panditharatna E, Kilburn LB, Aboian MS, Kambhampati M, Gordish-Dressman H, Magge SN, Gupta N, Myseros JS, Hwang EI, Kline C, et al. (2018). Clinically Relevant and Minimally Invasive Tumor Surveillance of Pediatric Diffuse Midline Gliomas Using Patient-Derived Liquid Biopsy. Clinical Cancer Research 24, 5850–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault S, Ramaswamy V, Achrol AS, Chao K, Liu TT, Shih D, Remke M, Schubert S, Bouffet E, Fisher PG, et al. (2014). MRI Surrogates for Molecular Subgroups of Medulloblastoma. American Journal of Neuroradiology 35, 1263–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V, Remke M, Bouffet E, Bailey S, Clifford SC, Doz F, Kool M, Dufour C, Vassal G, and Milde T (2016). Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathologica 131, 821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho Y-J, Shih DJ, Luu B, Dubuc AM, and Northcott PA (2013). Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. The Lancet Oncology 14, 1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson GW, Rudneva VA, Buchhalter I, Billups CA, Waszak SM, Smith KS, Bowers DC, Bendel A, Fisher PG, Partap S, et al. (2018). Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. The Lancet Oncology 19, 768–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubnitz JE, Inaba H, Dahl G, Ribeiro RC, Bowman WP, Taub J, Pounds S, Razzouk BI, Lacayo NJ, and Cao X (2010). Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. The Lancet Oncology 11, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabel M, Fleischhack G, Tippelt S, Gustafsson G, Doz F, Kortmann R, Massimino M, Navajas A, Von Hoff K, and Rutkowski S (2016). Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study. Journal of Neuro-Oncology 129, 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siravegna G, Marsoni S, Siena S, and Bardelli A (2017). Integrating liquid biopsies into the management of cancer. Nature Reviews Clinical Oncology 14, 531–548. [DOI] [PubMed] [Google Scholar]
- Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, et al. (2012). Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, and Bettegowda C (2017). Applications of DNA-Based Liquid Biopsy for Central Nervous System Neoplasms. The Journal of Molecular Diagnostics 19, 24–34. [DOI] [PubMed] [Google Scholar]
- Wang Y, Springer S, Zhang M, McMahon KW, Kinde I, Dobbyn L, Ptak J, Brem H, Chaichana K, Gallia GL, et al. (2015). Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proceedings of the National Academy of Sciences 112, 9704–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood B, Wu D, Crossley B, Dai Y, Williamson D, Gawad C, Borowitz MJ, Devidas M, Maloney KW, and Larsen E (2018). Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood 131, 1350–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code generated during this study is available through Github at https://github.com/kylessmith/cfdna. Sequencing data used in the manuscript have been deposited to European Genome-Phenome Archive (EGAS00001005592) and are publicly available as of the date of publication. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.