

Abstract
Cell lines used in bioproduction are routinely engineered to improve their production efficiency. Numerous strategies, such as random mutagenesis, RNA interference screens, and transcriptome analyses have been employed to identify effective engineering targets. A genome-wide small interfering RNA screen previously identified the CASP8AP2 gene as a potential engineering target for improved expression of recombinant protein in the HEK293 cell line. Here, we validate the CASP8AP2 gene as an engineering target in HEK293 cells by knocking it out using CRISPR/Cas9 genome editing and assessing the effect of its knockout on recombinant protein expression, cell growth, cell viability, and overall gene expression. HEK293 cells lacking CASP8AP2 showed a seven-fold increase in specific expression of recombinant luciferase and a 2.5-fold increase in specific expression of recombinant SEAP, without significantly affecting cell growth and viability. Transcriptome analysis revealed that the deregulation of the cell cycle, specifically the upregulation of the cyclin-dependent kinase inhibitor 2A (CDKN2A) gene, contributed to the improvement in recombinant protein expression in CASP8AP2 deficient cells. The results validate the CASP8AP2 gene is a viable engineering target for improved recombinant protein expression in the HEK293 cell line.
Keywords: caspase 8-associated protein 2, CRISPR/Cas9, cyclin-dependent kinase inhibitor 2A, recombinant protein, RNA-seq analysis
1 |. INTRODUCTION
Improved expression of recombinant proteins from mammalian cells is the desired goal being pursued through many different strategies. The main focus has been on optimizing growth and culture parameters (De Jesus & Wurm, 2011) and on modifying the properties of the producing cells (Fischer et al., 2015). The gene targets for cell modification are usually selected based on their known biological functions and the desired phenotypic outcome. For example, proapoptotic and antiapoptotic genes have been manipulated to extend the longevity of cultured cells (Baek et al., 2017; Cost et al., 2010); and genes involved in sugar transport have been manipulated to improve the metabolic efficiency of the producing cells (Inoue et al., 2010; Paredes et al., 1999). However, this approach is limited to genes with known function, whose altered expression may achieve the desired phenotype. In addition, genetic redundancy or other system-level robustness can nullify attempted modifications (Ekholm & Reed, 2000). As a result, novel approaches for identifying engineering targets are needed.
In our ongoing research, we employ noncoding RNA, microRNA, and small interfering RNA (siRNA) to identify genes whose altered expression can affect cell growth and recombinant protein expression (Inwood, Betenbaugh et al., 2018; Inwood, Buehler et al., 2018; Xiao et al., 2015; Xiao et al., 2016). Genome-wide RNA interference screens can identify targets which were not considered as engineering candidates before (Druz et al., 2013; Klanert et al., 2019; Xiao et al., 2016) while avoiding the effects of genetic redundancy and other system-level controls.
In a genome-wide siRNA screen, the caspase 8-associated protein 2 (CASP8AP2) gene was identified as a potential engineering target for improved recombinant protein expression. The knockdown of its messenger RNA (mRNA) by siRNA led to improved recombinant protein expression without significantly affecting cell growth and viability (Xiao et al., 2016). CASP8AP2 encodes a protein known as “FLICE-associated large protein” (FLASH), which is involved in numerous cellular processes. In Fas-mediated apoptosis, FLASH is part of the death-inducing signaling complex (DISC) assembled following the binding of the death receptor Fas to its ligand FasL. The DISC assembly activates procaspase 8 to form proteolytic caspase 8, which triggers the extrinsic apoptotic cascade (Imai et al., 1999). FLASH, in addition to its role in apoptosis, is also involved in replication-dependent histone mRNA maturation and transcriptional regulation of histone genes (Barcaroli et al., 2006; Hummon et al., 2012; Yang et al., 2009), in the S-phase progression of the cell cycle, and in the activation of the transcription factors cMYB and nuclear factor kappa B (Alm-Kristiansen et al., 2008; Barcaroli et al., 2006; Jun et al., 2005).
In this study, the positive effect of CASP8AP2 inactivation on recombinant protein expression was validated by knocking out this gene in HEK293 cells expressing luciferase using the CRISPR/Cas9 genome editing. The engineered cell line was evaluated for its recombinant protein expression and its growth parameters. The effect of CASP8AP2 knockout on global gene transcription was also investigated using comparative RNA-seq analysis.
2 |. MATERIALS AND METHODS
2.1 |. Cell line
HEK293 stably expressing the luciferase gene of the firefly, Photinus pyralis under the cytomegalovirus (CMV) promoter (CMV-Luc2-Hygro HEK293; ID #CAS140901; Promega). This cell line will be referred to in this study as the parental cell line.
2.2 |. Cell culture
Cells were grown adherent in Dulbecco’s modified Eagle medium (Cat #11995-040; Gibco) and supplemented with 10% fetal bovine serum (Cat #S11150H; Atlanta Biologicals), 100 units/ml of penicillin, and 100 μg/ml of streptomycin (Cat #15140-122; Gibco). The cultures were incubated in a humidity-controlled incubator at 37°C and 5% CO2.
2.3 |. CRISPR/Cas9 constructs
Two plasmids encoding paired guide RNAs (gRNAs) targeting the 10th exon of the CASP8AP2 gene, designed by transOMIC Technologies, were used in this study. The plasmids also encode carbenicillin and zeocin resistance genes and Zoanthus sp. green fluorescence.
Cat #TEDH-1011889, referred to in the text as plasmid 889,
gRNA-a: AAACGTAGAGTGTCAGCTGATGTGCGGAAG;
gRNA-b: ACTGGCTGTGGTGATGGTGAACCAAGGATA.
Cat #TEDH-1011892 referred to in the text as plasmid 892,
gRNA-a: CCTCCATAAGTGAACATATCTTGGGGGAAG;
gRNA-b: CTCGACTGTGTGACCTTTCTCCAGGGGTCT.
Plasmid encoding Cas9 nuclease, transOMIC (Cat #TECC1006), referred to in the text as Cas9 plasmid.
2.4 |. Transfections of parental and CASP8AP2 mutant cell lines
Twenty-four-well plates were seeded with 200,000 cells per well. The seeded cells were transfected 24 h later with the relevant plasmid(s) as follows:
For the CRISPR/Cas9 plasmid expression: Cells were cotransfected with 500 ng per well of plasmid 892 and 500 ng per well of Cas9 plasmid or with 500 ng per well of plasmid 889 and 500 ng per well of Cas9 plasmid.
For secreted alkaline phosphatase (SEAP) expression: Cells were transfected with 500 ng per well of the pSELECT-zeo-SEAP plasmid, encoding the embryonic SEAP driven by an EF-1α/HTLV composite promoter (Cat #psetz-seap; InvivoGen).
For cyclin-dependent kinase inhibitor 2A (CDKN2A) expression: cells were transfected with 500 ng per well of plasmid pCMV p16 INK4A (Plasmid #10916; Addgene), a gift from Bob Weinberg (Medema et al., 1995).
Lipofectamine® 2000 transfection reagent (Cat #11668-019; Invitrogen) was used in all transfections, following the manufacturer’s protocol. After 2 days, cell culture media were replaced with selection media: culture media supplemented with 1 μg/ml puromycin and 10 μg/ml blasticidin for CASP8AP2 CRISPR/Cas9 transfected cells; zeocin 200 μg/ml for SEAP transfected cells; G418 400 μg/ml for CDKN2A transfected cells.
2.5 |. Isolation of single cells
After the CASP8AP2 CRISPR/Cas9 transfected cell pool was propagated, single colonies were isolated using BD FACSAria™ III (BD Biosciences) green fluorescence-based flow cytometry into 96-well plates. Single clones were propagated in selection media.
2.6 |. Quantification of SEAP activity
Culture supernatant samples from SEAP transfected cells were collected on Days 3 and 5 posttransfection. Alkaline phosphatase activity in the culture supernatant was quantified using the Quanti-Blue colorimetric assay (Cat #rep-qb1; InvivoGen), cells were enumerated using CEDEX HiREs cell analyzer/counter (Cat #7766; Roche), and specific SEAP activity determined.
2.7 |. Sequencing of the target CASP8AP2 genomic locus
Total genomic DNA was isolated using the DNeasy Kit (Cat #69506; Qiagen), following the manufacturer’s protocol. The genomic region flanking the CRISPR target site of the CASP8AP2 gene was amplified using Phusion® High-Fidelity PCR Master Mix with HF Buffer (Cat #MO531S; NEB) and the following primer sets:
For 889 CRISPR-transfected cells:
forward primer—CTGCCAAATCTGGAAAAGGA;
reverse primer—CAGAATCTGAGTTACTTTTAAACTTCG;
forward primer—GTGAAGAGCCCATTTGTGGT;
reverse primer—TCATGATCTACCTGCCTCAGC;
forward primer—CTAGCACTTAGGGAGGCTGA;
reverse primer—ATCAGGTCTTTTATTCCATTTGTT
For 892 CRISPR-transfected cells:
forward primer—CAAAGACTGAAAGCAAAAGTTCG;
reverse primer—ACCACAAATGGGCTCTTCAC.
Amplicons were run on ethidium bromide agarose gel, the DNA was extracted using the QIAquick® Gel Extraction Kit (Ref #28704; QIA-GEN) and sequenced using capillary DNA sequencing. Sequences were aligned with the parental sequence using the EMBOSS Water (European Bioinformatics Institute) Pairwise Sequence Alignment program.
2.8 |. Luciferase activity assay
Cell viability and luciferase activity were determined using CellTiter-Glo and One-Glo luciferase luminescent assays (Cat #G7570 and Cat #E6110, respectively; Promega). Plates were read in a SpectraMax®microplate reader (Molecular Devices), at an integration time of 250 ms.
2.9 |. Quantitative polymerase chain reaction analysis
Real-time quantitative polymerase chain reaction (RT-qPCR) was completed using the SYBR GREEN protocol. Total RNA was extracted from parental cells and CASP8AP2 knockout cells using the RNeasy kit (Cat #74101; QIAGEN). Complementary DNA (cDNA) was synthesized from total RNA using the Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Cat #K1642; Thermo Fisher Scientific). Twenty nanograms of synthesized cDNA were amplified in a qPCR using the following primers: CASP8AP2: forward primer—AGAGAAGAGGATGGGAAGATTG, reverse primer—GCATGGCATC AACTTCCTTAG; Luc: forward primer—GTGGTGTGCAGCGAGAAT AG, reverse primer—CGCTCGTTGTAGATGTCGTTAG; CDKN2A forward primer—TACTGAGGAGCCAGCGTCTA, reverse primer—AGCACCACCAGCGTGTC. The threshold and threshold cycle (Ct) values were determined automatically by RQ Manager™ software (Applied Biosystems) using default parameters. The comparative cycle threshold (2−ΔΔCt) method was used to analyze the expression levels of genes examined in this study. The abundance of each gene transcript was normalized to the expression of glyceraldehyde phosphate dehydrogenase (GAPDH) or β-actin (ACTB) and expressed in arbitrary units. The relative quantification of gene expression was performed in triplicates for each sample.
2.10 |. Luciferase western blot
Parental and engineered cells in confluent six-well plates were lysed using radioimmunoprecipitation assay cell lysis buffer (Cat #89900; Thermo Fisher Scientific) supplemented with 1X protease and phosphatase inhibitor (Cat #1861281; Thermo Fisher Scientific), and total protein was quantified at A280 using Nanodrop Onec (Thermo Fisher Scientific). Cell lysate samples containing 340 μg of total protein and 100 ng of recombinant firefly luciferase (Cat #ab100961; Abcam) were electrophoresed on a 4%–12% bis-tris gel (Cat #NP0322BOX; Thermo Fisher Scientific). The separated proteins were transferred onto a nitrocellulose membrane and luciferase was detected using mouse anti-luciferase polyclonal antibodies (Cat #PA1-179; Thermo Fisher Scientific)/horseradish peroxidase-conjugated goat anti-mouse antibodies (Cat #474-1806; KPL). Mouse anti-β-actin monoclonal antibodies (Cat #A2228; Sigma) were used to detect β-actin in identical samples. The signal was developed using SuperSignal® West Pico Chemiluminescent substrate (Cat #OD187429; Thermo Fisher Scientific) and visualized in a LAS-4000 Mini Luminescent Image Analyzer (Cat #28955810; GE Healthcare). The intensity of the Western blot bands was determined using ImageJ software (NIH) and the luciferase/actin ratio and fold increase was reported.
2.11 |. Growth characterization determination
Four T-25 flasks were inoculated with 7 × 105 cells in 5 ml culture media and incubated as described above. The cells were enumerated each day for 4 days using the CEDEX HiREs counter (Cat #7766; Roche). Glucose and lactate concentrations were quantified using the YSI 2900 bio-analyzer (Yellow Springs Instrument Co.).
2.12 |. Cell cycle analysis
The proportion of cells in different cell cycle phases was evaluated using flow cytometry (Beckman Coulter FC500; Beckman Coulter). Cells were cultured asynchronously or synchronized by double thymidine block as previously described (Chen & Deng, 2018) and collected at 0, 2, 6, and 29 h after release from the second thymidine block. Cells (1 × 106 cells/ml) were then briefly washed twice with phosphate-buffered saline (PBS) and resuspended in 300 μl PBS in a microtube. Seven hundred microliters of 100%, −20°C high-grade ethanol was added and the tube inverted several times. Cells were stored at −20°C until required for flow cytometric analysis. Propidium iodide (PI) staining was carried out using the FxCycle™PI/RNase Staining Solution (Cat #F10797; Life Technologies), following the manufacturer’s protocol. The cells were analyzed using a FACSCalibur flow cytometer (Becton Dickinson), counting 10,000 events for each sample. Data were analyzed using FlowJo software (Tree Star).
2.13 |. Fas-mediated apoptosis induction and caspase 8 activity assay
Cells in the culture at a confluency of 80%–90% were treated with anti-Fas antibodies at a concentration of 500 ng/ml (Cat #ab133619; Abcam) and incubated as described above for 4 h. The cells were then harvested by trypsinization and washed with PBS. Untreated cells were used as controls.
The FLICE/Caspase-8 Colorimetric Assay kit (Cat #K113-25; BioVision) was used to quantify caspase 8 activity in treated and untreated control cells using the manufacturer’s protocol, assaying 100 ng total protein per well in a 96-well plate, and read in a SpectraMax® microplate reader.
2.14 |. RNA-seq analysis
Cell culture samples from biological triplicate experiments of CASP8AP2 mutant and parental cell lines were harvested at 80%–90% confluence and total RNA was extracted using Qiagen RNeasy Kit (Cat #74101). RNA quality was assessed using Agilent 2100 Bioanalyzer with RNA 6000 Nano Kit (Agilent Technologies). Samples with an RNA integrity number of 8 or higher were processed to generate libraries for mRNA sequencing following the Illumina® TruSeq Stranded mRNA Sample Preparation Guide. cDNA libraries of six samples were loaded onto each lane of a rapid-run flow cell on a HiSeq2500 sequencer to generate paired-end sequencing reads (Illumina) for 100 × 2 cycles.
The fastq files were generated from bcl files, which is the primary output of Illumina HiSeq when using the bcl2fastq module on Biowulf (NIH’s high performing computation system). The fastq files were processed with PartekFlow (Partek Inc.) for bioinformatic analysis. The raw reads were preprocessed by trimming bases from the 3′-end to remove the low-quality reads. The QA/QC of the subsequent reads were checked to ensure the quality. The reads were aligned to recent transcript model hg38 (Ensembl transcript release 92) using the STAR—2.5.3a algorithm, which mapped 203,376 transcripts and 56,852 genes. The transcriptome was quantified using the Partek E/M algorithm. The differences in the sample due to technical variations, sequencing depth, gene length, and composition were minimized by performing normalization using the transcripts per million method, adding +1, and compressing data taking log2. The principal components of the gene expression values in each population/sample of biological triplicates were closer to each other and were significantly distant from the compared samples. Statistical tests were performed on the biological triplicate data to predict the statistically significant differentially expressed genes. Analysis of variance (ANOVA) was used to compare CASP8AP2 deleted cells with the parental cells and to calculate the fold change (FC) values. Statistically significant genes (p ≤ .05 and the FC cut-off <1.5>) were further evaluated for their biological significance. Gene expression data were exported from PartekFlow and imported into ingenuity pathway analysis (IPA; QIAGEN; www.qiagen.com/ingenuity) for pathway investigation. The gene ontology, overrepresented canonical pathways, regulatory networks, and upstream regulators were analyzed using the IPA knowledgebase. Based on the change in gene expression in the parental and mutant samples, pathways were predicted and ranked based on their Z-score (−log [p]).
The Fastq file and processed data file were submitted to the NCBI GEO database with GEO accession number GSE143808.
3 |. RESULTS
3.1 |. Creating and validating CASP8AP2 knockout in HEK293 cells using CRISPR/Cas9 genome editing
The CASP8AP2 gene was knocked out in HEK293 cells constitutively expressing luciferase by using CRISPR/Cas9 genome editing, and a single clone was isolated by fluorescence-activated cell sorting. Analysis of the gRNA target locus in the selected clone (892–7) showed the presence of a biallelic heterozygous mutation, with one allele having a consecutive 91 basepair insertion, while the other allele had a nonconsecutive deletion of a similar length in the tenth exon of the gene. The CASP8AP2 gene knockout in the 892–7 clone was confirmed by qPCR analysis, which showed a 20-fold reduction in the abundance of CASP8AP2 mRNA in the knockout cells when primers targeting exon 10 were used for amplification (Figure 1a). The functional activity of FLASH, the protein encoded by CASP8AP2, was tested by apoptosis assay in which the activity of caspase 8 was quantified 4 h after inducing Fas-mediated apoptosis in parental- and CASP8AP2-deficient cells, by using anti-Fas antibodies. As shown in Figure 1b, apoptosis induction increased caspase 8 activity by 150% in the parental cell line, while caspase 8 activity in the 892–7 clone remained unchanged, confirming the absence of functional FLASH in the 892–7 clone.
FIGURE 1.
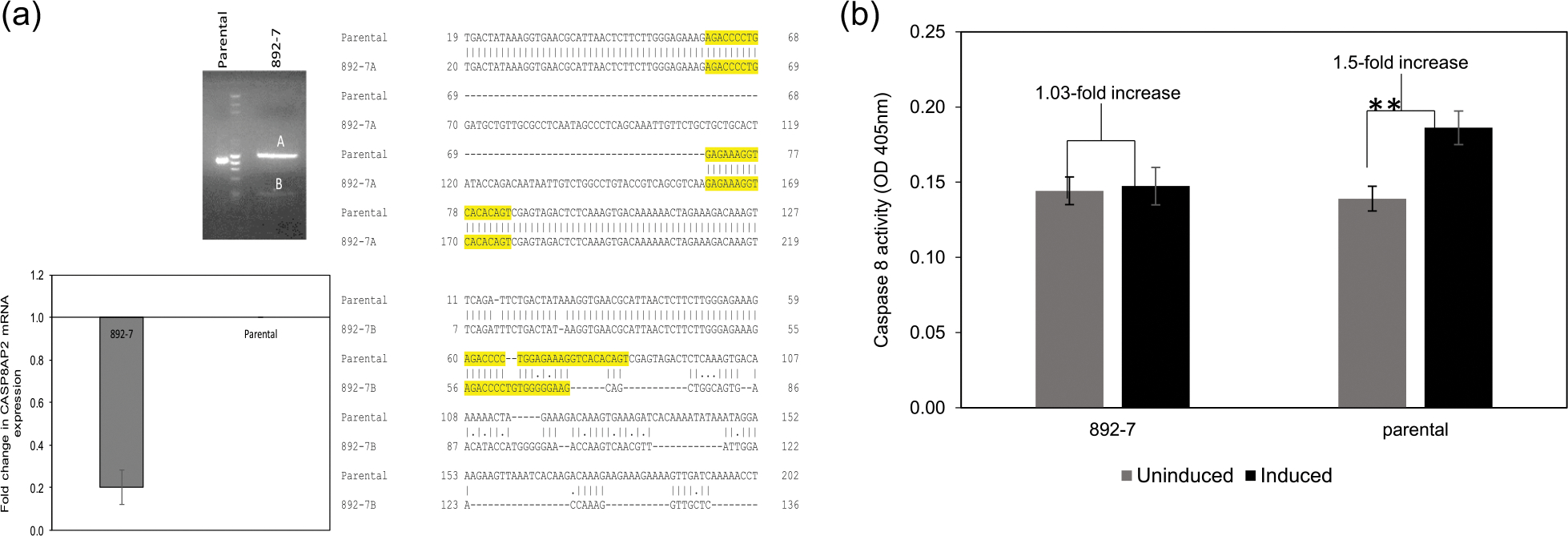
Knockout of the CASP8AP2 gene. (a) Agarose gel electrophoresis of polymerase chain reaction (PCR) amplified CASP8AP2 locus in the parental and the 892–7 cell lines, and EMBOSS WATER sequence alignment of both alleles with the parental sequence. Guide RNA (gRNA) target sequence is highlighted. GAPDH-normalized qPCR analysis of CASP8AP2 messenger RNA (mRNA) abundance in parental and CASP8AP2 knockout clone, 892–7, using primers targeting the gRNA locus for amplification. (b) Caspase 8 activity in cell lysates of control (uninduced) and apoptosis-induced (induced) 892–7 and parental cells (**p < .001)
3.2 |. Effect of CASP8AP2 knockout on recombinant protein expression in the HEK293 cell line
The effect of CASP8AP2 knockout on the constitutive expression of recombinant luciferase and the transient expression of SEAP is shown in Figure 2. Compared with the parental cell line the clone deficient in the CASP8AP2 gene, 892–7, showed a 7.4 average fold increase in specific luciferase activity (Figure 2a), which was also associated with 1.6-fold higher abundance of luciferase mRNA (Figure 2b) and a 6.9-fold higher luciferase protein (Figure 2c). The CRISPR CASP8AP2 knockout pool from which the 892–7 clone was isolated (892 pool), had on average a 1.3-fold increase in luciferase activity compared with the parental cell line (Figure S3).
FIGURE 2.
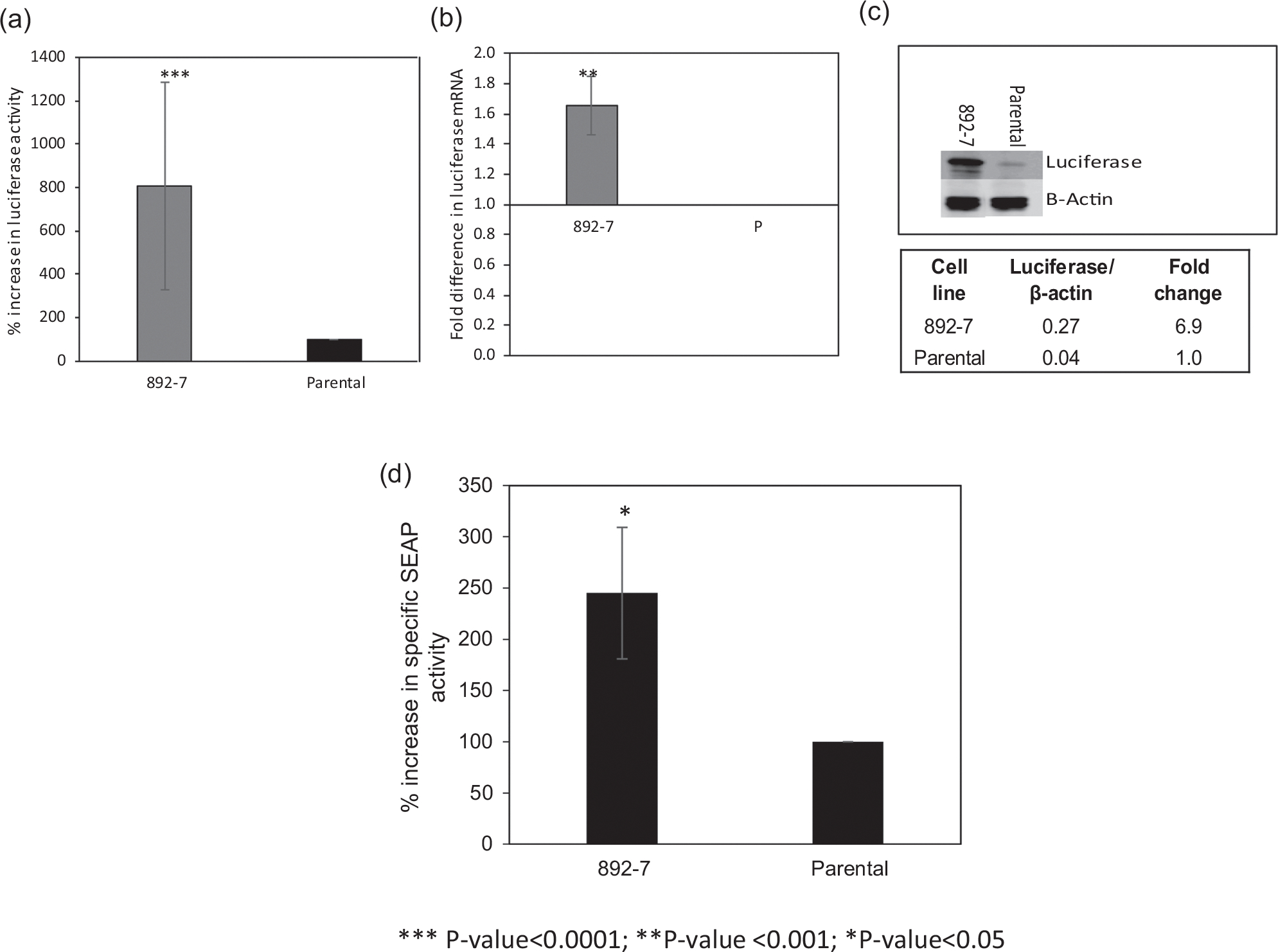
Luciferase and secreted alkaline phosphatase (SEAP) production from the parental cell line and the CASP8AP2 knockout clone, 892–7. (a) Luciferase enzymatic activity (***p < .0001). (b) GAPDH-normalized luciferase messenger RNA (mRNA) abundance determined using real-time quantitative polymerase chain reaction (** p < .001). (c) Luciferase expression by Western blot analysis. (d) Alkaline phosphatase activity in the cell culture supernatant of cells transfected with a plasmid encoding the SEAP gene (*p < .05). Results represent the average of at least three biological replicates, and error bars represent the standard deviation
Transient expression of recombinant SEAP from the parental cell line and the CASP8AP2-deficient clone, 892–7, transfected with plasmid encoding SEAP is shown in Figure 2d. Specific SEAP activity in the culture supernatant of the CASP8AP2 knockout clone, 892–7, was 2.5-fold higher than that of the parental cell line.
3.3 |. Effect of CASP8AP2 knockout on growth, metabolism, and cell cycle progression
Growth characteristics and luciferase production from parental and CASP8AP2 knockout clone (892–7) are shown in Figure 3a,b. The growth of the two cell lines for 4 days was comparable, with average doubling times of 24 h for the parental cell line and 26 h for the CASP8AP2 knockout clone (Figure 3a). Glucose consumption and lactate excretion were similar, with slightly higher glucose consumption (on average 0.23 g/L more) and lower lactate excretion (on average 0.28 g/L less) in the parental cell line (results not shown). Specific luciferase production during the 4-day growth period was sixfold to ninefold higher in the knockout clone, compared with the parental cells (Figure 3b). Flow cytometric cell cycle analysis on asynchronous cells showed that compared with the parental cell line, the CASP8AP2 knockout cell line had a 9% higher proportion of cells in the G0/G1 phase and a 6% lower proportion of cells in the G2/M phase. The proportion of cells in the S-phase was only slightly different between the two cell lines (Figure 4a–c). These results were confirmed in thymidine synchronized cells in which the 892–7 clone had 17.1%, 22.7%, 2.6%, and 11.7% more cells in the G0/G1 phase than the parental cell line at 0, 2, 6, and 29 h, respectively, after releasing from thymidine block. The percentage of cells in the G2/M phase was higher in the parental cell line at all time points (Figure 4d).
FIGURE 3.
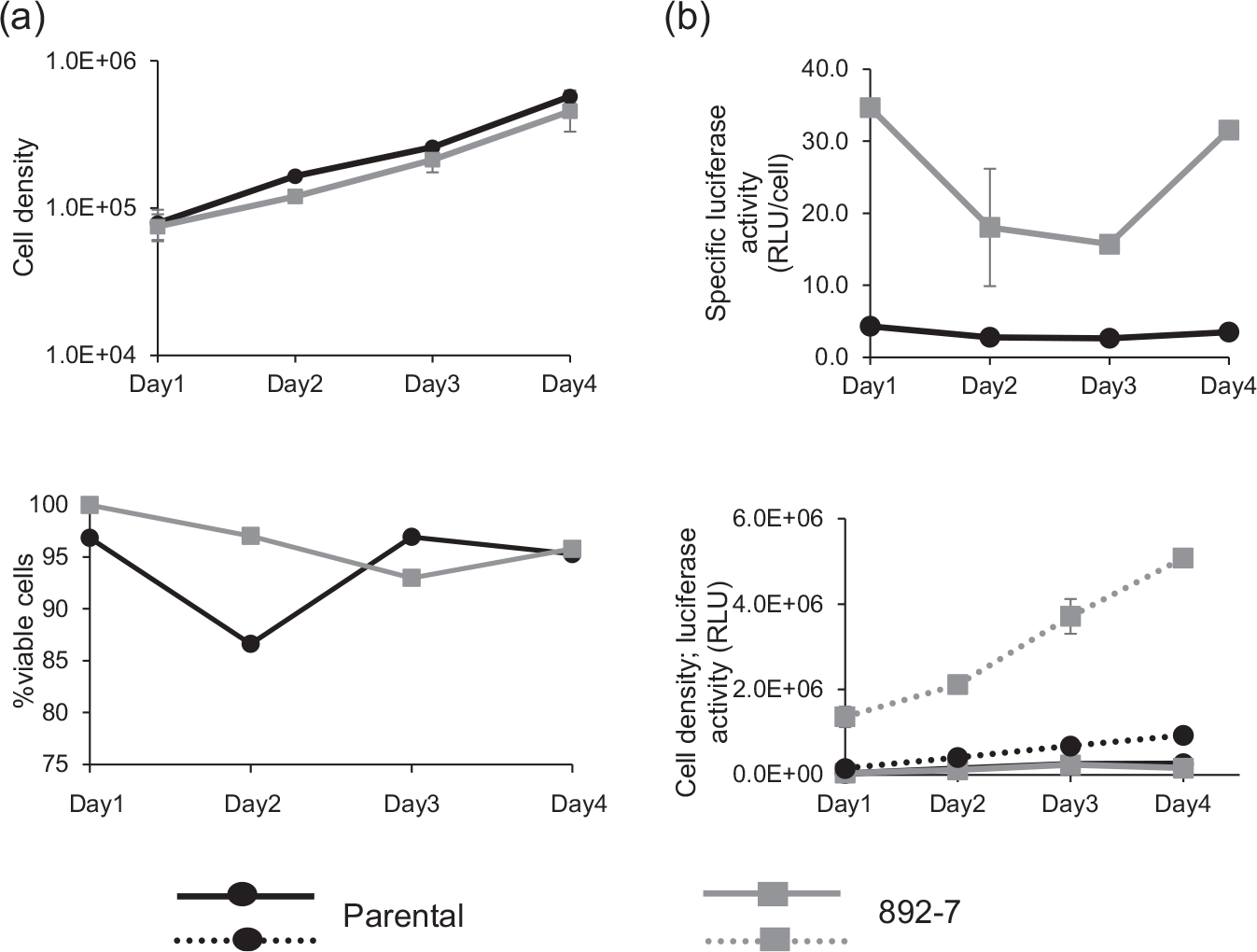
Growth and luciferase production in the 892–7 clone and parental cell line for 4 days growth period. (a) Cell density and viability. (b) Specific and total luciferase production. Graphs are representative of three separate growth and production experiments, and error bars represent the standard deviation
FIGURE 4.
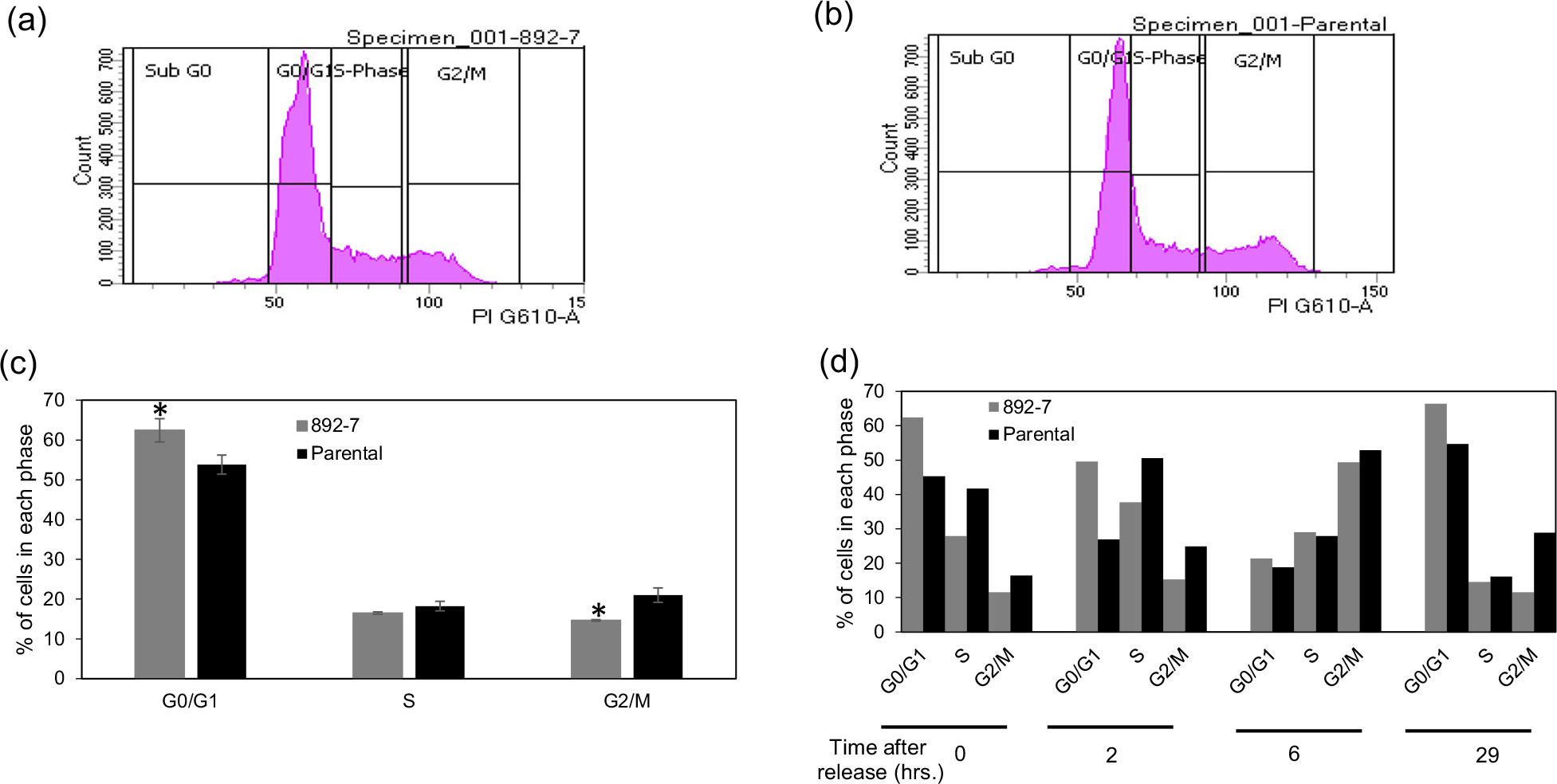
The effect of CASP8AP2 knockout on cell cycle progression by FACS-based, propidium iodide cell cycle analysis. (a) Knockout clone. (b) Parental cells. (c) Percentages of cells in the G1, S, or G2/M in asynchronous cultures calculated using BD FACS DIVA software. Results represent the average of three analyses, and error bars represent the standard deviation (*p < .05). (d) Parental and 892–7 cells were treated with thymidine to synchronize the cell cycle. After release from the thymidine block, the cells were harvested at the indicated time points and cell cycle progression was analyzed using a cytometer
3.4 |. Identifying genes associated with CASP8AP2 knockout by RNA sequencing analysis
To investigate the downstream effects of CASP8AP2 deletion, complete transcriptomic RNA-seq data were generated from the parental and the CASP8AP2 knockout cells. The RNA-seq analysis is summarized in Table 1. By alignment of the reads with the recent human transcript model hg38 (Ensemble transcript release 92); with a minimum overlap of 100%, 203,376 transcripts and 56,852 genes were mapped. From these genes, 3216 significant differentially expressed genes were identified by applying ANOVA to the mapped CASP8AP2 knockout and the parental genes, using a filter with p ≤ .05 and FC cut-off of <1.5>. Evaluation of these genes by IPA identified several overrepresented canonical pathways. Among the top 25 differentially expressed pathways, six were found to be involved in cell cycle regulation. They are: (1) cell cycle control of chromosomal replication, (2) HIPPO signaling, (3) cell cycle G2/M DNA damage checkpoint regulation, (4) DNA methylation and transcriptional repression signaling, (5) cell cycle G1/S checkpoint regulation, and (6) p53 signaling. Additional significantly affected pathways were the eukaryotic initiation factor 2 (EIF2) signaling pathway, protein ubiquitination pathway, regulation of eukaryotic initiation factor 4 (eIF4), p70 S6 kinase, and the mammalian target of rapamycin signaling pathway (Figure 5). The most highly differentially expressed gene family was the replication-dependent histone genes. All, except two, were upregulated in the CASPASAP2-deleted cell line, with 72% having a 1.5-fold or higher mRNA abundance (Figure S1).
TABLE 1.
Summary of raw and processed RNA-seq reads generated from CASP8AP2 mutant clone, 892–7, and parental RNA samples
| Sample name | Total reads (10e6) | Total alignments (10e6) | Aligned (%) | Coverage(%) | Avg. coverage depth |
|---|---|---|---|---|---|
| CASP8AP2 mutant | 17.94 | 40.83 | 95.19 | 5.11 | 25.31 |
| CASP8AP2 mutant | 74.56 | 16.42 | 94.67 | 10.16 | 51.34 |
| CASP8AP2 mutant | 18.95 | 42.17 | 95.31 | 5.51 | 24.28 |
| Parental | 10.53 | 23.9 | 97.98 | 3.46 | 21.87 |
| Parental | 18.85 | 42.85 | 97.56 | 4.60 | 29.60 |
| Parental | 15.78 | 34.73 | 97.68 | 5.26 | 21.03 |
FIGURE 5.
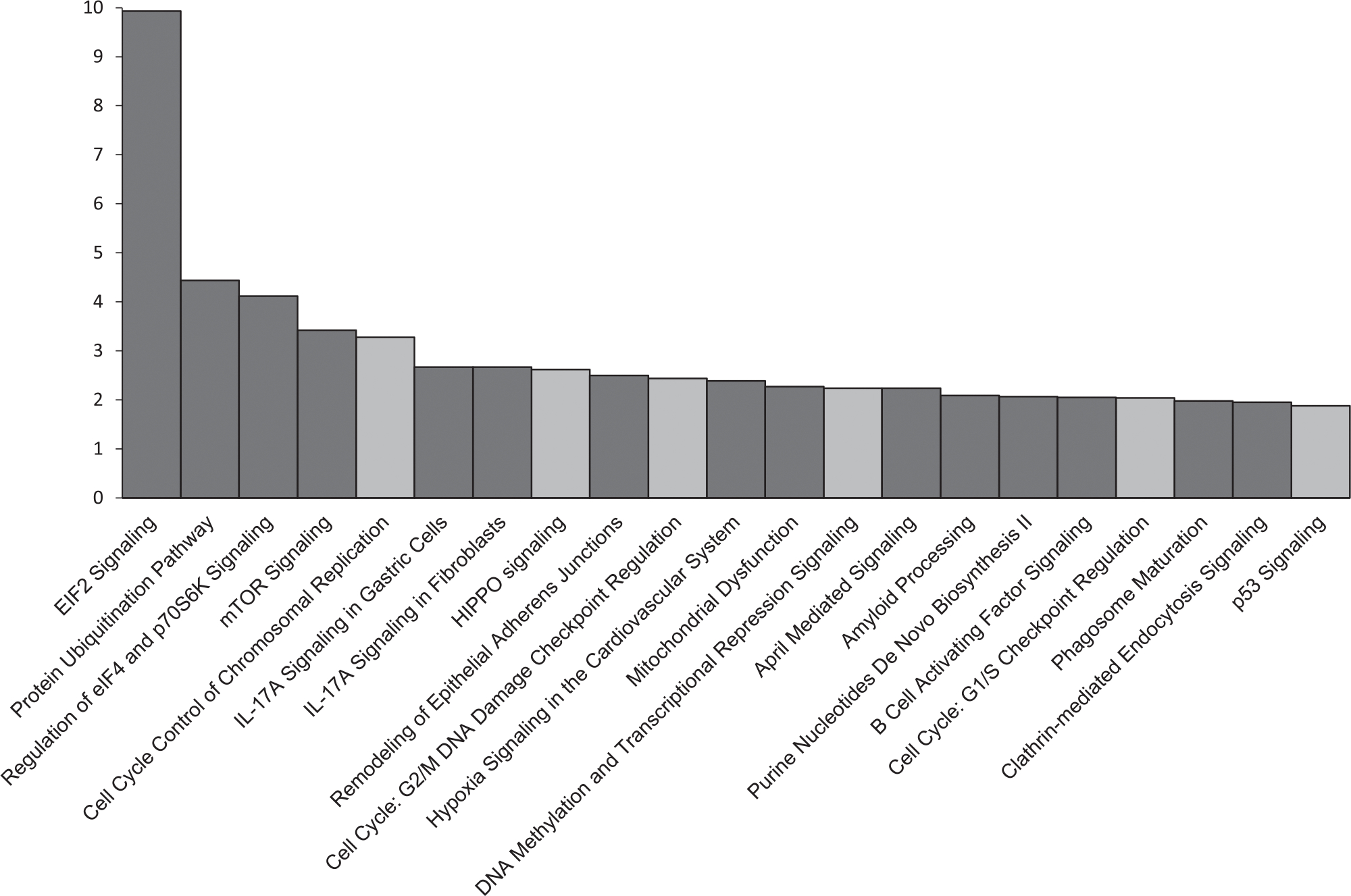
Pathway ranking based on Z-score following enrichment analysis of the differentially expressed genes between the CASP8AP2 knockout and the parental cell lines. Gray bars represent pathways involved in the cell cycle
3.5 |. Identification of CDKN2A gene as an effector of improved recombinant protein expression
As was shown above (Figure 4), the CASP8AP2-deficient cell line exhibited slow cell cycle progression at the G0/G1 phase. Since slower cell cycle progression is known to improve recombinant protein expression (Sunley & Butler, 2010), the differentially expressed genes involved in cell cycle progression were evaluated for their involvement in the improved recombinant protein expression in the CASP8AP2-deleted cell line. RNA-seq transcriptomic analysis identified nine genes involved in cell cycle progression that were at least twofold differentially expressed between the parental and the CASP8AP2-deleted cell lines (Figure 6). Validation of the RNA-seq data using qPCR led to the selection of the HDAC10 and the CDKN2A genes for further investigation. The CDKN2A mRNA was 4-fold and 2.4-fold more abundant, and the HDAC10 mRNA was 3-fold and 4-fold more abundant in the CASP8AP2-deleted clone, by RNA-seq and qPCR analyses, respectively (Figure 7a,b). The effect of overexpressing these two genes on luciferase expression was tested by transfecting the parental and the CASP8AP2-deleted cell lines with plasmids encoding them. Overexpression of HDAC10 showed no improvement in luciferase expression (data not shown) in either cell line. Overexpression of CDKN2A resulted in 2.5-fold higher luciferase activity in the transfected parental cells compared with the nontransfected parental cells and 1.5-fold higher luciferase activity in the CASP8AP2-deleted 892–7 cells compared with the nontransfected 892–7 cells (Figure 7c). The differential effect of the overexpression of CDKN2A on luciferase activity between the parental cell line and the CASP8AP2-deleted clone (in which the CDKN2A gene is upregulated), suggests that the CDKN2A gene is instrumental in the improved expression of luciferase in the CASP8AP2-deleted clone. The overexpression of the CDKN2A gene did not significantly affect cell growth and viability (Figure 7d).
FIGURE 6.
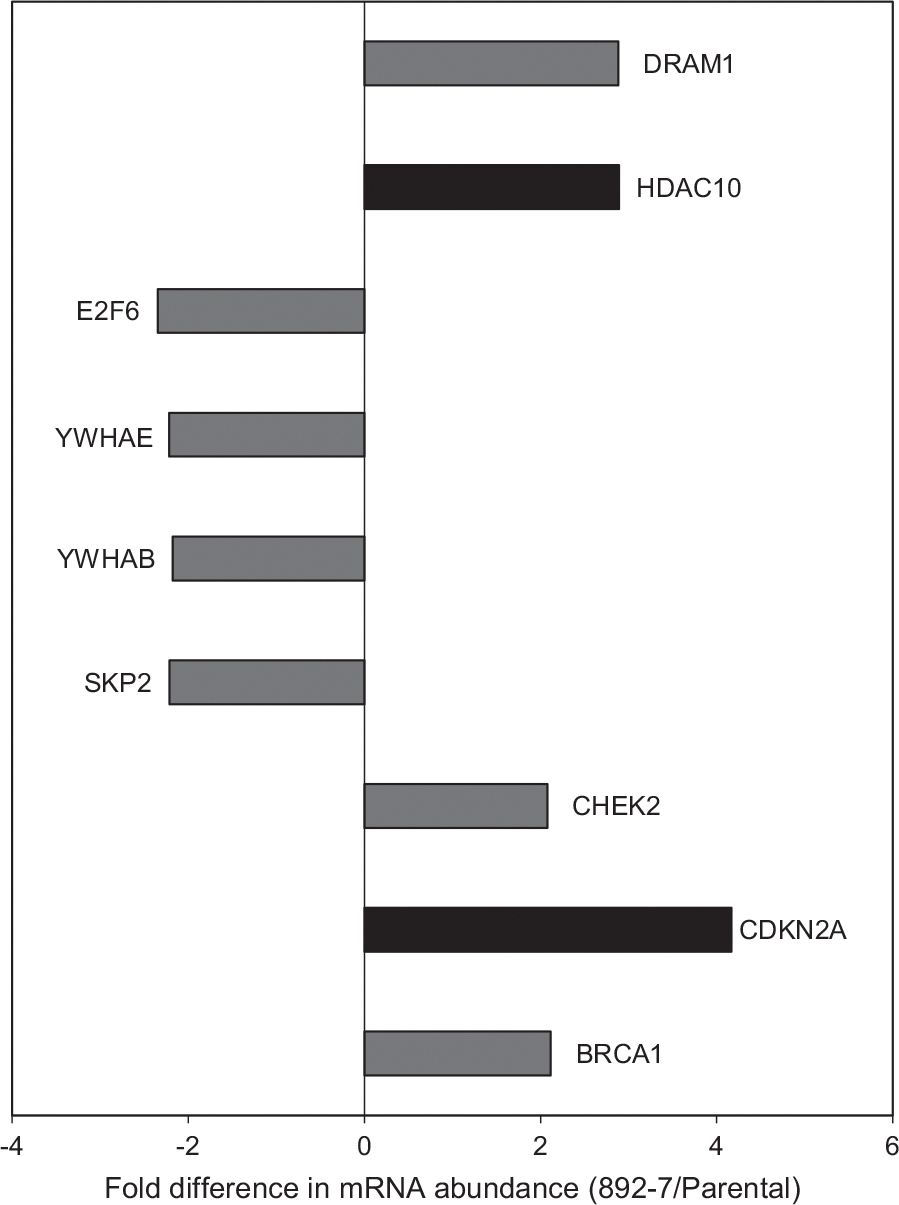
RNA-seq analysis of genes involved in the cell cycle. Differentially expressed cell cycle-related genes with more than the 1.5-fold difference in messenger RNA (mRNA) abundance between the mutant clone, 892–7, and the parental cell line with p < .05
FIGURE 7.
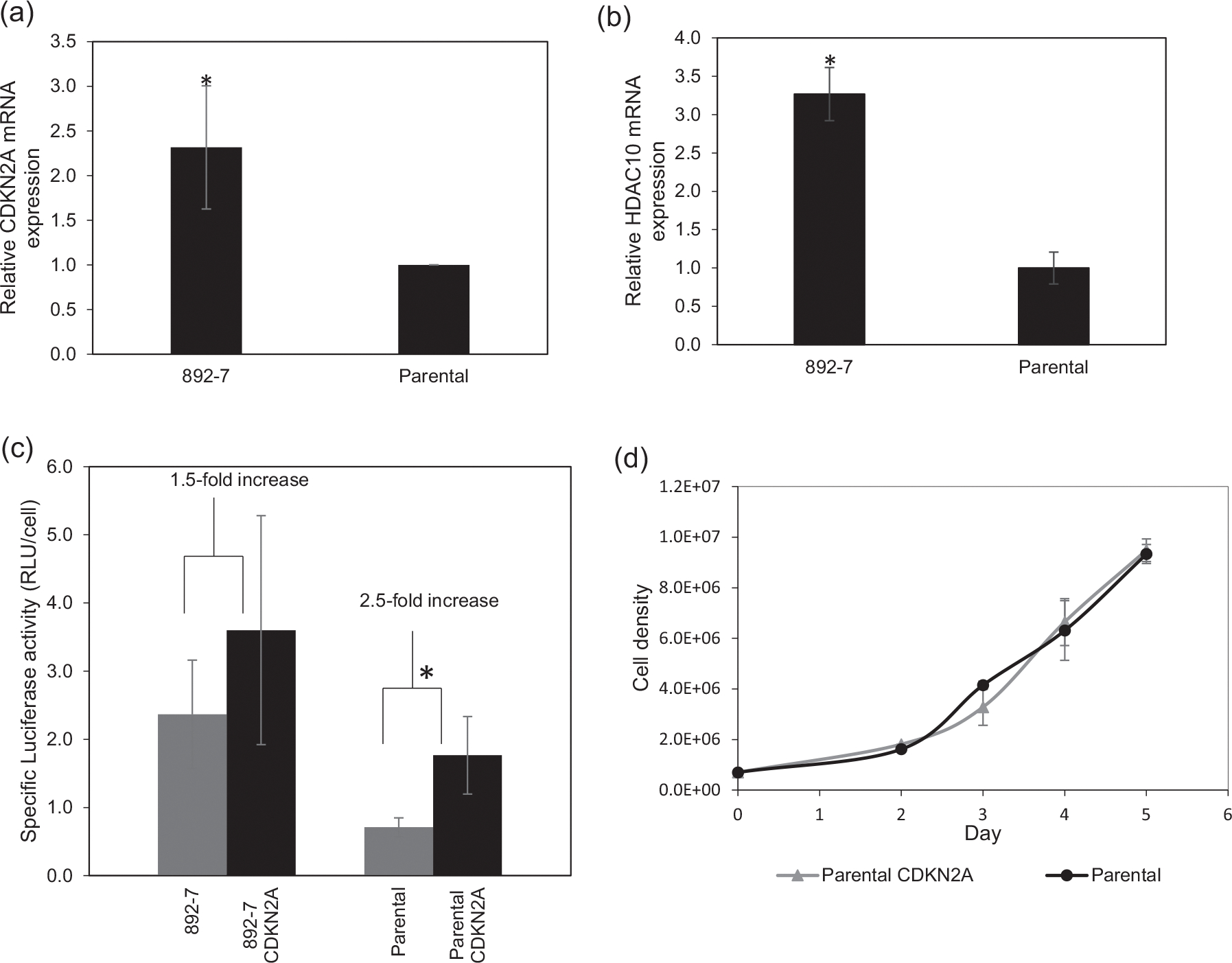
(a and b) Relative CDKN2A and HDAC10 expression in parental and CASP8AP2 knockout HEK293 cells determined in a GAPDH-normalized real-time quantitative polymerase chain reaction. (c) Effect of overexpressing CDKN2A on luciferase activity in parental (parental CDKN2A) and CASP8AP2 knockout (892–7 CDKN2A) HEK293 cells (*p < .05). (d) Effect of CDKN2A overexpression on cell growth of the parental cell line. Results represent the average of at least three biological replicates, with error bars representing standard deviation
4 |. DISCUSSION
The improved luciferase expression from the CASP8AP2 knockout HEK293 cells validates the information previously obtained from the genome-wide siRNA high-throughput screen that identified this gene as a promising engineering target for improving recombinant protein expression (Xiao et al., 2016). The expression of recombinant luciferase from the CASP8AP2 knockout clone was approximately sevenfold higher than its expression in the parental cells. This study also identifies the CDKN2A gene, which was upregulated in the CASP8AP2 knockout cell line (Figure 6c), as involved in the higher luciferase expression. The CASP8AP2-deleted clone showed a consistently higher proportion of cells in the G0/G1 phase and a lower proportion of cells in the G2/M phase, suggesting a G0/G1 cell cycle arrest (Figure 4).
Growth arrest at the G0/G1 phase, where the cells demonstrate higher metabolic activity, is a known strategy for improving specific productivity in mammalian cell lines (Du et al., 2015; Hendrick et al., 2001; Ibarra et al., 2003). In this phase, the cells are larger and actively express genes responsible for ribosome biosynthesis and protein translation (Bi et al., 2004; Carvalhal et al., 2003; Dez & Tollervey, 2004). Strategies for enhancing recombinant protein production based on growth arrest include overexpression of cyclin-dependent kinase inhibitors (p21Cip1 or p27Kip1; Bi et al., 2004; Carvalhal et al., 2003; Fussenegger et al., 1997; Meents et al., 2002), the addition of cytostatic chemical agents such as sodium butyrate or dimethyl sulfoxide (Hendrick et al., 2001; Hunt et al., 2002; Hwa Chang et al., 2002; Liu et al., 2001), and maintaining mild hypothermic culture conditions (Ahn et al., 2008; Fox et al., 2004; Nam et al., 2008). However, improved specific production using these approaches is associated with reduced cell growth and requires a careful balancing of growth with protein expression to achieve overall improvements in recombinant protein yield (Sunley & Butler, 2010). The knockout of the CASP8AP2 presented here is likely an attractive alternative for improved recombinant protein expression mediated by growth arrest at the G0/G1 phase since it offers improvements in recombinant protein expression with minimal effect on cell growth. A possible reason for this behavior is the association of CASP8AP2 knockout with the altered expression of two apoptosis regulators: the upregulation of the antiapoptotic gene XIAP, a potent inhibitor of apoptosis that inhibits caspase 3, 7, and 9; and the downregulation of DIABLO, its proapoptotic inhibitor (Figure S1).
The potential for utilizing CDKN2A as an engineering target for improved recombinant protein production is also highlighted in this study. The protein encoded by the CDKN2A gene, P16inka, is an inhibitor of cyclin-dependent kinases 4 and 6 that play pivotal roles in cell cycle G1 phase progression. Overexpressing CDKN2A in parental cells led to a 2.5-fold increase in luciferase expression while its overexpression in the CASP8AP2 knockout cells led to an additional 1.5-fold increase, an indication that this gene is likely responsible for the overall higher expression of luciferase in CASP8AP2 knockout cells. Previous studies that focused on overexpression of p21cip1 and p27kip1 (encoded by CDKN1A and CDKN1B genes, respectively), cyclin-dependent kinase inhibitors of cyclin E-CDK and A-CDK complexes, demonstrate two-to five-fold higher recombinant protein expression (Bi et al., 2004; Carvalhal et al., 2003; Fussenegger et al., 1997) and up to 10–30-fold, when coexpressed with the differentiation factor C/EBPα and the antiapoptotic protein Bcl-xL (Mazur et al., 1998). The overexpression of CDKN2A in parental cells expressing luciferase reported in this study led to a 2.5-fold increase in recombinant luciferase expression, which is within the range of the reported increases in recombinant protein expression prompted by the singular overexpression of p21Cip1 or p27Kip1. We, therefore, suggest that CASP8AP2 knockout causes the upregulation of cyclin-dependent kinase 2A which, in turn, leads to cell cycle arrest at the G0/G1 phase, and to the improvement of recombinant protein expression. A schematic presentation of the proposed role of CDKN2A is shown in Figure 8.
FIGURE 8.
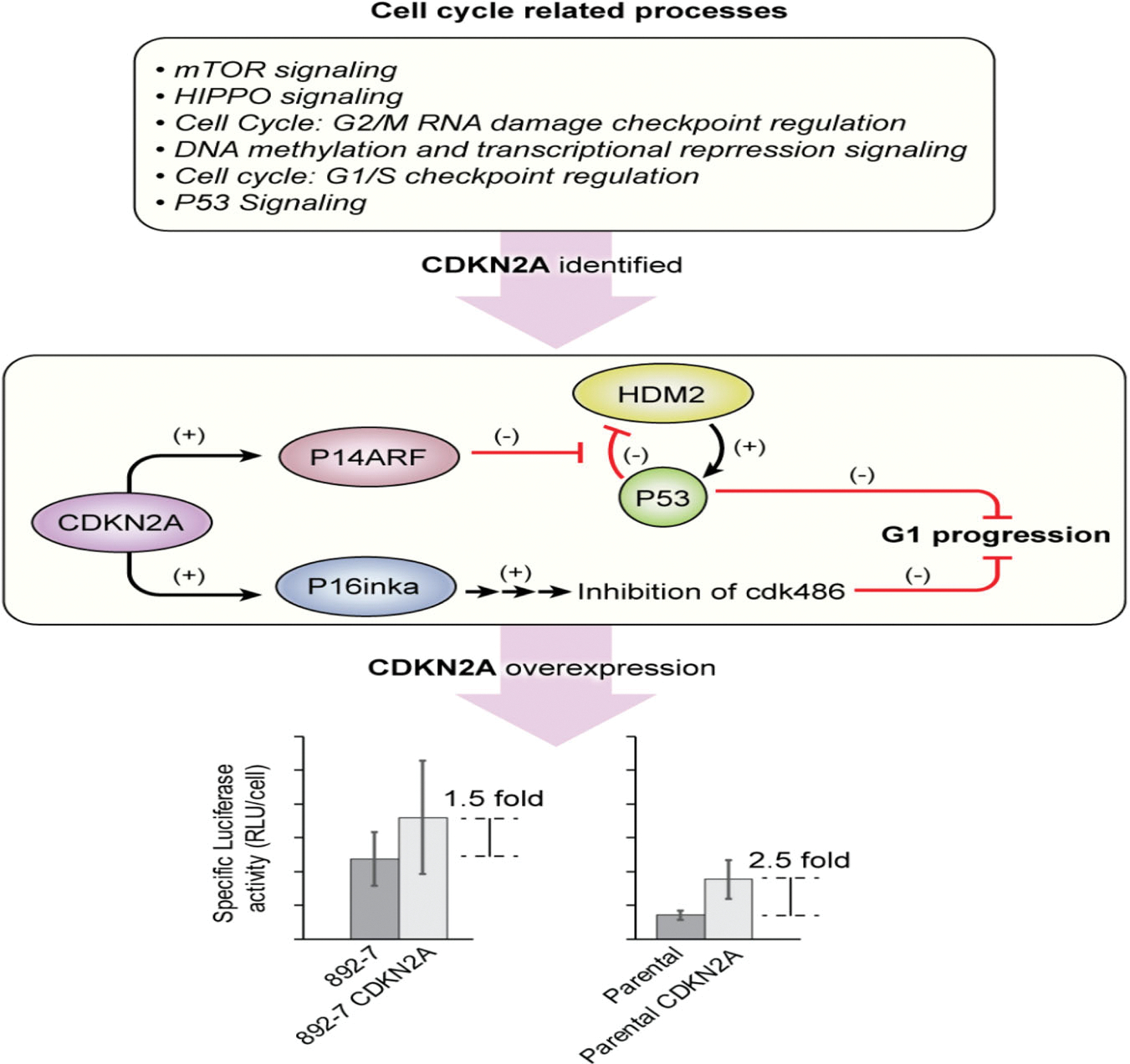
Identification of the CDKN2A gene as an effector for recombinant protein expression in the CASP8AP2 deficient 892–7 clone
Overexpressing the CDKN2A gene in parental cells improved luciferase expression by an average of 2.5-fold, which is significantly less than the seven-fold increase driven by CASP8AP2 knockout in the 892–7 clone. Although this difference can be the result of the additional burden of overexpression of CDKN2A, it is possible that the upregulation of the CDKN2A gene is only one of many genes whose altered expression is responsible for the observed phenotype in the 892–7 clone. Future studies concentrating on identifying these genes will provide more information on the mechanism involved.
The improvement in luciferase expression in the 892–7 clone was observed at both the transcriptional and translational levels (Figure 1a). The EIF2 signaling pathway which was highly differentially expressed between the two cell lines might be involved in the increased translation of recombinant luciferase. Among the differentially expressed genes of this pathway were the eukaryotic initiation factor 4A (EIF4A), and the activating transcription factor 5 (ATF5) that were upregulated 3.3- and 3.5-fold, respectively, in the 892–7 clone, whilst the phosphatase 1 catalytic subunit alpha (PPP1CA) was downregulated 3.5-fold. The EIF4A gene encodes a helicase that impacts translation by unwinding double-stranded RNA enabling the binding of ribosomal 40S units. Ribosomal proteins, which are involved in protein synthesis were highly differentially expressed, with a similar number of them upregulated or downregulated in the 892–7 clone (16 vs. 15). Other members of the eukaryotic initiation factor family (EIF2A, 2B, 3, 5) were only marginally up- or downregulated. Since there is no apparent pattern among the differentially expressed genes involved in protein biosynthesis, further investigation will be needed to understand their role in the improved expression of luciferase.
It was previously reported that RNAi CASP8AP2 knockdown caused cell cycle arrest in the S-phase with a consequent reduction in cell growth and viability in different cell lines, including the HEK293 cell line, (Barcaroli et al., 2006; Hummon et al., 2012; Kiriyama et al., 2009), and in vivo lethality observed in mouse embryos deficient in the CASP8AP2 gene (De Cola et al., 2012; Minamida et al., 2014). Cell cycle arrest at the S-phase was accompanied by suppression of the nonpolyadenylated replication-dependent histone genes. In the present study, the knockout of the CASP8AP2 gene did not significantly affect cell cycle S phase progression and cell growth and viability. This difference could be due to the mode of inactivation of the CASP8AP2 gene: CRISPR knockout in our study versus RNAi knockdown in the previous studies. Since cells can carry out compensatory changes when a gene is knocked out to diminish potentially lethal effects (which is not usually the case with knockdowns; Bunton-Stasyshyn et al., 2019), we speculate that the upregulation of poly-adenylated, noncanonical variants of the replication-dependent histones in the 892–7 clone (Figure S2), is a way for the cells to compensate for the suppression of canonical replication-dependent histones as a result of CASP8AP2 knockout. Reduced synthesis of replication-dependent histones has been shown to be responsible for the S-phase cell cycle arrest and eventual cell death when CASP8AP2 was knocked down by RNA interference (Bongiorno-Borbone et al., 2010). Another study in which the CASP8AP2 gene was knocked out in embryonic stem cells agrees with our findings, with no significant changes in cell growth and viability observed in the CASP8AP2 knockout cells (Minamida et al., 2014).
CASP8AP2 has been associated with multiple cellular functions. Its critical role is associated with Fas-mediated apoptosis, which was confirmed in this study by the absence of caspase 8 activation following the induction of Fas-mediated apoptosis in the CASP8AP2 knockout clone (Figure 2b). The finding that CASP8AP2 knockout led to the upregulation of noncanonical replication-dependent histone genes (Figure S2) supports the findings of Hummon et al. (2012), where there was a significant enrichment of histone transcripts in CASP8AP2-deficient colorectal cancer cells. The work presented here recommends not just a tool for improved recombinant protein expression but also suggests a possible mechanism for the improved phenotype.
Supplementary Material
ACKNOWLEDGMENTS
The work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) at the National Institutes of Health. The authors thank Dr. Je Nie Phue from the Center for Biologics Evaluation and Research, U.S. Food and Drug Administration, Silver Spring, MD, USA for assisting with the RNA-seq analysis, and the NIH Library for manuscript assistance.
Footnotes
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
REFERENCES
- Ahn WS, Jeon JJ, Jeong YR, Lee SJ, & Yoon SK (2008). Effect of culture temperature on erythropoietin production and glycosylation in a perfusion culture of recombinant CHO cells. Biotechnology and Bioengineering, 101(6), 1234–1244. 10.1002/bit.22006 [DOI] [PubMed] [Google Scholar]
- Alm-Kristiansen AH, Saether T, Matre V, Gilfillan S, Dahle O, & Gabrielsen OS (2008). FLASH acts as a co-activator of the transcription factor c-Myb and localizes to active RNA polymerase II foci. Oncogene, 27(34), 4644–4656. 10.1038/onc.2008.105 [DOI] [PubMed] [Google Scholar]
- Baek E, Noh SM, & Lee GM (2017). Anti-apoptosis engineering for improved protein production from CHO cells. Methods in Molecular Biology, 1603, 71–85. 10.1007/978-1-4939-6972-2_5 [DOI] [PubMed] [Google Scholar]
- Barcaroli D, Bongiorno-Borbone L, Terrinoni A, Hofmann TG, Rossi M, Knight RA, & De Laurenzi V (2006). FLASH is required for histone transcription and S-phase progression. Proceedings of the National Academy of Sciences of the United States of America, 103(40), 14808–14812. 10.1073/pnas.0604227103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi JX, Shuttleworth J, & Al-Rubeai M (2004). Uncoupling of cell growth and proliferation results in enhancement of productivity in p21CIP1-arrested CHO cells. Biotechnology and Bioengineering, 85(7), 741–749. 10.1002/bit.20025 [DOI] [PubMed] [Google Scholar]
- Bongiorno-Borbone L, De Cola A, Barcaroli D, Knight RA, Di Ilio C, Melino G, & De Laurenzi V (2010). FLASH degradation in response to UV-C results in histone locus bodies disruption and cell-cycle arrest. Oncogene, 29(6), 802–810. 10.1038/onc.2009.388 [DOI] [PubMed] [Google Scholar]
- Bunton-Stasyshyn RKA, Wells S, & Teboul L (2019). When all is not lost: Considering genetic compensation in laboratory animals. Lab Animal, 48(10), 282–284. 10.1038/s41684-019-0397-4 [DOI] [PubMed] [Google Scholar]
- Carvalhal AV, Marcelino I, & Carrondo MJ (2003). Metabolic changes during cell growth inhibition by p27 overexpression. Applied Microbiology and Biotechnology, 63(2), 164–173. 10.1007/s00253-003-1385-5 [DOI] [PubMed] [Google Scholar]
- Chen G, & Deng X (2018). Cell synchronization by double thymidine block. Bio Protocol, 8(17), e2994. 10.21769/BioProtoc.2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cost GJ, Freyvert Y, Vafiadis A, Santiago Y, Miller JC, Rebar E, & Gregory PD (2010). BAK and BAX deletion using zinc-finger nucleases yields apoptosis-resistant CHO cells. Biotechnology and Bioengineering, 105(2), 330–340. 10.1002/bit.22541 [DOI] [PubMed] [Google Scholar]
- De Cola A, Bongiorno-Borbone L, Bianchi E, Barcaroli D, Carletti E, Knight RA, & De Laurenzi V (2012). FLASH is essential during early embryogenesis and cooperates with p73 to regulate histone gene transcription. Oncogene, 31(5), 573–582. 10.1038/onc.2011.274 [DOI] [PubMed] [Google Scholar]
- De Jesus M, & Wurm FM (2011). Manufacturing recombinant proteins in kg-ton quantities using animal cells in bioreactors. European Journal of Pharmaceutics and Biopharmaceutics, 78(2), 184–188. 10.1016/j.ejpb.2011.01.005 [DOI] [PubMed] [Google Scholar]
- Dez C, & Tollervey D (2004). Ribosome synthesis meets the cell cycle. Current Opinion in Microbiology, 7(6), 631–637. 10.1016/j.mib.2004.10.007 [DOI] [PubMed] [Google Scholar]
- Druz A, Chen YC, Guha R, Betenbaugh M, Martin SE, & Shiloach J (2013). Large-scale screening identifies a novel microRNA, miR-15a-3p, which induces apoptosis in human cancer cell lines. RNA Biology, 10(2), 287–300. 10.4161/rna.23339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Treiber D, McCarter JD, Fomina-Yadlin D, Saleem RA, McCoy RE, Reddy P (2015). Use of a small molecule cell cycle inhibitor to control cell growth and improve specific productivity and product quality of recombinant proteins in CHO cell cultures. Biotechnology and Bioengineering, 112(1), 141–155. 10.1002/bit.25332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekholm SV, & Reed SI (2000). Regulation of G1 cyclin-dependent kinases in the mammalian cell cycle. Current Opinion in Cell Biology, 12(6), 676–684. 10.1016/S0955-0674(00)00151-4 [DOI] [PubMed] [Google Scholar]
- Fischer S, Handrick R, & Otte K (2015). The art of CHO cell engineering: A comprehensive retrospect and future perspectives. Biotechnology Advances, 33(8), 1878–1896. 10.1016/j.biotechadv.2015.10.015 [DOI] [PubMed] [Google Scholar]
- Fox SR, Patel UA, Yap MG, & Wang DI (2004). Maximizing interferon-gamma production by Chinese hamster ovary cells through temperature shift optimization: Experimental and modeling. Biotechnology and Bioengineering, 85(2), 177–184. 10.1002/bit.10861 [DOI] [PubMed] [Google Scholar]
- Fussenegger M, Mazur X, & Bailey JE (1997). A novel cytostatic process enhances the productivity of Chinese hamster ovary cells. Biotechnology and Bioengineering, 55(6), 927–939. [DOI] [PubMed] [Google Scholar]
- Hendrick V, Winnepenninckx P, Abdelkafi C, Vandeputte O, Cherlet M, Marique T, & Werenne J (2001). Increased productivity of recombinant tissular plasminogen activator (t-PA) by butyrate and shift of temperature: A cell cycle phases analysis. Cytotechnology, 36(1–3), 71–83. 10.1023/A:1014088919546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummon AB, Pitt JJ, Camps J, Emons G, Skube SB, Huppi K, & Caplen NJ (2012). Systems-wide RNAi analysis of CASP8AP2/FLASH shows transcriptional deregulation of the replication-dependent histone genes and extensive effects on the transcriptome of colorectal cancer cells. Molecular Cancer, 11, 1. 10.1186/1476-4598-11-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt L, Batard P, Jordan M, & Wurm FM (2002). Fluorescent proteins in animal cells for process development: Optimization of sodium butyrate treatment as an example. Biotechnology and Bioengineering, 77(5), 528–537. 10.1002/bit.10108 [DOI] [PubMed] [Google Scholar]
- Hwa Chang K, Hwa Park J, Hyung Lee Y, Ho Kim J, Ok Chun H, Hak Kim J, & Sik Chung I (2002). Dimethylsulfoxide and sodium butyrate enhance the production of recombinant cyclooxygenase 2 in stably transformed Drosophila melanogaster S2 cells. Biotechnology Letters, 24(16), 1353–1359. 10.1023/A:1019841829667 [DOI] [Google Scholar]
- Ibarra N, Watanabe S, Bi JX, Shuttleworth J, & Al-Rubeai M (2003). Modulation of cell cycle for enhancement of antibody productivity in perfusion culture of NS0 cells. Biotechnology Progress, 19(1), 224–228. 10.1021/bp025589f [DOI] [PubMed] [Google Scholar]
- Imai Y, Kimura T, Murakami A, Yajima N, Sakamaki K, & Yonehara S (1999). The CED-4-homologous protein FLASH is involved in Fas-mediated activation of caspase-8 during apoptosis. Nature, 398(6730), 777–785. 10.1038/19709 [DOI] [PubMed] [Google Scholar]
- Inoue Y, Tsukamoto Y, Yamanaka M, Nakamura S, Inoue A, Nishino N, & Kawahara H (2010). Efficient production of recombinant IgG by metabolic control and co-expression with GLUT5 in a fructose-based medium. Cytotechnology, 62(4), 301–306. 10.1007/s10616-010-9289-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inwood S, Betenbaugh MJ, Lal M, & Shiloach J (2018). Genome-wide high-throughput RNAi screening for identification of genes involved in protein production. Methods in Molecular Biology, 1850, 209–219. 10.1007/978-1-4939-8730-6_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inwood S, Buehler E, Betenbaugh M, Lal M, & Shiloach J (2018). Identifying HIPK1 as target of miR-22–3p enhancing recombinant protein production from HEK 293 cell by using microarray and HTP siRNA screen. Biotechnology Journal, 13(2), 1700342. 10.1002/biot.201700342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun J-I, Chung C-W, Lee H-J, Pyo J-O, Lee KN, Kim N-S, & Jung Y-K (2005). Role of FLASH in caspase-8-mediated activation of NF-κB: Dominant-negative function of FLASH mutant in NF-κB signaling pathway. Oncogene, 24(4), 688–696. 10.1038/sj.onc.1208186 [DOI] [PubMed] [Google Scholar]
- Kiriyama M, Kobayashi Y, Saito M, Ishikawa F, & Yonehara S (2009). Interaction of FLASH with arsenite resistance protein 2 is involved in cell cycle progression at S phase. Molecular and Cellular Biology, 29(17), 4729–4741. 10.1128/mcb.00289-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klanert G, Fernandez DJ, Weinguny M, Eisenhut P, Buhler E, Melcher M, & Borth N (2019). A cross-species whole genome siRNA screen in suspension-cultured Chinese hamster ovary cells identifies novel engineering targets. Scientific Reports, 9(1), 8689. 10.1038/s41598-019-45159-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C-H, Chu I-M, & Hwang S-M (2001). Enhanced expression of various exogenous genes in recombinant Chinese hamster ovary cells in presence of dimethyl sulfoxide. Biotechnology Letters, 23(20), 1641–1645. 10.1023/a:1012466112116 [DOI] [Google Scholar]
- Mazur X, Fussenegger M, Renner WA, & Bailey JE (1998). Higher productivity of growth-arrested Chinese hamster ovary cells expressing the cyclin-dependent kinase inhibitor p27. Biotechnology Progress, 14(5), 705–713. 10.1021/bp980062h [DOI] [PubMed] [Google Scholar]
- Medema RH, Herrera RE, Lam F, & Weinberg RA (1995). Growth suppression by p16ink4 requires functional retinoblastoma protein. Proceedings of the National Academy of Sciences of the United States of America, 92(14), 6289–6293. 10.1073/pnas.92.14.6289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meents H, Enenkel B, Werner RG, & Fussenegger M (2002). p27Kip1-mediated controlled proliferation technology increases constitutive sICAM production in CHO-DUKX adapted for growth in suspension and serum-free media. Biotechnology and Bioengineering, 79(6), 619–627. 10.1002/bit.10322 [DOI] [PubMed] [Google Scholar]
- Minamida Y, Someda M, & Yonehara S (2014). FLASH/casp8ap2 is indispensable for early embryogenesis but dispensable for proliferation and differentiation of ES cells. PLoS One, 9(9), e108032. 10.1371/journal.pone.0108032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam JH, Zhang F, Ermonval M, Linhardt RJ, & Sharfstein ST (2008). The effects of culture conditions on the glycosylation of secreted human placental alkaline phosphatase produced in Chinese hamster ovary cells. Biotechnology and Bioengineering, 100(6), 1178–1192. 10.1002/bit.21853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes C, Prats E, Cairo JJ, Azorin F, Cornudella L, & Godia F (1999). Modification of glucose and glutamine metabolism in hybridoma cells through metabolic engineering. Cytotechnology, 30(1–3), 85–93. 10.1023/A:1008012518961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunley K, & Butler M (2010). Strategies for the enhancement of recombinant protein production from mammalian cells by growth arrest. Biotechnology Advances, 28(3), 385–394. 10.1016/j.biotechadv.2010.02.003 [DOI] [PubMed] [Google Scholar]
- Xiao S, Chen YC, Betenbaugh MJ, Martin SE, & Shiloach J (2015). MiRNA mimic screen for improved expression of functional neurotensin receptor from HEK293 cells. Biotechnology and Bioengineering, 112(8), 1632–1643. 10.1002/bit.25567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Chen YC, Buehler E, Mandal S, Mandal A, Betenbaugh M, & Shiloach J (2016). Genome-scale RNA interference screen identifies antizyme 1 (OAZ1) as a target for improvement of recombinant protein production in mammalian cells. Biotechnology and Bioengineering, 113(11), 2403–2415. 10.1002/bit.26017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Burch BD, Yan Y, Marzluff WF, & Dominski Z (2009). FLASH, a proapoptotic protein involved in activation of caspase-8, is essential for 3′ end processing of histone pre-mRNAs. Molecular Cell, 36(2), 267–278. 10.1016/j.molcel.2009.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.