

Abstract
Multiple sclerosis is associated with Epstein–Barr virus (EBV) infection, B‐cell dysfunction, gut dysbiosis, and environmental and genetic risk factors, including female sex. A disease model incorporating all these factors remains elusive. Here, we hypothesise that EBV‐infected memory B cells (MBCs) migrate to gut‐associated lymphoid tissue (GALT) through EBV‐induced expression of LPAM‐1, where they are subsequently activated by gut microbes and/or their products resulting in EBV reactivation and compartmentalised anti‐EBV immune responses. These responses involve marginal zone (MZ) B cells that activate CD4+ T‐cell responses, via HLA‐DRB1, which promote downstream B‐cell differentiation towards CD11c+/T‐bet+ MBCs, as well as conventional MBCs. Intrinsic expression of low‐affinity B‐cell receptors (BCRs) by MZ B cells and CD11c+/T‐bet+ MBCs promotes polyreactive BCR/antibody responses against EBV proteins (e.g. EBNA‐1) that cross‐react with central nervous system (CNS) autoantigens (e.g. GlialCAM). EBV protein/autoantigen‐specific CD11c+/T‐bet+ MBCs migrate to the meningeal immune system and CNS, facilitated by their expression of CXCR3, and induce cytotoxic CD8+ T‐cell responses against CNS autoantigens amplified by BAFF, released from EBV‐infected MBCs. An increased abundance of circulating IgA+ MBCs, observed in MS patients, might also reflect GALT‐derived immune responses, including disease‐enhancing IgA antibody responses against EBV and gut microbiota‐specific regulatory IgA+ plasma cells. Female sex increases MZ B‐cell and CD11c+/T‐bet+ MBC activity while environmental risk factors affect gut dysbiosis. Thus, EBV infection, B‐cell dysfunction and other risk factors converge in GALT to generate aberrant B‐cell responses that drive pathogenic T‐cell responses in the CNS.
Keywords: CD11c+/T‐bet+ memory B cells, Epstein–Barr virus, gut‐associated lymphoid tissue, marginal zone B cells, multiple sclerosis
A novel and comprehensive hypothesis for the pathogenesis of multiple sclerosis that incorporates immunological mechanisms and genetic and environmental risk factors is proposed. Activation of Epstein–Barr virus (EBV)‐infected memory B cells (MBCs) residing in gut‐associated lymphoid tissue results in lytic EBV infection that induces compartmentalised immune responses by marginal zone B cells and CD4+ T cells and production of CD11c+/T‐bet+ memory B cells, influenced by genetic and environmental risk factors. EBV‐specific B‐cell receptors and antibodies produced by these B‐cell subpopulations are cross‐reactive with CNS autoantigens and the CD11c+/T‐bet+ MBCs drive CD8+ T‐cell responses against CNS autoantigens.
Introduction
Multiple sclerosis (MS) is an immune‐mediated inflammatory disease of the central nervous system (CNS) that results in demyelination of neurons and neurological disability. Multiple risk factors for developing MS have been identified, 1 , 2 including female sex; genetic variations impacting the function of the immune system, 3 particularly alleles of genes encoding human leucocyte antigens (HLAs) of which HLA‐DRB1*1501 is the strongest genetic risk factor 4 ; chronic infection with Epstein–Barr virus (EBV) 5 ; variation of the gut microbiome; low ultraviolet radiation (UVR) exposure; low vitamin D levels; and smoking cigarettes. While past research on the immunopathology of MS has focussed on CD4+ T cells, there is growing awareness that CD8+ T cells and B cells, and the effect of EBV infection on these cells, are central to the immunopathology underlying MS. 6 We have examined various aspects of B‐cell dysfunction 7 , 8 , 9 , 10 and the relationship of gut microbiome‐derived short‐chain fatty acids (SCFAs) with immune dysfunction 11 in patients with early MS and identified abnormalities that led to the hypothesis proposed here; that EBV infection, B‐cell dysfunction and other risk factors converge in gut‐associated lymphoid tissue (GALT) to generate aberrant B‐cell responses that drive pathogenic T‐cell responses in the CNS (Figure 1).
Figure 1.
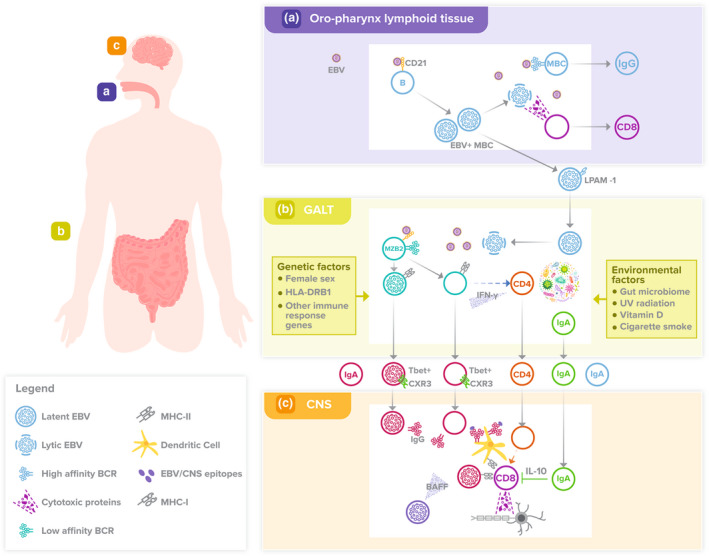
Proposed model of MS disease pathogenesis. (a) During primary EBV infection, a reservoir of EBV‐infected MBCs is established in lymphoid tissue, particularly in the oro‐pharynx. Reactivation of latent EBV infection in MBCs responding to oropharyngeal infections leads to lytic EBV infection, which induces an EBV‐specific CD8+ T‐cell and antibody response by circulating T cells and B cells expressing high affinity BCRs. Active EBV infection in MBCs during primary EBV infection, and possibly during subsequent reactivations of infection, induces LPAM‐1 expression on EBV‐infected MBCs and their migration to GALT. (b) Episodic reactivation of latent EBV infection in MBCs residing in GALT by gut microbes via BCRs and/or pathogen recognition receptors and/or SCFAs leads to lytic EBV infection, which induces a compartmentalised EBV‐specific immune response. This response includes MZB2 cells, some of which may also become infected by EBV. Low‐affinity BCRs on MZB2 cells promote an antibody response against EBV proteins, such as EBNA‐1, that is polyreactive and cross‐reacts with CNS autoantigens, such as GlialCAM. CD4+ T cells are activated by MBZ2 cells via HLA‐DRB1 and ‘autoproliferate’, including in blood, as well as migrate to the CNS. These CD4+ T cells produce IFN‐γ, which facilitates a CD11c+/T‐bet+ MBC differentiation pathway amongst both EBV‐infected and ‐uninfected EBV/autoantigen‐specific MZB2 cells. Gut microbiota‐specific IgA+ B cells and/or plasma cells are also produced in GALT and migrate to the CNS. In addition, IgA+ cMBCs and T‐bet+ MBCs accumulate in blood, possibly as a result of repeated EBV reactivation in GALT, and may generate EBV‐specific IgA antibody responses that interfere with EBV‐specific IgG antibody responses. The major genetic and environmental factors associated with an increased risk of developing MS affect these processes in GALT. (c) Both EBV‐infected and ‐uninfected EBV protein/CNS autoantigen‐specific CD11c+/T‐bet+ IgG+ MBCs migrate to the meningeal immune system within the dura, and subsequently to the CNS, facilitated by cell‐surface expression of CXCR3. At sites of inflammation in the CNS, these B cells mediate a B‐cell/antibody response against EBV proteins (e.g. EBNA‐1) and CNS autoantigens (e.g. GlialCAM). This drives an autoantigen‐specific CD8+ T‐cell response by CD11c+/T‐bet+ IgG+ MBCs acting as APCs themselves and/or by generating production of IgG antibodies that form immune complexes with autoantigens and deliver them to cDCs, which cross‐present antigens to, and activate, CD8+ T cells. Autoantigen‐specific CD8+ T cells mediate a cytotoxic cellular immune response against antigens of myelin and/or glial cells. These activities are augmented by BAFF produced by EBV‐infected CD11c+/T‐bet+ MBCs or cMBCs or both. Gut microbe‐specific IgA+ plasma cells may modulate CD8+ T‐cell activation via IL‐10.
Recently, there have been several important developments in understanding the relationship between EBV infection and the immunopathogenesis of MS. 12 , 13 , 14 However, the hypothesis presented here, coupled with published evidence from this and other laboratories, provides a deeper and clearer insight into the mechanisms by which EBV infection may drive MS development.
EBV‐induced dysfunction of memory B cells is central to the immunopathogenesis of MS
The peri‐vascular and parenchymal inflammatory infiltrates associated with demyelination in brains of MS patients are characterised by an abundance of CD8+ T cells, which predominantly have characteristics of resident memory T (Trm) cells, and B cells. 6 , 15 , 16 While uncertainty remains about interactions between CD8+ T cells and B cells, 6 B cells clearly play a fundamental role in the inflammatory process. This became apparent over 40 years ago with the description of oligoclonal immunoglobulins in the CSF of MS patients. More recently, the contribution of B cells has been demonstrated by the pronounced therapeutic effect of B‐cell depletion therapies. 17 Most recently, it has been reported that CSF oligoclonal immunoglobulins are enriched for antibodies reactive with EBV proteins, particularly EBV nuclear antigen‐1 (EBNA‐1), reflecting the expansion of oligoclonal B cells producing an antibody response against EBNA‐1. 12 Because of antigenic cross‐reactivity, some of these antibodies also recognise GlialCAM, a cell adhesion molecule expressed by astrocytes and oligodendrocytes in the CNS, in a subset of patients. Furthermore, plasma levels of IgG antibodies to GlialCAM were significantly higher in three independent groups of MS patients compared with controls. 12 These findings provide a mechanistic explanation for the numerous reports linking MS with EBV infection, 5 culminating in recent reports that serological evidence of EBV infection is present in 100% of MS patients 18 and that EBV infection acquired in late adolescence or early adulthood is associated with a 32‐fold increased risk of developing MS. 13 Furthermore, the particular significance of antibodies to EBNA‐1 in MS immunopathogenesis is illustrated by reports that high serum levels of anti‐EBNA‐1 IgG antibodies are not only associated with an increased risk of MS but synergise with other risk factors to increase that risk and predict conversion from clinically isolated syndrome suggestive of MS (CIS) to MS, whereas antibodies to other EBV proteins do not. 19 , 20 , 21
Epstein–Barr virus is a human gamma‐herpesvirus that is transmitted from one person to another primarily by saliva and infects both epithelial cells and B cells, the latter via CD21 (complement receptor 2). Following primary EBV infection, a reservoir of latently infected B cells is established in lymphoid tissue, particularly in the oro‐pharynx, 22 from where EBV‐infected B cells may migrate to GALT and associated lymph nodes. 23 This migration is facilitated by EBV‐induced expression of the ‘gut homing molecule’ LPAM‐1 (integrin α4/β7) (Figure 1). EBV primarily establishes persistent infection of IgD−CD27+ B cells (conventional memory B cells [cMBCs]). However, EBV DNA is also detectable to a lesser degree in CD27+IgD+ B cells, 24 which form the major component of the IgM+ MBC subpopulation and exhibit characteristics of circulating marginal zone (MZ) B cells. 25 Infection of B cells by EBV leads to the production of various EBV proteins, including the cell‐surface molecules latent membrane protein (LMP) 1 and 2, as well as viral transcription factors, such as EBNA‐1. Together, these molecules modulate B‐cell function to keep the infected MBC alive and evade innate and adaptive immune responses. 26 Such immune evasion strategies first become apparent during primary EBV infection when EBV‐induced B‐cell dysfunction causes a decreased abundance of circulating MBCs and impaired EBV‐specific neutralising antibody responses. 27 This appears to reflect impaired B‐cell proliferation and survival associated with increased expression of Fas (CD95) on B cells and increased plasma levels of Fas ligand, as well as increased plasma levels of B‐cell‐activating factor (BAFF) but decreased levels of a proliferation‐inducing ligand (APRIL), associated with reciprocal changes in B‐cell expression of their receptors.
Approximately 95% of adults are infected by EBV with the majority of infections occurring during childhood. The risk of MS is highest in individuals who experience symptomatic primary EBV infection, usually presenting as infectious mononucleosis (IM), in adolescence or early adulthood. 28 IM is not observed in younger children, 29 , 30 possibly reflecting differences in the immune system associated with young age, such as Th2‐skewedness, more abundant regulatory T cells and a smaller pool of memory T cells, rather than differences in plasma EBV DNA loads, which are comparable in children with primary EBV infection and IM patients. 30 , 31 As tonsillar B cells of IM patients strongly express the β7 integrin component of LPAM‐1, it has been proposed that EBV establishes a reservoir of latently infected B cells in GALT during primary infection to evade EBV‐specific CD8+ T‐cell responses. 23 In chronic EBV infection, EBV‐specific circulating CD8+ memory T (CD8+ Tm) cells and CD8+ Trm cells normally control EBV replication in epithelial cells and MBCs of the oropharynx. 32 By contrast, EBV‐specific CD8+ T‐cell responses may be less effective in GALT because gut CD8+ T cells are predominantly intra‐epithelial and lamina propia CD8+ Trm cells. 31 , 33 In addition, MS patients exhibit progressively less effective circulating CD8+ Tm cell responses against EBV‐infected B cells, related to a decreased abundance and exhaustion of EBV‐specific CD8+ Tm cells. This is associated with skewing of the EBV‐specific immune response towards latent phase antigens and increased production of IgG antibodies to EBNA‐1. 34
Disease‐associated MBCs in MS patients exhibit heterogeneous immunophenotypic and functional characteristics
A large proportion of B cells infiltrating the brain of MS patients are CD27+ MBCs and express the EBV proteins LMP‐1 and ‐2 on their surface. 35 These cells also produce BAFF, likely as a result of the transcriptional regulation by LMP‐1. 36 Analyses of CSF cells from MS patients have provided some insight into the derivation of these B cells. An unsupervised analysis of cells detected by mass cytometry demonstrated a CD27+ B‐cell population that was clearly associated with MS, although represented < 1.5% of total cells. 37 Notably, these B cells expressed the chemokine receptor CXCR3, which facilitates cell trafficking to tissues and is only expressed by about one‐third of circulating MBCs. 38 In a separate study, analysis of single‐cell transcriptomes and immunoglobulin sequences in defined B‐cell subpopulations demonstrated that clonally expanded, somatically hypermutated IgG1+ and IgM+ B cells were associated with inflammation, blood–brain barrier breakdown and intrathecal immunoglobulin synthesis. 39 Furthermore, when compared with blood, transcripts upregulated in CSF switched MBCs (IgD−CD27+) and plasmablasts included those for the Th‐1‐associated transcription factor T‐bet, as well as CXCR3, providing further evidence that disease‐associated MBCs might differ from the majority of circulating MBCs. In addition, somatically hypermutated (antigen experienced) IgM+ B cells had a phenotype resembling MZ B cells. Together, these observations provided evidence that MBCs associated with CNS immunopathology in MS might, wholly or in part, differ from cMBCs.
One of the first abnormalities of circulating B cells recognised in MS patients was expansion of age‐associated B cells (ABCs). 40 While ABCs were initially considered to be activated and senescent MBCs, it is now clear that they represent a distinct subpopulation of MBCs, commonly referred to as ‘atypical MBCs’, which exhibit phenotypic and functional characteristics that distinguish them from cMBCs. These characteristics include activation via TLR7 and 9 as well as B‐cell receptors (BCRs) and differentiation under the influence of T cells and Th1‐associated cytokines, such as IFN‐γ. 41 , 42 , 43 In addition, they express a distinctive pattern of molecules that determine their functional characteristics, particularly T‐bet and the integrin molecule CD11c (integrin alpha X). 41 , 44 , 45 Furthermore, transcriptome analysis of ‘atypical MBCs’ has demonstrated high expression of CD18 (integrin β2). 42 The CD11c/CD18 complex is a major adhesion molecule on antigen presenting cells (APCs), particularly conventional dendritic cells (cDCs). 46 ‘Atypical MBCs’ express high levels of CXCR3 in health and disease, 47 reflecting the regulation of CXCR3 expression by T‐bet, 48 and low levels of CD21, while CD27 expression is variable. 41 Thus, whereas CD21−/T‐bet+ MBCs usually exhibit a double negative (DN; CD27−/IgD−) phenotype, differentiation towards plasma cells is associated with high CD27 expression. 49 Several other names are used to describe B‐cell subpopulations that are identical to, or substantially overlap with, ‘atypical MBCs’ 50 , 51 , 52 , 53 , 54 (Table 1), including CD11c+/T‐bet+ MBCs which, in our opinion, best describes these cells with regard to important functional characteristics and is how we will refer to them moving forward.
Table 1.
Various names used to describe CD11c+/T‐bet+ memory B cells
| Name | Context in which described |
|---|---|
| Age‐associated B cells | Originally described in aged mice and subsequently shown to be increased in autoimmune disease in mice and humans 45 |
| Tissue‐like MBCs | HIV‐1 infection 50 |
| Atypical MBCs | Chronic infectious diseases, including HIV‐1 infection, malaria and tuberculosis 51 |
| Double negative type 2 (DN2) B cells | Systemic lupus erythematosus (SLE) 52 |
| CD21low B cells | Common variable immunodeficiency disorder 53 |
| CD11c+, T‐bet+ or CD11c+/T‐bet+ MBCs | Autoimmune diseases, including SLE, rheumatoid arthritis and systemic sclerosis, as well as infectious diseases, including HIV‐1 infection and malaria 54 |
Production of CD11c+/T‐bet+ MBCs is part of the normal B‐cell response to infection by various pathogens, most comprehensively described for viruses and Plasmodium sp., and to viral vaccines. 41 , 55 , 56 Furthermore, data from studies in mice suggest that CD11c+/T‐bet+ MBCs contribute to the control, as opposed to prevention, of virus infections, including infection by EBV‐like gamma‐herpesvirus 68. 57 , 58 In view of the T‐cell dependence of CD11c+/T‐bet+ MBC differentiation, 43 the functional effects of these cells likely occur in concert with those of effector T cells, particularly CD8+ T cells. There is also accumulating evidence indicating that CD11c+/T‐bet+ MBCs contribute to autoantibody responses and that activation of these cells via TLR7 (the gene of which is on the X chromosome) may in part explain the female predominance of some autoimmune diseases. 52 , 59 Indeed, ABCs were first observed in aged female mice predisposed to autoimmune disease. 59 Furthermore, B‐cell‐specific depletion of T‐bet in a mouse model of SLE not only resulted in decreased disease manifestations but also resulted in reduced levels of germinal centre B cells and plasmablasts, suggesting that T‐bet+ B cells may be precursors of autoantibody‐producing plasmablasts. B cell‐specific T‐bet depletion was also associated with decreased T‐cell activation. 60
Although circulating CD11c+/T‐bet+ MBCs are increased in some MS patients, 40 their contribution to MS immunopathology has been unclear. Recently, van Langelaar et al. 61 demonstrated that patients with advanced MS exhibited an accumulation of CXCR3+ B cells in the CSF, meninges and brain and that circulating CXCR3+ B cells expressed T‐bet, the level of which correlated with the level of CXCR3 expression. These CXCR3(T‐bet)+ B cells also expressed IgG. CXCR3(T‐bet)+ IgG+ B cells were less abundant in blood than the CNS but their abundance increased after natalizumab therapy, which blocks α4 integrin and prevents migration of cells expressing this molecule, providing evidence that CXCR3(T‐bet)+ IgG+ B cells migrate from blood to the CNS of MS patients. The high transmigration capacity of CXCR3(T‐bet)+ IgG+ B cells was confirmed by ex vivo studies. 61 In addition, ex‐vivo cell culture studies demonstrated that T‐cell‐derived IFN‐γ increased the expression of T‐bet in B cells of MS patients to a greater degree than in controls and that this correlated strongly with CXCR3 expression. 61 Together, these findings provided compelling evidence that CD11c+/T‐bet+ MBCs are more abundant than normal in the CNS of MS patients, to where they have migrated from blood facilitated by their expression of CXCR3.
Preliminary evidence suggests that CXCR3(T‐bet)+ IgG+ B cells are induced by reactivation of EBV infection, as EBV DNA load correlated with the frequency of circulating CXCR3+ cMBCs in MS patients who had received an autologous haematopoietic stem cell transplant, which increases the risk of EBV reactivation. 62 Supportive evidence is provided by a study of the single‐cell transcriptome of primary B cells infected with EBV ex vivo, which demonstrated that EBV infection simulates antigen‐induced activation and differentiation of B cells, including production of ‘atypical MBCs’ expressing CXCR3, FcRL4 and T‐bet. 63 In mice, CD11c+ B cells exhibit potent APC activity for T cells, associated with higher expression of HLA‐DR and the costimulatory molecules CD80 and CD86 as well as more stable interactions with T cells, compared with follicular B cells. 64 Therefore, CD11c+/T‐bet+ MBCs may be involved in MS pathogenesis through increased numbers related to EBV replication, increased migratory capacity to the CNS via CXCR3 expression and by acting as APCs for pathogenic T cells (Figure 1).
While most B cells in the meninges, peri‐vascular spaces and brain parenchyma adjacent to areas of demyelination are IgG+, IgA+ cells are also present and predominantly have the characteristics of plasma cells. 65 These cells presumably produce the IgA that increases in CSF during relapses of MS. 65 Probstel et al. 65 also reported that a subset of these cells are gut microbiota‐specific IgA+ plasma cells that express IL‐10 and might play an immunoregulatory role in MS.
Reactivation of EBV in GALT may induce pathogenic MBC responses
As evidence mounts that CD11c+/T‐bet+ MBCs may be important contributors to MS immunopathology in the CNS, evidence is also emerging that B‐cell dysfunction induced by EBV infection may arise in GALT. Within GALT and associated mesenteric lymph nodes, 23 EBV‐infected MBCs are exposed to microbial SCFAs, which have been shown to exert stimulatory and inhibitory effects on EBV replication, 66 as well as to antigenic stimulation by gut microbes via BCR‐dependent and ‐independent mechanisms. 67 We propose that MBC activation and corresponding reactivation of EBV infection induces compartmentalised immune responses against EBV. The GALT consists of multifollicular lymphoid tissues, such as Peyer's patches of the small intestine, isolated lymphoid follicles of the small and large intestines, the appendix, caecal and colonic patches and rectal lymphoid tissues. 67 These lymphoid tissues contain diverse immune cells involved in the initation and propagation of immune responses, including CD4+ T cells, IgM+ MBCs such as MZ B cells, as well as both IgA+ and IgG+ MBCs. IgA+ MBCs are particularly prominent because of the importance of IgA antibodies in regulating microbial growth in the intestines. Furthermore, IgA+ B cells and/or plasma cells derived from GALT are an important component of the gut‐meningeal immune axis that protects the CNS from gut‐derived infections. 68 , 69 MZ B cells, which differentiate from IgMhi T2 transitional B cells, circulate between the spleen MZ and GALT, where they occupy a micro‐anatomical niche independently of cMBCs. 67 , 70 Two subpopulations of MZ B cells have recently been defined in GALT and spleen, referred to as MZB1 and MZB2 cells. 71 MZB2 cells exhibit characteristics that suggest they contribute to antiviral responses and, importantly, express HLA‐DRB1 in contrast to MZB1 cells. 71 DN MBCs have also been demonstrated in GALT but it is unclear whether they are CD11c+/T‐bet+ MBCs. 71 Studies undertaken in mice and humans have demonstrated that CD11c+/T‐bet+ MBCs migrate to MZ regions of the spleen after interacting with T follicular helper (Tfh) cells in lymphoid follicles 44 , 72 and it is possible that, like MZ B cells, they also migrate to GALT. In support of this proposal is the finding that rotavirus infection of the gastro‐intestinal tract induces circulating antigen‐specific MBCs that are skewed towards CD27− IgG+ MBCs, as well as MZ B cells. 73 , 74
To gain further insight into circulating MBC dysfunction in patients with early MS, we investigated the characteristics of MBCs expressing BCRs with different immunoglobulin isotypes. 10 We demonstrated an increased abundance of IgA+ MBCs, which has been confirmed in an independent study of patients with more advanced MS. 75 The IgA+ MBCs in our study consisted of one major and two smaller subpopulations that were all more abundant compared with controls. One of the smaller subpopulations consisted of CD27− IgA+ MBCs, which are enriched amongst gut bacteria‐specific B cells, 76 and may be precursors of the gut microbiota‐specific IgA+ plasma cells proposed to exert an immunoregulatory role in MS patients. 65 The major subpopulation had characteristics of cMBCs (CD27+/CD24hi) while the other smaller subpopulation consisted of T‐bet+/CD21low IgA+ MBCs. The increased abundance of these subpopulations also suggested an accumulation of MBCs from mucosal B‐cell responses, possibly from repeated episodes of EBV reactivation. In support of this proposal are reports that patients with naso‐pharyngeal carcinoma (NPC), a classical EBV‐induced cancer of mucosal tissue, exhibit an increased frequency of IgA+ B cells in the tumor microenvironment and blood, 77 , 78 although a direct relationship remains to be established. Furthermore, a strong relationship between high serum levels of IgA antibodies to EBV proteins and both the development of, and a poor outcome from, NPC has been reported. 79 , 80 Preliminary evidence suggests that IgA antibodies to EBV proteins may have a pathogenic effect in EBV‐associated diseases, including interference with antibody‐dependent cellular cytotoxicity (ADCC) of EBV‐infected cells mediated by IgG antibodies. 79 , 81 Similar mechanisms have been reported to explain the adverse effect of IgA anti‐HIV‐1 gp120 on ADCC responses in people with HIV‐1 infection. 82 The circulating IgA+ MBCs that are more abundant in MS patients might therefore be part of separate immune responses against gut microbiota or EBV that have opposite effects on disease pathogenesis (Figure 1). It is possible that the increased abundance of circulating IgA+ MBCs in EBV‐associated diseases is an effect of EBV infection itself, as expression of EBV LMP‐1 on B cells induces class switch recombination (CSR) of immunoglobulin heavy chain (IGHC) gene DNA, potentially through increased expression of BAFF and APRIL. 36
In addition to the increased abundance of circulating IgA+ MBC, further evidence that B‐cell dysfunction in patients with early MS arises in GALT is provided by our observations on abnormalities of circulating MZ B cells. First, female patients exhibited decreased expression of the inhibitory Fcγ receptor (FcγR) FcγRIIb on circulating MZ B cells, as well as on naïve B cells, which was associated with serological evidence of EBV reactivation. 9 Second, EBV reactivation was associated with an increased abundance of circulating T‐bet+/CD21low IgM+ MBCs and CXCR3+ IgM+ MBCs as well as increased expression of HLA‐DR on IgM+ MBC in all patients. 10 Based on these findings and those of multiple studies in mice and humans implicating MZ B cells in producing autoantibody responses, 83 we propose that MZ B‐cell activation and dysfunction associated with EBV reactivation in GALT may be determinants of pathogenic B‐cell responses in MS (Figure 1). MZ B cells express BCRs with low affinity for antigens, 84 which would confer greater polyreactivity with antigens. 85 While this may be advantageous in ‘frontline’ B‐cell responses against pathogens, including gut microbes, it increases the likelihood of BCR and antibody cross‐reactivity with autoantigens and induction of autoantibody responses. 86 Indeed, monoclonal IgG antibodies to HIV‐1 gp140 derived from intestinal B cells of people with HIV infection exhibit low affinity, high polyreactivity and cross‐reactivity with autoantigens. 87 Impaired regulation of MZ B‐cell activation via decreased FcγRIIB expression in female patients with MS would compound these abnormalities by impairing peripheral tolerance mechanisms. 88 In addition, murine T‐bet+ IgM+ MBCs, which also express CXCR3, generate multilineage effector B cells 89 and maintain long‐term antibody responses. 90 It is therefore possible that the circulating T‐bet+/CD21low and CXCR3+ IgM+ MBCs we observed to be associated with EBV reactivation are precursors of the CXCR3(T‐bet)+ IgG+ B cells implicated in the immunopathogenesis of brain lesions in advanced MS 61 and of the circulating T‐bet+/CD21low IgA+ MBCs that we reported are increased in patients with early MS. 10 However, further studies are required to experimentally establish such a link. Infection of CD11c+/T‐bet+ MBCs by EBV is likely to occur at the MZ B‐cell stage because these cells express large amounts of CD21. 83 Notably, CpG DNA ligation of TLR9 induces terminal differentiation of MZ B cells into autoantibody‐producing cells, 91 which may contribute to EBV‐induced MZ B‐cell dysfunction. Therefore, we propose that MZ B cells expressing polyreactive BCRs are primed by the microbiome and/or lytic EBV infection in the GALT and differentiate into CD11c+/T‐bet+ MBCs.
EBV‐induced activation and dysfunction of B cells in GALT may induce B‐cell responses that facilitate pathogenic CD8 + T‐cell responses in the CNS
While debate continues about the role of CD8+ T cells in CNS lesions of MS patients, 92 compelling arguments have been made that they mediate glial cell damage and/or demyelination, although uncertainty remains concerning their antigen reactivity. 6 Whether the CD8+ T‐cell response is primarily against neurons, other CNS cell‐types, or against EBV‐infected cells, with bystander damage to neurons, remains uncertain. Indeed, there has been a robust debate as to whether or not EBV proteins or genomes are detectable in brain lesions of MS patients. 93 In our view, there is convincing evidence of EBV in inflammatory cells, including B cells, 94 , 95 as well as evidence that CD8+ T cells reactive with multiple lytic and latent phase proteins of EBV and exhibiting a cytotoxic phenotype are co‐located with B cells in CNS lesions of patients with advanced MS. 94 However, it could not be determined whether these CD8+ T cells were EBNA‐1‐specific because only the most immunodominant EBNAs (3A and 3C) were included in the pentamer probes used. Nevertheless, the demonstration of cross‐reactive B‐cell responses against EBNA‐1 and GlialCAM in patients with MS 12 provides support for a hypothesis that EBV‐infected MBCs, including CD11c+/T‐bet+ MBCs, generate B‐cell responses against both EBV proteins and glial cell and/or myelin proteins that could drive pathogenic CD8+ T‐cell responses. Thus, EBV‐infected B cells are capable of cross‐presenting antigens to CD8+ T cells, facilitated by increased expression of class I and II MHC molecules and the costimulatory molecules CD40, CD80 and CD86, as well as activation of the cross‐presentation machinery. 96 This process is likely more robust in CD11c+/T‐bet+ MBCs because they express molecules typically found on APCs (CD11c/CD18) and exhibit potent APC activity in mice. 64 In addition, IgG antibodies to antigens, such as EBNA‐1/GlialCAM, might enhance CD8+ T‐cell responses against these molecules, and possibly other CNS autoantigens through epitope spreading, 97 via cDCs. As exemplified by HIV‐1 98 , 99 and murine cytomegalovirus 100 infections, antibodies can act synergistically with CD8+ T cells in immune responses against viruses, which is likely to occur via binding of antigen–antibody complexes to FcγRs on cDCs and cross‐presentation of antigens to CD8+ T cells. 101 , 102 The latter mechanism would likely be augmented by an IgG antibody response generated from CD11c+/T‐bet+ MBCs as they are skewed towards IgG3, as well as IgG1, production. 103 , 104 , 105 IgG3 binds more avidly to cell‐surface FcγRs than other IgG subclasses 106 and also elicits an antiviral response by binding to the intra‐cellular Fc receptor TRIM21, 107 which when bound by immune complexes in DCs, promotes antigen cross‐presentation and stimulation of CD8+ T cells. 108 Furthermore, in murine models of SLE, CD11c+/T‐bet+ MBCs cause aberrant differentiation of Tfh cells resulting in impaired affinity maturation of antibody responses. 109 Our observation that serum levels of IgG2, which is encoded by the third IGHC gene block (IgG2, IgG4 and IgA) and a product of high levels of CSR in MBCs, 105 are decreased in patients with early MS 7 raises the possibility that expansion of CD11c+/T‐bet+ MBCs may also impair Tfh cell regulation of B‐cell CSR in patients with MS. Together, these effects of CD11c+/T‐bet+ MBCs might explain our observations that higher serum IgG3 levels predict the rate of progression of early MS 7 and that the abundance of IgG3+ MBC is increased in some MS patients. 110
Another mechanism by which MBCs might activate T cells was proposed by Jelcic et al., 111 who demonstrated that circulating MBCs from patients with MS drive the proliferation of autologous CD4+ T cells (referred to as autoproliferation). This mechanism was HLA‐DR‐dependent and most notable in patients carrying HLA‐DR15. ‘Autoproliferative’ CD4+ T cells exhibited a Th1 phenotype, including production of IFN‐γ, and it was proposed that they contribute to autoimmune responses in the CNS. Notably, the degree of CD4+ T‐cell ‘autoproliferation’ positively correlated with the frequency of circulating unswitched MBCs only, implying that CD4+ T‐cell proliferation was driven by IgM+ MBCs, which are predominantly MZ B cells. Furthermore, the same research group demonstrated that CD4+ T‐cell responses against self‐peptides, presented by HLA‐DR15 on B cells, may be cross‐reactive with EBV peptides. 112 Given that MZ B cells can potently stimulate CD4+ T cells, 113 our observations that reactivation of EBV infection in patients with early MS is associated with increased expression of HLA‐DR on IgM+ MBC 10 and decreased expression of FcγRIIb on circulating MZ B cells in females 9 raises the possibility that EBV‐induced activation and/or dysregulation of MZ B cells may contribute to CD4+ T‐cell ‘autoproliferation’. Furthermore, IFN‐γ produced by ‘autoproliferative’ CD4+ T cells might promote a pathway of CD11c+/T‐bet+ MBC differentiation from MZ B cells 43 (Figure 1). It should be noted that ‘autoproliferative’ CD4+ T cells were most frequent during disease remission but this likely reflects the ‘brain‐homing’ characteristics of these cells during disease relapse. 111
EBV‐induced dysregulation of the BAFF/BAFF‐R pathway may contribute to pathogenic B‐cell responses that drive CD8 + T‐cell responses
Primary EBV infection is associated with increased production of BAFF and decreased expression of BAFF‐R on B cells, 27 likely reflecting the effect of LMP‐1 on BAFF production by B cells 36 and BAFF‐R shedding resulting from BAFF ligation. 114 Our analysis of circulating MBCs in patients with early MS demonstrated decreased BAFF‐R expression on multiple B‐cell subpopulations and was related to serum BAFF levels. 10 EBV‐induced dysregulation of the BAFF/BAFF‐R pathway may therefore persist in early MS, although we did not observe a relationship between BAFF‐R expression and EBV reactivation. 10 The role that the BAFF/BAFF‐R pathway plays in the immunopathogenesis MS is unclear, as highlighted by the failure of BAFF inhibitor therapy to prevent relapses of MS in clinical trials. 115 However, there is robust evidence that CSF BAFF levels and CSF/serum BAFF indices are decreased in MS 37 , 116 and that CSF/serum BAFF indices correlate inversely with intrathecal IgG synthesis, 117 suggesting BAFF utilisation by CNS and/or meningeal B cells. Evidence from other diseases, such as malaria, 118 suggests that the effect of BAFF on CNS and/or meningeal B cells may impact CD11c+/T‐bet+ MBCs to a greater degree than cMBCs. In controlled human malaria infection, an increased abundance of circulating CD11c+/T‐bet+ MBCs, increased plasma BAFF levels and reduced expression of BAFF‐R on B cells were observed and BAFF levels correlated with proliferation of CD11c+/T‐bet+ MBCs but not cMBCs. 118 In addition, in patients with SLE, inhibition of BAFF activity by belimumab, a monoclonal antibody to BAFF (also known as Blys), led to a decline in circulating CD11c+/T‐bet+ MBCs (defined as IgD−/CD27− or CD11c+/CD21−), as well as naïve B cells but not cMBCs (IgD−/CD27+). 119 Interestingly, murine T‐bet+ IgM+ MBCs express larger amounts of BAFF‐R compared with follicular B cells. 89 Given that BAFF enhances the APC activity of B cells, 120 it is possible that BAFF produced by EBV‐infected MBCs 35 or astrocytes 121 augments the effects of pathogenic B cells on CD8+ T‐cell responses in the CNS (Figure 1). The effect of belimumab therapy is currently being evaluated in patients with MS (ClinicalTrials.gov identifier: NCT04767698).
The increased risk of MS development in females may be related to sex‐specific B‐cell responses against EBV
MS is 2–3 times more common in females than in males. 122 , 123 It is well‐established that females respond more strongly to viral infections than males and this also applies to EBV, as we have recently reviewed. 124 As a result, EBNA‐1‐specific IgG antibody levels are generally higher in females than in males 125 , 126 and this likely contributes to some of the increased risk of MS in females. Sex‐related differences in the function of CD11c+/T‐bet+ MBCs and MZ B cells might also contribute. Data from animal models suggest that CD11c+/T‐bet+ MBCs are more prominent in females than in males, particularly during ageing when only female mice display an expansion of these cells. 59 A recent study has further identified that CD11c+/T‐bet+ MBCs are functionally different in females compared with males, particularly in their ability to mount an interferon gene signature, and this difference was related to the development of SLE in female mice. 127 The mechanism by which CD11c+/T‐bet+ MBCs are expanded in females remains unclear but likely relates to TLR7 signalling being a driving pathway for CD11c+/T‐bet+ MBC induction and TLR7 expression being higher in females because of deficient X chromosome inactivation. 45 Support for this argument is provided by the recent report that TLR7 gain‐of‐function mutations are a cause of SLE. 128 The location of CXCR3 on the X chromosome and its susceptibility to X chromosome inactivation escape 129 may also result in higher CXCR3 expression on CD11c+/T‐bet+ MBCs in females. In our studies, low expression of FcγRIIb was only observed on MZ and naive B cells in females. 9 Healthy females have lower expression of FcγRIIb on B cells than males 130 and oestrogens increase the abundance of MZ B cells. 83 Hyper‐responsiveness and increased abundance of MZ B cells might therefore contribute to dysregulation of downstream B‐cell subsets following class switching and female predisposition to autoimmune diseases, including MS.
Potential effects of changes in gut microbiome, low sun exposure, low vitamin D levels and cigarette smoking on EBV‐induced B‐cell dysfunction in GALT
Environmental risk factors for MS, such as an altered gut microbiome, low sun exposure, low vitamin D levels and cigarette smoking must also be considered in a hypothesis that implicates EBV‐infected MBC within GALT in the immunopathogenesis of MS (Figure 1). Here, these risk factors will be considered as potential regulators of EBV infection and immune cell interactions in GALT.
Gut microbiota
The gut microbiota plays a homeostatic role in regulating intestinal barrier integrity and cellular interactions in GALT. Soluble products of gut microbes, particularly SCFAs, may alter B‐cell activity 131 , 132 and contribute to modulating other immune cells in GALT. 133 , 134 This laboratory has shown, like others, 135 that serum propionate levels are lower in patients with CIS or MS than in healthy controls. 11 Furthermore, serum levels of propionate positively correlated with the frequencies of circulating Tfh and T follicular regulatory cells. Serum levels of butyrate correlated positively with frequencies of IL‐10‐producing B cells but negatively with frequencies of cMBCs, while acetate levels correlated negatively with TNF production by polyclonally‐activated circulating total B cells. Thus, levels of serum SCFAs were associated with changes in circulating immune cells that can be implicated in regulation of immune cell function in GALT and the development of MS. Furthermore, the findings of other investigators suggest that there is a link between changes in the gut microbiota and dysregulation of MZ B cells associated with autoimmunity. 83
Antigenic cross‐reactivity between gut microbes and autoantigens has been proposed as a mechanism of inducing autoreactive T cells in several autoimmune diseases. 136 In addition, SCFAs produced by gut microbes may exert stimulatory or inhibitory effects on EBV reactivation in GALT MBCs, particularly in the colon where acetate, propionate and butyrate are produced in large amounts by bacteria. 66 However, the relationship between plasma levels of SCFAs and their abundance in the colon, and the effect of relative amounts of each SCFA on EBV reactivation, require further investigations.
Low sunlight exposure
A latitude gradient for MS risk has been recognised for decades, with a higher prevalence of MS recorded in populations living at higher latitudes. 137 A recent study has also suggested a relationship between low sun exposure and MS severity. 138 Multiple mechanisms of immune suppression after skin exposure to sunlight and its component ultraviolet B (UVB) radiation have been proposed and many altered immune cell types and soluble mediators have been described as potential contributors to systemic immunosuppression. 139 , 140 , 141 A recent study by our group suggested that narrowband UVB phototherapy primed the participant's B cells to be less responsive to activation through TLR7. 8 Direct induction of type I IFNs through sun exposure could be another mechanism of UV‐induced immune modulation of MS. 138
In a recent report, low sun exposure acted synergistically with high EBNA‐1 antibody levels in its association to increased MS risk 142 supporting the submitted hypothesis. The authors proposed that the two risk factors for MS (EBV infection and low sun exposure) re‐enforced the actions of each other. In addition, the proportion of EBV positive individuals is positively associated with latitude independently of MS status. 143
Exposure of skin to suberythemal narrowband UVB phototherapy can also modulate the gut microbiome. 144 In healthy individuals receiving three exposures to narrowband UVB radiation over a single week, there was increased diversity of the gut microbiome and the relative abundance of Firmicutes and Proteobacteria increased in their faecal samples; the Bacteroidetes phyla were reduced by UVB. 144 In mice chronically exposed to suberythemal broadband UVR, similar faecal changes in Firmicutes and Bacteroidetes have been reported. 145
The impact of these UVR‐induced changes has been difficult to interpret for patients with MS, for whom there have been multiple varied and confusing disease‐associated changes reported in their microbiome. 11 In the context of gut microbiota, families within Firmicutes, such as Lachnospiraceae and Ruminococcaceae, are generally considered health promoting. 146 Despite indirect evidence, further studies are required to determine whether any UVB radiation‐induced changes to the microbiome of MS patients might regulate reactivation of EBV infection and/or EBV‐specific immune responses and EBV‐induced immune dysregulation in the GALT.
Low vitamin D levels
Low vitamin D levels are a risk factor for MS. 147 However, it is much debated whether they are merely a biomarker of low sun exposure or the causal agent in the development of immune‐driven diseases associated with low sun exposure. Supplementation studies of vitamin D for patients with MS have cast doubt on the role of reduced vitamin D levels per se 147 but more studies are required. Of interest, reduced serum levels of EBNA‐1 antibodies have been reported in vitamin D‐supplemented MS patients. 148 , 149 Although the active form of vitamin D (1,25(OH)2 vitamin D) has proven direct effects on immune cells in vitro, 150 in studies of mice, vitamin D obtained by diet induced a different gut microbiome to that measured in those obtaining their vitamin D by broadband UVR exposure. 145 , 151 By contrast, there was evidence that UVR‐induced vitamin D was responsible for microbiome changes in individuals receiving UVB phototherapy. 144 It remains possible that UVR regulates the gut microbiome, and we propose immune cell reactivity in the GALT, by multiple pathways that may be dependent on, and independent of, vitamin D actions.
Cigarette smoke
Smoking cigarettes is a well‐established risk factor for MS. 152 Recently, the impact of smoking on EBV reactivation and circulating levels of anti‐EBNA IgG and IgA has been investigated. 153 These and other studies 154 suggest that smoking may promote EBV reactivation and increase serum EBNA‐1 IgG antibody levels. Furthermore, a history of IM and smoking appears to interact to increase the risk of MS. 153 Cigarette smoking also affects the composition of the gut microbiome 155 and may interact with other factors affecting the gut microbiome in MS patients. Smoking induces an expansion of circulating cMBCs, including IgA+ cells, 156 , 157 but this effect may be transient because circulating B‐cell profiles are not different in smokers and non‐smokers. 157 Expression of CD11c or T‐bet by B cells does not differ in smokers compared with non‐smokers. 158
Conclusions and future perspectives
Our hypothesis brings together the major genetic and environmental risk factors for developing MS into a single disease model (Figure 1) and proposes mechanisms to explain altered B‐cell phenotypes, the production of B‐cell and antibody responses against EBV proteins that cross‐react with CNS autoantigens, the contribution of CD4+ T cells, and the concurrence of CD8+ T cells and B cells in CNS inflammatory lesions. It also opens new avenues of research on MS immunopathogenesis and therapy. As shown in Table 2, all three types of MBCs likely contribute to the immunopathogenesis of MS, but through different mechanisms. As all express CD20, monoclonal anti‐CD20 antibody therapies would deplete all subpopulations. We suggest that investigation of EBV‐specific immune responses arising in GALT may be as enlightening as investigations of gut microbiota‐specific immune responses have been, specifically those involving MZ B cells, CD11c+/T‐bet+ IgG+ MBCs and IgA+ MBCs. Furthermore, the possibility that all GALT‐associated B cells contribute to the gut‐meningeal immune axis 69 in health and disease should be considered. Such investigations may lead to more targeted therapies for modulating B‐cell dysfunction. For example, therapeutic inhibition of adenosine receptor 2a on CD11c+/T‐bet+ MBCs 159 is potentially a means of decreasing pathogenic B‐cell responses without impairing global B‐cell function. Future investigations of the gut microbiota and/or SCFAs in studies of MS pathogenesis should include analyses of the relationship with EBV reactivation and activation of circulating MBCs, particularly MZ B cells. Such investigations may identify new biomarkers to assess interactions of EBV with B cells and reveal novel therapeutic targets to limit EBV‐associated immunopathology in the CNS.
Table 2.
Major subpopulations of human circulating memory B cells, highlighting important functional characteristics, proposed mechanisms of involvement in the immunopathogenesis of multiple sclerosis and known effects of MS therapies on them
| Marginal zone B cells | CD11c+/T‐bet+ MBCs | Conventional MBCs | |
|---|---|---|---|
| Distinguishing and important immunophenotypes | CD19+, CD20+, CD27+, IgDlow, IgMhi | CD19+, CD20+, CD27+/−, IgD−, CXCR3+, CD11c+ | CD19+, CD20+, CD27+, IgD− |
| Subpopulations | MZB1 and MZB2 | IgG+, IgA+, (IgM‐only) | IgM‐only, IgG+, IgA+ |
| Primary functions |
Initiating B‐cell and antibody responses (‘first responders’) Long‐term Bcell memory |
Controlling infections in concert with effector T cells (mainly established for viruses and Plasmodium sp.) | Preventing infections through memory responses that produce plasmablasts and long‐lived plasma cells and antibodies |
| Important functional characteristics |
Microbe recognition by low‐affinity BCRs and various pathogen recognition molecules APC function for CD4+ T cells via HLA molecules MBZ2 cells express HLA‐DBR1 and exhibit antiviral characteristics |
Microbe recognition by BCRs and TLR7/9 Probable APCs for T cells, involving CD11c/CD18 Antibody responses skewed towards IgG3, enhancing cDC activation and antigen presentation to CD8+ T cells |
Microbe recognition by high affinity BCRs Production of antibodies with high affinity for antigens and functional diversity of Fc regions through immunoglobulin isotype switching of B cells |
| Proposed involvement in the immunopathogenesis of MS |
Initiate a B‐cell response against EBV proteins, such as EBNA‐1, in GALT that is cross‐reactive with autoantigens, such as GlialCAM Possibly activate ‘autoproliferative’ CD4+ T cells, as well as pathogenic B cells in CSF |
EBNA‐1/GlialCAM‐specific IgG+ cells:
IgA+ cells possibly produce IgA anti‐EBV that interferes with IgG antibodies. |
EBV‐infected cells possibly produce BAFF in CNS IgA+ cells possibly produce IgA anti‐EBV that interferes with IgG antibodies. |
| Demonstrated effect of MS therapies 1 , 160 , 161 , 162 | Depletion by anti‐CD52 (ATB), anti‐CD20 (OLB), IFN‐β and Cladribine a |
Inhibition of adhesion to receptors in CNS by anti‐α4‐integrin (NLB) Depletion by MS therapies? b |
Inhibition of adhesion to receptors in CNS and possibly GALT by anti‐α4‐integrin (NLB) Depletion by anti‐CD52 (ATB) anti‐CD20 (OLB), IFN‐β, DMF, GTA, Cladribine and Fingolimod |
ATB, alemtuzumab; DMF, dimethyl fumarate; GTA, glatiramer acetate; NLB, natalizumab; OLB, ocrelizumab.
Based on data for ‘unswitched’ CD27+ B cells.
Insufficient information on depletion of this subpopulation by MS therapies but probable that anti‐CD20 (OLB), at least, does this.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Jonatan Leffler: Investigation; writing – review and editing. Stephanie Trend: Investigation; writing – review and editing. Prue H Hart: Funding acquisition; investigation; project administration; supervision; writing – review and editing. Martyn A French: Conceptualization; project administration; supervision; writing – original draft; writing – review and editing.
Acknowledgments
The authors acknowledge the Telethon Kids Institute communication team for assistance with the illustration as well as financial support from MSWA (formerly known as Multiple Sclerosis Society of Western Australia).
References
- 1. Ebers GC. Environmental factors and multiple sclerosis. Lancet Neurol 2008; 7: 268–277. [DOI] [PubMed] [Google Scholar]
- 2. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol 2017; 13: 25–36. [DOI] [PubMed] [Google Scholar]
- 3. International Multiple Sclerosis Genetics Consortium . Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019; 365: eaav7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hollenbach JA, Oksenberg JR. The immunogenetics of multiple sclerosis: a comprehensive review. J Autoimmun 2015; 64: 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bar‐Or A, Pender MP, Khanna R et al. Epstein‐Barr virus in multiple sclerosis: theory and emerging immunotherapies. Trends Mol Med 2020; 26: 296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Veroni C, Aloisi F. The CD8 T cell‐Epstein‐Barr virus‐B cell trialogue: a central issue in multiple sclerosis pathogenesis. Front Immunol 2021; 12: 665718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trend S, Jones AP, Cha L et al. Higher serum immunoglobulin G3 levels may predict the development of multiple sclerosis in individuals with clinically isolated syndrome. Front Immunol 2018; 9: 1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Trend S, Leffler J, Cooper MN et al. Narrowband UVB phototherapy reduces TNF production by B‐cell subsets stimulated via TLR7 from individuals with early multiple sclerosis. Clin Transl Immunology 2020; 9: e1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trend S, Leffler J, Teige I et al. FcgammaRIIb expression is decreased on naive and marginal zone‐like B cells from females with multiple sclerosis. Front Immunol 2020; 11: 614492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leffler J, Trend S, Ward NC et al. Circulating memory B cells in early multiple sclerosis exhibit increased IgA cells, globally decreased BAFF‐R expression and an EBV‐related IgM+ cell signature. Front Immunol 2022; 13: 812317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trend S, Leffler J, Jones AP et al. Associations of serum short‐chain fatty acids with circulating immune cells and serum biomarkers in patients with multiple sclerosis. Sci Rep 2021; 11: 5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lanz TV, Brewer RC, Ho PP et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022; 603: 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bjornevik K, Cortese M, Healy BC et al. Longitudinal analysis reveals high prevalence of Epstein‐Barr virus associated with multiple sclerosis. Science 2022; 375: 296–301. [DOI] [PubMed] [Google Scholar]
- 14. Attfield KE, Jensen LT, Kaufmann M, Friese MA, Fugger L. The immunology of multiple sclerosis. Nat Rev Immunol 2022. Epub ahead of print. 10.1038/s41577-022-00718-z [DOI] [PubMed] [Google Scholar]
- 15. Fransen NL, Hsiao CC, van der Poel M et al. Tissue‐resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain 2020; 143: 1714–1730. [DOI] [PubMed] [Google Scholar]
- 16. Machado‐Santos J, Saji E, Troscher AR et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue‐resident CD8+ T lymphocytes and B cells. Brain 2018; 141: 2066–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Margoni M, Preziosa P, Filippi M, Rocca MA. Anti‐CD20 therapies for multiple sclerosis: current status and future perspectives. J Neurol 2022; 269: 1316–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abrahamyan S, Eberspacher B, Hoshi MM et al. Complete Epstein‐Barr virus seropositivity in a large cohort of patients with early multiple sclerosis. J Neurol Neurosurg Psychiatry 2020; 91: 681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mescheriakova JY, van Nierop GP, van der Eijk AA, Kreft KL, Hintzen RQ. EBNA‐1 titer gradient in families with multiple sclerosis indicates a genetic contribution. Neurol Neuroimmunol Neuroinflamm 2020; 7: e872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hedstrom AK, Huang J, Michel A et al. High levels of Epstein‐Barr virus nuclear Antigen‐1‐specific antibodies and infectious mononucleosis act both independently and synergistically to increase multiple sclerosis risk. Front Neurol 2019; 10: 1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lünemann JD, Tintoré M, Messmer B et al. Elevated Epstein‐Barr virus‐encoded nuclear antigen‐1 immune responses predict conversion to multiple sclerosis. Ann Neurol 2010; 67: 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hudnall SD, Ge Y, Wei L, Yang NP, Wang HQ, Chen T. Distribution and phenotype of Epstein‐Barr virus‐infected cells in human pharyngeal tonsils. Mod Pathol 2005; 18: 519–527. [DOI] [PubMed] [Google Scholar]
- 23. Delecluse S, Tsai MH, Shumilov A et al. Epstein‐Barr virus induces expression of the LPAM‐1 integrin in B cells in vitro and in vivo . J Virol 2019; 93: e01618‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Souza TA, Stollar BD, Sullivan JL, Luzuriaga K, Thorley‐Lawson DA. Influence of EBV on the peripheral blood memory B cell compartment. J Immunol 2007; 179: 3153–3160. [DOI] [PubMed] [Google Scholar]
- 25. Seifert M, Kuppers R. Human memory B cells. Leukemia 2016; 30: 2283–2292. [DOI] [PubMed] [Google Scholar]
- 26. Hatton OL, Harris‐Arnold A, Schaffert S, Krams SM, Martinez OM. The interplay between Epstein‐Barr virus and B lymphocytes: implications for infection, immunity, and disease. Immunol Res 2014; 58: 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Panikkar A, Smith C, Hislop A et al. Impaired Epstein‐Barr virus‐specific neutralizing antibody response during acute infectious mononucleosis is coincident with global B‐cell dysfunction. J Virol 2015; 89: 9137–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheik‐Ali S. Infectious mononucleosis and multiple sclerosis ‐ updated review on associated risk. Mult Scler Relat Disord 2017; 14: 56–59. [DOI] [PubMed] [Google Scholar]
- 29. Rickinson AB, Long HM, Palendira U, Munz C, Hislop AD. Cellular immune controls over Epstein‐Barr virus infection: new lessons from the clinic and the laboratory. Trends Immunol 2014; 35: 159–169. [DOI] [PubMed] [Google Scholar]
- 30. Jayasooriya S, de Silva TI, Njie‐jobe J et al. Early virological and immunological events in asymptomatic Epstein‐Barr virus infection in African children. PLoS Pathog 2015; 11: e1004746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity 2018; 48: 202–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Woon HG, Braun A, Li J et al. Compartmentalization of total and virus‐specific tissue‐resident memory CD8+ T cells in human lymphoid organs. PLoS Pathog 2016; 12: e1005799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bartolome‐Casado R, Landsverk OJB, Chauhan SK et al. Resident memory CD8 T cells persist for years in human small intestine. J Exp Med 2019; 216: 2412–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pender MP, Csurhes PA, Burrows JM, Burrows SR. Defective T‐cell control of Epstein‐Barr virus infection in multiple sclerosis. Clin Transl Immunology 2017; 6: e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Serafini B, Severa M, Columba‐Cabezas S et al. Epstein‐Barr virus latent infection and BAFF expression in B cells in the multiple sclerosis brain: implications for viral persistence and intrathecal B‐cell activation. J Neuropathol Exp Neurol 2010; 69: 677–693. [DOI] [PubMed] [Google Scholar]
- 36. He B, Raab‐Traub N, Casali P, Cerutti A. EBV‐encoded latent membrane protein 1 cooperates with BAFF/BLyS and APRIL to induce T cell‐independent Ig heavy chain class switching. J Immunol 2003; 171: 5215–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johansson D, Rauld C, Roux J et al. Mass cytometry of CSF identifies an MS‐associated B‐cell population. Neurol Neuroimmunol Neuroinflamm 2021; 8: e943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Muehlinghaus G, Cigliano L, Huehn S et al. Regulation of CXCR3 and CXCR4 expression during terminal differentiation of memory B cells into plasma cells. Blood 2005; 105: 3965–3971. [DOI] [PubMed] [Google Scholar]
- 39. Ramesh A, Schubert RD, Greenfield AL et al. A pathogenic and clonally expanded B cell transcriptome in active multiple sclerosis. Proc Natl Acad Sci USA 2020; 117: 22932–22943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Claes N, Fraussen J, Vanheusden M et al. Age‐associated B cells with proinflammatory characteristics are expanded in a proportion of multiple sclerosis patients. J Immunol 2016; 197: 4576–4583. [DOI] [PubMed] [Google Scholar]
- 41. Knox JJ, Myles A, Cancro MP. T‐bet+ memory B cells: generation, function, and fate. Immunol Rev 2019; 288: 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holla P, Dizon B, Ambegaonkar AA et al. Shared transcriptional profiles of atypical B cells suggest common drivers of expansion and function in malaria, HIV, and autoimmunity. Sci Adv 2021; 7: eabg8384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Keller B, Strohmeier V, Harder I et al. The expansion of human T‐bethighCD21low B cells is T cell dependent. Sci Immunol 2021; 6: eabh0891. [DOI] [PubMed] [Google Scholar]
- 44. Johnson JL, Rosenthal RL, Knox JJ et al. The transcription factor T‐bet resolves memory B cell subsets with distinct tissue distributions and antibody specificities in mice and humans. Immunity 2020; 52: 842–855.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Phalke S, Rivera‐Correa J, Jenkins D, Flores Castro D, Giannopoulou E, Pernis AB. Molecular mechanisms controlling age‐associated B cells in autoimmunity. Immunol Rev 2022; 307: 79–100. [DOI] [PubMed] [Google Scholar]
- 46. Sándor N, Lukácsi S, Ungai‐Salánki R et al. CD11c/CD18 dominates adhesion of human monocytes, macrophages and dendritic cells over CD11b/CD18. PLoS One 2016; 11: e0163120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chang LY, Li Y, Kaplan DE. Hepatitis C viraemia reversibly maintains subset of antigen‐specific T‐bet+ tissue‐like memory B cells. J Viral Hepat 2017; 24: 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kallies A, Good‐Jacobson KL. Transcription factor T‐bet orchestrates lineage development and function in the immune system. Trends Immunol 2017; 38: 287–297. [DOI] [PubMed] [Google Scholar]
- 49. Louis K, Bailly E, Macedo C et al. T‐bet+CD27+CD21− B cells poised for plasma cell differentiation during antibody‐mediated rejection of kidney transplants. JCI Insight 2021; 6: e148881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moir S, Ho J, Malaspina A et al. Evidence for HIV‐associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV‐infected viremic individuals. J Exp Med 2008; 205: 1797–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Portugal S, Obeng‐Adjei N, Moir S, Crompton PD, Pierce SK. Atypical memory B cells in human chronic infectious diseases: an interim report. Cell Immunol 2017; 321: 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jenks SA, Cashman KS, Zumaquero E et al. Distinct effector B cells induced by unregulated toll‐like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity 2018; 49: e726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rakhmanov M, Keller B, Gutenberger S et al. Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate‐like B cells. Proc Natl Acad Sci USA 2009; 106: 13451–13456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Karnell JL, Kumar V, Wang J, Wang S, Voynova E, Ettinger R. Role of CD11c+ T‐bet+ B cells in human health and disease. Cell Immunol 2017; 321: 40–45. [DOI] [PubMed] [Google Scholar]
- 55. Sutton HJ, Aye R, Idris AH et al. Atypical B cells are part of an alternative lineage of B cells that participates in responses to vaccination and infection in humans. Cell Rep 2021; 34: 108684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mouat IC, Horwitz MS. Age‐associated B cells in viral infection. PLoS Pathog 2022; 18: e1010297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, Marrack P. T‐box transcription factor T‐bet, a key player in a unique type of B‐cell activation essential for effective viral clearance. Proc Natl Acad Sci USA 2013; 110: e3216–e3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barnett BE, Staupe RP, Odorizzi PM et al. Cutting edge: B cell‐intrinsic T‐bet expression is required to control chronic viral infection. J Immunol 2016; 15: 1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rubtsov AV, Rubtsova K, Fischer A et al. Toll‐like receptor 7 (TLR7)‐driven accumulation of a novel CD11c+ B‐cell population is important for the development of autoimmunity. Blood 2011; 118: 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, Marrack P. B cells expressing the transcription factor T‐bet drive lupus‐like autoimmunity. J Clin Invest 2017; 127: 1392–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van Langelaar J, Rijvers L, Janssen M et al. Induction of brain‐infiltrating T‐bet‐expressing B cells in multiple sclerosis. Ann Neurol 2019; 86: 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. van Langelaar J, Wierenga‐Wolf AF, Samijn JPA et al. The association of Epstein‐Barr virus infection with CXCR3+ B‐cell development in multiple sclerosis: impact of immunotherapies. Eur J Immunol 2021; 51: 626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. SoRelle ED, Dai J, Reinoso‐Vizcaino NM et al. Time‐resolved transcriptomes reveal diverse B cell fate trajectories in the early response to Epstein‐Barr virus infection. Cell Rep 2022; 40: 111286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rubtsov AV, Rubtsova K, Kappler JW, Jacobelli J, Friedman RS, Marrack P. CD11c‐expressing B cells are located at the T cell/B cell border in spleen and are potent APCs. J Immunol 2015; 195: 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Probstel AK, Zhou X, Baumann R et al. Gut microbiota‐specific IgA+ B cells traffic to the CNS in active multiple sclerosis. Sci Immunol 2020; 5: eabc7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gorres KL, Daigle D, Mohanram S, Miller G. Activation and repression of Epstein‐Barr virus and Kaposi's sarcoma‐associated herpesvirus lytic cycles by short‐ and medium‐chain fatty acids. J Virol 2014; 88: 8028–8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Morbe UM, Jorgensen PB, Fenton TM et al. Human gut‐associated lymphoid tissues (GALT); diversity, structure, and function. Mucosal Immunol 2021; 14: 793–802. [DOI] [PubMed] [Google Scholar]
- 68. Fitzpatrick Z, Frazer G, Ferro A et al. Gut‐educated IgA plasma cells defend the meningeal venous sinuses. Nature 2020; 587: 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Di Marco Barros R, Fitzpatrick Z, Clatworthy MR. The gut‐meningeal immune axis: priming brain defense against the most likely invaders. J Exp Med 2022; 219: e20211520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhao Y, Uduman M, Siu JHY et al. Spatiotemporal segregation of human marginal zone and memory B cell populations in lymphoid tissue. Nat Commun 2018; 9: 3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Siu JHY, Pitcher MJ, Tull TJ et al. Two subsets of human marginal zone B cells resolved by global analysis of lymphoid tissues and blood. Sci Immunol 2022; 7: eabm9060. [DOI] [PubMed] [Google Scholar]
- 72. Song W, Antao OQ, Condiff E et al. Development of Tbet‐ and CD11c‐expressing B cells in a viral infection requires T follicular helper cells outside of germinal centers. Immunity 2022; 55: 290–307.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rojas OL, Narváez CF, Greenberg HB, Angel J, Franco MA. Characterization of rotavirus specific B cells and their relation with serological memory. Virology 2008; 380: 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Narváez CF, Feng N, Vásquez C et al. Human rotavirus‐specific IgM memory B cells have differential cloning efficiencies and switch capacities and play a role in antiviral immunity in vivo . J Virol 2012; 86: 10829–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Marsh‐Wakefield F, Juillard P, Ashhurst TM et al. Peripheral B‐cell dysregulation is associated with relapse after long‐term quiescence in patients with multiple sclerosis. Immunol Cell Biol 2022; 100: 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Berkowska MA, Schickel JN, Grosserichter‐Wagener C et al. Circulating human CD27−IgA+ memory B cells recognize bacteria with polyreactive Igs. J Immunol 2015; 195: 1417–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li W, Wang L, Shen C, Xu T, Chu Y, Hu C. Radiation therapy‐induced reactive oxygen species specifically eliminates CD19+IgA+ B cells in nasopharyngeal carcinoma. Cancer Manag Res 2019; 11: 6299–6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ai P, Li Z, Jiang Y et al. Tumor microenvironment contributes to Epstein‐Barr virus anti‐nuclear antigen‐1 antibody production in nasopharyngeal carcinoma. Oncol Lett 2017; 14: 2458–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Xu J, Ahmad A, Blagdon M et al. The Epstein‐Barr virus (EBV) major envelope glycoprotein gp350/220‐specific antibody reactivities in the sera of patients with different EBV‐associated diseases. Int J Cancer 1998; 79: 481–486. [DOI] [PubMed] [Google Scholar]
- 80. Coghill AE, Hildesheim A. Epstein‐Barr virus antibodies and the risk of associated malignancies: review of the literature. Am J Epidemiol 2014; 180: 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mathew GD, Qualtiere LF, Neel HB 3rd, Pearson GR. IgA antibody, antibody‐dependent cellular cytotoxicity and prognosis in patients with nasopharyngeal carcinoma. Int J Cancer 1981; 27: 175–180. [DOI] [PubMed] [Google Scholar]
- 82. Ruiz MJ, Ghiglione Y, Falivene J et al. Env‐specific IgA from viremic HIV‐infected subjects compromises antibody‐dependent cellular cytotoxicity. J Virol 2016; 90: 670–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Palm AE, Kleinau S. Marginal zone B cells: from housekeeping function to autoimmunity? J Autoimmun 2021; 119: 102627. [DOI] [PubMed] [Google Scholar]
- 84. Tjiam MC, Fernandez S, French MA. Characterising the phenotypic diversity of antigen‐specific memory B cells before and after vaccination. Front Immunol 2021; 12: 738123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Shehata L, Maurer DP, Wec AZ et al. Affinity maturation enhances antibody specificity but compromises conformational stability. Cell Rep 2019; 28: 3300–3308.e4. [DOI] [PubMed] [Google Scholar]
- 86. Dimitrov JD, Planchais C, Roumenina LT, Vassilev TL, Kaveri SV, Lacroix‐Desmazes S. Antibody polyreactivity in health and disease: statu variabilis. J Immunol 2013; 191: 993–999. [DOI] [PubMed] [Google Scholar]
- 87. Planchais C, Kök A, Kanyavuz A et al. HIV‐1 envelope recognition by polyreactive and cross‐reactive intestinal B cells. Cell Rep 2019; 27: 572–585.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Palm AK, Friedrich HC, Mezger A et al. Function and regulation of self‐reactive marginal zone B cells in autoimmune arthritis. Cell Mol Immunol 2015; 12: 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kenderes KJ, Levack RC, Papillion AM, Cabrera‐Martinez B, Dishaw LM, Winslow GM. T‐bet+ IgM memory cells generate multi‐lineage effector B cells. Cell Rep 2018; 24: 824–837.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Winslow GM, Papillion AM, Kenderes KJ, Levack RC. CD11c+ T‐bet+ memory B cells: immune maintenance during chronic infection and inflammation? Cell Immunol 2017; 321: 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Guerrier T, Youinou P, Pers J‐O, Jamin C. TLR9 drives the development of transitional B cells towards the marginal zone pathway and promotes autoimmunity. J Autoimmun 2012; 39: 173–179. [DOI] [PubMed] [Google Scholar]
- 92. Mockus TE, Munie A, Atkinson JR, Segal BM. Encephalitogenic and regulatory CD8 T cells in multiple sclerosis and its animal models. J Immunol 2021; 206: 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tyler KL. The enigmatic links between Epstein‐Barr virus infection and multiple sclerosis. J Clin Invest 2022; 132: e160468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Serafini B, Rosicarelli B, Veroni C, Mazzola GA, Aloisi F. Epstein‐Barr virus‐specific CD8 T cells selectively infiltrate the brain in multiple sclerosis and interact locally with virus‐infected cells: clue for a virus‐driven immunopathological mechanism. J Virol 2019; 93: e00980‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Veroni C, Serafini B, Rosicarelli B, Fagnani C, Aloisi F. Transcriptional profile and Epstein‐Barr virus infection status of laser‐cut immune infiltrates from the brain of patients with progressive multiple sclerosis. J Neuroinflammation 2018; 15: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Morandi E, Jagessar SA, 't Hart BA, Gran B. EBV infection empowers human B cells for autoimmunity: role of autophagy and relevance to multiple sclerosis. J Immunol 2017; 199: 435–448. [DOI] [PubMed] [Google Scholar]
- 97. Tuohy VK, Yu M, Yin L et al. The epitope spreading cascade during progression of experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol Rev 1998; 164: 93–100. [DOI] [PubMed] [Google Scholar]
- 98. Niessl J, Baxter AE, Mendoza P et al. Combination anti‐HIV‐1 antibody therapy is associated with increased virus‐specific T cell immunity. Nat Med 2020; 26: 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Veenhuis RT, Garliss CC, Bailey JR, Blankson JN. CD8 effector T cells function synergistically with broadly neutralizing antibodies to enhance suppression of HIV infection. Front Immunol 2021; 12: 708355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Pardieck IN, van Duikeren S, Veerkamp DMB et al. Dominant antiviral CD8+ T cell responses empower prophylactic antibody‐eliciting vaccines against cytomegalovirus. Front Immunol 2022; 13: 680559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. van Montfoort N, Mangsbo SM, Camps MGM et al. Circulating specific antibodies enhance systemic cross‐priming by delivery of complexed antigen to dendritic cells in vivo . Eur J Immunol 2012; 42: 598–606. [DOI] [PubMed] [Google Scholar]
- 102. Bournazos S, Corti D, Virgin HW, Ravetch JV. Fc‐optimized antibodies elicit CD8 immunity to viral respiratory infection. Nature 2020; 588: 485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Obeng‐Adjei N, Portugal S, Holla P et al. Malaria‐induced interferon‐γ drives the expansion of Tbethi atypical memory B cells. PLoS Pathog 2017; 13: e1006576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Knox JJ, Buggert M, Kardava L et al. T‐bet+ B cells are induced by human viral infections and dominate the HIV gp140 response. JCI Insight 2017; 2: e92943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. de Jong BG, IJspeert H, Marques L et al. Human IgG2‐ and IgG4‐expressing memory B cells display enhanced molecular and phenotypic signs of maturity and accumulate with age. Immunol Cell Biol 2017; 95: 744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol 2014; 5: 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Foss S, Jonsson A, Bottermann M et al. Potent TRIM21 and complement‐dependent intracellular antiviral immunity requires the IgG3 hinge. Sci Immunol 2022; 7: eabj1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ng PML, Kaliaperumal N, Lee CY et al. Enhancing antigen cross‐presentation in human monocyte‐derived dendritic cells by recruiting the intracellular fc receptor TRIM21. J Immunol 2019; 202: 2307–2319. [DOI] [PubMed] [Google Scholar]
- 109. Zhang W, Zhang H, Liu S et al. Excessive CD11c+Tbet+ B cells promote aberrant TFH differentiation and affinity‐based germinal center selection in lupus. Proc Natl Acad Sci USA 2019; 116: 18550–18560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Marsh‐Wakefield F, Ashhurst T, Trend S et al. IgG3+ B cells are associated with the development of multiple sclerosis. Clin Transl Immunology 2020; 9: e01133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Jelcic I, Al Nimer F, Wang J et al. Memory B cells activate brain‐homing, autoreactive CD4+ T cells in multiple sclerosis. Cell 2018; 175: 85–100.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wang J, Jelcic I, Mühlenbruch L et al. HLA‐DR15 molecules jointly shape an autoreactive T cell repertoire in multiple sclerosis. Cell 2020; 183: 1264–1281.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Schriek P, Ching AC, Moily NS et al. Marginal zone B cells acquire dendritic cell functions by trogocytosis. Science 2022; 375: eabf7470. [DOI] [PubMed] [Google Scholar]
- 114. Smulski CR, Kury P, Seidel LM et al. BAFF‐ and TACI‐dependent processing of BAFFR by ADAM proteases regulates the survival of B cells. Cell Rep 2017; 18: 2189–2202. [DOI] [PubMed] [Google Scholar]
- 115. Kappos L, Hartung HP, Freedman MS et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo‐controlled, double‐blind, phase 2 trial. Lancet Neurol 2014; 13: 353–363. [DOI] [PubMed] [Google Scholar]
- 116. Puthenparampil M, Miante S, Federle L et al. BAFF is decreased in the cerebrospinal fluid of multiple sclerosis at clinical onset. J Neuroimmunol 2016; 297: 63–67. [DOI] [PubMed] [Google Scholar]
- 117. Puthenparampil M, Federle L, Miante S et al. BAFF index and CXCL13 levels in the cerebrospinal fluid associate respectively with intrathecal IgG synthesis and cortical atrophy in multiple sclerosis at clinical onset. J Neuroinflammation 2017; 14: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Scholzen A, Teirlinck AC, Bijker EM et al. BAFF and BAFF receptor levels correlate with B cell subset activation and redistribution in controlled human malaria infection. J Immunol 2014; 192: 3719–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ramskold D, Parodis I, Lakshmikanth T et al. B cell alterations during BAFF inhibition with belimumab in SLE. EBioMedicine 2019; 40: 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Yiwen Z, Shilin G, Yingshi C et al. Efficient generation of antigen‐specific CTLs by the BAFF‐ activated human B lymphocytes as APCs: a novel approach for immunotherapy. Oncotarget 2016; 7: 77732–77748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Krumbholz M, Theil D, Derfuss T et al. BAFF is produced by astrocytes and up‐regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med 2005; 201: 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Orton S‐M, Herrera BM, Yee IM et al. Sex ratio of multiple sclerosis in Canada: a longitudinal study. Lancet Neurol 2006; 5: 932–936. [DOI] [PubMed] [Google Scholar]
- 123. Trojano M, Lucchese G, Graziano G et al. Geographical variations in sex ratio trends over time in multiple sclerosis. PLoS One 2012; 7: e48078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Leffler J, Trend S, Gorman S, Hart PH. Sex‐specific environmental impacts on initiation and progression of multiple sclerosis. Front Neurol 2022; 13: 835162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ford JL, Stowe RP. Racial‐ethnic differences in Epstein‐Barr virus antibody titers among U.S. children and adolescents. Ann Epidemiol 2013; 23: 275–280. [DOI] [PubMed] [Google Scholar]
- 126. Wagner H‐J, Hornef M, Teichert H‐M, Kirchner H. Sex difference in the serostatus of adults to the Epstein‐Barr virus. Immunobiology 1994; 190: 424–429. [DOI] [PubMed] [Google Scholar]
- 127. Ricker E, Manni M, Flores‐Castro D et al. Altered function and differentiation of age‐associated B cells contribute to the female bias in lupus mice. Nat Commun 2021; 12: 4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Brown GJ, Canete PF, Wang H et al. TLR7 gain‐of‐function genetic variation causes human lupus. Nature 2022; 605: 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Jiwrajka N, Anguera MC. The X in seX‐biased immunity and autoimmune rheumatic disease. J Exp Med 2022; 219: e20211487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Prokopec KE, Rhodiner M, Matt P, Lindqvist U, Kleinau S. Down regulation of Fc and complement receptors on B cells in rheumatoid arthritis. Clin Immunol 2010; 137: 322–329. [DOI] [PubMed] [Google Scholar]
- 131. Kim M, Qie Y, Park J, Kim CH. Gut microbial metabolites fuel host antibody responses. Cell Host Microbe 2016; 20: 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Sanchez HN, Moroney JB, Gan H et al. B cell‐intrinsic epigenetic modulation of antibody responses by dietary fiber‐derived short‐chain fatty acids. Nat Commun 2020; 11: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Silva YP, Bernardi A, Frozza RL. The role of short‐chain fatty acids from gut microbiota in gut‐brain communication. Front Endocrinol (Lausanne) 2020; 11: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Smith PM, Howitt MR, Panikov N et al. The microbial metabolites, short‐chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013; 341: 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Duscha A, Gisevius B, Hirschberg S et al. Propionic acid shapes the multiple sclerosis disease course by an immunomodulatory mechanism. Cell 2020; 180: e1016. [DOI] [PubMed] [Google Scholar]
- 136. Garabatos N, Santamaria P. Gut microbial antigenic mimicry in autoimmunity. Front Immunol 2022; 13: 873607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Simpson S Jr, Wang W, Otahal P, Blizzard L, van der Mei IAF, Taylor BV. Latitude continues to be significantly associated with the prevalence of multiple sclerosis: an updated meta‐analysis. J Neurol Neurosurg Psychiatry 2019; 90: 1193–1200. [DOI] [PubMed] [Google Scholar]
- 138. Ostkamp P, Salmen A, Pignolet B et al. Sunlight exposure exerts immunomodulatory effects to reduce multiple sclerosis severity. Proc Natl Acad Sci USA 2021; 118: e2018457118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ullrich SE, Byrne SN. The immunologic revolution: photoimmunology. J Invest Dermatol 2012; 132: 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hart PH, Norval M, Byrne SN, Rhodes LE. Exposure to ultraviolet radiation in the modulation of human diseases. Annu Rev Pathol 2019; 14: 55–81. [DOI] [PubMed] [Google Scholar]
- 141. Bernard JJ, Gallo RL, Krutmann J. Photoimmunology: how ultraviolet radiation affects the immune system. Nat Rev Immunol 2019; 19: 688–701. [DOI] [PubMed] [Google Scholar]
- 142. Hedstrom AK, Huang J, Brenner N et al. Low sun exposure acts synergistically with high Epstein‐Barr nuclear antigen 1 (EBNA‐1) antibody levels in multiple sclerosis etiology. Eur J Neurol 2021; 28: 4146–4152. [DOI] [PubMed] [Google Scholar]
- 143. Disanto G, Pakpoor J, Morahan JM et al. Epstein‐Barr virus, latitude and multiple sclerosis. Mult Scler 2013; 19: 362–365. [DOI] [PubMed] [Google Scholar]
- 144. Bosman ES, Albert AY, Lui H, Dutz JP, Vallance BA. Skin exposure to narrow band ultraviolet (UVB) light modulates the human intestinal microbiome. Front Microbiol 2019; 10: 2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Ghaly S, Kaakoush NO, Lloyd F et al. Ultraviolet irradiation of skin alters the faecal microbiome independently of vitamin D in mice. Nutrients 2018; 10: 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Truax AD, Chen L, Tam JW et al. The inhibitory innate immune sensor NLRP12 maintains a threshold against obesity by regulating gut microbiota homeostasis. Cell Host Microbe 2018; 24: e366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Smolders J, Torkildsen O, Camu W, Holmoy T. An update on vitamin D and disease activity in multiple sclerosis. CNS Drugs 2019; 33: 1187–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Najafipoor A, Roghanian R, Zarkesh‐Esfahani SH, Bouzari M, Etemadifar M. The beneficial effects of vitamin D3 on reducing antibody titers against Epstein–Barr virus in multiple sclerosis patients. Cell Immunol 2015; 294: 9–12. [DOI] [PubMed] [Google Scholar]
- 149. Rolf L, Muris AH, Mathias A et al. Exploring the effect of vitamin D3 supplementation on the anti‐EBV antibody response in relapsing‐remitting multiple sclerosis. Mult Scler 2018; 24: 1280–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Charoenngam N, Holick MF. Immunologic effects of vitamin D on human health and disease. Nutrients 2020; 12: 2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ghaly S, Kaakoush NO, Hart PH. Effects of UVR exposure on the gut microbiota of mice and humans. Photochem Photobiol Sci 2020; 19: 20–28. [DOI] [PubMed] [Google Scholar]
- 152. Jafari N, Hintzen RQ. The association between cigarette smoking and multiple sclerosis. J Neurol Sci 2011; 311: 78–85. [DOI] [PubMed] [Google Scholar]
- 153. Hedstrom AK, Huang J, Brenner N et al. Smoking and Epstein‐Barr virus infection in multiple sclerosis development. Sci Rep 2020; 10: 10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Xu FH, Xiong D, Xu YF et al. An epidemiological and molecular study of the relationship between smoking, risk of nasopharyngeal carcinoma, and Epstein‐Barr virus activation. J Natl Cancer Inst 2012; 104: 1396–1410. [DOI] [PubMed] [Google Scholar]
- 155. Prakash A, Peters BA, Cobbs E et al. Tobacco smoking and the fecal microbiome in a large, multi‐ethnic cohort. Cancer Epidemiol Biomarkers Prev 2021; 30: 1328–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Brandsma CA, Hylkema MN, Geerlings M et al. Increased levels of (class switched) memory B cells in peripheral blood of current smokers. Respir Res 2009; 10: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Piaggeschi G, Rolla S, Rossi N et al. Immune trait shifts in association with tobacco smoking: a study in healthy women. Front Immunol 2021; 12: 637974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Martos SN, Campbell MR, Lozoya OA et al. Single‐cell analyses identify dysfunctional CD16+ CD8 T cells in smokers. Cell Rep Med 2020; 1: 100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Levack RC, Newell KL, Cabrera‐Martinez B et al. Adenosine receptor 2a agonists target mouse CD11c+T‐bet+ B cells in infection and autoimmunity. Nat Commun 2022; 13: 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Baker D, Marta M, Pryce G, Giovannoni G, Schmierer K. Memory B cells are major targets for effective immunotherapy in relapsing multiple sclerosis. EBioMedicine 2017; 16: 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Rizzo F, Giacomini E, Mechelli R et al. Interferon‐β therapy specifically reduces pathogenic memory B cells in multiple sclerosis patients by inducing a FAS‐mediated apoptosis. Immunol Cell Biol 2016; 94: 886–894. [DOI] [PubMed] [Google Scholar]
- 162. Ceronie B, Jacobs BM, Baker D et al. Cladribine treatment of multiple sclerosis is associated with depletion of memory B cells. J Neurol 2018; 265: 1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]