

Abstract
Background and Objectives
Follicular helper T (Tfh) cells play a critical role in protective immunity helping B cells produce antibodies against foreign pathogens and are likely implicated in the pathogenesis of various autoimmune diseases. The purpose of this study was to investigate the role of Tfh cells in the pathogenesis of multiple sclerosis (MS).
Methods
Using flow cytometry, we investigated phenotype, prevalence, and function of Tfh cells in blood and CSF from controls and patients with relapsing-remitting MS (RRMS) and primary progressive MS (PPMS). In addition, an in vitro blood-brain barrier coculture assay of primary human astrocytes and brain microvascular endothelial cells grown in a Boyden chamber was used to assess the migratory capacity of peripheral Tfh cells.
Results
This study identified 2 phenotypically and functionally distinct Tfh cell populations: CD25− Tfh cells (Tfh1-like) and CD25int Tfh cells (Tfh17-like). Whereas minor differences in Tfh cell populations were found in blood between patients with MS and controls, we observed an increased frequency of CD25− Tfh cells in CSF of patients with RRMS and PPMS and CD25int Tfh cells in patients with RRMS, compared with controls. Increasing frequencies of CSF CD25− Tfh cells and the CD25− Tfh/Tfr ratio scaled with increasing IgG index in patients with RRMS. Despite an increased prevalence of intrathecal Tfh cells in patients with MS, no difference in the migratory capacity of circulating Tfh cells was observed between controls and patients with MS. Instead, CSF concentrations of CXCL13 scaled with total counts of Tfh and Tfr cell subsets in the CSF.
Discussion
Our study indicates substantial changes in intrathecal Tfh dynamics, particularly in patients with RRMS, and suggests that the intrathecal inflammatory environment in patients with RRMS promotes recruitment of peripheral Tfh cells rather than the Tfh cells having an increased capacity to migrate to CNS.
Multiple sclerosis (MS) is a chronic immune-mediated demyelinating disease of the CNS characterized by immune cell-mediated damage of nervous tissue.1,2 A hallmark of MS is intrathecal antibody production, but despite intense investigation, the mechanism remains unresolved.3 Although antibody production is naturally dependent on B cells, specific subsets of T cells are also involved. Follicular helper T (Tfh) cells are a subset of peripheral lymphoid tissue T cells primarily located in germinal centers where they function to promote B-cell differentiation to antibody-producing plasma cells.4-6 Conversely, follicular regulatory T cells (Tfr) function by suppressing the Tfh-induced B-cell response.7 Phenotypically, both Tfh and Tfr cells express CD4 and the lymphoid follicle-homing chemokine receptor CXCR5, and their expression of programmed cell death protein 1 (PD-1) is induced by cognate antigen activation.8,9 Since Tfh and Tfr cells exhibit opposite functions in regulating humoral immune responses, their balance is important for maintaining immune homeostasis.10 Imbalances between Tfh and Tfr cells promote dysregulated antibody production and may contribute to the development of autoimmunity.11 Recently, the discovery of circulating Tfh cells has allowed investigation of their involvement in the pathogenesis of several autoimmune disorders.12 Furthermore, it has been shown that analogous to nonfollicular Th cells, circulating Tfh cells can be classified according to the expression of CXCR3 and CCR6 into Tfh1 (CXCR3+CCR6−), Tfh17 (CXCR3−CCR6+), Tfh2 (CXCR3−CCR6−), and Tfh17.1 (CXCR3+CCR6+) cells producing different cytokines that exert distinct B-cell helper activity.12-14
Previous studies have observed higher frequencies of circulating Tfh cells in patients with relapsing-remitting MS (RRMS) compared with healthy controls and that Tfh cells correlate positively with disease severity and progression.15-17 In addition, an imbalance in the blood Tfh/Tfr cell ratio has been reported in patients with RRMS. This ratio was found to scale with intrathecal immunoglobulin synthesis, as assessed by the immunoglobulin G (IgG) index.10 Furthermore, studies have shown an increased intrathecal level of the chemokine ligand of CXCR5 and CXCL13 in patients with RRMS, suggesting that Tfh cells are more prone to migrate into CSF.18-21 We, therefore, hypothesize that Tfh cells play a similar B-cell priming role in the inflamed CNS in addition to helping a B-cell response in peripheral lymphoid tissue. To investigate this hypothesis, we analyzed Tfh cells and their activity in blood and CSF samples from patients with RRMS and primary progressive MS (PPMS).
Methods
Study Participants
In this case-control observational study, we included 35 treatment-naive patients with RRMS (mean age 36 years; range 21–62), 21 untreated patients with PPMS (mean age 55 years; range 44–62), 14 symptomatic controls (mean age 34 years; range 20–60), and 22 healthy controls (mean age 45; range 26–74). All patients and controls were recruited at the Danish MS Center. In addition to blood from all test participants, CSF was collected from 25 patients with RRMS, 17 patients with PPMS, and 10 of the symptomatic controls.
An exploratory cohort of 10 healthy controls donating blood and CSF was included at the Neurogenetics Clinic, Danish Dementia Research Center, Rigshospitalet. The healthy controls were huntingtin gene expansion negative family members of Huntington's disease gene expansion carriers. Due to a possible center variation, data from this cohort have not been used for direct comparison with the other cohorts.
The patients with MS were diagnosed according to McDonald 2017 criteria.22,23 Patients with RRMS were treatment-naive, and patients with PPMS were clinically inactive and untreated for at least 6 months before sampling and had never received any immune cell-depleting therapies. In addition, all patients were included at least 1 month after last steroid treatment. Healthy controls included had no autoimmune, neurologic, or chronic illness. Symptomatic controls were defined as individuals with neurologic symptoms but with no objective clinical or abnormal paraclinical findings (e.g., no oligoclonal bands, normal albumin quotient, and normal white blood cell count in the CSF) to define any neurologic disease.24
Clinical and paraclinical data of the 35 patients with RRMS included median EDSS 2 (range 0–4.5), mean disease duration 1 year (range 0–25), IgG oligoclonal bands observed in 33 patients, and relapses reported in 29 patients 12 months before sampling (of whom 16 were reported within the past 3 months). Clinical and paraclinical data of the 21 patients with PPMS include median EDSS 4 (range 2–6.5), mean disease duration 10 years (range 2–24), IgG oligoclonal bands observed in 20 patients, and no relapses reported 6 months before sampling.
Study Protocols Approval, Registrations, and Patient Consent
All included individuals gave informed, written consent. Ethical approvals were granted for all investigative procedures (Protocol Nos.: H-16047666 and KF-01-314009).
Blood and Cerebrospinal Fluid Samples
Venous blood was drawn, and peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation (Lymphoprep, Axis-Shield, Oslo, Norway) and washed twice in cold phosphate-buffered saline containing 2 mM EDTA. Parallel with blood, 10 mL of CSF was collected in polypropylene tubes on ice and instantly centrifuged for 10 minutes at 400 g for isolation of cells. Then, CSF cells were immediately stained and analyzed by flow cytometry.
Flow Cytometry Analyses of Freshly Isolated Cells
For flow cytometry analyses of freshly isolated cells, a minimum of 350,000 PBMCs and 3,000 CSF cells were incubated with a FcR-blocking reagent (Miltenyi Biotec, Bergisch Gladbach, Germany) and stained with fluorochrome-conjugated antibodies against surface molecules of interest. For Tfh cell phenotyping, the following antigens were assessed: CD3 (AF488; UCHT1), CD127 (BV421; A01905), CD25 (PE; M-A251), PD-1 (BV605; EH12.2H7), CXCR5 (PE/Cy7; J252D4), CCR7 (AF674; G043H7), CD45RA (FITC; H100), CD69 (APC; FN50), CXCR3 (APC; G025H7), CCR2 (PerCP/Cy5.5; K036C2), CCR4 (APC; L291H4), CCR6 (PerCP/Cy5.5; G034E3), melanoma cell adhesion molecule 1 (BV421; P1H12), CD49d (APC; 9F10), SLAMF5 (FITC; MZ18-21F6) all from BioLegend (San Diego, CA, USA), HLA-DR (APC; L203) from R&D Systems (Minneapolis, MS), and CD4 (APC/AF750; S3.5) from Invitrogen (Waltham, MA). The corresponding isotype controls were included where appropriate. Data were acquired on a FACS Canto II flow cytometer, and data analysis was performed using FlowJo software (Tree Star, Inc., Ashland, OR).
Intracellular Cytokine and Chemokine Staining
For analysis of Tfh cell cytokine and chemokine production, cryopreserved PBMCs from 12 patients with RRMS and 12 healthy controls were thawed and cultured in culture medium Roswell Park Memorial Institute medium (RPMI) 1640/5% human AB serum (Invitrogen, Carlsbad, CA)/penicillin/streptomycin (50 U/mL) (Gibco, Waltham, MA) in 24-well flat-bottomed plates (1.1 × 106 cells/well). Cells were either left unstimulated or stimulated for 30 minutes at 37°C, 5% CO2 with Dynabeads Human T-Activator CD3/CD28 (Gibco) at a bead to cell ratio of 1:3 after which 5 μg/mL brefeldin A (Sigma-Aldrich) was added to the cell culture and the cells incubated for an additional 6 hours. Subsequently, cells were surface stained with fluorochrome-conjugated antibodies against TCRαβ (APC/Cy7; IP26), CD4 (PerCP/Cy5.5; OKT4), CD127 (BV421; A01905), CD25 (PE; M-A251), PD-1 (BV605; EH12.2H7), and CXCR5 (PE/Cy7; J252D4) all from BioLegend. Cells were then fixed, permeabilized (fixation and permeabilization wash buffer from BioLegend), and stained intracellularly with fluorochrome-conjugated antibodies specific for IFN-γ (APC; B27), IL-17 (AF488; BL168), IL-4 (AF488; MP4-25D2), and IL-21 (AF647; 3A3-N2) or corresponding isotypes from BioLegend, and CXCL10 (AF488; 33036), CXCL13 (APC; 53610) or corresponding isotypes from R&D systems. Data were acquired on a FACS Canto II flow cytometer, and data analysis was performed using FlowJo software.
Single-Molecule Array
CXCL13 concentrations were measured in cryopreserved CSF from 25 patients with RRMS, 17 patients with PPMS, and 10 symptomatic controls. Samples were thawed on ice, and CXCL13 was measured by single-molecule array (Simoa) using the commercially available CXCL13 Discovery Assay kit (Cat. No. 102635) from Quanterix (Billarica, MA) and an SR-X instrument (Quanterix). All samples were measured in duplicates with a mean intra-assay CV of 4.9%.
Blood-Brain Barrier Assay
The membranes of transwell inserts (CellQuart 12-well cell culture inserts with 3.0 µm pore polyester membrane; SABEU, Northeim, Germany) were coated on both sides with 20 µg/mL human fibronectin (Sigma, Merck, MO) at 37°C, 5% CO2 for 2 hours. Fibronectin was then removed, and the transwells were left to dry for 45 minutes. Hereafter, the transwells were inverted, and 106.000 human brain astrocyte (95,000 astrocytes/cm2, Gibco, ThermoFisher) in astrocyte growth medium (Gibco, ThermoFisher) applied to the abluminal side of the membranes and incubated at 37°C, 5% CO2 for 3 hours. The transwell inserts were then reverted into the normal position, and warm astrocyte growth medium was added to the wells and the luminal side of the transwell insert and further incubated for 3 days at 37°C, 5% CO2. All astrocyte growth medium was then carefully removed not to disturb the abluminal layer of astrocytes, and 106,000 human brain microvascular endothelial cells (HBMEC; 95.000 HBMEC/cm2, PeloBiotech, Martinsried, Germany) in HBMEC medium (PeloBiotech) applied to the luminal side of the transwell inserts. HBMEC medium was added to the wells and incubated for an additional 2 days, where astrocytes on the abluminal side and HBMEC on the luminal side of the insert reached a confluent monolayer. The quality of the coculture was monitored by measuring the transendothelial electrical resistance (TEER) value. Transmigration assay was performed when a stable TEER-value of approximately 40 Ω × cm2 was reached when background was subtracted.
To induce inflammation of the in vitro blood-brain barrier (BBB), 100 U/mL TNF-α and IFN-γ (R&D systems) in HBMEC medium were added to the wells 24 hours before starting the transmigration assay. After this, the luminal side of the transwell membrane was carefully emptied and the transwell moved to a new 12-well plate. 700,000 T cells in RPMI/2% B-27 (Gibco, ThermoFisher) from 12 treatment-naive patients with RRMS and 12 healthy controls were transferred to the luminal side of the membrane. T cells were purified from thawed PBMCs using a human T-cell isolation kit from STEMCELL Technologies (Vancouver, Canada). RPMI/2% B-27 with or without 1,000 ng/mL CXCL13 (BioLegend) and 1,000 ng/mL CXCL10 (R&D Systems) were added to the wells and incubated for 5 hours at 37°C, 5% CO2. As a control 700,000 T cells were additionally added to a well without a transwell insert. After migration, a standardized number of flow-count fluorophores (Beckman Coulter, CA) were added to each well containing either control T cells or migrated T cells. Migrated cells plus flow-count fluorophores were harvested and stained for flow cytometry, as previously described using fluorochrome-conjugated antibodies against CD3 (APC/Cy7; UCHT1), CD4 (PerCP/Cy5.5; OKT4), CD127 (APC; A01905), CD25 (PE; M-A251), PD-1 (BV605; EH12.2H7), and CXCR5 (AF488; J252D4) all from BioLegend, and the number and percentage of migrated cells calculated
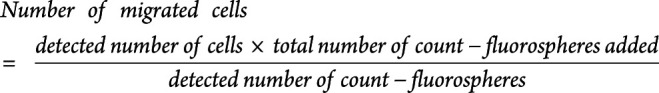 |
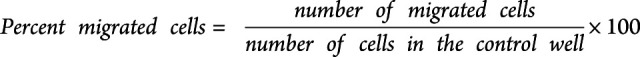 |
Statistics
Statistical analyses were performed using GraphPad Prism 7 software (GraphPad software Inc, La Jolla, CA). The Kruskal-Wallis test followed by a post hoc Dunn multiple comparison test to compare T-cell subpopulations between healthy controls, symptomatic controls, and patients with RRMS and PPMS in blood and CSF. The Wilcoxon paired signed rank test was applied for comparison of T-cell subpopulations between blood and CSF of symptomatic controls, patients with RRMS, and patients with PPMS, respectively. The Mann-Whitney test was applied for comparison of T-cell cytokines, adhesion molecules, chemokine receptors, and migration capacity between healthy controls and patients with RRMS. Correlations were assessed by Spearman rank correlation analysis. A p-value < 0.005 was considered statistically significant and <0.05 as suggestive of significance.25
STROBE Guidelines
For this article, the STROBE reporting guidelines for observational studies was used.26
Data Availability
Data are available in anonymized form and can be shared by request form any qualified investigator. Sharing requires approval of a data transfer agreement by the Danish Data Protection Agency.
Results
Two Phenotypically Different Tfh Cell Populations
In this study, we defined Tfh cells as CD4+CD127+CXCR5+ T cells and their corresponding regulatory counterpart, Tfr cells as CD4+CD127−CD25hiCXCR5+ T cells. Furthermore, we defined active Tfh and Tfr cells as PD-1+ (Figure 1A). Using this gating strategy, a CD25− and a CD25int Tfh cell population became evident (Figure 1A). To investigate a possible phenotypical difference between CD25− and CD25int Tfh cells, various surface markers on freshly isolated PBMCs from 12 healthy controls were measured by flow cytometry. This showed that approximately half of the CD25− Tfh cell population were naive cells (CD45RA+CCR7+), and half were central memory cells (CD45RA−CCR7+) in contrast to CD25int Tfh cells representing a population primarily of central memory T cells (Figure 1B, Table 1). In addition, a lower frequency of CD25− Tfh cells expressed the chemokine receptor CCR2 and suggestively significantly lower frequency of the adhesion molecule MCAM-1 compared with CD25int Tfh cells (Table 1). Similar results were found for PD-1+ Tfh cells (Table 1). We also examined the expression of the early activation marker CD69, the late activation marker HLA-DR, the adhesion molecule CD49d, a part of the VLA-4 molecule, and the chemokine receptor CCR4 and SLAMF5 involved in Tfh-B-cell interaction on Tfh cells. However, no differences were observed between the 2 Tfh subsets (Table 1).
Figure 1. Two Phenotypically Different Tfh Cell Populations.

(A–C) Gating strategy of Tfh and Tfr cells. A.a-c: Gating strategy includes a CD3/CD4 gate in a dot plot of single-cell lymphocytes (A.a), a CD127/CD25 dot plot for gating of CD127+CD25−, CD127+CD25int, and CD127-CD25hi (A.b), and a PD-1/CXCR5 dot plot for Tfh and Tfr cells (A.c). B.a-c: Flow cytometry dot plot gating example of the 3 differentiation stages of Tfh cells in blood (B.a); naive (CD45RA + CCR7+), central memory (CM) (CD45RACCR7+), effector memory (EM) (CD45RACCR7-) and pie charts showing the distribution of naive, CM, and EM cells within CD25− Tfh (B.b) and CD25int Tfh (B.c) cell compartments. C.a-c: Dot plot gating example of the 3 Tfh subpopulations in blood: Tfh1 (CCR6-CXCR3+), Tfh17 (CCR6+CXCR3-), and Tfh17.1 (CCR6+CXCR3+) (C.a) and pie charts showing the distribution of Tfh1, Tfh17, and Tfh17.1 cells within CD25− Tfh (C.b) and CD25int Tfh (C.c) cell compartments. D.a-b: Frequencies of CD25− Tfh and CD25int Tfh cells producing IFN-γ and IL-17 (D.a), and IL-4 and CXCL10 (D.b). Tfh = follicular helper T; Tfr = follicular regulatory T cells.
Table 1.
Tfh Subpopulation Phenotyping

We next examined the expression of CXCR3 and CCR6 on CD25− and CD25int Tfh cells, dividing them into Tfh1 (CXCR3+CCR6−), Tfh17 (CXCR3−CCR6+), and Tfh17.1 (CXCR3+CCR6+) subpopulations representing different T-helper functions.12-14 This showed that CD25− and PD-1+CD25− Tfh cells significantly represented the Tfh1 phenotype and that CD25int and PD-1+CD25int Tfh cells significantly represented the Tfh17 phenotype. The Tfh17.1 phenotype was suggestively significantly higher in PD-1+CD25int compared with PD-1+CD25− Tfh cells (Figure 1C, Table 1).
Elucidating a potential functional difference between CD25− and CD25int Tfh cells, we assessed cytokine production of cryopreserved PBMCs from 12 healthy controls stimulated with anti-CD3/CD28 beads. This showed no difference of CXCL13 and IL-21 production between Tfh cell subpopulations. However, IFN-γ and CXCL10 production was significantly higher in CD25− and PD-1+CD25− Tfh cells, whereas IL-17 and IL-4 production was significantly higher in CD25int and PD-1+CD25int Tfh cells (Figure 1D, Table 1). See eFigure 1, links.lww.com/NXI/A733 for cytokine and chemokine gating examples.
Minor Differences of Tfh and Tfr Cells in the Blood of Patients With MS and Controls
To explore a possible implication of Tfh cells in the pathogenesis of MS, we compared the prevalence of peripheral Tfh and Tfr cells between patients with RRMS, patients with PPMS, symptomatic controls, and healthy controls. This showed no difference in either of the peripheral Tfh or Tfr cell populations between groups, except a suggestively significant increase in peripheral CD25− Tfh cells in patients with RRMS compared with healthy controls and a suggestive increase in PD-1+CD25int Tfh cells in blood from patients with PPMS compared with healthy controls (p < 0.05; Figure 2, A–C). In addition, we analyzed absolute counts of blood Tfh and Tfr cells and found no differences between groups, except a lower number of CD25− Tfh cells in blood from patients with PPMS compared with RRMS (eFigure 2, links.lww.com/NXI/A733). We further investigated the Tfh/Tfr cell ratio in the blood and found no difference between the groups analyzed (data not shown).
Figure 2. Enrichment of Tfh and Tfr Cells in the CSF of Patients With MS.

The prevalence of CD25− Tfh cells (A.a) and PD-1+ CD25− Tfh cells (A.b), CD25int Tfh cells (B.a) and PD-1+ CD25int Tfh cells (B.b), and Tfr cells (C.a) and PD-1+ Tfr cells (C.b), in blood and CSF from patients with RRMS, patients with PPMS, symptomatic controls (SC), and healthy controls (HC). The mean value is shown for all groups analyzed. ***p < 0.0001; **p < 0.005; *p < 0.05. MS = multiple sclerosis; PPMS = progressive MS; RRMS = relapsing-remitting MS; Tfh = follicular helper T; Tfr = follicular regulatory T cells.
Enrichment of Tfh and Tfr Cells in the CSF of Patients With MS
Next, we measured the prevalence of Tfh and Tfr cells in the CSF of patients with RRMS, patients with PPMS, and symptomatic controls. This showed enrichment of CD25− Tfh cells in CSF compared with blood in symptomatic controls and patients with RRMS and PPMS (p < 0.0001; Figure 2A). The same was observed for PD-1+CD25− Tfh cells in patients with RRMS and PPMS (p < 0.0001; Figure 2A). By contrast, we found a reduced frequency of CD25int and PD-1+CD25int Tfh cells in the CSF of symptomatic controls (p < 0.05) and patients with PPMS (p < 0.0001; p < 0.005) compared with blood (Figure 2B).
As observed in symptomatic controls, we found an enrichment of CD25− and PD-1+CD25− Tfh cells in the CSF compared with blood in the exploratory cohort of 10 healthy controls (eFigure 3A, links.lww.com/NXI/A733). The frequencies of CD25int Tfh and Tfr cells were reduced in CSF compared with blood in healthy controls, whereas frequencies of PD-1+CD25int Tfh and PD-1+ Tfr were similar in blood and CSF (eFigure 3B,C, links.lww.com/NXI/A733).
Comparing frequencies of intrathecal Tfh cells between groups showed an increased frequency of CD25− and PD-1+CD25− Tfh cells in patients with RRMS (p < 0.0001; p < 0.05) and PPMS (p < 0.05) compared with symptomatic controls (Figure 2A). Comparing CD25int and PD-1+CD25int Tfh cells between groups only showed a suggestive increase in the CSF of patients with RRMS compared with symptomatic controls (p < 0.05; Figure 2B).
These observations indicate changes in the intrathecal Tfh dynamics, particularly in patients with RRMS. This was further supported by the observation of an increased intrathecal frequency of Tfr and PD-1+ Tfr cells in patients with RRMS compared with symptomatic controls (p < 0.005, p < 0.05; Figure 2C). Conversely, the frequency of Tfr cells was decreased in CSF compared with blood in patients with PPMS and symptomatic controls (p < 0.0001; Figure 2C), but not in patients with RRMS. No differences in intrathecal Tfh/Tfr cell ratios between groups were found (data not shown).
Intrathecal CD25− Tfh Cells Correlate With the IgG Index
We next considered a possible association between intrathecal Tfh cells and the IgG index. This showed that high levels of IgG index scaled with increased frequencies of CD25− Tfh cells (p = 0.025; Figure 3A), PD-1+CD25− Tfh cells (p = 0.012; Figure 3B), and the PD-1+CD25− Tfh/Tfr cell ratio (p = 0.031; Figure 3C) of patients with RRMS.
Figure 3. IgG Index Correlate With Tfh and Tfr Cells and Tfh/Tfr Cell Ratio.

(A) Correlation between IgG index and percentage of CD25− Tfh cells in the CSF from patients with RRMS. (B) Correlation between IgG index and percentage of PD-1+CD25− Tfh cells in the CSF from patients with RRMS. (C) Correlation between IgG index and PD-1+CD25− Tfh/Tfr ratio in CSF from patients with RRMS. RRMS = relapsing-remitting MS; Tfh = follicular helper T; Tfr = follicular regulatory T cells.
Intrathecal Counts of Tfh and Tfr Cells Correlate With CXCL13 CSF Concentration in RRMS
Considering the pronounced CSF enrichment of Tfh and Tfr cells in patients with RRMS, we analyzed a possible association between Tfh and Tfr cell count and the CSF concentration of the chemoattractant CXCL13. CSF levels of CXCL13 were significantly higher in patients with RRMS (median 9.45 pg/mL, interquartile range (IQR) 6.3 pg/mL) than in symptomatic controls (median 0.5 pg/mL, IQR 0.19 pg/mL, p < 0.0001), and suggestively higher in patients with PPMS (median 3.6 pg/mL, IQR 3 pg/mL) than in SC (p = 0.01). Furthermore, we found that in patients with RRMS, CSF CXCL13 concentrations scaled with total counts of all the Tfh and Tfr cell subsets studied in CSF (Figure 4, A–C).
Figure 4. CXCL13 CSF Concentrations Correlate With Intrathecal Tfh and Tfr Cell Counts.

Correlation between CXCL13 CSF concentrations and total CSF counts of CD25− Tfh cells (A.a) and PD-1+ CD25− Tfh cells (A.b), CD25int Tfh cells (B.a) and PD-1+ CD25int Tfh cells (B.b), and Tfr cells (C.a) and PD1+ Tfr cells (C.b) from patients with RRMS. RRMS = relapsing-remitting MS; Tfh = follicular helper T; Tfr = follicular regulatory T cells.
Equal Migration Capacity of Tfh Cells From Healthy Controls and Patients With RRMS
We next investigated the CNS migratory potential of peripheral Tfh cells from patients with RRMS and healthy controls. For this, we applied purified T cells to an in vitro BBB coculture assay of primary human astrocytes and primary HBMEC grown in a Boyden chamber. In this experiment, T cells were left to migrate on both a noninflamed and an inflamed BBB (prestimulated with TNF-α and IFN-γ for 24 hours) using the chemokines CXCL13 and CXCL10 as chemoattractants. This showed no difference in absolute number and frequencies of migrated CD25−, CD25int, PD-1+CD25−, or PD-1+CD25int Tfh cells between healthy controls and patients with RRMS in any of the BBB assay conditions applied (Figure 5, A–D).
Figure 5. Equal In Vitro Migration of Tfh Cells From Patients With RRMS and Healthy Controls.

Absolute number and frequency of CD25− Tfh cells (A.a-b), CD25int Tfh cells (B.a-b), PD-1+ CD25− Tfh cells (C.a-b), and PD-1+ CD25int Tfh cells (D.a-b) migrated across an in vitro human BBB either noninflamed (-TNF-α/IFN-γ) or inflamed (+TNF-α/IFN-γ) and either with or without a chemoattractant (+/− CXCL13/CXCL10) applied to the assay. Compared cells are from patients with RRMS and healthy controls (HC). The mean value is shown for all groups analyzed. RRMS = relapsing-remitting MS; Tfh = follicular helper T.
Discussion
In this study, we investigated the involvement of Tfh cells in MS. Several studies have reported dysregulated levels of Tfh and Tfr cells in blood from patients with MS suggesting their importance in disease pathogenesis.6,17,27 However, there are discrepancies between studies reporting both elevated and similar frequencies of circulating Tfh cells in patients with RRMS compared with healthy controls.10,14,15,28 This may at least partially relate to differing definitions of Tfh cells.
In this study, we considered that the definition of CD4+ Tfh cells based exclusively on CXCR5 expression includes a fraction of T cells with regulatory functions. Therefore, to separate effector Tfh cells (CD127+) from the regulatory Tfr cells (CD127−CD25hi), we included the markers CD127 and CD25. Thorough phenotyping of CD4+CD127+CXCR5+ Tfh cells revealed 2 distinct Tfh populations: a CD25− and a CD25int Tfh population. These were further subdivided according to their activation status based on PD-1 expression. The CD25− Tfh cell population was composed of approximately 45% naive and 45% central memory T cells, while CD25int Tfh cells were mainly central memory T cells, suggesting that CD25int Tfh cells to a greater extent represent an antigen-primed subset of Tfh cells.
Proportional differences of the Tfh subsets Tfh1, Tfh17, and Tfh17.1, defined according to the expression of the chemokine receptors CXCR3 and CCR6, have been associated with autoimmunity.12-14 Tfh17 cells were described as efficient inducers of naive B-cell production of IgG and IgA in contrast to Tfh1 lacking the capacity to help B cells.12 Here, we found that CD25− and PD-1+CD25− Tfh cells were enriched for Tfh1 phenotype cells producing significantly higher levels of IFN-γ and CXCL10 than CD25int and PD-1+CD25int Tfh cells, which conversely expressed a Tfh17 phenotype producing higher levels of IL-17 and the Th2 cytokine IL-4. The observation that CD25int Tfh cells represent a Tfh17 phenotype was further supported by our findings of a significant increase in expression of the chemokine receptor CCR2 and the adhesion molecule MCAM-1 on CD25int Tfh cells, both predominantly expressed on Th17 cells.29-32
In this study, we considered the implication of Tfh cells in MS pathogenesis. First, we assessed the prevalence of the 4 Tfh populations in blood from healthy controls, symptomatic controls, and patients with RRMS and PPMS. In contrast to previous studies, we found no differences in the peripheral Tfh cell populations or their regulatory counterpart Tfr cells, between groups except for a suggestively significant increase in CD25− Tfh cells in patients with RRMS compared with healthy controls and a suggestive increase in PD-1+CD25int Tfh cells in blood from patients with PPMS compared with healthy controls. Circulating blood Tfr cells were previously observed to be significantly reduced in patients with RRMS compared with healthy controls and also had a clearly reduced suppressive function.33,34 However, we found no difference in frequencies of circulating Tfr cells between groups.
A single-cell RNA sequencing study recently reported increased frequencies of Tfh cells in CSF from a small group of patients with RRMS compared with healthy controls, suggesting these as key mediators of B-cell recruitment and expansion in CSF.35 In agreement, we found a significant CSF enrichment of CD25− Tfh cells compared with blood in healthy and symptomatic controls and patients with RRMS and PPMS. Similarly, we found a significant CSF enrichment of PD-1+CD25− Tfh cells, however only in patients with RRMS and PPMS and healthy controls. When comparing Tfh cells in CSF, we found that both CD25− and PD-1+CD25− Tfh cells were significantly increased in patients with MS compared with symptomatic controls. Therefore, our data indicate an increased presence of CD25− Tfh cells in the CSF of patients with MS in contrast to CD25int Tfh cells. The increase in CSF CD25− Tfh1 cells may be due to their expression of CXCR3, which is notoriously linked to cell recruitment to CXCL10-expressing tissue, as the CSF of patients with MS. The observed low abundance of CD25int Tfh17 cells in the CSF may be due to diminished recruitment of this antibody-promoting Tfh subset from the blood or result from their migration into the meninges, the center of Tfh:B-cell interaction, or into the brain parenchyma. Where CD25int Tfh17 cells likely participate in MS pathogenesis through B-cell activation and antibody production, the importance of the observed increased frequency and activity (PD1+) of intrathecal CD25− Tfh1 cells is more speculative. CD25− Tfh1 cells may contribute to MS pathogenesis through B-cell recruitment in response to their CXCL13 production or through microglia activation in response to their IFN-γ production. CD25− Tfh1 cells and particularly PD-1+CD25− Tfh cells produce high amounts of IFN-γ, a cytokine that enhances the production of superoxides and expression of FcR on microglia cells.36 Microglia are a population of cells in the CNS considered immune guards capable of performing a potent inflammatory response.37 In patients with MS, microglia increase their expression of FcγR, receptors that detect and bind IgG antibodies. FcγR:IgG binding on microglia leads to a release of inflammatory mediators, oxidative burst, phagocytosis, neurotoxicity, and regulation of B-cell activation and antibody production.36 It is possible that CD25− Tfh1 and CD25int Tfh17 cells cooperate in B-cell activation and nervous tissue damage. This hypothesis is supported by our finding of a correlation between intrathecal CD25− and PD-1+CD25− Tfh1 cells and the IgG index.
Considering that the Tfh cells were increased in CSF and that this was more pronounced in patients with RRMS compared with symptomatic controls, we investigated whether the Tfh cells of patients with RRMS had a greater potential to migrate across a human in vitro BBB. We found that Tfh cells of patients with RRMS and healthy controls migrated equally well across the BBB when exposed to the same conditions. This suggests that the enrichment of Tfh cells observed in vivo in the CSF might depend on the concentration of intrathecal chemoattractants. Previous studies have shown increased concentrations of the CXCR5 and CXCR3 chemokine ligands CXCL13 and CXCL10 in CSF from patients with MS compared with healthy controls.18-21,38,39 We confirm significantly higher CXCL13 CSF levels, especially in patients with RRMS, and furthermore, we found that CXCL13 levels correlated with intrathecal counts of Tfh and Tfr cell subsets. Collectively, these data indicate that the enhanced recruitment of both Tfh and Tfr cells to the CSF observed in patients with RRMS likely is a result of the increased concentrations of CXCL13 rather than an increased migratory potential of Tfh and Tfr cells.
With this study, we investigated the role of Tfh cells in the pathogenesis of MS. We provide evidence that Tfh cells are related to the MS-associated intrathecal inflammation. We found enrichment of Tfh cells in symptomatic controls; however, this was even more pronounced in patients with MS. Despite increased frequencies of Tfh cells in CSF, we found no differences in the migratory capacity between Tfh cells from patients with RRMS and healthy controls. Instead, the increased number of CSF Tfh cells in patients with MS might be attributed to increased CXCL13 concentrations in the CSF. In addition, we found correlations between IgG index and frequencies of CSF-resident CD25− Tfh cells as well as CD25− Tfh/Tfr ratio in patients with RRMS, suggesting a dysregulated Tfr function important for the Tfh activity. These findings suggest that Tfh cells possess significant potential for migration across the BBB and that they are likely implicated in the intrathecal antibody production observed in MS.
Acknowledgment
The authors highly acknowledge Lisbeth Stolpe for her excellent technical assistance.
Glossary
- BBB
blood-brain barrier
- HBMEC
human brain microvascular endothelial cells
- IgG
immunoglobulin G
- IFN
interferon
- IL
interleukin
- MCAM-1
melanoma cell adhesion molecule 1
- MS
multiple sclerosis
- PBMCs
peripheral blood mononuclear cells
- PD-1
programmed cell death protein 1
- PPMS
progressive MS
- RPMI
Roswell Park Memorial Institute medium
- RRMS
relapsing-remitting MS
- TEER
transendothelial electrical resistance
- Tfh
follicular helper T
- Tfr
follicular regulatory T cells
Appendix. Authors
Contributor Information
Jacob Talbot, Email: jacob.lando.talbot@regionh.dk.
Helene Højsgaard Chow, Email: helene.hoejsgaard.chow@regionh.dk.
Malene Bredahl Hansen, Email: malene.bredahl.hansen.01@regionh.dk.
Sophie Buhelt, Email: sophie.buhelt.01@regionh.dk.
Sebastian Herich, Email: s.herich@yahoo.de.
Nicholas Schwab, Email: nicholas.schwab@ukmuenster.de.
Marie Nathalie Nickelsen Hellem, Email: marie.nathalie.nickelsen.hellem@regionh.dk.
Joergen Erik Nielsen, Email: joergen.erik.nielsen.01@regionh.dk.
Finn Sellebjerg, Email: sellebjerg@dadlnet.dk.
Marina Rode von Essen, Email: marina.rode.von.essen@regionh.dk.
Study Funding
This study was supported by the Aase and Ejner Danielsen foundation and the Carl and Ellen Hertz' grant for Danish medical and nature science. Parts of this study were funded by the German Research Council (DFG) through SFB-CRC TR128 ‟Initiating/Effector vs Regulatory Mechanisms in Multiple Sclerosis—Progress towards Tackling the Disease,” project B01 to N.S.
Disclosure
R. Holm Hansen declares no conflict of interest; J. Talbot reports non-financial support from Biogen and Sanofi Genzyme outside the submitted work; H. Højsgaard Chow reports non-financial support from Merck, non-financial support from Teva, non-financial support from Biogen, and non-financial support from Roche, outside the submitted work; M. Bredahl Hansen declares no conflict of interest; S. Herich declares no conflict of interest; N. Schwab received travel support from Biogen and Novartis, as well as research support from Biogen and Roche outside the submitted work; S. Buhelt declares no conflict of interest; M.N. Nickelsen Hellem declares no conflict of interest; J.E. Nielsen declares no conflict of interest; F. Sellebjerg has served on scientific advisory boards for, served as consultant for, received support for congress participation or received speaker honoraria from Alexion, Biogen, Bristol Myers Squibb, Merck, Novartis, Roche, and Sanofi Genzyme and his laboratory has received research support from Biogen, Merck, Novartis, Roche, and Sanofi Genzyme; M. Rode von Essen declares no conflict of interest. Go to Neurology.org/NN for full disclosures.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502-1517. doi: 10.1016/S0140-6736(08)61620-7 [DOI] [PubMed] [Google Scholar]
- 2.Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15(9):545-558. doi: 10.1038/nri3871 [DOI] [PubMed] [Google Scholar]
- 3.Disanto G, Morahan JM, Barnett MH, Giovannoni G, Ramagopalan SV. The evidence for a role of B cells in multiple sclerosis. Neurology. 2012;78(11):823-832. doi: 10.1212/WNL.0b013e318249f6f0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol. 2012;8(6):337-347. doi: 10.1038/nrrheum.2012.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinstein JS, Herman EI, Lainez B, et al. TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol. 2016;17(10):1197-1205. doi: 10.1038/ni.3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X, Ge R, Chen H, et al. Follicular helper CD4(+) T cells, follicular regulatory CD4(+) T cells, and inducible costimulator and their roles in multiple sclerosis and experimental autoimmune encephalomyelitis. Mediators Inflamm. 2021;2021:2058964. doi: 10.1155/2021/2058964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linterman MA, Pierson W, Lee SK, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17(8):975-982. doi: 10.1038/nm.2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol. 2010;11(6):535-542. doi: 10.1038/ni.1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H. PD-1 controls follicular T helper cell positioning and function. Immunity. 2018;49(2):264-274.e4. doi: 10.1016/j.immuni.2018.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puthenparampil M, Zito A, Pantano G, et al. Peripheral imbalanced TFH/TFR ratio correlates with intrathecal IgG synthesis in multiple sclerosis at clinical onset. Mult Scler. 2019;25(7):918-926. doi: 10.1177/1352458518779951 [DOI] [PubMed] [Google Scholar]
- 11.Dong L, He Y, Cao Y, et al. Functional differentiation and regulation of follicular T helper cells in inflammation and autoimmunity. Immunology. 2021;163(1):19-32. doi: 10.1111/imm.13282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morita R, Schmitt N, Bentebibel SE, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34(1):108-121. doi: 10.1016/j.immuni.2010.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cunill V, Clemente A, Lanio N, et al. Follicular T cells from smB(-) common variable immunodeficiency patients are skewed toward a Th1 phenotype. Front Immunol. 2017;8:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunill V, Massot M, Clemente A, et al. Relapsing-remitting multiple sclerosis is characterized by a T follicular cell pro-inflammatory shift, reverted by dimethyl fumarate treatment. Front Immunol. 2018;9:1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo J, Zhao C, Wu F, et al. T follicular helper-like cells are involved in the pathogenesis of experimental autoimmune encephalomyelitis. Front Immunol. 2018;9:944. doi: 10.3389/fimmu.2018.00944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gharibi T, Hosseini A, Marofi F, et al. IL-21 and IL-21-producing T cells are involved in multiple sclerosis severity and progression. Immunol Lett. 2019;216:12. doi: 10.1016/j.imlet.2019.09.003 [DOI] [PubMed] [Google Scholar]
- 17.Romme Christensen J, Bornsen L, Ratzer R, et al. Systemic inflammation in progressive multiple sclerosis involves follicular T-helper, Th17- and activated B-cells and correlates with progression. PLoS One. 2013;8(3):e57820. doi: 10.1371/journal.pone.0057820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwanowski P, Losy J, Kramer L, Wojcicka M, Kaufman E. CXCL10 and CXCL13 chemokines in patients with relapsing remitting and primary progressive multiple sclerosis. J Neurol Sci. 2017;380:22-26. doi: 10.1016/j.jns.2017.06.048 [DOI] [PubMed] [Google Scholar]
- 19.Khademi M, Kockum I, Andersson ML, et al. Cerebrospinal fluid CXCL13 in multiple sclerosis: a suggestive prognostic marker for the disease course. Mult Scler. 2011;17(3):335-343. doi: 10.1177/1352458510389102 [DOI] [PubMed] [Google Scholar]
- 20.Krumbholz M, Theil D, Cepok S, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain. 2006;129(pt 1):200-211. doi: 10.1093/brain/awh680 [DOI] [PubMed] [Google Scholar]
- 21.Novakova L, Axelsson M, Malmestrom C, et al. NFL and CXCL13 may reveal disease activity in clinically and radiologically stable MS. Mult Scler Relat Disord. 2020;46:102463. doi: 10.1016/j.msard.2020.102463 [DOI] [PubMed] [Google Scholar]
- 22.McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50(1):121-127. doi: 10.1002/ana.1032 [DOI] [PubMed] [Google Scholar]
- 23.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173. doi: 10.1016/S1474-4422(17)30470-2 [DOI] [PubMed] [Google Scholar]
- 24.Teunissen C, Menge T, Altintas A, et al. Consensus definitions and application guidelines for control groups in cerebrospinal fluid biomarker studies in multiple sclerosis. Mult Scler. 2013;19(13):1802-1809. doi: 10.1177/1352458513488232 [DOI] [PubMed] [Google Scholar]
- 25.Amrhein V, Greenland S, Johannesson M, et al. Redefine statistical significance. Nat Hum Behav. 2018;2(1):6. doi: 10.1038/s41562-017-0189-z [DOI] [PubMed] [Google Scholar]
- 26.von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP, STROBE Initiative. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet. 2007;370(9596):1453-1457. doi: 10.1016/S0140-6736(07)61602-X [DOI] [PubMed] [Google Scholar]
- 27.Qiu H, Wu H, Chan V, Lau CS, Lu Q. Transcriptional and epigenetic regulation of follicular T-helper cells and their role in autoimmunity. Autoimmunity. 2017;50(2):71-81. doi: 10.1080/08916934.2017.1284821 [DOI] [PubMed] [Google Scholar]
- 28.Haque R, Kim Y, Park K, et al. Altered distributions in circulating follicular helper and follicular regulatory T cells accountable for imbalanced cytokine production in multiple sclerosis. Clin Exp Immunol. 2021;205(1):75-88. doi: 10.1111/cei.13596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192(6):899-905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuziel WA, Morgan SJ, Dawson TC, et al. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci U S A. 1997;94(22):12053-12058. doi: 10.1073/pnas.94.22.12053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider-Hohendorf T, Rossaint J, Mohan H, et al. VLA-4 blockade promotes differential routes into human CNS involving PSGL-1 rolling of T cells and MCAM-adhesion of TH17 cells. J Exp Med. 2014;211(9):1833-1846. doi: 10.1084/jem.20140540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.von Essen MR, Hellem MNN, Vinther-Jensen T, et al. Early intrathecal T helper 17.1 cell activity in Huntington disease. Ann Neurol. 2020;87(2):246-255. doi: 10.1002/ana.25647 [DOI] [PubMed] [Google Scholar]
- 33.Schwarz A, Schumacher M, Pfaff D, et al. Fine-tuning of regulatory T cell function: the role of calcium signals and naive regulatory T cells for regulatory T cell deficiency in multiple sclerosis. J Immunol. 2013;190(10):4965-4970. doi: 10.4049/jimmunol.1203224 [DOI] [PubMed] [Google Scholar]
- 34.Dhaeze T, Peelen E, Hombrouck A, et al. Circulating follicular regulatory T cells are defective in multiple sclerosis. J Immunol. 2013;195(3):832-840. doi: 10.4049/jimmunol.1500759. [DOI] [PubMed] [Google Scholar]
- 35.Schafflick D, Xu CA, Hartlehnert M, et al. Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nat Commun. 2020;11(1):247. doi: 10.1038/s41467-019-14118-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pryce G, Baker D. Oligoclonal bands in multiple sclerosis; Functional significance and therapeutic implications. Does the specificity matter? Mult Scler Relat Disord. 2018;25:131-137. doi: 10.1016/j.msard.2018.07.030 [DOI] [PubMed] [Google Scholar]
- 37.Gogoleva VS, Drutskaya MS, Atretkhany KSN. The role of microglia in the homeostasis of the central nervous system and neuroinflammation [in Russian]. Mol Biol (Mosk). 2019;53(5):790-798. doi: 10.1134/S0026898419050057 [DOI] [PubMed] [Google Scholar]
- 38.Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci U S A. 1999;96(12):6873-6878. doi: 10.1073/pnas.96.12.6873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hassanshahi G, Roohi MA, Esmaeili SA, Pourghadamyari H, Nosratabadi R. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. 1999;103(6):807. doi: 10.1172/JCI5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available in anonymized form and can be shared by request form any qualified investigator. Sharing requires approval of a data transfer agreement by the Danish Data Protection Agency.