

Keywords: cytokines, organ failure, severe pancreatitis, systemic inflammatory response syndrome
Abstract
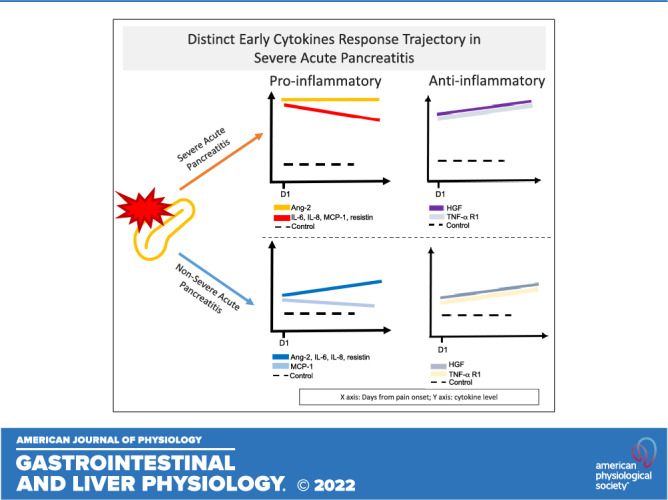
Severe acute pancreatitis (SAP) is associated with substantial morbidity and mortality. Several cytokines have been identified to have pathophysiological significance in SAP, but studies characterizing their early trajectories are lacking. Here we characterize the early trajectories of seven key cytokines associated with SAP and compare them with non-SAP subjects. Five proinflammatory cytokines (angiopoietin-2, interleukin-6, interleukin-8, monocyte chemoattractant protein-1, resistin) and two anti-inflammatory cytokines (hepatocyte growth factor, and soluble tumor necrosis factor-α receptor-1A) were measured in a prospective cohort of acute pancreatitis subjects (2012–2016) at the time of enrollment and then every 24 h for 5 days or until discharge. The cytokines’ levels and trajectories were calibrated based on date of pain onset and were compared between healthy controls and three severity categories (mild, moderate, and severe). The cohort (n = 170) consisted of 27 healthy controls, 65 mild, 38 moderate, and 40 SAP. From day 1 of symptom onset, SAP subjects exhibited significantly higher levels of both pro- and anti-inflammatory cytokines compared with non-SAP and healthy subjects. But in SAP subjects, all proinflammatory cytokines' levels trended downward after day 2 (except for a flat slope for angiopoeitin-2) whereas for non-SAP subjects, the trajectory was upward: this trajectory difference between SAP versus non-SAP subjects resulted in narrowing of the differences initially seen on day 1 for proinflammatory cytokines. For anti-inflammatory cytokines, the trajectories were uniformly upward for both SAP and non-SAP subjects. Proinflammatory cytokine response is an early and time-sensitive event in SAP that should be accounted for when designing future biomarker studies and/or therapeutic trials.
NEW & NOTEWORTHY In this study, we showed that the proinflammatory cytokine response in SAP is an early event, with subsequent downregulation of proinflammatory cytokines beginning at day 1 of symptom onset. Our findings underscore the importance of enrolling subjects very early in the disease course when conducting studies to investigate early immune events of SAP; this current study also serves as an important reference for the design of future biomarker studies and therapeutic trials in SAP.
INTRODUCTION
Acute pancreatitis (AP) is a common acute inflammatory condition of the pancreas with significant morbidity and mortality (1, 2). Severe acute pancreatitis (SAP) is defined as persistent organ failure (POF) lasting ≥ 48 h (3, 4). At least 80% of SAP patients requires intensive care unit admission, and up to 30% expire from organ failure (OF) (5, 6). The pathophysiology of SAP is still poorly understood, and there is no effective drug therapy (7). Clinical observations suggest that SAP typically begins with the systemic inflammatory response syndrome (SIRS), (i.e., the “cytokine storm”), followed by capillary leak syndrome and eventual remote dysfunction in one or more of vital organs including the kidneys, lungs, or the heart (8). Therefore, the host’s unabated early immune response to pancreatic injury is a key step in the development of SAP (9).
We and other investigators have previously identified several cytokines1 and chemokines that are strongly associated with SAP (9–11). These include proinflammatory cytokines [angiopoietin-2 (Ang-2), interleukin-6 (IL-6), interleukin-8 (IL-8), monocyte chemoattractant protein-1 (MCP-1), and resistin] and anti-inflammatory cytokines [hepatocyte growth factor (HGF) and sTNF-α receptor 1 A (sTNFα-R1)] among others (12, 13). Each of the cytokines has robust animal model evidence relevant to important pathways in POF, such as endothelial destabilization and permeability (Ang-2 and IL-8) (14), chemotaxis of innate immune cells (IL-8 and MCP-1) (15), amplification of inflammatory response (IL-6 and resistin) (16–18), and suppression of immune response (i.e., anti-inflammatory properties; HGF and sTNFα-R1) (16, 19). Data on cytokine temporal trends have been used for clinical trials and informed mechanistic studies for other acute inflammatory diseases such as severe sepsis and COVID-19 (20–22). But very few investigations focused on the temporal trajectory of these cytokines in human AP, were limited by small sample size (23, 24) and were conducted before the development of a valid AP severity classification system (23, 24). Characterization of temporal trajectories of AP-associated cytokine signatures not only help design immune therapy trials but can serve as an excellent future reference for future biomarker studies in SAP. The aim of this study was to characterize the baseline levels of these cytokines and their trajectories over time in a well-phenotyped cohort of AP subjects and healthy controls.
METHODS
Patient Population
The pancreatitis-associated risk of organ failure (PROOF) is an observational study conducted at the University of Pittsburgh Medical Center enrolling hospitalized subjects with AP. For this study, subjects were enrolled within 24 h of their presentation. Direct admissions or subjects transferred from the outside hospital within 24 h of presentation were included in the study. The PROOF protocol was approved by the Institutional Review Board (IRB) of the University of Pittsburgh (PRO00000496) and submitted to the United States National Library of Medicine (ClinicalTrials.gov Identifier: NCT03075605). The clinical protocol and cohort characteristics have been previously described (11, 25, 26). We followed the prospective-specimen-collection, retrospective-blinded-evaluation approach to accord with the recommended biomarker study design (27).
Detailed demographic, radiographic, laboratory, and clinical data were collected throughout the hospitalization. Serial blood samples were collected from enrollment and for each 24-h period thereafter up to 120 h after admission or discharge as available. Serial healthy control samples were also collected serially for three time points over 4 days (covered by the umbrella IRB). Serum samples were obtained from 170 subjects on predetermined time points, processed within 2 h from collection by an experienced technician in the laboratory of investigators D.C.W. and G.I.P. following a standardized laboratory manual of operations, and then stored at −80°C freezers.
Subjects were classified post hoc by the severity of AP using the Revised Atlanta Criteria (RAC) (6). Categories included mild AP (MAP), moderately severe AP (MSAP), and severe AP (SAP). Defining criterion for SAP is the presence of persistent organ failure [POF: lasting ≥ 48 h; OF defined by the Modified Marshall Scoring System (28)]. The MSAP group included subjects who develop either local (peri)pancreatic complications (i.e., acute fluid collections and/or necrosis) and/or exacerbation of a comorbid condition and/or transient OF (lasting less than 48 h). The MAP group included subjects with none of the above criteria.
Laboratory Methods
The cytokine serum concentrations were measured and analyzed using Meso Scale Discovery (MSD) MULTI-SPOT Assay System (MESO QuickPlex SQ 120 instrument). The biomarkers included Ang-2, HGF, IL-6, IL-8, MCP-1, resistin, and sTNFα-R1. All were measured on three separate assay kits, the V-Plex Human Pro-inflamatory for IL-6, IL-8, and MCP-1 and two custom human duplex kits combining Ang2 and HGF on one, and resistin and sTNFα-R1 on the other, purchased from MSD (Gaithersburg, MD). Samples aliquots were batch analyzed to minimize repeating thawing before analysis and processed per manufacturer’s protocol. With the use of MSD Discovery Workbench analysis software v4.0, standard curves were formed by fitting electrochemiluminescence signal from calibrators to a 4-parameter logistic model with a 1/y2 weighting from which concentrations of test samples were estimated. R2 values for the fitted curves were >0.95 for all markers.
Samples were run in duplicates with median intra-assay variability range being 2.70%–9.56% and median interassay variability range of 7.44%–43.48% for all seven assays. The lowest detected concentrations for cytokines were 9.96 pg/mL (Ang2), 10.4 pg/mL (HGF), 0.024 pg/mL (IL-6), 0.018 pg/mL (IL-8), 0.10 pg/mL (MCP-1), 0.096 pg/mL (Resistin), and 1.75 pg/mL (sTNFα-R1). Of the 469 samples, two samples showed cytokine levels below the detection limits for Ang-2 and HGF and were removed from the analysis. In addition, 19 Ang2 and three IL-8 samples were above the standard curve. All extremely high samples values were obtained from subjects with SAP and were kept in the analysis.
The samples were run in two separate batches approximately 2 yr apart, 28% in 2015 and 72% in 2017. As two of the MSD plates were a custom design and there was a possibility of sample degradation over the intervening time period (29, 30), 42 samples were run in both batches and compared with create a correction factor and minimize any bias due to batch or lot effects. Values above the standard curve were not included for the creation of the correction factor.
At the time of data analysis, we found that the complete run for the original 2015 resistin and sTNFα-R1 levels could not be fully replicated. Thus, all samples (n = 416) were used for measurement of resistin and sTNFα-R1 in a final, combined run with no need for batch correction. All other cytokines were scaled to the 2017 values. The mean ratio and thus correction factors between 2017 and 2015 were as follows: IL-6 = 0.825, IL-8 = 0.909, MCP1 = 1.152, Ang-2 = 0.143, and HGF = 1.545. The correlation coefficient for each comparison was >0.975.
Statistical Analysis
The cytokine trajectories were calibrated based on date of pain onset: date of pain onset was defined as “1” for any measurement up to 24 h from pain onset, and the value was incremented by one for each 24-h period thereafter in our analysis up to day 5. Descriptive statistics are presented as proportions for categorical and as means ± standard deviation or median (interquartile range) for continuous data. Each cytokine distribution was assessed for normality using graphical methods and the Shapiro–Wilks test. Correlation between continuous measures was assessed using Spearman’s correlation. Comparisons in discrete baseline characteristics between the AP subjects and controls were performed using the χ2 test. Comparisons for continuous variables between the AP subjects and controls and within subset of subjects and controls were through the Mann–Whitney U test. To adjust for multiple comparisons, the Hommel’s procedure was adopted for calculating adjusted P values.
Because of the skewness of the cytokine distributions, each cytokine value was transformed using a logarithmic transformation to approximate a normal distribution. Cytokine time courses were modeled using a linear mixed model with random intercepts for each subject and fixed effects controlling for age, sex, and body mass index (BMI). An interaction term between time and severity was used to compare the different trajectories based on severity as defined by the RAC (6). Visual inspection of residual plots did not reveal any obvious deviations from homoscedasticity or normality. P values were obtained by likelihood ratio tests of the full model with the effect in question against the model without the effect in question. All statistical tests were run using R and the lme4 package to perform a linear mixed effects analysis (31).
RESULTS
Baseline Characteristics
A total of 170 subjects consisting of 27 healthy controls and 143 AP subjects provided 471 samples for this study. The AP cohort included 65 MAP, 38 MSAP, and 40 SAP subjects (Table 1). The exact number of subjects and samples for each cytokine analysis differed based on the amount of serum available and the sample results meeting QC metrics. Supplemental Table S1 (all Supplemental material is available at https://doi.org/10.6084/m9.figshare.19578811.v1) shows the distribution of samples across the 5 days of the study for each subject group and each immunoassay.
Table 1.
Baseline and clinical characteristics of the cohort
| Variable | Value | Controls (n = 27) | Mild (n = 65) | Moderate (n = 38) | Severe (n = 40) | P Value |
|---|---|---|---|---|---|---|
| Age | Median [IQR] | 42 [32.5, 52.0] | 49 [36, 67] | 47 [31.5, 59.8] | 57 [38.8, 67.2] | 0.039 |
| BMI | 27.5 [25.1, 29.8] | 27.8 [24.6, 32.9] | 32.8 [27.5, 36.5] | 32.6 [27.7, 36.3] | 0.006 | |
| Sex | Male | 8 (29.6) | 26 (40.0) | 16 (42.1) | 25 (62.5) | 0.040 |
| Race | White | 24 (88.9) | 61 (93.8) | 35 (92.1) | 38 (95.0) | |
| African American | 1 (3.7) | 3 (4.6) | 1 (2.6) | 2 (5.0) | ||
| Other | 2 (7.4) | 1 (1.5) | 2 (5.1) | 0 (0.0) | 0.540 | |
| Etiology | Biliary | 30 (46.2) | 15 (39.5) | 21 (52.5) | 0.003 | |
| Post-ERCP | 19 (29.2) | 5 (13.2) | 1 (2.5) | |||
| Idiopathic | 7 (10.8) | 5 (13.2) | 3 (7.5) | |||
| Hypertriglyceridemia | 2 (3.1) | 8 (21.1) | 7 (17.5) | |||
| Alcohol | 1 (1.5) | 4 (10.5) | 5 (12.5) | |||
| Other | 6 (9.2) | 1 (2.6) | 3 (7.5) | |||
| AP episode | Sentinel | 41 (63.1) | 25 (67.6) | 31 (77.5) | 0.569 | |
| Transfer status | Yes | 21 (32.3) | 22 (57.9) | 33 (82.5) | <0.001 | |
| Time to transfer, days | Median [IQR] | 2 [1, 2] | 2 [1, 2.2] | 2 [1, 2] | 0.809 | |
| Severity prediction score | APACHE Score | Median [IQR] | 6 [3, 8] | 6 [4, 9] | 11.5 [9.0, 16.2] | <0.001 |
| BISAP | 1 [0, 1] | 1 [1, 2] | 3 [2.0, 3.2] | <0.001 | ||
| Ranson’s | 1 [1, 2] | 2 [2, 3] | 6 [3.8, 7.0] | <0.001 | ||
| SIRS | 1 [0, 2] | 2 [1, 2] | 2 [1, 3] | <0.001 | ||
| SIRS+ | 17 (26.2) | 27 (71.1) | 31 (77.5) | <0.001 | ||
| Pancreatic necrosis | Present | 0 (0.0) | 21 (58.3) | 22 (59.5) | <0.001 | |
| Organ failure | Present | 0 (0.0) | 9 (24.3) | 40 (100.0) | <0.001 | |
| Death | Yes | 0 (0.0) | 0 (0.0) | 9 (22.5) | <0.001 | |
| Total LOS, days | Median [IQR] | 5 [3, 6] | 9 [6, 14] | 27 [19, 46] | <0.001 | |
χ2 test for discrete values and Mann–Whitney U test for continuous variables. AP, acute pancreatitis; BISAP, Bedside Index for Severity of Acute Pancreatitis; BMI, body mass index; IQR, interquartile range; LOS, length of stay; SIRS, systemic inflammatory response syndrome.
Table 1 shows the baseline and clinical characteristics of the study cohort. Control subjects were significantly younger (42 vs. 50 yr), had a lower BMI (27.5 vs. 30.4), and were predominantly female (70.4% vs. 53.1%) compared with the AP cohort, but their baseline characteristics were similar to mild AP cohort. Among the AP subjects, older age, male gender, higher BMI, and certain etiologies were associated with increased severity (Table 1). With alcoholic and hypertriglyceridemia etiologies representing a higher proportion of MSAP and SAP, whereas post-endoscopic retrograde cholangiopancreatography (ERCP) etiology was more common among MAP. The actual values of the cytokines across groups over time before log transformation and statistical modeling are given in Supplemental Table S2, A–C. Results of the linear mixed models (post log transformation and statistical modeling) that compared the cytokines’ trajectories between MAP, MSAP, and SAP are given in Supplemental Tables S3 and S4.
Cytokine Levels over Days 1–5
To evaluate the dynamic changes in serum cytokine levels over time, we used linear mixed effects models controlling for age, sex, and BMI. The results of the models can be seen in Tables 2 and 3 and graphically in Figs. 1 and 2. These figures were plotted with x-axis representing elapsed number of days between time of pain onset and sample collection (instead of time elapsed since day of presentation), which would most accurately depict the chronology of cytokine response in relation to disease onset. Age (Ang-2, IL-6, IL-8 and sTNFα-R1), sex (IL-6), and BMI (Ang-2) each showed significant effects on many models so they were kept in all models. These models were computed on a log scale with each estimate being exponentiated to get the true level.
Table 2.
Linear mixed model results for IL-6, IL-8, and MCP-1 using Control subjects as baseline
| IL-6 |
IL-8 |
MCP-1 |
||||
|---|---|---|---|---|---|---|
| Coefficient | Estimates | P | Estimates | P | Estimates | P |
| (Intercept) | −1.57 (−2.56–−0.57) |
0.002 | 1.14 (0.46–1.82) |
0.001 | 5.13 (4.67–5.59) |
<0.001 |
| Age | 0.01 (0.00–0.02) |
0.032 | 0.01 (0.00–0.01) |
0.025 | 0.00 (−0.00–0.01) |
0.149 |
| Male | 0.43 (0.06–0.79) |
0.023 | 0.15 (−0.09–0.39) |
0.219 | 0.09 (−0.08–0.26) |
0.299 |
| Body mass index | 0.01 (−0.02–0.03) |
0.518 | 0.01 (−0.01–0.03) |
0.249 | 0.01 (−0.00–0.02) |
0.226 |
| Time from pain onset (days) | 0.01 (−0.15–0.16) |
0.947 | 0.00 (−0.14–0.14) |
0.980 | −0.04 (−0.12–0.03) |
0.275 |
| AP severity | ||||||
| MAP | 2.01 (1.33–2.69) |
<0.001 | 1.17 (0.65–1.69) |
<0.001 | 0.64 (0.32–0.96) |
<0.001 |
| MSAP | 3.82 (3.00–4.64) |
<0.001 | 1.22 (0.58–1.86) |
<0.001 | 0.61 (0.22–1.00) |
0.002 |
| SAP | 6.28 (5.45–7.10) |
<0.001 | 3.56 (2.93–4.20) |
<0.001 | 1.85 (1.45–2.24) |
<0.001 |
| Time by AP severity interaction | ||||||
| MAP | 0.01 (−0.19–0.20) |
0.940 | −0.05 (−0.23–0.12) |
0.570 | −0.13 (−0.23–−0.03) |
0.008 |
| MSAP | −0.18 (−0.39–0.04) |
0.112 | 0.00 (−0.19–0.20) |
0.969 | −0.03 (−0.14–0.08) |
0.641 |
| SAP | −0.30 (−0.50–−0.09) |
0.005 | −0.28 (−0.47–−0.10) |
0.003 | −0.08 (−0.18–0.03) |
0.147 |
| Random effects | ||||||
| σ2 | 0.52 | 0.43 | 0.13 | |||
| τ00 | 1.09 Person.ID | 0.38 Person.ID | 0.21 Person.ID | |||
| ICC | 0.68 | 0.47 | 0.62 | |||
| N | 165 Person.ID | 165 Person.ID | 165 Person.ID | |||
| Observations | 439 | 439 | 438 | |||
| Marginal R2 /Conditional R2 | 0.689/0.900 | 0.527/0.748 | 0.514/0.815 | |||
All cytokine concentrations were transformed on a log scale before mixed model estimation. All models use the control series as baseline. A significant result in AP severity denotes a log scale shift from baseline control levels for that category. A significant result in Time by AP Severity denotes a significant change (±) in slope from the baseline control slope. Significance P values with P < 0.05 are in bold for emphasis. IL-8, interleukin-8; IL-6, interleukin-6; MCP-1, monocyte chemoattractant protein-1; MAP, mild acute pancreatitis; MSAP, moderately severe acute pancreatitis; SAP, severe acute pancreatitis.
Table 3.
Linear mixed model results for Ang-2, HGF, resistin, and sTNFα-R1 using controls as baseline
| Ang-2 |
HGF |
Resistin |
sTNFα-R1 |
|||||
|---|---|---|---|---|---|---|---|---|
| Coeffcient | Estimates | P | Estimates | P | Estimates | P | Estimates | P |
| (Intercept) | 8.07 (7.46–8.67) |
<0.001 | 6.14 (5.43–6.86) |
<0.001 | 6.78 (6.16–7.39) |
<0.001 | 5.85 (5.37–6.34) |
<0.001 |
| Age | 0.01 (0.00–0.02) |
0.004 | 0.01 (−0.00–0.01) |
0.184 | 0.01 (−0.00–0.01) |
0.054 | 0.01 (0.01–0.02) |
<0.001 |
| Male | 0.03 (−0.20–0.27) |
0.779 | −0.05 (−0.33–0.23) |
0.740 | 0.10 (−0.12–0.33) |
0.362 | 0.10 (−0.08–0.29) |
0.268 |
| Body mass index | 0.02 (0.00–0.03) |
0.042 | 0.00 (−0.02–0.02) |
0.724 | 0.01 (−0.00–0.03) |
0.145 | 0.01 (−0.01–0.02) |
0.247 |
| Time from pain onset [days] | 0.03 (−0.00–0.06) |
0.082 | 0.00 (−0.05–0.05) |
0.928 | −0.01 (−0.10–0.08) |
0.811 | −0.00 (−0.05–0.05) |
0.927 |
| AP severity | ||||||||
| MAP | 0.21 (−0.13–0.56) |
0.225 | 0.27 (−0.13–0.67) |
0.185 | 0.04 (−0.36–0.45) |
0.834 | 0.49 (0.20–0.78) |
0.001 |
| MSAP | 0.28 (−0.10–0.66) |
0.152 | 0.79 (0.34–1.23) |
0.001 | 0.96 (0.47–1.44) |
<0.001 | 0.59 (0.26–0.93) |
0.001 |
| SAP | 1.74 (1.35–2.13) |
<0.001 | 1.92 (1.46–2.39) |
<0.001 | 2.01 (1.51–2.52) |
<0.001 | 1.37 (1.01–1.73) |
<0.001 |
| Time by AP severity interaction | ||||||||
| MAP | 0.04 (−0.07–0.16) |
0.466 | −0.03 (−0.09–0.03) |
0.351 | ||||
| MSAP | −0.04 (−0.16–0.09) |
0.589 | 0.02 (−0.05–0.09) |
0.636 | ||||
| SAP | −0.18 (−0.31–−0.05) |
0.006 | 0.08 (0.01–0.14) |
0.033 | ||||
| Random effects | ||||||||
| σ2 | 0.10 | 0.31 | 0.18 | 0.05 | ||||
| τ00 | 0.53 Person.ID | 0.66 Person.ID | 0.35 Person.ID | 0.26 Person.ID | ||||
| ICC | 0.84 | 0.68 | 0.66 | 0.83 | ||||
| N | 171 Person.ID | 171 Person.ID | 135 Person.ID | 135 Person.ID | ||||
| Observations | 455 | 455 | 412 | 412 | ||||
| Marginal R2 /Conditional R2 | 0.467 / 0.916 | 0.349 / 0.791 | 0.414 / 0.800 | 0.575 / 0.926 | ||||
All cytokine concentrations were transformed on a log scale before mixed model estimation. All models use the control series as baseline, A significant result in AP severity denotes a log scale shift from baseline control levels for that category. A significant result in Time by AP Severity denotes a significant change (±) in slope from the baseline control slope. Significance P values with P < 0.05 are in bold for emphasis. HGF and Ang-2 showed no significant interaction between time and severity, thus the interaction was excluded from the model. MAP, mild acute pancreatitis; MSAP, moderately severe acute pancreatitis; SAP, severe acute pancreatitis. Ang-2, angiopoietin-2; HGF, hepatocyte growth factor; TNF-α R1, soluble TNF-α receptor 1.
Figure 1.

Trajectories of interleukin-8 (IL-8), interleukin-6 (IL-6), and monocyte chemoattractant protein-1 ( MCP-1) during the first 5 days of acute pancreatitis (AP) symptom onset. The trajectories with 95% confidence intervals (shaded gray) of cytokine concentrations in pg/mL for IL-8 (A), IL-6 (B), and MCP-1 (C) over time. Time points 1–5 are days after pain onset. Comparison between subsets with Severe AP (red), Moderately Severe AP (gold), Mild AP (blue), and Control (black).
Figure 2.

Trajectories of angiopoietin-2 (Ang-2), hepatocyte growth factor (HGF), resistin, and soluble TNF-α receptor 1 (sTNF-α R1) during the first 5 days of acute pancreatitis (AP) symptom onset. The trajectories with 95% confidence intervals (shaded gray) of biomarker concentrations in pg/mL for Ang-2 (A), HGF (B), and resistin (C), and sTNFα-R1 (D) over time. Time points 1–5 are days after pain onset. Comparison between subsets with Severe AP (acute pancreatitis; red), Moderately Severe AP (gold), Mild AP (blue), and Control (black).
Patterns of Cytokine Trajectories in Subjects That Develop SAP versus Non-SAP Subjects
As expected, all seven cytokines’ levels were significantly upregulated in AP subjects compared with controls. The following patterns of differing trajectories were observed (Figs. 1 and 2).
Rapidity of cytokine upregulation in AP differed by cytokine type and severity when compared with healthy controls.
Early upregulation occurred in SAP subjects for all seven cytokines, as indicated by their significantly higher day 1 levels when compared with healthy controls. Similarly early and rapid upregulation occurred in MAP and MSAP subjects for IL-6, IL-8, MCP-1, and TNFα-R1, but Ang-2 and HGF upregulation occurred slower over time (i.e., levels of Ang-2 and HGF did not diverge from healthy controls until after day 2 for MAP and MSAP). For resistin, early and rapid upregulation was seen in MSAP but a slower upregulation in MAP where its levels started to diverge from healthy controls in day 2.
Association between the initial magnitude of cytokine upregulation and AP severity exhibited two distinct patterns.
Pattern 1.
IL-6, HGF, and resistin upregulation progressively correspond to the three progressive severity classifications of AP, which would allow prediction of all three severity categories (i.e., cytokine levels in SAP > MSAP > MAP).
Pattern 2.
Ang-2, IL-8, MCP-1, and sTNFα-R1 cytokine upregulation corresponded to AP in a binary manner, which would predict SAP from non-SAP but not between the non-SAP groups (i.e., cytokine levels in SAP > non-SAP, but similar levels between MAP and MSAP).
Trajectories of the cytokines differed by cytokine type and AP severity.
In SAP subjects, the anti-inflammatory cytokine (HGF and sTNFα-R1) levels exhibited an upward slope respectively over time in contrast to a downward slope seen in proinflammatory cytokine levels (IL-6, IL-8, MCP-1, and resistin) except Ang-2, which stayed at a similar level in all time points. In MAP and MSAP subjects; however, both pro- and anti-inflammatory cytokines’ levels remained either similar (IL-6) or increased over time (Ang-2, IL-8, and resistin; HGF and sTNFα-R1) except for MCP-1, which remained stable in MSAP and decreased over time in MAP.
Correlation between Cytokine Levels
The seven cytokine levels were significantly (P < 0.001) correlated with one another on their transformed log scale across all samples as seen in Fig. 3. Spearman’s correlation coefficients (CC) ranged from 0.51 through 0.75. Not all correlations were linear but all were positively correlated. Highest correlations were found between IL-6 and IL-8, MCP-1, resistin and sTNF-α R1, between Ang-2 and sTNF-α R1 and between IL-8 and MCP-1 (CC 0.72–0.75).
Figure 3.
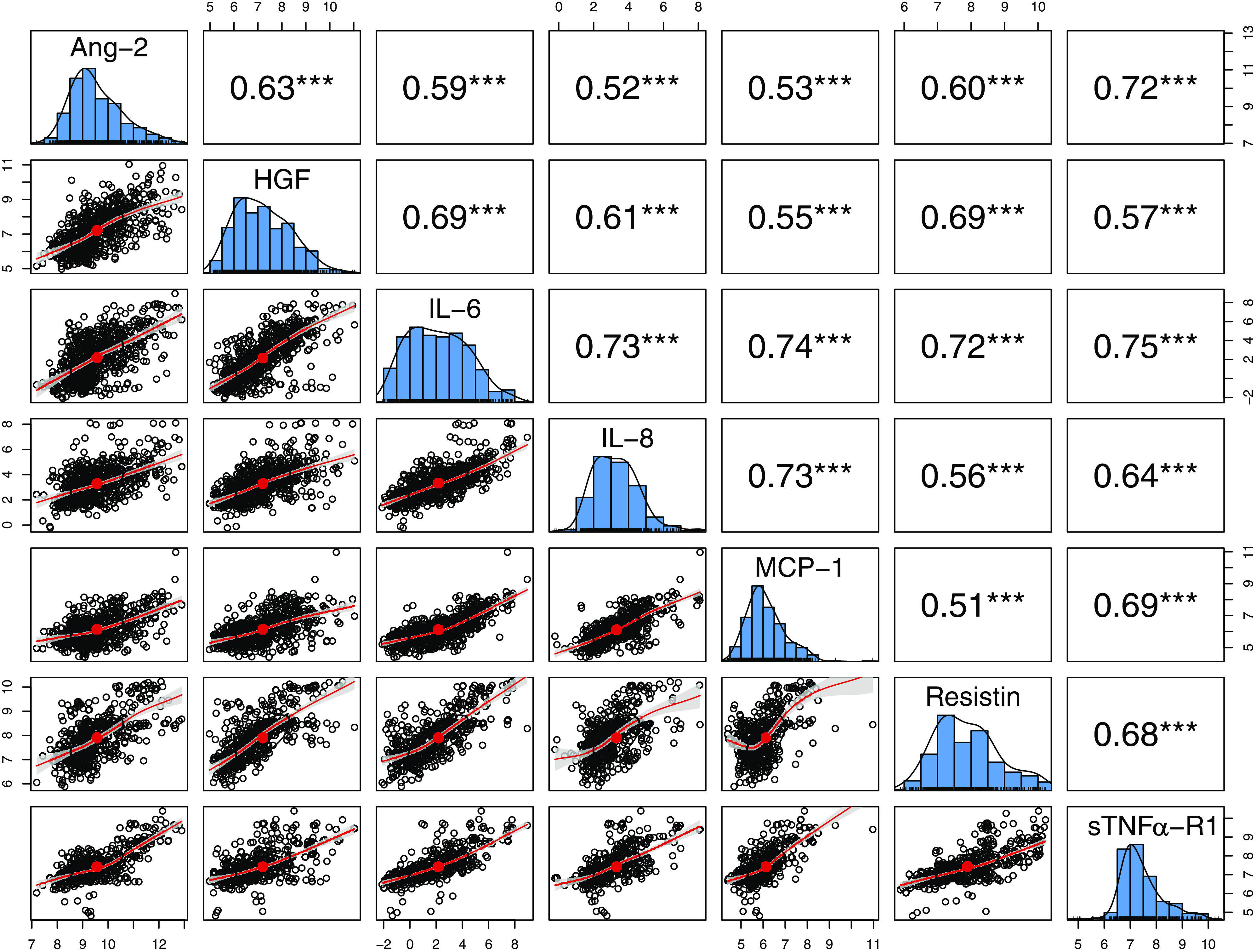
Summary of correlations between angiopoietin-2 (Ang-2), hepatocyte growth factor (HGF), interleukin-6 (IL-6), interleukin-8 (IL-8), monocyte chemoattractant protein-1 (MCP-1), resistin, and soluble TNF-α receptor 1 (sTNFα-RI). Pairwise correlation matrix for all biomarkers measured across all time points and all subjects. Cytokine levels are plotted on a log scale. ***Spearman’s correlations presented in the top right with all correlations statistically significant at P < 0.001.
DISCUSSION
To our knowledge, the present study is the first to model the early time course of seven key cytokines strongly associated with SAP in a well-phenotyped human cohort of AP subjects and healthy controls. Our findings confirm several of the observations about the cytokine response seen in the animal models, but also offer new insights.
As expected, admission levels of all seven cytokines were significantly higher in SAP versus non-SAP subjects. But when comparing MAP versus MSAP subjects, admission levels differed only for IL-6 and resistin and not the other five cytokines. These observations consequently have implications for each cytokine’s ability to discriminate between the three severity categories. We found that admission Ang-2, IL-8, and MCP-1 levels, while discriminating those that eventually developed SAP from non-SAP, fail to distinguish the MSAP from the MAP group. This may indicate that Ang-2, IL-8, and MCP-1 may have distinct roles in POF pathways (the defining criterion of SAP) in comparison to transient OF and pancreatic necrosis, which are defining criteria for MSAP. In contrast, admission IL-6 and resistin levels discriminated all three severity categories (i.e., levels were highest in SAP, higher in MSAP, and lowest in MAP), suggesting potential contributory roles for mediating both local (pancreatic necrosis) and systemic complications (OF and exacerbation of comorbid condition). Given the crucial role of Ang-2 and IL-8 in regulating endothelial permeability and their association with vascular leak syndrome in other disease models such as trauma, our findings support the prominent role of capillary permeability in mediating POF as previously described (14, 15, 32). IL-6 and MCP-1 may exert their deleterious systemic effects by activated immune cells in the pulmonary system such as neutrophils and monocytes via ischemia-reperfusion events and/or direct lung tissue injury (33–35). Further studies are warranted to investigate how these cytokines’ pathways interact with each other to mediate endothelial dysfunction and systemic organ injury in SAP (8).
In line with animal models of SAP, our findings support the hypothesis that immune response (both pro- and anti-inflammatory) to pancreatic injury occurs rapidly in human SAP, with a greater magnitude of cytokine increase in SAP compared with non-SAP. Of interest, the proinflammatory cytokines (Ang-2, IL-6, IL-8, MCP-1, and resistin) started to decline or plateau after day 1 for SAP. This downward trajectory over time suggests that the proinflammatory cytokine response likely peaks within the first 24 h of disease onset in SAP. Previous findings in studies of post-ERCP pancreatitis support this hypothesis. Chen et al. trended cytokine levels starting before the ERCP to 24 h after the procedure (36). Both IL-6 and IL-8 levels started to increase compared with controls as early as 8 h after the procedure and ∼4 h after onset of symptoms and reached their highest levels at 24 h from the ERCP (time of pancreatic injury) (36). The proinflammatory cytokine early peak is explained by mechanisms of their release upon pancreatic injury. IL-6 and MCP-1 are secreted by injured acinar cells and IL-8 is among the earliest chemokines that recruit and activate circulating neutrophils but also regulates endothelial permeability (37). Resistin, mainly found in the local adipose tissues, is also likely released early in the disease course as lipase digests the adipose tissues surrounding the pancreas (38, 39). Ang-2 is a regulator of endothelial permeability, and its release likely starts locally from the endothelial cells within the pancreatic microvascular system triggered by hypoxia, TNF-α, turbulent flow, and thrombin that typically occur upon pancreatic injury (19, 40, 41). The exact factors (i.e., genetic, demographic, lifestyle, and/or etiology) that determine the magnitude of their release remain however unclear.
A novel finding of our study, discordant from the conventional thinking, is the downregulation of the proinflammatory cytokine response over time in SAP subjects (i.e., downward slope instead of a flat slope) in contrast to milder grades of AP (i.e., MAP and MSAP). This downregulation may be a result of increasing effects of regulatory immune cells and cytokines that dampen the cytokine release but also potentially indicate that the dysregulated immune response may not be the sole driver of organ dysfunction. We postulate that additional downstream events, such as capillary leak syndrome, ensues following the cytokines’ peak (triggered by the proinflammatory response), perpetuating the pancreatic injury and mediating organ dysfunction via splanchnic hypoperfusion and pulmonary edema (8). Nevertheless, our findings need to be confirmed in future studies. Contrary to the expected decline, MAP and MSAP subjects exhibited either an upward slope (Ang-2 and resistin) or plateauing of cytokine levels (IL-6 and 8) except for MCP-1, which rapidly declined in MAP. Given that MAP and MSAP subjects recovered and were discharged after a median stay of 5 and 9 days in the hospital, respectively, these upward trajectories up to day 5 are intriguing and possibly suggest that complete resolution of systemic inflammation lags clinical recovery.
The anti-inflammatory cytokine (HGF and sTNF-α R1) levels showed an upward trend over time in all three AP categories. Our observation confirms findings of other studies that investigated the association of HGF and IL-10, which is another anti-inflammatory cytokine with disease severity in AP and COVID-19 (42–44). HGF is known to downregulate potent proinflammatory cytokines, such as IL-1β, IL-6 and TNF-α, increase production of anti-inflammatory cytokine IL-10 and promote organ regeneration (45, 46). sTNF-α R1 is a soluble form (i.e., extracellular) of the transmembrane receptor to TNF-α, which is found in most nucleated cells including immune cells, and exerts inhibitory effect on TNF-α by preventing its binding to the cellular TNF receptors (19, 47, 48). This pattern likely reflects the immune system’s attempt (possibly unsuccessful) to regulate inflammation and stop the immunopathology (49, 50).
Our findings indicate that AP is a time-sensitive disease similar to other acute disease models such as acute trauma, stroke, and myocardial infarction (51–53). We recently reported in an international, multicenter prospective cohort study of more than 1,500 AP subjects that 92% of all systemic organ injury in SAP develops within the first week of disease (3). In the context of a therapeutic trial design in AP, if reduction of a cytokine-peak is to be chosen as an end point, the time window for targeting the immune response may be short, less than 24 h from symptom onset. Our findings also have relevance beyond providing cytokines’ endpoints when designing immune therapy trials in SAP. The cytokines’ signatures and their trajectory characterized in our study will serve as a robust reference for future biomarker studies in SAP and for subject selection in therapeutic trials in SAP when bedside cytokine measurement becomes routinely available.
This study has many strengths. First, it has a robust and prospective cohort design with a sample size of 144 subjects and 27 controls resulting in the analysis of 455 serum samples. This is the only study in human AP to include seven of the most studied cytokines. Second, our choice to use a linear mixed model for analysis of the temporal trends is robust to the missingness of the cohort. Third, we utilized the Revised Atlanta Classification for defining disease severity, a validated prognostic classification system, widely accepted in the research community, that minimizes the misclassification bias of outcomes. Fourth, we utilized the prospective-specimen-collection, retrospective-blinded evaluation design, where appropriate blinding was implemented to minimize reporting bias and enhance scientific rigor.
Few limitations need to be mentioned. Despite extensive efforts, the median time of the first blood collection was 40 h from symptom onset with the interquartile range of 24–54 h with only few subjects contributing to every sample time point. This illustrates the difficulty in enrolling AP subjects in research studies within hours from the onset of their disease. Because of this limitation, our data cannot exactly pinpoint when these cytokines peak over the first 12 h of pain onset. Future efforts should focus on enrolling subjects even earlier, ideally in the Emergency Department. Second, our study is limited to the group of seven cytokines without performing any cytometric and functional analyses of the immune system. However, our investigation was designed as a steppingstone to inform a more comprehensive analysis of the immune response to pancreatic injury, which can now be guided by our findings. Nine out of the 40 SAP subjects did not mount a positive SIRS response. This could reflect presence of ascertainment bias in its assessment and/or limitation of SIRS score in consistently capturing presence of cytokine storm in SAP. In support of the latter possibility, SAP prediction studies have shown limitations of a negative SIRS state in ruling out SAP (54).
In conclusion, we confirmed that proinflammatory cytokine response in SAP subjects occurs early in the disease course (within 24 h), but there is gradual downregulation of these cytokines over the next 5 days. These early cytokines predicted eventual severity classification of the RAC in post hoc analysis. In addition, upregulation of certain cytokines suggests that complete resolution of systemic inflammation lags clinical recovery in MAP and MSAP subjects. Taken together, these observations will serve as an excellent reference for future biomarker studies and design of therapeutic trials in SAP.
SUPPLEMENTAL DATA
Supplemental Tables S1–S4: https://doi.org/10.6084/m9.figshare.19578811.v1.
GRANTS
This research was funded by the United States Department of Defense Congressionally Directed Medical Research Programs (CDMRP) Awards W81XWH-14-1-0376 (to D.C.W.) and W81XWH-17-1-0502 (to D.C.W.), The National Institutes of Health Grants DK061451 (to D.C.W.), and the Department of Veterans Affairs Merit Review I01CX000272 (to G.I.P.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.C.W. and G.I.P. conceived and designed research; P.J.G., P.P., K.S., A.P., and G.I.P. performed experiments; P.J.G., P.J.L., P.P., A.P., C.S., D.C.W., and G.I.P. analyzed data; P.J.G., G.I.P., P.J.L., P.H., C.S., A.L.-H., and D.C.W. interpreted results of experiments; P.J.G. prepared figures; P.J.G., G.I.P., and P.J.L. drafted manuscript; P.J.G., P.J.L., P.H., C.S., D.C.W., and G.I.P. edited and revised manuscript; P.J.L., D.C.W., and G.I.P. approved final version of manuscript.
Footnotes
For simplicity, we use the term “cytokines” in a loose and inclusive way in this manuscript.
REFERENCES
- 1. Petrov MS, Yadav D. Global epidemiology and holistic prevention of pancreatitis. Nat Rev Gastroenterol Hepatol 16: 175–184, 2019. doi: 10.1038/s41575-018-0087-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med 354: 2142–2150, 2006. doi: 10.1056/NEJMcp054958. [DOI] [PubMed] [Google Scholar]
- 3. Machicado JD, Gougol A, Tan X, Gao X, Paragomi P, Pothoulakis I, Talukdar R, Kochhar R, Goenka MK, Gulla A, Gonzalez JA, Singh VK, Ferreira M, Stevens T, Barbu ST, Nawaz H, Gutierrez SC, Zarnescu NO, Capurso G, Easler JJ, Triantafyllou K, Pelaez-Luna M, Thakkar S, Ocampo C, de-Madaria E, Cote GA, Wu BU, Conwell DL, Hart PA, Tang G, Papachristou GI. Mortality in acute pancreatitis with persistent organ failure is determined by the number, type, and sequence of organ systems affected. United Eur Gastroenterol J 9: 139–149, 2021. doi: 10.1002/ueg2.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Machicado JD, Gougol A, Stello K, Tang G, Park Y, Slivka A, Whitcomb DC, Yadav D, Papachristou GI. Acute pancreatitis has a long-term deleterious effect on physical health related quality of life. Clin Gastroenterol Hepatol 15: 1445–1443.e2, 2017. doi: 10.1016/j.cgh.2017.05.037. [DOI] [PubMed] [Google Scholar]
- 5. Sternby H, Bolado F, Canaval-Zuleta HJ, Marra-Lopez C, Hernando-Alonso AI, Del-Val-Antonana A, Garcia-Rayado G, Rivera-Irigoin R, Grau-Garcia FJ, Oms L, Millastre-Bocos J, Pascual-Moreno I, Martinez-Ares D, Rodriguez-Oballe JA, Lopez-Serrano A, Ruiz-Rebollo ML, Viejo-Almanzor A, Gonzalez-de-la-Higuera B, Orive-Calzada A, Gomez-Anta I, Pamies-Guilabert J, Fernandez-Gutierrez-Del-Alamo F, Iranzo-Gonzalez-Cruz I, Perez-Munante ME, Esteba MD, Pardillos-Tome A, Zapater P, de-Madaria E. Determinants of severity in acute pancreatitis: a nation-wide multicenter prospective cohort study. Ann Surg 270: 348–355, 2019. doi: 10.1097/SLA.0000000000002766. [DOI] [PubMed] [Google Scholar]
- 6. Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, Sarr MG, Tsiotos GG, Vege SS, Group APCW; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis–2012: revision of the Atlanta classification and definitions by international consensus. Gut 62: 102–111, 2013. doi: 10.1136/gutjnl-2012-302779. [DOI] [PubMed] [Google Scholar]
- 7. Abu-El-Haija M, Gukovskaya AS, Andersen DK, Gardner TB, Hegyi P, Pandol SJ, Papachristou GI, Saluja AK, Singh VK, Uc A, Wu BU. Accelerating the drug delivery pipeline for acute and chronic pancreatitis: summary of the working group on drug development and trials in acute pancreatitis at the National Institute of Diabetes and Digestive and Kidney Diseases Workshop. Pancreas 47: 1185–1192, 2018. doi: 10.1097/MPA.0000000000001175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Komara NL, Paragomi P, Greer PJ, Wilson AS, Breze C, Papachristou GI, Whitcomb DC. Severe acute pancreatitis: capillary permeability model linking systemic inflammation to multiorgan failure. Am J Physiol Gastrointest Liver Physiol 319: G573–G583, 2020. doi: 10.1152/ajpgi.00285.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nieminen A, Maksimow M, Mentula P, Kyhälä L, Kylänpää L, Puolakkainen P, Kemppainen E, Repo H, Salmi M. Circulating cytokines in predicting development of severe acute pancreatitis. Crit Care 18: R104, 2014. doi: 10.1186/cc13885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Whitcomb DC, Muddana V, Langmead CJ, Houghton FDJ, Guenther A, Eagon PK, Mayerle J, Aghdassi AA, Weiss FU, Evans A, Lamb J, Clermont G, Lerch MM, Papachristou GI. Angiopoietin-2, a regulator of vascular permeability in inflammation, is associated with persistent organ failure in patients with acute pancreatitis from the United States and Germany. Am J Gastroenterol 105: 2287–2292, 2010. doi: 10.1038/ajg.2010.183. [DOI] [PubMed] [Google Scholar]
- 11. Langmead C, Lee PJ, Paragomi P, Greer P, Stello K, Hart PA, Whitcomb DC, Papachristou GI. A novel 5-cytokine panel outperforms conventional predictive markers of persistent organ failure in acute pancreatitis. Clin Transl Gastroenterol 12: e00351, 2021. doi: 10.14309/ctg.0000000000000351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vasseur P, Devaure I, Sellier J, Delwail A, Chagneau-Derrode C, Charier F, Tougeron D, Tasu JP, Rabeony H, Lecron JC, Silvain C. High plasma levels of the pro-inflammatory cytokine IL-22 and the anti-inflammatory cytokines IL-10 and IL-1ra in acute pancreatitis. Pancreatology 14: 465–469, 2014. doi: 10.1016/j.pan.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 13. Sennello JA, Fayad R, Pini M, Gove ME, Ponemone V, Cabay RJ, Siegmund B, Dinarello CA, Fantuzzi G. Interleukin-18, together with interleukin-12, induces severe acute pancreatitis in obese but not in nonobese leptin-deficient mice. Proc Natl Acad Sci USA 105: 8085–8090, 2008. doi: 10.1073/pnas.0804091105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Akwii RG, Sajib MS, Zahra FT, Mikelis CM. Role of angiopoietin-2 in vascular physiology and pathophysiology. Cells 8: 471, 2019. doi: 10.3390/cells8050471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Allen TC, Kurdowska A. Interleukin 8 and acute lung injury. Arch Pathol Lab Med 138: 266–269, 2014. doi: 10.5858/arpa.2013-0182-RA. [DOI] [PubMed] [Google Scholar]
- 16. Zhang H, Neuhofer P, Song L, Rabe B, Lesina M, Kurkowski MU, Treiber M, Wartmann T, Regner S, Thorlacius H, Saur D, Weirich G, Yoshimura A, Halangk W, Mizgerd JP, Schmid RM, Rose-John S, Algul H. IL-6 trans-signaling promotes pancreatitis-associated lung injury and lethality. J Clin Invest 123: 1019–1031, 2013. doi: 10.1172/JCI64931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hirota M, Nozawa F, Okabe A, Shibata M, Beppu T, Shimada S, Egami H, Yamaguchi Y, Ikei S, Okajima T, Okamoto K, Ogawa M. Relationship between plasma cytokine concentration and multiple organ failure in patients with acute pancreatitis. Pancreas 21: 141–146, 2000. doi: 10.1097/00006676-200008000-00006. [DOI] [PubMed] [Google Scholar]
- 18. Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol 115: 911–919, 2005. doi: 10.1016/j.jaci.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 19. Lee PJ, Papachristou GI. New insights into acute pancreatitis. Nat Rev Gastroenterol Hepatol 16: 479–496, 2019. doi: 10.1038/s41575-019-0158-2. [DOI] [PubMed] [Google Scholar]
- 20. Ling L, Chen Z, Lui G, Wong CK, Wong WT, Ng RWY, Tso EYK, Fung KSC, Chan V, Yeung ACM, Hui DSC, Chan PKS. Longitudinal cytokine profile in patients with mild to critical COVID-19. Front. Immunol 12: 763292, 2021. doi: 10.3389/fimmu.2021.763292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kellum JA, Kong L, Fink MP, Weissfeld LA, Yealy DM, Pinsky MR, Fine J, Krichevsky A, Delude RL, Angus DC, Investigators For The G. Understanding the inflammatory cytokine response in pneumonia and sepsis: results of the genetic and inflammatory markers of sepsis (GenIMS) study. Arch Intern Med 167: 1655–1663, 2007. doi: 10.1001/archinte.167.15.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Del Valle DM, Kim-Schulze S, Huang H-H, Beckmann ND, Nirenberg S, Wang B, Lavin Y, Swartz TH, Madduri D, Stock A, Marron TU, Xie H, Patel M, Tuballes K, Van Oekelen O, Rahman A, Kovatch P, Aberg JA, Schadt E, Jagannath S, Mazumdar M, Charney AW, Firpo-Betancourt A, Mendu DR, Jhang J, Reich D, Sigel K, Cordon-Cardo C, Feldmann M, Parekh S, Merad M, Gnjatic S. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med 26: 1636–1643, 2020. doi: 10.1038/s41591-020-1051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giroir BP. Pancreatitis, cytokines, and SIRS: deja vu all over again?. Crit Care Med 27: 680–681, 1999. doi: 10.1097/00003246-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 24. Mayer J, Rau B, Gansauge F, Beger HG. Inflammatory mediators in human acute pancreatitis: clinical and pathophysiological implications. Gut 47: 546–552, 2000. doi: 10.1136/gut.47.4.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koutroumpakis E, Slivka A, Furlan A, Dasyam AK, Dudekula A, Greer JB, Whitcomb DC, Yadav D, Papachristou GI. Management and outcomes of acute pancreatitis patients over the last decade: a US tertiary-center experience. Pancreatology 17: 32–40, 2017. doi: 10.1016/j.pan.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 26. Koutroumpakis E, Wu BU, Bakker OJ, Dudekula A, Singh VK, Besselink MG, Yadav D, Mounzer R, van Santvoort HC, Whitcomb DC, Gooszen HG, Banks PA, Papachristou GI. Admission hematocrit and rise in blood urea nitrogen at 24 h outperform other laboratory markers in predicting persistent organ failure and pancreatic necrosis in acute pancreatitis: a post hoc analysis of three large prospective databases. Am J Gastroenterol 110: 1707–1716, 2015. doi: 10.1038/ajg.2015.370. [DOI] [PubMed] [Google Scholar]
- 27. Pepe MS, Feng Z, Janes H, Bossuyt PM, Potter JD. Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: standards for study design. J Natl Cancer Inst 100: 1432–1438, 2008. doi: 10.1093/jnci/djn326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marshall JC, Cook DJ, Christou NV, Bernard GR, Sprung CL, Sibbald WJ. Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med 23: 1638–1652, 1995. doi: 10.1097/00003246-199510000-00007. [DOI] [PubMed] [Google Scholar]
- 29. Kenis G, Teunissen C, De Jongh R, Bosmans E, Steinbusch H, Maes M. Stability of interleukin 6, soluble interleukin 6 receptor, interleukin 10 and CC16 in human serum. Cytokine 19: 228–235, 2002. [PubMed] [Google Scholar]
- 30. De Paoli P. Bio-banking in microbiology: from sample collection to epidemiology, diagnosis and research. FEMS Microbiol Rev 29: 897–910, 2005. doi: 10.1016/j.femsre.2005.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2021. http://www.r-project.org/. [Google Scholar]
- 32. Biffl WL, Moore EE, Moore FA, Carl VS, Franciose RJ, Banerjee A. Interleukin-8 increases endothelial permeability independent of neutrophils. J Trauma 39: 98–98, 1995. doi: 10.1097/00005373-199507000-00013. [DOI] [PubMed] [Google Scholar]
- 33. Gross V, Andreesen R, Leser HG, Ceska M, Liehl E, Lausen M, Farthmann EH, Scholmerich J. Interleukin-8 and neutrophil activation in acute pancreatitis. Eur J Clin Invest 22: 200–203, 1992. doi: 10.1111/j.1365-2362.1992.tb01826.x. [DOI] [PubMed] [Google Scholar]
- 34. Bhatia M, Neoptolemos JP, Slavin J. Inflammatory mediators as therapeutic targets in acute pancreatitis. Curr Opin Investig Drugs 2: 496–501, 2001. [PubMed] [Google Scholar]
- 35. Zhou G-X, Zhu X-J, Ding X-L, Zhang H, Chen J-P, Qiang H, Zhang H-F, Wei Q. Protective effects of MCP-1 inhibitor on a rat model of severe acute pancreatitis. Hepatobiliary Pancreat Dis Int 9: 201–207, 2010. [PubMed] [Google Scholar]
- 36. Chen C-C, Wang S-S, Lu R-H, Lu C-C, Chang F-Y, Lee S-D. Early changes of serum proinflammatory and anti-inflammatory cytokines after endoscopic retrograde cholangiopancreatography. Pancreas 26: 375–380, 2003. doi: 10.1097/00006676-200305000-00011. [DOI] [PubMed] [Google Scholar]
- 37. Lugea A, Waldron RT, Mareninova OA, Shalbueva N, Deng N, Su H-Y, Thomas DD, Jones EK, Messenger SW, Yang J, Hu C, Gukovsky I, Liu Z, Groblewski GE, Gukovskaya AS, Gorelick FS, Pandol SJ. Human pancreatic acinar cells: proteomic characterization, physiologic responses, and organellar disorders in ex vivo pancreatitis. Am J Pathol 187: 2726–2743, 2017. doi: 10.1016/j.ajpath.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Acharya C, Navina S, Singh VP. Role of pancreatic fat in the outcomes of pancreatitis. Pancreatology 14: 403–408, 2014. doi: 10.1016/j.pan.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schaffler A, Hamer O, Dickopf J, Goetz A, Landfried K, Voelk M, Herfarth H, Kopp A, Buchler C, Scholmerich J, Brunnler T. Admission resistin levels predict peripancreatic necrosis and clinical severity in acute pancreatitis. Am J Gastroenterol 105: 2474–2484, 2010. doi: 10.1038/ajg.2010.278. [DOI] [PubMed] [Google Scholar]
- 40. Dumnicka P, Maduzia D, Ceranowicz P, Olszanecki R, Drożdż R, Kuśnierz-Cabala B. The interplay between inflammation, coagulation and endothelial injury in the early phase of acute pancreatitis: clinical implications. Int J Mol Sci 18: 354, 2017. doi: 10.3390/ijms18020354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sack KD, Kellum JA, Parikh SM. The angiopoietin-tie2 pathway in critical illness. Crit Care Clin 36: 201–216, 2020. doi: 10.1016/j.ccc.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ueda T, Takeyama Y, Hori Y, Nishikawa J, Yamamoto M, Saitoh Y. Hepatocyte growth factor in assessment of acute pancreatitis: comparison with C-reactive protein and interleukin-6. J Gastroenterol 32: 63–70, 1997. doi: 10.1007/BF01213298. [DOI] [PubMed] [Google Scholar]
- 43. Ueda T, Takeyama Y, Toyokawa A, Kishida S, Yamamoto M, Saitoh Y. Significant elevation of serum human hepatocyte growth factor levels in patients with acute pancreatitis. Pancreas 12: 76–83, 1996. doi: 10.1097/00006676-199601000-00010. [DOI] [PubMed] [Google Scholar]
- 44. Perreau M, Suffiotti M, Marques-Vidal P, Wiedemann A, Levy Y, Laouénan C, Ghosn J, Fenwick C, Comte D, Roger T, Regina J, Vollenweider P, Waeber G, Oddo M, Calandra T, Pantaleo G. The cytokines HGF and CXCL13 predict the severity and the mortality in COVID-19 patients. Nat Commun 12: 4888, 2021. doi: 10.1038/s41467-021-25191-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ueda T, Takeyama Y, Hori Y, Shinkai M, Takase K, Goshima M, Yamamoto M, Kuroda Y. Hepatocyte growth factor increases in injured organs and functions as an organotrophic factor in rats with experimental acute pancreatitis. Pancreas 20: 84–93, 2000. doi: 10.1097/00006676-200001000-00012. [DOI] [PubMed] [Google Scholar]
- 46. Boros P, Miller CM. Hepatocyte growth factor: a multifunctional cytokine. Lancet 345: 293–295, 1995. doi: 10.1016/s0140-6736(95)90279-1. [DOI] [PubMed] [Google Scholar]
- 47. Sendler M, Dummer A, Weiss FU, Kruger B, Wartmann T, Scharffetter-Kochanek K, van Rooijen N, Malla SR, Aghdassi A, Halangk W, Lerch MM, Mayerle J. Tumour necrosis factor alpha secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut 62: 430–439, 2013. doi: 10.1136/gutjnl-2011-300771. [DOI] [PubMed] [Google Scholar]
- 48. Porteu F, Nathan C. Shedding of tumor necrosis factor receptors by activated human neutrophils. J Exp Med 172: 599–607, 1990. doi: 10.1084/jem.172.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Waetzig GH, Rosenstiel P, Arlt A, Till A, Bräutigam K, Schäfer H, Rose-John S, Seegert D, Schreiber S. Soluble tumor necrosis factor (TNF) receptor-1 induces apoptosis via reverse TNF signaling and autocrine transforming growth factor-beta1. FASEB J 19: 91–93, 2005. doi: 10.1096/fj.04-2073fje. [DOI] [PubMed] [Google Scholar]
- 50. Van Zee KJ, Kohno T, Fischer E, Rock CS, Moldawer LL, Lowry SF. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necrosis factor alpha in vitro and in vivo. Proc Natl Acad Sci USA 89: 4845–4849, 1992. doi: 10.1073/pnas.89.11.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lansky AJ, Goto K, Cristea E, Fahy M, Parise H, Feit F, Ohman EM, White HD, Alexander KP, Bertrand ME, Desmet W, Hamon M, Mehran R, Moses J, Leon M, Stone GW. Clinical and angiographic predictors of short- and long-term ischemic events in acute coronary syndromes: results from the Acute Catheterization and Urgent Intervention Triage strategY (ACUITY) trial. Circ Cardiovasc Interv 3: 308–316, 2010. doi: 10.1161/CIRCINTERVENTIONS.109.887604. [DOI] [PubMed] [Google Scholar]
- 52. Krumholz HM, Merrill AR, Schone EM, Schreiner GC, Chen J, Bradley EH, Wang Y, Wang Y, Lin Z, Straube BM, Rapp MT, Normand SL, Drye EE. Patterns of hospital performance in acute myocardial infarction and heart failure 30-day mortality and readmission. Circ Cardiovasc Qual Outcomes 2: 407–413, 2009. doi: 10.1161/CIRCOUTCOMES.109.883256. [DOI] [PubMed] [Google Scholar]
- 53. Kotwal RS, Howard JT, Orman JA, Tarpey BW, Bailey JA, Champion HR, Mabry RL, Holcomb JB, Gross KR. The effect of a golden hour policy on the morbidity and mortality of combat casualties. JAMA Surg 151: 15–24, 2016. doi: 10.1001/jamasurg.2015.3104. [DOI] [PubMed] [Google Scholar]
- 54. Mounzer R, Langmead CJ, Wu BU, Evans AC, Bishehsari F, Muddana V, Singh VK, Slivka A, Whitcomb DC, Yadav D, Banks PA, Papachristou GI. Comparison of existing clinical scoring systems to predict persistent organ failure in patients with acute pancreatitis. Gastroenterology 142: 1476, 2012. doi: 10.1053/j.gastro.2012.03.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables S1–S4: https://doi.org/10.6084/m9.figshare.19578811.v1.