

Abstract
The forkhead box M1 (FoxM1) protein, a transcription factor, plays critical roles in regulating tumor growth and drug resistance, while cellular FLICE-inhibitory protein (c-FLIP), an anti-apoptotic regulator, is involved in the ubiquitin–proteasome pathway. In this study, we investigated the effects of c-FLIP on the expression and ubiquitination levels of FoxM1 along with drug susceptibility in non-small-cell lung cancer (NSCLC) cells. We first showed that the expression levels of FoxM1 and c-FLIP were increased and positively correlated (R2 = 0.1106, P < 0.0001) in 90 NSCLC samples. The survival data from prognostic analysis demonstrated that high expression of c-FLIP and/or FoxM1 was related to poor prognosis in NSCLC patients and that the combination of FoxM1 and c-FLIP could be a more precise prognostic biomarker than either alone. Then, we explored the functions of c-FLIP/FoxM1 in drug resistance in NSCLC cell lines and a xenograft mouse model in vivo. We showed that c-FLIP stabilized FoxM1 by inhibiting its ubiquitination, thus upregulated the expression of FoxM1 at post-transcriptional level. In addition, a positive feedback loop composed of FoxM1, β-catenin and p65 also participated in c-FLIP–FoxM1 axis. We revealed that c-FLIP promoted the resistance of NSCLC cells to thiostrepton and osimertinib by upregulating FoxM1. Taken together, these results reveal a new mechanism by which c-FLIP regulates FoxM1 and the function of this interaction in the development of thiostrepton and osimertinib resistance. This study provides experimental evidence for the potential therapeutic benefit of targeting the c-FLIP–FoxM1 axis for lung cancer treatment.
Keywords: non-small-cell lung cancer, c-FLIP, FoxM1, ubiquitination, thiostrepton, osimertinib
Introduction
Forkhead box M1 (FoxM1) is an important member of the forkhead box transcription factor family. Abnormal expression of FoxM1 exists in a variety of human cancers and is involved in tumor initiation, progression, invasion, metastasis, angiogenesis and drug resistance [1]. FoxM1 is reported to be a biomarker of resistance to PI3Kα inhibitors in ER+ breast cancer [1] and may be a therapeutic target in EGFR TKI-resistant cells because it drives resistance to EGFR inhibition [2], suggesting that targeting FoxM1 could be a viable strategy for the treatment of cancer. Further understanding of the regulatory mechanisms of FoxM1 may identify more reliable prognostic or diagnostic biomarkers as well as help to develop rational treatment strategies to overcome drug resistance.
FoxM1 expression is regulated at multiple levels, and its modulation is crucial for FoxM1 activity and cancer development. First, there are several classical regulatory elements in the core promoter regions of the FoxM1 gene. CCCTC-binding factor (CTCF), glioma-associated oncogene homolog 1 (Gli1), signal transducer and activator of transcription 3 (STAT3) and E2F are reported to directly bind to the FoxM1 promoter region and activate FoxM1 transcription [3–5]. Additionally, the FoxM1 protein can potentially bind to its own promoter region to form a FoxM1 positive autoregulatory loop [6]. Second, post-translational modifications of FoxM1, such as phosphorylation, ubiquitination, SUMOylation, acetylation and methylation, affects the activation and function of FoxM1 [6]. Therefore, regulating the FoxM1 ubiquitination process to target its stability could be a potential strategy for inactivation of FoxM1. Its degradation is mediated by E3 ubiquitin ligases, such as TRIM6, RNF168/RNF8, APC/C-Cdh1 and FBXL2, in cancer cells [7–10], and its stability is regulated by deubiquitinases, including USP5, USP21, USP22, OTUB1, FAM188B and UCHL3, in carcinoma [11–14].
Cellular FLIP (c-FLIP) is an inhibitor of apoptosis initiated by death receptor ligation [15]. In addition to its involvement in death receptor-mediated apoptosis, c-FLIP has been shown to be a multifunctional protein, modulating processes, characteristics, and pathways such as autophagy, necrosis, inflammation, endoplasmic reticulum morphology, the ubiquitin-proteasome system and the NF-κB pathway [16]. It has been reported that c-FLIP enhances Wnt signaling by inhibiting the ubiquitination of β-catenin [17, 18] and modulates tumor proliferation by reducing GLT1 protein degradation under low-glucose conditions [16]. An in-depth study proved that c-FLIP is prone to aggregation and impairs ubiquitin-proteasome system (UPS) function, thereby increasing the expression of various short-lived proteins [15].
Owing to its function in the UPS, c-FLIP may also regulate the ubiquitination of other proteins, such as FoxM1. To test this hypothesis, we examined the effects of c-FLIP on the expression and ubiquitination levels of FoxM1 along with drug susceptibility in NSCLC cell lines. Here, we found that c-FLIP resulted in the upregulation of FoxM1, formed an immune complex with FoxM1 to regulate its ubiquitination level, and modulated the responses to thiostrepton and osimertinib by deubiquitinating and stabilizing FoxM1 in vivo and in vitro. Furthermore, a high protein expression level of c-FLIP was observed in lung cancer tissues and was associated with high expression of FoxM1 and negatively correlated with survival in lung cancer patients. These findings indicate that the c-FLIP-FoxM1 axis may play a crucial role in the pathogenesis of lung cancers. Targeting c-FLIP/FoxM1 could be a potential strategy for controlling lung cancer progression.
Materials and methods
Cell lines
The human NSCLC cell lines H1975, H157, H460, A549, H157-Lac-Z, and H157-FLIPL were stored in our laboratory. PC-9 cells and the corresponding osimertinib-resistant (PC-9/OR) cells as well as HCC827 cells and the corresponding osimertinib-resistant (HCC827/OR) cells were provided by Dr. P.Y. Shi (Xi’an Jiaotong University) [19]. All cells were maintained in our laboratory in RPMI-1640 medium containing 10% (v/v) fetal calf serum (Gibco BRL, Waltham, MA, USA), penicillin (100 U/mL), and streptomycin (100 μg/mL) (North China Pharmaceutical Inc, Shijiazhuang, China) in an incubator containing 5% CO2 at 37 °C.
Plasmids, transfection and treatments
siRNAs were purchased from JTS Scientific and RiboBio. All siRNA transfections were performed using Lipofectamine 3000 (Thermo Fisher, Waltham, MA, USA) following the manufacturer’s instructions. The human FoxM1-3HA expression vector was obtained from Miaolingbio (Wuhan, China). The GFP-c-FLIP expression plasmid was constructed by inserting a synthetic oligonucleotide encoding human c-FLIP into the KpnI and XhoI sites of pEGFP-N1 (forward primer: 5ʹ-CCGCTCGAGATGTCTGCTGAAGTCATCC-3ʹ; reverse primer: 5ʹ-CGGGGTACCGTTGTGTAGGAGAGGATAAGTTTC-3ʹ). All constructs generated from PCR products were sequenced. All plasmid transfections into cells were performed using Lipofectamine 3000 following the manufacturer’s instructions.
MG132 (5 μM; MedChemExpress, Monmouth Junction, USA) and cycloheximide (40 μg/mL; Selleck Chemicals, Houston, TX, USA) were used for the indicated durations. Thiostrepton (MedChemExpress, Monmouth Junction, USA) and osimertinib (Selleck Chemicals, Houston, TX, USA) were used at different concentrations for the indicated durations. The natural compound library for high-throughput screening was purchased from TargetMol (Shanghai, China).
Establishment of stable knockout cell lines
FoxM1 knockout cell lines were generated using CRISPR/Cas9 gene editing technology. The gRNA sequences targeting the FoxM1 gene were designed with an online gRNA design tool (https://zlab.bio/guide-design-resources). Two coupled single-guide RNAs (sgRNAs) were individually cloned into the Cas9-gRNA vector (SyngenTech, Beijing, China) according to the manufacturer’s instructions. After the inserted sequences were confirmed by DNA sequencing, Cas9-gRNA plasmids were transfected into H157 cells. Twenty-four hours later, the transfected cells were selected with puromycin (1 μg/mL) for 2 days, and single cells were isolated by limiting dilution in a 96-well plate. Furthermore, the isolated cell colonies were cultured and verified by immunoblotting.
Cell viability assays
Cell viability was evaluated using an MTT assay (Solarbio, Beijing, China) or a CCK8 assay (Meilunbio, Beijing, China). Cells were seeded in a 96-well plate at a density of 5 × 103 cells/well. After 24 h, the cells were treated with the indicated concentrations of reagents for 48 h. For the MTT assay, cells were treated with MTT solution (10 μL, 5 mg/mL) for 4 h. Subsequently, the supernatant was discarded. Formazan crystals were solubilized with 150 μL of DMSO. The absorbance was measured at 570 nm using a microplate reader (Thermo Fisher, Waltham, MA, USA). For the CCK8 assay, CCK8 reagent (10 μL/well) was added and incubated for 2 h, and the absorbance was measured using a microplate reader (450 nm). Cell viability was determined by comparing the absorbance of the wells containing reagent-treated cells with that of the wells containing control cells. All experiments were performed with triplicate samples and were repeated at least 3 times.
Colony formation assay
H157 cells and HCC827 cells were seeded into 12-well plates at 300 and 500 cells per well, respectively. After culture for 24 h, the H157 and HCC827 cell lines were treated with osimertinib for 8 and 13 days, respectively. After 15 min of fixation with 4% paraformaldehyde, colonies were visualized by staining with 0.1% crystal violet for 30 min. The number of colonies/well was counted and analyzed under a light microscope.
Western blot analysis and immunoprecipitation
Cells were lysed with RIPA buffer, and protein concentrations were determined using a BCA kit (Thermo Fisher, Waltham, MA, USA). Equal amounts of protein in cell lysates were separated on 8%-10% gels and transferred to PVDF membranes (Merck Millipore, St. Louis, MO, USA). The membranes were blocked in 5% milk/TBST for 1 h and incubated with primary antibodies at 4 °C overnight. The membranes were then washed with TBST and incubated with secondary antibodies at room temperature for 1 h, and immunoreactions were then detected with ECL reagent (Merck Millipore, St. Louis, MO, USA). Quantification of band intensities was carried out with ImageJ software. IP was performed according to a standard protocol described previously [20]. Antibodies specific for the following proteins were used: FoxM1 (cat no. 20459), p-FoxM1 (cat. no. 14655), β-catenin (cat. no. 8480 s), HA (cat. no. 3724), p65 (cat. no. 8242), and Ub (cat. no. 3933), all obtained from Cell Signaling Technologies (Danvers, MA, USA); actin (cat. no. AC026), obtained from ABclonal (Wuhan, China); c-FLIP (cat. no. ALX-804-961), obtained from Enzo (New York, NY, USA); and GAPDH (cat. no. AP0066), obtained from Bioworld (Nanjing, China).
RNA extraction and quantitative RT-PCR
Total RNA was extracted using a Total RNA Purification Kit according to the manufacturer’s instructions (Feijie, Shanghai, China). RNA quality was assessed using a U-Drop spectrophotometer (UlabX, USA). cDNA was synthesized using ReverTra Ace qPCR RT Master Mix (Toyobo Co, Tokyo, Japan). The mRNA levels of FoxM1, c-FLIP, actin and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were measured by quantitative RT-PCR (qRT-PCR) using SYBR Premix ExTaq (Toyobo Co, Tokyo, Japan) and a LightCycler 96 system (Roche, Basel, Switzerland). The following primers were used: human c-FLIP, 5ʹ-TGCTCTTTTTGTGCCGGGAT-3ʹ (forward) and 5ʹ-CGACAGACAGCTTACCTCTTTC-3ʹ (reverse); human FoxM1, 5ʹ-GGAGCAGCGACAGGTTAAGG-3ʹ (forward) and 5ʹ-GTTGATGGCGAATTGTATCATGG-3ʹ (reverse); human actin, 5ʹ-AACCGCGAGAAGATGACCCAG-3ʹ (forward) and 5ʹ-GGATAGCACAGCCTGGATAGCAA-3ʹ (reverse); and human GAPDH, 5ʹ-ACAACTTTGGTATCGTGGAAGG-3ʹ (forward) and 5ʹ-GCCATCACGCCACAGTTTC-3ʹ (reverse). GAPDH or actin was used for normalization. The data were analyzed using the ΔΔCt method, and each assay was performed in at least triplicate.
In vivo experiments
Female BALB/c nude mice (6–8 weeks old) were purchased from SPF Biotechnology (Beijing, China) and were maintained under specific pathogen-free conditions. All animal experiments were conducted in accordance with the institutional guidelines, which were in compliance with national and international laws and policies. H157-Lac-Z and H157-c-FLIP cells were inoculated into the right axillae of mice (three mice per group) at a concentration of 6 × 106 cells/0.2 mL PBS per mouse, and the mice were then fed for 24 days. Next, the tumors were removed, cut into 2 × 2 × 2 mm3 pieces, suspended in normal PBS and transplanted into the right axillae of wild-type female BALB/c nude mice. The next day, the mice were randomly divided into four groups (six nude mice in each group): the H157-Lac-Z control group administered PBS (200 μL/mouse, every four days), the H157-Lac-Z experimental group administered thiostrepton (50 mg/kg every four days), the H157-c-FLIP control group administered PBS, and the H157- c-FLIP experimental group administered thiostrepton. Mice in all groups were treated via i.p. injection. The body weights and tumor volumes were recorded every four days. The formula used to calculate tumor volumes has been mentioned previously [21]. On Day 21, the mice were sacrificed. The tumors were excised, fixed with 10% formalin and subjected to hematoxylin and eosin (H&E) staining and immunohistochemical analysis as described previously [20].
Tissue microarray and immunohistochemical (IHC) analysis
A tissue microarray was obtained from Outdo Biotech (Shanghai, China). IHC analysis of FoxM1 and c-FLIP was performed on lung cancer samples in the tissue microarray. IHC analysis was performed as previously described [20]. The anti-FoxM1 antibody was used at a 1:50 dilution, and the anti-c-FLIP antibody was used at a 1:25 dilution. The staining index was calculated based on the percentage of positive cells and the staining intensity. The staining intensity was scored as 0 (no staining), 1 (weak staining), 2 (moderate staining), or 3 (strong staining). Protein expression correlations were analyzed using GraphPad Prism (version 6, USA).
Statistical analysis
Unless otherwise specified, all experiments included a minimum of three independent replicates. Statistical tests (all two-tailed) were performed using GraphPad Prism 8 software. In the graphs, the error bars indicate the mean ± SD values, and statistical significance was set at P < 0.05 (in the figures, *P < 0.05; **P < 0.01; ***P < 0.001; and ns, nonsignificant).
Results
FoxM1 and c-FLIP are overexpressed and correlated in human lung cancer
To investigate the clinical significance of c-FLIP and FoxM1 in lung cancer, we evaluated the expression of c-FLIP and FoxM1 via immunohistochemical staining in a tissue microarray containing 90 samples and matched paracancerous tissues from primary human lung adenocarcinoma patients (Supplementary Fig. 1a, b). The proportions of tissues with high c-FLIP expression and high FoxM1 expression in the microarray were 78.9% (71/90) and 51.1% (46/90), respectively. High FoxM1 expression was observed more frequently in tissues with high c-FLIP expression than in tissues with low c-FLIP expression (60.5% vs. 15.8%). Representative images of tissues with high and low c-FLIP and FoxM1 expression are shown in Fig. 1a. There was a significant positive correlation between the expression levels of c-FLIP and FoxM1 (R2 = 0.1106, P < 0.0001, Fig. 1c) according to Pearson’s correlation analysis. In further analysis, the relationships between c-FLIP/FoxM1 expression levels and clinical outcomes were evaluated. High c-FLIP expression was found to be significantly associated with worse survival (P = 0.0305), and similar findings were observed in FoxM1-positive tumors (P = 0.0436) (Fig. 1d, e). Furthermore, combined high expression of c-FLIP and FoxM1 was more significantly associated with worse survival in lung cancer patients than combined low expression of c-FLIP and FoxM1 (P = 0.0141) (Fig. 1f). Together, these results suggested that both c-FLIP and FoxM1 were highly expressed in clinical lung cancer samples, supporting the critical role of c-FLIP and FoxM1 in lung cancers.
Fig. 1. FoxM1 expression is positively correlated with c-FLIP expression in human NSCLC.

a Representative images of immunohistochemical staining for FoxM1 and c-FLIP in human lung cancer samples and adjacent noncancer tissues. Scale bar: 200 μm. b, c The relationship between c-FLIP and FoxM1 expression in human lung cancer and adjacent lung tissues was analyzed using Pearson correlation analysis according to the quantification of IHC staining. d Survival analysis of patients with high (c-FLIP High) or low expression of c-FLIP (c-FLIP Low). e Survival analysis of patients with high (FoxM1 High) or low (FoxM1 Low) expression of FoxM1. f Survival analysis of patients with high (c-FLIP/FoxM1 High) or low (c-FLIP/FoxM1 Low) expression of both c-FLIP and FoxM1.
FoxM1 expression is positively regulated by c-FLIP
To validate the correlation between c-FLIP and FoxM1 expression, we examined the expression levels of these two proteins in several human NSCLC cell lines (HCC827, A549, H157, PC9, H1975, and H460) by Western blot analysis. The results showed a marked positive correlation between c-FLIP and FoxM1 protein expression (Fig. 2a, b, c). In addition, as shown in Fig. 2a, these cell lines were divided into the higher c-FLIP/FoxM1 expression group (HCC827, A549 and H157) and the lower c-FLIP/FoxM1 expression group (PC9, H1975, and H460). It has been reported that phosphorylation of FoxM1 is crucial for its activity [6]; thus, we examined the phosphorylation of FoxM1 in tandem with its protein expression level. In our study, the level of phosphorylated FoxM1 was consistent with its total protein level in lung cancer cells (Fig. 2a), which suggested the association of c-FLIP with the function of FoxM1.
Fig. 2. c-FLIP regulates FoxM1 expression in human NSCLC.

a–c The expression levels of the c-FLIP and FoxM1 proteins were analyzed in different lung cancer cell lines. Correlations between the c-FLIP and FoxM1 protein levels were determined using correlation analysis. d Protein levels of FoxM1 and c-FLIP in H460 cells transfected with control vector (pEGFP-N1) or the c-FLIP plasmid. e Protein levels of FoxM1 and c-FLIP in H157 cells stably expressing c-FLIP and control H157 cells. f Protein levels of FoxM1 and c-FLIP in H460 cells transfected with control vector (pcDNA3.1) or the FoxM1 plasmid. g The expression of FoxM1 and c-FLIP at the protein level was evaluated by Western blotting in the FoxM1 knockout and NC H157 cell lines. h The expression of FoxM1 and c-FLIP at the protein level was evaluated by Western blotting in the c-FLIP knockdown and NC cell lines.
To identify the relationship between c-FLIP and FoxM1, cells were transfected with plasmids expressing c-FLIP or FoxM1. The Western blot results showed that transient overexpression of c-FLIP clearly increased the FoxM1 protein expression and phosphorylation levels, which were confirmed in a stable H157 cell line with constitutive c-FLIP expression (Fig. 2d and e, Supplementary Fig. 2a, b). However, there was no change in the protein level of c-FLIP after FoxM1 overexpression in H460 cells (Fig. 2f and Supplementary Fig. 2c). Then, we generated FoxM1 knockout H157 cells using CRISPR/Cas9 gene editing technology and observed the same phenomenon in these FoxM1 knockout cells (Fig. 2g, Supplementary Fig. 2d, e).
Then, four pairs of siRNAs targeting c-FLIP were designed and synthesized. Western blot analysis was used to verify the knockdown efficiency in HCC827 cells (Supplementary Fig. 2f). After c-FLIP expression was knocked down by a pair of siRNAs in three cell lines with comparatively high c-FLIP levels, the levels of FoxM1 and p-FoxM1 were markedly reduced (Fig. 2h and Supplementary Fig. 5g, h, i), consistent with our previous observation (Fig. 2d, e). These data demonstrated that FoxM1 expression and activation were regulated by c-FLIP.
c-FLIP regulates the sensitivity of NSCLC cells to thiostrepton and osimertinib via FoxM1
There is accumulating evidence that c-FLIP induces a drug resistance phenotype via various mechanisms, and FoxM1 mediates drug resistance in gastric cancers and B-lymphoblastic leukemia [22, 23]. We compared the sensitivity of H157 cells stably transfected with Lac-Z or c-FLIP to a panel of small molecule compounds and found that c-FLIP-overexpressing H157 cells displayed a resistance phenotype to various agents (Supplementary Fig. 3a), including thiostrepton (TST), a potent molecular drug targeting FoxM1[24].
As our results indicated that FoxM1 is a key target of c-FLIP, we confirmed the importance of c-FLIP in regulating cell sensitivity to TST. We first assessed the viability of several lung cancer cell lines after TST treatment. Among these cell lines, A549 and HCC827 cells exhibited comparatively high tolerance to TST, and PC9, H157, and H1975 cells exhibited comparatively low tolerance (Fig. 3a). This pattern was consistent with the protein expression levels of c-FLIP and FoxM1 (Fig. 2a, b). Next, we overexpressed c-FLIP in H460 cells via c-FLIP plasmids. The results of the CCK8 assay showed that overexpression of c-FLIP significantly protected H460 cells from the cytotoxic effect of TST (Fig. 3b), consistent with the result observed in H157 cells stably expressing c-FLIP (Fig. 3c).
Fig. 3. c-FLIP promotes resistance to TST by upregulating FoxM1.

a Different lung cancer cells were treated with TST at the indicated concentration for 48 h. Cell viability was evaluated by a CCK8 assay. b H460 cells transfected with pEGFP-c-FLIP or empty control expression vector (pEGFP-N1) were treated with TST at the indicated concentration for 48 h. Cell viability was evaluated by a CCK8 assay. c Stable H157-Lac-Z and c-FLIP cells were treated with TST at the indicated concentrations for 48 h. Cell viability was evaluated by a CCK8 assay. d FoxM1 knockout and control H157 cells were transfected with pEGFP-c-FLIP or control expression vector (pEGFP-N1) for 24 h and were then treated with TST for 48 h. Cell viability was evaluated by a CCK8 assay. e, f FoxM1 knockout and control H157 cells were transfected with pEGFP-c-FLIP or control expression vector (pEGFP-N1) for 72 h. Cell lysates were collected and analyzed by immunoblotting. ImageJ software was used to calculate protein band densities. *P < 0.05, **P < 0.01 vs corresponding control.
To determine whether c-FLIP affects the susceptibility of cells to TST in a FoxM1-dependent manner, c-FLIP expression plasmids were used to transiently transfect FoxM1 knockout or negative control H157 cells. FoxM1 was efficiently decreased in the knockout cells compared with the control cells. In addition, ectopic expression of c-FLIP dramatically increased the levels of FoxM1 and p-FoxM1 in control cells but resulted in no clear changes in FoxM1 knockout cells (Fig. 3e, f). Consistent with this finding, FoxM1 knockout inhibited the antitumor effects of TST in vitro (Fig. 3d, Supplementary Fig. 3b). Ectopic expression of c-FLIP did not influence the antitumor activity of TST in FoxM1 knockout cells compared with control cells (Fig. 3d). These results demonstrated that c-FLIP increased resistance to TST in lung carcinoma cells via its regulatory effect on FoxM1.
As one of the third-generation of EGFR mutant-selective TKIs, osimertinib (AZD9291) has been approved for the treatment of metastatic EGFR T790M mutation-positive NSCLC. Previous studies have shown that FoxM1 can enhance the resistance of HCC827 cells to osimertinib [2]. Our results showed that at the indicated concentrations, osimertinib significantly decreased the colony formation capacity of H157 and HCC827 cells (Fig. 4a–c). Overexpression of c-FLIP significantly enhanced cell viability after osimertinib treatment in H157 cells (Fig. 4d). Similarly, FoxM1 depletion decreased the viability of H157 cells after osimertinib treatment (Fig. 4e). Therefore, these findings indicated that c-FLIP also augmented resistance to osimertinib in NSCLC cells through FoxM1.
Fig. 4. c-FLIP promotes resistance to osimertinib via FoxM1.

a–c Colony formation ability was evaluated using a colony formation assay. H157 and HCC827 cells were treated with osimertinib in complete medium for 10–14 days. The colonies were counted and imaged. d Stable H157-Lac-Z and c-FLIP cells were treated with osimertinib. Cell viability was evaluated by a CCK8 assay. e A CCK8 assay was used to evaluate the effect of osimertinib on the growth of negative control and FoxM1 knockout H157 cells. f Immunoblotting showed the protein levels of FoxM1 and c-FLIP in PC9 and PC9/OR cells. g The viability of PC9 and PC9/OR cells upon osimertinib treatment was evaluated by a CCK8 assay. h The viability of PC9 and PC9/OR cells upon TST treatment was evaluated by a CCK8 assay. The data presented are from three independent experiments (*P < 0.05 vs. the control group; **P < 0.01 vs. the control group).
In osimertinib-resistant PC9 (PC9/OR) and HCC827 (HCC827/OR) cells, abnormal elevation of FoxM1 expression was detected (Fig. 4f, g, Supplementary Fig. 3c, d). Further investigation indicated that PC9/OR cells were more resistant to TST (Fig. 4h). These data also showed the key role of FoxM1 in cells resistant to TST and osimertinib.
c-FLIP attenuates the antitumor activity of TST in a murine xenograft model
We further evaluated the effect of c-FLIP on TST treatment in nude mice bearing H157-Lac-Z and H157-c-FLIP xenografts. The body weights of TST-treated mice and vehicle (DMSO)-treated control mice did not differ (Fig. 5d). TST administration resulted in significant decreases in tumor volume and weight compared with those in the DMSO-treated control group in mice bearing H157 lac-Z xenografts, but only a slight decrease was observed between TST- and DMSO-treated mice bearing H157-c-FLIP xenografts (Fig. 5a–c). These findings indicated that c-FLIP could be essential for determining the chemotherapeutic efficacy of TST in lung cancer even in vivo.
Fig. 5. Overexpression of c-FLIP attenuates the sensitivity of lung cancer to TST in vivo.

a Tumor growth curves. Tumor volumes at each time point are presented as the mean ± SD values. n = 6. b Tumor weights at the time of sacrifice of mice in each group. c Morphological images of each tumor xenograft resected from nude mice in each group. d Body weights of nude mice in each group. n = 6. e HE and immunohistochemical staining of c-FLIP and FoxM1 in tumor tissue from mice. Scale bars = 100 μm. f The bar diagram shows the relative quantitative data as evaluated by densitometry. *P < 0.05 and **P < 0.01 compared with the control group.
Furthermore, the immunohistochemical staining data revealed that the levels of c-FLIP and FoxM1 expression were decreased in the two groups of mice treated with TST compared with the corresponding DMSO-treated mice (Fig. 5e, f). Taken together, these data suggested that c-FLIP and FoxM1 might be used as biomarkers for selecting lung cancer patients who may respond to and benefit from TST treatment.
c-FLIP interacts with FoxM1 and upregulates its protein level
FoxM1 expression can be regulated at the transcriptional and post-transcriptional levels. We next examined the mRNA expression levels of c-FLIP and FoxM1 in several lung cancer cell lines by qRT-PCR. Among these cells, only A549 cells showed significantly high mRNA levels of c-FLIP and FoxM1, while only H1975 cells showed significantly low mRNA levels of c-FLIP and FoxM1 (Fig. 6a). Correlation analysis of quantitative mRNA expression data showed that there was a positive correlation (R2 = 0.8613) between the mRNA expression levels of c-FLIP and FoxM1 (Fig. 6b), which was validated in H157 cells stably expressing c-FLIP (Fig. 6c). Unexpectedly, in H157 and A549 cells transiently expressing c-FLIP, the results of qRT-PCR showed that only c-FLIP mRNA level was significantly increased; there was no change in the FoxM1 mRNA expression level (Supplementary Fig. 4a, b). As the FoxM1 protein can potentially bind to the FoxM1 promoter region, enhancing its own transcription [6], it was speculated that c-FLIP might directly regulate FoxM1 expression at the protein level but not at the mRNA level.
Fig. 6. c-FLIP binds with FoxM1 to regulate its protein expression.

a The c-FLIP and FoxM1 mRNA expression levels were analyzed in human lung cancer cell lines. b Correlation analysis of c-FLIP and FoxM1 mRNA expression in lung cancer cells. P values are shown in the figures. c c-FLIP and FoxM1 mRNA expression levels in control H157 cells and H157 cells with stable c-FLIP overexpression were measured by qRT-PCR. d, e Western blot analysis was used to examine FoxM1 and c-FLIP protein levels in HCC827 cells transfected with si-NC or si-c-FLIP and treated with cycloheximide (CHX) at different time points. The quantitative FoxM1 protein levels were normalized to those of GAPDH. f Western blot analysis was used to examine the protein levels of c-FLIP and FoxM1 in HCC827 cells with or without si-c-FLIP transfection following proteasome inhibition with MG132 for 4 h. g, h FoxM1 was immunoprecipitated (IP) from control H157 cells and H157 cells with stable c-FLIP overexpression after treatment with MG132 (5 μM) for 4 h. Immunoblotting for ubiquitin, FoxM1 and c-FLIP was performed. The relative ubiquitination levels of FoxM1 were normalized to the expression levels of FoxM1. i c-FLIP was immunoprecipitated from stable c-FLIP-overexpressing H157 cells. Immunoblot analysis was performed with the indicated antibodies. j HCC827 cells were transfected with control siRNA and two c-FLIP siRNAs for 48 h. Protein expression was evaluated by immunoblot analysis with the indicated antibodies. k Control H157 cells and H157 cells with stable c-FLIP overexpression were harvested and subjected to WB with the indicated antibodies. A549 cells were transfected with control siRNA and siRNAs targeting FoxM1 (l), β-catenin (m) or p65 (n) for 48 h. Protein expression was evaluated by immunoblot analysis with the indicated antibodies. Statistical significance was determined by two-tailed Student’s t test. *P < 0.05, **P < 0.01 vs corresponding control.
Considering that the changes in the phosphorylation level of FoxM1 were mainly caused by changes in the protein level, we focused on the protein level of FoxM1 (Fig. 2d, e, g, h). Then, the protein stability of FoxM1 in the presence and absence of c-FLIP knockdown was assessed by a cycloheximide chase assay. FoxM1 was more stable in NC siRNA-treated HCC827 cells than in c-FLIP siRNA-treated cells (Fig. 6d, e). Moreover, downregulation of FoxM1 in c-FLIP knockdown cells was partially reversed after treatment with the proteasome inhibitor MG132 (Fig. 6f and Supplementary Fig. 5a). To determine the involvement of c-FLIP in the regulation of the FoxM1 protein, the interaction of these two proteins and FoxM1 ubiquitination were assessed by immunoprecipitation and Western blot analysis. The results showed that c-FLIP interacted with FoxM1 in H157 cells and that c-FLIP overexpression significantly decreased the ubiquitination level of FoxM1 (Fig. 6g, h, i). Based on this finding, the protective role of c-FLIP in FoxM1 proteasomal degradation was suggested.
A positive feedback loop composed of FoxM1, NF-κB p65 and β-catenin was discovered in chronic myelogenous leukemia and is critical to the self-renewal capacity of leukemia stem cells [25]. We confirmed this feedback loop in A549 cells using siRNAs targeting these three genes (Fig. 6l, m, n) and then investigated the participation of this feedback loop in the regulation of FoxM1 by c-FLIP by Western blot analysis. c-FLIP knockdown decreased the protein levels of FoxM1, β-catenin and p65 (Fig. 6j and Supplementary Fig. 5b). Higher protein levels of only FoxM1 and p65 were detected in H157 cells stably overexpressing c-FLIP (Fig. 6k and Supplementary Fig. 5c). No interaction of c-FLIP with either p65 or β-catenin was detected in H157 cells (Fig. 6i), which further demonstrated that c-FLIP regulates FoxM1 expression via the FoxM1/p65/β-catenin feedback loop but not via protein interactions. Taken together, these findings suggested that c-FLIP might regulate FoxM1 through the FoxM1/β-catenin/p65 feedback loop via FoxM1 and p65.
FoxM1 and c-FLIP are targets of TST and osimertinib
TST has been studied in the oncology field for its ability to target FoxM1 [24, 26]. Our data confirmed that TST reduced the expression of FoxM1 at both the mRNA and protein levels and also reduced its phosphorylation level (Fig. 7a–e, Supplementary Fig. 6a–e). In addition, we found that exposure to TST reduced the mRNA and protein levels of c-FLIP in A549 and H157 cells in a time- and dose-dependent manner (Fig. 7a, b, d, e). It is worth noting that TST was able to reduce the c-FLIP protein expression level in A549 cells transiently expressing c-FLIP (Fig. 7c and Supplementary Fig. 6e) in which c-FLIP transcription was controlled by the CMV promoter. These data indicated that c-FLIP was also a target of TST at both the mRNA and protein levels.
Fig. 7. TST and osimertinib decrease the expression levels of c-FLIP and FoxM1.

a A549 and H157 cells were treated with the indicated concentrations of TST. Twenty-four hours after treatment, cell lysates were subjected to immunoblotting. b A549 and H157 cells were treated with TST as indicated. Immunoblot analysis was performed. c A549 cells were transfected with the c-FLIP plasmid for 24 h and were then treated with TST at the indicated concentrations. The protein expression of FoxM1 and c-FLIP was measured by Western blotting after 24 h of treatment. d, e A549 and H157 cells were treated with TST as indicated. The mRNA levels of FoxM1 and c-FLIP were determined by qRT-PCR. f H157 and HCC827 cells were exposed to different concentrations of osimertinib, and the indicated proteins were analyzed by Western blotting. The data are presented as the mean ± SD of triplicate analyses. *P < 0.05 and **P < 0.01.
As a third-generation EGFR inhibitor, osimertinib has been reported to enhance TRAIL-induced apoptosis by targeting c-FLIP [27]. We examined the effect of osimertinib on c-FLIP and FoxM1 protein expression in H157 and HCC827 cells. A decrease in FoxM1 and c-FLIP expression was observed in both osimertinib-treated cell lines through Western blot analysis, which proved that osimertinib exerted an antitumor effect by targeting FoxM1 and c-FLIP (Fig. 7f and Supplementary Fig. 6f).
Discussion
FoxM1 is a key transcription factor in cancer initiation, progression and drug resistance and could potentially be a useful molecular biomarker and therapeutic target for the elimination of cancer cells [28, 29]. In this study, FoxM1 and c-FLIP expression were found to be correlated in human lung cancer tissues and NSCLC cell lines, and the combination of high c-FLIP and FoxM1 expression predicts poorer prognosis in lung cancer patients. c-FLIP binds to FoxM1 and induces its expression by inhibiting its degradation, which results in cellular resistance to TST and osimertinib. Our results also show that overexpression of c-FLIP increases FoxM1 and p65 expression and that c-FLIP knockdown decreases FoxM1, p65 and β-catenin expression. Furthermore, siFoxM1, sip65 or siβ-catenin reduced the expression of the corresponding protein. In previous studies, c-FLIP has been linked to the activation of p65 [30, 31], and there exists a regulatory loop between p65 and FoxM1/β-catenin [25]. Thus, we hypothesize that c-FLIP regulates FoxM1 by two possible mechanisms. First, c-FLIP interacts with FoxM1 to protect it from degradation. Second, c-FLIP positively regulates p65 expression and increases the FoxM1 protein level via the p65/FoxM1/β-catenin feedback loop (Fig. 8).
Fig. 8. Schematic diagram of the proposed model.
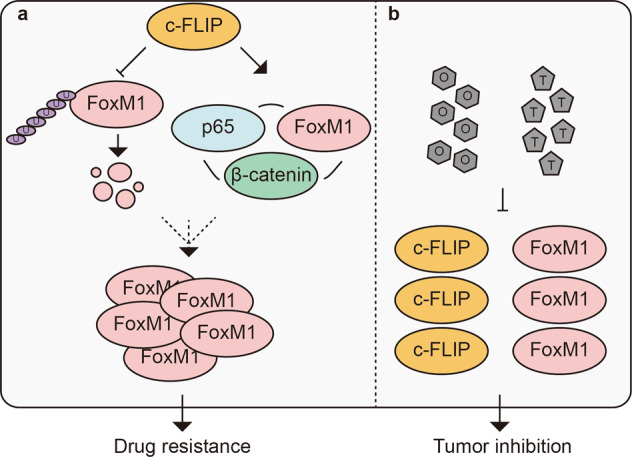
a c-FLIP binds to FoxM1 to prevent FoxM1 protein degradation mediated by ubiquitin-proteasome pathway. c-FLIP enhances the FoxM1/p65/β-catenin positive feedback loop by upregulating FoxM1 and p65 to further increase FoxM1 expression. b TST and osimertinib decrease c-FLIP and FoxM1 expression to inhibit lung cancer growth.
It has been reported that UCHL3 regulates the deubiquitination and stabilization of FoxM1. The authors discovered that knockdown of UCHL3 in BxPC-3 cells dramatically reduced the FoxM1 protein level without affecting the FoxM1 mRNA level [14]. As a putative deubiquitinase, FAM188B regulates FoxM1 by deubiquitinating it, and FAM188B knockdown decreases both the mRNA and protein levels of FoxM1 [11]. c-FLIP has been reported to regulate the ubiquitination of several proteins. In our study, the mRNA level of FoxM1 was elevated in H157 cells stably overexpressing c-FLIP (Fig. 6c) but not in H157 and A549 cells transiently expressing c-FLIP (Supplementary Fig. 6a, b). As FoxM1 was regulated at the transcriptional level via positive feedback, we suspected that not enough time was allowed for changes in the FoxM1 mRNA level to occur in A549 and H157 cells transfected with the c-FLIP plasmid for 24 h because of the time lag effect. Thus, the FoxM1 mRNA level was elevated only in NSCLC cell lines with comparatively high c-FLIP expression and stable overexpression of c-FLIP.
An elevated level of c-FLIP has been reported in many different cancer types and is often correlated with poor prognosis [32]. It has been reported to protect lung cancer cells from the effects of erlotinib through modulation of p65 activity [31] and is related to survival and sensitivity to docetaxel, TRAIL, oxaliplatin and bicalutamide in CRPC cells [33]. In this study, we uncovered a previously unrecognized role of c-FLIP in regulating TST and osimertinib resistance in lung cancer cells. We observed that elevated expression of c-FLIP diminished the sensitivity of the cells to TST and osimertinib through upregulation of FoxM1. These findings revealed a new mechanism of drug resistance and confirmed the key role of c-FLIP and FoxM1 in tumor therapy.
TST is a cyclic oligopeptide antibiotic that is naturally produced by several Streptomyces species. Recently, TST was shown to exhibit antitumor activity in human cancer cells through inhibition of FoxM1 [34]. In addition, it has been reported to reprogram tumor-associated macrophages toward an M1-like phenotype in mice and exhibit potent antitumor activity [35]. Moreover, TST could act as an autophagy inducer to enhance anticancer immune responses to oxaliplatin in immunocompetent mice [36]. Here, we found a new mechanism of TST in cancer therapy in which TST induced downregulation of c-FLIP, which was tightly connected to apoptosis (Fig. 7a–e). In turn, the decrease in the c-FLIP expression level also altered the sensitivity of NSCLC cells to TST, suggesting that c-FLIP suppression is an important event contributing to the antitumor activity of TST. In the investigation of cell sensitivity to TST, we found that cells with higher levels of c-FLIP and FoxM1 expression tended to be resistant to TST, and vice versa. However, the relationship was not strictly reciprocal (Fig. 3a). Although the expression levels of c-FLIP and FoxM1 were two important factors determining cell sensitivity to TST in our present research, further study on the molecular mechanisms of TST is needed to provide new insights into tumor therapy.
Although the protein levels of c-FLIP and FoxM1 were higher in HCC827 cells with mutant EGFR than in H157 cells with wild-type EGFR, osimertinib exhibited more potent inhibitory effects on HCC827 cells (Fig. 4a, b, c). In addition, osimertinib induced a sharper decrease in FoxM1 expression in HCC827 cells than in H157 cells (Fig. 7f). These data indicated that the EGFR mutation status was the main factor determining the cellular response to osimertinib and that the expression level of FoxM1 then tended to influence drug sensitivity in NSCLC cells.
In summary, our present work identifies a new function of c-FLIP in regulating FoxM1 expression by deubiquitinating FoxM1 and enhancing the p65/FoxM1/β-catenin feedback loop, thereby causing NSCLC cells to become resistant to TST and osimertinib (Fig. 8). This discovery increases the possibility of considering the combination of c-FLIP and FoxM1 expression as a predictive biomarker for sensitivity and suggests that the combination of c-FLIP and FoxM1 may be a potential therapeutic target in lung cancer therapy.
Supplementary information
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81702934), CAMS Innovation Fund for Medical Sciences (CIFMS, 2019-I2M-1-003, 2021-I2M-1-030), and Beijing Natural Science Foundation (7202132, 7192041).
Author contributions
SZC designed the project. WDW and SZC designed the experiments and wrote the paper. WDW performed the research. YS and JN assisted with the murine experiments. CW, AMW, and GJL assisted with the cell-based high-throughput drug screen. LS offered the stable H157-Lac-Z and c-FLIP cells.
Competing interests
The authors declare no competing interests.
Footnotes
The original online version of this article was revised: The ‘HCC827’ in the sentence of Results section ‘Among these cells, only HCC827 cells showed significantly high mRNA levels of c-FLIP and FoxM1, while only H1975 cells showed significantly low mRNA levels of c-FLIP and FoxM1 (Fig. 6a)’ should be changed to ‘A549’ according to the original data. For Fig6c, the color of the columns in c-FLIP should be marked in orange. The reason for this error is that when editing Fig.6c in AI software, the color of the columns in c-FLIP was omitted.
Change history
7/23/2024
A Correction to this paper has been published: 10.1038/s41401-024-01343-3
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-022-00905-7.
References
- 1.Ros S, Wright AJ, D’Santos P, Hu DE, Hesketh RL, Lubling Y, et al. Metabolic imaging detects resistance to PI3Kalpha inhibition mediated by persistent FOXM1 expression in ER+ breast cancer. Cancer Cell. 2020;38:516–33e9. 10.1016/j.ccell.2020.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nilsson MB, Sun H, Robichaux J, Pfeifer M, McDermott U, Travers J, et al. A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Sci Transl Med. 2020;12:eaaz4589. [DOI] [PMC free article] [PubMed]
- 3.Zhang B, Zhang Y, Zou X, Chan AW, Zhang R, Lee TK, et al. The CCCTC-binding factor (CTCF)-forkhead box protein M1 axis regulates tumour growth and metastasis in hepatocellular carcinoma. J Pathol. 2017;243:418–30. 10.1002/path.4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shang R, Wang M, Dai B, Du J, Wang J, Liu Z, et al. Long noncoding RNA SLC2A1-AS1 regulates aerobic glycolysis and progression in hepatocellular carcinoma via inhibiting the STAT3/FOXM1/GLUT1 pathway. Mol Oncol. 2020;14:1381–96. 10.1002/1878-0261.12666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang D, Hu G, Du Y, Zhang C, Lu Q, Lv N, et al. Aberrant activation of hedgehog signaling promotes cell proliferation via the transcriptional activation of forkhead Box M1 in colorectal cancer cells. J Exp Clin Cancer Res. 2017;36:23. 10.1186/s13046-017-0491-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liao GB, Li XZ, Zeng S, Liu C, Yang SM, Yang L, et al. Regulation of the master regulator FOXM1 in cancer. Cell Commun Signal. 2018;16:57. 10.1186/s12964-018-0266-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng S, Zhou C, Wang Y, Li H, Sun Y, Shen Z. TRIM6 promotes colorectal cancer cells proliferation and response to thiostrepton by TIS21/FoxM1. J Exp Clin Cancer Res. 2020;39:23. 10.1186/s13046-019-1504-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kongsema M, Zona S, Karunarathna U, Cabrera E, Man EP, Yao S, et al. RNF168 cooperates with RNF8 to mediate FOXM1 ubiquitination and degradation in breast cancer epirubicin treatment. Oncogenesis. 2016;5:e252. 10.1038/oncsis.2016.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li LQ, Pan D, Chen H, Zhang L, Xie WJ. F-box protein FBXL2 inhibits gastric cancer proliferation by ubiquitin-mediated degradation of forkhead box M1. FEBS Lett. 2016;590:445–52. 10.1002/1873-3468.12071 [DOI] [PubMed] [Google Scholar]
- 10.Chen Z, Li L, Xu S, Liu Z, Zhou C, Li Z, et al. A Cdh1-FoxM1-Apc axis controls muscle development and regeneration. Cell Death Dis. 2020;11:180. 10.1038/s41419-020-2375-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi YE, Madhi H, Kim H, Lee JS, Kim MH, Kim YN, et al. FAM188B expression is critical for cell growth via FOXM1 regulation in lung cancer. Biomedicines. 2020;8:465. [DOI] [PMC free article] [PubMed]
- 12.Ning Z, Wang A, Liang J, Xie Y, Liu J, Feng L, et al. USP22 promotes the G1/S phase transition by upregulating FoxM1 expression via beta-catenin nuclear localization and is associated with poor prognosis in stage II pancreatic ductal adenocarcinoma. Int J Oncol. 2014;45:1594–608. 10.3892/ijo.2014.2531 [DOI] [PubMed] [Google Scholar]
- 13.Li XY, Wu HY, Mao XF, Jiang LX, Wang YX. USP5 promotes tumorigenesis and progression of pancreatic cancer by stabilizing FoxM1 protein. Biochem Biophys Res Commun. 2017;492:48–54. 10.1016/j.bbrc.2017.08.040 [DOI] [PubMed] [Google Scholar]
- 14.Song Z, Li J, Zhang L, Deng J, Fang Z, Xiang X, et al. UCHL3 promotes pancreatic cancer progression and chemo-resistance through FOXM1 stabilization. Am J Cancer Res. 2019;9:1970–81. [PMC free article] [PubMed] [Google Scholar]
- 15.Ishioka T, Katayama R, Kikuchi R, Nishimoto M, Takada S, Takada R, et al. Impairment of the ubiquitin-proteasome system by cellular FLIP. Genes Cells. 2007;12:735–44. 10.1111/j.1365-2443.2007.01087.x [DOI] [PubMed] [Google Scholar]
- 16.Lei S, Yang J, Chen C, Sun J, Yang L, Tang H, et al. FLIP(L) is critical for aerobic glycolysis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2016;35:79. 10.1186/s13046-016-0358-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naito M, Katayama R, Ishioka T, Suga A, Takubo K, Nanjo M, et al. Cellular FLIP inhibits beta-catenin ubiquitylation and enhances Wnt signaling. Mol Cell Biol. 2004;24:8418–27. 10.1128/MCB.24.19.8418-8427.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakagiri S, Murakami A, Takada S, Akiyama T, Yonehara S. Viral FLIP enhances Wnt signaling downstream of stabilized beta-catenin, leading to control of cell growth. Mol Cell Biol. 2005;25:9249–58. 10.1128/MCB.25.21.9249-9258.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi P, Oh YT, Deng L, Zhang G, Qian G, Zhang S, et al. Overcoming acquired resistance to AZD9291, a third-generation EGFR inhibitor, through modulation of MEK/ERK-dependent Bim and Mcl-1 degradation. Clin Cancer Res. 2017;23:6567–79. 10.1158/1078-0432.CCR-17-1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang WD, Shang Y, Li Y, Chen SZ. Honokiol inhibits breast cancer cell metastasis by blocking EMT through modulation of Snail/Slug protein translation. Acta Pharmacol Sin. 2019;40:1219–27. 10.1038/s41401-019-0240-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ni J, Wang X, Shang Y, Li Y, Chen S. CD13 inhibition augments DR4-induced tumor cell death in a p-ERK1/2-independent manner. Cancer Biol Med. 2021;18:569–86. [DOI] [PMC free article] [PubMed]
- 22.Li X, Qiu W, Liu B, Yao R, Liu S, Yao Y, et al. Forkhead box transcription factor 1 expression in gastric cancer: FOXM1 is a poor prognostic factor and mediates resistance to docetaxel. J Transl Med. 2013;11:204. 10.1186/1479-5876-11-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Consolaro F, Basso G, Ghaem-Magami S, Lam EW, Viola G. FOXM1 is overexpressed in B-acute lymphoblastic leukemia (B-ALL) and its inhibition sensitizes B-ALL cells to chemotherapeutic drugs. Int J Oncol. 2015;47:1230–40. 10.3892/ijo.2015.3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hegde NS, Sanders DA, Rodriguez R, Balasubramanian S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat Chem. 2011;3:725–31. 10.1038/nchem.1114 [DOI] [PubMed] [Google Scholar]
- 25.Jin B, Wang C, Li J, Du X, Ding K, Pan J. Anthelmintic niclosamide disrupts the interplay of p65 and FOXM1/beta-catenin and eradicates leukemia stem cells in chronic myelogenous leukemia. Clin Cancer Res. 2017;23:789–803. 10.1158/1078-0432.CCR-16-0226 [DOI] [PubMed] [Google Scholar]
- 26.Hardy LR, Pergande MR, Esparza K, Heath KN, Onyuksel H, Cologna SM, et al. Proteomic analysis reveals a role for PAX8 in peritoneal colonization of high grade serous ovarian cancer that can be targeted with micelle encapsulated thiostrepton. Oncogene. 2019;38:6003–16. 10.1038/s41388-019-0842-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi P, Zhang S, Zhu L, Qian G, Ren H, Ramalingam SS, et al. The yhird-generation EGFR inhibitor, osimertinib, promotes c-FLIP degradation, enhancing apoptosis including TRAIL-induced apoptosis in NSCLC cells with activating EGFR mutations. Transl Oncol. 2019;12:705–13. 10.1016/j.tranon.2019.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nandi D, Cheema PS, Jaiswal N, Nag A. FoxM1: Repurposing an oncogene as a biomarker. Semin Cancer Biol. 2018;52:74–84. 10.1016/j.semcancer.2017.08.009 [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Li W, Zhao Y, Kang FW, Zheng X, et al. Members of FOX family could be drug targets of cancers. Pharmacol Ther. 2018;181:183–96. 10.1016/j.pharmthera.2017.08.003 [DOI] [PubMed] [Google Scholar]
- 30.Luebke T, Schwarz L, Beer YY, Schumann S, Misterek M, Sander FE, et al. c-FLIP and CD95 signaling are essential for survival of renal cell carcinoma. Cell Death Dis. 2019;10:384. 10.1038/s41419-019-1609-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471:523–6. 10.1038/nature09870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shirley S, Micheau O. Targeting c-FLIP in cancer. Cancer Lett. 2013;332:141–50. 10.1016/j.canlet.2010.10.009 [DOI] [PubMed] [Google Scholar]
- 33.McCourt C, Maxwell P, Mazzucchelli R, Montironi R, Scarpelli M, Salto-Tellez M, et al. Elevation of c-FLIP in castrate-resistant prostate cancer antagonizes therapeutic response to androgen receptor-targeted therapy. Clin Cancer Res. 2012;18:3822–33. 10.1158/1078-0432.CCR-11-3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiler SME, Pinna F, Wolf T, Lutz T, Geldiyev A, Sticht C, et al. Induction of chromosome instability by activation of yes-associated protein and Forkhead box M1 in liver cancer. Gastroenterology. 2017;152:2037–-51e22. 10.1053/j.gastro.2017.02.018 [DOI] [PubMed] [Google Scholar]
- 35.Hu G, Su Y, Kang BH, Fan Z, Dong T, Brown DR, et al. High-throughput phenotypic screen and transcriptional analysis identify new compounds and targets for macrophage reprogramming. Nat Commun. 2021;12:773. 10.1038/s41467-021-21066-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Xie W, Humeau J, Chen G, Liu P, Pol J, et al. Autophagy induction by thiostrepton improves the efficacy of immunogenic chemotherapy. J Immunother Cancer 2020;8:e000462. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.