

This cross-sectional study investigates if genetic diagnoses in patients with epilepsy are associated with changes in clinical management and subsequent outcomes.
Key Points
Question
How frequently do genetic diagnoses in patients with epilepsy change clinical management, and what are the subsequent patient outcomes?
Findings
In this cross-sectional study among 418 patients with epilepsy who received a genetic diagnosis, 208 (49.8%) had clinical management changes. Of 167 patients with follow-up information, treatment changes were associated with improved patient outcomes in 125 patients (74.9%); the most common improvement was a reduction or elimination of seizures (108 of 167 patients [64.7%]).
Meaning
These findings suggest the use of genetic testing to guide clinical management of patients with epilepsy to improve patient outcomes.
Abstract
Importance
It is currently unknown how often and in which ways a genetic diagnosis given to a patient with epilepsy is associated with clinical management and outcomes.
Objective
To evaluate how genetic diagnoses in patients with epilepsy are associated with clinical management and outcomes.
Design, Setting, and Participants
This was a retrospective cross-sectional study of patients referred for multigene panel testing between March 18, 2016, and August 3, 2020, with outcomes reported between May and November 2020. The study setting included a commercial genetic testing laboratory and multicenter clinical practices. Patients with epilepsy, regardless of sociodemographic features, who received a pathogenic/likely pathogenic (P/LP) variant were included in the study. Case report forms were completed by all health care professionals.
Exposures
Genetic test results.
Main Outcomes and Measures
Clinical management changes after a genetic diagnosis (ie, 1 P/LP variant in autosomal dominant and X-linked diseases; 2 P/LP variants in autosomal recessive diseases) and subsequent patient outcomes as reported by health care professionals on case report forms.
Results
Among 418 patients, median (IQR) age at the time of testing was 4 (1-10) years, with an age range of 0 to 52 years, and 53.8% (n = 225) were female individuals. The mean (SD) time from a genetic test order to case report form completion was 595 (368) days (range, 27-1673 days). A genetic diagnosis was associated with changes in clinical management for 208 patients (49.8%) and usually (81.7% of the time) within 3 months of receiving the result. The most common clinical management changes were the addition of a new medication (78 [21.7%]), the initiation of medication (51 [14.2%]), the referral of a patient to a specialist (48 [13.4%]), vigilance for subclinical or extraneurological disease features (46 [12.8%]), and the cessation of a medication (42 [11.7%]). Among 167 patients with follow-up clinical information available (mean [SD] time, 584 [365] days), 125 (74.9%) reported positive outcomes, 108 (64.7%) reported reduction or elimination of seizures, 37 (22.2%) had decreases in the severity of other clinical signs, and 11 (6.6%) had reduced medication adverse effects. A few patients reported worsening of outcomes, including a decline in their condition (20 [12.0%]), increased seizure frequency (6 [3.6%]), and adverse medication effects (3 [1.8%]). No clinical management changes were reported for 178 patients (42.6%).
Conclusions and Relevance
Results of this cross-sectional study suggest that genetic testing of individuals with epilepsy may be materially associated with clinical decision-making and improved patient outcomes.
Introduction
Genetic etiologies are responsible for seizures in up to 40% of children and 23% of adults with epilepsy.1,2,3,4,5,6,7 Early use of genetic testing for diagnosis and clinical management has been proposed; however, recommendations for genetic testing in individuals with epilepsy are currently limited.8,9,10,11 Additionally, estimates of how often a molecular diagnosis has clinically actionable implications have varied widely, from 60% in a single-center study12 to 20% in a multicenter study,13 although more recent studies estimate 50%.6,14,15 Studies of treatment approaches based on genetic diagnoses have shown improved outcomes in patients with epilepsy.1,5,7,12,16,17,18,19
Our goal was to perform a large-scale, real-world characterization of how often and in which ways genetic testing was associated with clinical management and outcomes in a broad sample of more than 400 adults and children with epilepsy who were referred for genetic testing.
Methods
Study Population and Design
This study was reviewed by the WCG institutional review board and was granted a waiver of authorization and considered exempt because no personal health information was shared on the case report forms (CRFs). Patients of any age were eligible for the study if their health care professionals (HCPs) had ordered genetic testing for epilepsy through Invitae between March 18, 2016, and August 3, 2020, and their genetic test results indicated pathogenic (P) or likely pathogenic (LP) variant(s) in at least 1 gene on the panel. In May 2020, HCPs were invited by email with a link to a CRF (Qualtrics) that remained open through November 2020. The following information was requested: HCP practice characteristics, patients’ clinical information related to the genetic test, subsequent clinical management decisions, and patient outcomes (eMethods in the Supplement). Information from the patients’ test requisition forms at the time of testing (ie, age at testing, sex, self-reported race and ethnicity) and genetic test results were also analyzed. The following race and ethnicity groups were included: Asian, Black, Hispanic, White, multiracial, and other (ie, Arab, Ashkenazi Jewish, Bangladeshi, Kenyan, Mediterranean, Native American, Pacific Islander, Persian, Polish, Punjabi, and Slavic). Patients consented to genetic testing at time of order and opted into research.
Outcomes
For each HCP-completed CRF, the patient was confirmed to have received testing through Invitae based on the test requisition form and deidentified patient identification. Only completed CRF data fields for unique patients with a definitive genetic diagnosis, as confirmed by verifying the genetic test report, were included (eMethods in the Supplement). Free-text responses and/or conflicting CRF responses were reviewed, standardized and reconciled, and coded by 2 authors (A.M., D.M.). For the final study cohort, data were aggregated and evaluated using descriptive statistics and reported according to Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guidelines.20
We assessed response rates in 2 ways to assess ascertainment bias. First, we calculated the overall and mean proportions of eligible patients who had a CRF completed by the HCP when there were 2 or more eligible patients (ie, the HCP response rate). Second, the cohort of patients for whom HCPs had filled out CRFs (study cohort) was compared with all eligible patients with definitive genetic diagnoses without a CRF (nonresponder cohort) to assess differences in sex and age, time lapsed from genetic testing to CRF invitation, and the diagnostic gene. Clinical actionability of genes with diagnostic findings were assessed based on previous literature.6,15
To assess patient outcomes after changes in clinical management (if reported), responses to question 19 on the CRF were assessed, which was designed to capture multiple options related to qualitative changes in seizure frequency, adverse effects, and other changes in condition. HCPs could also report no information on outcomes (ie, not enough time passed to evaluate outcome or yet to follow up with the patient). All patients with a reported outcome (positive or negative) were included in the analyses. In addition, the proportion of patients with any positive outcome or with a reduction/elimination in seizures was calculated.
Statistical Analysis
A quantitative measurement of changes in seizure frequency was assessed in patients for whom a clinical presentation of epilepsy was indicated (question 9) and for whom clinical management was changed due to genetic test results (question 20). The Kolmogorov-Smirnov nonparametric test assessed differences in distribution. Statistical significance was set at a 2-sided P value < .05. Of note, this analysis included patients who may have been reported not to have follow-up available or not enough time for follow-up per responses to question 19.
Results
Characteristics of Responding HCPs and Patients
We contacted 1567 HCPs with 3572 eligible patients. One-hundred sixty-eight HCPs completed CRFs for 479 patients. Sixty-one responses were excluded due to duplicate or incomplete responses (25 responses), nondiagnostic findings (26 responses), invalid patient identifiers (9 responses), and exome sequencing (1 response), leaving a final cohort of 418 unique patients from 415 families (11.7% response rate) (eFigure 1 in the Supplement). Median (IQR) patient age at the time of testing was 4 (1-10) years of age, with an age range of 0 to 52 years; 53.8% (n = 225) were female individuals, and 193 (46.2%) were male individuals. Patients identified with the following race and ethnic groups: 15 Asian (3.6%), 18 Black (4.3%), 81 Hispanic (19.4%), 220 White (52.6%), 30 multiracial (7.2%), and 14 other (3.3%).
On average, each HCP completed CRFs for 2.5 patients (range, 1-13 patients); 83 (49.4%) reported on behalf of 1 patient, 32 (19.0%) for 2 patients, and 53 (31.5%) for 3 or more patients. The clinical specialties of the 168 HCPs were genetics (38 [22.6%]), pediatric neurology (29 [17.3%]), neurology (26 [15.5%]), epilepsy (13 [7.7%]), internal medicine (1 [0.6%]), or multiple specialties (61 [36.3%]). The mean (SD) time from test order to CRF completion was 595 (368) days (range, 27-1673 days; median [IQR], 507 [304-832] days).
Demographic characteristics of the study cohort are reported in the Table. Approximately two-thirds of patients (287 of 418 [68.7%]) were 7 years or younger at the time of genetic testing (Figure 1A). The majority of patients (378 of 418 [90.4%]) were reported to have a clinical presentation of epilepsy, with seizure onset and a clinical diagnosis occurring most frequently in infancy (Figure 1A). A small portion of patients was tested several years after their clinical diagnoses (Figure 1B).
Table. Patient Characteristics.
| Characteristic | Patients, No. (%) |
|---|---|
| No. | 418 |
| Age at testing, y | |
| 0-7 | 287 (68.7) |
| 8-17 | 90 (21.5) |
| ≥18 | 41 (9.8) |
| Age at testing, median (IQR) [range], y | 4 (1-10) [0-52] |
| Sex | |
| Male | 193 (46.2) |
| Female | 225 (53.8) |
| Race and ethnicity | |
| Asian | 15 (3.6) |
| Black | 18 (4.3) |
| Hispanic | 81 (19.4) |
| Multiracial | 30 (7.2) |
| White | 220 (52.6) |
| Othera | 14 (3.3) |
| Unknown | 40 (9.6) |
| How the patient was referred to the HCP | |
| Self | 56 (13.4) |
| Another clinicianb | 351 (84.0) |
| Other | 3 (0.7) |
| Unknown | 8 (1.9) |
| Reason for genetic testing | |
| Clinical presentation of epilepsy | 378 (90.4) |
| Clinical presentation suspicious for genetic etiology | 6 (1.4) |
| Suspected genetic syndrome | 5 (1.2) |
| Family history | 4 (1.0) |
| Multiple reasons | 12 (2.9) |
| Other/unknown | 13 (3.1) |
| HCP practice type | |
| Academic center | 114 (27.3) |
| Children’s hospital | 134 (32.1) |
| Private practice | 56 (13.4) |
| Community hospital | 22 (5.3) |
| Multiplec | 92 (22.0) |
| Country of HCP referral | |
| US | 307 (73.4) |
| Ukraine | 28 (6.7) |
| Chile | 24 (5.7) |
| Romania | 11 (2.6) |
| Peru | 10 (2.4) |
| Mexico | 8 (1.9) |
| Australia | 5 (1.2) |
| Israel | 4 (1.0) |
| New Zealand | 4 (1.0) |
| Lebanon | 3 (0.7) |
| Malaysia | 3 (0.7) |
| Other countriesd | 11 (2.6) |
Abbreviation: HCP, health care provider.
Includes Arab, Ashkenazi Jewish, Bangladeshi, Kenyan, Mediterranean, Native American, Pacific Islander, Persian, Polish, Punjabi, and Slavic.
Includes referrals from the following specialties: pediatrician (189 [53.8%]), neurologist (75 [21.4%]), hospital consult (49 [14.0%]), family medicine (14 [4.0%]), epilepsy specialist (12 [3.4%]), physician assistant (3 [0.8%]), genetics (2 [0.6%]), neonatologist (1 [0.3%]), psychiatrist (1 [0.3%]), and unknown (5 [1.4%]).
Includes combinations of the following practice types: children’s hospital and academic center (56 patients); private practice and children’s hospital (20 patients); private practice, children’s hospital, and academic center (11 patients); private practice and academic center (2 patients); academic center and community hospital (1 patient); children’s hospital, academic center, and community hospital (1 patient); and children’s hospital and community hospital (1 patient).
Includes the following countries: Canada (2 patients), Croatia (2 patients), Ecuador (2 patients), Pakistan (2 patients), Brazil (1 patient), Dominican Republic (1 patient), and Thailand (1 patient).
Figure 1. Age When Seizures Began, at Clinical Diagnosis, and at Time of Genetic Testing.

A, Age at seizure initiation and age of clinical diagnosis were available if respondents indicated that the reason for testing was a clinical presentation of epilepsy. Age was unknown or not applicable for seizure onset (n = 40) and clinical diagnosis (n = 42). B, Violin plots reporting the difference between age at clinical diagnosis and age at time of genetic testing.
Ascertainment Bias
Among the 168 HCPs who completed a CRF, 84 were invited to provide information for more than 1 eligible patient (400 patients in total). A CRF was completed for 313 patients (78.3%), and the mean response rate per clinician was 83.6%.
In addition, the study cohort (418 patients) was compared with all patients with a genetic diagnosis without a completed CRF (2992 patients). No significant differences in sex, age, or lapse from time of testing to CRF request were observed (eTable 1 in the Supplement). Both groups had in common 7 of the 10 genes that yielded the most genetic diagnoses (ie, SCN1A, PRRT2, KCNQ2, MECP2, DEPDC5, PCDH19, and STXBP1). The other genes with frequent genetic diagnoses included SCN2A, NPRL3, and CDKL5 in the nonresponder cohort and TSC2, KCNT1, and SYNGAP1 in the study cohort (eTable 2 in the Supplement). The proportion of patients with genetic findings in genes with established clinical management changes was similar between the study (291 of 422 [69.0%]) and nonresponder (2026 of 3018 [67.1%]) cohorts (eTables 1 and 2 in the Supplement).6,15
Clinical Actionability of Genetic Test Results
Diagnostic findings were found across 76 genes (eTable 3 in the Supplement), and 26 genes (34%) are reported in the literature as being clinically actionable (eTable 2 in the Supplement). HCPs reported that genetic diagnoses led to changed clinical management (question 13) for half of all patients (208 of 418 [49.8%]) (Figure 2A), the majority of whom (170 [81.7%]) had changes implemented within 3 months of receiving the genetic result (question 18). The genes with the most frequent genetic diagnoses that had the highest rates of reported clinical management changes were TSC2 (78.6%), SCN1A (75.0%), MECP2 (62.5%), PCDH19 (60.0%), and KCNQ2 (52.6%) (eTable 4 in the Supplement). The most common changes (question 14) were adding a new medication (78 [21.7%]), initiating medication (51 [14.2%]), being referred to a specialist (48 [13.4%]), monitoring for extraneurological disease (46 [12.8%]), or stopping a medication (42 [11.7%]) (Figure 2B). Among the 48 patients referred to a specialist, the most common referrals were for genetics specialists (11 [22.9%]), cardiologists (9 [18.8%]), and ophthalmologists (5 [18.8%]) (eFigure 2 in the Supplement). Clinical management changes were similar regardless of age at testing, though fewer adults (1 [5.6%]) than pediatric patients (50 [26.3%]) across all age groups were reported to start a new medication (eFigure 3 in the Supplement).
Figure 2. Reported Clinical Actions After a Definitive Genetic Diagnosis.

A, Respondents reported whether the genetic finding influenced a change in clinical management of the patient. B, Those who indicated “yes” selected all of the changes that were implemented or recommended (a patient could have >1 recommendation reported and is counted in each recommendation category). C, Those who indicated “no” reported the reason best describing why no changes were made.
No changes in clinical management were reported for 178 patients (42.6%) (Figure 2A). The most common reason (86 [48.3%]) was that the patient’s current management plan was already consistent with recommendations associated with the genetic test result, noted in question 15 in the eMethods in the Supplement (Figure 2C). Other common reasons were that the genetic test result was not immediately informative for clinical management (45 [25.2%]) or that it could be useful for informing future treatment possibilities (34 [19.1%]) (eg, to monitor a patient’s prognosis).
Patient Outcomes After Changes to Clinical Management
When clinical management was changed after genetic testing, HCPs were asked about patient outcomes (208 of 418 [49.8%]) in question 19. Of the 208 patients, follow-up information was not available for 41 of them (20.2%) because not enough time had passed (in 11 patients) or follow-up with the HCP had not yet occurred (in 30 patients).
The mean (SD) follow-up period from genetic testing to CRF completion among the 167 patients with available outcomes was 584 (365) days. A positive outcome was reported for 125 individuals (74.9%). Among patients with diagnostic findings in genes that were most common to have outcomes reported, positive outcome rates after treatment changes were highest for SCN1A (35 of 42 [83.3%]), PRRT2 (12 of 13 [92.3%]), TSC2 (8 of 9 [88.9%]), KCNQ2 (8 of 8 [100%]), and DEPDC5 (7 of 8 [87.5%]) (Figure 3; eTable 4 in the Supplement). A reduction or elimination of seizures was reported in 108 patients (64.7%) when clinical management was altered due to genetic testing results. Decreases in the severity of other clinical signs were also reported in 37 patients (22.2%) as indicated by free text, including improvements in behavior, development, alertness, school performance, or movement issues. Eleven patients (6.6%) experienced decreases in medication adverse effects.
Figure 3. Patient Outcomes.
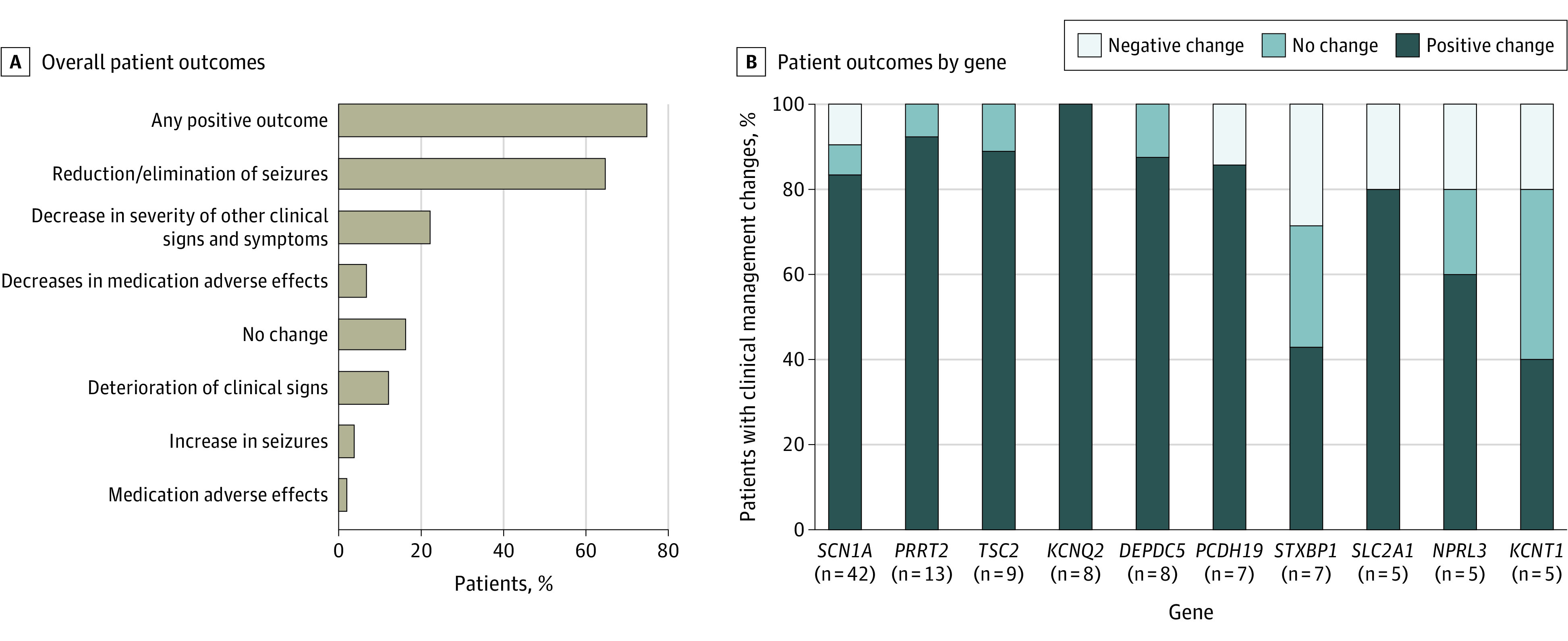
A, Overall patient outcomes after treatment changes were reported in question 13 of the case report form (eMethods in the Supplement). B, Patient outcomes after treatment changes were grouped according to positive outcomes, no change in outcomes, or negative outcomes and analyzed by the gene with diagnostic findings. Outcomes associated with the 11 genes with the highest frequency of diagnostic findings with reported follow-up after treatment changes are shown here. Outcomes for all genes with diagnostic findings that had clinical management changes reported and outcomes available are summarized in eTable 3 in the Supplement.
Seizure frequency before and after clinical management changes were compared among 175 patients who had a clinical presentation of epilepsy reported before genetic testing and who had clinical management changes due to the genetic diagnosis. Overall, seizure frequency decreased in 113 patients (64.6%), did not change for 40 patients (22.9%), and increased for 22 patients (12.6%) (Figure 4). Seizure frequency significantly decreased after genetic result-based management changes (P = 4.60 × 10−9).
Figure 4. Changes in Seizure Frequency After Changes in Clinical Management.

Individuals were grouped by their initial reported seizure frequency (daily, weekly, monthly, annually, or no seizures) before genetic testing and treatment changes on the y-axis, based on health care professional–reported responses to question 9 from the case report form (CRF). The reported seizure frequencies after treatment changes are represented on the x-axis, based on health care professional–reported responses to question 20 from the CRF. Only individuals who had a response provided for both pretreatment and posttreatment were included in this analysis.
Sixteen patients had recommended dietary changes (3, ketogenic diet; 1, modified Atkins diet; 12, not specified); 11 of 16 also had additional clinical management changes. Of 12 patients with sufficient follow-up to report on outcomes (KCNT1, SCN1A, SCN8A, SLC2A1), 8 had additional management changes. Nine patients (75.0%) reported improved seizure control, and 3 patients (25.0%) reported worsening seizures.
The association of age at diagnosis and age at time of testing on patient outcomes was also assessed. Seizure frequency decreased regardless of whether the patients were children (163, aged <18 years) or adults (12, aged ≥18 years) when clinical changes were made (eFigure 4 in the Supplement). In total, 41 patients were clinically diagnosed with epilepsy before the age of 3 years and experienced a substantial delay (of at least 10 years) before genetic testing provided a definitive diagnosis; 19 patients (46.3%) had their clinical management adjusted. Among 15 patients with follow-up information, 9 (60.0%) experienced better seizure control, and none experienced a deterioration in their seizure control.
A small number of patients had reported worsening of outcomes, including deterioration of clinical signs (20 [12.0%]), increase in seizures (6 [3.6%]), or adverse medication effects (3 [1.8%]). Among patients with diagnostic findings in genes that were most common to have outcomes reported, negative outcome rates after treatment changes were highest in STXBP1 (2 of 7 [28.6%]) and PCDH19 (1 of 7 [14.3%]).
Discussion
In this large, retrospective, real-world, international cross-sectional analysis, we found that a definitive genetic diagnosis was associated with changes in clinical management in approximately half the patients with epilepsy, consistent with previous reports.6,15 Although these studies used panel testing, an analysis of diagnostic findings for patients with epilepsy using exome sequencing reported that the majority of findings were in the same genes tested by panels,21 suggesting that the results from this report may be applicable to other comprehensive genetic testing modalities for epilepsy. In this study, the majority of patients with management changes was associated with a consequent reduction or elimination of seizures. Among the other half of patients, for whom clinical management was not changed, HCPs reported that genetic test results would have been beneficial, but treatment was already consistent with the gene finding (48.3%) or that they could be informative in future clinical decision-making (44.3%). Strikingly, these reasons account for more than 90% of patients without changes to their clinical management, suggesting that the genetic diagnoses were still informative.
Genetic testing in epilepsy has additional demonstrated benefits beyond informing diagnoses and improving clinical management. It can clarify diagnostic uncertainty (eg, when diagnostic criteria are unclear), especially in young children such as infants with Dravet syndrome.22 When genetic testing was used as a first-line tool for epilepsy, time to diagnosis decreased by 98%, and health care costs decreased by 70%.23 Further, well-controlled seizures result in reduced average annual overall health care costs for all types of seizures ($23 238 in 2007 to $13 839 in 2009) and improved quality of life compared with uncontrolled seizures.24,25,26 This highlights potential annual savings in care of $9399, inflation-adjusted to $11 644 in 2021, that might be realized if an accurate diagnosis leads to a change in treatment that controls seizures.25 These observations, together with the finding that genetic testing for epilepsy is a cost-effective strategy in patients without an etiology,27 demonstrate the benefits of standardizing the use of genetic testing for epilepsy.
Though historically limited to patients with a high index of suspicion due to cost and other barriers,28,29,30,31 genetic testing is now far more accessible due to advances in DNA sequencing technologies. Although genetic testing for epilepsy is not yet standard of care, some professional societies have recently published guidelines that advocate genetic testing after other types of diagnostic tests and referrals to specialists.10 The paucity of evidence demonstrating direct clinical benefit from genetic testing has precluded the development of explicit guidance for clinicians to routinely use genetic testing in the diagnosis and management of individuals with epilepsy. Data from this study may inform more extensive, case-controlled, prospective interventional studies to determine precision therapies based on the genetic etiology of a patient’s seizures.
Beneficial outcomes after genetic testing were observed in patients of all ages in this study. Many (72 patients) were reported to be seizure free after management changes based on questions 19 or 20, whereas some of these individuals had genetic findings associated with self-limiting seizures (eg, PRRT2), others had findings in which the management change likely contributed to better seizure control (eg, γ-aminobutyric acid modulator for TSC2; sodium channel blockers for SCN2A; pyridoxine for ALDH7A1) (eTable 5 in the Supplement). Although a causal relationship could not be established based on the data reported here due to limitations of the survey design, the findings do suggest that a positive genetic test result may inform treatment decisions that are directly associated with patient outcomes. A reduction in seizures was also observed regardless of age, demonstrating the lifelong association of a genetic diagnosis for treatment decisions and subsequent outcomes. It is increasingly clear that genetic testing provides necessary information to confirm clinical diagnoses and that early use in the diagnostic journey may be associated with management and outcomes.
Whereas precision medicine in oncology has advanced rapidly, making clinical trials and genetics-informed treatment available to patients with cancer, a similar transformation in rare hereditary diseases is still at its early stages. The number of genetics-informed approaches to treating epilepsy is limited, as most antiseizure medications are prescribed based on seizure type.18 However, an understanding of the underlying genetic etiology of a patient’s seizure disorder can inform the selection of the best antiseizure medication based on its mechanism of action.32 The first uses of precision medicine for epilepsy were diet modifications for seizures caused by biochemical defects,33,34,35,36,37,38 and these modifications were associated with positive outcomes in most patients. Recent advances in precision medicine based on specific genetic etiologies, including enzyme replacement therapies and other types of gene or gene-product targeted therapies, are now available or in clinical trials for individuals diagnosed with CLN2 Batten disease, Dravet syndrome, spinal muscular atrophy, Duchenne muscular dystrophy, and sickle cell disease.39,40,41,42,43,44,45,46,47 As additional gene-specific therapies become available for epilepsy, genetic testing will be increasingly necessary to quickly identify those who may benefit from them.
Limitations
Retrospective CRF-based studies pose certain limitations. The responding HCPs may or may not have been the primary HCP managing patient care. Another concern is ascertainment bias, in which HCPs could have been more likely to respond if they had patients with changes in management or with positive clinical outcomes. Although ascertainment bias cannot be ruled out, we have some reassurance that our findings could be representative because the response rate was high among HCPs who had more than 1 eligible patient, and the characteristics between the study and nonresponder cohorts, including the representation of diagnostic genes with established clinical management changes, were similar. Another limitation is the varying time between genetic testing and CRF completion, resulting in limited or no follow-up information for some patients. The design of the CRF only allowed for collection of clinical management changes and outcomes at a single time point, prohibiting deeper analysis of whether specific clinical management changes occurred at once or over the course of the follow-up period and whether these changes were directly linked to outcomes. For some disorders, such as those associated with PCDH19 and STXBP1, there is a natural worsening or improvement of seizures through the natural history of that disease and/or a lack of reported best practices; therefore, we should be cautious when interpreting causality for these gene-specific findings. Lastly, the CRF design precluded the ability to compare outcomes with control cohorts, such as patients who had a diagnostic genetic finding but no clinical management changes or patients without genetic testing who were treated based on standard clinical practices. Future studies that collect and compare outcomes in these cohorts may provide additional clarity on the benefits of genetic testing.
Conclusions
Based on this large cross-sectional study of clinician-reported outcomes in patients with epilepsy, our findings address a long-standing question about the clinical benefits of genetic testing for epilepsy. Just as clinical trials and case-controlled studies were designed more than 20 years ago to assess the safety and efficacy of early epilepsy treatments based on seizure type, renewed efforts should focus on understanding how current and emerging therapies can improve patient outcomes based on the known genetic etiology of a patient’s disease. The data from our study suggest the development of recommendations related to the use of genetic testing in the clinical evaluation of all individuals with epilepsy to potentially guide clinical decision-making, improve patient outcomes, and save health care dollars.
eMethods.
eTable 1. Demographic Information for Study Cohort and Nonresponder Cohort
eTable 2. Frequency of Positive Findings by Gene Among the Study Cohort (Responders) and the Nonresponder Cohort
eTable 3. Genes With Definitive Genetic Diagnoses in This Study Cohort
eTable 4. Patient Outcomes After Treatment Changes by Gene With Diagnostic Finding
eTable 5. Summary of Treatment Changes Noted for Patients With Seizures That Resolved
eFigure 1. Flowchart of Cohort Development
eFigure 2. Types of Referrals to Specialists
eFigure 3. Clinical Management Changes by Age
eFigure 4. Frequency of Seizures Before and After Changes in Clinical Management in Pediatric and Adult Cohorts
References
- 1.Borlot F, Regan BM, Bassett AS, Stavropoulos DJ, Andrade DM. Prevalence of pathogenic copy number variation in adults with pediatric-onset epilepsy and intellectual disability. JAMA Neurol. 2017;74(11):1301-1311. doi: 10.1001/jamaneurol.2017.1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shellhaas RA, Wusthoff CJ, Tsuchida TN, et al. ; Neonatal Seizure Registry . Profile of neonatal epilepsies: characteristics of a prospective US cohort. Neurology. 2017;89(9):893-899. doi: 10.1212/WNL.0000000000004284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berg AT, Coryell J, Saneto RP, et al. Early-life epilepsies and the emerging role of genetic testing. JAMA Pediatr. 2017;171(9):863-871. doi: 10.1001/jamapediatrics.2017.1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindy AS, Stosser MB, Butler E, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. 2018;59(5):1062-1071. doi: 10.1111/epi.14074 [DOI] [PubMed] [Google Scholar]
- 5.Borlot F, de Almeida BI, Combe SL, Andrade DM, Filloux FM, Myers KA. Clinical utility of multigene panel testing in adults with epilepsy and intellectual disability. Epilepsia. 2019;60(8):1661-1669. doi: 10.1111/epi.16273 [DOI] [PubMed] [Google Scholar]
- 6.Truty R, Patil N, Sankar R, et al. Possible precision medicine implications from genetic testing using combined detection of sequence and intragenic copy number variants in a large cohort with childhood epilepsy. Epilepsia Open. 2019;4(3):397-408. doi: 10.1002/epi4.12348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johannesen KM, Nikanorova N, Marjanovic D, et al. Utility of genetic testing for therapeutic decision-making in adults with epilepsy. Epilepsia. 2020;61(6):1234-1239. doi: 10.1111/epi.16533 [DOI] [PubMed] [Google Scholar]
- 8.Ottman R, Hirose S, Jain S, et al. Genetic testing in the epilepsies–report of the ILAE Genetics Commission. Epilepsia. 2010;51(4):655-670. doi: 10.1111/j.1528-1167.2009.02429.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Committee on Bioethics; Committee on Genetics, and; American College of Medical Genetics and; Genomics Social; Ethical; Legal Issues Committee . Ethical and policy issues in genetic testing and screening of children. Pediatrics. 2013;131(3):620-622. doi: 10.1542/peds.2012-3680 [DOI] [PubMed] [Google Scholar]
- 10.Jain P, Andrade D, Donner E, et al. Development of criteria for epilepsy genetic testing in Ontario, Canada. Can J Neurol Sci. 2019;46(1):7-13. doi: 10.1017/cjn.2018.341 [DOI] [PubMed] [Google Scholar]
- 11.Nabbout R, Kuchenbuch M. Impact of predictive, preventive and precision medicine strategies in epilepsy. Nat Rev Neurol. 2020;16(12):674-688. doi: 10.1038/s41582-020-0409-4 [DOI] [PubMed] [Google Scholar]
- 12.Hoelz H, Herdl C, Gerstl L, et al. Impact on clinical decision-making of next-generation sequencing in pediatric epilepsy in a tertiary epilepsy referral center. Clin EEG Neurosci. 2020;51(1):61-69. doi: 10.1177/1550059419876518 [DOI] [PubMed] [Google Scholar]
- 13.Balestrini S, Chiarello D, Gogou M, et al. Real-life survey of pitfalls and successes of precision medicine in genetic epilepsies. J Neurol Neurosurg Psychiatry. 2021;92(10):1044-1052. doi: 10.1136/jnnp-2020-325932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Møller RS, Hammer TB, Rubboli G, Lemke JR, Johannesen KM. From next-generation sequencing to targeted treatment of nonacquired epilepsies. Expert Rev Mol Diagn. 2019;19(3):217-228. doi: 10.1080/14737159.2019.1573144 [DOI] [PubMed] [Google Scholar]
- 15.McKnight D, Bristow SL, Truty RM, et al. Multigene panel testing in a large cohort of adults with epilepsy: diagnostic yield and clinically actionable genetic findings. Neurol Genet. 2021;8(1):e650. doi: 10.1212/NXG.0000000000000650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ream MA, Mikati MA. Clinical utility of genetic testing in pediatric drug-resistant epilepsy: a pilot study. Epilepsy Behav. 2014;37:241-248. doi: 10.1016/j.yebeh.2014.06.018 [DOI] [PubMed] [Google Scholar]
- 17.Symonds JD, Zuberi SM, Johnson MR. Advances in epilepsy gene discovery and implications for epilepsy diagnosis and treatment. Curr Opin Neurol. 2017;30(2):193-199. doi: 10.1097/WCO.0000000000000433 [DOI] [PubMed] [Google Scholar]
- 18.Perucca E. Antiepileptic drugs: evolution of our knowledge and changes in drug trials. Epileptic Disord. 2019;21(4):319-329. [DOI] [PubMed] [Google Scholar]
- 19.Symonds JD, Zuberi SM, Stewart K, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. 2019;142(8):2303-2318. doi: 10.1093/brain/awz195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP; STROBE Initiative . The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet. 2007;370(9596):1453-1457. doi: 10.1016/S0140-6736(07)61602-X [DOI] [PubMed] [Google Scholar]
- 21.Snoeijen-Schouwenaars FM, van Ool JS, Verhoeven JS, et al. Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia. 2019;60(1):155-164. doi: 10.1111/epi.14618 [DOI] [PubMed] [Google Scholar]
- 22.Hattori J, Ouchida M, Ono J, et al. A screening test for the prediction of Dravet syndrome before 1 year of age. Epilepsia. 2008;49(4):626-633. doi: 10.1111/j.1528-1167.2007.01475.x [DOI] [PubMed] [Google Scholar]
- 23.Oates S, Tang S, Rosch R, et al. Incorporating epilepsy genetics into clinical practice: a 360° evaluation. NPJ Genom Med. 2018;3:13. doi: 10.1038/s41525-018-0052-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harden CL, Maroof DA, Nikolov B, et al. The effect of seizure severity on quality of life in epilepsy. Epilepsy Behav. 2007;11(2):208-211. doi: 10.1016/j.yebeh.2007.05.002 [DOI] [PubMed] [Google Scholar]
- 25.Cramer JA, Wang ZJ, Chang E, et al. Health care utilization and costs in adults with stable and uncontrolled epilepsy. Epilepsy Behav. 2014;31:356-362. doi: 10.1016/j.yebeh.2013.09.046 [DOI] [PubMed] [Google Scholar]
- 26.Toledo M, Brandt C, Quarato PP, et al. Long-term safety, efficacy, and quality of life during adjunctive brivaracetam treatment in patients with uncontrolled epilepsy: an open-label follow-up trial. Epilepsy Behav. 2021;118:107897. doi: 10.1016/j.yebeh.2021.107897 [DOI] [PubMed] [Google Scholar]
- 27.Sánchez Fernández I, Loddenkemper T, Gaínza-Lein M, Sheidley BR, Poduri A. Diagnostic yield of genetic tests in epilepsy: a meta-analysis and cost-effectiveness study. Neurology. 2019;92(5):e418-e428. doi: 10.1212/WNL.0000000000006850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klitzman R, Chung W, Marder K, et al. Attitudes and practices among internists concerning genetic testing. J Genet Couns. 2013;22(1):90-100. doi: 10.1007/s10897-012-9504-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ream MA, Patel AD. Obtaining genetic testing in pediatric epilepsy. Epilepsia. 2015;56(10):1505-1514. doi: 10.1111/epi.13122 [DOI] [PubMed] [Google Scholar]
- 30.Chambers CP, Echenique F, Saito K. Testing theories of financial decision-making. Proc Natl Acad Sci U S A. 2016;113(15):4003-4008. doi: 10.1073/pnas.1517760113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kutscher EJ, Joshi SM, Patel AD, Hafeez B, Grinspan ZM. Barriers to genetic testing for pediatric Medicaid beneficiaries with epilepsy. Pediatr Neurol. 2017;73:28-35. doi: 10.1016/j.pediatrneurol.2017.04.014 [DOI] [PubMed] [Google Scholar]
- 32.Bayat A, Bayat M, Rubboli G, Møller RS. Epilepsy syndromes in the first year of life and usefulness of genetic testing for precision therapy. Genes (Basel). 2021;12(7):1051. doi: 10.3390/genes12071051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lefevre F, Aronson N. Ketogenic diet for the treatment of refractory epilepsy in children: a systematic review of efficacy. Pediatrics. 2000;105(4):E46. doi: 10.1542/peds.105.4.e46 [DOI] [PubMed] [Google Scholar]
- 34.Bergqvist AGC, Schall JI, Gallagher PR, Cnaan A, Stallings VA. Fasting versus gradual initiation of the ketogenic diet: a prospective, randomized clinical trial of efficacy. Epilepsia. 2005;46(11):1810-1819. doi: 10.1111/j.1528-1167.2005.00282.x [DOI] [PubMed] [Google Scholar]
- 35.Henderson CB, Filloux FM, Alder SC, Lyon JL, Caplin DA. Efficacy of the ketogenic diet as a treatment option for epilepsy: meta-analysis. J Child Neurol. 2006;21(3):193-198. doi: 10.2310/7010.2006.00044 [DOI] [PubMed] [Google Scholar]
- 36.Neal EG, Chaffe H, Schwartz RH, et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 2008;7(6):500-506. doi: 10.1016/S1474-4422(08)70092-9 [DOI] [PubMed] [Google Scholar]
- 37.Levy RG, Cooper PN, Giri P. Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst Rev. 2012;(3):CD001903. doi: 10.1002/14651858.CD001903.pub2 [DOI] [PubMed] [Google Scholar]
- 38.Kossoff EH, Wang HS. Dietary therapies for epilepsy. Biomed J. 2013;36(1):2-8. doi: 10.4103/2319-4170.107152 [DOI] [PubMed] [Google Scholar]
- 39.Schulz A, Ajayi T, Specchio N, et al. ; CLN2 Study Group . Study of intraventricular cerliponase alfa for CLN2 disease. N Engl J Med. 2018;378(20):1898-1907. doi: 10.1056/NEJMoa1712649 [DOI] [PubMed] [Google Scholar]
- 40.Fortunato F, Rossi R, Falzarano MS, Ferlini A. Innovative therapeutic approaches for Duchenne muscular dystrophy. J Clin Med. 2021;10(4):820. doi: 10.3390/jcm10040820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi E, Koo T. CRISPR technologies for the treatment of Duchenne muscular dystrophy. Mol Ther. 2021;29(11):3179-3191. doi: 10.1016/j.ymthe.2021.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376(5):429-439. doi: 10.1056/NEJMoa1611770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niihara Y, Miller ST, Kanter J, et al. ; Investigators of the Phase 3 Trial of l-Glutamine in Sickle Cell Disease . A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. 2018;379(3):226-235. doi: 10.1056/NEJMoa1715971 [DOI] [PubMed] [Google Scholar]
- 44.Vichinsky E, Hoppe CC, Ataga KI, et al. ; HOPE Trial Investigators . A Phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 2019;381(6):509-519. doi: 10.1056/NEJMoa1903212 [DOI] [PubMed] [Google Scholar]
- 45.Mercuri E, Darras BT, Chiriboga CA, et al. ; CHERISH Study Group . Nusinersen vs sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625-635. doi: 10.1056/NEJMoa1710504 [DOI] [PubMed] [Google Scholar]
- 46.Finkel RS, Mercuri E, Darras BT, et al. ; ENDEAR Study Group . Nusinersen vs sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-1732. doi: 10.1056/NEJMoa1702752 [DOI] [PubMed] [Google Scholar]
- 47.An Open-Label Study to Investigate the Safety of Single and Multiple Ascending Doses in Children and Adolescents With Dravet Syndrome. ClinicalTrials identifier: NCT04442295. Updated March 4, 2022. Accessed January 7, 2022. https://www.clinicaltrials.gov/ct2/show/NCT04442295
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods.
eTable 1. Demographic Information for Study Cohort and Nonresponder Cohort
eTable 2. Frequency of Positive Findings by Gene Among the Study Cohort (Responders) and the Nonresponder Cohort
eTable 3. Genes With Definitive Genetic Diagnoses in This Study Cohort
eTable 4. Patient Outcomes After Treatment Changes by Gene With Diagnostic Finding
eTable 5. Summary of Treatment Changes Noted for Patients With Seizures That Resolved
eFigure 1. Flowchart of Cohort Development
eFigure 2. Types of Referrals to Specialists
eFigure 3. Clinical Management Changes by Age
eFigure 4. Frequency of Seizures Before and After Changes in Clinical Management in Pediatric and Adult Cohorts