

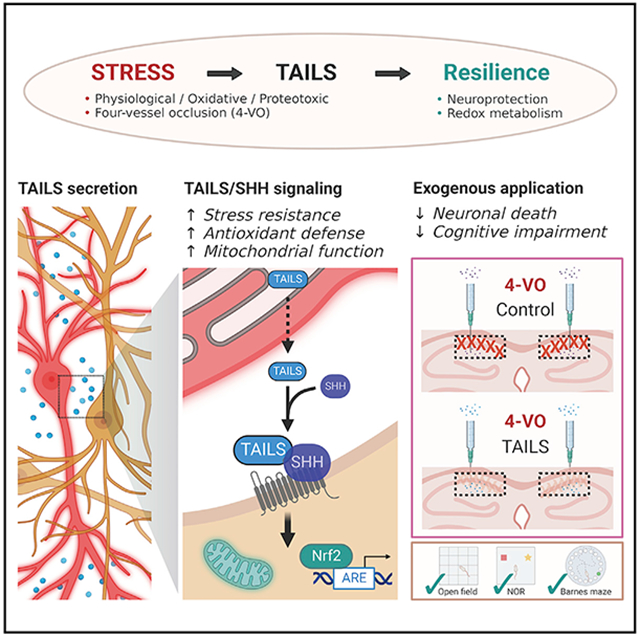
SUMMARY
Cells possess several conserved adaptive mechanisms to respond to stress. Stress signaling is initiated to reestablish cellular homeostasis, but its effects on the tissue or systemic levels are far less understood. We report that the secreted luminal domain of the endoplasmic reticulum (ER) stress transducer CREB3L2 (which we name TAILS [transmissible activator of increased cell livability under stress]) is an endogenous, cell non-autonomous activator of neuronal resilience. In response to oxidative insults, neurons secrete TAILS, which potentiates hedgehog signaling through direct interaction with Sonic hedgehog (SHH) and its receptor PTCH1, leading to improved antioxidant signaling and mitochondrial function in neighboring neurons. In an in vivo model of ischemic brain injury, administration of TAILS enables survival of CNS neurons and fully preserves cognitive function in behavioral tests. Our findings reveal an SHH-mediated, cell non-autonomous branch of cellular stress signaling that confers resilience to oxidative stress in the mature brain, providing protection from ischemic neurodegeneration.
Graphical Abstract
In brief
Whether and how neurons can communicate stress to other neurons is poorly understood. Chung et al. demonstrate that oxidative stress in neurons leads to secretion of cell non-autonomous stress signaling protein. This protein, TAILS, induces neuronal resilience against cell stress in recipient cells in an SHH-dependent manner.
INTRODUCTION
Cells have developed several mechanisms to react to stress elicited by extrinsic cues or by disturbances of protein synthesis and secretion. A group of these mechanisms, which share as a common, pivotal element, phosphorylation of the elongation initiation factor 2α (eIF2α), is called the integrated stress response (ISR) (Harding et al., 2003; Donnelly et al., 2013; Costa-Mattioli and Walter, 2020). Activation of the ISR allows cells to adapt to a changed environment and reestablish or maintain health. In the central nervous system (CNS), the ISR is crucial during long-term memory formation (Sharma et al., 2020) and central in many cognitive and neurodegenerative disorders (Costa-Mattioli and Walter, 2020). Although the majority of studies of the ISR have focused on its intracellular, cell-autonomous effects (Diaz-Hung and Hetz, 2022), there is evidence of the existence of cell non-autonomous stress signaling (Taylor et al., 2014; Zanetti et al., 2016; Mahadevan et al., 2011; Smith et al., 2020). Despite demonstrations of cell non-autonomous stress signaling in the Caenorhabditis elegans nervous system and in cancer and of secreted proinflammatory molecules in response to astrocytic cell stress, it remains an open question whether neurons can initiate transmissible stress signaling (Sprenkle et al., 2019; van Ziel et al., 2020).
Among the many effectors of the ISR is a group of transmembrane endoplasmic reticulum (ER) stress transducers, the old astrocyte specifically induced substance (OASIS) family of transcription factors (Kondo et al., 2011). The N-terminal, cytoplasmic domains of OASIS family members comprise basic leucine zipper domain (bZIP) transcription factors, whereas their ER-luminal domains share no significant homologies among each other or with any other proteins. Cleavage by site 1 and/or site 2 proteases (S1Ps and S2Ps, respectively) releases the transcription factors from the ER. A peculiarity is that the ER-luminal domains of OASIS, CREB3L2, and CREB4 can be secreted from the ER to the extracellular space as soluble proteins, and the secreted luminal domain of CREB3L2 promotes cell proliferation of chondrocytes and axon growth in dorsal root ganglion neurons via interaction with Indian hedgehog (IHH) and Sonic hedgehog (SHH), respectively (Saito et al., 2014; McCurdy et al., 2019).
The generation of the secreted ER-luminal domain of CREB3L2 as a by-product of cell stress signaling and its interaction with hedgehog molecules are suggestive of a potential role as a cell non-autonomous stress signal. Although identified as a morphogen (Nüsslein-Volhard and Wieschaus, 1980), SHH also acts as an axon guidance molecule, mitogen, and regulator of astrocyte function (Charron et al., 2003; Wechsler-Reya and Scott, 1999; Garcia et al., 2010). A role of SHH in neuroprotection, especially against oxidative stress, has been discussed (Yao et al., 2016) and connected to SHH’s ability to increase autophagy and mitochondrial mass in hippocampal neurons (Yao et al., 2017; Petralia et al., 2013). Administration of exogenous SHH or of a Smoothened (SMO) agonist partially prevented neurodegeneration and improved functional recovery in rodent models of focal ischemia (Bambakidis et al., 2012; Huang et al., 2013; Chechneva et al., 2014). In contrast, the role of endogenous SHH as a neuroprotectant and especially whether its level and activity in the mature nervous system are sufficient to functionally rescue neurons from oxidative stress-induced degeneration remain unaddressed.
Here we report that the luminal domain of CREB3L2 (hereafter referred to as TAILS [transmissible activator of increased cell livability under stress]) increases the resilience of neurons to cell stress in vitro and in vivo by potentiating endogenous SHH signaling. The cell stress-induced generation and secretion of TAILS demonstrates the existence of a cell non-autonomous branch of the ISR in the CNS.
RESULTS
TAILS is a survival factor for neurons
During an unrelated experiment, we observed that cells constitutively expressing the secreted C-terminal domain of the ER stress transducer CREB3L2 (McCurdy et al., 2019) exhibited remarkable robustness to various kinds of stress. To distinguish this effect from the functionally unrelated transcription factor CREB3L2, encoded by the N-terminal part of a common precursor protein, we now refer to the secreted luminal domain as TAILS. Motivated by our initial observation, we treated rat primary hippocampal neurons with TAILS-enriched or control conditioned medium and challenged the neurons with 4-hydroxynonenal (HNE), an end product of lipid peroxidation that is a second messenger of oxidative stress (Esterbauer et al., 1991). HNE treatment resulted in a significant increase in TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling)-positive neurons that was greatly reduced by the presence of TAILS-conditioned medium (Figures 1A and 1B). Likewise, TAILS prevented the increase of cleaved (i.e., active) forms of the apoptosis markers caspase-3 and poly(ADP-ribose) polymerase (PARP) in HNE-treated mixed hippocampal and cortical neurons (Figures 1C and 1D). In each assay, the neuroprotective effect of the TAILS-conditioned medium was exclusively detected in HNE-treated neurons without affecting control cells. Next, we purified recombinant TAILS protein from conditioned medium of stably transfected HEK293 cells. Using viability of HNE-treated hippocampal neurons as the readout, we established a half-maximal effective concentration (EC50) for TAILS of approximately 150 nM (Figure 1E). Treatment of HNE-treated neurons with purified recombinant TAILS resulted in a concentration-dependent decrease in cleaved caspase-3 level (Figures 1F and 1G). Conversely, blocking endogenous TAILS using a neutralizing antibody greatly reduced neuronal resilience against HNE, as seen in a marked rise in cleaved caspase-3 (Figures 1H and 1I). Consistent with the caspase-3 results, cell death in HNE-treated neurons was reduced by recombinant TAILS but elevated upon addition of the anti-TAILS antibody (Figures 1J-1M). Our results demonstrate that TAILS is necessary and sufficient to increase survival of neurons under HNE-induced stress.
Figure 1. TAILS is a stress-generated survival factor for neurons.

(A) Cell death analysis by TUNEL assay of rat hippocampal neurons treated with HNE (10 μM), TAILS-conditioned medium (CM), or vehicle controls.
(B) Quantification of (A). Means ± SEM (n = 4 biological replicates; 1,022–2,067 neurons per experiment). two-way ANOVA, Tukey’s multiple comparisons test. ***p < 0.001.
(C) Western blot analysis for molecular markers of cell death in HNE-treated neurons co-treated with TAILS CM. FL, full-length; clv, cleaved.
(D) Quantification of (C). Mean ± SEM (n = 4 biological replicates). two-way ANOVA, Tukey’s multiple comparisons test. **p < 0.01, ***p < 0.001.
(E) Dose-response curve of cell viability in hippocampal neurons treated with recombinant TAILS proteins. Non-linear modeling was used to determine the EC50 (149.4 nM) of cell viability of neurons under treatment with varying concentrations of recombinant TAILS. Mean ± SEM (n = 5 replicates).
(F) Western blot for clv CASP3 in neurons treated with recombinant TAILS protein (50 ng mL−1).
(G) Quantification of (F). Mean ± SEM (n = 3–6 biological replicates). two-way ANOVA, Holm-Sidak’s multiple comparisons test. ***p < 0.001.
(H) Western blot for clv CASP3 in neurons treated with TAILS or GFP antibody. +, antibody-treated at 0.1 μg mL−1; ++, antibody-treated at 0.2 μg mL−1.
(I) Quantification of (H). Antibody-treated at 0.2 μg mL−1. Means ± SEM (n = 4 biological replicates). two-way ANOVA, Holm-Sidak’s multiple comparisons test. *p < 0.05; **p = 0.0041.
(J) Cell death analysis of HNE-treated neurons co-treated with recombinant TAILS protein by TUNEL assay.
(K) Quantification of (J). Means ± SEM (n = 4 biological replicates; 494–964 neurons per experiment). two-way ANOVA, Tukey’s multiple comparisons test. ***p < 0.001.
(L) Cell death analysis by TUNEL assay in neurons co-treated with anti-TAILS or anti-GFP antibodies upon HNE challenge.
(M) Quantification of (L). Means ± SEM (n = 4 biological replicates; 543–937 neurons per experiment). two-way ANOVA, Tukey’s multiple comparisons test. **p < 0.01, ***p < 0.001. Scale bars, 20 μm.
TAILS is generated and secreted in response to various stressors
Based on the finding that blockade of endogenous TAILS renders neurons more susceptible to cell stress, we next investigated its presence in the mature CNS. TAILS was easily detected in the papain-digested fraction of rat brain comprising neurons and glia as well as in a preparation of extracellular matrix (Figure 2A). The four-vessel occlusion (4-VO) model of global ischemia induces selective death of CA1 hippocampal pyramidal neurons with around 48 h of delayed onset (Pulsinelli et al., 1982). Western blot analysis of CA1 regions from 4-VO animals revealed a sharp spike of TAILS levels 3 h after surgery, followed by a gradual return to the baseline (Figures 2B and 2C). Oxidative stress is a hallmark of many neurodegenerative diseases (Barnham et al., 2004), and indeed, TAILS levels were elevated in prefrontal cortices from individuals diagnosed with Alzheimer’s disease (AD); one of the non-AD controls (Figures 2D and 2E, lane 1, gold) was an acute hypoxic-ischemic (H-I) encephalopathy case and displayed high TAILS levels compared with other non-AD cases (Figures 2D and 2E). The transcription factor CREB3L2, which is necessarily co-generated with TAILS, does not act neuroprotectively in our experimental paradigm; rather, its expression results in decreased cell viability (Figure 2F). The finding that TAILS levels are elevated under pathological conditions (Figures 2B-2E) led us to ask how TAILS generation is controlled by cell stress. We developed a peroxidase activity-based TAILS secretion assay, which revealed that TAILS secretion is prevented by brefeldin A, an inhibitor of ER-Golgi apparatus protein transport (Helms and Rothman, 1992), and monensin, an inhibitor of trans-Golgi function (Griffiths et al., 1983; Figures 2G and S1A). A HEK293 cell line stably expressing the full-length CREB3L2/TAILS precursor protein with a horseradish peroxidase tag on its C terminus (full-length horseradish peroxidase [FL-HRP]) was challenged with various inducers of cell stress—oxidative (HNE), ER (tunicamycin [TN]), and lysosomal (chloroquine [CQ])—and the culture media were probed for the presence of HRP-tagged TAILS (Figures 2G and S1B). Secreted TAILS levels were significantly increased by all tested stressors, and co-treatment with nelfinavir, an S2P inhibitor that prevents cleavage of TAILS from CREB3L2 (McCurdy et al., 2019), abolished the effect, albeit at varying degrees for each stressor (Figure 2H). Given the effect size of HNE in this assay and the marked spike of TAILS in ischemic brain samples, we decided to focus on oxidative stress and HNE in the following experiments. HNE treatment in neurons revealed a temporal pattern in TAILS secretion, characterized by a 3-fold increase 4 h after HNE treatment, followed by attenuation to the baseline at 12 h (Figures 2I and 2J), similar to the findings in 4-VO animals (Figures 2B and 2C). Induction of oxidative stress with hydrogen peroxide (H2O2) and 3-morpholinosydnonimine (SIN-1) similarly promoted TAILS secretion in neurons (Figure 2J). We also validated the stress-induced generation and secretion of endogenous TAILS by immunoprecipitation from the supernatant of hippocampal neurons (Figure 2K). HNE treatment raised TAILS levels in the supernatant by about 30%. Together with our previous finding that translation and cleavage of CREB3L2 is part of the unfolded protein response (UPR) (McCurdy et al., 2019), these results demonstrate that generation and secretion of TAILS is part of the cellular stress response in vitro and in the CNS.
Figure 2. TAILS is generated and secreted under stress or pathological conditions.

(A) Western blot of TAILS in papain-dissociated rat brain. The TAILS band is indicated by an arrow.
(B) Western blot analysis of TAILS protein levels in micro-dissected CA1 tissues of rats after the 4-VO procedure. cyt, cytosolic.
(C) Quantification of TAILS protein levels in (B). Means ± SEM (sham, n = 5; 3-h 4-VO, n = 5; 6-h 4-VO, n = 3; 24-h 4-VO, n = 2; 48-h 4-VO, n = 3). One-way ANOVA, Tukey’s multiple comparisons test. *p = 0.0464.
(D) Western blot analysis of CREB3L2 and TAILS in prefrontal cortices from individuals diagnosed with Alzheimer’s disease (AD). H-I, acute hypoxic-ischemic (underlined, gold).
(E) Quantification of (D). Means ± SEM (non-AD, n = 4; AD, n = 7). Unpaired t test, one-tailed. #p = 0.0672, *p = 0.0434 without H-I sample (gold).
(F) Assessment of the CREB3L2 transcription factor on neuronal viability. Means ± SEM (n = 3 biological replicates; 38–39 wells per condition). One-way ANOVA, Tukey’s multiple comparisons test. **p = 0.008.
(G) Scheme and immunoblotting of the TAILS secretion assay (left). S2P, site 2 protease; HRP, horseradish peroxidase. Immunoblot of FL- and TAILS-HRP expression in HEK293T cells and colorimetric validation of HRP activity in culture media (right). A non-specific band and TAILS-HRP are indicated by an asterisk and an arrow, respectively.
(H) Quantification of TAILS secretion in HEK293T cells treated with oxidative (HNE, 10 μM), ER (TN, 2 μM), and lysosomal (CQ [chloroquine], 10 μM) stress inducers. Means ± SEM (n = 6 biological replicates). One-way ANOVA, Tukey’s multiple comparisons test. #p < 0.1, **p < 0.01, ***p < 0.001.
(I) TCA precipitation of secreted TAILS-HRP from hippocampal neurons expressing FL- or TAILS-HRP.
(J) Time course analysis of the TAILS secretion assay in hippocampal neurons under oxidative stress induced by HNE (10 μM), hydrogen peroxide (H2O2, 200 μM), and SIN-1 (200 μM). Means ± SEM (n = 4 biological replicates). One-way ANOVA, Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001.
(K) Immunoblot comparison of endogenous TAILS proteins immunoprecipitated from neuronal culture media to His-purified recombinant TAILS protein (3 μg). TAILS is indicated by an arrow.
(L) Western blot analysis of ISR pathway components in neurons challenged with HNE (10 μM).
(M) Quantification of (L). Means ± SEM (n = 5–6 biological replicates). Paired t test, two-tailed. *p = 0.0445 (p-eIF2α) and p = 0.0408 (ATF4).
(N) Western blot analysis of ISR pathway components in neurons treated with TAILS-containing or control conditioned medium.
(O) Quantification of (N). Means ± SEM (n = 5 biological replicates). One-way repeated-measures ANOVA, Tukey’s multiple comparisons test.
TAILS does not activate cell stress signaling
Having found that TAILS is generated as part of the cellular response to stress and improves survival of receiving stressed cells, we wondered whether TAILS would activate the ISR pathway in neurons. We first confirmed that HNE activates ISR in hippocampal neurons, as assessed by the increased levels of p-eIF2α and ATF4 (Figures 2L and 2M). However, the levels of p-eIF2α and ATF4 remained unaltered after recombinant TAILS was added to neurons (Figures 2N and 2O). These results reveal that TAILS does not transmit ISR horizontally between cells but functions through another pathway.
TAILS confers neuronal resilience by potentiating canonical Hh signaling
A potential interaction between TAILS and hedgehog proteins has been reported for two different cell culture models (Saito et al., 2014; McCurdy et al., 2019). In the postnatal brain, neurons are the primary source of SHH and express its receptors Patched (PTCH) and SMO (Alvarez-Buylla and Ihrie, 2014; Petralia et al., 2011), although canonical SHH signaling is inactive in non-stressed postnatal neurons (Garcia et al., 2010). To test whether TAILS interacts with SHH in the brain, we employed two independent approaches: co-immunoprecipitation (coIP) and proximity ligation assay (PLA), which provides spatial resolution for TAILS/SHH binding events. CoIP from rat brain lysate revealed interaction between TAILS and the N-terminally processed SHH ligand (SHH-N) (Figure 3A). PLA visualized a notable abundance of TAILS/SHH-N binding events in the stratum radiatum layer of the CA1 region of the hippocampus (Figure 3B). SHH-N is not the rate-limiting factor for TAILS/SHH-N formation because significantly more SHH proteins co-immunoprecipitated with TAILS after HNE treatment (Figures 3C and 3D). Increased TAILS/SHH-N binding was confirmed by IP with an anti-SHH antibody from the conditioned media of H2O2 and SIN-1 treated neurons (Figure S2). Analysis of a single-nucleus RNA sequencing database (Yang et al., 2022) revealed that neurons are the main source and recipient of Hh signaling components in the hippocampus (Figure S3), justifying our focus on neuronal effects of TAILS. Next, we investigated whether TAILS was an endogenous regulator of Hh signaling in neurons. Exogenous application of TAILS increased SHH-N/PTCH1 ligand-receptor binding (Figures 3E and 3F) and activated the downstream SHH effector GLI (Figures 3G and 3H) in hippocampal neurons. A deletion mutant of TAILS (ΔBD), lacking the proposed SHH/PTCH1 interaction site (Saito et al., 2014) and neither affected SHH-N/PTCH1 interaction nor activated Hh signaling (Figures 3E-3H). It also failed to increase neuronal resilience in neurons exposed to oxidative stress inducers (Figure 3I). The dose-response curve of TAILS with GLI activity as readout confirmed that TAILS activates the Hh pathway at nanomolar concentrations (Figure S4A). These results demonstrate that greater TAILS levels result in increased activation of canonical Hh signaling and that TAILS neuroprotective effects depend entirely on its SHH/PTCH1 interaction domain.
Figure 3. TAILS facilitates SHH signaling and increases neuronal resilience.

(A) CoIP of TAILS and SHH from rat cortex. TAILS is indicated by an arrow.
(B) Visualization of TAILS/SHH-N interaction in the mouse hippocampus by PLA. s.o., stratum oriens; s.p., stratum pyramidale; s.r., stratum radiatum. Scale bars, 200 μm (left) and 50 μm (right).
(C) CoIP of SHH-N by TAILS from culture medium of hippocampal neurons treated with HNE (10 μM) for 6 h. h.c., heavy chain; l.c., light chain. TAILS is indicated by an arrow.
(D) Quantification of co-immunoprecipitated SHH protein by TAILS in (C). Means ± SEM (n = 4 biological replicates). Paired t test, two-tailed. *p = 0.0191.
(E) Detection of SHH-N/PTCH1 binding by PLA in hippocampal neurons after adding recombinant TAILS, the deletion mutant protein (ΔBD), or a control. Scale bar, 20 μm.
(F) Quantification of PLA reactions per square millimeter in (E). Means ± SEM (n = 3 biological replicates; 48–66 optical fields per condition). One-way ANOVA, Tukey’s multiple comparisons test. **p = 0.0012, ***p = 0.0006.
(G) Visualization of GLI activity in neurons expressing the 7×GLI-GFP reporter after adding recombinant TAILS, ΔBD, or a control. Indexed color image, 16-color lookup table (LUT). Scale bar, 50 μm.
(H) Quantification of GFP fluorescence intensity in (G). Means ± SEM (n = 4 biological replicates; 151–192 optical fields per condition). One-way ANOVA, Tukey’s multiple comparisons test. ***p < 0.001.
(I) Measurement of cell viability in hippocampal neurons treated with recombinant TAILS, ΔBD, or a control under oxidative stress induced by HNE (10 μM), H2O2 (200 μM), and SIN-1 (200 μM). Quantified cell viability values were normalized to the non-stressed control condition. Means ± SEM (n = 6 biological replicates). One-way repeated-measures ANOVA, Tukey’s multiple comparisons test. *p < 0.05.
(J) Detection of TAILS/PTCH1 binding by PLA in HNE-stressed hippocampal neurons treated with the SHH inhibitor robotnikinin (20 μM) or left untreated. Scale bar, 20 μm.
(K) Means ± SEM (n = 5 biological replicates; 29–35 optical fields per condition). One-way ANOVA, Tukey’s multiple comparisons test. *p = 0.0209; ***p = 0.001.
(L) Measurement of cell viability in HNE-stressed hippocampal neurons co-treated with recombinant TAILS in the presence or absence of robotnikinin (20 μM). Quantified cell viability values were normalized to the non-stressed control condition. Means ± SEM (n = 6 biological replicates). two-way ANOVA, Tukey’s multiple comparisons test. **p < 0.01, ***p = 0.001.
(M) Detection of SHH-N/PTCH1 binding by PLA in HNE-stressed hippocampal neurons treated with an antibody against TAILS or GFP (0.2 μg mL−1). Scale bar, 20 μm.
(N) Quantification of (M). Means ± SEM (n = 5 biological replicates; 77–97 optical fields per condition). One-way ANOVA, Tukey’s multiple comparisons test. *p = 0.0209, ***p = 0.001.
(O) Visualization of GLI activity in HNE-stressed neurons expressing 7×GLI-GFP reporter with anti-TAILS or anti-GFP antibodies (0.2 μg mL−1). Indexed color image, 16-color LUT. Scale bar, 50 μm.
(P) Quantification of GFP fluorescence intensity in (O). Means ± SEM (n = 4 biological replicates; 212–241 optical fields per condition). One-way ANOVA, Tukey’s multiple comparisons test. **p = 0.0026, ***p = 0.0002.
(Q) Measurement of cell viability in HNE-stressed neurons co-treated with the SMO agonist purmorphamine (PUR; 5 μM) under TAILS-neutralizing or control conditions. Means ± SEM (n = 4 biological replicates). two-way ANOVA, Tukey’s multiple comparisons test. #p < 0.1, *p < 0.05, ***p < 0.001.
(R) Scheme of the TAILS-mediated Hh pathway activation proposed in this study. Inhibitors of extracellular TAILS and SHH-N—a TAILS-neutralizing antibody and robotnikinin, respectively—are indicated in red and the SHH pathway agonist PUR in green.
See also Figures S2-S4.
TAILS/SHH-N signaling is upregulated in response to stress
Next, we tested the effects of TAILS on Hh signaling in neurons under stress conditions. HNE-stressed neurons showed increased formation of TAILS/PTCH1 binding compared with the control (Figures 3J and 3K). Addition of the SHH inhibitor robotnikinin, which blocks SHH upstream of its binding to PTCH1 (Stanton et al., 2009), reduced TAILS/PTCH1 binding to the baseline level, demonstrating that TAILS interaction with SHH is required for its binding to PTCH1. Western blot analysis of cleaved caspase-3 showed that the TAILS-induced protection of cell death against HNE toxicity was prevented by co-treatment with robotnikinin (Figure S4B). Robotnikinin abolished the TAILS-mediated rescue of cell viability in stressed neurons (Figure 3L).
Having observed that addition of TAILS enhanced SHH signaling in HNE-stressed neurons, we next investigated the effect of its loss. The frequency of SHH/PTCH1 binding events in stressed neurons was reduced upon addition of the TAILS-neutralizing antibody (Figures 3M and 3N). Evaluation of the downstream SHH transducer SMO and effector GLI confirmed that stress-induced activation of Hh signaling is prevented in the presence of the TAILS-blocking antibody (Figures 3O, 3P, and S4C). To exclude any off-target effects of the antibody, we confirmed that activation of SMO by its agonist purmorphamine was sufficient to increase cell viability in neurons even in the presence of the TAILS-blocking antibody (Figure 3Q). These results establish that the interaction between TAILS and SHH-N is necessary for the pro-survival effect of TAILS (Figure 3R).
TAILS stabilizes the antioxidant transcription factor NRF2
HNE-induced toxicity in neurons is largely due to oxidative stress, which manifests as formation of reactive oxygen species (ROS) (Kruman et al., 1997), and addition of recombinant TAILS to HNE-treated neurons prevented the rise in ROS (Figures 4A and 4B). Next, we wanted to determine how TAILS affected ROS levels and examined the master regulator of redox homeostasis, the transcription factor NRF2. Under homeostatic conditions, NRF2 associates with KEAP1, a component of an E3 ligase complex, leading to its ubiquitination and proteasomal degradation. Under oxidative stress conditions, KEAP1 dissociates from NRF2, leading to stabilization and nuclear translocation of NRF2, which protects against oxidative damage (Itoh et al., 1999). Therefore, treatment with the proteasome inhibitor MG132 can reveal the amount of NRF2 destined for degradation. NRF2/KEAP1 association significantly increased in MG132-treated neurons, and this effect was reduced by 54%, indicating the known stabilization of NRF2 under cell stress conditions. Remarkably, in TAILS-treated stressed neurons, MG132 did not affect the amount of NRF2/KEAP1 association, indicating that virtually all NRF2 is stabilized (Figures 4C and 4D). To directly test the requirement of NRF2 for TAILS-induced resilience, we used the NRF2 inhibitor ML385 (Singh et al., 2016) and found that recombinant TAILS failed to increase cell viability in HNE-stressed neurons that were co-treated with ML385 (Figure 4E). These data reveal that TAILS signals through stabilization of NRF2.
Figure 4. TAILS signaling strengthens NRF2-dependent antioxidant defense.

(A) Detection of ROS by CellROX in HNE-stressed hippocampal neurons treated with recombinant TAILS (50 ng mL−1) or a control. Indexed color image, 16-color LUT. Scale bar, 100 μm.
(B) Quantification of (A). Means ± SEM (n = 6 biological replicates). One-way ANOVA, Tukey’s multiple comparisons test. **p < 0.01.
(C) Detection of NRF2-KEAP1 interaction by PLA in hippocampal neurons. Proteasome activity was blocked by treatment with MG132 (10 μM) for 1 h. Scale bar, 25 μm.
(D) Quantification of (C). Means ± SEM (n = 4 biological replicates; 371–573 neurons per condition). One-way ANOVA, Tukey’s multiple comparisons test. ***p < 0.001.
(E) Measurement of cell viability in hippocampal neurons treated with the NRF2 inhibitor ML385. Means ± SEM (n = 4 biological replicates). two-way ANOVA, Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001.
(F) Western blot detection of ARE-driven GFP expression in Neuro2a cells transfected with the 8×ARE-GFP reporter.
(G) Quantification of (F). Means ± SEM (n = 6 biological replicates). One-way ANOVA, Tukey’s multiple comparisons test. **p < 0.05.
(H and I) qRT-PCR analysis of GSS (H) and NFE2L2 mRNA levels in HNE-stressed hippocampal neurons treated with recombinant TAILS or a control. Ratio-paired t test, one-tailed. *p < 0.05.
(J) Detection and quantification of redox status as reduced to oxidized (GSH/GSSG) ratio. Means ± SEM (n = 8 biological replicates). two-way ANOVA, Tukey’s multiple comparisons test. *p =0.0116, ***p<0.0001.
(K) Scheme of the proposed TAILS-mediated SHH signaling and antioxidant defense system.
TAILS signaling activates the NRF2-dependent antioxidant defense system
To investigate whether NRF2 stabilization resulted in its increased transcriptional activity, we employed a GFP reporter for the antioxidant response element (8×ARE-GFP), which is a proxy measure for expression of several antioxidant genes (Wyler et al., 2019). Neuro2a cells transfected with the 8×ARE-GFP reporter showed elevated GFP level in HNE-treated cells co-treated with recombinant TAILS (Figures 4F and 4G). Among the target genes of NRF2 is glutathione synthetase (GSS), one of two key enzyme synthesis pathways for the principal intracellular antioxidant glutathione (GSH) (Cuadrado et al., 2019). Quantitative real-time PCR results revealed upregulated expression of GSS in presence of recombinant TAILS in hippocampal neurons treated with HNE (Figure 4H). Transcript levels for NFE2L2, the gene encoding NRF2, were unaltered (Figure 4I), again indicating that TAILS activates the antioxidant pathway primarily through NRF2 stability. To test whether the upregulation of antioxidant genes through TAILS affected the redox status in hippocampal neurons, we measured the ratio between reduced glutathione (GSH) and oxidized GSH (glutathione disulfide, GSSG), GSH/GSSG, a well-established marker for oxidative stress. Treatment with recombinant TAILS significantly increased the ratio of GSH/GSSG in neurons under oxidative stress (Figure 4J). Our results demonstrate that TAILS enhances antioxidant defense in neurons under stress by facilitating transcriptional activation of ARE-driven genes via increasing NRF2 stability (Figure 4K).
TAILS increases mitochondrial mass and respiration in stressed neurons
We hypothesized that TAILS/SHH-N signaling enhances the mitochondrial capacity for catabolism and detoxification of reactive molecules in neurons under oxidative stress. Mitochondrial mass was increased by recombinant TAILS in HNE-stressed hippocampal neurons (Figures 5A and 5B). In line with our observation, a previous study has revealed that activation of Hh signaling increases mitochondrial abundance and activity in hippocampal neurons (Yao et al., 2017). To investigate whether the increased mitochondrial mass positively translates to improved function, we assessed mitochondrial respiration by Seahorse XF Cell Mito Stress Test (Figures 5C and 5D). Through sequential injection of specific inhibitors for electron transport chain components—oligomycin (ATP synthase inhibitor), FCCP (an uncoupling agent), and rotenone/antimycin A (complex I/III inhibitors)—changes in oxygen consumption rates of neurons were measured for analysis. Key parameters of mitochondrial function—basal and maximal respiration levels and ATP production—were significantly improved in HNE-stressed neurons treated with recombinant TAILS (Figures 5E-5G). Our results demonstrate that TAILS increases neuronal resilience, at least in part, by enhancing mitochondrial respiration.
Figure 5. TAILS increases mitochondrial function in HNE-stressed neurons.

(A) Western blot analysis of cyt and mitochondrial fractions from TAILS-treated neurons.
(B) Quantification of TOM20 in (A). Means ± SEM (n = 3 biological replicates). two-way ANOVA, Tukey’s multiple comparisons test. ns, not significant. *p = 0.0471.
(C) Mitochondrial stress test profile of control hippocampal neurons. Key parameters of mitochondrial respiration are illustrated in colored boxes.
(D) Mitochondrial respiration of HNE-treated neurons incubated with TAILS or a control. Oxygen consumption rates (OCRs) were calculated per 10,000 hippocampal neurons. Electron transport chain components inhibited by sequential injection of oligomycin (OLIG; 6 μM, an ATP synthase inhibitor), FCCP (3 μM, an uncoupling agent), and rotenone/antimycin A (ROT/AA; 0.5 μM, complex I/III inhibitors).
(E–G) Quantitative comparisons of basal respiration (E), maximal respiration (F), and ATP production (G) in HNE-treated neurons incubated with TAILS or a control. Means ± SEM (n = 7 biological replicates). Paired t test, one-tailed. *p < 0.05, **p < 0.01.
TAILS rescues CA1 neurons in a 4-VO model of global ischemia
To test the effect of TAILS on neuronal resilience to oxidative stress in vivo, we selected the 4-VO model of global ischemia. In 4-VO, selective neuronal death occurs in the CA1 region of the hippocampus, where we detected frequent TAILS/SHH-N interactions (Figure 3B), and we saw a prominent increase in TAILS protein in 4-VO samples (Figures 2B and 2C). Two separate cohorts of experimental animals were processed for histology analysis to measure neuronal death or for behavioral tests to assess cognitive impairment (Figure 6A). For histology analysis of neuronal death, adult rats were subjected to 4-VO or sham operation, during which intrahippocampal injections of TAILS or the inactive control protein ΔBD were made in the right hemisphere for direct comparisons within each brain. For animals prepared for behavioral tests, intrahippocampal injections were made bilaterally. Seven days after the surgery, significantly more toluidine blue-positive live CA1 neurons were detected in the TAILS-injected hemisphere of the 4-VO animal (Figures 6B and 6C). Fluoro-Jade staining selectively labels degenerating neurons and has been used previously in the 4-VO model of global ischemia for assessment of neuronal death (Hwang et al., 2017; Noh et al., 2012). Consistent with quantification of toluidine blue staining, 4-VO-induced death of CA1 pyramidal neurons was prevented specifically in the TAILS-injected hemisphere (Figures 6D and 6E). With Fluoro-Jade and toluidine blue staining, administration of TAILS rescued CA1 neuron survival to levels statistically not distinguishable from the sham-operated animals. The combined histology-based assessments of degenerating and live neurons provide in vivo evidence of greatly enhanced neuronal resilience after TAILS administration.
Figure 6. Administration of TAILS rescues 4-VO pathology.

(A) Timeline scheme of ischemic surgery, molecular assays, and behavior tests.
(B–E) Histology analysis of pyramidal neurons in the hippoampal CA1 region (indicated by arrow heads) in animals subjected to 4-VO or sham operation, administered a TAILS or ΔBD (100 ng μL−1, 3 μL per hemisphere) unilateral intrahippocampal injection. Two-way ANOVA, Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001.
(B and C) Brain sections stained with toluidine blue (B) and quantification of toluidine blue-stained live cells (C). Scale bars, 500 μm and 50 μm (magnified).
(D and E) Brain sections labeled with Fluoro-Jade (D) and quantification of Fluoro-Jade-positive degenerating cells (E). Scale bars, 500 μm and 50 μm (magnified).
(F and G) Behavioral assessment of locomotion and cognitive deficits in animals subjected to 4-VO or sham operation, administered TAILS or ΔBD (100 ng μL−1, 3 μL per hemisphere).
(F) Measurement of animal locomotion by open field test shows that animals subjected to 4-VO or sham operation have no locomotion deficits.
(G) Assessment of non-spatial memory by NOR task. Discrimination index is defined as ([novel object investigation time — familiar object investigation time]/[total investigation time] × 100). One-way ANOVA, Tukey’s multiple comparisons test. *p < 0.05.
(H and I) Measurement of hippocampus-dependent spatial memory by Barnes maze task.
(H) Primary latency time to reach the escape hole measured during the training trials for learning. Two-way mixed-effect ANOVA, Sidak’s multiple comparisons test. ***p < 0.001.
(I) Latency time measured during the test for retrieval of spatial information. One-way ANOVA, Tukey’s multiple comparisons test. **p < 0.01.
See also Figure S5.
Rescue of behavioral deficits caused by 4-VO
We next wanted to determine whether the TAILS-induced resilient neurons in the 4-VO animals remain functionally integrated in their neural circuits. CA1 pyramidal neurons are pivotal for cognitive functions, particularly spatial learning and memory, and severe deficits in cognition and learning after ischemic damage in the brain have been reported in humans and rodents (Nys et al., 2005; Oksala et al., 2009; Hartman et al., 2005). CA1 neuronal death in the 4-VO model of global ischemia manifests after about 48 h with delayed onset and is induced until 7 days after the surgery. The experimental animals were subjected to behavioral tests starting on post-operation day 8 to ensure that the cognitive function of 4-VO animals was tested after CA1 neuronal death. First, animals were subjected to the open field test on post-operation day 8, solely to assess the locomotor activity, impairment of which could become a critical confounding effect on animal behavior in all behavioral tests. Animal locomotion was unaltered by 4-VO surgery or TAILS/ΔBD administration (Figure 6F). The habituated 4-VO or sham animals were then subjected to the novel object recognition (NOR) task for testing non-spatial memory. We found that TAILS administration rescued the decreased preferential exploratory behavior for a novel object over the familiar in the 4-VO animals (Figure 6G). The animals from each experimental group displayed comparable traveled distance during the NOR tasks, indicating that locomotor activity did not affect the exploratory behaviors (Figure S5). To test hippocampus-dependent spatial reference memory, the animals were subjected to the Barnes maze task, in which the animals learned the location of an escape hole through trial and error over 5-day training, and spatial memory retention was assessed by the latency time to find an escape. The ΔBD-injected (i.e., control) 4-VO animals demonstrated significant impairment in spatial learning; in sharp contrast, the 4-VO animals injected with TAILS exhibited prominent improvement in learning the task to the degree of cognitive performance by the sham-operated animals (Figures 6H and 6I). Exogenous application of TAILS reversed the 4-VO-induced loss of CA1 pyramidal neurons and the consequent cognitive decline. Our data reveal a function of TAILS as a cell non-autonomous, cell stress-responsive neuroprotectant.
DISCUSSION
Stress response pathways such as the UPR or ISR are adaptive responses intended to reestablish homeostasis (Ron and Walter, 2007; Costa-Mattioli and Walter, 2020). In multicellular organisms, homeostasis is controlled and coordinated at the cellular, tissue, and systemic level (Sala et al., 2017), leading to the question of whether cells can communicate stress among each other. There are several examples of UPR-induced cell-to-cell signaling. Long-term activation of the UPR in the ER of C. elegans neurons or specific glia cells is sufficient to induce ER UPR in the intestine and other distal tissues, leading to increased stress resistance and life span (Taylor and Dillin, 2013; Frakes et al., 2020). Comparably, in human tumor cells, induction of ER stress has been shown to lead to activation of the UPR in macrophages and dendritic cells, in turn leading to their activation and T cell suppression, respectively (Mahadevan et al., 2011, 2012). In both examples, the precise identity of the extracellular signal(s) remains unknown. Although these examples demonstrate direct transmission of the ER UPR between cell types and tissues, we find that ER UPR-dependent secretion of TAILS does not lead to activation of the ER UPR in recipient cells (eIF2α is not activated). Rather, we found that TAILS leads to pre-conditioning of the endogenous antioxidant machinery. Another difference is that previous examples of non-cell-autonomous stress signaling were restricted to specific donor and recipient cell pairs (Frakes et al., 2020; Williams et al., 2014), whereas, at least in principle, the mechanism described here just requires CREB3L2/TAILS expression in the stressed cells and Hh responsiveness in the receiving cell. The mechanism described here seems to be non-selective for specific stressors and/or cell types and might constitute a ubiquitous, broad-spectrum, fast-acting alarm system.
Besides the identification of TAILS as a resilience factor in the CNS, we also confirmed previous reports proposing a function of SHH as a neuroprotectant in the mature brain (Yao et al., 2017). Components of the Hh signaling pathway remain expressed in the postnatal brain, raising the question of its post-development functions (Rivell et al., 2019). Previously, a neuroprotective effect of the SMO agonist SAG has been documented in hippocampal neurons (Yao et al., 2017), and activation of the Hh pathway has been investigated as a therapeutic target in middle cerebral artery occlusion-induced focal ischemia (Sims et al., 2009; Huang et al., 2013; Chechneva et al., 2014). However, it remained unclear whether endogenous SHH had any effect on neuronal survival under stress conditions. Our findings unveil a molecular mechanism by which neurons secrete an SHH enhancer to potentiate Hh signaling as an immediate response to stress. We found that stress-evoked Hh signaling in response to oxidative stress orchestrates activation of molecular defense mechanisms, particularly strengthening antioxidant and mitochondrial function in neurons, suggesting yet another function of SHH as a part of an adaptive stress mechanism in the mature CNS. The canonical Hh signaling pathway is inactive in neurons under homeostatic conditions, and addition of TAILS to non-stressed neurons did not have any effect in our experiments. Only under conditions of stress, when TAILS and SHH are simultaneously increased, do they activate Hh signaling in neurons. This dual-factor mechanism might be part of safeguarding the cell against erroneous activation of Hh signaling under non-stress conditions.
Prevention of neuronal degeneration and hippocampus-dependent cognitive deficits in the 4-VO model of global ischemia suggests administration of TAILS as a potential therapeutic strategy to strengthen endogenous neuroprotective Hh signaling under pathological conditions of acute (e.g., stroke) and chronic (e.g., AD) oxidative damages. Different from administration of SHH or SAG, TAILS acts only on cells that already receive endogenous SHH signaling. By targeting only cells in areas with active Hh signaling, TAILS administration might limit undesirable side effects of aberrant Hh pathway activation, including tumorigenesis (Samkari et al., 2015). Our finding that TAILS is secreted in response to various stressors and that its levels in the CNS are significantly upregulated in several pathological conditions, including hypoxic ischemia, global ischemia, or AD, point to the possibility that TAILS might have utility as a biomarker for neuronal stress.
We discovered a cell non-autonomous signaling pathway that is activated in neurons in response to cell stress and confers enhanced resilience and mitochondrial function to recipient cells. Our results establish TAILS as a stress-regulated modulator of protective neuronal Shh signaling and provide evidence of intercellular communication of cell stress in the mammalian nervous system.
Limitations of the study
The specific mechanism by which TAILS-enhanced SHH signaling strengthens mitochondrial respiration and cellular antioxidant defense has not been fully described. More studies are needed to determine whether a delayed application of TAILS after the 4-VO procedure still has protective effects.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ulrich Hengst (uh2112@cumc.columbia.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
This paper analyzes existing, publicly available data. The accession number for the single-cell profiling of hippocampus and superior frontal cortex from 25 hippocampus and cortex samples across 17 control and AD patients is listed in the key resources table and the data were accessed via Human BBB portal (https://twc-stanford.shinyapps.io/human_bbb) (Yang et al., 2022).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CREB3L2 C terminus | Abcam | ab102989; RRID:AB_10711653 |
| Rabbit polyclonal anti-CREB3L2 C terminus | Abcepta | AP9654b; RRID:AB_2544158 |
| Rabbit polyclonal anti-CREB3L2 N terminus | MilliporeSigma | HPA015068; RRID:AB_1847224 |
| Mouse monoclonal Alexa Fluor 488 anti-βIII-Tubulin (TUJ1 clone) | BioLegend | 801203; RRID:AB_2564757 |
| Mouse monoclonal anti-SHH (E-1) | Santa Cruz | sc-365112; RRID:AB_10709580 |
| Rabbit polyclonal anti-PTCH1 | Proteintech | 17520-1-AP; RRID:AB_2176561 |
| Mouse monoclonal anti-SMO (E-5) | Santa Cruz | sc-166685; RRID:AB_2239686 |
| Rabbit polyclonal anti-GLI1 (V812) | Cell Signaling Technology | 2534; RRID:AB_2294745 |
| Rabbit polyclonal anti- GFP | Abcam | ab290; RRID:AB_303395 |
| Rabbit polyclonal anti-cleaved CASP3 | Cell Signaling Technology | 9661; RRID:AB_2341188 |
| Rabbit polyclonal anti-PARP | Cell Signaling Technology | 9542; RRID:AB_2160739 |
| Mouse monoclonal anti-β-Actin (AC-15) | Abcam | ab6276; RRID:AB_2223210 |
| Mouse monoclonal anti-GAPDH (6C5) | Santa Cruz | sc-32233; RRID:AB_627679 |
| Rabbit monoclonal anti-p-eIF2α | Cell Signaling Technology | 3398; RRID:AB_2096481 |
| Rabbit monoclonal anti-ATF4 | Cell Signaling Technology | 11815; RRID:AB_2616025 |
| Rabbit monoclonal anti-NRF2 (D1Z9C) | Cell Signaling Technology | 12721; RRID:AB_2715528 |
| Mouse monoclonal anti-KEAP1 | Proteintech | 60027-1-lg; RRID:AB_2132623 |
| Mouse monoclonal anti-TOM20 (2F8.1) | MilliporeSigma | MABT166; RRID:AB_2890986 |
| Bacterial and virus strains | ||
| Stellar competent cells | Takara | 636766 |
| Biological samples | ||
| See Table S1. A complete list of all human brain samples used in this study | New York Brain Bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DMSO | MilliporeSigma | D8418 |
| Nelfinavir | MilliporeSigma | CDS021783 |
| Robotnikinin | Thermo Fisher Scientific | 50464659 |
| 4-Hydroxynonenal (HNE) | MilliporeSigma | 3932041MG |
| Purmorphamine | MilliporeSigma | 5402235MG |
| MG132 | Cayman Chemical | 133407-82-6 |
| SIN-1 | MilliporeSigma | 567028 |
| Hydrogen peroxide | MilliporeSigma | MAK092C |
| Chloroquine diphosphate | Tocris Bioscience | 4109/50 |
| Tunicamycin | MilliporeSigma | 50-457-00001 |
| ΔBD (recombinant protein) | His-tag purification | N/A |
| TAILS (recombinant protein) | His-tag purification | N/A |
| Critical commercial assays | ||
| Luna Universal Probe One-Step RT-qPCR Kit | NEB | E3006L |
| Duolink In Situ Detection Reagents (Red) | MilliporeSigma | DUO92008 |
| Duolink In Situ Detection Reagents (FarRed) | MilliporeSigma | DUO92013 |
| Duolink In Situ PLA Probe anti-Mouse PLUS | MilliporeSigma | DUO92001 |
| Duolink In Situ PLA Probe anti-Rabbit MINUS | MilliporeSigma | DUO92005 |
| In Situ Cell Death Detection Kit (TMR red) | Roche | 12156792910 |
| Peroxidase Activity Assay Kit | MilliporeSigma | MAK092 |
| CellROX (Green) | Invitrogen | C10444 |
| AlamarBlue Cell Viability Assay | G-Biosciences | 786922 |
| GSH/GSSG-Glo Glutathione Assay | Promega | V6611 |
| Seahorse XFp FluxPak | Agilent Technologies | 103022-100 |
| Seahorse XFp Cell Mito Stress Test Kit | Agilent Technologies | 103010-100 |
| Capturem His-Tagged Purification Kit | Takara | 635710 |
| CyQUANT Cell Proliferation Assay | Thermo Fisher Scientific | C7026 |
| CalFectin Transfection Reagent | SignaGen Laboratories | SL100478 |
| Deposited data | ||
| Single-nucleus RNA-sequencing database | Yang et al., 2022 | GSE163577; https://twc-stanford.shinyapps.io/human_bbb |
| Experimental models: Cell lines | ||
| Human: HEK293T | ATCC | CRL-3216 |
| Mouse: Neuro2a | ATCC | CCL-131 |
| Human: HEK293T (constitutive TAILS secretion) | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Rat: Hsd:Sprague Dawley SD (pregnant) | Envigo | N/A |
| Rat: Hsd:Sprague Dawley SD (adult male) | Charles River Laboratories | N/A |
| Oligonucleotides | ||
| Gss (Rn00564188_m1) | Thermo Fisher Scientific | 4331182 |
| Nfe2l2 (Rn00821258_g1) | Thermo Fisher Scientific | 4331182 |
| Rpl13 (Rn00582415_m1) | Thermo Fisher Scientific | 4331182 |
| Gss (Rn00564188_m1) | Thermo Fisher Scientific | 4331182 |
| Recombinant DNA | ||
| 7xGLI-GFP | Addgene | 110494 |
| 8xARE-GFP | Addgene | 134910 |
| pCMV-erHRP (N175S mutant) | Addgene | 79909 |
| C-terminal CREB3L2 (His-tagged) | McCurdy et al., 2019 | N/A |
| TAILS (BiP sequence, His-tagged) | This paper | N/A |
| ΔBD (binding domain deletion mutant; BiP sequence, His-tagged) | This paper | N/A |
| CREB3L2 (HRP-tagged) | This paper | N/A |
| Software and algorithms | ||
| ImageJ (FIJI) | Schindelin et al., 2012 | RRID:SCR_002285 |
| ANY-Maze | Stoelting | RRID:SCR_014289 |
| GraphPad Prism 9.4 | GraphPad Software | RRID:SCR_002798 |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary culture of rat embryonic neurons
Hippocampi were dissected from embryonic day (E)16–18 rat embryos obtained from pregnant Sprague-Dawley dams (Envigo). All animal procedures were approved by the Institutional Animal Care and Use Committee at Columbia University. Cell dissociation was performed using TrypLE Express enzyme. Neurons (40,000 and 70,000 cells per cm2 for immunocytochemistry and immunoblotting, respectively) were plated on 0.1 mg mL−1 poly-D-lysine (MilliporeSigma)- and 2 mg mL−1 laminin (Bio-Techne)-coated substrates and grown in Neurobasal supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific), 2 mM L-glutamine, 1 mM sodium pyruvate, and antibiotics (50 U ml−1 penicillin-streptomycin) and placed in a humified atmosphere of 5% CO2 at 37°C. After 4–6 hours, the medium was changed to Neurobasal containing 1 × B27 and 2 mM L-glutamine. Subsequent medium changes (half volumes) were performed at 2 days in vitro (DIV) and thereafter every 3–5 days. To prevent glial cell proliferation, medium changes included 10 mM 5-flurodeoxyuridine and uridine (MilliporeSigma) after DIV2. Neuronal cultures were grown in a 37°C, 5% CO2 humidified atmosphere until DIV12–14. For Western blot and immunoprecipitation, co-cultures of primary cortical and hippocampal neurons were used. Unless otherwise mentioned, hippocampal neurons were subjected to experimental treatments at DIV16–18; and the co-cultures were used after DIV12.
Cell culture
HEK293T (human embryonic kidney-derived) and Neuro2a (mouse neuroblastoma) cells, were cultured in DMEM medium supplemented with 10% and 1% FBS, respectively, and 2 mM L-glutamine in a humified atmosphere of 5% CO2 at 37°C. Cells were passaged at 80%–90% confluency and transfected at 50%–60% confluency.
Human brain tissue
Human brain tissues were obtained through the New York Brain Bank at Columbia University according to the institutional guidelines and policies. Neuropathological evaluation for each autopsy case includes scores for the CERAD, Braak, and NIA-Reagan diagnoses (Table S1). Cerebral amyloid angiopathy was assessed according to the Vonsattel grading. The obtained tissue samples were derived from Brodmann area (BA) 9/10 of the dorsolateral prefrontal cortex.
METHOD DETAILS
Generation of DNA constructs
Generation of the full-length and luminal domain of CREB3L2 plasmids containing a poly-histidine tag was described in prior literature (McCurdy et al., 2019). In the plasmid encoding the luminal domain of CREB3L2, a signal sequence of BiP (AAG CTC TCC CTG GTG GCC GCG ATG CTG CTG CTG CTC AGC GCG GCG CGG GCC) was inserted following the start codon to construct the TAILS-His plasmid. Histidine tags from the full-length CREB3L2 (FL-His) and TAILS-His were replaced with a horseradish peroxidase (HRP) variant subcloned from pCMV-erHRP (Addgene) to generate FL-HRP and TAILS-HRP constructs for a peroxidase activity-based TAILS secretion assay. The HRP variant was codon-optimized for improved expression in mammalian system and introduced with N175S mutation for increased stability against heat and hydrogen peroxide (Joesch et al., 2016).
Transfection
Primary hippocampal neurons
Primary hippocampal neurons were transfected at DIV1–2 by using calcium phosphate transfection kit (Thermo Fisher Scientific) with few modifications from the manufacturer’s instruction to minimize toxicity. Briefly, a fine precipitate from DNA/CaCl2 and 2× HBS mixture was gently applied to cells plated on coverslips in a dropwise manner. After 1-h transfection in a humified atmosphere of 5% CO2 at 37°C, cells were exposed to a DMSO shock (10% in culture medium) for 2.5 min and washed with acidified medium, followed by 2–3 additional wash with pre-warmed phosphate buffered saline (PBS). In general, 1–2 mg DNA per 12-mm diameter cover-slip was used for calcium phosphate-based transfection in neurons.
HEK293T and Neuro2a
HEK293T or Neuro2a cells were plated 2 days prior, and transfection was carried out at 70%–80% confluency using CalFectin DNA transfection reagent (SignaGen Laboratories) following the manufacturer’s instructions. For each well of 6-well plate, 1 mg DNA was transfected and scaled accordingly for different culture formats.
Lentiviral transduction
Lentivirus was produced by transfecting the lentiviral plasmid (10 mg) with the packaging (pCMV-dR8.91; 7 mg) and envelope plasmids (pCMV-VSV-G; 3 mg) in HEK293T cells plated in a 100-mm plate at 80% confluency. Medium was changed to Neurobasal medium supplemented with B27 and L-glutamine at 6-h post-transfection and the virus-containing medium was collected 24–36 h later. Viruses were passed through a 0.45 μm PES filter to remove cell debris and frozen at −80°C in aliquots. The viral titers were measured with qPCR lentivirus titration kit (Applied Biological Materials). For generation of stable cell lines, HEK293T cells transduced with lentivirus at an MOI of 20 were subjected to antibiotic selection after 5–7 days post-transduction.
Stable cell line generation
For generation of a TAILS-expressing clone, HEK293T cells were transfected with a plasmid encoding the luminal domain of CREB3L2 preceded by a signal sequence of BiP and followed by a hexa-histidine tag for constitutive induction of TAILS secretion. Transfected cells were manually diluted into single cells and seeded in 96-well plate. Once cells reached ~30% confluency, antibiotic selection was performed with 100 mg mL−1 Zeocin until confluency reached over 80%. The Zeocin-resistant cells were passaged to 6-well plates in duplicates—one for expansion and one for immunoblot validation—and the clone with the highest TAILS expression was maintained sequentially in 200 and 500 mg mL−1 Zeocin for three passages each before stocks for the stable cell line were made. Stable cells expressing HRP-tagged full-length CREB3L2 proteins were generated by lentiviral transduction. Like the clone generation, stable cell colonies were established through Zeocin selection.
Preparation of conditioned medium
HEK293T cells stably integrated with the histidine-tagged luminal domain of CREB3L2 were used to generate TAILS-enriched conditioned media. For collection of conditioned media, the TAILS-secreting cells were plated in 100-mm plates at least two passages after antibiotic selection to avoid possible contamination of culture medium with debris from dead cells. At ~80% confluency cells were gently washed with PBS and added with 6 mL Neurobasal medium supplemented with B27 and L-glutamine for 12–16 h. Conditioned medium was collected and centrifuged at 3,000 xg for 20 min. Supernatant was then passed through a 0.45 μm PES filter and treated to neurons or aliquots of stocks were made and stored at −80°C. Control conditioned medium was prepared through identical procedure, in parallel, using regular HEK293T cells. For application to primary neurons, conditioned media were added at 20% of total volume per well.
His-tagged recombinant protein purification
TAIL-enriched conditioned media were passed through Amicon Ultra 50K centrifuge tubes (MilliporeSigma) to remove debris and large-sized constituents from FBS. Filtrates were collected and concentrated using Amicon Ultra 3K centrifuge tubes (MilliporeSigma) for histidine-tag purification. Pore size of Amicon Ultra centrifuge devices were determined empirically for the highest specificity of purified product. Recombinant TAILS protein with poly-histidine tag was purified using a Capturem His-tagged purification kit (Takara) and protein concentration was measured by micro-BCA protein assay (Thermo Fisher Scientific). The protein size of the purified product was confirmed by Western blot.
TAILS secretion assay
HEK293T and primary hippocampal neurons were transfected with a construct for full-length CREB3L2/TAILS precursor protein, C-terminally tagged with HRP (FL-HRP). Lentivirally transduced HEK293T cells were maintained without antibiotics for at least two passages before used for the assay. Treatments were made at 70–80% confluency and culture media were collected through identical procedure as described for preparation of conditioned medium. After removing debris, peroxidase activity assay (MilliporeSigma) was performed, and the level of TAILS secretion was measured. For measurement of TAILS-HRP secretion in neurons, primary hippocampal neurons were transduced with FL-HRP lentiviral particles at DIV1 and the culture media were subjected to the secretion assay at DIV13–15. Collected culture media from HEK293T and hippocampal neurons were subjected to peroxidase activity assay (Sigma) following the manufacturer’s instruction. Samples were transferred to a black 96-well plate with clear bottom for measurements of absorbance at 570 nm in a microplate reader (Tecan).
TCA precipitation of secreted proteins
Conditioned media were concentrated over Amicon Ultra 30K filters and incubated on ice for 10 min with 20% TCA v/v added. Precipitated proteins were pelleted by 5-min centrifugation at 8,000 x g. Pellets were washed with 200 μL ice-cold acetone three times and dried before resuspended in 2x Laemmli sample buffer (Bio-Rad) supplemented with 50 mM DTT for immunoblot analysis.
Human brain sample processing
Protein extracts were prepared from 30–50 mg of tissue in ice-cold RIPA buffer supplemented with protease and phosphatase inhibitors (Roche) using a Dounce homogenizer. The amount of lysis buffer was determined accordingly to the tissue weight (300 mL per ~5 mg of tissue). Samples were incubated in RIPA buffer for 2 h at 4°C under constant rotation with a 10-min sonication (Branson 1510). Lysates were centrifuged at 10,000 x g for 20 min in cold, and supernatants were mixed with 2x Laemmli buffer containing DTT. Extracts were heated at 95°C for 5 min before Western blot analysis.
Western blot analysis
For protein isolation from cells, each well was washed in HBSS and collected in ice-cold RIPA buffer supplemented with protease and phosphatase inhibitors (Roche). Brain tissue was obtained from the pregnant Sprague-Dawley dam (Envigo) sacrificed for primary culture of embryonic neurons, and immediately processed for lysis on ice. Protein extracts were vortexed every 10 min and kept in ice for 30 min, followed by centrifugation at 15,000 xg for 20 min at 4°C. Supernatants were collected and sampled in Laemmli sample buffer containing 50 mM dithiothreitol (DTT) and heated at 95°C for 5 min. Samples were resolved by SDS-PAGE using the NuPAGE electrophoresis system (Thermo Fisher Scientific), followed by wet transfer (Bio-Rad) onto 0.2 μm pore-sized nitrocellulose membranes (GE Healthcare). Electroblotted membranes were incubated for 1 h in blocking buffer consisting of 5% BSA in Tris-based saline with 0.1% Tween 20 (TBS-T). Primary antibodies were diluted in the blocking buffer for overnight incubation at 4°C. For immunodetection, blots were incubated with their respective HRP-conjugated secondary antibodies diluted in TBS-T with 2% skim milk for 1 h at room temperature. Blots were washed three times with TBS-T between each step. Images were acquired using the KwikQuant Imager (Kindle Biosciences) and quantified using Fiji/ImageJ (Schindelin et al., 2012).
For preparation of brain lysates of 4-VO and sham animals, brains were removed from ischemic and sham animals, followed by hippocampal dissection by transversely slicing the dorsal hippocampus (1 mm) with a McIlwain tissue chopper (Cavey Laboratory Engineering Co.). The CA1 subfield was rapidly micro-dissected and snap-frozen in liquid nitrogen. Proteins were extracted in lysis buffer (50 mM Tris-HCl, pH 8.0, 1% NP-40, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate and a cocktail of protease and phosphatase inhibitors (Sigma)).
Immunoprecipitation
Cell/tissue lysates
Immunoprecipitation of TAILS was carried out by overnight incubation at 4°C with antibody against the luminal domain of full-length CREB3L2/TAILS precursor protein, or the appropriate control IgG antibody species. Samples were prepared in lysis buffer containing Triton X-100—0.2% for primary-cultured neurons and 1% for brain tissues—supplemented with protease and phosphatase inhibitors (Roche). In parallel, M-280 Sheep Anti-Rabbit IgG Dynabeads (Thermo Fisher Scientific) were blocked with PBS containing 0.1% BSA. Immunoprecipitates were captured with the pre-blocked Dynabeads (40 μL for cell lysate, and 60 μL for tissue lysate) and washed four times with PBS—first twice with 0.1% Triton X-100 and the rest without. Immunoprecipitates were eluted from beads with 0.2 M glycine buffer (pH 2.5) and heated 70°C for 10 min. The pH of the eluate was adjusted by adding 1 M Tris (pH 7.5) and beads were discarded after spin-down. Supernatants were collected and mixed with 2x Laemmli sample buffer (Bio-Rad) supplemented with 50 mM DTT for immunoblot analysis.
TAILS-enriched conditioned medium
Culture or conditioned medium from hippocampal neurons was collected and sequentially centrifuged at 2,000 x g and 10,000 x g for 20 min each at 4°C to remove debris. The debris-free media—supplemented with protease and phosphatase inhibitors (Roche)—were incubated overnight with a primary antibody against the luminal domain of full-length CREB3L2/TAILS precursor protein, or the appropriate control IgG antibody species. For immunoprecipitation from 10 mL conditioned medium, 40 μL pre-blocked Dynabeads were added. Beads were added with 2× Laemmli sample buffer (Bio-Rad) supplemented with 50 mM DTT and heated at 85°C for 10 min for protein denaturation. Supernatants were collected for immunoblot analysis.
Immunocytochemistry
Primary embryonic neurons were cultured as described above and fixed for 10 min at room temperature in pre-warmed PBS solution containing 4% paraformaldehyde (PFA) and 2% sucrose. After washing with PBS, cells were incubated in a PBS-based blocking buffer containing 3% heat-inactivated goat serum, 2% bovine serum albumin (BSA), 0.2% Triton X-100 for 30 min at room temperature. Coverslips were incubated overnight at 4°C with primary antibodies diluted in the blocking buffer. Neurons were washed with PBS and incubated with fluorophore-conjugated Alexa Fluor secondary antibodies and Alexa Fluor 488-conjugated βIII-tubulin antibody (BioLegend) in the dark for 1 h at room temperature. Samples were mounted in ProLong Diamond Antifade with DAPI (Invitrogen) and air-dried in dark overnight.
Cell death analysis by TUNEL
Cell death in primary neuronal culture was assessed by labeling of DNA strand breaks by in situ cell death detection kit (TMR red; Roche) in accordance with the manufacturer’s instructions. Cells were fixed in pre-warmed 4% PFA, 2% sucrose PBS solution for 10 min at room temperature. Cells were washed in PBS and incubated in a 5% bovine serum albumin (BSA), 0.2% Triton X-100, PBS-based blocking buffer. For selective analysis of neuronal death, cells were immunolabeled with an Alexa Fluor 488-conjugated neuron-specific βIII-tubulin antibody (BioLegend), followed by nuclear staining with DAPI in ProLong Diamond Antifade Mountant (Thermo Fisher Scientific). For analysis, only TUNEL-positive nuclei were counted from βIII-tubulin-positive neurons.
Proximity ligation assay
Primary neuronal cultures
For detection of protein-protein interaction, hippocampal neurons plate on coverslips were fixed and immunolabeled with primary antibodies as described in immunocytochemistry. Following the overnight incubation with primary antibodies, proximity ligation assay (PLA) was performed using Duolink in situ Red Detection reagents (MilliporeSigma) according to the manufacturer’s guidelines. In short, neurons were labeled with PLUS (anti-mouse) and MINUS (anti-rabbit) probes which were hybridized and amplified for visualization of the protein complex. PLA was followed by counterstaining with Alexa Fluor 488-conjugated βIII-tubulin antibody (BioLegend) and coverslips were mounted on glass-bottom dishes (MatTek Corporation) using Duolink in situ mounting medium with DAPI (MilliporeSigma). All incubations were performed in a humidity chamber at 37°C. Images were acquired using a Zeiss Axio Observer.Z1 microscope and Zen Blue 2.1 software (Zeiss). PLA puncta for TAILS/SHH-N, TAILS/PTCH1 and SHH-N/PTCH1 complexes were counted and normalized to the area of βIII-tubulin-labeled neurites; PLA puncta for NRF2/KEAP1 complex were counted and normalized to the area of the soma.
Brain tissues
Frozen fixed mouse brain sections were performed with a heat-induced antigenic epitope retrieval prior to PLA. Sections mounted on slides were thawed and washed in TBS while 0.01 M sodium citrate buffer (0.05% Tween 20, pH 6.0) was heated in a pressure cooker. Epitope unmasking was performed in the boiling sodium citrate buffer for 3 min, followed by TBS wash. Slides were incubated in a TBS-based blocking/permeabilization solution containing 6% heat-inactivated goat serum, 4% BSA, 0.2% Triton X-100 for 1 h at room temperature. PLA steps were carried out equivalently to as described above for primary neuronal cultures, followed by counterstaining with Alexa Fluor 488-conjugated βIII-tubulin antibody (BioLegend). To avoid relatively strong autofluorescence in the yellow/red spectral region, Duolink in situ FarRed Detection reagents (MilliporeSigma) were used for PLA in brain tissues.
Analysis of GFP-based reporters
Two GFP-based reporters—7xGLI-GFP and 8xARE-GFP for assessment of hedgehog signaling and antioxidant gene expression, respectively—were utilized in this study. For fluorescence microscopy-based analysis, primary hippocampal neurons plated on coverslips were transfected at DIV1–2 using calcium phosphate-based method as described above. Transfected neurons were fixed with PFA on DIV15 and counterstained with Alexa Fluor 594-conjugated βIII-tubulin antibody (BioLegend) in for 1 h at room temperature. Samples were mounted in ProLong Diamond Antifade with DAPI (Invitrogen) and stored at 4°C no longer than 2 days before fluorescence microscopy. For assessing the dose-response of GLI activity, Neuro2a cells were seeded in black 96-well plates with clear bottom and subjected to experiments on DIV15. Background was determined by measurements from non-transfected neurons from the same plate. All measurements were normalized to DNA content per well using the CyQUANT assay (Thermo Fisher Scientific). GFP expression was also confirmed by immunoblotting the samples of Neuro2a cells expressing the reporter constructs.
Fluorescence microscopy
Samples for immunocytochemistry, PLA, TUNEL staining, and GFP-based reporter analyses were subjected to fluorescence microscopy. Images for immunocytochemistry and PLA (in neurons) and TUNEL-stained cell death analysis were acquired using a 40× oil objective on an Axio Observer.Z1 inverted microscope (Zeiss) equipped with an AxioCam MRm Rev 3. camera. PLA puncta within the region of interest (ROI) were counted manually and normalized to ROI area. For image generation for PLA in brain sections and GLI activity-dependent expression of GFP in neurons, a LSM800 confocal microscope (Zeiss) with a 40x or 63x oil objective was employed. For tissue PLA, images were acquired in Z stacks and tiles to generate comprehensive images of greater depth and larger field of view. PLA puncta within the ROI were counted and normalized by ROI area using Fiji/ImageJ software.
Cell viability analysis
For assessment of cell viability, primary hippocampal neurons seeded in black 96-well plates with clear bottom were stained with alamarBlue (G-Biosciences), a fluorescent indicator for cellular metabolic activity. For each well, 10 μL of alamarBlue dye was added and neurons were incubated for 1 h at 37°C. Cell viability was measured at 560/590 nm (excitation/emission) in a fluorescence microplate reader (Tecan). Background was determined by measurements from wells prepared in parallel but were added with 10 μL culture medium instead. All measurements were normalized to DNA content per well using the CyQUANT assay (Thermo Fisher Scientific).
Glutathione redox assay
We measured both reduced (GSH) and oxidized (GSSG) glutathione for ratiometric analysis of glutathione redox metabolism using a luminescent-based GSH/GSSG-Glo assay (Promega). Primary hippocampal neurons were seeded in white 96-well plates with clear bottom and cultured for 15 days and subjected to the assay in accordance with the manufacturer’s instructions. Briefly, total and GSSG-converted GSH levels were separately measured from two identical configurations. Based on these measures, the levels of GSH and GSSG were calculated and normalized to DNA content per well quantified by using the CyQUANT assay (Thermo Fisher Scientific).
Mitochondrial fractionation
Neuro2a cells were plated in 100-mm plates for centrifugation-based crude fractionation of mitochondria. Cells were collected in ice-cold fractionation buffer (10 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 20 mM HEPES, pH 7.4) and mechanically lysed in syringe through 27-gauge needles. Samples were centrifuged at 800 g for 5 min to pellet nuclei. Supernatant, the cytosolic fraction, was subjected to 10,000 g centrifugation for 5 min to pellet mitochondria fraction. The mitochondria-containing pellet was washed in ice-cold PBS and centrifuged at 10,000 g for 5 min twice and resuspended in TBS with 0.1% SDS supplemented with protease and phosphatase inhibitors (Roche). All centrifugations were done at 4°C and samples were kept on ice throughout the procedure.
Mitochondrial respiration measurements
To assess mitochondrial respiration, oxygen consumption rates (OCR) were measured in hippocampal neurons using a Seahorse XFp Analyzer with Cell Mito Stress Kit (Agilent Technologies) in accordance with the manufacturer’s instructions. Medium composition and inhibitor concentrations were determined following a published guideline for assessing mitochondrial function in primary neurons (Connolly et al., 2018). Briefly, dissociated embryonic hippocampal neurons were plated at a density of 100,000 cells/well in Seahorse XFp mini-plates pre-coated with poly-D-lysine and laminin, and cultured for 15 days before treatment. On the day of the assay (DIV15-DIV18), cells were washed and equilibrated in experimental buffer consisting of low-buffered bicarbonate-free XF DMEM medium (pH 7.4) supplemented with 10 mM glucose and 2 mM sodium pyruvate for 1 h at 37°C with no CO2. Baseline rates were measured at 37°C before the sequential injection of oligomycin (6 μM), FCCP (3 μM), and rotenone/antimycin A cocktail (0.5 μM). Basal OCR levels were determined by subtracting the non-mitochondrial respiration rate from the last respiration rate measured before the oligomycin injection. ATP production was measured by subtracting the proton leak rate from the basal respiration rate. Maximum respiration was calculated by subtracting the non-mitochondrial respiration rate from the maximum rate measurement after FCCP injection. All measurements were normalized to DNA content per well using the CyQUANT assay (Thermo Fisher Scientific).
Four-vessel occlusion (4-VO) model of global ischemia
Age-matched adult male Sprague Dawley rats (Charles River Laboratories) weighing 150–200 g were subjected to transient global ischemia by means of the 4-VO method as previously described (Hwang et al., 2017; Noh et al., 2012). For sham operation, all surgical procedures were performed except that the carotid arteries were not occluded. Severity of ischemia was assessed by pupil dilation. Animals exhibiting complete loss of the righting reflex and dilation of the pupils, after occlusion were scored from 1 to 4 (based on the pupil size; 4 is maximum) every minute until the end of occlusion (10 min), then the animal scored more than 17 in total for 10 min were used for the experiments. Those few animals showing obvious behavioral manifestations (abnormal vocalization when handled, generalized convulsions, hypoactivity), or losing >20% body weight through 3–7 days after surgery were excluded from the study. All rats were housed in standard Plexiglass cages under a 12-h light/dark cycle. Food and water were provided ad libitum. Animal care and handling procedures were approved by the Institutional Animal Care and Use Committee of Creighton University in accordance with National Institutes of Health guidelines.
Stereotaxic injections
For neuronal death assessment, recombinant TAILS (100 ng μL−1) or ΔBD (100 ng μL−1) were administered to rats by stereotaxic injection into the right hippocampal CA1 (3.0 mm posterior and 3.0 mm lateral to bregma, 2.6 mm ventral from dura) using a 33-gauge needle and a Hamilton syringe driven by a Quintessential Stereotaxic Injector (Stoelting) at a flow rate of 0.5 μL min~1 (total 3 μL). The needle was left in place for an additional 5 min and then gently withdrawn. For behavioral assays, TAILS (100 ng μL−1) or ΔBD (100 ng μL−1) were administered bilaterally to a position defined by the following coordinates: 3.0 mm posterior and ± 3.0 mm lateral to bregma, 2.6 mm ventral from dura. Animals were then subjected to 4-VO and sham surgery immediately after injection and used for histology and behavioral assays.
Histology analysis of neuronal death
Neuronal cell loss was assessed by histological analysis in brain sections from animals sacrificed at 7 days after sham or 4-VO surgery. Animals were perfused using 0.9% saline with heparin followed by ice-cold 4% PFA in 0.1M PBS (pH, 7.4). Brains were removed and immersed in fixative. Coronal sections (20 μm) were cut at the level of the dorsal hippocampus with a cryostat and processed for staining with Fluoro-Jade C or Toluidine Blue (MilliporeSigma). Images were viewed through a Zeiss Axiolab 5 fluorescence (Fluor-Jade), or light microscope (Toluidine Blue), and acquired with a Zeiss Axiocam 208 digital camera equipped with Zen 3.0 Pro imaging software at 5x and 20x magnification. The images were then processed for representative figures and cell counting. The number of pyramidal neurons per 710 μm length of medial CA1 were counted in 4–5 rats per treatment group (3–4 images per section, 4 sections per rat). All analysis was conducted with a blind test.
Behavioral tests
Open field test
To examine the effects of 4-VO on locomotor ability, the open field test was performed at 8 days after sham or 4-VO surgery. Animals were individually placed in the center of a plexiglass open field arena (90 cm × 90 cm × 40 cm) for 10 min. Overall locomotion in the arena was recorded by a ceiling-mounted camera and analyzed using an ANY-maze video tracking software (Stoelting) to measure the total distance travelled by each animal.
Novel object recognition
The novel object recognition test was conducted in the same arena used for the open field test to facilitate the animals to habituate to the novel environment. Following the open field test, animals were subjected to a 10-min training session at 9 days and a 10-min acquisition session was conducted at 10 days after sham or 4-VO surgery. Animals were exposed with two identical objects placed in adjacent corners of the arena equally distant from the walls. 24 h after training session, animals were placed into the arena containing the familiar object and a new object placed at the same locations as before the interval. Exploration behaviors were recorded and analyzed using an ANY-maze video tracking software (Stoelting). Exploration was defined as physical proximity within 2 cm towards an object.
Barnes maze
For assessment of hippocampus-dependent cognitive function, animals were tested in the Barnes maze at 11–17 days after surgery. The maze is a 90 cm-elevated circular apparatus with 120-cm diameter and contains 20 holes with 10-cm diameter—only one hole with an escape room—equally spaced around the perimeter with the spatial cues. At 11 days after surgery, animals were given a 5-min habituation session, allowed to explore the Barnes maze platform. During both learning and memory tests, animals were placed in the center and a strong light (1,000 lux) served as aversive stimuli to search for the escape. For learning tests, animals were daily trained two trials with a 30-min interval for 5 days (post-operation day 12–16). 24 h after the last learning session, a memory test was performed in the maze with an escape room and its spatial cues altogether rotated 180°. The primary latency time to reach the escape hole was measured during the learning and memory tests using an ANY-maze video tracking software (Stoelting).
QUANTIFICATION AND STATISTICAL ANALYSIS
Each experiment was conducted in at least three biological replicates unless otherwise indicated. For comparisons of two groups, a paired or unpaired t test was performed depending on experimental design. When comparing three independent groups, a one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test was performed. For comparisons of three or more groups encompassing two independent variables, two-way ANOVA with Tukey’s multiple comparisons test was performed. Details of statistical test used for analysis are described in figure legends. Data were analyzed with Prism 9.4 software (GraphPad). For all statistical tests, statistical significance is defined at p < 0.05.
For dose-response curve analysis, half maximal effective concentration (EC50) was calculated as a normalized percentage of the maximum values measured in control wells without treatment. Results were transformed using a non-linear regression with Prism 9.4 software (GraphPad).
Supplementary Material
Highlights.
Neurons secrete a facilitator of hedgehog signaling (TAILS) in response to stress
TAILS/SHH-N activates cell non-autonomous resilience mechanisms in neurons
TAILS prevents neuronal death and cognitive impairment in a global ischemia model
ACKNOWLEDGMENTS
C.G.R. was supported by the Alzheimer’s Association (AARF-17-533511 and AARF-17-533511-RAPID), E.P.M. was supported by the National Science Foundation Graduate Research Fellowship Program, J.-Y.H. was supported by the LB692 Nebraska Biomedical Research Development Fund and a Cognitive Neuroscience of Development and Aging Center Pilot Award funded by the National Institute of General Medical Sciences (P20GM130447), and U.H. was supported by grants from the National Institute of Neurological Disorders and Stroke and the National Institute on Aging (R01NS121337 and R01NS109607).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111488.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alvarez-Buylla A, and Ihrie RA (2014). Sonic hedgehog signaling in the postnatal brain. Semin. Cell Dev. Biol 33, 105–111. 10.1016/j.semcdb.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bambakidis NC, Petrullis M, Kui X, Rothstein B, Karampelas I, Kuang Y, Selman WR, LaManna JC, and Miller RH (2012). Improvement of neurological recovery and stimulation of neural progenitor cell proliferation by intrathecal administration of Sonic hedgehog. J. Neurosurg 116, 1114–1120. 10.3171/2012.1.JNS111285. [DOI] [PubMed] [Google Scholar]
- Barnham KJ, Masters CL, and Bush AI (2004). Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov 3, 205–214. 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- Charron F, Stein E, Jeong J, McMahon AP, and Tessier-Lavigne M (2003). The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell 113, 11–23. 10.1016/s0092-8674(03)00199-5. [DOI] [PubMed] [Google Scholar]
- Chechneva OV, Mayrhofer F, Daugherty DJ, Krishnamurty RG, Bannerman P, Pleasure DE, and Deng W (2014).A Smoothened receptor agonist is neuroprotective and promotes regeneration after ischemic brain injury. Cell Death Dis. 5, e1481. 10.1038/cddis.2014.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly NMC, Theurey P, Adam-Vizi V, Bazan NG, Bernardi P, Bolaños JP, Culmsee C, Dawson VL, Deshmukh M, Duchen MR, et al. (2018). Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 25, 542–572. 10.1038/s41418-017-0020-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, and Walter P (2020). The integrated stress response: from mechanism to disease. Science 368, eaat5314. 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, Attucks OC, Franklin S, Levonen AL, Kensler TW, and Dinkova-Kostova AT (2019). Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov 18, 295–317. 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- Díaz-Hung ML, and Hetz C (2022). Proteostasis and resilience: on the interphase between individual’s and intracellular stress. Trends Endocrinol. Metab 33, 305–317. 10.1016/j.tem.2022.02.003. [DOI] [PubMed] [Google Scholar]
- Donnelly N, Gorman AM, Gupta S, and Samali A (2013). The eIF2α kinases: their structures and functions. Cell. Mol. Life Sci 70, 3493–3511. 10.1007/s00018-012-1252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, and Zollner H (1991). Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med 11, 81–128. 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- Frakes AE, Metcalf MG, Tronnes SU, Bar-Ziv R, Durieux J, Gildea HK, Kandahari N, Monshietehadi S, and Dillin A (2020). Four glial cells regulate ER stress resistance and longevity via neuropeptide signaling in C. elegans. Science 367, 436–440. 10.1126/science.aaz6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia ADR, Petrova R, Eng L, and Joyner AL (2010). Sonic hedgehog regulates discrete populations of astrocytes in the adult mouse forebrain. J. Neurosci 30, 13597–13608. 10.1523/JNEUROSCI.0830-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G, Quinn P, and Warren G (1983). Dissection of the Golgi complex. I. Monensin inhibits the transport of viral membrane proteins from medial to trans Golgi cisternae in baby hamster kidney cells infected with Semliki Forest virus. J. Cell Biol 96, 835–850. 10.1083/jcb.96.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Hartman RE, Lee JM, Zipfel GJ, and Wozniak DF (2005). Characterizing learning deficits and hippocampal neuron loss following transient global cerebral ischemia in rats. Brain Res. 1043, 48–56. 10.1016/j.brainres.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Helms JB, and Rothman JE (1992). Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature 360, 352–354. 10.1038/360352a0. [DOI] [PubMed] [Google Scholar]
- Huang SS, Cheng H, Tang CM, Nien MW, Huang YS, Lee IH, Yin JH, Kuo TBJ, Yang CCH, Tsai SK, and Yang DI (2013). Anti-oxidative, anti-apoptotic, and pro-angiogenic effects mediate functional improvement by sonic hedgehog against focal cerebral ischemia in rats. Exp. Neurol 247, 680–688. 10.1016/j.expneurol.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Hwang JY, Gertner M, Pontarelli F, Court-Vazquez B, Bennett MVL, Ofengeim D, and Zukin RS (2017). Global ischemia induces lysosomal-mediated degradation of mTOR and activation of autophagy in hippocampal neurons destined to die. Cell Death Differ. 24, 317–329. 10.1038/cdd.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, and Yamamoto M (1999). Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86. 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joesch M, Mankus D, Yamagata M, Shahbazi A, Schalek R, Suissa-Peleg A, Meister M, Lichtman JW, Scheirer WJ, and Sanes JR (2016). Reconstruction of genetically identified neurons imaged by serial-section electron microscopy. Elife 5, e15015. 10.7554/eLife.15015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Saito A, Asada R, Kanemoto S, and Imaizumi K (2011). Physiological unfolded protein response regulated by OASIS family members, transmembrane bZIP transcription factors. IUBMB Life 63, 233–239. 10.1002/iub.433. [DOI] [PubMed] [Google Scholar]
- Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, and Mattson MP (1997). Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J. Neurosci 17, 5089–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan NR, Anufreichik V, Rodvold JJ, Chiu KT, Sepulveda H, and Zanetti M (2012). Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8(+) T cell priming. PLoS One 7, e51845. 10.1371/journal.pone.0051845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, and Zanetti M (2011).Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc. Natl. Acad. Sci. USA 108, 6561–6566. 10.1073/pnas.1008942108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurdy EP, Chung KM, Benitez-Agosto CR, and Hengst U (2019). Promotion of axon growth by the secreted end of a transcription factor. Cell Rep. 29, 363–377.e5. 10.1016/j.celrep.2019.08.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM, Bennett MVL, and Zukin RS (2012). Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc. Natl. Acad. Sci. USA 109, E962–E971. 10.1073/pnas.1121568109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nüsslein-Volhard C, and Wieschaus E (1980). Mutations affecting segment number and polarity in Drosophila. Nature 287, 795–801. 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- Nys GMS, van Zandvoort MJE, de Kort PLM, van der Worp HB, Jansen BPW, Algra A, de Haan EHF, and Kappelle LJ (2005). The prognostic value of domain-specific cognitive abilities in acute first-ever stroke. Neurology 64, 821–827. 10.1212/01.WNL.0000152984.28420.5A. [DOI] [PubMed] [Google Scholar]
- Oksala NKJ, Jokinen H, Melkas S, Oksala A, Pohjasvaara T, Hietanen M, Vataja R, Kaste M, Karhunen PJ, and Erkinjuntti T (2009). Cognitive impairment predicts poststroke death in long-term follow-up. J. Neurol. Neurosurg. Psychiatry 80, 1230–1235. 10.1136/jnnp.2009.174573. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Schwartz CM, Wang YX, Kawamoto EM, Mattson MP, and Yao PJ (2013). Sonic hedgehog promotes autophagy in hippocampal neurons. Biol. Open 2, 499–504. 10.1242/bio.20134275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Schwartz CM, Wang YX, Mattson MP, and Yao PJ (2011). Subcellular localization of Patched and Smoothened, the receptors for Sonic hedgehog signaling, in the hippocampal neuron. J. Comp. Neurol 519, 3684–3699. 10.1002/cne.22681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, and Plum F (1982). Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol 11, 491–498. 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Rivell A, Petralia RS, Wang YX, Clawson E, Moehl K, Mattson MP, and Yao PJ (2019). Sonic hedgehog expression in the postnatal brain. Biol. Open 8, bio040592. 10.1242/bio.040592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, and Walter P (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol 8, 519–529. 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Saito A, Kanemoto S, Zhang Y, Asada R, Hino K, and Imaizumi K (2014). Chondrocyte proliferation regulated by secreted luminal domain of ER stress transducer BBF2H7/CREB3L2. Mol. Cell 53, 127–139. 10.1016/j.molcel.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Sala AJ, Bott LC, and Morimoto RI (2017). Shaping proteostasis at the cellular, tissue, and organismal level. J. Cell Biol 216, 1231–1241. 10.1083/jcb.201612111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samkari A, White J, and Packer R (2015). SHH inhibitors for the treatment of medulloblastoma. Expert Rev. Neurother 15, 763–770. 10.1586/14737175.2015.1052796. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, Sood R, Khlaifia A, Eslamizade MJ, Hung TY, Lou D, Asgarihafshejani A, Lalzar M, Kiniry SJ, Stokes MP, et al. (2020). eIF2alpha controls memory consolidation via excitatory and somatostatin neurons. Nature 586, 412–416. 10.1038/s41586-020-2805-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JR, Lee SW, Topalkara K, Qiu J, Xu J, Zhou Z, and Moskowitz MA (2009). Sonic hedgehog regulates ischemia/hypoxia-induced neural progenitor proliferation. Stroke 40, 3618–3626. 10.1161/STRO-KEAHA.109.561951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Venkannagari S, Oh KH, Zhang YQ, Rohde JM, Liu L, Nimmagadda S, Sudini K, Brimacombe KR, Gajghate S, et al. (2016). Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem. Biol 11, 3214–3225. 10.1021/acschembio.6b00651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HL, Freeman OJ, Butcher AJ, Holmqvist S, Humoud I, Schätzl T, Hughes DT, Verity NC, Swinden DP, Hayes J, et al. (2020). Astrocyte unfolded protein response induces a specific reactivity state that causes non-cell-autonomous neuronal degeneration. Neuron 105, 855–866.e5. 10.1016/j.neuron.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenkle NT, Lahiri A, Simpkins JW, and Meares GP (2019). Endoplasmic reticulum stress is transmissible in vitro between cells of the central nervous system. J. Neurochem 148, 516–530. 10.1111/jnc.14642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton BZ, Peng LF, Maloof N, Nakai K, Wang X, Duffner JL, Taveras KM, Hyman JM, Lee SW, Koehler AN, et al. (2009). A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat. Chem. Biol 5, 154–156. 10.1038/nchembio.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Berendzen KM, and Dillin A (2014). Systemic stress signalling: understanding the cell non-autonomous control of proteostasis. Nat. Rev. Mol. Cell Biol 15, 211–217. 10.1038/nrm3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, and Dillin A (2013). XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153, 1435–1447. 10.1016/j.cell.2013.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ziel AM, Wolzak K, Nöolle A, Hoetjes PJ, Berenjeno-Correa E, van Anken E, Struys EA, and Scheper W (2020). No evidence for cell-to-cell transmission of the unfolded protein response in cell culture. J. Neurochem 152, 208–220. 10.1111/jnc.14856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, and Scott MP (1999). Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22, 103–114. 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Williams KW, Liu T, Kong X, Fukuda M, Deng Y, Berglund ED, Deng Z, Gao Y, Liu T, Sohn JW, et al. (2014). Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab. 20, 471–482. 10.1016/j.cmet.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyler E, Franke V, Menegatti J, Kocks C, Boltengagen A, Praktiknjo S, Walch-Rückheim B, Bosse J, Rajewsky N, Grässer F, et al. (2019). Single-cell RNA-sequencing of herpes simplex virus 1-infected cells connects NRF2 activation to an antiviral program. Nat. Commun 10, 4878. 10.1038/s41467-019-12894-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, Losada PM, Chen MB, Schaum N, Khoury N, et al. (2022).A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603, 885–892. 10.1038/s41586-021-04369-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao PJ, Manor U, Petralia RS, Brose RD, Wu RTY, Ott C, Wang YX, Charnoff A, Lippincott-Schwartz J, and Mattson MP (2017). Sonic hedgehog pathway activation increases mitochondrial abundance and activity in hippocampal neurons. Mol. Biol. Cell 28, 387–395. 10.1091/mbc.E16-07-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao PJ, Petralia RS, and Mattson MP (2016). Sonic hedgehog signaling and hippocampal neuroplasticity. Trends Neurosci. 39, 840–850. 10.1016/j.tins.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti M, Rodvold JJ, and Mahadevan NR (2016). The evolving paradigm of cell-nonautonomous UPR-based regulation of immunity by cancer cells. Oncogene 35, 269–278. 10.1038/onc.2015.108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. The accession number for the single-cell profiling of hippocampus and superior frontal cortex from 25 hippocampus and cortex samples across 17 control and AD patients is listed in the key resources table and the data were accessed via Human BBB portal (https://twc-stanford.shinyapps.io/human_bbb) (Yang et al., 2022).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CREB3L2 C terminus | Abcam | ab102989; RRID:AB_10711653 |
| Rabbit polyclonal anti-CREB3L2 C terminus | Abcepta | AP9654b; RRID:AB_2544158 |
| Rabbit polyclonal anti-CREB3L2 N terminus | MilliporeSigma | HPA015068; RRID:AB_1847224 |
| Mouse monoclonal Alexa Fluor 488 anti-βIII-Tubulin (TUJ1 clone) | BioLegend | 801203; RRID:AB_2564757 |
| Mouse monoclonal anti-SHH (E-1) | Santa Cruz | sc-365112; RRID:AB_10709580 |
| Rabbit polyclonal anti-PTCH1 | Proteintech | 17520-1-AP; RRID:AB_2176561 |
| Mouse monoclonal anti-SMO (E-5) | Santa Cruz | sc-166685; RRID:AB_2239686 |
| Rabbit polyclonal anti-GLI1 (V812) | Cell Signaling Technology | 2534; RRID:AB_2294745 |
| Rabbit polyclonal anti- GFP | Abcam | ab290; RRID:AB_303395 |
| Rabbit polyclonal anti-cleaved CASP3 | Cell Signaling Technology | 9661; RRID:AB_2341188 |
| Rabbit polyclonal anti-PARP | Cell Signaling Technology | 9542; RRID:AB_2160739 |
| Mouse monoclonal anti-β-Actin (AC-15) | Abcam | ab6276; RRID:AB_2223210 |
| Mouse monoclonal anti-GAPDH (6C5) | Santa Cruz | sc-32233; RRID:AB_627679 |
| Rabbit monoclonal anti-p-eIF2α | Cell Signaling Technology | 3398; RRID:AB_2096481 |
| Rabbit monoclonal anti-ATF4 | Cell Signaling Technology | 11815; RRID:AB_2616025 |
| Rabbit monoclonal anti-NRF2 (D1Z9C) | Cell Signaling Technology | 12721; RRID:AB_2715528 |
| Mouse monoclonal anti-KEAP1 | Proteintech | 60027-1-lg; RRID:AB_2132623 |
| Mouse monoclonal anti-TOM20 (2F8.1) | MilliporeSigma | MABT166; RRID:AB_2890986 |
| Bacterial and virus strains | ||
| Stellar competent cells | Takara | 636766 |
| Biological samples | ||
| See Table S1. A complete list of all human brain samples used in this study | New York Brain Bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DMSO | MilliporeSigma | D8418 |
| Nelfinavir | MilliporeSigma | CDS021783 |
| Robotnikinin | Thermo Fisher Scientific | 50464659 |
| 4-Hydroxynonenal (HNE) | MilliporeSigma | 3932041MG |
| Purmorphamine | MilliporeSigma | 5402235MG |
| MG132 | Cayman Chemical | 133407-82-6 |
| SIN-1 | MilliporeSigma | 567028 |
| Hydrogen peroxide | MilliporeSigma | MAK092C |
| Chloroquine diphosphate | Tocris Bioscience | 4109/50 |
| Tunicamycin | MilliporeSigma | 50-457-00001 |
| ΔBD (recombinant protein) | His-tag purification | N/A |
| TAILS (recombinant protein) | His-tag purification | N/A |
| Critical commercial assays | ||
| Luna Universal Probe One-Step RT-qPCR Kit | NEB | E3006L |
| Duolink In Situ Detection Reagents (Red) | MilliporeSigma | DUO92008 |
| Duolink In Situ Detection Reagents (FarRed) | MilliporeSigma | DUO92013 |
| Duolink In Situ PLA Probe anti-Mouse PLUS | MilliporeSigma | DUO92001 |
| Duolink In Situ PLA Probe anti-Rabbit MINUS | MilliporeSigma | DUO92005 |
| In Situ Cell Death Detection Kit (TMR red) | Roche | 12156792910 |
| Peroxidase Activity Assay Kit | MilliporeSigma | MAK092 |
| CellROX (Green) | Invitrogen | C10444 |
| AlamarBlue Cell Viability Assay | G-Biosciences | 786922 |
| GSH/GSSG-Glo Glutathione Assay | Promega | V6611 |
| Seahorse XFp FluxPak | Agilent Technologies | 103022-100 |
| Seahorse XFp Cell Mito Stress Test Kit | Agilent Technologies | 103010-100 |
| Capturem His-Tagged Purification Kit | Takara | 635710 |
| CyQUANT Cell Proliferation Assay | Thermo Fisher Scientific | C7026 |
| CalFectin Transfection Reagent | SignaGen Laboratories | SL100478 |
| Deposited data | ||
| Single-nucleus RNA-sequencing database | Yang et al., 2022 | GSE163577; https://twc-stanford.shinyapps.io/human_bbb |
| Experimental models: Cell lines | ||
| Human: HEK293T | ATCC | CRL-3216 |
| Mouse: Neuro2a | ATCC | CCL-131 |
| Human: HEK293T (constitutive TAILS secretion) | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Rat: Hsd:Sprague Dawley SD (pregnant) | Envigo | N/A |
| Rat: Hsd:Sprague Dawley SD (adult male) | Charles River Laboratories | N/A |
| Oligonucleotides | ||
| Gss (Rn00564188_m1) | Thermo Fisher Scientific | 4331182 |
| Nfe2l2 (Rn00821258_g1) | Thermo Fisher Scientific | 4331182 |
| Rpl13 (Rn00582415_m1) | Thermo Fisher Scientific | 4331182 |
| Gss (Rn00564188_m1) | Thermo Fisher Scientific | 4331182 |
| Recombinant DNA | ||
| 7xGLI-GFP | Addgene | 110494 |
| 8xARE-GFP | Addgene | 134910 |
| pCMV-erHRP (N175S mutant) | Addgene | 79909 |
| C-terminal CREB3L2 (His-tagged) | McCurdy et al., 2019 | N/A |
| TAILS (BiP sequence, His-tagged) | This paper | N/A |
| ΔBD (binding domain deletion mutant; BiP sequence, His-tagged) | This paper | N/A |
| CREB3L2 (HRP-tagged) | This paper | N/A |
| Software and algorithms | ||
| ImageJ (FIJI) | Schindelin et al., 2012 | RRID:SCR_002285 |
| ANY-Maze | Stoelting | RRID:SCR_014289 |
| GraphPad Prism 9.4 | GraphPad Software | RRID:SCR_002798 |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.