

Abstract
Background:
Among children, sex-specific differences in disease prevalence, age of onset, and susceptibility have been observed in health conditions including asthma, immune response, metabolic health, some pediatric and adult cancers, and psychiatric disorders. Epigenetic modifications such as DNA methylation may play a role in the sexual differences observed in diseases and other physiological traits.
Methods:
We performed a meta-analysis of the association of sex and cord blood DNA methylation at over 450,000 CpG sites in 8,438 newborns from 17 cohorts participating in the Pregnancy And Childhood Epigenetics (PACE) Consortium. We also examined associations of child sex with DNA methylation in older children ages 5.5 to 10 years from 8 cohorts (n=4,268).
Results:
In newborn blood, sex was associated at Bonferroni level significance with differences in DNA methylation at 46,979 autosomal CpG sites (p<1.3 x 10-7) after adjusting for white blood cell proportions and batch. Most of those sites had lower methylation levels in males than in females. Of the differentially methylated CpG sites identified in newborn blood, 68% (31,727) met look-up level significance (p<1.1 x 10-6) in older children and had methylation differences in the same direction.
Conclusions:
This is a large-scale meta-analysis examining sex differences in DNA methylation in newborns and older children. Expanding upon previous studies, we replicated previous findings and identified additional autosomal sites with sex-specific differences in DNA methylation. Differentially methylated sites were enriched in genes involved in cancer, psychiatric disorders, and cardiovascular phenotypes.
Keywords: DNA methylation, sex, children, cord blood, EWAS
1. Introduction
There is a growing body of literature demonstrating that the in utero environment can impact health later in life [1–5]. DNA methylation is a commonly studied epigenetic mark that can influence gene expression without change in DNA sequence and is one mechanism through which early-life exposures might contribute to the developmental origins of disease [6]. Exposures to chemicals during pregnancy such as those found in tobacco smoke and air pollution, as well as perinatal characteristics such as birth weight and gestational age, have been associated with differences in umbilical cord blood DNA methylation [7–11]. Furthermore, site-specific differential methylation of cord blood DNA has also been associated with later-life health outcomes including asthma and insulin sensitivity [12–14].
In addition to exposures and health outcomes, inter-individual differences in DNA methylation levels are also impacted by sex. Among females, DNA methylation plays an important role in X-chromosome inactivation [15]. Prior studies have shown sex to be associated with DNA methylation measured in blood at birth (umbilical cord blood), [16–20] in older children [19,21,22], and in adults [21,23–26] as well as in placenta [20,27,28]. As expected, there are widespread differences in DNA methylation levels between sexes at X chromosome CpG sites; however, these studies also reported significant differences in methylation of autosomes [18,21]. Furthermore, autosomal sites differentially methylated between sexes were enriched in genes involved in pathways related to RNA splicing, DNA repair, the nervous system and behavior [18,21]. Most prior studies have been limited in sample size, with fewer than 200 subjects. It is likely that a much larger meta-analysis would improve power to identify CpG sites with smaller DNA methylation differences between males and females at birth, a critical developmental period. Significant sex differences in disease prevalence, age of onset, and susceptibility across the life course have been observed for various conditions such as asthma and allergies, immune response, metabolic health, some pediatric and adult cancers, and psychiatric disorders [29–35]. Therefore, identifying the differences in DNA methylation between males and females may highlight the genes that play an active role in the biological mechanisms involved in sex-dependent differences impacting health.
We performed a meta-analysis of associations between sex and DNA methylation in newborn blood samples and conducted a follow-up meta-analysis in blood from older children in multiple cohorts. We also investigated enrichment of sex-associated differential methylation in specific biological pathways and diseases.
2. Material and methods
2.1. Participating cohorts
PACE consists of over 40 international birth and child cohorts with a goal of performing coordinated Epigenome Wide Association Studies (EWAS) followed by meta-analysis to understand relationships between methylation and both exposures and child health outcomes [36]. Twenty-one independent cohorts contributed data to this study. We included 8,438 newborns from 17 cohorts in the analysis of newborn blood DNA methylation and sex (ALSPAC, CHAMACOS, CHS, EARLI, EXPOsOMICS, GECKO, Gen3G, Generation R, GOYA, INMA, IOW F2, MoBa1, MoBa2, MoBa3, NEST, PREDO, Viva). For the child methylation analysis, we included 4,268 children from eight cohorts (ALSPAC, BAMSE, CHAMACOS, CHOP, Generation R, HELIX, IOW F1, Viva). Detailed methods on the individual cohorts participating in the cord blood and child analyses are provided in the supplementary methods. All cohorts obtained written informed consent from participants prior to data collection which was approved by local ethics committees.
2.2. Methylation Measurement and Quality Control
DNA methylation was measured using the Illumina Infinium HumanMethylation450 BeadChip [37] in all but one study (child blood samples from the IOWF1 cohort) that used the Illumina Infinium Methylation EPIC BeadChip. DNA from newborn or child blood samples underwent bisulfite conversion using the EZ-96 DNA Methylation kit (Zymo Research Corporation, Irvine, USA). Methylation quality control and normalization was conducted at the cohort level, as described in the supplementary material. β-values representing proportion of methylation at each CpG site (ranging from 0 = completely unmethylated to 1 = completely methylated) were used as the methylation outcome. In order to minimize the influence of outlier methylation values, β-values more extreme than 3 times the interquartile range below the 25th percentile or above the 75th percentile were removed prior to all cohort analyses.
2.3. Sex descriptive
As part of quality control, each cohort checked for sex-mismatches using the getSex function in the R package minfi [38] and sex mismatches were removed prior to individual cohort analyses. The number of participants for each cohort excludes sex mismatches.
2.4. Cohort Specific Statistical Analyses
Each cohort performed independent EWAS according to a common analysis plan. Each cohort used recorded child sex with females as the reference group. Each study used batch covariates most appropriate for their cohort (e.g. principal components or plate number) or a method such as ComBat [39]. Cell composition was estimated using estimateCellCounts in the minfi R package [38]. For cord blood analyses, the ‘CordBlood’ reference data set [40] was used to estimate proportions of seven cell types (CD8+ T-cells, CD4+ T-cells, NK cells, B-cells, monocytes, granulocytes, and nucleated red blood cells), while older child models used the ‘Blood’ reference data set [41] which estimates proportions of six cell types (CD8+ T-cells, CD4+ T-cells, NK cells, B-cells, monocytes, and granulocytes). Cohorts were also given the option to adjust for genetic ancestry in their models, and this information is included in cohort specific methods.
Models were run using M-type multiple robust linear regression [rlm( ) in the MASS R package] [42] to control for potential heteroscedasticity and influential outliers in the methylation data. In the primary cord blood analysis, the exposure was sex with the outcome of newborn methylation β-values, adjusting for seven estimated cell counts and batch covariates. In the primary older child models, the exposure was sex with the outcome of child methylation β-values, adjusting for six estimated cell counts, age of the child at blood draw, and batch covariates.
2.5. Meta-analysis
All cohorts submitted the results of their cohort level EWAS to the Children’s Environmental Health Laboratory (N. Holland-PI) at the University of California, Berkeley, USA. We then performed a fixed effects meta-analysis weighted by the inverse of the variance—using the software METAL [43]—for the main model, which adjusted for seven cell-type proportions and batch. Shadow meta-analyses were conducted independently by L. Küpers at the University of Bristol, UK to verify results. All further analyses were conducted in R version 3.5.2 [44]. Since all but one study utilized the 450K BeadChip array (child blood samples from the IOWF1 cohort use the EPIC BeadChip) only probes present on the 450K BeadChip array were included in the analysis. We excluded SNP control probes (n = 65). The majority of cohorts included probes mapping to the X and Y chromosomes; however, some cohorts were only able to provide results for autosomal probes leaving a total sample size of N = 8,438 for autosomes, and N = 5,213 for subjects with data for sex chromosomes. Filtering of previously identified cross-reactive probes [45] was performed during processing of meta-analysis results. For autosomal probes, this left a total of 390,810 CpG sites measured for association with sex at birth in the meta-analysis.
We adjusted for multiple hypothesis testing using the stringent Bonferroni method, and considered CpG sites with Bonferroni adjusted p-values < 0.05 significant (e.g. 1.3 x 10-7 for 390,810 tests for autosomes in newborns and 1.1x10-6 for 46,979 tests for lookup level correction in children). Summary statistics from the genome-wide DNA methylation meta-analysis are available at figshare (10.6084/m9.figshare.14228927).
To distinguish the relative contribution of these two explanations, we used one cohort (GOYA) as a reference in which to identify a group of CpGs with no true biological signal (P > 0.2) and recalculated λ values for this subset of CpGs in the other cohorts.
2.6. Enrichment Analyses
Before enrichment analysis was performed, CpG sites were annotated to nearby genes using the IlluminaHumanMethylation450kanno.ilmn12.hg19 package. We defined I2>50% as reflective of a high level of between-study heterogeneity [46] and restricted enrichment analysis to those CpGs with I2≤ 50%, which included 17,243 CpGs (in 8,059 genes) for the cord blood analysis and 10,436 CpGs (in 5,572 genes) for the older child analysis.
Using these genes, we performed enrichment analyses at two different levels: pathways and diseases. Detection of KEGG pathway database [47] over-representation against a universal Homo Sapien background was assessed by hypergeometric tests [48] using the gometh() function in the package missMethyl [49]. This function, which was designed specifically for gene set enrichment analysis of methylation data, minimizes bias due to the uneven distribution of probes in the Illumina 450K and EPIC BeadChip arrays. Hypergeometric over-representation was also performed against the DisGeNET [50] curated repository of gene-disease associations using the enrichDGN() function in the DOSE package [51]. A Bonferroni corrected cutoff of 0.05 was used for significance of pathways and diseases (e.g. 1.52 x 10-4 for 322 tests for pathways and 4.16 x 10-6 for 3,779 tests for diseases).
3. Results
3.1. Newborns
Results from 17 independent cohorts from the Pregnancy And Childhood Epigenetics (PACE) Consortium were included in the newborn meta-analysis (N=8,438). Newborn cohort sizes ranged from 53 to 1,319 participants. There was an even distribution of males (51%) and females (49%). Two cohorts, NEST and EARLI, performed separate models for European and non-European participants, resulting in two additional datasets (N=19 datasets total). The majority of datasets were made up of participants of European ancestry (N=15 datasets, N=7,576 participants). Other datasets included Hispanic, Mexican-American, African-American, and mixed ethnicities. A summary of the participating newborn cohorts and datasets is included in Table 1.
TABLE 1:
Characteristics of cohorts included in the meta-analysis of differences by sex in cord blood DNA methylation
| N | N-boy | N-girl | Ethnicity | |
|---|---|---|---|---|
| ALSPAC | 894 | 438 | 456 | European |
| CHAMACOS | 363 | 181 | 182 | Mexican-American |
| CHS | 248 | 101 | 147 | Hispanic and non-Hispanic white |
| EARLI (1) a | 82 | 45 | 37 | European |
| EARLI (2) a | 53 | 25 | 28 | Non-European |
| EXPOsOMICS b | 388 | 207 | 181 | European |
| GECKO | 252 | 135 | 117 | European |
| Gen3G | 176 | 81 | 95 | European |
| Generation R | 1,319 | 675 | 644 | European |
| GOYA | 518 | 260 | 258 | European |
| INMA | 385 | 197 | 188 | European |
| IOW F2 | 124 | 64 | 60 | European |
| MoBa1 | 1,051 | 560 | 491 | European |
| MoBa2 | 671 | 336 | 335 | European |
| MoBa3 | 249 | 124 | 125 | European |
| NEST (1) a | 198 | 100 | 98 | African American |
| NEST (2) a | 170 | 84 | 86 | European |
| PREDO | 812 | 429 | 383 | European |
| Viva | 485 | 254 | 231 | European |
|
| ||||
| Meta-analysis | 8,438 | 4296 (51%) | 4142 (49%) | |
|
| ||||
EARLI and NEST cohorts ran separate epigenome wide association studies for each ethnic population.
EXPOsoOMICS is composed of three cohorts: Rhea, Environage, and Piccolipiu.
The results of the individual cohort level newborn models are summarized in Supplemental Table 1. As expected, nearly all CpG sites (N=9,618, 99.8%) on the X chromosome were significantly differentially methylated between males and females. The average effect size (absolute value) expressed as a methylation beta value for differentially methylated CpG sites on the X chromosome was 0.18 (equals 18% methylation). In autosomes, there were a total of 46,979 Bonferroni significant sex-associated CpG sites out of a total of 390,810 autosomal CpG sites. The lambda (λ) value, a measure of p-value inflation, for the meta-analysis in autosomes only was 4.87. For cohort specific analyses, λ ranged from 1.18 to 2.67 with a sample-size weighted average of 1.91 for autosome only data. All Bonferroni significant CpGs for autosomes are presented in Supplemental Table 2.
The plot in Figure 1a shows sites in autosomes with lower methylation in males below the null line and sites with higher methylation in males above the null line. Differentially methylated CpGs were observed across all autosomes. The majority (67%) of sex-associated sites on autosomes had lower methylation in males than females (Figure 2). The CpG-specific differences in methylation levels between males and females were generally small with a median (interquartile range, IQR) difference in methylation of 0.62% (0.61%) for positive differences and 0.88% (1.1%) for negative differences (Figure 2a).
Figure 1.
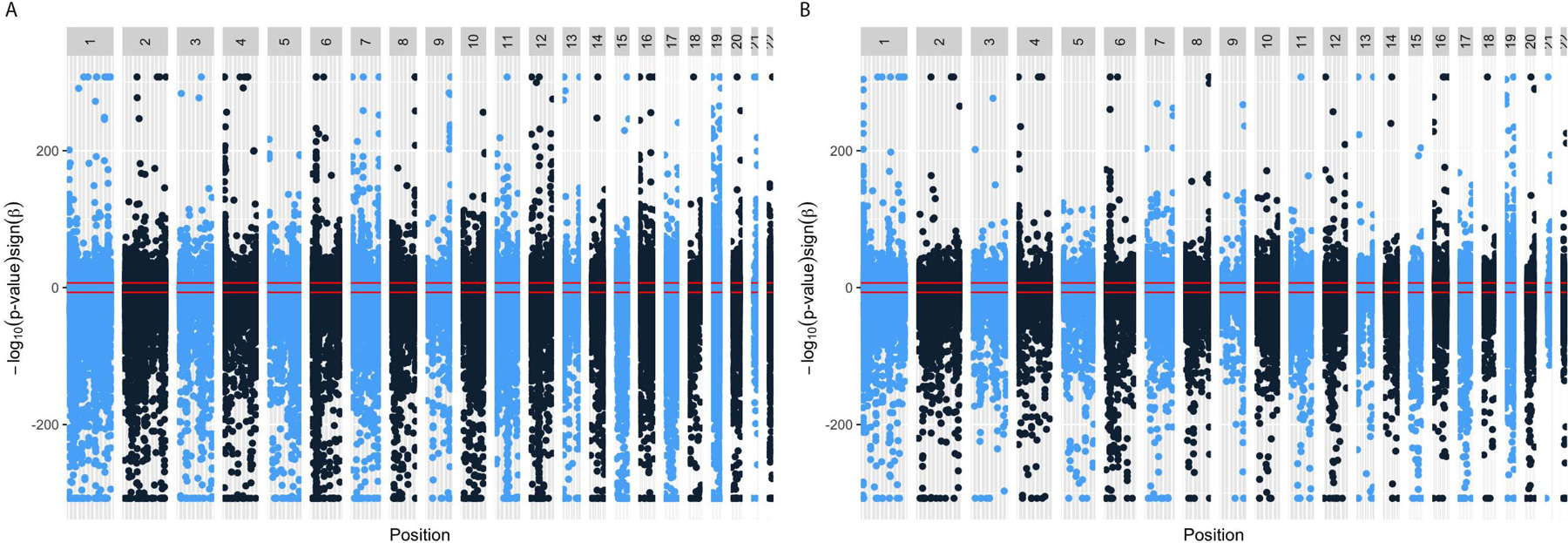
Plot of meta-analysis EWAS results of differential methylation by sex in (a) newborns and (b) children. Sites with higher methylation in males compared to females are plotted above the x-axis while those with lower methylation in males compared to females are shown below the x-axis. The red lines represent the Bonferroni thresholds for significant CpG sites in each direction (higher and lower methylation in males compared to females) The grey boxes indicate the chromosome and the plot colors vary between blue and black for visual clarity between different chromosomes. The varying widths of the chromosomes corresponds to the length of the chromosomes
Figure 2.
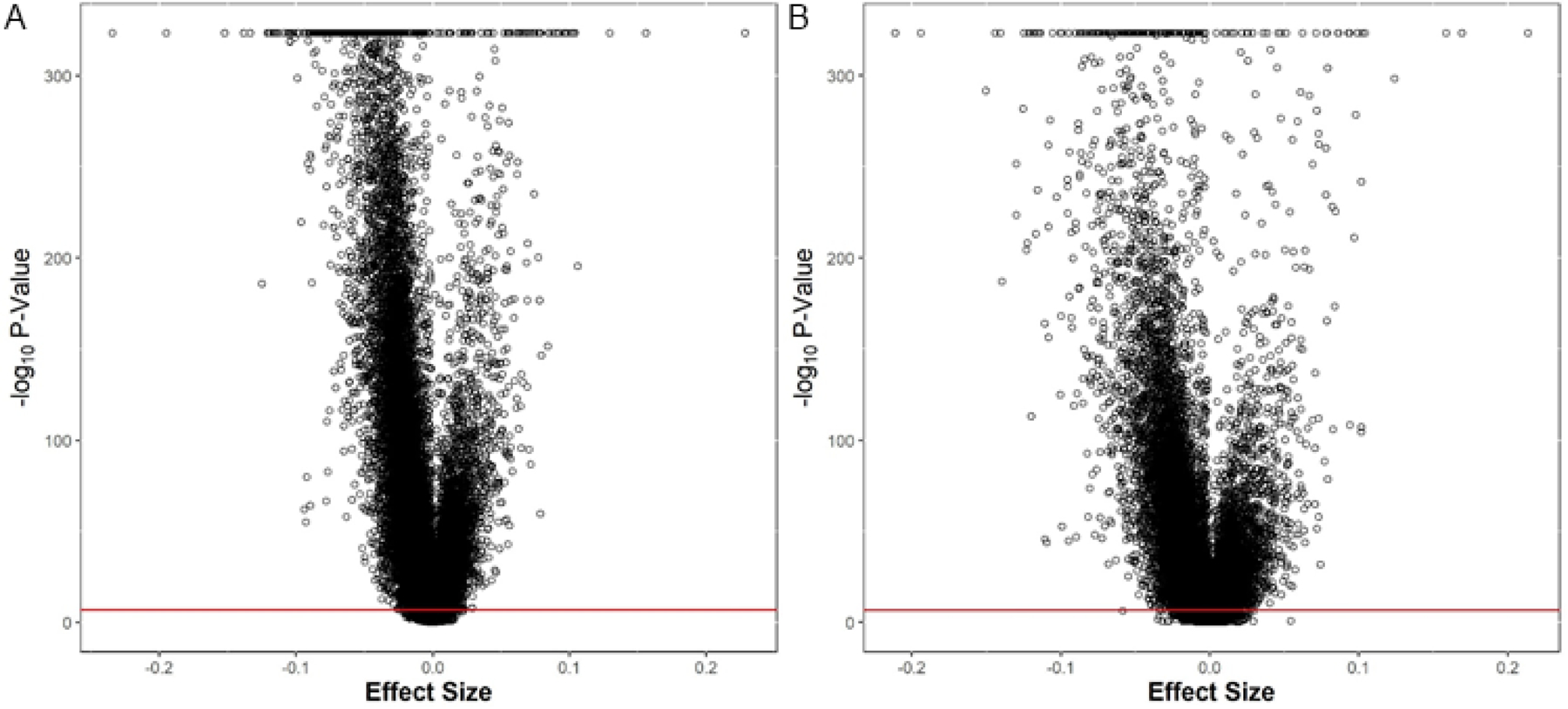
Volcano plots of meta-analysis EWAS results of differential methylation by sex in (a) newborns and (b) children. Absolute effect sizes ranged from 0 to 0.23 (methylation β-value). Overall, there were more CpG sites with lower methylation in males compared to females.
Among the top Bonferroni significant sites with effect sizes greater than 0.05 (absolute value) listed in Supplemental Table 2, cg26921482, which annotated to TBC1D24, had the largest effect size with mean methylation 23% lower in newborn males than females. Two CpGs also annotated to ZNF696 and had higher methylation in females than males. Additionally, some of the most differentially methylated CpGs annotated to lincRNA genes (LINC01347 and LINC00346) and a protein coding gene, PPP1R3G.
As expected, CpGs differentially methylated in relation to sex annotated to genes that were enriched for several biological processes and diseases. The top 15 KEGG pathways are summarized in Figure 3a with results sorted from most to least significant, and size of circles representing the number of genes included in that pathway. KEGG pathways fell into groups containing cancer, signaling, endocrine, addiction, and longevity. Only four enriched pathways of 322 tested were Bonferroni significant, including several signaling pathways. Disease enrichment analyses showed 83 significant diseases of the 3,779 tested. Genes to which significant CpG sites were annotated were significantly enriched in mental disorders as well as cancer-related outcomes (e.g. ductal carcinoma, neuroendocrine tumors, and thyroid neoplasms) and cardiovascular phenotypes (e.g. systemic arterial pressure, congenital heart defects, blood pressure) (Figure 4a).
Figure 3.
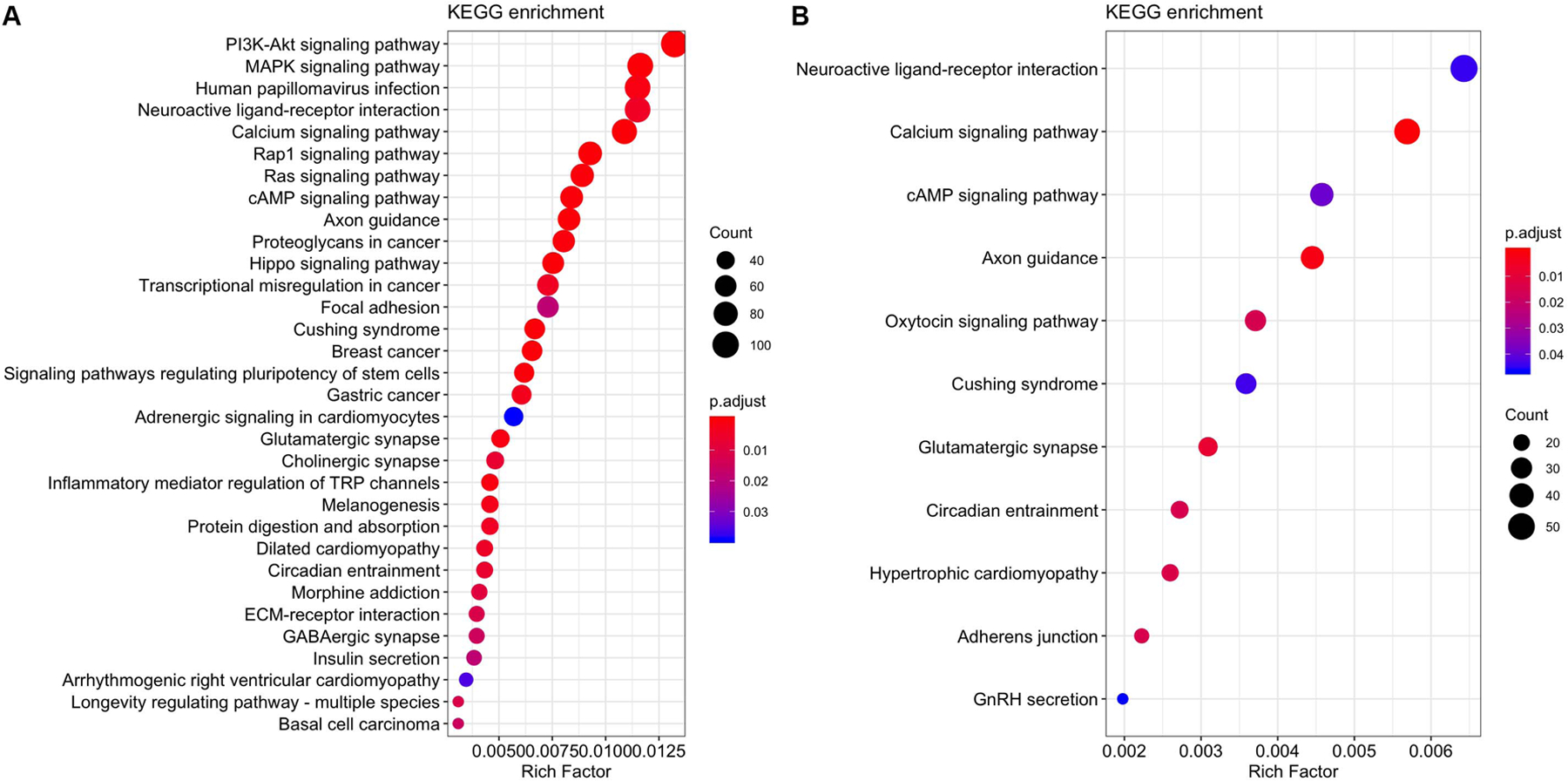
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways with enrichment differences between males and females in (a) newborn and (b) older children. The size of the circles represents the number of differentially methylated genes in a pathway.
Figure 4.
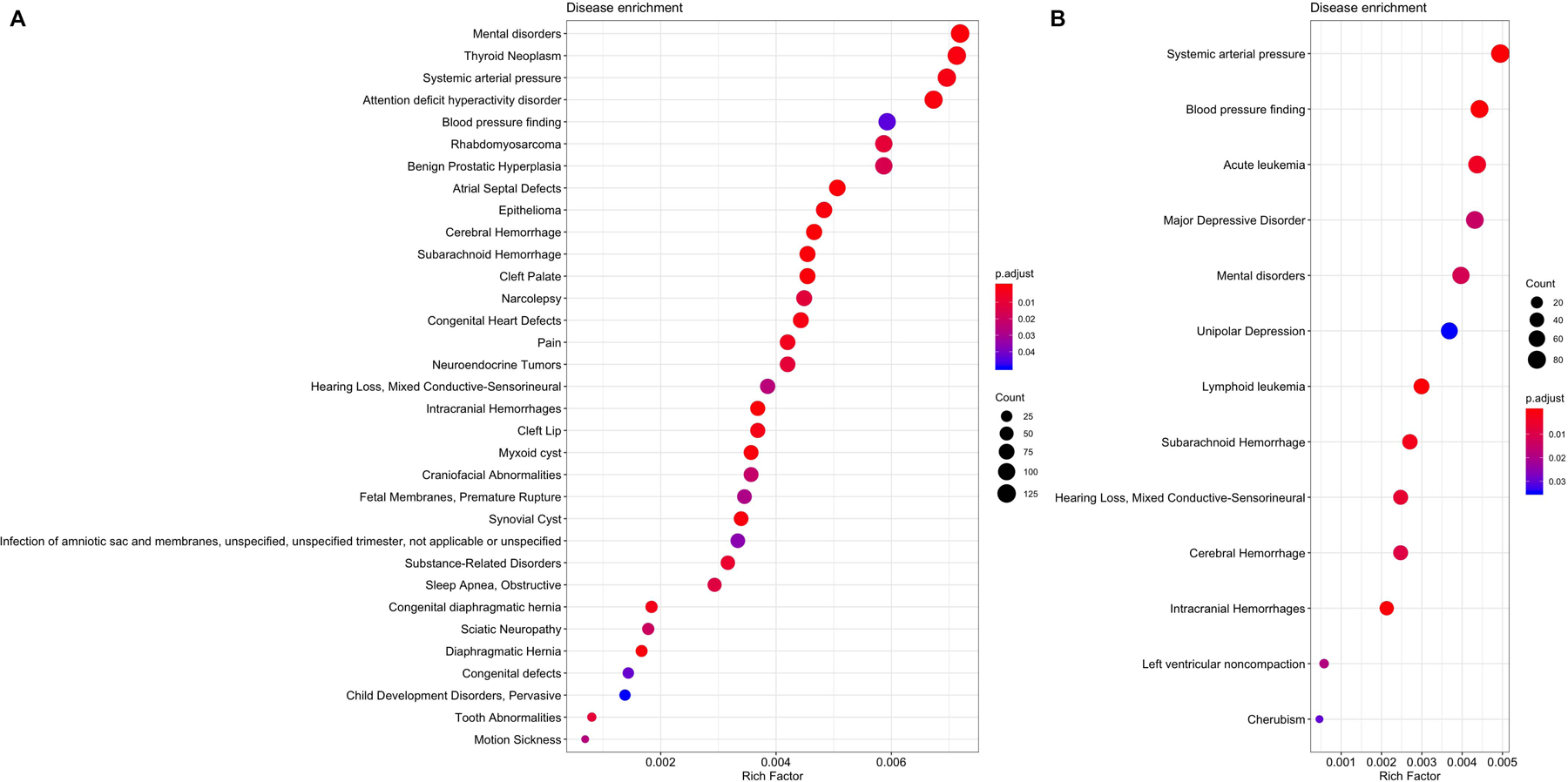
Diseases with significant enrichment differences between males and females in (a) newborns and (b) older children. Disease enrichment analysis was performed using DisGeNET. The size of the circles represent the number of differentially methylated genes in a pathway.
3.2. Children
For the analysis in older children, data from nine independent cohorts were meta-analyzed (N = 4,268) (Table 2). Child cohorts ranged from 234 to 1,053 participants with an average of 497 participants per cohort and had a similar distribution of males (53%) and females (47%). Similar to the newborn participants, the majority of child datasets contained participants of European ancestry (N=4,034 from 7 cohorts), with other contributions from a Mexican-American cohort. Lambdas for individual autosomal analyses ranged from 1.05 to 5.59 (Supplemental Table 3).
TABLE 2.
Top 10 Bonferroni-significant CpG sites ranked by effect size from the meta-analysis of sex in newborn blood
| probeID | Effect Sizea | Standard Error | P-Value | Direction of effect in each cohortb | Chr. | Position | Nearest Gene |
|---|---|---|---|---|---|---|---|
| cg26921482 | −0.2345 | 0.0013 | <5.0E-234 | -----------?------- | 16 | 2570283 | TBC1D24 |
| cg12691488 | 0.2287 | 8.00E-04 | <5.0E-234 | +++++++++++?+++++++ | 1 | 243053673 | LINC01347 |
| cg00157199 | −0.1951 | 0.0039 | <5.0E-234 | -----------?------- | 20 | 29551622 | FRG1BP |
| cg03151810 | 0.1559 | 0.0029 | <5.0E-234 | +++++++++++++++++++ | 8 | 144371745 | ZNF696 |
| cg11092486 | −0.1522 | 0.002 | <5.0E-234 | ------------------- | 6 | 5087604 | PPP1R3G |
| cg14815891 | −0.1522 | 0.0035 | <5.0E-234 | -----?------------- | 20 | 29611903 | FRG1BP |
| cg02282631 | −0.1381 | 0.0029 | <5.0E-234 | ------------------- | 5 | 42953543 | LOC648987 |
| cg14361252 | −0.1335 | 0.0028 | <5.0E-234 | -----------?-?----- | 13 | 111465457 | LINC00346 |
| cg11388673 | 0.1296 | 0.0026 | <5.0E-234 | +++++++++++++++++++ | 8 | 144371779 | ZNF696 |
| cg04737881 | −0.1251 | 0.0043 | 5.03E-181 | ------------------- | 4 | 183060943 | TENM3-AS1 |
negative effect size indicates lower methylation in boys compared to girls
For each cohort participating in the analysis, + indicates a positive direction of effect, - indicates a negative direction of effect, and ? indicates missing data for that CpG site in that specific cohort. The cohort order is: ALSPAC, CHAMACOS, CHS, EARLI (1), EARLI(2), EXPOsOMICS, GECKO, Gen3G, Generation R, GOYA, INMA, IOW F2, MoBa1, MoBa2, MoBa3, NEST(1), NEST(2), Predo, and Viva
Among older children, most of the X chromosome sites (9,313) were significant. For the autosomes, there were 40,219 Bonferroni significant sites associated with child sex (Supplemental Table 4). Like newborns, the majority of significant autosomal CpG sites had lower methylation levels in males than in females, which can be seen on the plot in Figure 1b. The effect size of differential methylation was small and similar to that in newborns with a median (IQR) difference in methylation of 0.62% (0.67%) among sites with significant positive differences and 0.94% (1.0%) among sites with significant negative differences (Figure 2b).
Among the top Bonferroni significant sites with effect sizes greater than 0.05 (absolute value), the CpG with the largest effect size (21% lower methylation in males than females) was cg12691488, which annotated to a lincRNA gene (LINC01347). Two significantly associated CpGs with lower methylation in males than females mapped to FRG1BP and three significantly associated CpGs with higher methylation in males than females mapped to ZNF696. Several significant CpGs mapped to PPP1R3G and had higher methylation in males compared to females.
The top 15 enriched KEGG pathways are summarized in Figure 3b with results sorted from lowest to highest p-value, and size of circles representing the number of genes included in that pathway. Of the 322 KEGG pathways tested, none were statistically significant after adjusting for multiple testing (Figure 3b). The top KEGG pathways predominantly belonged to groups associated with signaling, neuronal and endocrine functions. Of the 3,779 diseases tested, 35 had Bonferroni-corrected enrichment p-values lower than 0.05. Implicated genes are involved in blood pressure, hemorrhage, carcinomas, and mental disorders (Figure 4b).
3.3. Comparison of newborns and older children
There was considerable overlap between significant sites in newborns and children. Of the 46,979 CpG significant in newborns, 68% (31,850) met look-up level significance (p<1.1 x 10e-6 for 46,979 tests) in the smaller dataset of older children (Supplemental Table 5) demonstrating that many of these relationships with sex persisted over time. Of these overlapping sites, 99.6% (31,727) show methylation differences in the same direction. Additionally, there were 10,229 CpGs that were Bonferroni-significant (genome-wide significance, p<1.1 x 10-7) only in the older children, despite the smaller size of this dataset (Supplemental Table 6).
4. Discussion
This large-scale epigenome-wide meta-analysis involving multiple cohorts demonstrates widespread differences in methylation of autosomes between males and females at birth measured in newborn blood and most of these differences persist into later childhood. We report over 45,000 of the nearly 400,000 tested CpG sites to be significantly differentially methylated with small but consistently observed differences between males and females at birth and over 40,000 significant differences in older children with similar effect sizes. As expected, in both newborns and children, these differences were enriched in genes involved in a range of biological pathways important for development, but there was also sex-specific enrichment in cancer pathways and genes implicated in psychiatric disorders and cardiovascular phenotypes.
We compared our findings to prior studies investigating cord or peripheral blood methylation differences by sex (Supplemental Table 7). Only one prior meta-analysis by McCarthy et al. [21] has looked specifically at differential methylation between males and females, and this was assessed using the Illumina 27K chip. Although a few cohorts contributed cord-blood data, most of the cohorts included in their analysis used adult blood data. This study reported 187 significant autosomal CpGs, of which, in our newborn meta-analysis, we replicate 167 (90%) at genome-wide significance and same direction of effect and in our child meta-analysis, 164 (89%) at genome-wide significance and same direction of effect. Another study by Yousefi et al. [18] reported 3,031 CpGs with sex differences in cord-blood for a subset of the CHAMACOS population (which also contributed data to this meta-analysis). Our newborn meta-analysis replicated 2,762 (91%) of the Yousefi et al. significant CpGs with the same direction of effect, and our child meta-analysis replicated 2,709 (89%) significant CpGs in the same direction of effect. The newborn meta-analysis adds 44,107 autosomal CpGs not previously identified in studies focused on methylation differences by sex with increased sample size and after adjustment of cord blood cell-type heterogeneity. We also report 37,397 new autosomal CpG sites differentially methylated by sex in the blood of older children. Although we identified a large number of differentially methylated CpGs sites, it should be noted that for many of these sites, the effect sizes were quite small.
Interestingly, in both newborns and older children, about two-thirds of the significantly differentially methylated sites had lower methylation in males than females. In general, greater gene expression is observed with lower methylation when CpG sites are in the promoter region. Hypermethylation in females is expected in X chromosome CpG sites due to X chromosome inactivation in females; however, we found that differences in methylation by sex are not limited to sex-chromosomes and that the higher methylation pattern for females is also common outside of sex-chromosomes. Notably, the opposite trend has been observed in placenta, where the majority of differentially methylated sites in autosomes had higher methylation in males than females [27]. Our finding indicates that there are many autosomal CpG sites that are differentially methylated between males and females; however, most conventional EWAS adjust for sex allowing for these differences to be accounted for in sex-independent analyses.
The CpG with the largest negative association with newborn sex (lower methylation in males compared to females) annotated to TBC1D24, and was also significantly associated with sex in older children. TBC1D24 encodes for a protein involved in neuronal development and has also been associated with epilepsy and neurological disorders [52,53]; sex differences have been observed for both conditions [54,55]. The CpG with the largest positive association with child sex (lower methylation in females than males) was LINC01347, and was also significantly associated with newborn sex. Another significantly associated CpG site in both newborns and children also annotated to a lincRNA, LINC00346. Long intergenic noncoding RNAs (lncRNAs) affect gene expression through regulation of chromatin [56]. Differences in their expression levels have been associated with neuropsychiatric disorders (e.g. schizophrenia), cancers, and coronary artery disease [57–59]. Furthermore, differences in lincRNA expression profiles by sex have previously been observed [59,60].
Sex-specific differences have been observed in numerous diseases and studies show evidence for an epigenetic role in sexual dimorphism for disease [61]. Diseases with observed differences by sex include asthma [62], autoimmune and allergic diseases [32,63], cardiovascular diseases [64], and pediatric infectious diseases [31]. Early-life differences between males and females also suggests an underlying developmental component, especially in newborns who have had fewer lifetime exposures [65]. We report many biological pathways and diseases where the genes to which differentially methylated sites annotate are enriched in both newborns and children. While some differences are expected since the progression of development in females and males is naturally different, other differences may be related to disease risk. Some of the most significant disease pathways have been previously shown to differ between sexes. Our disease enrichment analysis included several psychiatric disorders, and studies have shown that anxiety disorders are more common and more severe in women [66]. In concordance with our findings, another recent study of differential methylation by sex in cord blood also reported significant enrichment of genes related to neurodevelopmental disorders [67]. Autism is diagnosed in males more often than females, and there are differences in the features of autism in each sex [68]. In children, genes involved in ADHD were significantly enriched for differentially methylated CpG sites. ADHD diagnoses are two-fold higher in males than females with different behaviors associated with the sexes [69]. A prior study in the CHAMACOS cohort also reported methylation differences between males and females in genes involved in neurological disorders [18]. Our data suggest that DNA methylation may represent one mechanism contributing to the developmental differences between males and females that impact sex-dependent differences in health.
We also observed inflated λ values in this meta-analysis, particularly for cohorts with larger sample sizes. λ, also referred to as the genomic inflation factor, is a measure of p-value inflation where a value > 1 implies some inflation of the observed test statistics. Such inflation could be due to the presence of residual confounding or to abundance of true biological signal, which is possible given the phenotype of interest and large number of associations identified even in autosomes. To distinguish the relative contribution of these two explanations (residual confounding or abundance of true biological signal), we used one cohort (GOYA) as a reference in which to identify a group of CpGs with no true biological signal (P > 0.2) and recalculated λ values for this subset of CpGs in the other cohorts. For all cohorts, newly calculated lambdas were closer to one but still somewhat inflated (Supplemental Tables 8 and 9) suggesting that abundance of true signal does contribute to but cannot entirely explain the inflated λ values observed.
Our study has several limitations and strengths. Although our study included cohorts of multiple ancestries, including European, Hispanic, and African American, most participants were of European ancestry. More work involving a larger number of non-European descent participants is needed to ensure generalizability of results. Individual cohorts used different normalization methods for methylation data; however, prior studies within the PACE consortium show little difference in final EWAS results from differently normalized data, so we do not expect this to strongly impact the final meta-analysis results [7]. Since this study did not assess if the methylation changes are impacting gene expression, we cannot confirm if these methylation differences extend to functional changes. These results warrant further follow-up to assess if these methylation changes do indeed impact gene expression in order to confirm the biological significance of these findings [2,70,71]. Although the gometh() function takes into account the bias introduced by the uneven distribution of probes in the 450K and EPIC BeadChip arrays in our pathway analyses, the hypergeometric over-representation analysis for disease enrichment (enrichDGN) does not. Further, our analyses were restricted to cord and peripheral blood. Sex-specific methylation in other tissues, such as placenta, may also have implications for disease across the life course.
We report novel findings of autosomal methylation differences between males and females using robust statistical models with a large sample size that was well-powered to assess small effect sizes. We used a cord blood reference dataset, which includes nucleated red blood cells to estimate and adjust for cell-type heterogeneity in newborns [72]. All cohorts ensured correct classification of sex prior to analyses using sex chromosome methylation data as a quality control measure. We also included analyses of methylation at two distinct time-points (newborns and older children) suggesting that methylation differences by sex at many of these CpG sites were relatively stable throughout childhood.
In summary, we observed numerous autosomal methylation differences in blood between males and females, which is likely to be important for normal sex-specific biological development. However, differentially methylated CpG sites were enriched in genes involved in diseases and pathways with differential prevalence between sexes. These findings may suggest that early life DNA methylation differences represent a potential mechanism contributing to regulation of differential disease prevalence by sex.
Supplementary Material
Supplemental Table 7. Other studies of cord or peripheral blood DNA methylation differences by sex
Supplemental Table 6. Bonferroni Significant CpG Sites from the meta-analysis of differences by sex in child blood DNA methylation that were not significant in the meta-analysis of newborns
Supplemental Table 8. Sensitivity analysis of λ in Newborn Cohorts
Supplemental Table 9. Sensitivity analysis of λ in Child Cohorts
Supplemental Table 1. Results of cohort specific EWAS of sex and cord blood DNA methylation
Supplemental Table 3. Results of cohort specific EWAS of sex and child blood DNA methylation
Supplemental Table 2. Bonferroni Significant CpG Sites from the meta-analysis of differences by sex in newborn blood DNA methylation
Supplemental Table 5. CpG Sites from the meta-analysis of differences by sex in child blood DNA methylation with look up level correction (based on cord blood meta-analysis)
Supplemental Table 4. Bonferroni Significant CpG Sites from the meta-analysis of differences by sex in child blood DNA methylation
Supplementary Methods. Methods, funding, and acknowledgements for each specific cohort used in the meta-analysis
TABLE 3:
Characteristics of cohorts included in the meta-analysis of differences by sex in child blood DNA methylation
| Mean Age (Yr) | N | N-boy | N-girl | Ethnicity | |
|---|---|---|---|---|---|
| ALSPAC | 7.5 | 959 | 479 | 480 | European |
| BAMSE | 8.3 | 450 | 250 | 200 | European |
| CHAMACOS | 9.1 | 234 | 109 | 125 | Mexican-American |
| CHOP | 5.5 | 360 | 172 | 188 | European |
| Generation R | 6.0 | 464 | 227 | 237 | European |
| HELIX | 8.0 | 1053 | 581 | 472 | European |
| IOW F1 | 10.0 | 289 | 186 | 103 | European |
| Viva | 7.9 | 459 | 241 | 218 | European |
|
| |||||
| Total | 4,268 | 2,245(53%) | 2,023(47%) | ||
TABLE 4.
Top 10 Bonferroni-significant CpG sites ranked by effect size from the meta-analysis of sex in child blood
| probeID | Effect Sizea | Standard Error | P-Value | Direction of effect in each cohortb | Chr. | Position | Nearest Gene |
|---|---|---|---|---|---|---|---|
| cg12691488 | 0.2137 | 0.001 | <5.0E-324 | +++++++++ | 1 | 243053673 | LINC01347 |
| cg00157199 | −0.2112 | 0.0051 | <5.0E-324 | --------- | 20 | 29551622 | FRG1BP |
| cg26921482 | −0.1938 | 0.0016 | <5.0E-324 | ------?-- | 16 | 2570283 | TBC1D24 |
| cg11388673 | 0.1692 | 0.0039 | <5.0E-324 | +++++++++ | 8 | 144371779 | ZNF696 |
| cg03151810 | 0.1588 | 0.0038 | <5.0E-324 | +++++++++ | 8 | 144371745 | ZNF696 |
| cg14361252 | −0.1503 | 0.0041 | 1.35E-286 | --?-?-?-- | 13 | 111465457 | LINC00346 |
| cg11092486 | −0.1439 | 0.0024 | <5.0E-324 | --------- | 6 | 5087604 | PPP1R3G |
| cg16810031 | −0.1404 | 0.0023 | <5.0E-324 | -?----?-- | 17 | 38024146 | ZPBP2 |
| cg14815891 | −0.1394 | 0.0048 | 3.41E-182 | -?------- | 20 | 29611903 | FRG1BP |
| cg01066472 | −0.1297 | 0.0041 | 1.28E-218 | --------- | 1 | 75591029 | LHX8 |
negative effect size indicates lower methylation in boys compared to girls
For each cohort participating in the analysis, + indicates a positive direction of effect, - indicates a negative direction of effect, and ? indicates missing data for that CpG site in that specific cohort. The cohort order is: ALSPAC, BAMSE, CHAMACOS, CHOP, Generation R, HELIX, IOW F1, and Viva.
Acknowledgements
For all studies, acknowledgements can be found in the Supplementary Material (Supplementary Methods).
Funding
For all studies, funding information can be found in the Supplementary Material (Supplementary Methods).
Abbreviations
- EWAS
Epigenome Wide Association Studies
- IQR
interquartile range
- lncRNAs
long intergenic noncoding RNAs
- PACE
Pregnancy And Childhood Epigenetics Consortium
References
- [1].Roseboom T, de Rooij S, Painter R The dutch famine and its long-term consequences for adult health, Early Hum. Dev 82 (2006) 485–491. [DOI] [PubMed] [Google Scholar]
- [2].Barker DJ In utero programming of chronic disease, Clin. Sci 95 (1998) 115–128. [PubMed] [Google Scholar]
- [3].Leonard SA, Rasmussen KM, King JC, Abrams B Trajectories of maternal weight from before pregnancy through postpartum and associations with childhood obesity, Am. J. Clin. Nutr 106 (2017) 1295–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kim GH, Berger K, Rauch S, Kogut K, Birgit CH, Antonia MC, Huen K, Eskenazi B, Holland N Association of prenatal urinary phthalate metabolite concentrations and childhood BMI and obesity, Pediatr. Res 82 (2017) 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Slopen N, Loucks EB, Appleton AA, Kawachi I, Kubzansky LD, Non AL, Buka S, Gilman SE Early origins of inflammation: An examination of prenatal and childhood social adversity in a prospective cohort study, Psychoneuroendocrinology 51 (2015) 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bianco-Miotto T, Craig JM, Gasser YP, van Dijk SJ, Ozanne SE Epigenetics and DOHaD: From basics to birth and beyond, J. Dev. Orig Health. Dis 8 (2017) 513–519. [DOI] [PubMed] [Google Scholar]
- [7].Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, Reese SE, Markunas CA, Richmond RC, Xu C, Küpers LK, Oh SS, Hoyo C, Gruzieva O, Söderhäll C, Salas LA, Baïz N, Zhang H, Lepeule J, Ruiz C, Ligthart S, Wang T, Taylor JA, Duijts L, Sharp GC, Jankipersadsing SA, Nilsen RM, Vaez A, Fallin MD, Hu D, Litonjua AA, Fuemmeler BF, Huen K, Kere J, Kull I, Munthe-Kaas M, Gehring U, Bustamante M, Saurel-Coubizolles M, Quraishi BM, Ren J, Tost J, Gonzalez JR, Peters MJ, Håberg SE, Xu Z, van Meurs JB, Gaunt TR, Kerkhof M, Corpeleijn E, Feinberg AP, Eng C, Baccarelli AA, Benjamin Neelon SE, Bradman A, Merid SK, Bergström A, Herceg Z, Hernandez-Vargas H, Brunekreef B, Pinart M, Heude B, Ewart S, Yao J, Lemonnier N, Franco OH, Wu MC, Hofman A, McArdle W, Van d.V., Falahi F, Gillman MW, Barcellos LF, Kumar A, Wickman M, Guerra S, Charles M, Holloway J, Auffray C, Tiemeier HW, Smith GD, Postma D, Hivert M, Eskenazi B, Vrijheid M, Arshad H, Antó JM, Dehghan A, Karmaus W, Annesi-Maesano I, Sunyer J, Ghantous A, Pershagen G, Holland N, Murphy SK, DeMeo DL, Burchard EG, Ladd-Acosta C, Snieder H, Nystad W, Koppelman GH, Relton CL, Jaddoe VWV, Wilcox A, Melén E, London SJ DNA methylation in newborns and maternal smoking in pregnancy: Genome-wide consortium meta-analysis, Am. J. Hum. Genet 98 (2016) 680–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gruzieva O, Xu CJ, Breton CV, Annesi-Maesano I, Antó JM, Auffray C, Ballereau S, Bellander T, Bousquet J, Bustamante M, Charles MA, de Kluizenaar Y, den Dekker HT, Duijts L, Felix JF, Gehring U, Guxens M, Jaddoe VV, Jankipersadsing SA, Merid SK, Kere J, Kumar A, Lemonnier N, Lepeule J, Nystad W, Page CM, Panasevich S, Postma D, Slama R, Sunyer J, Söderhäll C, Yao J, London SJ, Pershagen G, Koppelman GH, Melén E Epigenome-wide meta-analysis of methylation in children related to prenatal NO2 air pollution exposure, Environ. Health Perspect 125 (2017) 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gruzieva O, Xu CJ, Yousefi P, Relton C, Merid SK, Breton CV, Gao L, Volk HE, Feinberg JI, Ladd-Acosta C, Bakulski K, Auffray C, Lemonnier N, Plusquin M, Ghantous A, Herceg Z, Nawrot TS, Pizzi C, Richiardi L, Rusconi F, Vineis P, Kogevinas M, Felix JF, Duijts L, den Dekker HT, Jaddoe VWV, Ruiz JL, Bustamante M, Antó JM, Sunyer J, Vrijheid M, Gutzkow KB, Grazuleviciene R, Hernandez-Ferrer C, Annesi-Maesano I, Lepeule J, Bousquet J, Bergström A, Kull I, Söderhäll C, Kere J, Gehring U, Brunekreef B, Just AC, Wright RJ, Peng C, Gold DR, Kloog I, DeMeo DL, Pershagen G, Koppelman GH, London SJ, Baccarelli AA, Melén E Prenatal particulate air pollution and DNA methylation in newborns: An epigenome-wide meta-analysis, Environ. Health Perspect 127 (2019) 57012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Merid SK, Novoloaca A, Sharp GC, Küpers LK, Kho AT, Roy R, Gao L, Annesi-Maesano I, Jain P, Plusquin M, Kogevinas M, Allard C, Vehmeijer FO, Kazmi N, Salas LA, Rezwan FI, Zhang H, Sebert S, Czamara D, Rifas-Shiman SL, Melton PE, Lawlor DA, Pershagen G, Breton CV, Huen K, Baiz N, Gagliardi L, Nawrot TS, Corpeleijn E, Perron P, Duijts L, Nohr EA, Bustamante M, Ewart SL, Karmaus W, Zhao S, Page CM, Herceg Z, Jarvelin MR, Lahti J, Baccarelli AA, Anderson D, Kachroo P, Relton CL, Bergström A, Eskenazi B, Soomro MH, Vineis P, Snieder H, Bouchard L, Jaddoe VW, Sørensen TIA, Vrijheid M, Arshad SH, Holloway JW, Håberg SE, Magnus P, Dwyer T, Binder EB, DeMeo DL, Vonk JM, Newnham J, Tantisira KG, Kull I, Wiemels JL, Heude B, Sunyer J, Nystad W, Munthe-Kaas MC, Räikkönen K, Oken E, Huang RC, Weiss ST, Antó JM, Bousquet J, Kumar A, Söderhäll C, Almqvist C, Cardenas A, Gruzieva O, Xu CJ, Reese SE, Kere J, Brodin P, Solomon O, Wielscher M, Holland N, Ghantous A, Hivert MF, Felix JF, Koppelman GH, London SJ, Melén E Epigenome-wide meta-analysis of blood DNA methylation in newborns and children identifies numerous loci related to gestational age, Genome Med 12 (2020) 25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Küpers LK, Monnereau C, Sharp GC, Yousefi P, Salas LA, Ghantous A, Page CM, Reese SE, Wilcox AJ, Czamara D, Starling AP, Novoloaca A, Lent S, Roy R, Hoyo C, Breton CV, Allard C, Just AC, Bakulski KM, Holloway JW, Everson TM, Xu C, Huang R, van der Plaat DA,Wielscher M, Merid SK, Ullemar V, Rezwan FI, Lahti J, van Dongen J, Langie SAS, Richardson TG, Magnus MC, Nohr EA, Xu Z, Duijts L, Zhao S, Zhang W, Plusquin M, DeMeo DL, Solomon O, Heimovaara JH, Jima DD, Gao L, Bustamante M, Perron P, Wright RO, Hertz-Picciotto I, Zhang H, Karagas MR, Gehring U, Marsit CJ, Beilin LJ, Vonk JM, Jarvelin M, Bergström A, Örtqvist AK, Ewart S, Villa PM, Moore SE, Willemsen G, Standaert ARL, Håberg SE, Sørensen TIA, Taylor JA, Räikkönen K, Yang IV, Kechris K, Nawrot TS, Silver MJ, Gong YY, Richiardi L, Kogevinas M, Litonjua AA, Eskenazi B, Huen K, Mbarek H, Maguire RL, Dwyer T, Vrijheid M, Bouchard L, Baccarelli AA, Croen LA, Karmaus W, Anderson D, de Vries M, Sebert S, Kere J, Karlsson R, Arshad SH, Hämäläinen E, Routledge MN, Boomsma DI, Feinberg AP, Newschaffer CJ, Govarts E, Moisse M, Fallin MD, Melén E, Prentice AM, … Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birthweight, Nature Communications 10 (2019) 1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].den Dekker HT, Burrows K, Felix JF, Salas LA, Nedeljkovic I, Yao J, Rifas-Shiman S, Ruiz-Arenas C, Amin N, Bustamante M, DeMeo DL, Henderson AJ, Howe CG, Hivert MF, Ikram MA, de Jongste JC, Lahousse L, Mandaviya PR, van Meurs JB, Pinart M, Sharp GC, Stolk L, Uitterlinden AG, Anto JM, Litonjua AA, Breton CV, Brusselle GG, Sunyer J, Smith GD, Relton CL, Jaddoe VWV, Duijts L Newborn DNA-methylation, childhood lung function, and the risks of asthma and COPD across the life course, Eur. Respir. J 53 (2019) 10.1183/13993003.0179-2018. Print 2019 Apr. [DOI] [PubMed] [Google Scholar]
- [13].van Dijk SJ, Peters TJ, Buckley M, Zhou J, Jones PA, Gibson RA, Makrides M, Muhlhausler BS, Molloy PL DNA methylation in blood from neonatal screening cards and the association with BMI and insulin sensitivity in early childhood, Int. J. Obes. (Lond) 42 (2018) 28–35. [DOI] [PubMed] [Google Scholar]
- [14].Reese SE, Xu CJ, den Dekker HT, Lee MK, Sikdar S, Ruiz-Arenas C, Merid SK, Rezwan FI, Page CM, Ullemar V, Melton PE, Oh SS, Yang IV, Burrows K, Soderhall C, Jima DD, Gao L, Arathimos R, Kupers LK, Wielscher M, Rzehak P, Lahti J, Laprise C, Madore AM, Ward J, Bennett BD, Wang T, Bell DA, BIOS consortium, Vonk JM, Haberg SE, Zhao S, Karlsson R, Hollams E, Hu D, Richards AJ, Bergstrom A, Sharp GC, Felix JF, Bustamante M, Gruzieva O, Maguire RL, Gilliland F, Baiz N, Nohr EA, Corpeleijn E, Sebert S, Karmaus W, Grote V, Kajantie E, Magnus MC, Ortqvist AK, Eng C, Liu AH, Kull I, Jaddoe VWV, Sunyer J, Kere J, Hoyo C, Annesi-Maesano I, Arshad SH, Koletzko B, Brunekreef B, Binder EB, Raikkonen K, Reischl E, Holloway JW, Jarvelin MR, Snieder H, Kazmi N, Breton CV, Murphy SK, Pershagen G, Anto JM, Relton CL, Schwartz DA, Burchard EG, Huang RC, Nystad W, Almqvist C, Henderson AJ, Melen E, Duijts L, Koppelman GH, London SJ Epigenome-wide meta-analysis of DNA methylation and childhood asthma, J. Allergy Clin. Immunol 143 (2019) 2062–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Carrel L, Willard HF X-inactivation profile reveals extensive variability in X-linked gene expression in females, Nature 434 (2005) 400–404. [DOI] [PubMed] [Google Scholar]
- [16].Adkins RM, Thomas F, Tylavsky FA, Krushkal J Parental ages and levels of DNA methylation in the newborn are correlated, BMC Medical Genetics 12 (2011) 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Adkins RM, Krushkal J, Tylavsky FA, Thomas F Racial differences in gene-specific DNA methylation levels are present at birth, Birth Defects Res A Clin Mol Teratol 91 (2011) 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yousefi P, Huen K, Davé V, Barcellos L, Eskenazi B, Holland N Sex differences in DNA methylation assessed by 450 K BeadChip in newborns, BMC Genomics 16 (2015) 911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Suderman M, Simpkin A, Sharp G, Gaunt T, Lyttleton O, McArdle W, Ring S, Davey Smith G, Relton C Sex-associated autosomal DNA methylation differences are wide-spread and stable throughout childhood, bioRxiv (2017) 118265. [Google Scholar]
- [20].Bozack AK, Colicino E, Just AC, Wright RO, Baccarelli AA, Wright RJ, Lee AG Associations between infant sex and DNA methylation across umbilical cord blood, artery, and placenta samples, Epigenetics (2021) 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].McCarthy NS, Melton PE, Cadby G, Yazar S, Franchina M, Moses EK, Mackey DA, Hewitt AW Meta-analysis of human methylation data for evidence of sex-specific autosomal patterns, BMC Genomics 15 (2014) 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fuke C, Shimabukuro M, Petronis A, Sugimoto J, Oda T, Miura K, Miyazaki T, Ogura C, Okazaki Y, Jinno Y Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: An HPLC-based study, Ann. Hum. Genet 68 (2004) 196–204. [DOI] [PubMed] [Google Scholar]
- [23].Inoshita M, Numata S, Tajima A, Kinoshita M, Umehara H, Yamamori H, Hashimoto R, Imoto I, Ohmori T Sex differences of leukocytes DNA methylation adjusted for estimated cellular proportions, Biol. Sex. Differ 6 (2015) 11–7. eCollection 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu J, Morgan M, Hutchison K, Vince DC A study of the influence of sex on genome wide methylation, PLoS One 5 (2010) e10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Marco PB, Eske MD, Daniel JW, Strengman E, Janson E, Iris ES, René SK, Roel AO The relationship of DNA methylation with age, gender and genotype in twins and healthy controls, PLoS One 4 (2009) e6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Singmann P, Shem-Tov D, Wahl S, Grallert H, Fiorito G, Shin SY, Schramm K, Wolf P, Kunze S, Baran Y, Guarrera S, Vineis P, Krogh V, Panico S, Tumino R, Kretschmer A, Gieger C, Peters A, Prokisch H, Relton CL, Matullo G, Illig T, Waldenberger M, Halperin E Characterization of whole-genome autosomal differences of DNA methylation between men and women, Epigenetics Chromatin 8 (2015) 43–3. eCollection 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Martin E, Smeester L, Bommarito PA, Grace MR, Boggess K, Kuban K, Karagas MR, Marsit CJ, O’Shea TM, Fry RC Sexual epigenetic dimorphism in the human placenta: Implications for susceptibility during the prenatal period, Epigenomics 9 (2017) 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Inkster AM, Yuan V, Konwar C, Matthews AM, Brown CJ, Robinson WP A cross-cohort analysis of autosomal DNA methylation sex differences in the term placenta, Biology of Sex Differences 12 (2021) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dearden L, Bouret SG, Ozanne SE Sex and gender differences in developmental programming of metabolism, Mol. Metab 15 (2018) 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Arathimos R, Granell R, Henderson J, Relton CL, Tilling K Sex discordance in asthma and wheeze prevalence in two longitudinal cohorts, PLoS One 12 (2017) e0176293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Muenchhoff M, Goulder PJR Sex differences in pediatric infectious diseases, J. Infect. Dis 209 Suppl 3 (2014) 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Melén E, Bergström A, Kull I, Almqvist C, Andersson N, Asarnoj A, Borres MP, Georgellis A, Pershagen G, Westman M, van Hage M, Ballardini N Male sex is strongly associated with IgE-sensitization to airborne but not food allergens: Results up to age 24 years from the BAMSE birth cohort, Clin. Transl. Allergy 10 (2020) 15-w. eCollection 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Green T, Flash S, Reiss AL Sex differences in psychiatric disorders: What we can learn from sex chromosome aneuploidies, Neuropsychopharmacology 44 (2019) 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Capone I, Marchetti P, Ascierto PA, Malorni W, Gabriele L Sexual dimorphism of immune responses: A new perspective in cancer immunotherapy, Frontiers in Immunology 9 (2018) 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rubin JB, Lagas JS, Broestl L, Sponagel J, Rockwell N, Rhee G, Rosen SF, Chen S, Klein RS, Imoukhuede P, Luo J Sex differences in cancer mechanisms, Biology of Sex Differences 11 (2020) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Felix JF, Joubert BR, Baccarelli AA, Sharp GC, Almqvist C, Annesi-Maesano I, Arshad H, Baïz N, Bakermans-Kranenburg M, Bakulski KM, Binder EB, Bouchard L, Breton CV, Brunekreef B, Brunst KJ, Burchard EG, Bustamante M, Chatzi L, Cheng Munthe-Kaas M, Corpeleijn E, Czamara D, Dabelea D, Davey Smith G, De Boever P, Duijts L, Dwyer T, Eng C, Eskenazi B, Everson TM, Falahi F, Fallin MD, Farchi S, Fernandez MF, Gao L, Gaunt TR, Ghantous A, Gillman MW, Gonseth S, Grote V, Gruzieva O, Håberg SE, Herceg Z, Hivert M, Holland N, Holloway JW, Hoyo C, Hu D, Huang R, Huen K, Järvelin M, Jima DD, Just AC, Karagas MR, Karlsson R, Karmaus W, Kechris KJ, Kere J, Kogevinas M, Koletzko B, Koppelman GH, Küpers LK, Ladd-Acosta C, Lahti J, Lambrechts N, Langie SAS, Lie RT, Liu AH, Magnus MC, Magnus P, Maguire RL, Marsit CJ, McArdle W, Melén E, Melton P, Murphy SK, Nawrot TS, Nisticò L, Nohr EA, Nordlund B, Nystad W, Oh SS, Oken E, Page CM, Perron P, Pershagen G, Pizzi C, Plusquin M, Raikkonen K, Reese SE, Reischl E, Richiardi L, Ring S, Roy RP, Rzehak P, Schoeters G, Schwartz DA, Sebert S, Snieder H, Sørensen TIA, Starling AP, Sunyer J, Taylor JA, Tiemeier H, Ullemar V, Vafeiadi M, Van Ijzendoorn MH, Vonk JM, Vriens A, Vrijheid M, Wang P, Wiemels JL, Wilcox AJ, Wright RJ, Xu C, Xu Z, Yang IV, Yousefi P, Zhang H, Zhang W, Zhao S, Agha G, Relton CL, Jaddoe VWV, London SJ Cohort profile: Pregnancy and childhood epigenetics (PACE) consortium, Int. J. Epidemiol (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL High density DNA methylation array with single CpG site resolution, Genomics 98 (2011) 288–295. [DOI] [PubMed] [Google Scholar]
- [38].Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA Minfi: A flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays, Bioinformatics 30 (2014) 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Johnson WE, Li C, Rabinovic A Adjusting batch effects in microarray expression data using empirical bayes methods, Biostatistics 8 (2007) 118–127. [DOI] [PubMed] [Google Scholar]
- [40].B.K. Andrews SV. FlowSorted.CordBlood.450k: Illumina 450k data on sorted cord blood cells, R package version 1.12.0 (2019). [Google Scholar]
- [41].Jaffe AE FlowSorted.blood.450k: Illumina HumanMethylation data on sorted blood cell populations, R package version 1.22.0 (2019). [Google Scholar]
- [42].Venables WN, Ripley BD Modern Applied Statistics with S, Springer-Verlag, New York, 2002. [Google Scholar]
- [43].Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, Gottardo R, Hahne F, Hansen KD, Irizarry RA, Lawrence M, Love MI, MacDonald J, Obenchain V, Oleś AK, Pagès H, Reyes A, Shannon P, Smyth GK, Tenenbaum D, Waldron L, Morgan M Orchestrating high-throughput genomic analysis with bioconductor, Nature Methods 12 (2015) 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].C.T. R. R: A language and environment for statistical computing, 3.4.1 (2017).
- [45].Chen Y, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R Discovery of cross-reactive probes and polymorphic CpGs in the illumina infinium HumanMethylation450 microarray, Epigenetics 8 (2013) 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Higgins JP, Thompson SG Quantifying heterogeneity in a meta‐analysis, Stat. Med 21 (2002) 1539–1558. [DOI] [PubMed] [Google Scholar]
- [47].Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M KEGG as a reference resource for gene and protein annotation, Nucleic Acids Res 44 (2016) D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G GO:: TermFinder—open source software for accessing gene ontology information and finding significantly enriched gene ontology terms associated with a list of genes, Bioinformatics 20 (2004) 3710–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Maksimovic J, Oshlack A, Phipson B Gene set enrichment analysis for genome-wide DNA methylation data, Genome Biol 22 (2021) 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Piñero J, Bravo À, Queralt-Rosinach N, Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, García-García J, Sanz F, Furlong LI DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants, Nucleic Acids Res 45 (2017) D83–D839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yu G, Wang L, Yan G, He Q DOSE: An R/bioconductor package for disease ontology semantic and enrichment analysis, Bioinformatics 31 (2015) 608–609. [DOI] [PubMed] [Google Scholar]
- [52].Aprile D, Fruscione F, Baldassari S, Fadda M, Ferrante D, Falace A, Buhler E, Sartorelli J, Represa A, Baldelli P, Benfenati F, Zara F, Fassio A TBC1D24 regulates axonal outgrowth and membrane trafficking at the growth cone in rodent and human neurons, Cell Death Differ 26 (2019) 2464–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Balestrini S, Milh M, Castiglioni C, Lüthy K, Finelli MJ, Verstreken P, Cardon A, Stražišar BG, Holder JL Jr, Lesca G, Mancardi MM, Poulat AL, Repetto GM, Banka S, Bilo L, Birkeland LE, Bosch F, Brockmann K, Cross JH, Doummar D, Félix TM, Giuliano F, Hori M, Hüning I, Kayserili H, Kini U, Lees MM, Meenakshi G, Mewasingh L, Pagnamenta AT, Peluso S, Mey A, Rice GM, Rosenfeld JA, Taylor JC, Troester MM, Stanley CM, Ville D, Walkiewicz M, Falace A, Fassio A, Lemke JR, Biskup S, Tardif J, Ajeawung NF, Tolun A, Corbett M, Gecz J, Afawi Z, Howell KB, Oliver KL, Berkovic SF, Scheffer IE, de Falco FA, Oliver PL, Striano P, Zara F, Campeau PM, Sisodiya SM TBC1D24 genotype-phenotype correlation: Epilepsies and other neurologic features, Neurology 87 (2016) 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Loke H, Harley V, Lee J Biological factors underlying sex differences in neurological disorders, Int. J. Biochem. Cell Biol 65 (2015) 139–150. [DOI] [PubMed] [Google Scholar]
- [55].Reddy DS The neuroendocrine basis of sex differences in epilepsy, Pharmacology Biochemistry and Behavior 152 (2017) 97–104. [DOI] [PubMed] [Google Scholar]
- [56].Statello L, Guo C, Chen L, Huarte M Gene regulation by long non-coding RNAs and its biological functions, Nature Reviews Molecular Cell Biology (2020) 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Xu W, Zhou G, Wang H, Liu Y, Chen B, Chen W, Lin C, Wu S, Gong A, Xu M Circulating lncRNA SNHG11 as a novel biomarker for early diagnosis and prognosis of colorectal cancer, International Journal of Cancer 146 (2020) 2901–2912. [DOI] [PubMed] [Google Scholar]
- [58].Bitarafan S, Yari M, Broumand MA, Ghaderian SMH, Rahimi M, Mirfakhraie R, Azizi F, Omrani MD Association of increased levels of lncRNA H19 in PBMCs with risk of coronary artery disease, Cell Journal (Yakhteh) 20 (2019) 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fallah H, Azari I, Neishabouri SM, Oskooei VK, Taheri M, Ghafouri-Fard S Sex-specific up-regulation of lncRNAs in peripheral blood of patients with schizophrenia, Scientific Reports 9 (2019) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Majewska M, Lipka A, Paukszto L, Jastrzebski JP, Gowkielewicz M, Jozwik M, Majewski MK Preliminary RNA-seq analysis of long non-coding RNAs expressed in human term placenta, International Journal of Molecular Sciences 19 (2018) 1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ober C, Gilad Y, Loisel DA Sex-specific genetic architecture of human disease, Nature Reviews Genetics 9 (2008) 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shah R, Newcomb DC Sex bias in asthma prevalence and pathogenesis, Front. Immunol 9 (2018) 2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Whitacre CC Sex differences in autoimmune disease, Nat. Immunol 2 (2001) 777–780. [DOI] [PubMed] [Google Scholar]
- [64].Kander MC, Cui Y, Liu Z Gender difference in oxidative stress: A new look at the mechanisms for cardiovascular diseases, J. Cell. Mol. Med 21 (2017) 1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Uekert SJ, Akan G, Evans MD, Li Z, Roberg K, Tisler C, Dasilva D, Anderson E, Gangnon R, Allen DB, Gern JE, Lemanske RF Sex-related differences in immune development and the expression of atopy in early childhood, J. Allergy Clin. Immunol 118 (2006) 1375–1381. [DOI] [PubMed] [Google Scholar]
- [66].McLean Carmen P.|Asnaani Anu|Litz Brett T.|Hofmann Stefan G. Gender differences in anxiety disorders: Prevalence, course of illness, comorbidity and burden of illness, J. Psychiatr. Res 45 (2011) 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Maschietto M, Bastos LC, Tahira AC, Bastos EP, Euclydes VLV, Brentani A, Fink G, De Baumont A, Felipe-Silva A, Francisco RPV Sex differences in DNA methylation of the cord blood are related to sex-bias psychiatric diseases, Scientific Reports 7 (2017) 44547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Halladay AK, Bishop S, Constantino JN, Daniels AM, Koenig K, Palmer K, Messinger D, Pelphrey K, Sanders SJ, Singer AT, Taylor JL, Szatmari P Sex and gender differences in autism spectrum disorder: Summarizing evidence gaps and identifying emerging areas of priority, Molecular Autism 6 (2015) 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bauermeister JJ, Shrout PE, Chávez L, Rubio-Stipec M, Ramírez R, Padilla L, Anderson A, García P, Canino G ADHD and gender: Are risks and sequela of ADHD the same for boys and girls? Journal of Child Psychology and Psychiatry 48 (2007) 831–839. [DOI] [PubMed] [Google Scholar]
- [70].Essex MJ, Armstrong JM, Thomas Boyce W, Hertzman C, Lam LL, Sarah MAN, Kobor MS Epigenetic vestiges of early developmental adversity: Childhood stress exposure and DNA methylation in adolescence, Child Dev 84 (2013) 58–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Armstrong DA, Lesseur C, Conradt E, Lester BM, Marsit CJ Global and gene-specific DNA methylation across multiple tissues in early infancy: Implications for children's health research, Faseb J 28 (2014) 2088–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bakulski KM, Feinberg JI, Andrews SV, Yang J, Brown S, McKenney SL, Witter F, Walston J, Feinberg AP, Fallin MD DNA methylation of cord blood cell types: Applications for mixed cell birth studies, Epigenetics 11 (2016) 354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 7. Other studies of cord or peripheral blood DNA methylation differences by sex
Supplemental Table 6. Bonferroni Significant CpG Sites from the meta-analysis of differences by sex in child blood DNA methylation that were not significant in the meta-analysis of newborns
Supplemental Table 8. Sensitivity analysis of λ in Newborn Cohorts
Supplemental Table 9. Sensitivity analysis of λ in Child Cohorts
Supplemental Table 1. Results of cohort specific EWAS of sex and cord blood DNA methylation
Supplemental Table 3. Results of cohort specific EWAS of sex and child blood DNA methylation
Supplemental Table 2. Bonferroni Significant CpG Sites from the meta-analysis of differences by sex in newborn blood DNA methylation
Supplemental Table 5. CpG Sites from the meta-analysis of differences by sex in child blood DNA methylation with look up level correction (based on cord blood meta-analysis)
Supplemental Table 4. Bonferroni Significant CpG Sites from the meta-analysis of differences by sex in child blood DNA methylation
Supplementary Methods. Methods, funding, and acknowledgements for each specific cohort used in the meta-analysis