

Abstract
The amyloid aggregation of human islet amyloid polypeptide (hIAPP) is associated with pancreatic β-cell death in type 2 diabetes. The S20G substitution of hIAPP (hIAPP(S20G)), found in Japanese and Chinese, is more amyloidogenic and cytotoxic than wild-type hIAPP. The rat amylin (rIAPP) does not have aggregation propensity and cytotoxicity. Mounting evidence suggests that soluble low-molecular-weight amyloid oligomers formed during the early aggregation are more cytotoxic than mature fibrils. The self-assembly dynamics and oligomeric conformations remain unknown because the oligomers are heterogeneous and transient. The molecular mechanism of sequence-variation rendered dramatically different aggregation propensity and cytotoxicity is also elusive. Here, we investigated the oligomerization dynamics and conformations of the amyloidogenic hIAPP, hIAPP(S20G), and non-amyloidogenic rIAPP using atomistic discrete molecular dynamics (DMD) simulations. Our simulation results demonstrated that all three monomeric amylin peptides mainly adopted unstructured formation with partial dynamical helices near N-terminus. Relatively transient β-hairpin were more abundant in the hIAPP and hIAPP(S20G) than in rIAPP. The S20G substituting mutant of hIAPP altered the turn region of the β-hairpin motif resulting in more hydrophobic residue-pairwise contacts within the β-hairpin. Oligomerization dynamic investigation revealed that all the three peptides spontaneously accumulated into helix populated oligomers. The conformational conversion of forming β-sheet-rich oligomers was only observed in the hIAPP and hIAPP(S20G). The population of the high β-sheet content oligomers was enhanced by S20G substitution. Interestingly, both hIAPP and hIAPP(S20G) could form β-barrel formations, and the β-barrel propensity of hIAPP(S20G) was three times larger than hIAPP. No β-sheet-rich and β-barrel formations were observed in the rIAPP. Our direct observation of the correlation between β-barrel oligomer formation and cytotoxicity suggested that β-barrel might play a critically important role in the cytotoxicity of amyloidosis.
Introduction.
The pathological aggregation of human islet amyloid polypeptide (hIAPP, also known as amylin) forming insoluble amyloid deposits around pancreatic β-cells in the islets of Langerhans is the hallmark of type II diabetes (T2D)1–3. Human amylin is a 37-residue peptide hormone co-secreted with insulin by pancreatic β-cells for physiological glucose regulation4, 5. The self-assembly of hIAPP into amyloid fibrils features a typical nucleation–growth sigmoid curve, in which hIAPP monomers initially nucleate into soluble oligomers and β-rich aggregates followed by rapid elongation of proto-fibrils before reaching the saturation of mature fibrils6–9. The experimental determined hIAPP fibril structures are polymorphic but share a common parallel in-register cross-β-sheet core with the β-strands perpendicular to and the inter-strand hydrogen bonds parallel to the fibril axis10–15. Although the hIAPP fibrils have been found cytotoxic to pancreatic β-cells, accumulating evidence suggests that the transient and polymorphic oligomers formed during the early aggregation stage are much more cytotoxic8, 16–20. Therefore, characterizing the early oligomeric conformations and the nucleation dynamics proceed to the fibril structures of hIAPP becomes imperative to understanding the cytotoxicity mechanism.
Monomers of hIAPP are very flexible and described as an intrinsically disordered protein with an amidated C-terminus2, 6, 21, 22. N-terminal region, known as the lipid-membrane domain, mostly adopted helical formations stabilized by a conserved disulfide bond between Cys-2 and Cys-7 no matter under solution and membrane environments1–3, 22–25. The aggregation propensity of the N-terminal of hIAPP (residues 1–13) is dramatically weak26, 27 and hypothesized to be outside the fibril core in recent cryo-EM determined IAPP fibril structures10–12. Prior experimental and computational studies have shown the segments of hIAPP8-2025, 26, 28, hIAPP15-2519, 29, hIAPP19-2919, 29, and hIAPP22-2830, 31 could self-assemble into amyloid fibrillar deposits independent of the full-length polypeptide. In addition, residues from the above fragments also feature β-sheet structures in the experimental determined fibril models of hIAPP10–14, indicating the aggregation-prone regions of hIAPP should locate around residues 8-29. There is only one human amylin natural mutation with a serine to glycine substitution at position 20 (denoted as hIAPP(S20G)) found in Japanese and Chinese32, 33. The aggregation of hIAPP(S20G) is more rapid than the wild-type hIAPP and leads to an increase in β-cell death ratio and the risk of early-onset T2D19, 33–35. However, the mechanism of S20G mutation accelerated hIAPP fibrillization and associated with early onset T2D remains unknown. Apart from the amyloidogenic hIAPP (e.g., hIAPP and its S20G mutation), the non-amyloidogenic amylin species are also existent36, 37. For example, the rat amylin (denoted as rIAPP) with the six amino acids different from hIAPP (i.e., H18R, F23L, A25P, I26V, S28P, and S29P replacements) don’t form amyloid in vitro and in vivo, indicating residues 18-29 are crucial in the aggregation of amylin37, 38. In addition, the liquid-liquid phase separation (LLPS), which initiated hydrogenation and aggregation of hIAPP, was also observed in the rIAPP39. Albeit monomeric and small-size oligomeric (e.g., dimeric) conformations of hIAPP, hIAPP(S20G), and rIAPP have been extensively investigated in previous computational studies6, 36, 38, 40, 41. The molecular mechanism of these sequence-variation induced dramatically different aggregation propensity and cytotoxicity of IAPP remains to be established.
Numerous experimental studies of hIAPP aggregation kinetics have shown that the hIAPP monomers accumulated helical intermediates before forming β-sheet-rich aggregates1, 6, 9, 23, 25, 41. For example, enhancing helical conformations of hIAPP by negatively charged membranes accelerated the aggregation of hIAPP1–3, 22–25. A single point F15L substitution induced hIAPP monomer more helical and low β-sheet formations34. The F15L substitution resulted in more rapid fibrillization with shortened lag time34. The formation of helical intermediates accelerates hIAPP fibrillization, indicating that the helical accumulation might be on-pathway to amyloid assembly. Interestingly, De Carufel et al. found that preventing helical folding with non-helical hIAPP analogs promoted hIAPP aggregation42. In addition, preventing helical folding also enhanced the membrane perturbation and cytotoxicity of IAPP24. Prior ion-mobility spectrometry-mass spectrometry (IMS-MS) experiments and molecular dynamics (MD) simulations suggested that the β-hairpin structure might be the amyloidogenic precursor of hIAPP6, 38, 41, 43. These results indicated that the fibrillization of hIAPP might be irrelevant to their helical intermediates. However, the definite role of these helical intermediates in the aggregation and cytotoxicity of hIAPP remains ambiguous. The conformational conversion of these helical structures to β-sheet-rich aggregates is also unknown. Numerous experimental and computational studies revealed the toxic amyloid fragments (e.g. hIAPP19-29 and its S20G substitution29, hIAPP8-2025, 28, SOD128–3844, Aβ16-2245, 46, Aβ25-3547, and NACore48) and full-length peptides (e.g. Aβ1-4049, Aβ1-4250–52, and hIAPP1-376)) could form well-defined β-barrels as the aggregation intermediates before forming well-ordered cross-β fibrils but no β-barrel intermediates were detected in the fibrillization of the non-toxic amyloid fragment (hIAPP15-25 and its S20G substitution29, G33W and G33V substituting mutants of SOD144), indicating the β-barrel oligomers might serve as the toxic agent in amyloidosis.
To investigate the sequence-variation effects on the aggregation of amylin peptides, we investigated the oligomerization dynamics of hIAPP, hIAPP(S20G), and rIAPP with the number of simulated peptides one and five utilizing atomistic discrete molecular dynamics53, 54 (DMD) simulations with an implicit solvent model. Our simulation results demonstrated that all three monomeric amylin peptides mainly adopted unstructured formation with partial dynamical helices. A similar partially helical hIAPP structure was also observed in the experimental NMR measurement22. Relatively transient β-hairpin were more abundant in the hIAPP and hIAPP(S20G) than in rIAPP. The S20G substituting mutant of hIAPP altered the turn region of the β-hairpin motif resulting in more hydrophobic residue-pairwise contacts within the β-hairpin. Oligomerization dynamic analysis revealed that all the three peptides spontaneously accumulated into helix populated oligomers. The conformational conversion of forming β-sheet-rich oligomers after helical accumulation was only observed in the hIAPP and hIAPP(S20G). Oligomeric conformations of rIAPP were predominantly in helix, and β-sheet structures were extremely rare. The helix-to-β-sheet conformational conversion only observed in the aggregation of hIAPP and hIAPP(S20G), but not rIAPP, was consistent with prior the CD spectra measurements36. Conformational free energy landscape analysis revealed that S20G substitution enhanced the population of high β-sheet content formations. Interestingly, hIAPP and hIAPP(S20G) could form β-barrel formations, which were proposed as toxic oligomers of amyloid aggregation6, 49–52, 55, 56. The β-barrel propensity of hIAPP(S20G) was three times larger than hIAPP. Another recently experimental study also demonstrated that the barrel-like oligomers of hIAPP(S20G) were more abundant than hIAPP wild-type using transmission electron microscopy (TEM) imaging36. No β-barrel formations were observed in the rIAPP. Overall, our results not only uncover the nucleation mechanism of hIAPP and hIAPP(S20G) forming β-sheet-rich oligomers at the molecular level, but the observation of the correlation between forming β-barrel formation and cytotoxicity also supported that β-barrel might play an important role in the cytotoxicity of amyloidosis.
Materials and methods
Molecular systems used in simulations.
The sequence of hIAPP, hIAPP(S20G), and rIAPP were shown in Table 1. The initial structure of monomeric hIAPP (PDBid: 2L8657) and rIAPP(2KJ758) used in our simulation was taken from the protein data bank determined by NMR measurements. The starting structure of the hIAPP(S20G) monomer was constructed by S20G substitution using the PyMol mutagenesis based on the structure of hIAPP (PDBid: 2L8657). Two molecular systems with a number of simulated peptides of one and five were performed for each type of amylin molecule to investigate its nucleation dynamic and conformations. For the monomeric system of each amylin peptide, sixty independent simulations were performed starting from the same initial structure but with different velocities. The ionization-ion mobility spectrometry-mass spectrometry measurement revealed that the critical size of hIAPP oligomers adopted extended conformations was ~459. Thus, we chose a system size with a number of simulated peptides of 5 for the oligomerization simulation. For each amylin peptide, we also performed sixty independent DMD simulations with different initial configurations (i.e., coordinates and velocities). Five peptides were initially positioned randomly (both positions and orientations) within a 9.5 nm cubic simulation box with any minimum inter-molecular distances of no less than 1.5 nm. To avoid the potential biases from the initial states, the duration time of each independent DMD simulation was up to 1000 ns. Details of all the simulations are summarized in Table 2.
Table 1.
Amino acid sequences of hIAPP, hIAPP(S20G), and rIAPP used in our simulation. Each peptide with a Cys2-Cys7 intra-molecular disulfide bond.
Table 2.
Details of each molecular system, including the number of peptides, corresponding dimensions of the cubic simulation box, number of DMD runs, length of each DMD simulation, and accumulative total simulation times.
| Amylin | Peptide numbers | Box size (nm) | DMD runs | Simulation time (ns) | Total duration (μs) |
|---|---|---|---|---|---|
| hIAPP | 1 | 6.5 | 60 | 1000 | 60 |
| 5 | 9.5 | 60 | 1000 | 60 | |
| hIAPP(S20G) | 1 | 6.5 | 60 | 1000 | 60 |
| 5 | 9.5 | 60 | 1000 | 60 | |
| rIAPP | 1 | 6.5 | 60 | 1000 | 60 |
| 5 | 9.5 | 60 | 1000 | 60 |
DMD simulations.
All simulations were performed at 300K using the DMD algorithm with the Medusa force field53, 54, 60 benchmarked for the accurate prediction of protein stability change upon mutation and protein–ligand binding affinity60, 61. DMD is a unique molecular dynamics algorithm where stepwise functions model the continuous interaction potentials in classic molecular dynamics in DMD. The comprehensive descriptions of the atomistic DMD algorithm can be found in previous publications62, 63. The dynamics of the system in DMD are dictated by iteratively updating a series of collision events, predicting their new collisions with corresponding neighbors, and finding the next crash via quick sort algorithms. The sampling efficiency of DMD is significantly enhanced without frequent calculations of forces and accelerations (e.g., every ~1–2 fs) in traditional MD simulations. With significantly sampling efficiency, the DMD algorithm has been widely used to study protein folding and amyloid aggregation by both our group6, 8, 25, 51 and others64–66. The nonbonded parameters were adopted from the CHARMM19 force field67. Water was implicitly modelled using the EEF1 implicit solvation model68. A reaction-like algorithm was used to model the distance- and angle-dependent hydrogen bond63, 69. The screened electrostatic interactions were calculated using the Debye–Hückel approximation with the Debye length set to 10 Å, corresponding to ~100 mM NaCl (a widely used experimental condition in hIAPP aggregation35, 70). With significantly enhanced sampling efficiency and rapid computational speed, the DMD was widely used to study protein folding/aggregation63, 71 and protein–nanoparticle interactions18, 21, 48 both by our group and by others72, 73. DMD software is freely available to academic researchers at Molecules In Action (www.moleculesinaction.com). The units of mass, time, length, and energy used in our united-atom with implicit water model were 1 Da, ~50 fs, 1 Å, and 1 kcal/mol, respectively.
Analysis methods.
The secondary structure was calculated using the DSSP program74. The hydrogen bond was considered to be formed when the N…O distance was within 3.5 Å, and the N–H…O angle was more than 120°. According to prior protein folding studies75, 76, a pairwise residue contact was defined as the distance between the heavy atoms from the mainchain or sidechain of two non-sequential residues within 0.65 nm. Cluster analysis was performed using the Daura algorithm77 and a backbone root-mean-square-deviation (RMSD) cutoff of 0.55 nm. A two-dimensional (2D) free energy surface is constructed using –RT ln P(x, y), where P(x, y) is the probability of a conformation having a particular parameter value of x and y. If the β-strand segments of an oligomer could form a closed cycle with every β-strand connected by two β-strands neighbors through at least two hydrogen bonds, this oligomer was treated as a β-barrel oligomer6, 51.
Results and Discussion
Monomeric hIAPP, hIAPP(S20G), and rIAPP mainly adopted unstructured formations with partially dynamical helix structures, and transient β-sheets of hIAPP and hIAPP(S20G) monomers were more abundant than rIAPP.
Multiple long-timescale independent simulations were performed to investigate the monomeric conformational dynamics of hIAPP, hIAPP(S20G), and rIAPP. Time evolution of the secondary structure of every residue from each type of amylin peptide revealed that all the three peptides were dynamic with frequency conformational changes of forming dynamical helix and transient β-sheet structures (Fig. 1a–c). Overall, the transient structured formations of all the three amylin peptides were populated in helix, and dynamical β-sheet of amyloidogenic amylin (i.e., hIAPP or hIAPP(S20G)) were more abundant than the non-amyloidogenic amylin (i.e., rIAPP). The large conformational changes estimated by the time evolution of the secondary structures, the total number of hydrogen bonds, and the radius gyration (Fig. S1) suggested the simulation result should be independent of the initial structure. The equilibrium assessment was estimated by the time evolution of secondary structure content, the number of hydrogen bonds and contacts, and the radius gyration averaged over the total number of independent DMD simulations (Figs. S2&S3). There were not many noticeable changes during the last 500 ns, indicating all the systems were reasonably converged. Average secondary structure content showed that all the three peptides predominantly adopted unstructured formations (i.e., random coil and bend structures), structured formations were mostly in helical structures, and β-sheets were very rare (Fig. 1d). All three types of amylin monomer featured high helical propensity around residues 8-15. Prior NMR measurement also demonstrated that the solution hIAPP monomer mainly adopted unstructured formation with partial helix spanned residues 8–1722. Such partial helix formation near N-terminal structures of hIAPP and rIAPP was also observed in other MD simulations78, 79. Residues 16-23 of rIAPP were also populated in the helix (Fig. 1e). The weakly populated β-sheets were mainly formed by residues 16-28, and the β-sheet tendencies around the above region of hIAPP and hIAPP(S20G) were stronger than rIAPP. The top nine most populated conformations of each type of amylin showed that helical formation was mainly abundant around N-terminals, and partial β-hairpins were only observed in the amyloidogenic amylin peptides (Fig. 1f–h).
Fig. 1.
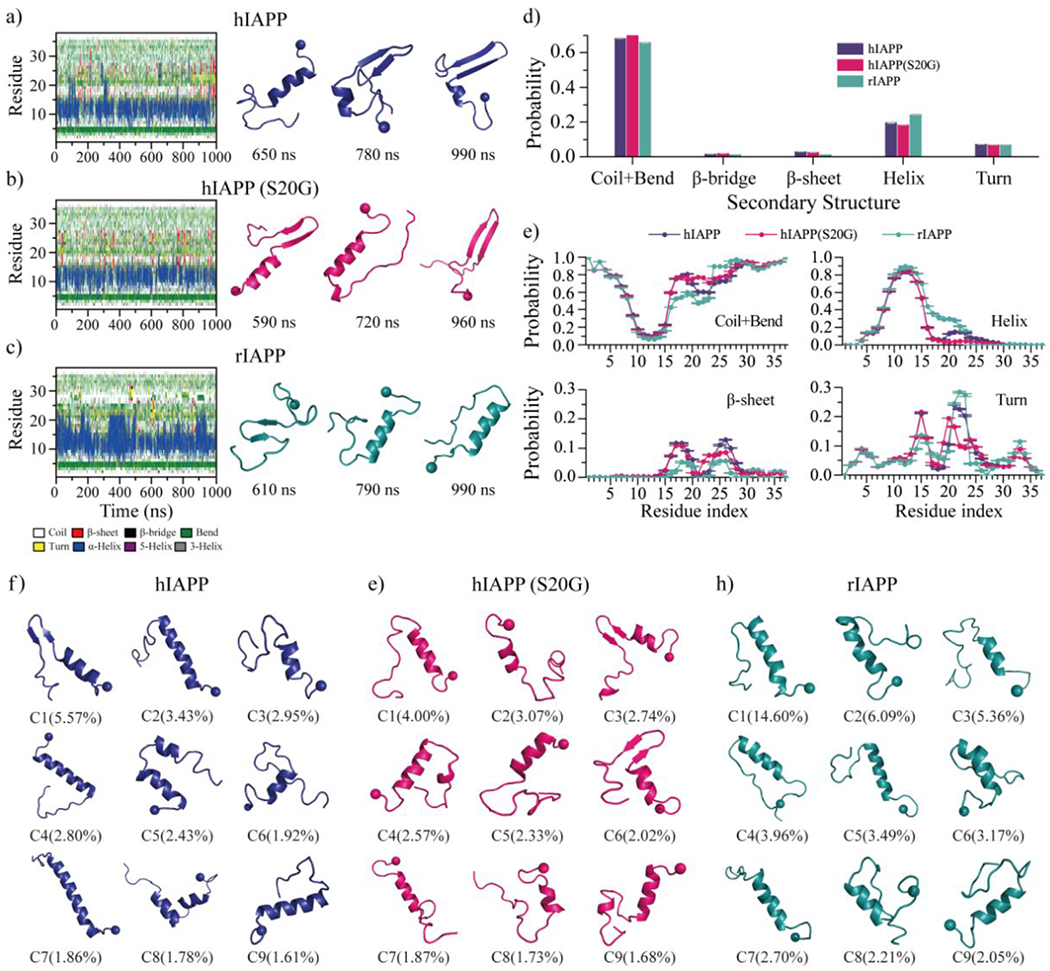
Conformational dynamics analysis of monomeric amylin peptide. a-c) Time evolution of the secondary structure for each residue from hIAPP, hIAPP(S20G), and rIAPP monomeric simulation is shown on the left panel. Dynamically ordered structures formed along the simulation trajectory (the time-stamped blow) are presented on the right. For each system, one 1000 ns DMD trajectory is randomly selected from sixty independent simulations. d) The average secondary structure contents of unstructured (coil and bend), β-sheet, helix, and turn conformation for each monomeric amylin peptide during last 500 ns. e) The average propensity of each residue from hIAPP, hIAPP(S20G), and rIAPP monomer adopting different secondary structures. f-h) Representative monomeric conformations of the top nine most-populated clusters of hIAPP, hIAPP(S20G), and rIAPP. The N-terminal Cα atom is highlighted as a bead.
The S20G substituting mutant of hIAPP altered the turn region of the β-hairpin motif resulting in more hydrophobic residue-pairwise contacts within the β-hairpin.
The conformation of each type of amylin monomer was analyzed by residue-pairwise contact frequency using the structures from saturation states (Fig. 2). The residue-pairwise contact formed by atoms from the main-chain demonstrated that both hIAPP and hIAPP(S20G) monomers featured a high tendency helical pattern along the diagonal around residues 8-19 (snapshots 1 shown in Fig. 2a&b), consistent with prior numerous computational and experimental studies6, 22, 25, 80. This helical region was also known as the membrane-binding helical domain24, 81, 82. The high propensity helical pattern of rIAPP ranged from residue 8 to residue 23, much longer than that of hIAPP and hIAPP(S20G) (Fig. 2c). We also observed a relatively weak β-hairpin contact pattern perpendicular to the diagonal in all three types of amylin monomers.
Fig. 2.
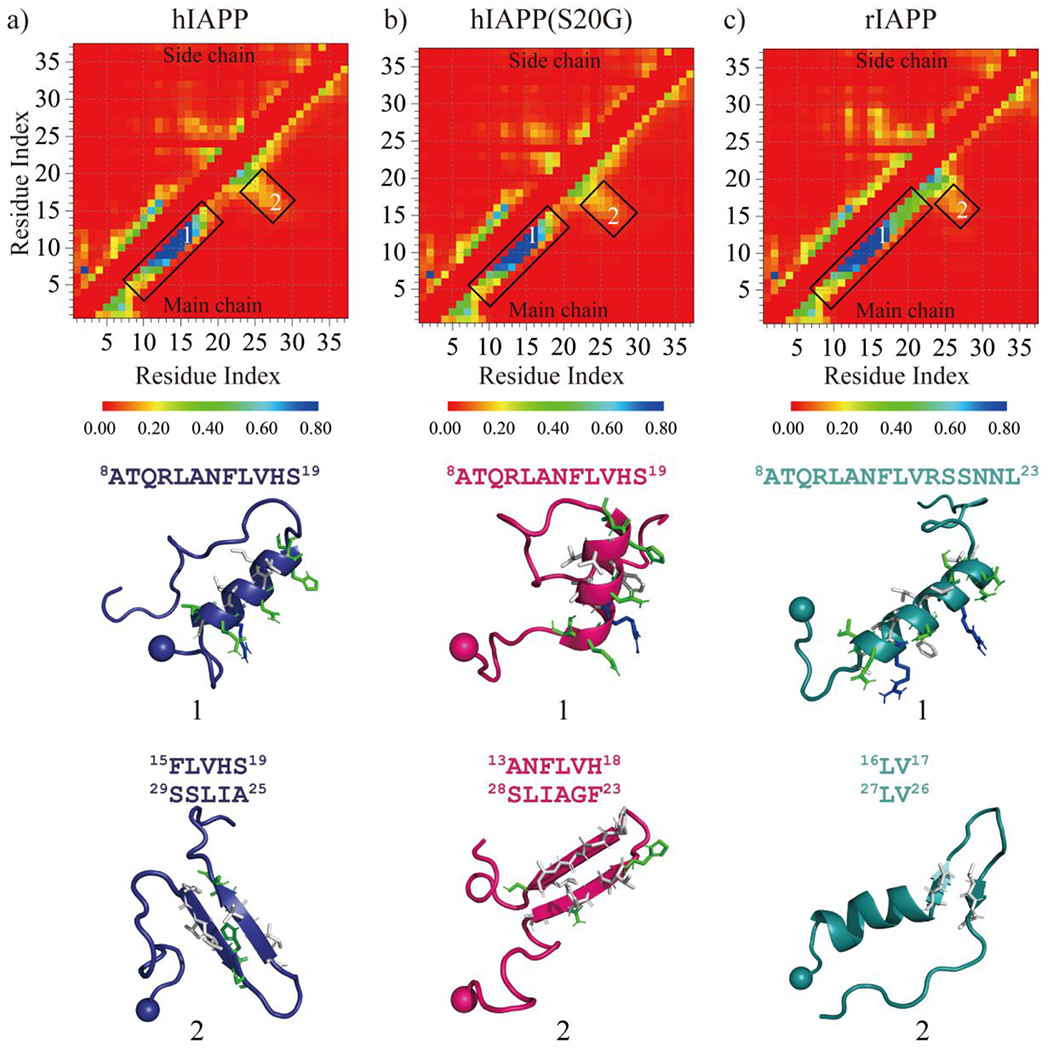
Residue-pairwise contact frequency of each monomeric amylin peptide. a-c) The residue-pairwise intramolecular contact frequency maps are computed between main-chain atoms (lower diagonal) and side-chain atoms (upper diagonal) based on the last 500 ns trajectories of sixty independent DMD simulations after reaching saturation state. The representative contact pattern labelled as 1&2 corresponding to the helical and β-hairpin formations highlighted by boxes in the contact frequency map is also presented. Side-chains within the helical and β-hairpin motif are shown as sticks and colored according to the residue type (hydrophobic in white and hydrophilic in green).
The β-strand within the β-hairpin of rIAPP was much shorter than that of hIAPP and hIAPP(S20G) (snapshots 2 in Fig. 2a&b). The S20G substitution altered the residue-pairwise contact of the β-hairpin (Figs. 1e&S4), which enhanced the residue-pairwise contact among hydrophobic residues within the β-hairpin formations by changing the turn region (snapshots 2 in Fig. 2).
The hIAPP and hIAPP(S20G) peptides could nucleate into β-sheet-rich oligomers via helical accumulation, but rIAPP peptides only accumulated into helical populated compact oligomers.
Sixty independent DMD simulations with five peptides were performed for each type of amylin peptide to investigate the oligomerization dynamic and conformations. Time evolution of each residue’s secondary structure, oligomer size each peptide aggregated into, and the number of hydrogen bonds and contacts showed all the three types of peptides readily nucleated into helical structure populated oligomers in less than 100 ns (Fig. 3). Some hIAPP and hIAPP(S20G) peptides converted into β-sheet structures after the helical accumulations (snapshots shown in Fig. 3a&b). The rIAPP peptides predominantly adopted helical formations, and β-sheet formations were drastically rare (Fig. 3c). The time evolution of the probability of each secondary structure (including unstructured formation, helix, and β-sheet), the total number of hydrogen bonds and contact, and the radius gyration for the five-peptide simulations of hIAPP, hIAPP(S20G), and rIAPP revealed that all three systems reached steady states during last 500 ns (Fig. S5). The equilibrium analysis of each molecular system with the five peptides simulation was further estimated by the time evolution of the averaged secondary content, the number of hydrogen bonds and contacts, and radius gyration over the sixty independent simulations (Figs. S6&S7). Only the saturated structures from the last 500 ns were used for the conformational analysis to avoid potential bias from the initial state. Time average β-sheet content in each independent trajectory during the last 500 ns displayed a high heterogeneity among the simulations of hIAPP and hIAPP(S20G) (Fig. 4a). For example, the first independent simulation of hIAPP and hIAPP(S20G) had much higher β-sheet content than the rest. The S20G substitution enhanced the hIAPP peptides to form more β-sheet populated formations. All the rIAPP independent simulations featured helix abundant and low β-sheet content formations (Fig. 4a&b). Representative snapshots of helices and β-sheets populated formations from the same trajectory suggested that some helical hIAPP and hIAPP(S20G) peptides converted into β-sheets after helical accumulations (Fig. 4c). The helix-to-β-sheet conformational conversion was only observed in the oligomerization of hIAPP and hIAPP(S20G) but not rIAPP, which was consistent with experimental CD spectra measurement36.
Fig. 3.
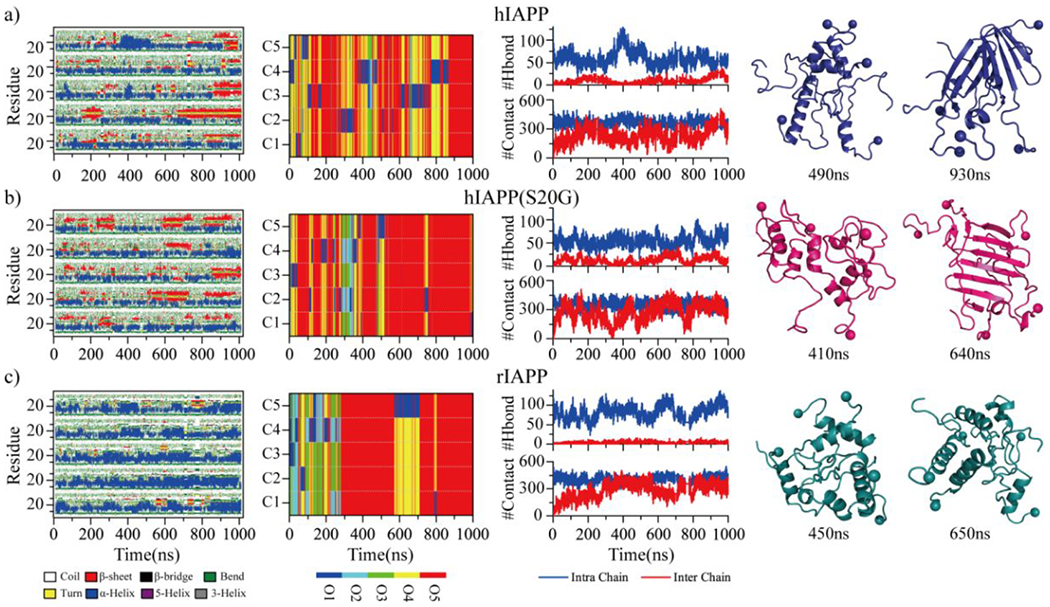
Oligomerization dynamics and conformational changes. The secondary structure of each residue (first column), the oligomer size into which a peptide aggregated (second column), the number of backbone hydrogen bonds and heavy contacts (third column) are presented as the function of simulation time in the representative five-peptide simulation trajectories of hIAPP a), hIAPP(S20G), and rIAPP c). Oligomer size and each peptide chain in the second column are described as O1 to O5 and C1 to C5, respectively. Two representative snapshots populated with helix or β-sheet formations along the simulation trajectories are presented to the right.
Fig. 4.
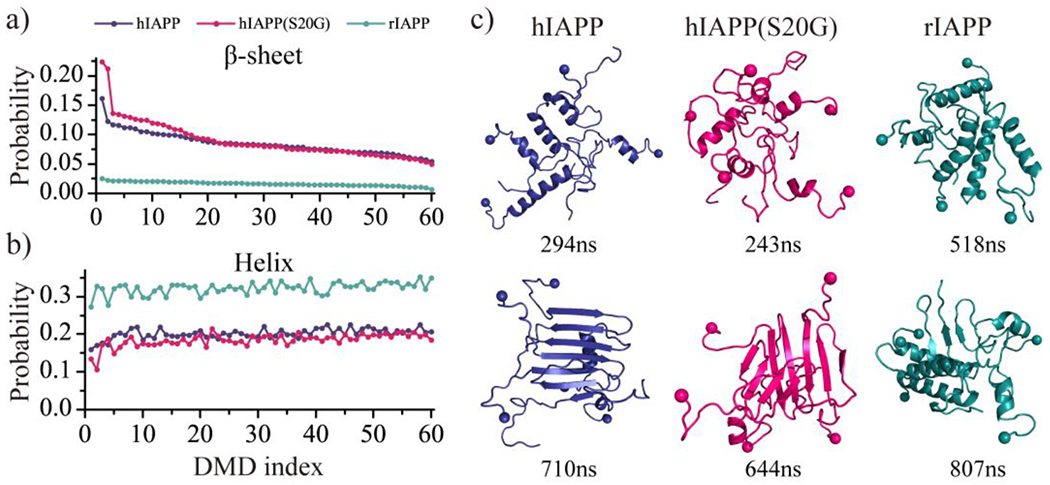
The time-average β-sheet and helix propensity of each independent simulation. The time-averaged β-sheet a) or helical b) probability is computed for each independent simulation during the last 500 ns. Simulation trajectories are sorted according to the averaged β-sheet content from high to low. c) One helix-rich and one β-sheet-rich structures from the same top-ranked trajectory are shown for each molecular system.
The average secondary structure propensities of the self-assemblies in each molecular system were also analyzed (Fig. 5a). The oligomers formed by all three types of amylin peptides were dominant with unstructured formations (more than 55%), and helical content is much larger than the β-sheet ratio. Compared with the monomeric simulations (Fig. 1d), the β-sheet propensity of hIAPP and hIAPP(S20G) peptides enhanced up to 9%, which was much larger than that in their monomers (less than 3%). In both one- and five-peptide simulations, the averaged β-sheet propensity of rIAPP was very rare (less than 2%), but the helix propensity of rIAPP (~32%) in the five-peptide simulation was much more than its monomeric simulation (~24%). These results indicated the oligomerization enhanced the hIAPP and hIAPP(S20G) peptides to form more β-sheets but promoted rIAPP to adopt more helices. The secondary structure propensity for each residue was also analyzed (Fig. 5b). Regardless of the system size, the helix and β-sheet abundant region of each amylin peptide obtained from the monomeric simulation (Fig. 1e) were similar to that in the oligomeric systems (Fig. 5b). Residues 16-28 of hIAPP and hIAPP(S20G) with a strong propensity of forming β-strand-turn-β-strand structure in the five-peptide oligomerization simulation were also observed in prior hIAPP dimer all-atom explicit REMD simulations41. In addition, residues 16-28 were present around the cross-β cores in previous experimentally-proposed amyloid fibril models of amylin15. Interestingly, the S20G substitution of hIAPP decreased the turn (i.e., loop) populations and boosted the β-sheet formations of residues 22-23 were consistent with a recent cryo-EM experimental study83. For example, the residues around 22-23 were described as β-strand in the hIAPP(S20G) fibril rather than the loop in the hIAPP fibril model 14, 83. The β-sheet regions obtained from our simulations were similar to the experimentally-determined fibrils14, 15, 83, indicating the interactions stabilizing amyloid fibrils were also present in stabilizing the oligomers of amyloidogenic amylin peptides.
Fig. 5.
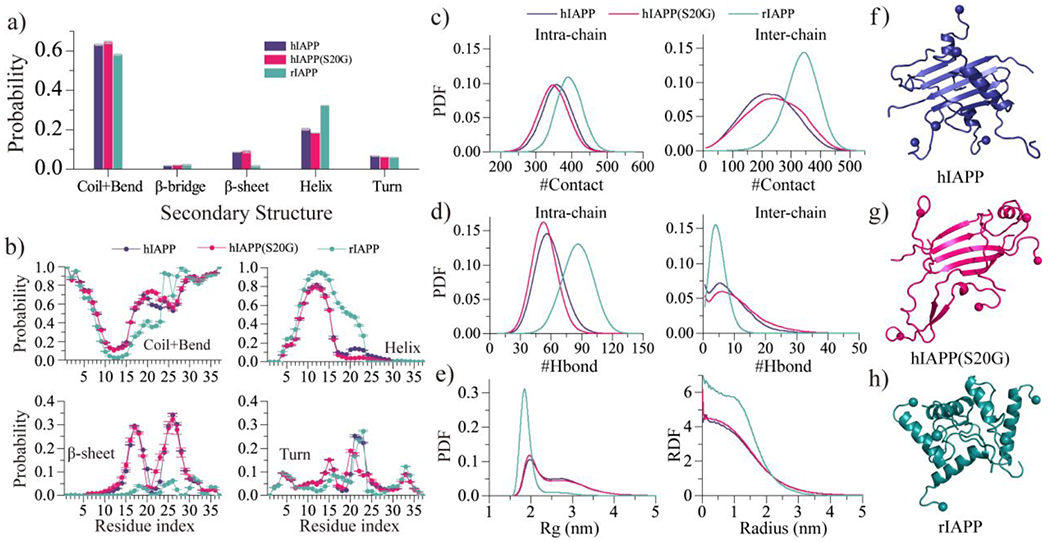
Oligomeric conformation analysis. a) The average secondary structure contents of unstructured (coil and bend), β-sheet, helix and turn conformation for the oligomers formed by hIAPP, hIAPP(S20G), and rIAPP peptides. b) The averaged propensity of every residue adopting coil and bend, helix, β-sheet, and bend conformations in five-peptide simulations for each type of amylin peptides. c-d) The probability distribution as a function of the number intrachain/interchain heavy-atom contact c) and backbone hydrogen bonds d). e) The probability distribution as a function of radius gyration (Rg) and radial distribution function (RDF) of Cα atoms for the self-assemblies formed by each peptide. f-h) The oligomeric formation of each type of amylin peptide. Only the last 500 ns trajectories from 60 independent simulations are used for the above conformational analysis.
Conformations of the oligomers formed by each type of amylin peptides were further analyzed through the probability distribution of the number of heavy atom contact and hydrogen bonds formed by main-chain atoms, radius gyration, and radial distribution function (RDF) of each residue’s Cα atoms (Fig. 5c–e). Oligomers of rIAPP were more populated with intramolecular hydrogen bonds and contacts than hIAPP and hIAPP(S20G) due to the oligomeric rIAPP being more helical than hIAPP and hIAPP(S20G) (Fig. 5f–h). The hIAPP and hIAPP(S20G) aggregates had more intermolecular hydrogen bonds than rIAPP because the self-assemblies of hIAPP and hIAPP(S20G) were abundant with intermolecular β-sheets (Fig. 5f–g). The S20G mutant promoted the amyloidogenic amylin to form more intermolecular contacts and hydrogen bond formations, indicating that the self-assembly propensity of hIAPP(S20G) should be more potent than hIAPP, which was consistent with prior numerous experimental studies19, 33–35. Radius gyration (Rg) distribution and radial distribution function (RDF) of Cα atoms revealed that conformations of rIAPP oligomers were more compact (Fig. 5e) than hIAPP and hIAPP(S20G).
Residue-pairwise contact frequency analysis for the self-assemblies of each type of amylin.
To better characterize the structure of each molecular self-assemblies, we computed the intra- and inter-chain contact probabilities between all residue pairs, shown as the contact frequency maps between main-chain atoms (Fig. 6) or side-chain atoms (Fig. S8). A high propensity intra-chain helical contact pattern along the diagonal was observed in all three molecular systems, and the helical pattern of rIAPP (residues 9-25) was much longer than hIAPP and hIAPP(S20G) (residues 9-16) (Fig. 6). Thus, residues of rIAPP displayed more intramolecular contact than hIAPP and hIAPP(S20G) (Fig. 5d). A weak intra-chain β-hairpin contact pattern perpendicular to the diagonal formed by residues 14-29 was only detected in the oligomeric hIAPP and hIAPP(S20G). The S20G mutant changed the turn region and increased the hydrophobic residue-pairwise contact with the β-hairpin (as we discussed in Fig. 2). The intermolecular contacts formed by main-chain atoms among residues 1-13 of hIAPP and hIAPP(S20G) peptides were too weak to be observed (Fig. 6a&b), indicating the aggregation propensity around this region was extremely weak, agreed with previous studies 26, 27. For example, Residues 1-13 were hypothesized to be outside the fibril core in recent cryo-EM determined IAPP fibril structures10–12 due to their conformations being too dynamic. Residues 13-30 of hIAPP and hIAPP(S20G) displayed a strong tendency to form intermolecular main-chain-main-chain contact suggesting that these residues played a critical in their fibrillization. Prior experimental and computational studies have shown the segment from residues 13-30 (e.g. hIAPP8-2025, 26, 28, hIAPP15-2519, 29, hIAPP19-29 19, 29, and hIAPP22-2830, 31) could directly self-assemble into amyloid fibril independent of the full-length polypeptide. The average number of intermolecular side-chain contacts of residues L12, F15, L16, V17, H18, F23, G24, A25, I26, and L27 from hIAPP and hIAPP(S20G) were larger than the remaining residues (Fig. S8a&b), indicating the fibrillization was mainly driven by the hydrophobic and aromatic interactions. The intermolecular main-chain contacts around residue 22 region of hIAPP(S20G) were a little larger than the hIAPP wild-type. The intermolecular residue-pairwise interactions for the main-chain atoms of residues 1-22 of rIAPP were dramatically weak, and high contact tendency regions were mainly spread around residues 23-30.
Fig. 6.
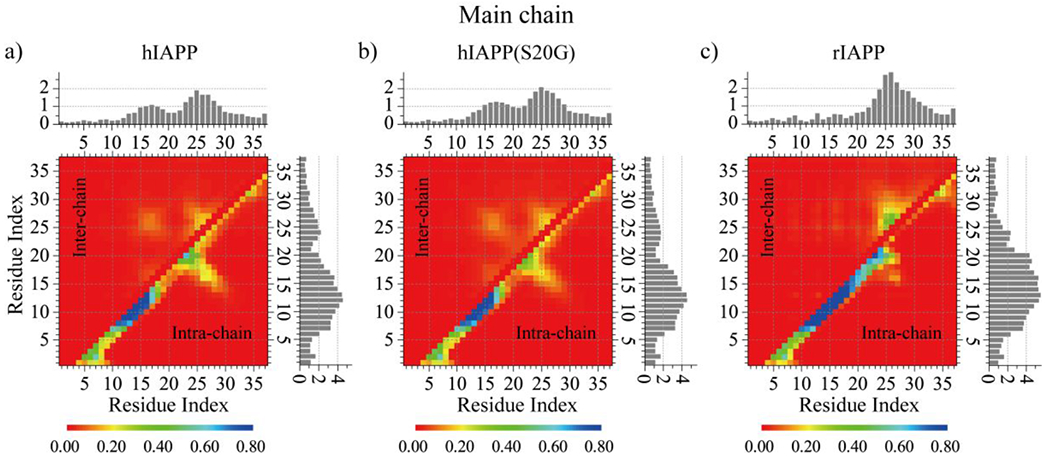
The residue-pairwise contact frequency of backbone atoms. The frequency of inter-chain (upper diagonal) and intra-chain (lower diagonal) inter-residue contact formed by backbone atoms are calculated by averaging over the last 500 ns trajectories of all independent simulations for hIAPP a), hIAPP(S20G) b), and rIAPP c). The total number of intra-chain (histograms to the right) and inter-chain (histograms on the top) contacts per residue is calculated by integrating the corresponding 2D contact probability map.
Conformational ensembles of hIAPP and hIAPP(S20G) oligomers were populated with β-sheet rich and extended conformations, while rIAPP self-assemblies predominantly adopted low β-sheet content compact formations.
To further characterize the structural ensemble of each repeat, we calculated the potential of mean force (PMF, the effective conformational free energy landscape) as a function of the radius of gyration (Rg) and β-sheet content using the last 500 ns DMD trajectories from sixty independent DMD simulations (Fig. 7). The free energy basins of hIAPP and hIAPP(S20G) were broad with Rg~1.8–3.5 nm and β-sheet content~0.03–0.2, indicating that the oligomeric conformations were very diverse (Fig. 7a&b). The hIAPP and hIAPP(S20G) aggregates with low β-sheet ratio (less than 0.10) were populated with the Rg ranging Rg~1.8–3.5 nm, but the β-sheet-rich aggregates (β-sheet probability more prominent than 0.1) were mainly adopting the compact formations with Rg less than 1.3 nm. Representative snapshots corresponding to the low energy regions labeled on the free energy surface confirmed that (Fig. 7a&b). In addition, the oligomers with β-sheet content greater than 20% formed by hIAPP(S20G) featured lower free energy than hIAPP, indicating the S20G substitution promoted the peptide to form more β-sheet formations. The probability distribution of helix and β-sheet content for each oligomer aggregated by hIAPP and hIAPP(S20G) further confirmed that (Fig. S9). However, the free energy landscape surface of rIAPP self-assemblies was completely different from those of hIAPP and hIAPP(S20G), with most rIAPP oligomers featuring small Rg values (1.7-2.1 nm) and relatively weak β-sheet content (0-0.06) (Figs. 7c&S9). Representative conformations of rIAPP oligomers with low free energy revealed that the rIAPP peptides accumulated into helix compact formations. Overall, conformational ensembles analysis revel oligomers of hIAPP and hIAPP(S20G) were much more populated with β-sheet formations than rIAPP, and the S20G substitution enhanced hIAPP to form more β-sheet content formations (Fig. S9).
Fig. 7.
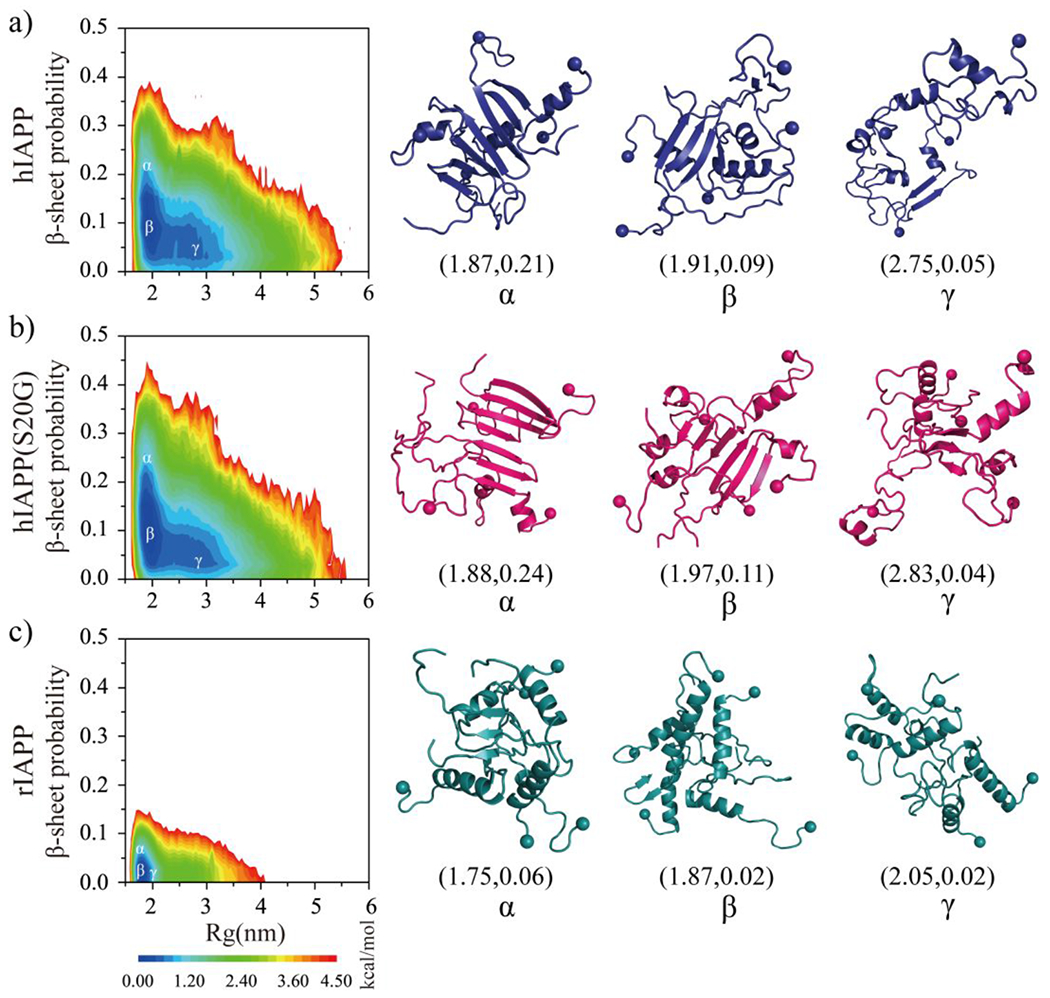
The conformational free energy landscape of each amylin peptide. The potential mean force as a function of the total number of radius gyration (Rg) and β-sheet content in self-assemblies of hIAPP a), hIAPP(S20G) b), and rIAPP c). Three representative structures labelled in the PMFs (α, β, γ) are shown on the right.
The S20G substitution significantly enhanced hIAPP to form β-barrel formations.
The β-barrel aggregates first observed in an 11-residue peptide derived from the slow-aggregating αB crystalline have been proposed as toxic oligomers of amyloid aggregation55 due to their well-defined structures and compatibility to the “amyloid-pore”56 hypothesis of amyloid toxicity44, 50. The formation of β-barrel oligomers by full-length Aβ peptides was supported by hydrogen exchange mass spectrometry, NMR measurement, cryo-EM essays, and computational simulations49–52. The correlation between the formation of β-barrel intermediates and the cytotoxicity in serials amyloid segments with contrastingly cytotoxicity (e.g., hIAPP15-25, hIAPP19-29, SOD128–38 and its G33W and G33V substitution, Aβ16-22, Aβ25-35, NACore) also supported β-barrels as the toxic oligomers in amyloidosis29, 44, 48. Nucleation of β-rich oligomers and β-barrels was also observed in the early aggregation simulation of hIAPP6. To investigate whether disease-associated S20G mutation of hIAPP could affect the population of β-barrels, the probability for each type of amylin peptide to form the β-barrel in every simulated system was also calculated (Fig. 8). Because β-barrel structures were also observed during the first 500 ns, all the whole 1000 ns trajectories were used for the β-barrel propensity analysis. No β-barrel formations were observed in the non-toxic rIAPP oligomerization simulations. The cytotoxic hIAPP and hIAPP(S20G) could form β-barrels. The β-barrel propensities of hIAPP(S20G), which was more toxic than hIAPP, were three times larger than hIAPP. Another recent TEM measurement also revealed that the barrel-like oligomers of hIAPP(S20G) were more abundant than hIAPP wild-type during the nucleation stage36. The structural stability of the β-barrel structure formed by either hIAPP or hIAPP(S20G) from DMD simulations was examined by all-atom explicit-solvent standard MD simulations at room temperature (Figs. S10&S11). Different force fields (including GROMOS9684, OPLS-AA85, AMBER99SB-ILDN86, and CHARMM36m87) were also tested with a duration time of each independent MD simulation up to 1 μs. Only small conformational changes were observed in the 1 μs MD simulations, which were estimated by the time evolution of the RMSD corresponding to the initial structure and the content of each secondary structure. The β-barrel structures were well dynamically maintained and underwent open-and-close dynamics during the course of traditional MD simulations, which was consistent with our DMD simulation results. Together with prior computation and experimental studies (summarized in Table S1)6, 25, 29, 44, 46, 48–52, our direct observation of the β-barrel formation during the self-assembly of toxic hIAPP and hIAPP(S20G) but not the nontoxic rIAPP indicated β-barrels as the common toxic intermediates in amyloid aggregation, which may serve as a novel target for the treatment of T2D.
Fig. 8.
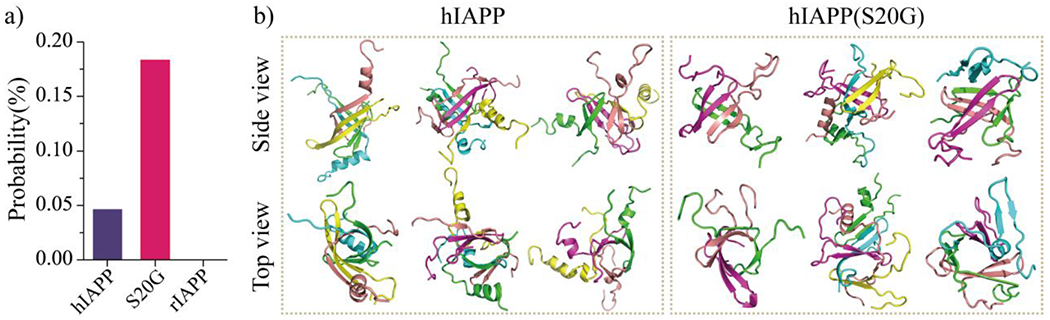
The population ensemble of β-barrel oligomers. a) The average probability of β-barrel formation formed by each type of amylin peptide is calculated using all the 1000 ns simulation trajectories for each type of peptide. b) Representative β-barrel structure formed by hIAPP and hIAPP(S20G) peptide. Three representative β-barrel structures formed by each type of amylin peptide are presented in two different views (side and top). Due to the β-barrel being extremely heterogeneous in structures, three representative β-barrels are randomly selected from the top-three most populated β-barrel trajectories.
The correlation between the formation of β-barrel intermediates and the cytotoxicity in the above discussion suggested that β-barrels might serve as common toxic oligomers in amyloidosis. Despite β-barrel pores formation causing membrane leakage was also supported by experimental evidence49, 50, 52, the nucleation of amyloid peptides along with conformational changes under membrane environments remains unknown. The S20G substitution effects on the hIAPP and lipid membrane interaction (e.g., membrane insertion) still need further study. Future work may include lipid membrane in the oligomerization simulation of amyloid peptides (e.g., hIAPP and hIAPP(S20G)) to investigate the membrane disruption and the early events of amyloid aggregation.
Conclusions.
In this study, we investigated the self-assembly dynamics and structures of hIAPP, hIAPP(S20G), and rIAPP using atomistic DMD simulations. Our simulation results demonstrated that all three monomeric amylin peptides were mainly adopted unstructured formations with frequently conformal changes, and dynamically ordered structures were populated with helix. The amyloidogenic hIAPP and hIAPP(S20G) peptides first accumulated into helix abundant conformations before converting into β-sheet-rich oligomers. The rIAPP spontaneously nucleated into helix dominant compact structures, and the β-sheet formation was very weak. The S20G substituting mutant enhanced hydrophobic residue-pairwise contact within the β-hairpin by changing the turn region of the β-hairpin motif. Conformational free energy landscape analysis revealed that S20G substitution enhanced the population of high β-sheet content structures. Interestingly, hIAPP and hIAPP(S20G) could form β-barrel formations, and the β-barrel propensity of hIAPP(S20G) was three times larger than hIAPP. No β-sheet-rich and β-barrel formations were observed in the rIAPP. Our direct observation of the correlation between β-barrel oligomer formation and cytotoxicity suggested that β-barrel might play a critically important role in the cytotoxicity of amyloidosis. These β-sheet-rich oligomers, especially the β-barrel oligomers, with well-defined structures might serve as a novel therapeutic target for T2D.
Supplementary Material
Acknowledgements.
This work was supported in part by the National Natural Science Foundation of China under Grant No. 11904189 (Yunxiang Sun), K. C. Wong Magna Fund in Ningbo University, China (Yunxiang Sun), NSF CBET-1553945 (Feng Ding), and NIH R35GM119691 (Feng Ding). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NSFC, NIH, and NSF.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic supplementary information (ESI) available: Figs. S1–S11 and Table S1.
References.
- 1.Milardi D, Gazit E, Radford SE, Xu Y, Gallardo RU, Caflisch A, Westermark GT, Westermark P, Rosa C and Ramamoorthy A, Chem Rev, 2021, 121, 1845–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saini RK, Goyal D and Goyal B, Chem Res Toxicol, 2020, 33, 2719–2738. [DOI] [PubMed] [Google Scholar]
- 3.Ke PC, Sani MA, Ding F, Kakinen A, Javed I, Separovic F, Davis TP and Mezzenga R, Chem Soc Rev, 2017, 46, 6492–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brender JR, Lee EL, Hartman K, Wong PT, Ramamoorthy A, Steel DG and Gafni A, Biophys J, 2011, 100, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abedini A and Schmidt AM, FEES Lett, 2013, 587, 1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun Y, Kakinen A, Xing Y, Pilkington EH, Davis TP, Ke PC and Ding F, Biochim Biophys Acta Mol Basis Dis, 2019, 1865, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iadanza MG, Jackson MP, Hewitt EW, Ranson NA and Radford SE, Nat Rev Mol Cell Biol, 2018, 19, 755–773. [DOI] [PubMed] [Google Scholar]
- 8.Faridi A, Sun Y, Okazaki Y, Peng G, Gao J, Kakinen A, Faridi P, Zhao M, Javed I, Purcell AW, Davis TP, Lin S, Oda R, Ding F and Ke PC, Small, 2018, 14, e1802825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saghir AE, Farrugia G and Vassallo N, Chem Phys Lipids, 2021, 234, 105010. [DOI] [PubMed] [Google Scholar]
- 10.Ragonis-Bachar P and Landau M, Curr Opin Struct Biol, 2021, 68, 184–193. [DOI] [PubMed] [Google Scholar]
- 11.Cao Q, Boyer DR, Sawaya MR, Ge P and Eisenberg DS, Nat Struct Mol Biol, 2020, 27, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roder C, Kupreichyk T, Gremer L, Schafer LU, Pothula KR, Ravelli RBG, Willbold D, Hoyer W and Schroder GF, Nat Struct Mol Biol, 2020, 27, 660–667. [DOI] [PubMed] [Google Scholar]
- 13.Sumner Makin O and Serpell LC, J Mol Biol, 2004, 335, 1279–1288. [DOI] [PubMed] [Google Scholar]
- 14.Luca S, Yau WM, Leapman R and Tycko R, Biochemistry, 2007, 46, 13505–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao Q, Boyer DR, Sawaya MR, Abskharon R, Saelices L, Nguyen BA, Lu J, Murray KA, Kandeel F and Eisenberg DS, Nat Struct Mol Biol, 2021, 28, 724–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abedini A, Plesner A, Cao P, Ridgway Z, Zhang J, Tu LH, Middleton CT, Chao B, Sartori DJ, Meng F, Wang H, Wong AG, Zanni MT, Verchere CB, Raleigh DP and Schmidt AM, Elife, 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kakinen A, Sun Y, Javed I, Faridi A, Pilkington EH, Faridi P, Purcell AW, Zhou R, Ding F, Lin S, Chun Ke P and Davis TP, Sci Bull (Beijing), 2019, 64, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang M, Sun Y, Cao X, Peng G, Javed I, Kakinen A, Davis TP, Lin S, Liu J, Ding F and Ke PC, Nanoscale, 2018, 10, 19995–20006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krotee P, Rodriguez JA, Sawaya MR, Cascio D, Reyes FE, Shi D, Hattne J, Nannenga BL, Oskarsson ME, Philipp S, Griner S, Jiang L, Glabe CG, Westermark GT, Gonen T and Eisenberg DS, Elife, 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez Camargo DC, Garg D, Buday K, Franko A, Rodriguez Camargo A, Schmidt F, Cox SJ, Suladze S, Haslbeck M, Mideksa YG, Gemmecker G, Aichler M, Mettenleiter G, Schulz M, Walch AK, Hrabe de Angelis M, Feige MJ, Sierra CA, Conrad M, Tripsianes K, Ramamoorthy A and Reif B, Chem Commun (Camb), 2018, 54, 5426–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faridi A, Sun Y, Mortimer M, Aranha RR, Nandakumar A, Li Y, Javed I, Kakinen A, Fan Q, Purcell AW, Davis TP, Ding F, Faridi P and Ke PC, Nano Res, 2019, 12, 2827–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez Camargo DC, Tripsianes K, Buday K, Franko A, Gobl C, Hartlmuller C, Sarkar R, Aichler M, Mettenleiter G, Schulz M, Boddrich A, Erck C, Martens H, Walch AK, Madl T, Wanker EE, Conrad M, de Angelis MH and Reif B, Sci Rep, 2017, 7, 44041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Apostolidou M, Jayasinghe SA and Langen R, J Biol Chem, 2008, 283, 17205–17210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo C, Cote S, Mousseau N and Wei G, J Phys Chem B, 2015, 119, 3366–3376. [DOI] [PubMed] [Google Scholar]
- 25.Sun Y, Kakinen A, Xing Y, Faridi P, Nandakumar A, Purcell AW, Davis TP, Ke PC and Ding F, Small, 2019, 15, e1805166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaikaran ET, Higham CE, Serpell LC, Zurdo J, Gross M, Clark A and Fraser PE, J Mol Biol, 2001, 308, 515–525. [DOI] [PubMed] [Google Scholar]
- 27.Andreetto E, Yan LM, Tatarek-Nossol M, Velkova A, Frank R and Kapurniotu A, Angew Chem Int Ed Engl, 2010, 49, 3081–3085. [DOI] [PubMed] [Google Scholar]
- 28.Wang L, Ilitchev AI, Giammona MJ, Li F, Buratto SK and Bowers MT, J Phys Chem B, 2016, 120, 11905–11911. [DOI] [PubMed] [Google Scholar]
- 29.Sun Y, Ge X, Xing Y, Wang B and Ding F, Sci Rep, 2018, 8, 10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Y, Wang B, Ge X and Ding F, Phys Chem Chem Phys, 2017, 19, 28414–28423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffmann W, Folmert K, Moschner J, Huang X, von Berlepsch H, Koksch B, Bowers MT, von Helden G and Pagel K, J Am Chem Soc, 2018, 140, 244–249. [DOI] [PubMed] [Google Scholar]
- 32.Cao P, Tu LH, Abedini A, Levsh O, Akter R, Patsalo V, Schmidt AM and Raleigh DP, J Mol Biol, 2012, 421, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meier DT, Entrup L, Templin AT, Hogan MF, Mellati M, Zraika S, Hull RL and Kahn SE, Diabetologia, 2016, 59, 2166–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tu LH and Raleigh DP, Biochemistry, 2013, 52, 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakagashira S, Hiddinga HJ, Tateishi K, Sanke T, Hanabusa T, Nanjo K and Eberhardt NL, Am J Pathol, 2000, 157, 2101–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi B, Kim NH, Jin GY, Kim YS, Kim YH and Eom K, Phys Chem Chem Phys, 2021, 23, 22532–22542. [DOI] [PubMed] [Google Scholar]
- 37.Hu R, Zhang M, Patel K, Wang Q, Chang Y, Gong X, Zhang G and Zheng J, Langmuir, 2014, 30, 5193–5201. [DOI] [PubMed] [Google Scholar]
- 38.Wu C and Shea JE, PLoS Comput Biol, 2013, 9, e1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pytowski L, Lee CF, Foley AC, Vaux DJ and Jean L, Proc Natl Acad Sci U S A, 2020, 117, 12050–12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang G, Zhao J, Yu X and Zheng J, Biochemistry, 2013, 52, 1089–1100. [DOI] [PubMed] [Google Scholar]
- 41.Mo Y, Lei J, Sun Y, Zhang Q and Wei G, Sci Rep, 2016, 6, 33076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Carufel CA, Quittot N, Nguyen PT and Bourgault S, Angew Chem Int Ed Engl, 2015, 54, 14383–14387. [DOI] [PubMed] [Google Scholar]
- 43.Dupuis NF, Wu C, Shea JE and Bowers MT, J Am Chem Soc, 2011, 133, 7240–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun Y, Huang J, Duan X and Ding F, J Chem Inf Model, 2021, 61, 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie L, Luo Y and Wei G, J Phys Chem B, 2013, 117, 10149–10160. [DOI] [PubMed] [Google Scholar]
- 46.Ge X, Sun Y and Ding F, Biochim Biophys Acta Biomembr, 2018, 1860, 1687–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Do TD, LaPointe NE, Nelson R, Krotee P, Hayden EY, Ulrich B, Quan S, Feinstein SC, Teplow DB, Eisenberg D, Shea JE and Bowers MT, J Am Chem Soc, 2016, 138, 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun Y, Kakinen A, Zhang C, Yang Y, Faridi A, Davis TP, Cao W, Ke PC and Ding F, Nanoscale, 2019, 11, 11933–11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan J, Han J, Borchers CH and Konermann L, Biochemistry, 2012, 51, 3694–3703. [DOI] [PubMed] [Google Scholar]
- 50.Wu J, Blum TB, Farrell DP, DiMaio F, Abrahams JP and Luo J, Angew Chem Int Ed Engl, 2021, 60, 18680–18687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun Y, Kakinen A, Wan X, Moriarty N, Hunt CPJ, Li Y, Andrikopoulos N, Nandakumar A, Davis TP, Parish CL, Song Y, Ke PC and Ding F, Nano Today, 2021, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Osterlund N, Moons R, Ilag LL, Sobott F and Graslund A, J Am Chem Soc, 2019, 141, 10440–10450. [DOI] [PubMed] [Google Scholar]
- 53.Urbanc B, Borreguero JM, Cruz L and Stanley HE, Methods Enzymol, 2006, 412, 314–338. [DOI] [PubMed] [Google Scholar]
- 54.Peng S, Ding F, Urbanc B, Buldyrev SV, Cruz L, Stanley HE and Dokholyan NV, Phys Rev E Stat Nonlin Soft Matter Phys, 2004, 69, 041908. [DOI] [PubMed] [Google Scholar]
- 55.Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, Cascio D, Glabe C and Eisenberg D, Science, 2012, 335, 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pannuzzo M, Alzheimers Dement, 2022, 18, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nanga RP, Brender JR, Vivekanandan S and Ramamoorthy A, Biochim Biophys Acta, 2011, 1808, 2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nanga RP, Brender JR, Xu J, Hartman K, Subramanian V and Ramamoorthy A, J Am Chem Soc, 2009, 131, 8252–8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Young LM, Cao P, Raleigh DP, Ashcroft AE and Radford SE, J Am Chem Soc, 2014, 136, 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yin S, Ding F and Dokholyan NV, Nat Methods, 2007, 4, 466–467. [DOI] [PubMed] [Google Scholar]
- 61.Yin S, Biedermannova L, Vondrasek J and Dokholyan NV, Journal of Chemical Information and Modeling, 2008, 48, 1656–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Proctor EA, Ding F and Dokholyan NV, Wires Comput Mol Sci, 2011, 1, 80–92. [Google Scholar]
- 63.Ding F, Tsao D, Nie H and Dokholyan NV, Structure, 2008, 16, 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bunce SJ, Wang Y, Stewart KL, Ashcroft AE, Radford SE, Hall CK and Wilson AJ, Sci Adv, 2019, 5, eaav8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brodie NI, Popov KI, Petrotchenko EV, Dokholyan NV and Borchers CH, Sci Adv, 2017, 3, e1700479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Emperador A and Orozco M, J Chem Theory Comput, 2017, 13, 1454–1461. [DOI] [PubMed] [Google Scholar]
- 67.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S and Karplus M, J Comput Chem, 1983, 4, 187–217. [Google Scholar]
- 68.Lazaridis T and Karplus M, Curr Opin Struct Biol, 2000, 10, 139–145. [DOI] [PubMed] [Google Scholar]
- 69.Ding F, Borreguero JM, Buldyrey SV, Stanley HE and Dokholyan NV, Proteins, 2003, 53, 220–228. [DOI] [PubMed] [Google Scholar]
- 70.Ridgway Z, Zhang X, Wong AG, Abedini A, Schmidt AM and Raleigh DP, Biochemistry, 2018, 57, 3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Y, Liu YY, Zhao WH and Sun YX, Int J Biol Macromol, 2021, 193, 1–7. [DOI] [PubMed] [Google Scholar]
- 72.Brodie NI, Popov KI, Petrotchenko EV, Dokholyan NV and Borchers CH, PLoS Comput Biol, 2019, 15, e1006859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xiao X, Wang Y, Seroski DT, Wong KM, Liu R, Paravastu AK, Hudalla GA and Hall CK, Sci Adv, 2021, 7, eabf7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kabsch W and Sander C, Biopolymers, 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 75.Pandey BP, Zhang C, Yuan X, Zi J and Zhou Y, Protein Sci, 2005, 14, 1772–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bahar I, Atilgan AR and Erman B, Fold Des, 1997, 2, 173–181. [DOI] [PubMed] [Google Scholar]
- 77.Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF and Mark AE, Angew Chem Int Edit, 1999, 38, 236–240. [Google Scholar]
- 78.Su XY, Wang K, Liu N, Chen JW, Li Y and Duan MJ, Proteins-Structure Function and Bioinformatics, 2019, 87, 541–550. [DOI] [PubMed] [Google Scholar]
- 79.Miller C, Zerze GH and Mittal J, J Phys Chem B, 2013, 117, 16066–16075. [DOI] [PubMed] [Google Scholar]
- 80.Laghaei R, Mousseau N and Wei G, J Phys Chem B, 2010, 114, 7071–7077. [DOI] [PubMed] [Google Scholar]
- 81.Dong X, Qiao Q, Qian Z and Wei G, Biochim Biophys Acta Biomembr, 2018, 1860, 1826–1839. [DOI] [PubMed] [Google Scholar]
- 82.Zhang M, Ren B, Liu Y, Liang G, Sun Y, Xu L and Zheng J, ACS Chem Neurosci, 2017, 8, 1789–1800. [DOI] [PubMed] [Google Scholar]
- 83.Gallardo R, Iadanza MG, Xu Y, Heath GR, Foster R, Radford SE and Ranson NA, Nat Struct Mol Biol, 2020, 27, 1048–1056. [DOI] [PubMed] [Google Scholar]
- 84.Oostenbrink C, Soares TA, van der Vegt NF and van Gunsteren WF, Eur Biophys J, 2005, 34, 273–284. [DOI] [PubMed] [Google Scholar]
- 85.Kaminski GA, Friesner RA, Tirado-Rives J and Jorgensen WL, Journal of Physical Chemistry B, 2001, 105, 6474–6487. [Google Scholar]
- 86.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO and Shaw DE, Proteins, 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmuller H and MacKerell AD Jr., Nat Methods, 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.