

Abstract
Background:
Asthma exacerbations are major causes of morbidity in urban children and have significant short- and long-term consequences. Eosinophilic phenotype-directed therapies reduce asthma exacerbations in adults, but limited data are available in children and diverse populations. Furthermore, the molecular mechanisms underlying exacerbations prevented and persistent with immune-based therapies are not well understood.
Methods:
Urban children and adolescents (6-17 years) living in disadvantaged neighborhoods (n=290) with exacerbation-prone asthma (2+ exacerbations in previous year) and blood eosinophils ≥150 cells/mm3 were randomized 1:1 to mepolizumab (6-11 years: 40 mg; 12-17 years: 100 mg) or placebo injections every 4 weeks added to guideline-based care for 52 weeks. The primary outcome was the number of asthma exacerbations treated with systemic corticosteroids. Mechanisms of treatment response were assessed using nasal transcriptomic modular analysis. [ClinicalTrials.gov: NCT03292588]
Findings:
The annualized rate of asthma exacerbations was 0.96 (95% confidence interval [CI] 0.78-1.17) with mepolizumab and 1.30 (95% CI, 1.08-1.57) with placebo (rate ratio 0.73; 95% CI, 0.56-0.96; p=0.027). There were no significant differences in secondary outcomes, including time to first exacerbation, lung function, or composite asthma severity index (CASI). Baseline nasal airway transcriptomics identified pathways differentially associated with exacerbation risk: 3 eosinophil-associated inflammatory pathways were significantly suppressed by mepolizumab therapy and related to exacerbation risk in placebo-treated participants (p=0.0012-0.015). In contrast, 5 epithelial-associated inflammatory pathways were uniquely related to exacerbation risk in mepolizumab-treated participants (p=0.0035-0.030).
Interpretation:
Phenotype-directed therapy with mepolizumab in urban children with exacerbation-prone eosinophilic asthma reduced exacerbations, but did not impact other asthma outcomes. Mepolizumab response was related to pre-treatment expression of specific airway inflammatory pathways. Our evaluation linking clinical responses to airway expression of inflammatory pathways demonstrates the importance of integrated mechanistic assessments of therapeutic responses, particularly in Black and Hispanic children, who are at greatest risk for morbidity and mortality from asthma.
Introduction
Asthma exacerbations are common events, particularly in urban school-aged children living in disadvantaged neighborhoods,(1) and have important short- and long-term sequelae.(2-4) While conventional therapies, such as daily inhaled corticosteroids (ICS) +/− long-acting beta agonists (LABA), often improve asthma control, a significant proportion of children and adolescents remain inadequately controlled and at significant risk of exacerbations.(5, 6) The identification of strategies to prevent asthma exacerbations remains an important unmet need, particularly for underserved urban Black and Hispanic children with exacerbation-prone disease.(7)
The emergence of biologic therapies for asthma provides opportunities for phenotype-directed approaches to asthma management. Elevated levels of eosinophils in the blood(8, 9) and airway(10) have been identified as biomarkers for increased asthma exacerbation risk. Targeted therapy with mepolizumab, a humanized monoclonal antibody directed at interleukin (IL)-5, significantly reduces exacerbations in adults with severe, exacerbation-prone asthma and blood eosinophils ≥150 cells/mm3.(11, 12) However, data in children, adolescents, and racially and ethnically diverse populations are limited.(7, 13, 14)
Exacerbations continue to occur, albeit at lower rates, despite the use of targeted biologic therapies, and the mechanisms underlying these variable treatment responses are not established. While mepolizumab markedly depletes blood eosinophils, the relative change in blood eosinophils does not associate with treatment efficacy and more precise biomarkers that predict response have not been determined. Patterns of inflammation in the airway may explain asthma mechanisms with greater detail and serve as more precise biomarkers for treatment responsiveness. Transcriptome profiling allows for an unbiased assessment of these inflammatory patterns. We previously identified upper airway transcriptome modules, which are networks of co-expressed and functionally related genes, associated with asthma exacerbations in urban children,(15) and hypothesized that mepolizumab would reduce exacerbations through suppression of eosinophil-associated airway transcriptome modules. We further hypothesized that airway transcriptome profiling would identify alternative mechanisms associated with reduced responses to therapy.
To address these gaps in asthma care and to gain insight into mepolizumab’s clinical and mechanistic effects,(16) we performed the Mechanisms Underlying Asthma Exacerbations Prevented and Persistent with Immune Based Therapy: A Systems Approach Phase 2 (MUPPITS-2) randomized controlled trial (NCT03292588) to assess the efficacy, safety and mechanisms of phenotype-directed therapy with mepolizumab added to guidelines-based care for urban children with exacerbation-prone eosinophilic asthma.
Methods
Study design
The MUPPITS-2 study was a randomized, double-blind, placebo-controlled, parallel group trial conducted at 9 urban study sites in the United States. The study was approved by a central Institutional Review Board (Western IRB). The protocol and statistical analysis plan are available in the online supplement.
Participants
We enrolled urban children and adolescents 6-17 years of age with exacerbation-prone asthma and blood eosinophils ≥150 cells/mm3. To qualify, participants were required to live in pre-specified low-income census tracts, have an MD-diagnosis of asthma for at least one year, ≥2 exacerbations treated with systemic corticosteroids in the prior year, and require twice daily treatment with at least fluticasone propionate 250 micrograms or equivalent (6-11 years) or at least fluticasone/salmeterol 250/50 micrograms or equivalent (12-17 years). Parents provided written informed consent and children provided assent.
Randomization and masking
Eligible participants were enrolled into a 4-week run-in period and study teams assumed management of their asthma care according to a guideline-based treatment algorithm.(17) After the 4-week run-in, eligible participants were randomized in a 1:1 ratio to receive blinded adjunctive therapy with either mepolizumab (6-11 years: 40 mg; 12-17 years: 100 mg) or matching placebo by subcutaneous injection every 4 weeks for 52 weeks.
Outcome Measures
The primary outcome was the number of severe asthma exacerbations treated with systemic corticosteroids during the blinded study treatment period. Systemic corticosteroids were started following consultation with a study-site clinician according to previously published criteria.(18) Secondary outcome measures, not adjusted for multiplicity, included time to first asthma exacerbation, changes in Composite Asthma Severity Index (CASI)(19), physician-patient global assessment, lung function (FEV1 % predicted), and adverse events. Exploratory outcomes included blood eosinophil counts and nasal lavage cell counts and transcriptomics in relationship to the number of exacerbations and effects of treatment to modulate immune inflammatory pathways.
Participant Characterization
Spirometry, Impulse oscillometry (IOS), and measurement of fractional exhaled nitric oxide (FeNO) were performed according to American Thoracic Society/European Respiratory Society guidelines. Peripheral blood eosinophil counts were determined by a central laboratory and study teams were blinded to results (Marshfield Hospital, Marshfield, WI). Total serum immunoglobulin E (IgE) and specific IgE to aeroallergens were quantified by a commercial laboratory (Viracor, Lee’s Summit, MO). Nasal lavage samples were collected and processed for RNA sequencing using a previously published protocol and analyzed with a validated module analysis framework (see details in Supplementary Methods).(15)
Statistical Analyses
The target sample size of 290 children, reduced from 320 due to lower than anticipated drop-out, provided power of ≥0.9 with a two-sided type 1 error rate of 0.05 if the relative exacerbation rate ratio of the mepolizumab vs. placebo treatment was <0.6. This calculation assumed an exacerbation rate of 1.2 events per year for the inferior treatment and allowed for 15% drop-out. The estimated rate ratios and corresponding 95% Confidence Interval (CI) for the primary and secondary analyses of exacerbation rates were calculated using a negative binomial model with an offset term accounting for per-participant follow-up time in the intention-to-treat population. The model adjusted for randomization variables: study site, number of exacerbations in year prior to study (2 or ≥3), peripheral blood eosinophils (< or ≥400 cells/uL), BMI (< or ≥95th percentile), total serum IgE (< or ≥540 kUA/L) and treatment dose (40 or 100 mg). An interaction term was included when testing the difference in rate ratios between those who fit the omalizumab FDA-approved dosing table and those who did not. Time to exacerbation survival function estimates were generated using the Kaplan-Meier method and comparison of survival curves by treatment group by means of a Cox regression model adjusted for these covariates. Continuous secondary and exploratory endpoints were estimated using mixed model for repeated measures (and generalized additive mixed model for exacerbation seasonality), which included treatment arm as a fixed effect, and adjusted for randomization variables, with weeks specified as an unstructured covariance matrix for the repeated effects. With a mixed-model, no imputation of missing data was needed, as the estimates were unbiased under the missing at random assumption. The interaction term of weeks and treatment were used to estimate the least-square mean for each treatment group at each week, and the difference in least-square means was used to compare mepolizumab and placebo. All analyses presented are for the intention-to-treat population, clinical analyses were performed with SAS Version 9.4 (SAS Institute Inc, NC, USA) and mechanistic analyses with R Version 4.1.0 (R Foundation for Statistical Computing, Vienna, Austria). Notwithstanding, all figures were constructed using R package ggplot2. Non-normally distributed data were log-transformed prior to statistical analysis. Description of methods used in the nasal transcriptome analyses are provided in the Supplementary Methods.
Role of the funding source
The study was funded by the National Institute of Allergy and Infectious Diseases (NIAID) and an unrestricted grant from GlaxoSmithKline. The protocol was approved by the Inner-City Asthma Consortium steering committee, protocol review committee, data safety monitoring board, and single IRB. Study medication (mepolizumab) was donated by GlaxoSmithKline, which had the opportunity to comment on the study design, but had no role in the data collection, analysis, or interpretation. The authors are responsible for the study design, data collection, interpretation, manuscript preparation, and decision to submit.
Results
The trial was conducted from November 7, 2017 to April 20, 2021. Of 390 children who attended a screening visit, 290 were randomized to mepolizumab (n=146) or placebo (n=144) injections added to guideline-based care (Figure S1). There were 42 participants who terminated the trial early after randomization (mepolizumab (n=20) and placebo (n=22)). Baseline characteristics were similar between treatment groups (Table 1): 57% male, 70% Black race, 25% Hispanic ethnicity, and 37% had a BMI≥95th percentile. Asthma and type-2 inflammatory characteristics included mean FEV1% predicted 93%, FEV1/FVC 0.76, FEV1 % reversibility 14%, median blood eosinophils 430 cells/mm3, FeNO 34 ppb, and total serum IgE 404 kUA/L. Baseline asthma therapy was fluticasone/salmeterol 500/50 micrograms twice daily or equivalent for 58% of participants, and the mean number of oral corticosteroid courses in the year prior to study entry was 2.8.
Table 1.
Participant Characteristics
| Variables | Overall N = 2901 |
Mepolizumab N = 1461 |
Placebo N = 1441 |
|---|---|---|---|
| Demographics | |||
| Site | |||
| Boston | 21 (7%) | 11 (7%) | 10 (7%) |
| Chicago | 26 (9%) | 13 (9%) | 13 (9%) |
| Cincinnati | 31 (11%) | 17 (12%) | 14 (10%) |
| Dallas | 36 (12%) | 18 (12%) | 18 (12%) |
| Denver | 26 (9%) | 13 (9%) | 13 (9%) |
| Detroit | 41 (14%) | 19 (13%) | 22 (15%) |
| New York | 39 (13%) | 19 (13%) | 20 (14%) |
| St. Louis | 29 (10%) | 14 (10%) | 15 (10%) |
| Washington DC | 45 (15%) | 23 (16%) | 22 (15%) |
| Age (yr.) | 10.0 (9.0 - 13.0) | 10.0 (9.0 - 13.0) | 11.0 (9.0 - 13.0) |
| Sex (Male) | 167 (57%) | 76 (52%) | 91 (62%) |
| Race | |||
| Black/African American | 206 (70%) | 107 (73%) | 99 (67%) |
| White | 31 (11%) | 15 (10%) | 16 (11%) |
| More than One Race | 27 (9%) | 12 (8%) | 15 (10%) |
| Unknown or not reported | 27 (9%) | 12 (8%) | 15 (10%) |
| Other | 3 (1%) | 1 (1%) | 2 (1%) |
| Ethnicity (Hispanic or Latino) | 72 (24%) | 35 (24%) | 37 (25%) |
| Total monthly household income | |||
| Less than $1,250 / month | 114 (39%) | 54 (37%) | 60 (41%) |
| $1,251 - $2,500 / month | 100 (34%) | 47 (32%) | 53 (36%) |
| More than $2,501 / month | 75 (26%) | 43 (29%) | 32 (22%) |
| Don't know | 5 (1.7%) | 3 (2.0%) | 2 (1.4%) |
| Caretaker completed high school | 250 (86%) | 124 (85%) | 126 (87%) |
| One or more smokers in home | 85 (29%) | 37 (25%) | 48 (33%) |
| Baseline Clinical Characteristics | |||
| Weight (kg) | 47 (35 - 66) | 46 (32 - 67) | 48 (36 - 65) |
| Body-Mass-Index Percentile | 88 (63 - 98) | 87 (64 - 98) | 88 (61 - 98) |
| Body-Mass-Index > 95th percentile | 106 (37%) | 52 (36%) | 54 (38%) |
| FeNO (ppm) | 34 (18 - 64) | 29 (16 - 62) | 35 (20 - 66) |
| FEV1% predicted | 93 (17) | 94 (16) | 92 (18) |
| FEV1/FVC | 0.76 (0.09) | 0.77 (0.09) | 0.74 (0.10) |
| FEV1 Reversibility (%) | 14 (13) | 13 (11) | 15 (15) |
| Any skin test positive | 263 (95%) | 135 (97%) | 128 (93%) |
| Total number of skin tests positive | 5 (2 - 7) | 5 (2 - 8) | 4 (2 - 7) |
| Any allergen with IgE ≥ 0.35 kU/L | 277 (96%) | 137 (94%) | 140 (97%) |
| Total number allergens with IgE ≥ 0.35 kU/L | 7 (4 - 11) | 7 (4 - 11) | 7 (4 - 11) |
| Eosinophils (cells/uL) | 430 (300 - 690) | 400 (300 - 655) | 455 (300 - 700) |
| Total Serum IgE (KUA/L) | 404 (172 - 1,008) | 390 (170 - 914) | 416 (174 - 1,112) |
| Baseline Asthma Characteristics | |||
| Composite Asthma Severity Index (CASI) | 6.9 (2.7) | 6.9 (2.8) | 7.0 (2.6) |
| Treatment Step | |||
| Step 3. Fluticasone 250 mcg bid | 35 (12%) | 18 (12%) | 17 (12%) |
| Step 4. Fluticasone 250 mcg bid plus LABA | 87 (30%) | 39 (27%) | 48 (33%) |
| Step 5. Fluticasone 500 mcg bid plus LABA | 169 (58%) | 90 (61%) | 79 (55%) |
| Number of exacerbations in year prior | |||
| 2 | 169 (58%) | 87 (59%) | 82 (57%) |
| 3 or more | 122 (42%) | 60 (41%) | 62 (43%) |
| Number of oral steroids burst in year prior | 2.8 (1.5) | 2.7 (1.4) | 2.8 (1.5) |
| Number of hospitalizations in year prior | |||
| 0 | 156 (54%) | 74 (50%) | 82 (57%) |
| 1 | 81 (28%) | 40 (27%) | 41 (28%) |
| 2 | 41 (14%) | 26 (18%) | 15 (10%) |
| 3 or more | 13 (4.5%) | 7 (4.8%) | 6 (4.2%) |
n (%); Median (IQR) or Mean (SD)
The primary outcome of the annualized rate of asthma exacerbations treated with systemic corticosteroids was 0.96 (95% confidence interval [CI] 0.78-1.17) with mepolizumab and 1.30 (95% CI, 1.08-1.57) with placebo (Rate Ratio 0.73; 95% CI, 0.56-0.96; p=0.027; Figure 1A).
Figure 1. Impact of Mepolizumab on Asthma Exacerbations.
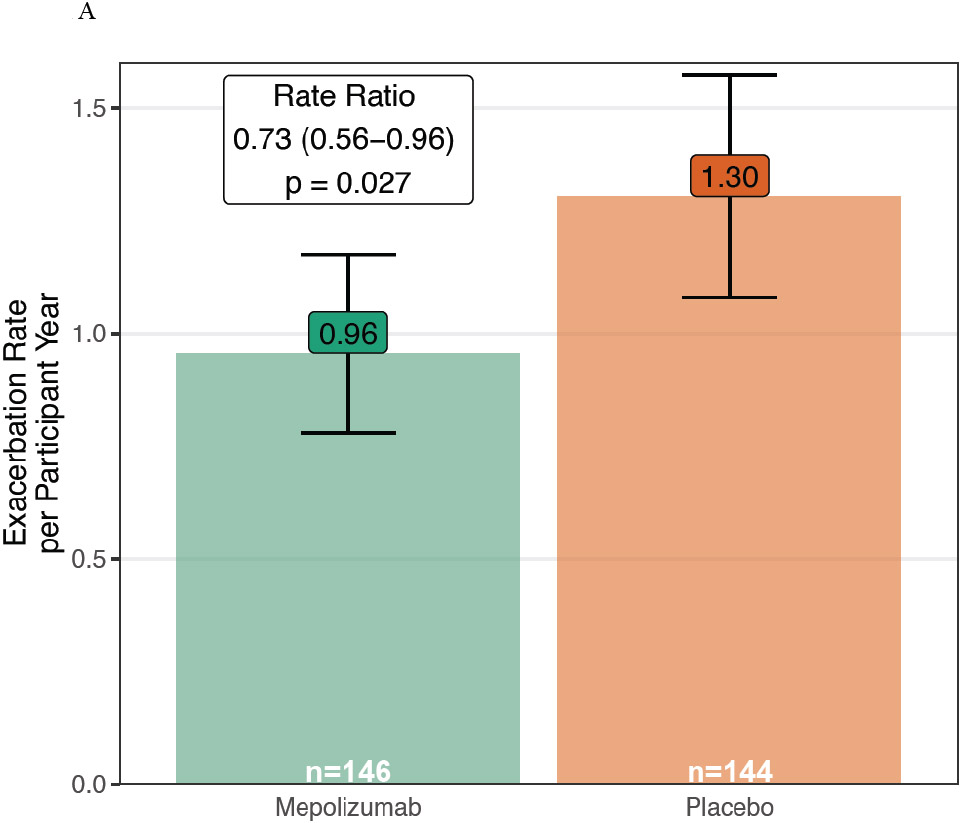
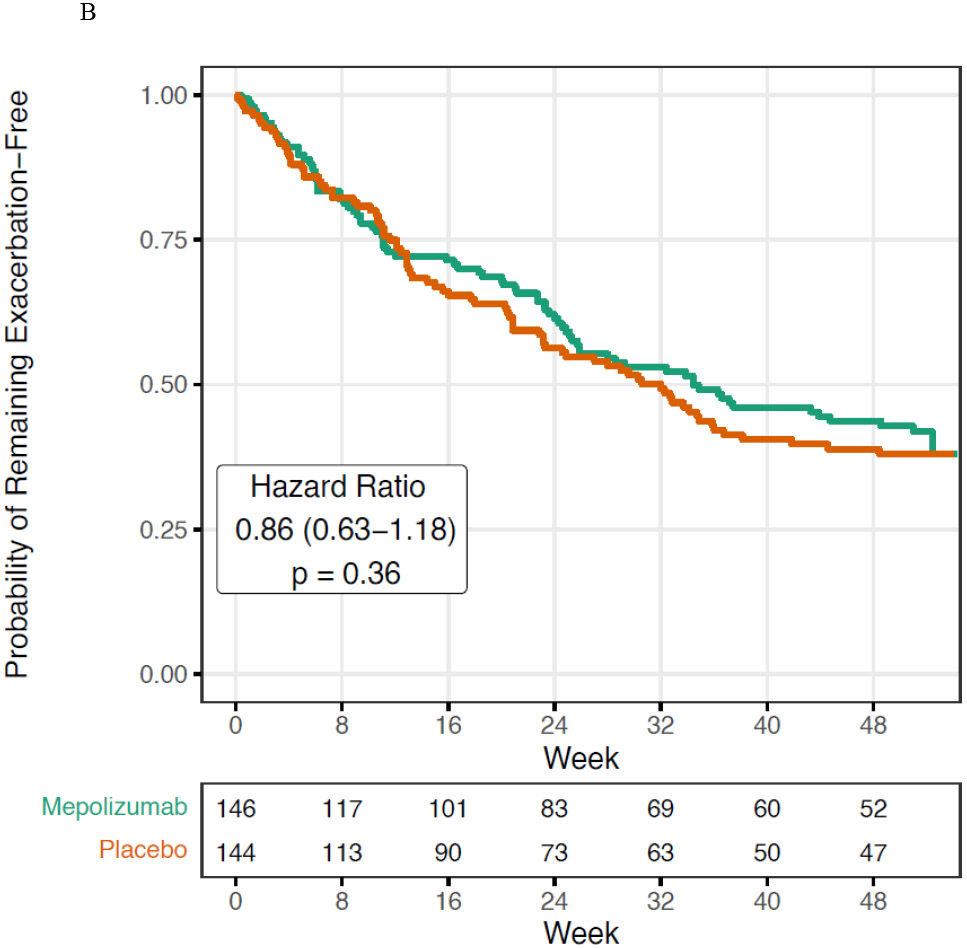
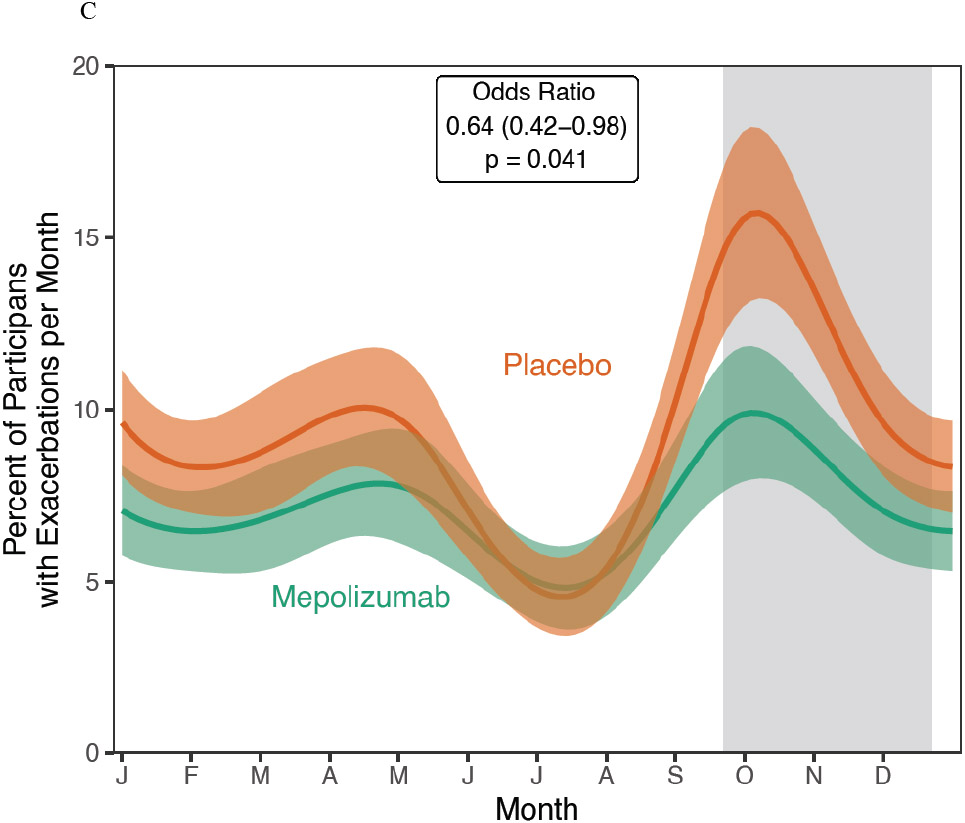
A. Asthma exacerbations were significantly reduced in the mepolizumab vs. placebo groups added to guideline-based care. B. The time to first asthma exacerbation was not significantly altered by mepolizumab. C. In a post hoc analysis, a seasonal pattern of asthma exacerbation was observed with a blunting of fall exacerbations in the mepolizumab treated participants.
The time to first asthma exacerbation was not significantly different between treatment groups (Hazard Ratio 0.86 (95% CI 0.63-1.18), p=0.36, Figure 1B). There were no between-group differences in FEV1% predicted, FEV1/FVC, or measures of IOS during the course of the study (Figure S2A-E). The CASI improved from baseline in both the mepolizumab and placebo groups during study follow up; however, there was no significant difference between treatment groups (Figure S3). Similarly, 85% of mepolizumab and 89% of placebo patients reported their asthma was moderately or significantly improved via the patient global assessment(12) at the end of study (Table S1, OR 0.72 (95% CI 0.42-1.24), p=0.24). Study clinicians completed the same assessment and reported moderate or significant improvement in 66% of mepolizumab and 71% of placebo participants (Supplemental Table S2, OR 1.01 (95% CI 0.62-1.64), p=0.97). When MUPPITS-2 was designed, omalizumab was the only approved biologic in children, so we included a secondary analysis based upon whether participants met FDA-approved omalizumab dosing criteria; the impact of mepolizumab on exacerbation risk did not differ based upon whether participants fit the omalizumab FDA-approved dosing table or not (interaction p-value 0.41).
Mepolizumab significantly reduced blood eosinophil counts, (Figure S4A, Difference at the end of the study −299 (95% CI −363, −235, p<0.0001) which remained unchanged in the placebo group. Similarly, nasal eosinophil percentage was significantly decreased in mepolizumab-treated participants, although to a lesser degree than blood eosinophils (Figure S4B, Difference at the end of the study −13.4 (95% CI −20.5, −6.3, p=0.0003). FeNO was not significantly changed by therapy (Figure S5).
As observed in prior studies in children, a post hoc analysis identified a strong seasonal exacerbation pattern was observed in the placebo group.(18, 20, 21) This seasonal pattern was significantly altered by mepolizumab (Figure 1C, p=0.0006), particularly with a blunting of the fall exacerbation peak (OR 0.64 (95% CI 0.42-0.98), p=0.041).
The majority of study follow up (79.1%) occurred prior to the COVID-19 pandemic (Table S3). The overall exacerbation rate was 58% lower during the COVID-19 pandemic; however, the relative reduction in the exacerbation rate observed with mepolizumab was not different prior to and during the pandemic.
Mepolizumab treatment was generally well tolerated; there were modest between-group differences in the adverse events reported by participants except for higher rates of injection-site reactions associated with mepolizumab (Table 2). Five episodes of anaphylaxis occurred (mepolizumab 3, placebo 2), none related to study therapy. One death occurred in the mepolizumab group during the trial due to an acute, severe asthma exacerbation leading to cardiopulmonary arrest.
Table 2.
Study Therapy Related Adverse Events (Incidence > 3%)
| Type of Event | Mepolizumab N = 146 n (%) |
Placebo N = 144 n (%) |
|---|---|---|
| Any Treatment Emergent AE | 42 (28.8) | 16 (11.1) |
| Injection site reactions | 19 (13.0) | 7 ( 4.9) |
| Skin and subcutaneous tissue disorders | 10 ( 6.8) | 1 ( 0.7) |
| Gastrointestinal disorders | 7 ( 4.8) | 3 ( 2.1) |
| Changes in Labs or Vital Signs | 5 ( 3.4) | 3 ( 2.1) |
| Nervous system disorders * | 7 ( 4.8) | 0 ( 0.0) |
Include headache, dizziness, and syncope
In pre-specified exploratory analyses, nasal lavage RNA-sequencing at baseline (week 0) and end of study (week 52) was utilized to identify molecular patterns associated with exacerbation risk and treatment responses. Whole genome transcriptome data were summarized using a previously defined and validated repertoire of 52 cell-associated nasal gene networks (modules), a subset of which were previously linked to risk for asthma exacerbations in urban children (Table S4).(15) Multivariate partial least squares regression (PLS-R) identified 12/52 modules for which expression values at baseline were related to exacerbation number during the course of the study in the placebo and/or mepolizumab treatment groups (Figure 2). Univariate negative binomial modeling of exacerbation rates as a function of baseline module expression yielded similar results (Table 3; Figure S6).
Figure 2. Nasal gene module expression levels are related to exacerbation rate and treatment.

Twelve gene expression modules measured from nasal lavage at the randomization visit, week 0, showed a significant relationship to the primary clinical outcome, the number of exacerbations during the course of the study, in either the placebo and/or mepolizumab treatment groups by multivariate partial least squares regression. Shown are the weights for each of the 12 modules and for each treatment group. Orange indicates a weight value for the placebo group and green for the mepolizumab group. A positive weight (indicated with a bar on the right of center) indicates a positive association with the number of exacerbations, a negative weight (left of center) indicates an inverse association. Weight values can range from 0-1 and in part reflect the relative importance of each module in modeling exacerbation number. Module annotations and cell associations are listed. Eight of these 12 modules showed differential expression from week 0 to week 52 in the mepolizumab group, whereas no modules were differentially expressed from week 0 to week 52 in the placebo group. Significance of differential expression is indicated. No significant change in expression is indicated by “ns”. Downward arrows indicate a decrease in expression, upward arrows an increase. The number of arrows indicates the significance level: one arrow, p<0.05; two arrows, p<0.01; 3 arrows, p<0.001. Further details about each module are in table S4.
Table 3:
Baseline Module Expression and Exacerbation Rates.
| Cell Association | Module Annotation | Mepolizumab N=125 |
Placebo N = 124 |
||
|---|---|---|---|---|---|
| RR | p-value | RR | p-value | ||
| Eosinophil | T2 inflammation | 1.10 (0.89-1.35) | 0.36 | 1.25 (1.05-1.5) | 0.015 |
| Eosinophil | Eicosanoid metabolism | 1.15 (0.84-1.57) | 0.37 | 1.62 (1.21-2.2) | 0.0012 |
| Eosinophil | Cytoplasmic proteins | 1.19 (0.77-1.82) | 0.42 | 1.74 (1.21-2.52) | 0.0027 |
| Eosinophil and Epithelium | Activation, mucus secretion | 1.29 (1.06-1.58) | 0.014 | 1.20 (1.00-1.44) | 0.039 |
| Epithelium | Keratinization, tight junctions | 1.32 (1.09-1.61) | 0.0053 | 1.11 (0.90-1.39) | 0.30 |
| Epithelium | EGFR signaling cell-cell adhesion | 1.34 (1.10-1.64) | 0.0049 | 1.10 (0.90-1.36) | 0.31 |
| Epithelium | Cilia function, IL33 response | 1.25 (1.04-1.51) | 0.020 | 1.11 (0.92-1.33) | 0.24 |
| Epithelium | Extracellular matrix production | 1.41 (1.12-1.77) | 0.0035 | 1.06 (0.86-1.32) | 0.58 |
| Epithelium | TGFB/SMAD3 cell differentiation | 1.36 (0.98-1.89) | 0.077 | 1.04 (0.74-1.45) | 0.83 |
| Epithelium | Tissue kallikreins, IL23/IL17 axis | 1.24 (1.02-1.51) | 0.030 | 1.03 (0.81-1.31) | 0.82 |
| Neutrophil | Neutrophil chemotaxis | 0.72 (0.51-1.02) | 0.062 | 0.70 (0.51-0.94) | 0.011 |
| Neutrophil | Type 1 IFN regulation | 0.76 (0.58-1.00) | 0.050 | 0.74 (0.58-0.95) | 0.0099 |
Shown are results of negative binomial regression of exacerbation number compared to baseline nasal lavage module expression on a log2 scale for each module depicted in Figure 2. Rate Ratios [RR] represent the estimated rate ratio for a one unit increase in log2 module expression (i.e. doubling of expression). Bolded values correspond to a p≤0.05. Modules are listed by their cell association and annotation. Further details about each module are in Table S4.
Baseline expression values of 3 eosinophil-associated modules functionally representative of T2 inflammation, eicosanoid metabolism, and cytoplasmic proteins, were associated with increased exacerbation risk in placebo, but not mepolizumab treated participants. Rate Ratios [RR]s, representing the estimated exacerbation rate ratio for a doubling of module expression of these modules, ranged from 1.25-1.74 in the placebo group (Table 3, p=0.0012-0.015). Furthermore, these 3 modules were significantly downregulated by mepolizumab from week 0 to week 52 (Figure 2, Table S5; Fold Changes [FC]s=0.77-0.90, p=0.00017-0.011), whereas they remained unchanged in the placebo group (p>0.05).
In contrast, baseline expression of 5 epithelial-associated modules were significantly related to exacerbation risk in the mepolizumab, but not placebo, group (RRs=1.24-1.41; p=0.0035-0.030); these modules represent multiple functional and inflammatory pathways of the epithelium including keratinization and tight junctions, epidermal growth factor receptor (EGFR) signaling and cell-cell adhesion, interleukin (IL)33 response and cilia function, extracellular matrix (ECM) production, and induction of tissue kallikreins and IL23/IL17 signaling. A sixth epithelial module identified by PLS-R, labeled transforming growth factor beta (TGFβ)/SMAD3 associated cell differentiation, showed a similar trend (RR=1.36; p=0.077). Five of these six modules were significantly upregulated by mepolizumab therapy (FCs=1.25-1.37, p=0.00016-0.0089); all were unchanged by placebo.
Uniquely, one module related to eosinophil activation and mucus hypersecretion (linked to both eosinophils and the epithelium) was positively associated with exacerbation risk in both treatment groups (RR=1.20-1.29, p=0.014-0.039) and was unchanged by therapy. Two modules related to type 1 IFN regulation and neutrophil chemotaxis were inversely associated with exacerbations in both groups (RR=0.70-0.76, p=0.0099-0.062) and were unchanged by therapy.
In addition to providing important mechanistic information, the nasal epithelial module expression values more accurately defined exacerbation risk and distinguished treatment effects than previously studied biomarkers including nasal cell percentages, blood eosinophils, pulmonary functions, and FeNO (Table S6). While nasal eosinophils numerically related to a increased exacerbation risk in the placebo group, and nasal epithelial cells were related to a increased exacerbation risk in the mepolizumab group, neither was statistically significant. Moreover, the eosinophil associated eicosanoid metabolism and cytoplasmic protein modules remained significantly associated with exacerbation numbers in the placebo group after adjustment for cell percentage (p<0.05); only the T2 inflammation module did not retain significance independent of cell percentage (p=0.26). Similarly, the epithelial associated keratinization and tight junctions, EGFR signaling and cell-cell adhesion, IL33 and cilia production, and ECM production modules remained significantly associated with the exacerbation number in the mepolizumab group after adjustment for cell percentage (p<0.05); the tissue kallikreins, IL23/IL17 axis module did not retain significance adjusted for cell percentages (p=0.12). Blood eosinophils (Figure S7), FEV1 % predicted, and FENO did not show significant relationships with exacerbations rates, while lower FEV1/FVC % predicted related to higher exacerbation rates in both groups (Table S6).
Discussion
Phenotype-directed therapy with mepolizumab added to guideline-based care reduced recurrent exacerbations in 6 to 17-year-old urban children with eosinophilic exacerbation-prone asthma. We did not observe significant between treatment differences in lung function as assessed by spirometry or impulse oscillometry, composite asthma severity index, or physician-patient global assessments. Notably, there were significant improvements in asthma control in both mepolizumab- and placebo-treated participants, highlighting the effectiveness of clinical-trial associated interventions that promote adherence to guideline-based care in high-risk children.
Our incorporation of longitudinal nasal transcriptomic analyses to more precisely characterize mepolizumab treatment responsiveness provided a significant advance to the understanding of mepolizumab’s effects on asthma exacerbations. These analyses provided critical insights into mechanisms underlying the response to mepolizumab therapy, and identified those inflammatory pathways that contribute to exacerbations despite reducing eosinophil-related inflammation. As anticipated, higher baseline expression of inflammatory pathways involving the eosinophil, inclusive of canonical T2 inflammation and eicosanoid metabolism, as well as eosinophil cytoplasmic proteins, identified risk for more exacerbations in the placebo group and better response to mepolizumab treatment. However, components of eosinophil activation associated with airway mucus hypersecretion persisted despite mepolizumab therapy and were associated with a continued exacerbation risk. These observations suggest that despite mepolizumab’s overall reduction in eosinophil numbers and T2 inflammation, refractory mechanisms of eosinophils and epithelium regulating mucins contribute to exacerbation risk and incomplete responses to mepolizumab. Indeed, our prior work has identified this pathway as key to asthma exacerbation progression and lower lung function.(15, 22, 23) Similarly, mucus overproduction has been established as having a key role in severe asthma in adults(24, 25) and is only partially related to airway eosinophil numbers.
A novel observation of our study was that elevated baseline expression of multiple non-T2 inflammatory pathways from the epithelium identified risk for exacerbations in the mepolizumab group, and expression of many of these pathways increased while on mepolizumab therapy. These epithelial pathways have been previously identified to underlie aspects of both viral and pollution triggered exacerbations in urban children(15, 26) and can occur independent of T2 inflammation. These observations suggest that when T2 inflammation is dampened with mepolizumab, epithelial inflammatory pathways can promote disease activity and may even show reciprocal elevation. Furthermore, adverse environmental exposures in urban children driving these epithelial inflammatory pathways may partially account for the limited therapeutic response observed in this population.
Additionally, these findings highlight the complexity of treating heterogeneous inflammatory pathways that exist in asthma, even among individuals meeting accepted criteria for an eosinophilic phenotype, and may help explain the persistent risk for exacerbations, albeit at lower rates, observed across studies of T2-targeted biologics. Perhaps most importantly, our results highlight key molecular pathways that can be considered for future interventional strategies, such as mucus hypersecretion, the kallikrein-kinin system, extracellular matrix overproduction, EGFR signaling, and TGF-β/SMAD signaling. Our data also highlight the need to tailor therapies towards each individual’s airway inflammatory profile, including the potential importance of targeting multiple inflammatory pathways and/or their proximal drivers.
Post hoc analyses identified significant seasonal variation in exacerbations with the most prominent mepolizumab treatment effect in the fall. The overall effects of mepolizumab therapy on exacerbations were more modest than expected despite similar substantial reductions in blood eosinophils to those in prior mepolizumab trials in adults and children,(11, 27) and also significant reductions, although to a lesser extent than blood, in nasal airway eosinophils. In contrast to our study, previous trials performed in adults, had low numbers of Black and Hispanic participants. Importantly, the social and environmental determinants of health facing disadvantaged urban Black and Hispanic children are unique relative to populations studied in prior mepolizumab trials. Furthermore, the pathways driving T2-high, eosinophilic asthma may differ between children and adults. For example, adult participants with childhood-onset vs. adult-onset eosinophilic asthma have been less responsive to benralizumab, another biologic therapy targeting IL-5.(16, 28) Our findings also raise the possibility that exacerbations in adults are more commonly driven by eosinophils whereas other inflammatory processes are instrumental in children.
Strengths of our study include diverse participants at greatest risk of asthma morbidity, predominantly Black and Hispanic children living in disadvantaged urban communities, an understudied population. Another study strength was high retention and adherence to injections despite a portion of the trial occurring during the COVID-19 pandemic. Further, integration of airway transcriptomic network analyses into this multi-site pediatric clinical trial enabled the identification of novel mechanisms of response and non-response to mepolizumab therapy.
A study limitation is the use of nasal airway samples as a proxy for lower airway disease; however this is mitigated by the direct relevance of these pathways to asthma outcomes.(15, 29, 30) The COVID-19 pandemic impacted the frequency of asthma exacerbations; however, this did not significantly impact the observed treatment effects of mepolizumab.
In conclusion, in urban children and adolescents with exacerbation-prone eosinophilic asthma, adjunctive therapy with mepolizumab reduced asthma exacerbations, but did not significantly impact other asthma outcomes. Airway transcriptomic analyses identified primarily eosinophil and epithelial associated molecular pathways underlying differential clinical responses to mepolizumab and have identified potential future targets to more precisely and effectively diminish disease burden from exacerbations in these high-risk youth. Our findings also highlight the importance of evaluating treatment responses for biologics, and other interventions, in Black and Hispanic children, populations often underrepresented in clinical trials and at greatest risk for morbidity and mortality from asthma.
Supplementary Material
Research in context.
Evidence before this study:
Black and Hispanic children living in urban environments in the US experience an excess burden of morbidity and mortality from asthma. Phenotype directed biologic therapies for eosinophilic asthma have revolutionized treatment for adults with severe disease, but data are lacking in children and diverse populations. Furthermore, precision biomarkers for responsiveness to these therapies are needed.
Added value of this study:
Eosinophil targeted therapy in urban children and adolescents living in disadvantaged neighborhoods significantly reduced asthma exacerbations, although to a lesser extent than previously observed in adults, but did not impact other asthma outcomes. Uniquely, we leveraged airway transcriptomic analyses to identify inflammatory pathways underlying differential responses to mepolizumab therapy in these children. Notably, multiple eosinophil and epithelial associated inflammatory pathways were identified as drivers of differential exacerbation risk in placebo and mepolizumab treated participants.
Implications of all the available evidence:
These findings highlight the importance of evaluating treatment responses for biologics and other interventions initially evaluated in adults in well-powered pediatric clinical trials. Furthermore, it is essential to evaluate treatment responses in Black and Hispanic individuals, those at greatest risk of morbidity and mortality from asthma and often underrepresented in clinical trials. Moreover, airway transcriptomic profiling provides detailed molecular insights into treatment response and non-response, thereby identifying essential novel biomarkers and disease mechanisms.
Acknowledgments:
Research reported in this publication was supported by NIH-NIAID under Award Numbers 5UM1AI114271, UM1AI160040, and UM2AI117870. Additional support was provided through Award Numbers UL1TRG01422, UL1RR025741, UL1TR000150, UL1TR001422, UL1 TR002535, UL1TR001876, and 5UL1TR001425-03 as well as Research reported in this publication was supported by NIH-NIAID under Award Numbers 5UM1AI114271 and UM2AI117870. Additional support was provided through Award Numbers UL1TRG01422, UL1RR025741, UL1TR000150, UL1TR001422, UL1 TR002535, UL1TR001876, and 5UL1TR001425-03, as well as an unrestricted grant from Glaxo Smith Kline. NIAID project scientists participated collaboratively in study design, data analysis and interpretation and writing of the report. The manuscript content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Funding:
The study was funded by the National Institute of Allergy and Infectious Diseases (NIAID) and an unrestricted grant from GlaxoSmithKline.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: All authors, with the exception of P. Becker, L. Gagalis and P. Gergen report grants from NIH/NIAID during the conduct of study. C. Visness, A. Calatroni, S. Wellford, L. Gagalis, P. Gergen and P. Becker have nothing to disclose outside the submitted work. C. Dutmer reports personal fees from Horizon Therapeutics and Enzyvant Therapeutics outside the submitted work. M. Altman reports personal fees from Sanofi-Regeneron outside the submitted work. W. Busse reports personal fees from Boston Scientific, Novartis, Glaxo SmithKline, Genentech, Sanofi/Genzyme, AstraZeneca, Teva, Regeneron and Elsevier outside the submitted work. M. Gill reports an honorarium for and support for travel to the 2017 AAAAI meeting during the conduct of study and monetary compensation from the American Academy of Pediatrics for her work teaching the biannual Pediatrics board review course, PREP The Course. K. Hershey reports grants from Adare, during the conduct of the study. D. Jackson reports personal fees from Novartis, Pfizer, Regeneron, AstraZeneca, Sanofi and Vifor Pharma, grants and personal fees from GlaxoSmithKline and grants from NIH/NHLBI, outside the submitted work. M. Kattan reports personal fees from Regeneron, outside the submitted work. R. Gruchalla reports government employment from Center for Biologics Evaluation and Research as well as personal fees from Consulting Massachusetts Medical Society, outside the submitted work. A. Liu reports personal fees from Phadia ThermoFisher as consulting honoraria, grants and non-financial support from ResMed/Propeller Health, non-financial support from Revenio, grants and personal fees from Avillion and personal fees from Labcorp, outside the submitted work. S. Teach reports grants from NIH/NICHD, NIH/NHLBI and EJF Philanthropies as well as personal fees from Uptodate, outside the submitted work. J. Pongracic reports provision of study drug for another asthma research study from Glaxo SmithKline and Boehringer-Ingelheim and provision of study drug for another asthma research study and for food allergy research studies from Genentech/Novartis, outside the submitted work.
Contributor Information
Prof Daniel J Jackson, Departments of Pediatrics and Medicine, University of Wisconsin School of Medicine and Public Health, Madison, WI.
Prof Leonard B Bacharier, Department of Pediatrics, Monroe Carell Jr Children’s Hospital at Vanderbilt, Nashville, TN.
Peter J Gergen, National Institute of Allergy and Infectious Diseases, Bethesda, MD.
Lisa Gagalis, National Institute of Allergy and Infectious Diseases, Bethesda, MD.
Agustin Calatroni, Rho Federal Systems Division, Inc., Durham, NC.
Stephanie Wellford, Rho Federal Systems Division, Inc., Durham, NC.
Prof Michelle A Gill, Department of Pediatrics, Washington University, St Louis, MO.
Jeffrey Stokes, Department of Pediatrics, Washington University in St Louis, St Louis, MO.
Prof Andrew H Liu, Pediatric Pulmonary and Sleep Medicine, Children’s Hospital Colorado and University of Colorado School of Medicine, Aurora, CO.
Prof Rebecca S Gruchalla, Department of Pediatrics, University of Texas Southwestern Medical Center, Dallas, TX.
Robyn T Cohen, Department of Pediatrics, Boston University School of Medicine, Boston, MA.
Melanie Makhija, Division of Allergy and Immunology, Ann and Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL.
Prof Gurjit K Khurana Hershey, Department of Pediatrics, University of Cincinnati College of Medicine; Division of Asthma Research, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH.
Prof George T O’Connor, Department of Medicine, Boston University School of Medicine, Boston, MA.
Prof Jacqueline A Pongracic, Division of Allergy and Immunology, Ann and Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL.
Michael G Sherenian, Department of Pediatrics, University of Cincinnati College of Medicine; Division of Asthma Research, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH.
Katherine Rivera-Spoljaric, Department of Pediatrics, Washington University School of Medicine and St Louis Children's Hospital, St Louis, MO.
Prof Edward M Zoratti, Department of Medicine, Henry Ford Health System, Detroit, MI.
Prof Stephen J Teach, Children’s National Hospital, George Washington University School of Medicine and Health Sciences, Washington, DC.
Prof Meyer Kattan, Columbia University College of Physicians and Surgeons, New York, NY.
Cullen M Dutmer, Allergy and Immunology, Children’s Hospital Colorado and University of Colorado School of Medicine, Aurora, CO.
Haejin Kim, Department of Medicine, Henry Ford Health System, Detroit, MI.
Carin Lamm, Department of Pediatrics, New York Columbia University Medical Center, New York, NY.
William J Sheehan, Children’s National Hospital, George Washington University School of Medicine and Health Sciences, Washington, DC.
R Max Segnitz, University of Washington, Seattle, WA.
Kimberly A Dill-McFarland, University of Washington, Seattle, WA.
Cynthia M Visness, Rho Federal Systems Division, Inc., Durham, NC.
Patrice M Becker, National Institute of Allergy and Infectious Diseases, Bethesda, MD.
Prof James E Gern, Departments of Pediatrics and Medicine, University of Wisconsin School of Medicine and Public Health, Madison, WI.
Prof Christine A Sorkness, School of Pharmacy, University of Wisconsin-Madison, Madison, WI.
Prof William W Busse, Department of Medicine, University of Wisconsin School of Medicine and Public Health, Madison, WI.
Matthew C Altman, University of Washington, Seattle WA; Benaroya Research Institute, Seattle WA.
References
- 1.Zahran HS, Bailey CM, Damon SA, Garbe PL, Breysse PN. Vital Signs: Asthma in Children - United States, 2001-2016. MMWR Morbidity and mortality weekly report. 2018;67(5):149–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Byrne PM, Pedersen S, Lamm CJ, Tan WC, Busse WW. Severe exacerbations and decline in lung function in asthma. Am J Respir Crit Care Med. 2009;179(1):19–24. [DOI] [PubMed] [Google Scholar]
- 3.O'Brian AL, Lemanske RF Jr., Evans MD, Gangnon RE, Gern JE, Jackson DJ Recurrent severe exacerbations in early life and reduced lung function at school age. J Allergy Clin Immunol. 2012;129(4):1162–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yao TC, Wang JY, Chang SM, Chang YC, Tsai YF, Wu AC, et al. Association of Oral Corticosteroid Bursts With Severe Adverse Events in Children. JAMA pediatrics. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pongracic JA, Krouse RZ, Babineau DC, Zoratti EM, Cohen RT, Wood RA, et al. Distinguishing characteristics of difficult-to-control asthma in inner-city children and adolescents. J Allergy Clin Immunol. 2016;138(4):1030–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zoratti EM, Krouse RZ, Babineau DC, Pongracic JA, O'Connor GT, Wood RA, et al. Asthma phenotypes in inner-city children. J Allergy Clin Immunol. 2016;138(4):1016–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez Burchard E, Borrell LN. Need for Racial and Ethnic Diversity in Asthma Precision Medicine. N Engl J Med. 2021;385(24):2297–8. [DOI] [PubMed] [Google Scholar]
- 8.Price DB, Rigazio A, Campbell JD, Bleecker ER, Corrigan CJ, Thomas M, et al. Blood eosinophil count and prospective annual asthma disease burden: a UK cohort study. The Lancet Respiratory medicine. 2015;3(11):849–58. [DOI] [PubMed] [Google Scholar]
- 9.Teach SJ, Gergen PJ, Szefler SJ, Mitchell HE, Calatroni A, Wildfire J, et al. Seasonal risk factors for asthma exacerbations among inner-city children. J Allergy Clin Immunol. 2015;135(6):1465–73 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green RH, Brightling CE, McKenna S, Hargadon B, Parker D, Bradding P, et al. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet. 2002;360(9347):1715–21. [DOI] [PubMed] [Google Scholar]
- 11.Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380(9842):651–9. [DOI] [PubMed] [Google Scholar]
- 12.Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, et al. Mepolizumab Treatment in Patients with Severe Eosinophilic Asthma. N Engl J Med. 2014. [DOI] [PubMed] [Google Scholar]
- 13.Yancey SW, Ortega HG, Keene ON, Bradford ES. Efficacy of add-on mepolizumab in adolescents with severe eosinophilic asthma. Allergy Asthma Clin Immunol. 2019;15:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta A, Ikeda M, Geng B, Azmi J, Price RG, Bradford ES, et al. Long-term safety and pharmacodynamics of mepolizumab in children with severe asthma with an eosinophilic phenotype. J Allergy Clin Immunol. 2019;144(5):1336–42 e7. [DOI] [PubMed] [Google Scholar]
- 15.Altman MC, Gill MA, Whalen E, Babineau DC, Shao B, Liu AH, et al. Transcriptome networks identify mechanisms of viral and nonviral asthma exacerbations in children. Nat Immunol. 2019;20(5):637–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brusselle GG, Koppelman GH. Biologic Therapies for Severe Asthma. N Engl J Med. 2022;386(2):157–71. [DOI] [PubMed] [Google Scholar]
- 17.Kercsmar CM, Sorkness CA, Calatroni A, Gergen PJ, Bloomberg GR, Gruchalla RS, et al. A computerized decision support tool to implement asthma guidelines for children and adolescents. J Allergy Clin Immunol. 2019;143(5):1760–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Busse WW, Morgan WJ, Gergen PJ, Mitchell HE, Gern JE, Liu AH, et al. Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. N Engl J Med. 2011;364(11):1005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wildfire JJ, Gergen PJ, Sorkness CA, Mitchell HE, Calatroni A, Kattan M, et al. Development and validation of the Composite Asthma Severity Index-an outcome measure for use in children and adolescents. J Allergy Clin Immunol. 2012;129(3):694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szefler SJ, Mitchell H, Sorkness CA, Gergen PJ, O'Connor GT, Morgan WJ, et al. Management of asthma based on exhaled nitric oxide in addition to guideline-based treatment for inner-city adolescents and young adults: a randomised controlled trial. Lancet. 2008;372(9643):1065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnston NW, Johnston SL, Norman GR, Dai J, Sears MR. The September epidemic of asthma hospitalization: school children as disease vectors. J Allergy Clin Immunol. 2006;117(3):557–62. [DOI] [PubMed] [Google Scholar]
- 22.Altman MC, Calatroni A, Ramratnam S, Jackson DJ, Presnell S, Rosasco MG, et al. Endotype of allergic asthma with airway obstruction in urban children. J Allergy Clin Immunol. 2021;148(5):1198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altman MC, Flynn K, Rosasco MG, Dapas M, Kattan M, Lovinsky-Desir S, et al. Inducible expression quantitative trait locus analysis of the MUC5AC gene in asthma in urban populations of children. J Allergy Clin Immunol. 2021;148(6):1505–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shrine N, Portelli MA, John C, Soler Artigas M, Bennett N, Hall R, et al. Moderate-to-severe asthma in individuals of European ancestry: a genome-wide association study. The Lancet Respiratory medicine. 2019;7(1):20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunican EM, Elicker BM, Gierada DS, Nagle SK, Schiebler ML, Newell JD, et al. Mucus plugs in patients with asthma linked to eosinophilia and airflow obstruction. The Journal of clinical investigation. 2018;128(3):997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med. 2014;190(12):1373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta A, Pouliquen I, Austin D, Price RG, Kempsford R, Steinfeld J, et al. Subcutaneous mepolizumab in children aged 6 to 11 years with severe eosinophilic asthma. Pediatr Pulmonol. 2019;54(12):1957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bleecker ER, Wechsler ME, FitzGerald JM, Menzies-Gow A, Wu Y, Hirsch I, et al. Baseline patient factors impact on the clinical efficacy of benralizumab for severe asthma. Eur Respir J. 2018;52(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poole A, Urbanek C, Eng C, Schageman J, Jacobson S, O'Connor BP, et al. Dissecting childhood asthma with nasal transcriptomics distinguishes subphenotypes of disease. J Allergy Clin Immunol. 2014;133(3):670–8 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kicic A, de Jong E, Ling KM, Nichol K, Anderson D, Wark PAB, et al. Assessing the Unified Airway Hypothesis in Children Via Transcriptional Profiling of the Airway Epithelium. J Allergy Clin Immunol. 2020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.