

Abstract
The dense network of capillaries composed of capillary endothelial cells (cECs) and pericytes lies in close proximity to all neurons, ideally positioning it to sense neuron- and glial-derived compounds that enhance regional and global cerebral perfusion. The membrane potential (VM) of vascular cells serves as the physiological bridge that translates brain activity into vascular function. In other beds, the ATP-sensitive K+ (KATP) channel regulates VM in vascular smooth muscle, which is absent in the capillary network. Here, with transgenic mice that expressed a dominant-negative mutant of the pore-forming Kir6.1 subunit specifically in brain cECs or pericytes, we demonstrated that KATP channels were present in both cell types and robustly controlled VM. We further showed that the signaling nucleotide adenosine acted through A2A receptors and the Gαs/cAMP/PKA pathway to activate capillary KATP channels. Moreover, KATP channel stimulation in vivo increased cerebral blood flow (CBF), an effect that was blunted by expression of the dominant-negative Kir6.1 mutant in either capillary cell type. These findings establish an important role for KATP channels in cECs and pericytes in the regulation of CBF.
INTRODUCTION
Normal brain function requires that every neuron receive an appropriate supply of oxygen, energy metabolites, and other nutrients through the cerebral vasculature. Cerebral blood flow (CBF) through the interconnected vascular network of surface arteries, penetrating arterioles, and the vast, complex mesh of capillaries is tuned by the precise and orchestrated action of neurovascular coupling mechanisms, which link blood flow to the metabolic needs of neurons. Capillaries account for a large majority of this vascular network and critically cover all brain regions (1), ideally positioning the capillary endothelial cells (cECs) and pericytes that constitute capillaries—and the astrocytic endfeet that completely cover them—in close contact with all neurons in the brain. In this context, we demonstrated that cECs can sense extracellular K+, a by-product of neural activity, through inward-rectifier K+ (Kir2.1) channels and respond by initiating a retrograde hyperpolarizing (electrical) signal that propagates upstream, causing dilation of feeding arterioles and thereby directing blood flow to the active brain region that initiated the signal (2).
Adenosine, a signaling nucleotide that arises through hydrolysis of astrocyte-released adenosine 5′-triphosphate (ATP) (3, 4) or that is directly released from neurons and glial cells in certain pathological settings (such as hypoxia or ischemia) (5–7), is a potent endogenous vasodilator that plays a central role in matching blood distribution to tissue metabolic demands (8). In many vascular systems, including the mesenteric circulation, the vasodilatory actions of adenosine are partly attributable to the activation of ATP-sensitive potassium (KATP) channels downstream of Gαs protein–coupled adenosine 2A (A2A) receptors (A2ARs). The resulting stimulation of adenylyl cyclase (AC) raises intracellular levels of cyclic adenosine 5′-monophosphate (cAMP); cAMP, in turn, binds to and activates cAMP-dependent protein kinase (PKA), which ultimately phosphorylates multiple sites in the KATP channel complex to promote its opening (9–12). The resulting KATP channel–mediated K+ efflux hyperpolarizes the cell membrane, closing voltage-gated Ca2+ channels and thereby inducing vessel dilation (13, 14).
KATP channels are potent regulators of the VM of vascular cells. In smooth muscle cells (SMCs), KATP channels are octamers of four pore-forming Kir6.1 subunits and four accessory sulfonylurea receptors (SUR2B) (15). Vascular KATP channel function is sensitive to the relative intracellular concentrations of ATP and adenosine 5’-diphosphate (ADP) ([ATP]/[ADP] ratio) (16). These channels are also stimulated by synthetic K+ channel openers (such as pinacidil) or endogenous vasodilators, including adenosine, vasoactive intestinal peptide, and calcitonin gene–related peptide (CGRP). They are inhibited by oral hypoglycemic sulfonylurea drugs such as glibenclamide (10, 13, 14, 17, 18), an antagonist of KATP channels whose selectivity has been established in multiple cell types, including pancreatic β cells, cardiac myocytes, and vascular SMCs (10). In the brain, however, SMCs may not be ideal sensors of endogenous vasoactive agents, specifically adenosine, because the density of SMC-containing arterioles is relatively low within the cortex. In contrast, neuron-juxtaposed brain cECs and pericytes express the genes encoding A2AR (Adora2a) and the vascular KATP channel subunits Kir6.1 (Kcnj8) and SUR2B (Abcc9). The high expression of Kcnj8 and Abcc9 in pericytes suggests that the corresponding Kir6.1/SUR2B-type KATP channel should be a major contributor to membrane currents (19–21). However, direct measurements of KATP channel currents in brain capillary pericytes and ECs are lacking.
Here, we demonstrated that the main cellular components of brain capillaries—ECs and pericytes—expressed functional KATP channels that influence resting VM. We further showed that capillary KATP channels responded to extracellular adenosine through the A2AR-Gαs-PKA pathway. Moreover, activation of KATP channels by adenosine or pinacidil in vivo increased CBF, and direct application of adenosine onto capillaries resulted in an elevation of capillary red blood cell (RBC) flux and upstream parenchymal arteriole dilation. Collectively, these findings establish the presence of functional KATP channels in the brain capillary network and demonstrate their potential physiological impact on brain hemodynamics.
RESULTS
Brain capillary cells express functional KATP channels
Multiple methodologies were used to ascertain whether brain capillaries express functional KATP channels, including patch-clamp electrophysiology on freshly isolated cECs and pericytes (Fig. 1A), VM measurements on pressurized retina preparations (Fig. 1B), and in vivo CBF measurements (Fig. 1, C and D). cECs, which are easily recognized by their characteristic tube-like shape, exhibited pronounced linear inward Kir2.1 currents at hyperpolarized potentials [K+ equilibrium potential (EK) = −23 mV at 60 mM external K+] (Fig. 1A). The identification of native pericytes was facilitated by the use of NG2-DsRed-BAC (neural/glial antigen 2–DsRed–bacterial artificial chromosome) transgenic mice, which express the DsRed fluorescent protein under control of the promoter/enhancer for chondroitin sulfate proteoglycan 4 (Cspg4), which encodes NG2 and is prominently expressed in SMCs and pericytes in the vasculature and in oligodendrocyte progenitor cells in the central nervous system. Freshly isolated pericytes were thus recognized as NG2-positive cells with protruding cell bodies and thin-stranded projections, the characteristic morphology of pericytes. We found that the current-voltage relationship of pericytes was unique and distinct from that of cECs and SMCs. When bathed in 60 mM K+, whole-cell currents in pericytes exhibited a nonlinear inward current and a modest outward current that was less than one-third of the SMC outward current at +40 mV (Fig. 1A and fig. S1, A and B). In contrast, cECs showed minimal outward currents positive to 0 mV.
Fig. 1. Experimental paradigm used to explore the expression of functional KATP channels in the capillary network.

(A) Schematic depiction of brain cEC and pericyte isolation for patch-clamp analysis. A small piece of brain somatosensory cortex was mechanically disrupted and filtered to yield capillary segments. Single capillary cells were released after enzymatic digestion and trituration. Because NG2-DsRed-BAC transgenic mice were used, pericytes were identified by DsRed fluorescence. Electrophysiological profiles of cECs and pericytes (black and red traces, respectively). (B) Illustration of the intact pressurized retina preparation (50 mmHg at the level of the ophthalmic artery) and measurement of VM in situ using sharp microelectrodes. The ophthalmic artery of an isolated mouse retina was cannulated, and the retina tissue was pinned down en face, allowing visualization of the entire superficial microvasculature and facilitating the impalement of a cell of interest (cEC or pericyte; inset). The phenotype of the impaled cell was confirmed by including iFluor 488–conjugated hydrazide in the sharp glass electrode. (C) CBF responses and blood pressure were monitored in the somatosensory cortex through a cranial window using laser Doppler flowmetry (LDF). (D) RBC flux and upstream parenchymal arteriole diameter were quantified in vivo using two-photon laser scanning microscopy (2PLSM). Scale bars, 10 μm.
Using the conventional whole-cell configuration of the patch-clamp technique, we tested whether the potent KATP channel opener pinacidil evokes KATP currents in isolated brain cECs and pericytes (Fig. 2, A to E). Cells were held at a VM of −70 mV, bathed in a 60 mM K+ solution, and dialyzed with a 140 mM K+ intracellular solution, which served to maximize the driving force for K+ and minimize currents through other channel types. Under these experimental conditions, K+ currents are inward (16). In cECs dialyzed with a low ATP concentration (0.1 mM) and a relatively high ADP concentration (0.1 mM), pinacidil increased whole-cell currents by an average of −26.1 ± 2.2 pA/pF (Fig. 2, B and F), an effect that was reversed by subsequent application of glibenclamide. PNU-37883, a putative selective antagonist of vascular KATP channels, also reversed pinacidil-induced currents in cECs dialyzed with the same [ATP]/[ADP] ratio (fig. S2A). Pinacidil-evoked, glibenclamide-sensitive currents in pericytes were ~2.6-fold greater than those in cECs under identical experimental conditions (Fig. 2, C and F). In contrast, glibenclamide-sensitive currents induced by pinacidil were very small in single cerebral pial artery SMCs and parenchymal arteriole SMCs (fig. S1, A and B). In SMCs from diverse vascular beds, including the mesenteric circulation, functional KATP channels require the presence of four Kir6.1 pore-forming subunits (15). We tested whether this was also the case in brain cECs and pericytes by using the Cre-Lox system to generate cell-specific dominant-negative (DN) Kir6.1 mice. In this model, a mutant Ki6.1-AAA subunit with a mutation in the pore (22) is expressed specifically in ECs (Cdh5 promoter) or pericytes (Pdgfrβ promoter) (fig. S3), resulting in nonfunctional KATP channels in the cell type of interest. In contrast to cells from cell-specific control (Cdh5-cre and Pdgfrβ-cre) mice, which exhibited robust increases in inward current in response to pinacidil that were completely eliminated by subsequent bath application of glibenclamide, none of the examined cECs isolated from Cdh5-DN-Kir6.1 mice (Fig. 2G) or pericytes isolated from Pdgfrβ-DN-Kir6.1 mice (Fig. 2, H and I) exhibited glibenclamide-sensitive currents in response to pinacidil. Despite this unresponsiveness, cECs and pericytes monitored at a holding potential of −70 mV exhibited very low baseline inward currents that were eliminated by subsequent addition of 100 μM Ba2+, a concentration selective for Kir2 channels. Glibenclamide-sensitive currents were intact in cECs isolated from pericyte-specific Pdgfrβ-DN-Kir6.1 mice and in pericytes isolated from EC-specific Cdh5-DN-Kir6.1 mice, reflecting the cellular specificity of each DN-Kir6.1 mouse line (fig. S4, A and B). Together, these findings indicate that the hyperpolarizing, glibenclamide-sensitive KATP current induced by pinacidil is primarily initiated in the capillary network and that the Kir6.1 subunit is a crucial element of this channel complex.
Fig. 2. Brain cECs and pericytes express functional KATP channels.

(A) Schematic diagram illustrating KATP channel stimulation by the synthetic opener pinacidil (PIN) and inhibition by the sulfonylurea glibenclamide (GLIB). (B and C) Representative traces showing GLIB (10 μM)–sensitive currents induced by PIN (10 μM) from a holding potential of −70 mV in freshly isolated cECs (B) (n = 9 cells per group) or pericytes (C) (n = 7 cells per group), recorded in the whole-cell configuration and dialyzed with 0.1 mM ATP/0.1 mM ADP. (D and E) Currents in cECs (black trace) and pericytes (red trace) recorded in the whole-cell configuration dialyzed with 3 mM ATP/0.1 mM ADP (n = 8 cECs or pericytes per group) (D) or in the perforated (cytoplasm intact) patch-clamp configuration (cECs, n = 8 cells per group; pericytes, n = 4 cells per group) (E). (F) Summary data showing PIN-induced current density in cECs and pericytes dialyzed with different ATP concentrations or recorded in the perforated patch configuration. Values are presented as means ± SEM (★P < 0.05; ★★P < 0.01; ★★★P < 0.001; Kruskal-Wallis test with post hoc Dunn’s test). (G and H) GLIB-sensitive currents in cECs (G) and pericytes (H) from Cdh5-DN-Kir6.1 and Pdgfrβ-DN-Kir6.1 mice, respectively (top), and from the corresponding Chd5-cre and Pdgfrβ-cre control mice (bottom), recorded in the whole-cell configuration and dialyzed with 0.1 mM ATP. BaCl2 (100 μM) was added at the end of experiments that used cells derived from Cdh5-DN-Kir6.1 and Pdgfrβ-DN-Kir6.1 mice (Cdh5-DN-Kir6.1, n = 8 cells; Chd5-cre, n = 7 cells; Pdgfrβ-DN-Kir6.1, n = 6 cells; Pdgfrβ-cre, n = 6 cells). (I) Summary data comparing PIN-induced current density in cECs and pericytes isolated from Cdh5-DN-Kir6.1 and Pdgfrβ-DN-Kir6.1 to that in the corresponding cell type from Cdh5-cre and Pdgfrβ-cre control mice. Values are presented as means ± SEM (★★P < 0.01; ★★★P < 0.001; Mann-Whitney test). Dotted lines in (B) to (E), (G), and (H) represent zero current level. For all conditions, external and internal K+ were 60 and 140 mM, respectively.
KATP channel activity is also influenced by the cytosolic [ATP]/[ADP] ratio. Specifically, KATP channels are activated by a low concentration of cytosolic ATP or by an elevated ADP concentration; thus, an increase in the [ATP]/[ADP] ratio would be expected to inhibit KATP channel activity. Consistent with this effect, KATP currents were ~5-fold smaller in cECs dialyzed with 3 mM ATP and 0.1 mM ADP ([ATP]/[ADP] ratio = 30) than those in cECs dialyzed with a 1:1 ratio. Similarly, raising the internal [ATP]/[ADP] ratio from 1 to 30 reduced the average KATP current in pericytes ~4.6-fold (Fig. 2, D and F). Last, measurement of whole-cell currents using the perforated patch configuration, in which the cytoplasmic composition remains undisturbed, revealed that currents evoked by pinacidil in intact cECs and pericytes were smaller than those observed after dialysis to create an internal [ATP]/[ADP] ratio of 30 (Fig. 2, E and F). These findings collectively support the idea that capillary cells express functional KATP channels that are activated by pinacidil and inhibited by glibenclamide, PNU-37883, and ATP. Furthermore, KATP channel currents in the absence of pinacidil (or other activating ligands) were too small to be detected in the perforated patch configuration, reflecting physiological intracellular [ATP]/[ADP] ratios.
Activation of KATP channels causes pronounced membrane potential hyperpolarization
The role of capillary KATP channels in controlling membrane potential was assessed in situ using intact pressurized retina preparations and sharp microelectrodes. In this preparation, the ophthalmic artery of an isolated retina is cannulated, and the retina tissue is pinned down en face, allowing visualization of the entire superficial microvascular network and enabling the impalement of cells of interest (Figs. 1B and 3, A to C). To determine whether we were recording from a pericyte (Fig. 3, D to F) or a cEC (Fig. 3, G to I), we included fluorescent iFluor 488–conjugated hydrazide in the glass microelectrode pipette. The resulting dye-filled cells (green) were identifiable on the basis of their morphology and were subsequently verified in a subset of experiments using NG2-DsRed mouse retinas, in which green dye can readily be visualized filling red pericytes (yellow in merged images; fig. S5, A to C) or underlying cECs (fig. S5, D to F). Note that, within our single recording duration (30 min to ~2 hours), dye from impaled cECs moved between adjacent cECs but demonstrated limited spread to overlying pericytes (Fig. 3I). In contrast, the dye remained predominantly in the cell body and projections of an impaled pericyte with no detectable movement to adjacent cECs or neighboring pericytes (Fig. 3, E and F). After the ophthalmic artery was cannulated and pressurized (to 50 mmHg), cells were impaled, pinacidil was applied, and VM was measured in cECs and pericytes using microelectrodes. In cECs, pinacidil induced a robust membrane hyperpolarization from a steady-state VM of −29.7 ± 1.9 to −73.9 ± 2.4 mV. Subsequent superfusion with glibenclamide depolarized endothelial VM to −24.7 ± 1.7 mV (Fig. 3J). Similar results were observed with pericytes, in which pinacidil hyperpolarized cells from −36.4 ± 2.3 to −82.4 ± 2.4 mV on average, an effect that was reversed by glibenclamide (−37.8 ± 4.2 mV) (Fig. 3K). These data indicate that activation of KATP channels in cECs and pericytes causes substantial membrane potential hyperpolarization.
Fig. 3. Activation of KATP channels causes membrane potential hyperpolarization.

(A) Representative wide-field image of the whole-mount en face pressurized retina preparation used for VM measurements. Rhodamine-isolectin was used to label the entire superficial microvasculature. (B and C) Higher-magnification images of the yellow inset in (A), depicting the vascular network stained with rhodamine-isolectin (B) and the impaled cell of interest (pericyte) filled with iFluor 488 hydrazide (C). (D) Stitched confocal z-stack image magnified from the yellow inset depicted in (A) and (B). (E) Confocal z-stack of the fill of a pericyte with iFluor 488 hydrazide with a sharp microelectrode. (F) Stitched z projection of spinning disk confocal images illustrating an impaled capillary pericyte in which fluorescent hydrazide labeling remained exclusively in the cell body and processes of the impaled cell. (G) Stitched confocal z-stack image of the microvascular network, labeled with rhodamine-isolectin. (H) Impaled cell of interest (cEC) filled with iFluor 488 hydrazide. (I) In impaled cECs, iFluor 488–conjugated hydrazide diffused to coupled adjacent cells, staining mainly cell bodies. Retinal tissue was stained with rhodamine-isolectin at the end of VM measurements. Scale bars, 1 mm (A), 100 μm (B and C), and 20 μm (D to I). (J and K) Representative VM recordings and summary data from cECs (J) (n = 3 to 10 cells per group) and pericytes (K) (n = 3 to 13 cells per group) in the pressurized (50 mmHg at the level of the ophthalmic artery) retina vasculature illustrating the robust hyperpolarization induced by pinacidil (PIN; 10 μM) and subsequent block by glibenclamide (GLIB; 20 μM). Data are presented as means ± SEM (★P < 0.05; ★★P < 0.01; Kruskal-Wallis test with post hoc Dunn’s test). Dashed line represents 0-mV baseline before impalement of the cell.
Adenosine activates capillary KATP channels through A2ARs and PKA
Adenosine is a potent endogenous vasodilator that has been reported to activate KATP channels through A2ARs and PKA in various vascular systems, including the mesenteric and coronary vasculature (Fig. 4A) (9, 23). To test whether brain capillary KATP channels respond to extracellular adenosine, we recorded currents in native cECs and pericytes dialyzed with an [ATP]/[ADP] ratio of 1. Adenosine (25 μM) robustly stimulated whole-cell currents in cECs (−17.2 ± 1.4 pA/pF) and pericytes (−59.5 ± 12.4 pA/pF) (Fig. 4B). In both cases, these currents were inhibited by glibenclamide (Fig. 4B) and PNU-37883 (fig. S2B), suggesting that the effects of adenosine in capillary cells are specifically attributable to the activation of KATP channels. Consistent with these findings, glibenclamide-sensitive currents were not detected in response to adenosine in cECs from Cdh5-DN-Kir6.1 mice or pericytes from Pdgfrβ-DN-Kir6.1 mice, whereas these currents were readily detected in cells from the corresponding control mice (fig. S6, A to C). In contrast, whole-cell currents in SMCs isolated from pial arteries or parenchymal arterioles were not affected by adenosine or glibenclamide (fig. S1, A and B). The metabolically stable analog of adenosine, 2-chloroadenosine (CADO), strongly evoked glibenclamide-sensitive currents in both cECs and pericytes (Fig. 4C), reaching comparable current density values at fivefold lower concentrations than required for adenosine. Glibenclamide-sensitive currents were also robustly activated in both capillary cell types by the endogenous vasodilator CGRP, which, similar to adenosine, binds to G protein–coupled receptors that signal through Gαs subunits and cAMP/PKA to activate KATP channels in the systemic vasculature (fig. S7, A and B) (14).
Fig. 4. Adenosine evokes KATP currents in cECs and pericytes through the A2AR and PKA signaling pathway.

(A) Schematic diagram illustrating how A2AR signaling through the Gαs-AC-PKA intracellular pathway activates KATP channels. The A2AR and PKA agonists or antagonists used are indicated. (B) Representative recordings showing the effects of adenosine (ADO; 25 μM) on glibenclamide (GLIB; 10 μM)–sensitive currents from native cECs (n = 7 cells) and pericytes (n = 7 cells). (C) Effects of the metabolically stable adenosine analog 2-chloroadenosine (CADO; 5 μM) on GLIB-sensitive currents in cECs (n = 9 cells) and pericytes (n = 6 cells per group). (D) Effects of the adenosine A2 receptor agonist CGS-21680 (CGS; 500 nM) on GLIB-sensitive currents evoked by in cECs (n = 8 cells) and pericytes (n = 7 cells). (E) Summary data showing the averaged GLIB-sensitive current density induced by ADO (25 μM), CADO (5 μM), and CGS-21680 (500 nM) in cECs and pericytes. (F) Original traces and summary data showing the effects of adenosine (25 μM) and pinacidil (PIN; 10 μM) in cECs (n = 8 cells; top) and pericytes (n = 8 cells; bottom) treated with the A2AR blocker ZM-241385 (30 nM). (G) Effects of the PKA inhibitor H-89 (1 μM) or PIN on GLIB-sensitive KATP currents induced by ADO or CSG-21680 in cECs (n = 7 cells) and pericytes (n = 7 cells). External K+ was 60 mM, and cells were dialyzed with 0.1 mM ATP, 0.1 mM ADP, and 140 mM K+. Membrane current was recorded at a VM of −70 mV. Dotted line represents zero current level. Data are presented as means ± SEM (★P < 0.05; ★★P < 0.01; ★★★P < 0.001; Kruskal-Wallis test with post hoc Dunn’s test).
To explore the mechanisms underlying capillary KATP channel activation by adenosine, we next pharmacologically manipulated A2AR activity, first using the selective A2AR agonist, CGS-21680 [2–4-(2-carboxethyl)-phenethylamino)-5′-N-ethylcarboxamidoadenosine hydrochloride]. We found that CGS-21680 induced glibenclamide-sensitive currents in cECs and pericytes with magnitudes comparable to those measured in response to adenosine or CADO exposure (Fig. 4, D and E). To further analyze the specific contributions of A2AR activation to KATP channel activity, we tested the effect of the potent A2AR antagonist, ZM-214385. Adenosine did not evoke currents in cECs or pericytes pretreated with extracellularly applied ZM-214385, whereas pretreatment with ZM-214385 did not significantly affect glibenclamide-sensitive currents induced by pinacidil, which directly opens KATP channels, in cECs (−20.7 ± 1.4 pA/pF) or pericytes (−62.8 ± 6.3 pA/pF) (Fig. 4F). Together, these findings indicate that adenosine acts on A2ARs to stimulate KATP channels in brain cECs and pericytes.
In many vascular beds, KATP activation through A2ARs is mediated by downstream signaling pathways involving stimulation of AC and elevation of intracellular levels of cAMP, which binds and activates PKA (Fig. 4A) (9). To explore coupling of A2ARs in brain capillaries to PKA signaling, we tested the effects of the membrane-permeant PKA inhibitor H-89. Pretreatment of cECs and pericytes with H-89 abolished both adenosine- and CGS-21680–induced glibenclamide-sensitive currents. In contrast, glibenclamide-sensitive currents evoked by the KATP activator pinacidil were not affected by H-89 treatment in cECs or pericytes (Fig. 4G). These data support a role for PKA in activation of KATP channels in the brain capillary network.
Adenosine receptor stimulation hyperpolarizes the membrane potential of capillary cells through KATP channels
To test the contribution of the A2AR-Gαs-PKA signaling pathway to the regulation of cEC or pericyte membrane potential through KATP channel activation, we used sharp microelectrodes and the pressurized retina preparation (50 mmHg applied at the level of the ophthalmic artery). Exposure to 5 μM adenosine robustly hyperpolarized the VM of cECs from −28.4 ± 2.6 to −56.4 ± 5.5 mV, a response that was not significantly increased by application of a higher concentration of adenosine (Fig. 5A). After washing off adenosine, a fivefold lower concentration of the stable adenosine analog CADO (1 μM) was capable of hyperpolarizing endothelial VM, and subsequent superfusion of glibenclamide induced a marked depolarization. Comparable findings were obtained in impaled pericytes (Fig. 5B); in these cells, 5 μM adenosine induced hyperpolarization to −60.4 ± 3.9 mV from a baseline VM of −33.6 ± 2.9 mV, and cumulative addition of adenosine (25 μM) did not further hyperpolarize VM. Similarly, 1 μM CADO induced a greater hyperpolarizing response that was reversed to resting VM values by glibenclamide. This series of experiments is consistent with the idea that the A2AR pathway is involved in activation of KATP channels and controlling capillary resting membrane potential.
Fig. 5. A2AR stimulation hyperpolarizes the membrane potential of capillary cECs and pericytes through KATP channels.

(A and B) Original VM recordings (left) and summary data (right) from cECs (A) (n = 3 to 7 cells per group) and pericytes (B) (n = 3 to 8 cells per group) in pressurized (50 mmHg at the level of the ophthalmic artery) retinal vasculature showing changes in VM induced by adenosine (ADO; 500 nM to 25 μM) or CADO (1 μM) superfused in the absence or presence of glibenclamide (GLIB; 20 μM). Note that ADO was washed off (~15 to 20 min) before CADO was superfused. Dashed line represents 0-mV baseline before impalement of the cell. Summary data illustrate the effects of ADO, CADO, and GLIB on the VM of cECs and pericytes. Data are presented as means ± SEM (★P < 0.05; ★★P < 0.01; Kruskal-Wallis test and Wilcoxon matched-pairs signed-rank test).
Activation of KATP channels increases CBF
Last, to examine the impact of KATP channel activity on CBF, we monitored in vivo changes in CBF in the mouse somatosensory cortex in response to surface application of the KATP agonists adenosine, CADO, or pinacidil, with or without glibenclamide, using laser Doppler flowmetry (Fig. 1C). Superfusion of adenosine at a relatively low concentration (5 μM) that was sufficient to induce a robust VM hyperpolarization in capillary cells (Fig. 5, A and B) increased CBF by 18.2 ± 4.5%, when measured after 25 min (Fig. 6A), although this response was highly variable among mice and was sometimes undetectable. The metabolically stable adenosine analog CADO potently enhanced CBF, increasing it by 99.3 ± 13.5% at a lower concentration (1 μM). Raising the concentration of superfused adenosine to 50 μM similarly evoked a robust increase in CBF (87.4 ± 6.9%), an effect that was greatly attenuated (relative increase, 17.2 ± 8.1%) by glibenclamide pretreatment (Fig. 6, A and B). The cell specificity of capillary KATP channel activity was assessed by monitoring CBF in Cdh5-DN-Kir6.1 and Pdgfrβ-DN-Kir6.1 mice. In contrast to Cdh5-cre and Pgdfrβ-cre control mice, which exhibited robust adenosine-induced CBF increases (66.7 ± 6.0 and 64.6 ± 10.6%, respectively), adenosine-induced responses were significantly blunted in both Cdh5-DN-Kir6.1 (4.8 ± 1.6%) (Fig. 6, C and D) and Pdgfrβ-DN-Kir6.1 (12.0 ± 2.0%) mice (Fig. 6, E and F). Pinacidil application similarly increased CBF responses by 60.0 ± 3.5%, and this effect was significantly blunted by pretreatment with glibenclamide, which reduced the relative increase to 16.6 ± 4.5% (fig. S8, A and B). Note that glibenclamide alone did not affect resting CBF (Fig. 6B). Last, we tested microvascular zone–specific aspects of the ability of adenosine-induced KATP channel activation to evoke upstream arteriolar dilation and CBF increases in vivo (Fig. 1D). To this end, we stimulated fourth-order brain capillaries (fourth branch measured from the feeding arteriole) with adenosine (50 μM) and monitored capillary RBC flux and upstream parenchymal arteriole diameter through a cranial window using two-photon laser scanning microscopy. Fluorescein isothiocyanate (FITC)–labeled dextran was introduced into the circulation of anesthetized mice to enable visualization of the brain microcirculation and allow contrast imaging of RBCs, and tetramethyl rhodamine isothiocyanate (TRITC)–labeled dextran was included in the picospritzing pipette to visualize the spatial coverage of the ejected solution containing adenosine (Fig. 6G). Pressure ejection of adenosine provoked a robust increase (~3-fold) in capillary RBC flux (Fig. 6H and fig. S9, A and B) and upstream arteriole diameter (~11% dilation; Fig. 6I and fig. S9, C to E). Collectively, these findings highlight the ability of adenosine and pinacidil to effectively increase CBF through activation of capillary KATP channels (Fig. 7, A to D).
Fig. 6. Activation of KATP channels increases CBF.
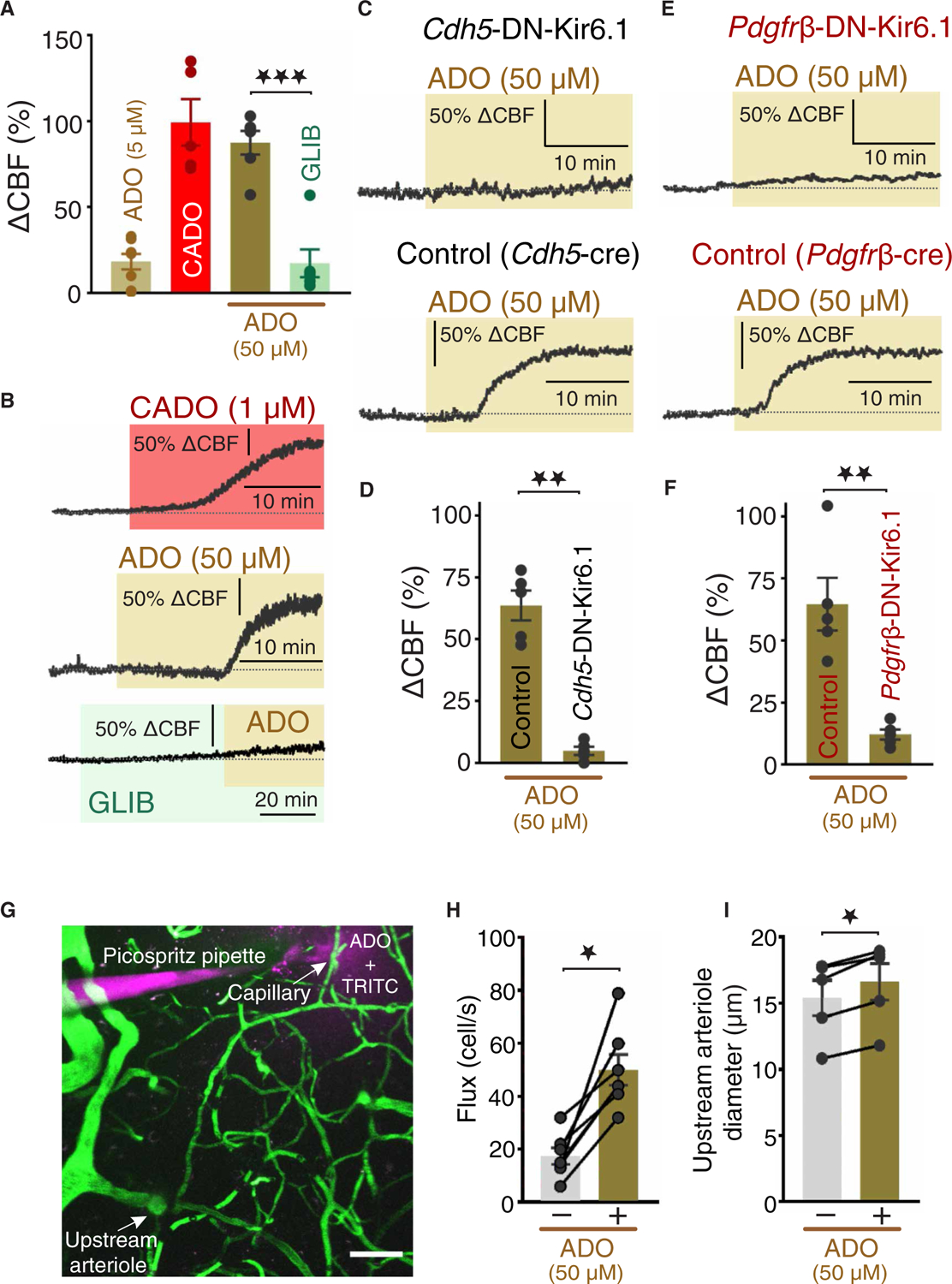
CBF responses in the somatosensory cortex were continuously monitored through a cranial window using laser Doppler flowmetry. (A and B) Summary data (A) and original traces (B) illustrating changes in CBF induced by a low concentration of adenosine (ADO; 5 μM) or CADO (1 μM) or a higher concentration of ADO (50 μM) superfused in the absence or presence of glibenclamide (GLIB; 20 μM; n = 5 to 8 mice per group). (C and D) Original traces (C) and summary data (B) showing CBF responses induced by superfusion of ADO (50 μM) in Cdh5-cre control mice (n = 5 mice per group). (E and F) Original traces (E) and summary data (F) showing changes in CBF induced by superfusion of ADO in Pdgfrβ-DN-Kir6.1 and control (Pdgfrβ-cre) mice (n = 5 mice per group). (G) A pipette containing ADO (50 μM) plus TRITC-dextran in aCSF was placed next to a capillary, which was further line-scanned at 5 kHz during local pressure ejection of the pipette contents. (H and I) Summary data showing RBC flux responses (H) (n = 7 capillaries from three mice) and changes in arteriole diameter (I) (n = 5 capillaries from three mice) before and after ADO application. Data in (D), (F), (H), and (I) are presented as means ± SEM [★P < 0.05; ★★P < 0.01; (D) and (F), Mann-Whitney test; and (H) and (I), Wilcoxon matched-pairs signed-rank test].
Fig. 7. KATP channels in capillaries and their impact on CBF.

(A) During normal brain activity or under pathophysiological conditions (hypoxia and ischemia), neurons and/or astrocytes release adenosine. (B) Adenosine, in turn, stimulates KATP channel activity through A2ARs and the Gαs-AC-PKA signaling pathway. (C) Activation of KATP channels causes profound hyperpolarization of cEC and pericyte membranes and (D) enhances CBF.
DISCUSSION
The dense, anastomosing capillary network represents the primary site of oxygen and nutrient exchange between the vasculature and neurons of the brain. Every neuron in the brain lies within ~10 to 20 μm of a neighboring capillary, an architectural relationship that favors intimate communication between neurons and capillaries (24). We have previously established the physiological relevance of this concept, demonstrating that capillaries act as a neural activity sensory web that responds to K+ released during neuronal activity through activation of Kir2.1 in cECs, which causes a retrograde, propagating hyperpolarizing signal that dilates upstream arterioles and increases blood flow to the active neuronal region (2). Capillaries are also en-wrapped by astrocytes and are thus similarly poised to respond to astrocyte-derived vasoactive modulators, in particular adenosine, an ATP hydrolysis product that is constantly released from astrocytes (25). A potential end target of adenosine is the KATP channel, which functionally links A2AR-GαsPCR (GαS protein–coupled receptor) signaling to vasodilation through cAMP/PKA in other vascular beds (10). However, whether the KATP channel is functionally expressed in brain pericytes or ECs was previously unknown. Although work from our laboratory has helped to define the biophysical signatures of ion channels in cECs (2, 26) and elucidate their influence on capillary function, the functional ion channel repertoire in these cells remains incompletely understood. These gaps in our knowledge are more pronounced for brain pericytes, in which endogenous ion channel protein expression and function have remained largely uninvestigated. In the present study, we explored the presence of KATP channels in capillary cells and assessed their potential impact on brain vascular function. Our electrophysiological findings demonstrated that brain capillary cells—both cECs and pericytes—had KATP channels. Direct electrophysiological recordings from single pericytes freshly isolated from brain capillary networks revealed current-voltage relationships distinct from those of other vascular cells, characterized by a robust, nonlinear, inward K+ current at hyperpolarized potentials and an outward current at depolarized potentials that was less prominent compared with that in SMCs (Fig. 1A). We further found that the endogenous vasoactive agent adenosine activated KATP channels in the capillary network by activating the A2AR-Gαs-AC-PKA signaling cascade. Last, activation of KATP channels in pericytes and cECs by adenosine increased CBF, and in vivo stimulation of brain capillaries with adenosine increased capillary RBC flux and upstream arteriole diameter. Collectively, our data provide multiple lines of evidence supporting the functional importance of the KATP channel in brain capillaries and underscore the potential of this channel to serve as a physiological bridge between neuronal/astrocyte activity and vascular function and thereby affect cerebral hemodynamics.
ATP-sensitive K+ channels play a central role in setting the membrane potential of vascular SMCs (10, 13). KATP channel opening increases K+ efflux, resulting in hyperpolarization, closure of voltage-gated Ca2+ channels, and subsequent vessel dilation (13, 14, 27). KATP channels are activated by various K+ channel openers, including pinacidil, cromakalim, and diazoxide, among others. These channels are also inhibited by oral hypoglycemic drugs, such as glibenclamide and tolbutamide (10, 28). Existing single-cell RNA sequencing data for mouse brain vascular cells indicate that both cECs and pericytes—especially the latter—express the genes Kcnj8 and Abcc9, which encode the KATP channel vascular subunits Kir6.1 and SUR2B, respectively (19–21). Despite the differences in the expression of mRNAs for KATP channel subunits between the two cell types (19), the difference in current density in the presence of pinacidil or adenosine was only ~3-fold (Fig. 2F). The membrane potential of both pericytes and cECs was fairly depolarized (−30 to −40 mV) relative to EK (about −100 mV), consistent with a low resting K+ permeability. Thus, glibenclamide did not affect membrane currents, membrane potential, or CBF in the absence of KATP channel activators. Our findings are in line with previous electrophysiological work performed in capillaries isolated from the heart (29) and in retinal pericytes (11, 30). Pinacidil-evoked K+ currents were also blocked by PNU-37883, a selective inhibitor of vascular KATP channels (31, 32). Collectively, these data provide support for the idea that brain cECs and pericytes have functional KATP channels with pharmacological and molecular properties similar to those of KATP channels in SMCs in the systemic vasculature (10).
An intact pressurized retina preparation that we developed (33, 34) provided the means to confirm the presence of KATP channels and experimentally test their contribution to membrane potential in intact capillaries under physiological pressure conditions and within a native environment that reflects their central nervous system context. We demonstrated that activation of KATP channels with pinacidil robustly hyperpolarized the membrane potential of both cECs and pericytes, effects that were reversed by the application of glibenclamide (Fig. 3, J and K). These findings are consistent with previous results obtained from impaled SMCs in pressurized rabbit arteries (10, 13), pericytes from the descending vasa recta of the kidney (35), and capillary fragments (29, 30, 36), highlighting the robust influence of KATP channels on membrane potential. Last, there were overt differences in the pattern of dye spread from impaled cECs compared with that from pericytes: Whereas dye spread rapidly from impaled EC to neighboring ECs, dye spread from impaled pericytes to underlying ECs was modest and localized. These differences in intercellular spread may reflect the unique electrical coupling properties of each cell type or heterogeneous gap junction expression between them (30, 37).
Vascular KATP channels are ideal downstream targets for adenosine, a potent endogenous regulator of blood flow. Adenosine in the brain is primarily derived from the breakdown of ATP released from astrocytic endfeet or directly liberated from astrocytes under conditions in which there is a mismatch between tissue blood/oxygen supply and demand, such as during hypoxic/ischemic conditions (25, 38). Because astrocytic endfeet are in intimate contact with capillaries, adenosine could conceivably have a major role in regulating KATP channels in cECs and pericytes, which express Adora2a, the gene encoding A2AR (19). Our demonstration that adenosine stimulates KATP channels in the capillary network through the A2AR-Gαs-AC-PKA signaling cascade suggests that capillaries are equipped with all the necessary molecular machinery for transducing glial-mediated stimuli into a subsequent electrical response (Fig. 4, A to G). This conclusion is reinforced by the potent hyperpolarizing effects of A2AR activation (by adenosine or the stable adenosine analog CADO) on the resting VM of capillary cells (Fig. 5, A and B). Consistent with this idea, we found that KATP channels in capillary cells were similarly activated by CGRP, another endogenous vasodilator that operates through the same intracellular pathway (14, 16). CGRP has been reported to play an important role in the development of episodic migraine attacks (39, 40) and their evolution into chronic migraine, a condition lacking effective therapeutic options. The KATP channel has been implicated in the pathogenesis of migraines in humans (41) and rodent models (42). In this context, our data open potentially new avenues for migraine treatment through the targeting of capillary KATP channels or upstream GαsPCRs.
That glial-derived mediators act through capillary KATP channels to serve an important physiological function is supported by our exploration of the dilatory impact of capillary KATP channels on brain blood flow in vivo. These studies, in which laser Doppler flowmetry was used to measure changes in CBF induced by either exogenous pinacidil or adenosine in the absence or presence of glibenclamide, revealed an increase in CBF upon exogenous pinacidil or adenosine exposure that was sensitive to glibenclamide. Using mice with cell-specific expression of DN-Kir6.1 to selectively eliminate KATP channel currents, we found that adenosine failed to increase CBF in both EC- and pericyte-specific Kir6.1 DN mice, suggesting that KATP channels are required in both cell types to elicit robust responses to adenosine (Fig. 6, A to F). In line with our results, multiple studies using topical application of high concentrations of adenosine or that pharmacologically dissected the A2AR signaling pathway in vivo have proposed that this vasodilator is involved in the regulation of CBF (43–45). Although difficult to determine, estimated basal adenosine levels are in the nanomolar range (7, 46), and under physiological conditions, low nanomolar levels (≥5 nM) of adenosine can stimulate high-affinity A2ARs in intact tissue preparations (11). Given that we used an adenosine concentration (50 μM) that is far above the estimated extracellular basal level (7, 46), it is possible that the signaling pathway described here is engaged primarily under metabolically compromised conditions characterized by increased levels of adenosine, such as hypoxia/ischemia (5–7), although perivascular levels of adenosine are likely higher. In addition, adenosine is a metabolically unstable molecule that can be rapidly metabolized by several mechanisms (47), which complicates determining effective physiological levels. However, much lower concentrations of the adenosine analog CADO, which has a similar affinity for A2ARs and is resistant to hydrolysis, induced strong responses, possibly due to delivery of a higher effective concentration at the site of activation.
We detected very small KATP currents in SMCs isolated from either brain pial (surface) arteries or parenchymal arterioles from mice. This observation contrasts with a body of literature reporting functional expression of these channels in various vascular bed types or smooth muscle from different species, including rabbit, rat, and cat (28, 48–50). Nonetheless, our findings are consistent with previous studies by us and others, showing little or no effect of diverse KATP channel openers or inhibitors on pressurized murine cerebral arteries and arterioles, findings that are distinct from those in the mesenteric circulation, where arterial diameter is affected by these drugs (51–53). These contrasting findings may reflect variability in channel expression among different species, different vascular beds, and/or location within the cerebrovasculature. Nevertheless, the absence of KATP channels in cerebrovascular SMCs supports the premise that adenosine-induced KATP channel–mediated hyperpolarization primarily originates in the capillary network and likely reflects a response to neural activity. Note that adenosine does dilate parenchymal arterioles and pial arteries independent of KATP channels (52), likely through a mechanism involving Ca2+ spark–mediated activation of large-conductance Ca2+-activated K+ channels (54). The density and intrinsic properties of pericytes are sex and zone specific within the microvasculature of the cerebral cortex and are altered in the aging brain (55), central factors that may affect capillary KATP channel function with physiological or pathophysiological consequences.
Collectively, our results provide support for a signal transduction pathway that links adenosine receptors to KATP channels in the capillary network and leads to hyperpolarization and increased CBF (Fig. 7). Thus, they provide a new theoretical foundation for understanding the electro-metabolic capillary control of brain blood flow as previously established in heart (56). Our study raises the exciting possibility that GαsPCR agonists such as adenosine and CGRP derived from neurons and astrocytes increase regional CBF through activation of KATP channels in cECs and pericytes.
MATERIALS AND METHODS
Experimental model and subject details
Adult (2- to 3-month-old) male C57BL/6J, NG2-DsRed (Jackson Laboratories, USA), Cdh5-DN-Kir6.1, and Pdgfrβ-DN-Kir6.1 mice were euthanized by intraperitoneal injection of sodium pentobarbital (100 mg/kg), followed by rapid decapitation, or were used for in vivo laser Doppler flowmetry experiments. Animals were housed on a 12-hour light/dark cycle with environmental enrichment and free access to food and water. All animals were used in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Vermont.
Generation of Cdh5-DN-Kir6.1 and Pdgfrβ-DN-Kir6.1 mice
EC-specific DN-Kir6.1 mice were generated by crossing Tg[CX1-eG-FP-Kir6.1-AAA] mice (22) with transgenic mice expressing tamoxifen-inducible Cre recombinase under control of the Cdh5 promoter (Cdh5-CreERT2) (fig. S3). Pericyte-specific DN-Kir6.1 mice were generated by crossing Tg[CX1-eGFP-Kir6.1-AAA] mice with mice in which cre expression is driven by the Pdgfrβ promoter (Pdgfrβ-CreERT2 mice) (fig. S3). Tg[CX1-eGFP-Kir6.1-AAA] and Cdh5-CreERT2 mice were provided by C. G. Nichols (Washington University School of Medicine, St. Louis, MO, USA; with permission of W. A. Coetzee, NYU Grossman School of Medicine, New York, NY, USA) and R. H. Adams (Max Planck Institute for Molecular Biomedicine, Munster, Germany), respectively; Pdgfrβ-CreERT2 mice were obtained from Jackson Laboratories. The Tg[CX1-eGFP-Kir6.1-AAA] transgene consists of the chicken β-actin (CX1) promoter, followed by a loxP-flanked enhanced green fluorescent protein (eGFP) gene containing a stop codon and a DN Kir6.1 pore mutant in which a Gly-Phe-Gly sequence was mutated to three alanines (Kir6.1-AAA). Transgenic mice with a GFP+/Cre− genotype express eGFP in all mammalian somatic cell types, whereas Cdh5 or Pdgfrβ Cre recombinase expression leads to eGFP excision from the transgene in a tissue-specific manner and activates the expression of mutant Kir6.1-AAA in ECs or pericytes, respectively.
Chemicals
Glibenclamide and CADO were purchased from Tocris Bioscience, and pinacidil was obtained from Cayman Chemicals. Unless otherwise noted, all other chemicals were obtained from Sigma-Aldrich.
Cell isolation
Individual cECs were isolated from C57BL/6J mouse brains by mechanical disruption of a small piece of brain somatosensory cortex (volume, ~10 to 20 mm3) using a Dounce homogenizer, as previously reported (2). Briefly, tissue was homogenized in ice-cold artificial cerebrospinal fluid (aCSF) consisting of 124 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 1.25 mM sodium phosphate buffer, 26 mM NaHCO3, and 4 mM glucose. Debris was removed by passing the homogenate through a 62-μm nylon mesh, and retained capillary fragments were enzymatically digested by incubating for 24 min at 37°C in an isolation solution composed of 55 mM NaCl, 80 mM Na-glutamate, 5.6 mM KCl, 2 mM MgCl2, 4 mM glucose, and 10 mM Hepes (pH 7.3) containing neutral protease (0.5 mg/ml; Worthington), elastase (0.5 mg/ml; Worthington), and 100 μM CaCl2. The sample was incubated with collagenase type I (0.5 mg/ml; Worthington) for an additional 2 min at 37°C. The suspension was filtered, and the remaining tissue was washed to remove enzymes and triturated with a fire-polished glass Pasteur pipette. Single cells and small capillary segments were stored in ice-cold isolation medium for use the same day (within ~5 hours).
Single capillary pericytes were isolated from NG2-DsRed mouse brains by mechanical dissociation using a papain-based Neural Tissue Dissociation kit (Miltenyi Biotec) as previously described (57). Briefly, a small piece of somatosensory cortex (~10 to 20 mm3 in volume) was chopped into small pieces with sharp scissors; transferred to isolation solution composed of 55 mM NaCl, 80 mM Na-glutamate, 5.6 mM KCl, 2 mM MgCl2, 4 mM glucose, and 10 mM Hepes (pH 7.3) containing enzyme 1 provided in the kit; and incubated for 17 min at 37°C. Enzyme 2 was added, and the brain suspension was mixed 10 times using a Pasteur pipette and incubated for 12 min at 37°C. The suspension was then passed 10 times through a 20-G needle and incubated for an additional 10 min. The suspension was filtered through a 62-μm nylon mesh and stored in ice-cold isolation solution. Cells were used within ~5 hours after dispersion.
Individual SMCs were obtained from cerebral pial arteries and arterioles isolated from a C57BL/6J mouse using an enzymatic and mechanical dissociation procedure. Briefly, pial and parenchymal artery segments were incubated in an isolation solution composed of 55 mM NaCl, 80 mM Na-glutamate, 5.6 mM KCl, 2 mM MgCl2, 4 mM glucose, and 10 mM Hepes (pH 7.3), containing papain (0.3 mg/ml; Worthington) and dithioerythritol (0.3 mg/ml; Sigma-Aldrich), for 14 min at 37°C; then transferred to a solution containing collagenase F (0.67 mg/ml), collagenase H (0.33 mg/ml; Sigma-Aldrich), and 100 μM CaCl2; and incubated for 5 min at 37°C. Single cells were released by triturating with a fire-polished glass Pasteur pipette and stored in ice-cold isolation medium for use the same day (within ~5 hours).
Electrophysiology
Patch-clamp electrophysiology was used to measure whole-cell currents in isolated cells. Currents were amplified using an Axopatch 200 amplifier, filtered at 1 kHz, digitized at 10 kHz, and stored on a computer for offline analysis with Clampfit 10.7 software (Molecular Devices). Whole-cell capacitance and series access resistance were measured using the cancellation circuity in the voltage-clamp amplifier. Electrophysiological analyses were performed in either the conventional or perforated whole-cell configuration. Patch pipettes were pulled from borosilicate microcapillary tubes (1.5-mm outer diameter and 1.17-mm inner diameter; Shutter Instruments, USA) and fire-polished (resistance, 3 to 5 megohms). The bath (external) solution contained 80 mM NaCl, 60 mM KCl, 1 mM MgCl2, 0.1 mM CaCl2, 10 mM glucose, and 10 mM Hepes (pH 7.4). For the conventional whole-cell configuration, pipettes were backfilled with a solution containing 102 mM KCl, 38 mM KOH, 10 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM EGTA, 10 mM glucose, 10 mM Hepes, and 0.1 mM ADP (pH 7.2). For perforated patch electrophysiology, the pipette solution was composed of 10 mM NaCl, 26.6 mM KCl, 110 mM K+ aspartate, 1 mM MgCl2, 10 mM Hepes, and amphotericin B (200 to 250 μg/ml), added freshly on the day of the experiment. Experiments were performed at negative membrane potentials (−70 mV), and the amplitude of K+ currents at hyperpolarized potentials was enhanced by raising external K+ to 60 mM. Under these conditions, K+ currents are inward and are detected as downward deflections. The mean capacitance of cECs, pericytes, pial SMCs, and parenchymal SMCs averaged 10.5 ± 0.2 (n = 126 cells), 10.2 ± 0.2 (n = 86 cells), 15.17 ± 0.42 (n = 18 cells), and 11.13 ± 0.5 (n = 14 cells), respectively. All experiments were performed at room temperature (~22°C).
Membrane potential measurements
VM was recorded from superficial cECs and pericytes using the whole-mount pressurized retina preparation. Implementation of the pressurized retina for studying microvascular reactivity has been described previously (33, 34). After euthanasia, the entire orbit, including the eye, optic nerve, and attached vasculature and muscles, was removed from the mouse. The retina, attached optic nerve, and ophtalmic artery were dissected in 4°C Ca2+-free buffer containing 119 mM NaCl, 3 mM KCl, 0 mM CaCl2, 3 mM MgCl2, 5 mM glucose, 26.2 mM NaHCO3, and 1 mM sodium phosphate buffer, bubbled with 95% O2/5% CO2 (pH 7.4). The isolated retina (with small sclera patch intact) was transferred to a perfusion chamber containing a fixed silicone platform. The ophthalmic artery was cannulated, and the retina was pinned (30-μm tungsten pins) en face using minor radial cuts to create a flattened surface. The entire preparation was perfused at 5 ml/min with a bath solution (36°C) containing 119 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM glucose, 26.2 mM NaHCO3, and 1 mM sodium phosphate buffer, bubbled with 95% O2/5% CO2 (pH 7.4). The retinal vasculature was pressurized to 50 mmHg (applied at the ophthalmic artery) with the same bath solution using a gravity column. After 15 min, perfusion was briefly stopped, and 170 μl of papain (4 U/ml in bath solution) was added dropwise from directly above the retina prep (bath volume, ~2.5 ml). Three minutes later, the papain solution was washed out by resuming perfusion. Pericytes and cECs on superficial distal capillaries (>4th order) of the retina were identified using bright-field microscopy. Cells were impaled using microelectrodes (pulled to ~200 to 300 megohms and filled with a 0.5 M KCl solution containing 3 μM iFluor 488 hydrazide; AAT Bioquest). In a subset of experiments, the entire superficial microvasculature was labeled using rhodamine-labeled Griffonia Simplicifolia Lectin I (GSL I) lectin (0.2 mg/ml, 10 min) after recordings. VM recordings were made using an Axoclamp-2A digital amplifier and HS-2 headstage (Molecular Devices). Signals were digitized and stored using Digidata 1322A and pCLAMP 9 software (Molecular Devices). Recordings satisfying the following criteria were considered successful: (i) steep negative deflection of VM upon impalement, (ii) steady resting VM for at least 1 min, and (iii) immediate return to 0 mV upon removal of the microelectrode. After VM recordings, the recorded cell type was confirmed by z-stack imaging of iFluor 488 hydrazide using an upright spinning disk confocal microscope (which was also used for bright-field location of cells).
Cranial window surgery and in vivo imaging of cerebral hemodynamics
A cranial window was implanted in mice anesthetized with isoflurane (5% induction and 2% maintenance), as previously described (2). Briefly, the skull was exposed and cleaned, and a stainless steel head plate was attached over the left hemisphere using a mixture of dental cement and superglue. After the head plate was secured to a holding frame, the somatosensory cortical territory of the brain was exposed by drilling a small circular (~2-mm diameter) section of the parietal bone of the skull and removing the underlying dura. To enable visualization of the cerebral vascular network and contrast imaging of RBCs, we injected ~150 μl of a solution (3 mg/ml) of FITC-dextran (molecular weight, 150 kDa) in saline through the retro-orbital sinus. After surgery, isoflurane anesthesia was replaced with α-chloralose (50 mg/kg, i.p.) and urethane (750 mg/kg, i.p.). Throughout the experiment, body temperature was maintained at 37°C using an electric heating pad, and the cranial window was continuously superfused with aCSF solution (37°C). A pipette was maneuvered into the exposed cortex to a point adjacent to a capillary downstream of an arteriole (fourth-order branch). Adenosine (50 μM) with TRITC-dextran (0.2 mg/ml; molecular weight, 150 kDa) was pressure-ejected (200 to 300 ms, 41.3 ± 6.9 kPa) directly onto the capillary. RBC flux was quantified by line scanning the capillary under study at 5 kHz. Arteriole diameter measurements were assessed using galvanometer scanning with the capillary and arteriole of interest in the imaging frame. Images were acquired with a Zeiss 20× Plan Apochromat 1.0 numerical aperture (NA) differential interference contrast (DIC) visible-infrared water-immersion objective mounted on an LSM 7 multiphoton microscope (Zeiss) coupled to a Chameleon Vision II Titanium-Sapphire pulsed infrared laser (Coherent). FITC and TRITC were excited at 820 nm, and emitted fluorescence was separated through 500- to 550- and 570- to 610-nm bandpass filters, respectively.
Laser Doppler flowmetry
Relative CBF was continuously monitored at the site of the cranial window using laser Doppler flowmetry, as previously described (58). Mice were anesthetized with isoflurane (5% induction and 2% maintenance). A catheter was inserted into the femoral artery to record mean arterial pressure. After surgery, isoflurane anesthesia was discontinued and replaced with α-chloralose (50 mg/kg, i.p.) and urethane (750 mg/kg, i.p.), and the depth of anesthesia was confirmed by testing corneal reflexes and motor responses to tail pinch. CBF was continuously recorded using a laser Doppler probe (Perimed) connected to a data acquisition system (LabChart Software; ADInstruments). The probe was positioned stereotaxically ~0.5 mm above the cortical surface at a site distant from visible pial arteries. Arterial pressure and CBF were equilibrated for 30 min before experimentation. Cerebrovascular reactivity was assessed by testing CBF responses before and after topical superfusion of aCSF solution containing the studied pharmacological agents. Blood pressure was monitored throughout the experiment, and body temperature was measured rec-tally and maintained at 37°C with an electric heating pad.
Quantification and statistical analysis
Patch-clamp and sharp electrode data were additionally analyzed using Clampfit 10.7 software (Molecular Devices), and CBF responses were analyzed using LabChart software (ADInstruments). RBC flux data were analyzed using ImageJ software as previously described (2). Briefly, RBC flux data were binned at 1-s intervals, and mean baseline flux data for summary figures were acquired by averaging the baseline (~6 s) for each measurement before pressure ejection of adenosine. The peak response was defined as the peak 1-s flux bin after delivery of adenosine within the remaining scanning period (~54 s). Arteriole dilation was assessed in ImageJ software by comparing luminal arteriole diameter before (frame averaged 6 s before pressure ejection) and after pressure ejection of adenosine (peak sustained 3-s response). Data are expressed as means ± SEM. In cases in which data were not normally distributed, statistical significance was determined with GraphPad Prism 8 software using nonparametric tests, including Kruskal-Wallis test, Mann-Whitney test, or Wilcoxon test, as specified in the figure legends. P values of <0.05 were considered statistically significant, and stars in figure panels denote significant differences. Sample size estimation was predicted on the basis of similar experiments performed previously in our laboratory.
Supplementary Material
Acknowledgments:
We thank T. Wellman and D. Enders for technical assistance.
Funding:
Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of NIH (F32HL152576 to N.R.K. and P20GM135007 to M.K.) and the American Heart Association (20POST35210155 and Career Development Award 856791 to A.M.). Support was also provided by the Million Dollar Bike Ride Grant Program (MDBR-21-101-CADASIL to M.K.), the Totman Medical Research Trust (to M.T.N.), and the European Union Horizon 2020 Research and Innovation Programme (grant agreement 666881, SVDs@target, to M.T.N.), as well as grants from the National Institute of Neurological Disorders and Stroke (NINDS) and National Institute on Aging (NIA) (R01-NS-110656 to M.T.N.), the National Institute of General Medical Sciences (NIGMS) (P20-GM-135007 to M.T.N.), and the National Heart, Lung, and Blood Institute (NHLBI) of NIH (R35-HL-140027 to M.T.N.).
Footnotes
SUPPLEMENTARY MATERIALS
www.science.org/doi/10.1126/scisignal.abl5405 Figs. S1 to S9
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The Tg[CX1-eGFP-Kir6.1-AAA] and Cdh5(PAC)-CreERT2 mouse lines require a material transfer agreement from NYU Grossman School of Medicine, USA and Cancer Research Technology Limited, UK, respectively.
REFERENCES AND NOTES
- 1.Gould IG, Tsai P, Kleinfeld D, Linninger A, The capillary bed offers the largest hemodynamic resistance to the cortical blood supply. J. Cereb. Blood Flow Metab 37, 52–68 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, Hill-Eubanks D, Nelson MT, Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci 20, 717–726 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunwiddie TV, Diao L, Proctor WR, Adenine nucleotides undergo rapid, quantitative conversion to adenosine in the extracellular space in rat hippocampus. J. Neurosci 17, 7673–7682 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S, ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron 40, 971–982 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Park KH, Rubin LE, Gross SS, Levi R, Nitric oxide is a mediator of hypoxic coronary vasodilatation. Relation to adenosine and cyclooxygenase-derived metabolites. Circ. Res 71, 992–1001 (1992). [DOI] [PubMed] [Google Scholar]
- 6.Latini S, Bordoni F, Pedata F, Corradetti R, Extracellular adenosine concentrations during in vitro ischaemia in rat hippocampal slices. Br. J. Pharmacol 127, 729–739 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Regan M, Adenosine and the regulation of cerebral blood flow. Neurol. Res 27, 175–181 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Berne RM, The role of adenosine in the regulation of coronary blood flow. Circ. Res 47, 807–813 (1980). [DOI] [PubMed] [Google Scholar]
- 9.Kleppisch T, Nelson MT, Adenosine activates ATP-sensitive potassium channels in arterial myocytes via A2 receptors and cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 92, 12441–12445 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quayle JM, Nelson MT, Standen NB, ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol. Rev 77, 1165–1232 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Li Q, Puro DG, Adenosine activates ATP-sensitive K+ currents in pericytes of rat retinal microvessels: Role of A1 and A2a receptors. Brain Res. 907, 93–99 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Quinn KV, Giblin JP, Tinker A, Multisite phosphorylation mechanism for protein kinase A activation of the smooth muscle ATP-sensitive K+ channel. Circ. Res 94, 1359–1366 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Standen NB, Quayle JM, Davies NW, Brayden JE, Huang Y, Nelson MT, Hyperpolarizing vasodilators activate ATP-sensitive K+ channels in arterial smooth muscle. Science 245, 177–180 (1989). [DOI] [PubMed] [Google Scholar]
- 14.Nelson MT, Huang Y, Brayden JE, Hescheler J, Standen NB, Arterial dilations in response to calcitonin gene-related peptide involve activation of K+ channels. Nature 344, 770–773 (1990). [DOI] [PubMed] [Google Scholar]
- 15.Li A, Knutsen RH, Zhang H, Osei-Owusu P, Moreno-Dominguez A, Harter TM, Uchida K, Remedi MS, Dietrich HH, Bernal-Mizrachi C, Blumer KJ, Mecham RP, Koster JC, Nichols CG, Hypotension due to Kir6.1 gain-of-function in vascular smooth muscle. J. Am. Heart Assoc 2, e000365 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quayle JM, Bonev AD, Brayden JE, Nelson MT, Calcitonin gene-related peptide activated ATP-sensitive K+ currents in rabbit arterial smooth muscle via protein kinase A. J. Physiol 475, 9–13 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson MT, Quayle JM, Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol 268, C799–C822 (1995). [DOI] [PubMed] [Google Scholar]
- 18.Zhang L, Bonev AD, Mawe GM, Nelson MT, Protein kinase A mediates activation of ATP-sensitive K+ currents by CGRP in gallbladder smooth muscle. Am. J. Physiol 267, G494–G499 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, Sun Y, Raschperger E, Räsänen M, Zarb Y, Mochizuki N, Keller A, Lendahl U, Betsholtz C, A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Sabbagh MF, Heng JS, Luo C, Castanon RG, Nery JR, Rattner A, Goff LA, Ecker JR, Nathans J, Transcriptional and epigenomic landscapes of CNS and non-CNS vascular endothelial cells. eLife 7, e36187 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ, An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malester B, Tong X, Ghiu I, Kontogeorgis A, Gutstein DE, Xu J, Hendricks-Munoz KD, Coetzee WA, Transgenic expression of a dominant negative KATP channel subunit in the mouse endothelium: Effects on coronary flow and endothelin-1 secretion. FASEB J. 21, 2162–2172 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Dart C, Standen NB, Adenosine-activated potassium current in smooth muscle cells isolated from the pig coronary artery. J. Physiol 471, 767–786 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Attwell D, Buchan AM, Charpak S, Lauritzen M, MacVicar BA, Newman EA, Glial and neuronal control of brain blood flow. Nature 468, 232–243 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haydon PG, Carmignoto G, Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev 86, 1009–1031 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Harraz OF, Longden TA, Eubanks DH, Nelson MT, PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. eLife 7, e38689 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knot HJ, Nelson MT, Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am. J. Physiol. - Hear. Circ. Physiol 269, H348–H355 (1995). [DOI] [PubMed] [Google Scholar]
- 28.Quayle JM, Bonev AD, Brayden JE, Nelson MT, Pharmacology of ATP-sensitive K+ currents in smooth muscle cells from rabbit mesenteric artery. Am. J. Physiol. - Cell Physiol 269, C1112–C1118 (1995). [DOI] [PubMed] [Google Scholar]
- 29.y Schnitzler MM, Derst C, Daut J, Preisig-Müller R, ATP-sensitive potassium channels in capillaries isolated from guinea-pig heart. J. Physiol 525, 307–317 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu D, Minami M, Kawamura H, Puro D, Electrotonic transmission within pericyte-containing retinal microvessels. Microcirculation 13, 353–363 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Wellman GC, Barrett-Jolley R, Köppel H, Everitt D, Quayle JM, Inhibition of vascular KATP channels by U-37883A: A comparison with cardiac and skeletal muscle. Br. J. Pharmacol 128, 909–916 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zambach SA, Cai C, Helms HCC, Hald BO, Dong Y, Fordsmann JC, Nielsen RM, Hu J, Lønstrup M, Brodin B, Lauritzen MJ, Precapillary sphincters and pericytes at first-order capillaries as key regulators for brain capillary perfusion. Proc. Natl. Acad. Sci. U.S.A 118, e2023749118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzales AL, Klug NR, Moshkforoush A, Lee JC, Lee FK, Shui B, Tsoukias NM, Kotlikoff MI, Hill-Eubanks D, Nelson MT, Contractile pericytes determine the direction of blood flow at capillary junctions. Proc. Natl. Acad. Sci. U.S.A 117, 27022–27033 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ratelade J, Klug NR, Lombardi D, Angelim MKSC, Dabertrand F, Domenga-Denier V, Salman RA-S, Smith C, Gerbeau J-F, Nelson MT, Joutel A, Reducing hypermuscularization of the transitional segment between arterioles and capillaries protects against spontaneous intracerebral hemorrhage. Circulation 141, 2078–2094 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z, Rhinehart K, Pallone TL, Membrane potential controls calcium entry into descending vasa recta pericytes. Am. J. Physiol 283, R949–R957 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Langheinrich U, Daut J, Hyperpolarization of isolated capillaries from guinea-pig heart induced by K+ channel openers and glucose deprivation. J. Physiol 502, 397–408 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovacs-Oller T, Ivanova E, Bianchimano P, Sagdullaev BT, The pericyte connectome: Spatial precision of neurovascular coupling is driven by selective connectivity maps of pericytes and endothelial cells and is disrupted in diabetes. Cell Discov. 6, 39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martín ED, Fernández M, Perea G, Pascual O, Haydon PG, Araque A, Ceña V, Adenosine released by astrocytes contributes to hypoxia-induced modulation of synaptic transmission. Glia 55, 36–45 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Edvinsson L, Haanes KA, Warfvinge K, Krause DN, CGRP as the target of new migraine therapies—Successful translation from bench to clinic. Nat. Rev. Neurol 14, 338–350 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Hargreaves R, Olesen J, Calcitonin gene-related peptide modulators—The history and renaissance of a new migraine drug class. Headache 59, 951–970 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Al-Karagholi MA-M, Hansen JM, Guo S, Olesen J, Ashina M, Opening of ATP-sensitive potassium channels causes migraine attacks: A new target for the treatment of migraine. Brain 142, 2644–2654 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Christensen SL, Munro G, Petersen S, Shabir A, Jansen-Olesen I, Kristensen DM, Olesen J, ATP sensitive potassium (KATP) channel inhibition: A promising new drug target for migraine. Cephalalgia 40, 650–664 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Ko KR, Ngai AC, Winn HR, Richard H, Roze W, Role of adenosine in regulation of regional cerebral blood flow in sensory cortex (1990); www.physiology.org/journal/ajpheart. [DOI] [PubMed]
- 44.Morii S, Ngai AC, Ko KR, Winn HR, Role of adenosine in regulation of cerebral blood flow: Effects of theophylline during normoxia and hypoxia. Am. J. Phys 253, H165–H175 (1987). [DOI] [PubMed] [Google Scholar]
- 45.Ibayashi S, Ngai AC, Meno JR, Winn HR, Effects of topical adenosine analogs and forskolin on rat pial arterioles in vivo. J. Cereb. Blood Flow Metab 11, 72–76 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Fredholm BB, Adenosine receptors as drug targets. Exp. Cell Res 316, 1284–1288 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Philis JW, Adenosine in the control of the cerebral circulation. Cerebrovasc. Brain Metab. Rev 1, 26–54 (1989). [PubMed] [Google Scholar]
- 48.Kleppisch T, Nelson MT, ATP-sensitive K+ currents in cerebral arterial smooth muscle: Pharmacological and hormonal modulation. Am. J. Physiol 269, H1634–H1640 (1995). [DOI] [PubMed] [Google Scholar]
- 49.Parsons AA, Ksoll E, Mackert JRL, Schilling L, Wahl M, Comparison of cromakalim-induced relaxation of potassium precontracted rabbit, cat, and rat isolated cerebral arteries. Naunyn Schmiedeberg’s Arch. Pharmacol 343, 384–392 (1991). [DOI] [PubMed] [Google Scholar]
- 50.Zimmermann PA, Knot HJ, Stevenson AS, Nelson MT, Increased myogenic tone and diminished responsiveness to ATP-sensitive K+ channel openers in cerebral arteries from diabetic rats. Circ. Res 81, 996–1004 (1997). [DOI] [PubMed] [Google Scholar]
- 51.McCarron JG, Quayle JM, Halpern W, Nelson MT, Cromakalim and pinacidil dilate small mesenteric arteries but not small cerebral arteries. Heart Circ. Physiol 261, H287–H291 (1991). [DOI] [PubMed] [Google Scholar]
- 52.Dabertrand F, Nelson MT, Brayden JE, Acidosis dilates brain parenchymal arterioles by conversion of calcium waves to sparks to activate BK channels. Circ. Res 110, 285–294 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mishra RC, Wulff H, Hill MA, Braun AP, Inhibition of myogenic tone in rat cremaster and cerebral arteries by SKA-31, an activator of endothelial KCa2.3 and KCa3.1 channels. J. Cardiovasc. Pharmacol 66, 118–127 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Porter VA, Bonev AD, Knot HJ, Heppner TJ, Stevenson AS, Kleppisch T, Lederer WJ, Nelson MT, Frequency modulation of Ca2+ sparks is involved in regulation of arterial diameter by cyclic nucleotides. Am. J. Phys 274, C1346–C1355 (1998). [DOI] [PubMed] [Google Scholar]
- 55.Berthiaume A-A, Hartmann DA, Majesky MW, Bhat NR, Shih AY, Pericyte structural remodeling in cerebrovascular health and homeostasis. Front. Aging Neurosci 10, 210 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao G, Joca HC, Nelson MT, Lederer WJ, ATP- and voltage-dependent electro-metabolic signaling regulates blood flow in heart. Proc. Natl. Acad. Sci 117, 7461–7470 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vanlandewijck M, Vanlandewijck M, Lebouvier T, Mäe MA, Nahar K, Betsholtz C, Primary isolation of vascular cells from murine brain for single cell sequencing. Protoc. Exch, doi: 10.1038/PROTEX.2017.159 (2018). [DOI] [Google Scholar]
- 58.Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, Nelson MT, Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc. Natl. Acad. Sci 107, 3811–3816 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The Tg[CX1-eGFP-Kir6.1-AAA] and Cdh5(PAC)-CreERT2 mouse lines require a material transfer agreement from NYU Grossman School of Medicine, USA and Cancer Research Technology Limited, UK, respectively.